
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 22 November 2021
Sec. Molecular Signalling and Pathways
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.762918
 Jessica D. Panes1†
Jessica D. Panes1† Paulina Saavedra1,2†
Paulina Saavedra1,2† Benjamin Pineda1
Benjamin Pineda1 Kathleen Escobar1,2
Kathleen Escobar1,2 Magdalena E. Cuevas1
Magdalena E. Cuevas1 Gustavo Moraga-Cid1
Gustavo Moraga-Cid1 Jorge Fuentealba1Coralia I. Rivas2Human Rezaei3,4,5
Jorge Fuentealba1Coralia I. Rivas2Human Rezaei3,4,5 Carola Muñoz-Montesino1*
Carola Muñoz-Montesino1*After the discovery of prion phenomenon, the physiological role of the cellular prion protein (PrPC) remained elusive. In the past decades, molecular and cellular analysis has shed some light regarding interactions and functions of PrPC in health and disease. PrPC, which is located mainly at the plasma membrane of neuronal cells attached by a glycosylphosphatidylinositol (GPI) anchor, can act as a receptor or transducer from external signaling. Although the precise role of PrPC remains elusive, a variety of functions have been proposed for this protein, namely, neuronal excitability and viability. Although many issues must be solved to clearly define the role of PrPC, its connection to the central nervous system (CNS) and to several misfolding-associated diseases makes PrPC an interesting pharmacological target. In a physiological context, several reports have proposed that PrPC modulates synaptic transmission, interacting with various proteins, namely, ion pumps, channels, and metabotropic receptors. PrPC has also been implicated in the pathophysiological cell signaling induced by β-amyloid peptide that leads to synaptic dysfunction in the context of Alzheimer’s disease (AD), as a mediator of Aβ-induced cell toxicity. Additionally, it has been implicated in other proteinopathies as well. In this review, we aimed to analyze the role of PrPC as a transducer of physiological and pathological signaling.
Prion was first proposed by Stanley Prusiner in 1982 as an infectious protein. This occurred in the context of a group of rare encephalopathies of unknown etiology in sheep and goats, characterized by abnormal trembling termed “scrapie” (Prusiner, 1982). Based on the experiments of ultraviolet irradiation of brain extracts of infected mice, a novel infectious component of low molecular weight was observed which did not depend on canonical transmission by nucleic acids and exhibited replicative and infective capacity (Alper et al., 1967; Griffith, 1967). Later, this infectious particle was isolated and corresponded to a 27–30 kDa protein, which was devoid of nucleic acids, and it was resistant to digestion by proteinase K, which was named “prion” (Bolton et al., 1982; Prusiner, 1982).
Later, several studies revealed that the ability of prion to propagate was related to an abnormally folded variant of prion protein (PrP), which is naturally expressed in mammals (Prusiner, 1982; Collinge, 2001). In this context, normally folded α-helix-enriched cellular prion protein (PrPC) can be converted into a scrapie protease-resistant form of PrP (PrPSc), requiring a cascade of conformational changes to form β-sheet-enriched conformation. Interestingly, PrPSc can propagate its own altered conformation using PrPC as a substrate, in a template replication process (Griffith, 1967; Lansbury, 1994).
PrPC is highly expressed in different neuronal and astrocytic cells of several central nervous system (CNS) areas, namely, amygdala, cerebellum, hypothalamus, occipital lobe, prefrontal cortex, and spinal cord (Su et al., 2004; Castle and Gill, 2017). It is also moderately or poorly expressed in non-neuronal cells, such as immune system, and endothelial and epithelial cells of colon, uterus, ovary, thyroid, and small intestine (Isaacs et al., 2006; Petit et al., 2013). During embryonic development, the high levels of Prnp messenger RNA (mRNA) have also been found in the CNS and peripheral nervous system (PNS) (Manson et al., 1992; Beringue et al., 2003; Lima et al., 2007; Castle and Gill, 2017).
PrP is a key mediator in several toxicity pathways in some neurodegenerative diseases (NDs), such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) (Prusiner, 2012; Urrea et al., 2017). In this review, we have focused on summarizing the current knowledge of PrPC as a sensor and key mediator of physiological significance, its role as a transducer in the amyloid cascade in AD, and its effect on other misfolding-related diseases.
Although PrPC has been implicated in synapse growth, neural plasticity, and memory, a unified variant of its on-target sites is still unknown. In fact, its mechanisms of action have been the subject of intense research for almost three decades. Despite this, there are fundamental questions that are yet to be solved: (1) What is the biological consequence of the association of PrPC with normal protein folding process? (2) What is the physiological role of PrPC interaction with channels? (3) How do PrP species act in association with other misfolded proteins? Based on the current knowledge of the function of PrPC, we have reviewed the physiological and pathological roles of PrPC signaling on synaptic function, providing a new angle to the putative role of PrP in health and disease.
PrPC is encoded by PRNP gene, located in chromosome 20 (in humans) or in chromosome 2 (in mice) (Chesebro et al., 1985; Sparkes et al., 1986). PrPC is a 210-residue glycoprotein attached to the cell surface by glycosylphosphatidylinositol (GPI) anchor (231–253 residues) (Stahl et al., 1987). Within the plasma membrane, PrPC is found at lipid rafts (also known as microdomains), enriched in cholesterol and sphingolipids (Simons and Gerl, 2010; Botto et al., 2014; Martellucci et al., 2020). Human PrP genomic cluster also contains the homologous genes PRND and PRNT of 55 kb, where PRND encodes for a Doppel (Dpl) protein of 179 residues and PRNT encodes three mRNA by alternative splicing, expressed exclusively in the testis (Premzl and Gamulin, 2007). PrP genomic family member also includes Shadoo protein, encoded by SPRN gene and located in the human chromosome 10 (Ciric and Rezaei, 2015).
The PrPC first moiety corresponds to a highly positively charged polybasic N-terminal region that is intrinsically disordered and flexible (Beland and Roucou, 2012). Some functions of the N-terminal domain are associated with protein–protein interactions, synaptic transmission, neuroprotection, and Cu2+- or Zn2+-mediated modulation (Beland and Roucou, 2012; Turnbaugh et al., 2012; Martellucci et al., 2020). Particularly, N-terminal PrPC contains a signal peptide (1–22 residues) and four functional regions, namely, two positively charged clusters (CC1 and CC2), an octarepeat (OR), and a hydrophobic domain (HD) (Beland and Roucou, 2012; Figure 1).
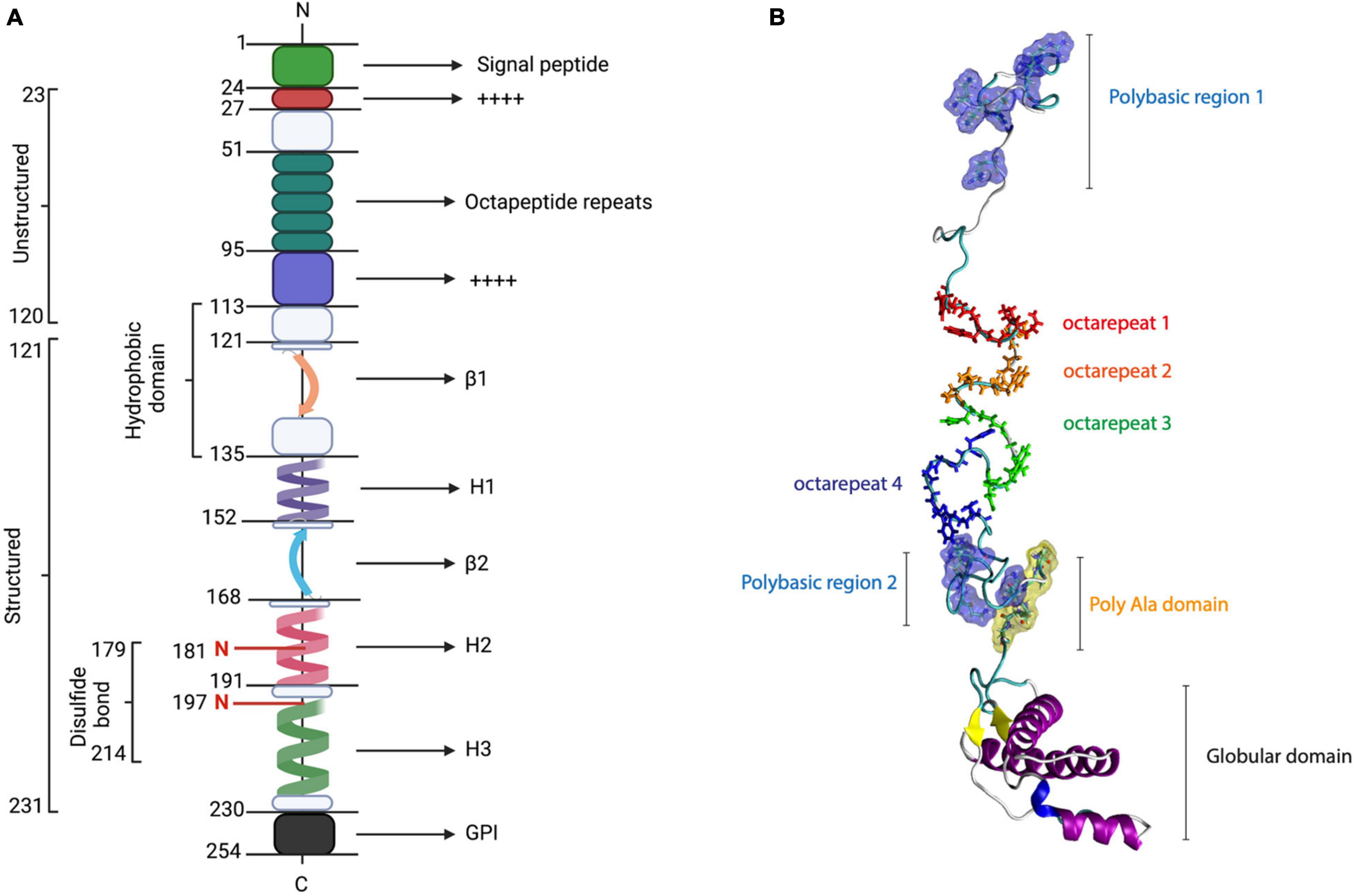
Figure 1. PrP structure. PrPC consists of 253 amino acids, which include the signal peptide (1–22), five octarepeat regions, a hydrophobic region (113–135), a disulfide bond between its cysteine residues 179 and 214, two N-glycosylation sites (residues 187 and 197), and a GPI anchor at its C-terminal. The structured conformation between amino acids 121 and 231 corresponds to a globular domain, which contains two β-sheets and three α-helices. (A) Linear representation of PrP sequence [modified from Acevedo-Morantes and Wille (2014)]. (B) PrP structure.
The PrPC CC1 domain (23–28 residues) has been associated with myelin homeostasis and PrPC α-folding stability (Martinez et al., 2015; Kuffer et al., 2016). The OR region (51–91 residues) consists of five octarepeat sequence repeats (PHGGGWGQ), enriched with glycine and histidine (His) residues, which contains several Cu2+- and Zn2+-binding units (Beland and Roucou, 2012; Wu et al., 2017). Currently, this region has been related to PrPC endoproteolysis, Cu2+ metabolism, and the initial steps of PrPC–PrPSc conversion (Lau et al., 2015).
In contrast, the PrPC CC2 region (100–109 residues)has been associated with lipid membranes, PrPC processing, and PrPC biogenesis (Kim et al., 2001; Wang et al., 2010; Martinez et al., 2015). In the core of the primary structure of PrPC, it has been described in the HD region (111–130 residues), which seems to have a neuroprotective role against neurotoxicity and myelotoxicity (Beland and Roucou, 2012; Gavin et al., 2020). Interestingly, HD seems to be relevant for the stabilization of PrPC homodimers, contributing to the prevention of prion conversion (Warwicker, 2000; Engelke et al., 2018).
As depicted in Figure 1, the C-terminal globular domain of PrPC is composed of three α-helix structures, two antiparallel β-sheets, and a GPI anchor (Heske et al., 2004; Sarnataro et al., 2017). Additionally, α-helices 2 and 3 are connected by a disulfide bond between cysteines 179 and 214, contributing to the stability of PrPC-folded state (Biasini et al., 2012). Another important function that was proposed for the C-terminal domain of PrPC is its neuroprotective activity against excitotoxicity mediated by Cu2+ coordination with N-terminal (Heske et al., 2004; Schilling et al., 2020). The globular domain of PrP is highly conserved among mammals (Salamat et al., 2013) and exhibits high structural similarity with the Dpl, sharing 25% identity (Ciric and Rezaei, 2015). This region is central in the conversion process; however, it allows certain changes, namely, insertions and deletions, in the C-terminal portion of H2 without affecting the conversion process (Salamat et al., 2012, 2013; Ciric and Rezaei, 2015; Munoz-Montesino et al., 2016, 2017).
Since the late 1990s, posttranslational modifications (PTMs) have been recognized as the main regulators of PrPC biosynthesis. The trafficking of PrPC precursor protein (253 residues) to the plasma membrane starts with the internalization into the endoplasmic reticulum (ER) by an N-terminal signal peptide (Heller et al., 2003; Chakrabarti et al., 2009; Miranzadeh Mahabadi and Taghibiglou, 2020). After translocation of ER, several PTM occurs to allow PrPC folding (23–231 residues), namely, C-terminal hydrophobic segment cleavage, C-terminal GPI anchor attachment, and the addition of different patterns of N-linked glycosylation (181 and 197 residues in humans, and 180 and 196 residues in mice), which can lead to a diglycosylated, monoglycosylated, or unglycosylated species (Choi, 1992; Zuegg and Gready, 2000; Miranzadeh Mahabadi and Taghibiglou, 2020).
Not all newly synthesized PrPC is translocated into the plasma membrane. There are two transmembrane (TM) PrPC topologies that are retained into ER or Golgi for proteasomal degradation, namely, the N-transmembrane (NtmPrPC) and cytosol transmembrane (CtmPrPC) (Hegde et al., 1998; Sarnataro et al., 2017). There are no precise physiological or pathological functions for NtmPrPC up to date (Westergard et al., 2007; Miranzadeh Mahabadi and Taghibiglou, 2020). In contrast, CtmPrPC has been associated with some neurodegenerative pathways, namely, PrPSc accumulation, ER stress, cell death, and neurodegeneration (Hegde et al., 1998; Crozet et al., 2008; Gavin et al., 2020).
Later, PrPC is transported through the Golgi apparatus to the trans-Golgi network (TGN), where several PTMs are found to be translocated finally to the plasma membrane, where it remains attached by its GPI anchor (Biasini et al., 2012; Miranzadeh Mahabadi and Taghibiglou, 2020). PrPC traffics through endocytic recycling compartment, mediated by clathrin-dependent mechanism, where it can be sorted in the plasma membrane for recycling or endolysosomal pathway for degradation (Chakrabarti et al., 2009; Marijanovic et al., 2009; Yim et al., 2015).
The proteolytic processing of PrPC has been the focus of numerous studies due to physiological or pathological significance of the cleavages, which is still uncertain. Normally, PrPC can be processed mainly by two proteolytic pathways. First, PrPC α-cleavage (that occurs at residues 110–111 or 111–112) generates a soluble ∼11 kDa fragment from PrPC N-terminal domain (N1), as well as a ∼16 kDa fragment from PrPC C-terminal region which remains attached to the plasma membrane (C1) (Mange et al., 2004; Liang and Kong, 2012). Second, PrPC β-cleavage releases a longer fragment that remains attached to the membrane (C2) of ∼18 kDa and a ∼9 kDa fragment from PrPC N-terminal domain (N2) (Biasini et al., 2012; Castle and Gill, 2017).
Although the regulatory role for C1 fragment production is unresolved, it seems that it negatively modulates key steps of PrP conversion process, namely, misfolding, replication, and fibrillization (Westergard et al., 2011; Campbell et al., 2013). Nevertheless, under experimental conditions, C1 lacking the C-terminal portion of H2 can be converted into a C1 prion by full-length spontaneous prion harboring the same deletion (Munoz-Montesino et al., 2020). Regarding C2 fragment, data strongly suggest that its accumulation would be a key product of the PrPC processing in prion replication (Dron et al., 2010). It is likely that C2 represents an important PrPSc phenotype-contributing factor during prion disease (Dron et al., 2010).
Although the precise function of PrPC at the cell surface is not completely understood, some researchers have proposed that it might be important in the nervous system, namely, the formation of synapses, neuronal viability, neuronal excitability, cell motility and neuronal growth, antiapoptotic effect, neurite adhesion, stress sensibility, and calcium homeostasis (Herms et al., 2000; Pantera et al., 2009; Carulla et al., 2011; Park et al., 2015; Wulf et al., 2017; Prado et al., 2020). Additionally, PrPC has been related to the immune system, namely, T-cell activation, the release of reactive oxygen species (ROS), monocyte maturation, and macrophage phagocytic activity (Isaacs et al., 2006; Miranzadeh Mahabadi and Taghibiglou, 2020). PrPC also participates in several signaling pathways that regulate innate immunity, namely, Akt, ERK-1/2, and NF-κB (Jeon et al., 2013).
To characterize the physiological function of PrPC, the initial strategy was to develop PrPC knockout (KO) mice. The first KOs developed were called Zurich and Npu, both of which did not show marked phenotypes. In both animals, the transmission by prions was completely prevented since the substrate for prion conversion, PrP, was absent (Bueler et al., 1992; Manson et al., 1994). Later, new models, namely, Zurich II, Ngsk, and Rcm0, developed late ataxia due to degeneration of Purkinje neurons (Sakaguchi et al., 1996). In these models, overexpression of Dpl was observed and it would be this protein that causes the death of this type of neurons due to neurotoxicity and not due to the lack of PrPC. Likewise, it has been established in vitro that overexpression of Dpl is toxic only when PrPC is not expressed; therefore, an interaction between both proteins is suggested to mediate toxicity phenomena (Sakudo et al., 2005).
Likewise, PrPC modulates growth factor receptor (EGFR) function in regulating cell cycle and growth (Llorens et al., 2013). Another function reported for PrPC is protection against oxidative stress. It has been determined that in SH-SY5Y neuroblastoma cells in which PrPC was overexpressed, there was greater resistance to oxidative stress than cells expressing endogenous levels and that this protection would be given by the N-terminal portion of PrPC (Zeng et al., 2003).
Even though the metal-binding relevance to PrPC study represents a challenge, a large number of studies support that PrPC could be involved in copper homeostasis due to its N-terminal unstructured portion. Two main regions are involved in the copper-binding ability of PrPC: first is the highly conserved octarepeat (OR) region (residues 60–91), where the His residues can bind up to four copper ions with high affinity, and second is the so-called non-OR region (residues 92–111), where two additional His residues are able to bind copper. This non-OR region is contiguous to a hydrophobic portion (residues 112–127) and is thought to be relevant during prion conversion (Giachin et al., 2015). Single His residue mutation in both OR and non-OR regions analyses has supported the idea of the critical role of copper-binding residues, suggesting also its role in regulating the function of PrPC in neuritogenesis and preserving the functional conformation of the protein, thus contributing to modulate prion conversion propensity (Nguyen et al., 2019). Therefore, copper binding might be relevant to both physiological and pathological roles of PrPC. Other roles associated with their interaction are endocytosis stimulation and trafficking, antioxidant effect, NMDA receptors modulation, and brain metal homeostasis (Salzano et al., 2019). Metal ion regulation in the CNS has also been related to NDs such as AD and PD (Salzano et al., 2019).
Finally, under physiological conditions in the nervous system, it has been reported that PrPC is mediating several functions such as cell growth, metal homeostasis, neuritic growth, the formation of lamellipodia, and synaptic transmission (Carulla et al., 2011; Llorens et al., 2013; Legname, 2017; Huang et al., 2018; Nguyen et al., 2019; Prado et al., 2020). The signaling pathways associated with PrPC neuronal growth-associated functions are achieved by its association with different proteins, such as NCAM and laminin, to promote neurite growth through the activation of Fyn kinase (Schmitt-Ulms et al., 2001; Santuccione et al., 2005). Also, it was determined that PrPC participates in myelin homeostasis in Schwann cells through interaction with its N-terminal through residues 23–33 with the GPCR 126 receptor on the surface of these cells (Kuffer et al., 2016). The role of PrPC in neuronal function is further discussed in the subsequent sections.
The normal physiological functions and cell behavior of PrPC, namely, neurite outgrowth, synaptogenesis, synaptic function, and neuroprotection, are not yet well understood. PrPC has been associated with several intracellular signaling pathways that modulate neuronal signal transduction and it participates in the organization of physiological brain networks, such as neuronal excitability, neuroprotection, neuritogenesis, neurotrophic function, and neuronal plasticity (Linden et al., 2008; Carulla et al., 2015; Castle and Gill, 2017; Linden, 2017). However, to understand how PrPC can regulate synaptic plasticity by neuronal activity, it is necessary to study the functional interaction of PrPC with transporters, ion pumps, ion channels, and metabotropic receptors expressed in neuronal cell surface (Table 1). We thus approached PrPC modulation in two key processes, namely, action potentials (APs) and postsynaptic potentials (PSPs), that coordinate the correct functioning of neuronal performance and the generation of a nerve impulse.
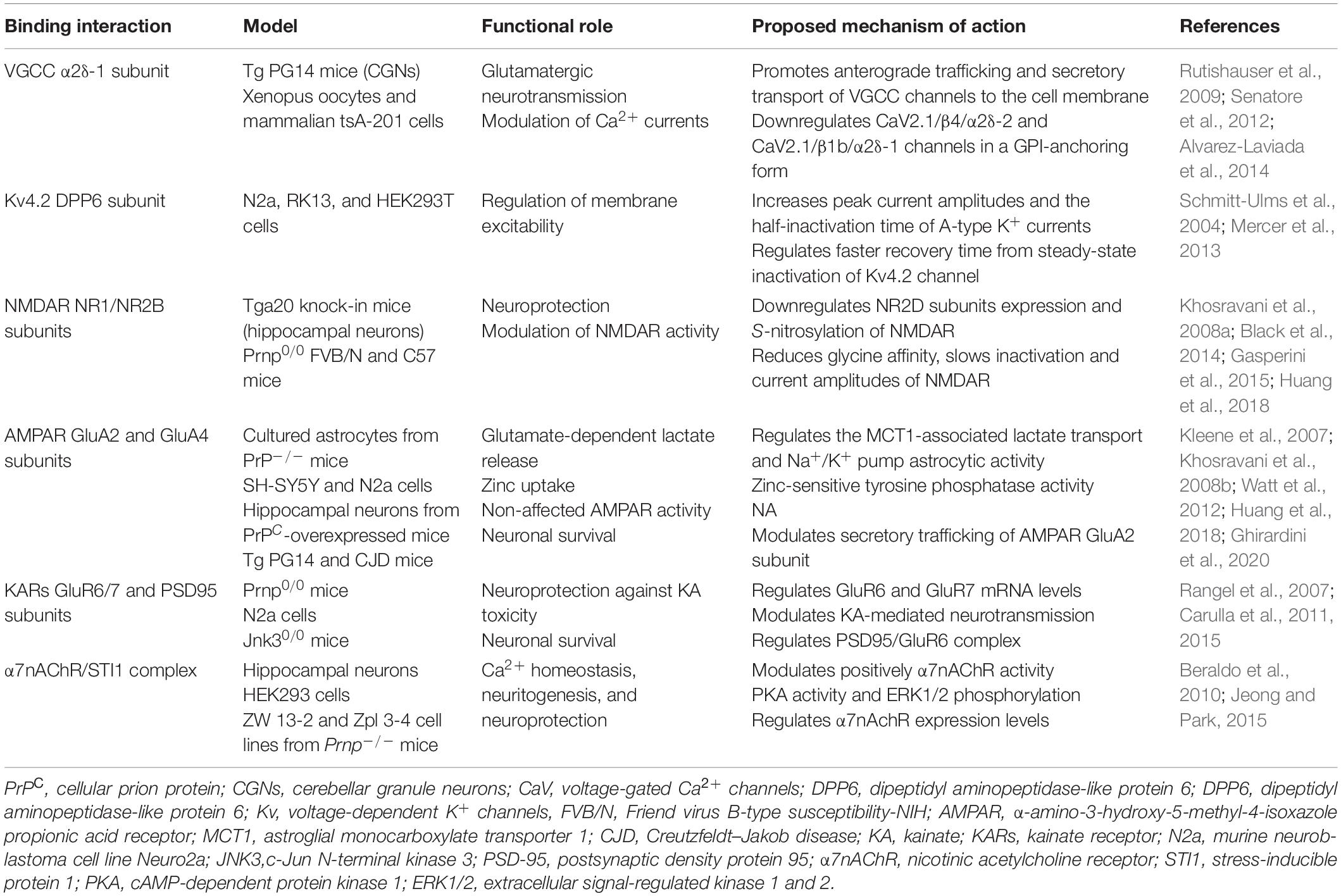
Table 1. Summary of the main effects of PrPC in synaptic function.
Collinge et al. (1994) established the role of PrP on neuronal excitability by electrophysiological studies in hippocampal pyramidal neurons and Purkinje cells, from non-transgenic (N-Tg) mice and conditional PrPC-null mice (Prnp0/0). Interestingly, the histopathological evaluation of Prnp0/0 did not exhibit significant variations with N-Tg mice, but it showed alterations on feedback mechanisms controlling frequency and patterning of neuronal firing, such as input resistance (Rinp), Ca2+-activated K+ current (IAHP), and afterhyperpolarization (AHP) current (Collinge et al., 1994; Colling et al., 1996; Herms et al., 2001; Mallucci et al., 2002).
One of the main modulators in the generation and shaping of APs is voltage-dependent calcium channels (VGCCs or CaV) (Llinas et al., 1976; Campiglio and Flucher, 2015). Electrophysiological and immunohistochemical studies have shown that PrPC is able to maintain neuronal excitability at the presynaptic level. This is achieved by stabilization and interaction with α2δ-1 auxiliary subunit of VGCC channels in a GPI anchor-dependent manner (Table 1; Rutishauser et al., 2009; Senatore et al., 2012; Alvarez-Laviada et al., 2014). Furthermore, co-expression of PrP with different Ca2+ channel subunits in Xenopus oocytes and mammalian tsA-201 cells has shown that PrP is able to modulate the amplitude peak of Ca2+ currents of the CaV2.1/β4/α2δ-2 and CaV2.1/β1b/α2δ-1 channels (Alvarez-Laviada et al., 2014). In contrast, in cerebellar granule neurons (CGN) of the transgenic mouse of PrP Tg (PG14), which synthesizes a misfolded mutant variant of PrP (PrPmut) that is partially retained in the ER, it was observed that PrPmut can impair α2δ-1 auxiliary subunit anterograde trafficking, reducing intracellular Ca2+ influx and glutamate transmission into the synaptic cleft (Senatore et al., 2012). Furthermore, PrPC modulates neuronal membrane excitability, synaptic integration of voltage threshold, and the repolarization process of the APs, mediated by their functional interaction with the Kv4.2 (voltage-gated K channels)/DPP6 (dipeptidyl aminopeptidase-like protein 6) complex at the neuronal cell surface (Schmitt-Ulms et al., 2004; Kim et al., 2008; Mercer et al., 2013). Electrophysiological studies in HEK293T cells transiently transfected with the Kv4.2/DPP6 channel complex have shown that PrPC is able to increase the amplitude peak and depolarizing potential of A-type K+ currents, as well as it shifts the activation curve of the Kv4.2 channels to more depolarized potentials in a DPP6-dependent form (Mercer et al., 2013). Further studies are needed to understand the link between PrPC, its misfolding, and the neuronal activity-dependent signaling pathways during the APs.
PrPC also participates in the regulation of excitatory postsynaptic responses through its functional interaction with ionotropic receptors, namely, N-methyl-D-aspartate receptor (NMDARs) (Khosravani et al., 2008a; You et al., 2012), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPARs) (Watt et al., 2012; Chater and Goda, 2014), kainate receptor (KARs) (Carulla et al., 2011), and α7 nicotinic acetylcholine receptors (α7nAChRs) (Zanata et al., 2002; Beraldo et al., 2010; Roffe et al., 2010; Table 1).
Increasing studies indicate that PrPC would be a key mediator in the maintenance of glutamatergic synapses, mediated by their interaction with NR1 and NR2 subunits of NMDAR (Khosravani et al., 2008a; You et al., 2012; Gasperini et al., 2015). It was observed that PrPC ablation induced an overexpression and S-nitrosylation of the NR2A and NR2B subunits of NMDAR, altering its kinetic properties. PrPC ablation induced a slow inactivation of the channel triggering an abnormal increase in neuronal excitability (Gasperini et al., 2015). Meanwhile, overexpression of mouse PrPC showed decreased activity of NMDAR (Maglio et al., 2004; Khosravani et al., 2008a; Gasperini et al., 2015; Huang et al., 2018). Additionally, recent studies have shown that the neuroprotective effects of PrPC associated with downregulation of NMDAR would occur in a Cu2+−dependent manner (Gasperini et al., 2015; Huang et al., 2018). More studies are needed to establish the interaction sites of PrPC in the modulation of NMDAR activity.
Regarding AMPA receptors, in vitro co-immunoprecipitation studies also revealed interactions with PrPC (Kleene et al., 2007; Watt et al., 2012; Huang et al., 2018). Interestingly, it has been observed that the increase in the formation of PrPC/AMPAR complex could exert neuroprotection in a Cu2+− and Zn2+−dependent manner, as well as AMPA-ergic activity (Watt et al., 2012; Huang et al., 2018). However, PrPC modulation does not induce significant changes in the amplitude or channel kinetics nor the long-term depression (LTD) maintenance (Khosravani et al., 2008a; Huang et al., 2018). Remarkably, the mutant variant of PrP could exert excitotoxicity mediated by intracellularly retained GluA2 AMPAR subunit (Ghirardini et al., 2020).
It has been postulated that PrPC has a neuroprotective function in association with KARs against neurotoxicity induced by kainite (KA), which induces neurodegeneration in presynaptic terminals (Carulla et al., 2011). Additionally, in vivo and in vitro evidence in Prnp0/0 mice indicated that PrPC can also regulate synaptic transmission and exert neuroprotection against KA toxicity, in a GPI anchoring-dependent manner (Rangel et al., 2007; Carulla et al., 2015). More studies are needed to determine the direct action of PrPC in channel kinetics and KARs activity, mediated by postsynaptic density protein 95 (PSD95) modulation.
Another postulated mechanism by which PrPC would exert neuroprotection and promote neuritogenesis is related to its association with α7nAChRs/stress-inducible protein 1 (STI1) complex at the cell membrane (Beraldo et al., 2010). Effectively, in several neuron cell lines, it has been observed that PrPC can upregulate several neuroprotective pathways, such as autophagic flux cAMP-dependent protein kinase 1 (PKA), and extracellular signal-regulated kinase 1 and 2 (ERK1/2) pathways, in a α7nAChRs-dependent manner (Beraldo et al., 2010; Jeong and Park, 2015).
The overall data suggest that PrPC would be acting as a new player in the regulation of glutamatergic and cholinergic neurotransmission. However, further research is needed to identify regions involved in the association of ionotropic receptors and PrPC, as well as the consequences of its disruption in the synaptic neurotransmission in a pathological context.
Alzheimer’s disease is a progressive disorder associated with cerebral cortex atrophy and irreversible loss of cortical neurons (Musiek and Schindler, 2013). AD is mainly characterized by an accumulation of amyloid-β (Aβ) plaques and phosphorylated Tau protein neurofibrillary tangles. The major plaque component is Aβ peptide made of 39–43 amino acids, which are derived from the amyloid precursor protein (APP) (Selkoe, 2001; Walsh and Selkoe, 2007). Aβ monomers are not toxic and do not interfere with the synapses, whereas small oligomers and larger aggregates are most likely to be the most toxic species, impairing synaptic plasticity (Legname and Scialo, 2020).
Several studies have related PrP with AD (Kellett and Hooper, 2009); however, the mechanism by which PrP affects the progression of the disease is not clear. Also, there is still controversy regarding whether or not PrPC is required for Aβ toxicity (Legname and Scialo, 2020). Therefore, we discussed the evidence for interaction of PrPC and Aβ and its role in mediating Aβ toxicity.
Aβ oligomers (AβOs)-induced neuronal toxicity is thought, at least partly, to be mediated by putative Aβ receptors. Among them, PrPC has emerged as an important potential receptor, due to its high affinity to the oligomeric form of the peptide (Laurén et al., 2009; Smith et al., 2019; Legname and Scialo, 2020). A cloning cDNA screening from a mouse brain library in order to find a protein that binds to AβOs (Aβ1-42) found that the only high-affinity binding protein was PrPC, an observation that has been further supported by other studies (Laurén et al., 2009; Corbett et al., 2020). In fact, in a systematic comparison of reported Aβ receptors, only PrPC, Nogo receptor 1 (NgR1), and leukocyte immunoglobulin-like receptor subfamily member 2 (LilrB2) showed direct binding to synthetic Aβ assemblies. Interestingly, binding with human AD brains-derived soluble AβOs revealed strong affinity only for PrPC, with a weak affinity for NgR1 and no detectable affinity for LilrB2 (Smith et al., 2019). Therefore, PrPC is most likely an Aβ-binding receptor.
In contrast to what was observed between PrPC and AβOs, experiments performed in vitro showed low-affinity interactions with Aβ monomers (Chen et al., 2010; Fluharty et al., 2013; Corbett et al., 2020). Solid-phase assays showed that there is neither interaction of monomeric Aβ1-42 with PrP23–231 nor full-length PrPC (Corbett et al., 2020). However, immunoassay studies have revealed that PrPC 23–39 and 93–119 can interact with monomeric Aβ1-42 (Kang et al., 2013). Reported sites of interaction between PrPC and different Aβ species are summarized in Table 2.
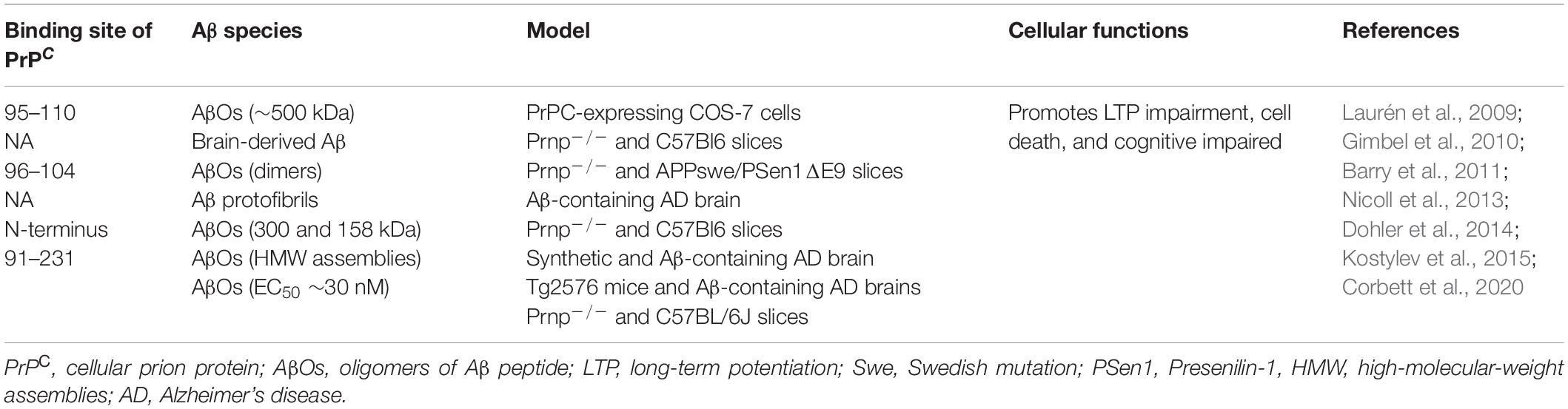
Table 2. Aβ–PrPC interaction sites.
Regarding the binding site in PrPC for AβOs, it was shown that the unstructured N-terminal domain was relevant for this interaction (Laurén et al., 2009). In fact, when anti-PrP antibodies were used to interfere with the interaction, only 6D11 (which binds to amino acids 93–109 in mouse PrP) blocked the binding between Aβ assemblies and PrPC with an IC50 of 1 nM (Laurén et al., 2009). In addition, the deletion of a similar region (95–105) impaired Aβ binding to PrPC (Laurén et al., 2009). Another site reported for this binding was the N-terminal basic amino acids 23–27 (KKRPK) in PrP (Legname and Scialo, 2020).
Protein misfolding and aggregation of Aβ peptide are key events in the onset of AD, especially AβOs, due its capacity to associate with the cell membrane and induce excitotoxicity (Puzzo et al., 2017; Cline et al., 2018). The main neurotoxic effects described for AβOs in AD are membrane disruption, synaptic failure, impaired LTP, and memory loss (Lambert et al., 1998; Cline et al., 2018). However, specific binding transducers of AβOs signals that mediate its neurotoxic effects are not yet clearly defined. Several works have postulated different interacting partners for Aβ assemblies in the cell membrane, namely, NMDAR (Rammes et al., 2018), APP (Puzzo et al., 2017), NgR1, nAChR, and PrPC (Fabiani and Antollini, 2019; Smith et al., 2019; Zhang et al., 2019).
As mentioned earlier, PrPC has been proposed as a high-affinity physiological receptor for soluble AβOs [see reviews Linden (2017) and Wiatrak et al. (2021)]. At present, in different animal AD models as well as in patients with AD, it has been established that PrPC could be one of the best specific binding partners for AβOs-mediated inhibition of LTP and cognitive defects in the early stages of AD (Kostylev et al., 2015; Smith and Strittmatter, 2017; Smith et al., 2019; Corbett et al., 2020). With this knowledge, it has been proposed that PrPC would play an important role in the onset of AD, occurring before clinical symptoms, such as movement and cognitive impairments associated with the late stages of the disease.
Laurén et al. (2009) have demonstrated that PrPC is a high-affinity receptor for AβOs, being amino acids 95–110 of PrPC involved in this interaction (Laurén et al., 2009). Solid-phase and ELISA-like assays showed further associations between AβOs and PrPC (EC50 ∼30 nM) (Corbett et al., 2020). In cellular and animal models of Aβ toxicity, PrPC was able to mediate impairment of synaptic plasticity, alteration in calcium transients, and reduction in the levels of synaptophysin (Riek et al., 1996; Laurén et al., 2009; Barry et al., 2011; Peters et al., 2015). Furthermore, these alterations can be rescued using antibodies that block the oligomer-binding site of Aβ in PrPC (6D11) (Riek et al., 1996; Laurén et al., 2009; Barry et al., 2011; Peters et al., 2015). Regarding the mechanism by which PrPC exerts its role as a receptor, it has been proposed that AβOs binding to PrP induces activation of Fyn, a Src kinase (SRK), through an undetermined TM partner (Figure 2; Malaga-Trillo and Ochs, 2016). After its activation, fyn phosphorylates NMDA receptor, which becomes transiently over-activated, producing excitotoxicity (Malaga-Trillo and Ochs, 2016). Fyn was already known to be relevant in the pathogenesis of AD, because it performs Tau phosphorylation. Tau is an axonal microtubule-associated protein, and phosphorylated Tau is the main constituent of neurofibrillary tangles in AD, which mediates Aβ toxicity at the post-synapse. The notion that AβO-induced Tau phosphorylation is mediated by PrPC comes from assays in human and mice brain, as well as analyses in primary neuron cultures, which show that soluble Aβ binds to a PrPC/Fyn complex and Prnp gene deletion uncouples AβOs and the Fyn/tau axis (Larson et al., 2012). Besides Tau phosphorylation, SRKs are able to regulate the stability at the neuronal plasma membrane of several synapse-relevant proteins as adhesion proteins and receptors (e.g., NMDAR, AMPAR, and GABAR) (Malaga-Trillo and Ochs, 2016). Therefore, PrPC–Fyn interaction might be directly involved in the pathological characteristics of AD. The signal transduction pathway generated by the interaction between AβOs and PrPC is depicted in Figure 2.
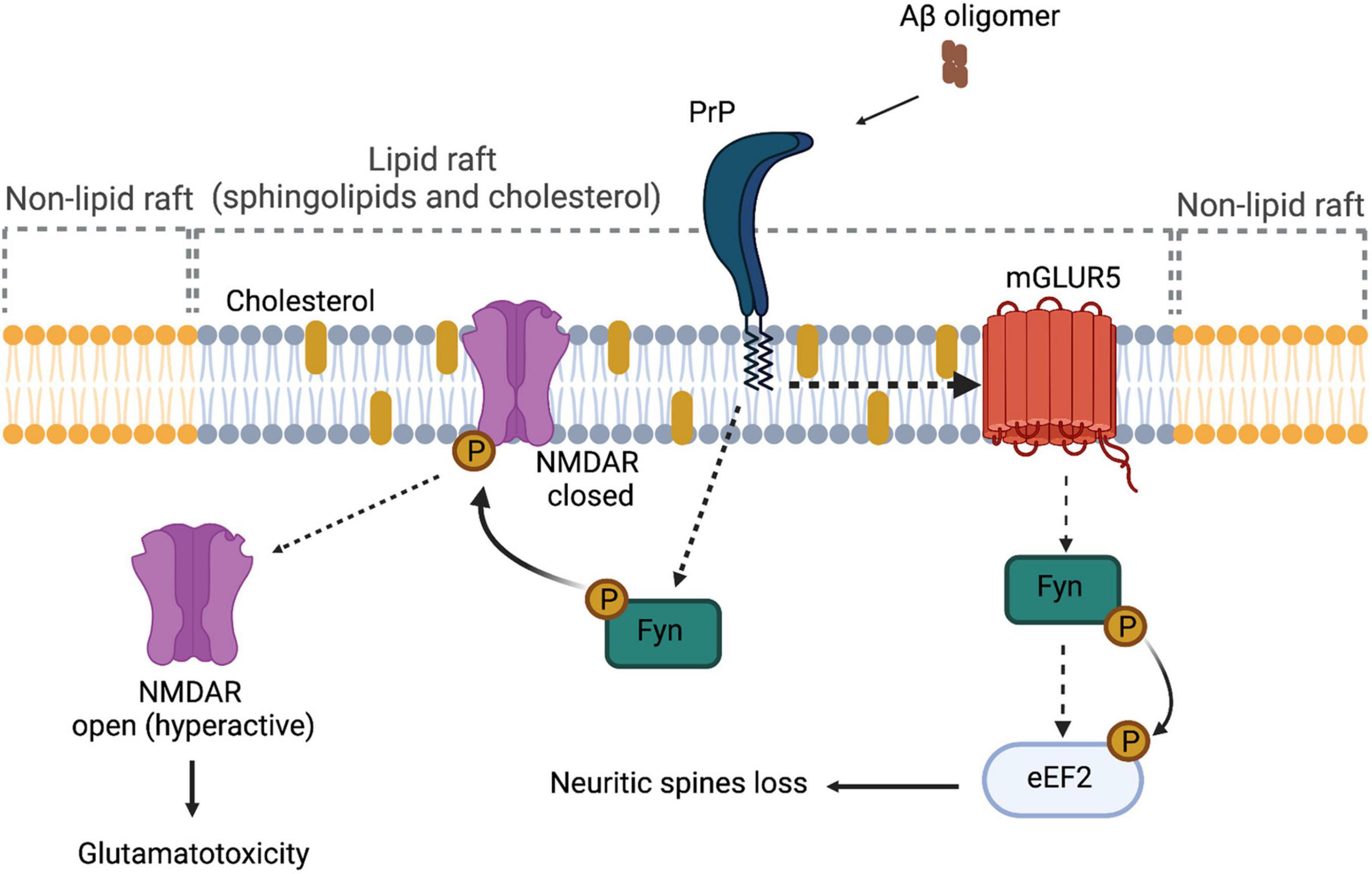
Figure 2. PrPC as a signal transducer. In the context of Alzheimer’s disease, the interaction between Aβ oligomers and PrPC affects receptors located on the plasmatic membrane, such as NMDAR and mGluR5. In the case of NMDAR, because of the interaction of Aβ oligomers and PrPC, the receptor is phosphorylated through Fyn, hyperactivating the channel and causing glutamatotoxicity. In the case of mGluR5, there is a direct interaction between PrPC and the receptor, causing the activation of Fyn kinase and promoting the phosphorylation of eEF2 and the consequent loss of neuritic spines.
Neurodegeneration caused by protein misfolding and aggregation is characterized by progressive neuronal dysfunction associated with deposition of insoluble aggregates from a misfolded protein (Legname and Scialo, 2020). As discussed earlier, in prion diseases, the appearance of PrPSc assemblies is involved in this process. In the case of AD, we have mentioned that AβOs, Aβ fibrils and plaques, and Tau tangles appear in the brain of patients with AD and that certain of these species are related to the neurotoxicity and neurodegeneration. In other proteinopathies, amyloids and deposits of other proteins, such as TAR DNA-binding protein 43 (TDP-43), α-synuclein (α-syn), and Tau, are found (Legname and Scialo, 2020; Scialo et al., 2021). In the following sections, how some of these proteins are interconnected with PrPC, and therefore, the role of PrP in the diseases linked to them are discussed.
Tauopathies are a group of diseases that have in common the deposition of abnormal tau in the nervous system. They comprise AD, Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, and primary age-related tauopathy, among others (Kovacs, 2015). Normal Tau, which is a microtubule-associated protein, plays a role in the stabilization of neuronal microtubules. In pathological conditions, tau undergoes phosphorylation and forms aggregates that are neurotoxic (Avila et al., 2004).
Regarding its relation to PrPC, in vitro and in vivo studies have found an association between PrPC and hyperphosphorylated tau forms, particularly with tau N-terminal region (De Cecco et al., 2020; Legname and Scialo, 2020). Electrophysiological experiments showed that antibodies against PrPC (6D11, MI-0131) could prevent LTP impairment induced by tau toxicity (Ondrejcak et al., 2018). At present, it has been reported that other antibodies against different epitopes of PrPC (POM 3, 4, 12) are able to impair the uptake of tau amyloid fibrils in mouse neuroblastoma cells (De Cecco et al., 2020). In contrast, it has been described that tau is a transcription regulator for PRPN gene in AD models (Lidon et al., 2020), linking both proteins in the progression of tauopathies.
The misfolding and accumulation of α-synuclein is involved in a group of pathologies known as synucleinopathies, such as PD, dementia with Lewy bodies (LBD), and multiple system atrophy (MSA). For instance, histopathological biomarker detected in patients with PD has been classically associated with abnormal deposits of α-syn, which mainly affects nigral dopaminergic system at the intracellular level, also called Lewy bodies (Kalia and Lang, 2015).
In these diseases, similar to other proteinopathies, fibrillar forms of α-syn spread from one cell to another. One of the mechanisms that this form of α-syn uses to enter cell is clathrin-dependent endocytosis, a process that requires the interaction with the TM protein lymphocyte-activation gene 3 (LAG3) (De Cecco and Legname, 2018). Other protein that was reported to be involved in the internalization of α-syn is PrPC (Aulic et al., 2017). Cells that express PrPC are able to internalize more amyloid α-syn fibrils compared to cells that do no express it; therefore, PrPC favors cell-to-cell transmission (Aulic et al., 2017). In contrast, when these cells are infected with prions, α-syn reduces prion replication, especially due to PrPC α cleavage, producing C1 and N1 that are neuroprotectors (Aulic et al., 2017).
Further analyses agreed on the connection between PrPC and α-syn: overexpression of PrPC in the striatum potentiates neurodegeneration, thereby altering α-syn propagation and toxicity. Electrophysiological and molecular approaches showed that antibodies against PrPC 6D11 could abolish LTP impairment, calcium dyshomeostasis, and cell degeneration induced by α-syn toxicity (Ferreira et al., 2017; Legname and Scialo, 2020).
Frontotemporal lobar degeneration (FTLD), a neurodegenerative syndrome in frontal and anterior temporal lobes (Rabinovici and Miller, 2010), and ALS, a motor neuron disorder characterized by degeneration in the upper and lower motor neurons (Prasad et al., 2019), are two distinct diseases that shared a histopathological hallmark: inclusion bodies composed of cytoplasmic deposits of the nuclear TDP-43 protein (Scialo et al., 2021). Under physiological conditions, TDP-43 is a transcriptional repressor that binds to chromosomally integrated TAR DNA. Nevertheless, a hyper-phosphorylated, ubiquitinated, and cleaved form of TDP-43 (pathological TDP-43) is the major disease protein in ubiquitin-positive, tau-, and α-synuclein-negative FTLD and in ALS (Mackenzie et al., 2011; Brauer et al., 2018).
It was observed in vitro that TDP-43 fibrils bind to recombinant PrPC. Also, in vitro, it was shown that full-length mouse (Mu)PrPC as well as human (Hu)PrPC act as a membrane receptor of TDP-43 in its fibrillar conformation, inducing the formation of intracytoplasmic aggregates and cell death (Scialo et al., 2021). In addition, the overexpression of PrPC in human and mouse cell lines was directly correlated with the internalization of TDP-43 fibrils. Increased internalization was associated with detrimental consequences in all PrP-overexpressing cell lines (Scialo et al., 2021).
As for other amyloids, treatment with TDP-43 fibrils induced a reduction in the accumulation of the misfolded form of PrPC, PrPSc, in cells chronically infected with prions. Our results expand the list of misfolded proteins whose uptake and detrimental effects are mediated by PrPC, which encompass almost all pathological amyloids involved in neurodegeneration (Scialo et al., 2021).
As we mentioned earlier, PrPC is mostly expressed in the brain. It is especially expressed in the hippocampus and it increases in the aging brain (Williams et al., 2004; Benvegnu et al., 2010). Aging, being the main risk factor for NDs (Wyss-Coray, 2016; Hou et al., 2019), can lead to cognitive impairment, affecting information processing and memory (Hedden and Gabrieli, 2004). Since PrPC has shown to participate in neuroprotection, metal homeostasis, and most probably as an antioxidant, it has been suggested that it may play a role in aging (Gasperini and Legname, 2014). In fact, in prion diseases, the function of PrPC is lost due to conversion into PrPSc and this event could also be related to the progression of the disease (Gasperini and Legname, 2014). Furthermore, the biochemical properties of PrPC are altered during aging (Gasperini and Legname, 2014). Even though it is likely that PrPC is involved in behavior and learning processes during aging, the analyses performed so far in PrPC KO mice are not conclusive, probably due to differences in mouse models and age (Gasperini and Legname, 2014). Zurich old KO mice exhibit alteration in nest building behavior and decline in associative learning compared to wild-type mice. At molecular level, mice lacking PrPC showed alterations in cytoskeletal proteins, due to the lower phosphorylation of the neurofilament heavy chain and reduction in B-tubulin III-positive neurons in the hippocampus (Gasperini and Legname, 2014; Schmitz et al., 2014). This might be related to neuronal structure changes due to the absence of PrPC and therefore a cellular explanation to behavioral abnormalities (Gasperini and Legname, 2014; Schmitz et al., 2014). Even though most studies suggest a role for PrPC in aging, more are still needed to better define this role.
Although PrPC studies started from a pathological context, such as prion diseases, in recent years, studies of its functions in physiological terms increased, especially in the nervous system where this protein participates in relevant functions in neural networks, from neurite growth to ion channel association. Despite its important role, it remains a challenge to determine why the lack PrPC does not show a relevant phenotype and how other proteins might compensate the absence of PrP.
Recently, the role of PrPC in AD has emerged as crucial, supported by several studies. As this protein does not present TM spans, its interaction with other TM proteins must be key for its role in mediating physiological and pathological phenomena. Since Fyn kinase is a protein involved in both physiological and pathological PrPC-mediated responses, more studies are needed to understand the differences in the signaling in both processes.
With the discovery that PrPC is the main receptor for AβOs, more studies are needed to determine whether PrP or other proteins in the pathological pathway might be a target for AD therapy and other NDs.
JDP and PS contributed equally to this work in the information search and in the preparation of the manuscript. CMM, JDP, PS, BP, KE, and MEC participated in the figure designed and information search. GMC, JF, HR, CIR, and CMM conducted the manuscript preparation and edited the text.
This study was supported by grants 1201496 (CIR), 1200908 (JF) and 1211095 (GMC) from the Fondo Nacional de Investigación Científica y Tecnológica (FONDECYT, Chile). The international cooperation has been possible thanks to Programa de Cooperación Científica ECOS-CONICYT grant C16S01.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
In memory of our colleague and friend, Danica Ciric who deceased on February 13, 2021.
Acevedo-Morantes, C. Y., and Wille, H. (2014). The structure of human prions: from biology to structural models-considerations and pitfalls. Viruses. 6, 3875–3892. doi: 10.3390/v6103875
Alper, T., Cramp, W. A., Haig, D. A., and Clarke, M. C. (1967). Does the agent of scrapie replicate without nucleic acid? Nature 214, 764–766. doi: 10.1038/214764a0
Alvarez-Laviada, A., Kadurin, I., Senatore, A., Chiesa, R., and Dolphin, A. C. (2014). The inhibition of functional expression of calcium channels by prion protein demonstrates competition with alpha2delta for GPI-anchoring pathways. Biochem. J. 458, 365–374. doi: 10.1042/BJ20131405
Aulic, S., Masperone, L., Narkiewicz, J., Isopi, E., Bistaffa, E., Ambrosetti, E., et al. (2017). Alpha-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci. Rep. 7:10050. doi: 10.1038/s41598-017-10236-x
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384.
Barry, A. E., Klyubin, I., Mc Donald, J.M., Mably, A. J., Farrell, M. A., Scott, M., et al. (2011). Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011
Beland, M., and Roucou, X. (2012). The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J. Neurochem. 120, 853–868. doi: 10.1111/j.1471-4159.2011.07613.x
Benvegnu, S., Poggiolini, I., and Legname, G. (2010). Neurodevelopmental expression and localization of the cellular prion protein in the central nervous system of the mouse. J. Comp. Neurol. 518, 1879–1891. doi: 10.1002/cne.22357
Beraldo, F. H., Arantes, C. P., Santos, T. G., Queiroz, N. G., Young, K., Rylett, R. J., et al. (2010). Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550. doi: 10.1074/jbc.M110.157263
Beringue, V., Mallinson, G., Kaisar, M., Tayebi, M., Sattar, Z., Jackson, G., et al. (2003). Regional heterogeneity of cellular prion protein isoforms in the mouse brain. Brain 126, 2065–2073.
Biasini, E., Turnbaugh, J. A., Unterberger, U., and Harris, D. A. (2012). Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103.
Black, S. A., Stys, P. K., Zamponi, G. W., and Tsutsui, S. (2014). Cellular prion protein and NMDA receptor modulation: protecting against excitotoxicity. Front. Cell Dev. Biol. 2:45. doi: 10.3389/fcell.2014.00045
Bolton, D. C., McKinley, M. P., and Prusiner, S. B. (1982). Identification of a protein that purifies with the scrapie prion. Science 218, 1309–1311. doi: 10.1126/science.6815801
Botto, L., Cunati, D., Coco, S., Sesana, S., Bulbarelli, A., Biasini, E., et al. (2014). Role of lipid rafts and GM1 in the segregation and processing of prion protein. PLoS One 9:e98344. doi: 10.1371/journal.pone.0098344
Brauer, S., Zimyanin, V., and Hermann, A. (2018). Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J. Neural. Transm. (Vienna) 125, 591–613. doi: 10.1007/s00702-018-1851-y
Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., DeArmond, S. J., et al. (1992). Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582. doi: 10.1038/356577a0
Campbell, L., Gill, A. C., McGovern, G., Jalland, C. M., Hopkins, J., Tranulis, M. A., et al. (2013). The PrP(C) C1 fragment derived from the ovine A136R154R171PRNP allele is highly abundant in sheep brain and inhibits fibrillisation of full-length PrP(C) protein in vitro. Biochim. Biophys. Acta 1832, 826–836. doi: 10.1016/j.bbadis.2013.02.020
Campiglio, M., and Flucher, B. E. (2015). The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J. Cell Physiol. 230, 2019–2031. doi: 10.1002/jcp.24998
Carulla, P., Bribian, A., Rangel, A., Gavin, R., Ferrer, I., Caelles, C., et al. (2011). Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 22, 3041–3054. doi: 10.1091/mbc.E11-04-0321
Carulla, P., Llorens, F., Matamoros-Angles, A., Aguilar-Calvo, P., Espinosa, J. C., Gavin, R., et al. (2015). Involvement of PrP(C) in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 5:11971. doi: 10.1038/srep11971
Castle, A. R., and Gill, A. C. (2017). Physiological functions of the cellular prion protein. Front. Mol. Biosci. 4:19. doi: 10.3389/fmolb.2017.00019
Chakrabarti, O., Ashok, A., and Hegde, R. S. (2009). Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 34, 287–295. doi: 10.1016/j.tibs.2009.03.001
Chater, T. E., and Goda, Y. (2014). The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell Neurosci. 8:401. doi: 10.3389/fncel.2014.00401
Chen, S., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi: 10.1074/jbc.M110.145516
Chesebro, B., Race, R., Wehrly, K., Nishio, J., Bloom, M., Lechner, D., et al. (1985). Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 315, 331–333. doi: 10.1038/315331a0
Ciric, D., and Rezaei, H. (2015). Biochemical insight into the prion protein family. Front. Cell Dev. Biol. 3:5. doi: 10.3389/fcell.2015.00005
Cline, E. N., Bicca, M. A., Viola, K. L., and Klein, W. L. (2018). The Amyloid-beta oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 64, S567–S610. doi: 10.3233/JAD-179941
Colling, S. B., Collinge, J., and Jefferys, J. G. (1996). Hippocampal slices from prion protein null mice: disrupted Ca(2+)-activated K+ currents. Neurosci. Lett. 209, 49–52. doi: 10.1016/0304-3940(96)12596-9
Collinge, J. (2001). Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24, 519–550.
Collinge, J., Whittington, M. A., Sidle, K. C., Smith, C. J., Palmer, M. S., Clarke, A. R., et al. (1994). Prion protein is necessary for normal synaptic function. Nature 370, 295–297. doi: 10.1038/370295a0
Corbett, G. T., Wang, Z., Hong, W., Colom-Cadena, M., Rose, J., Liao, M., et al. (2020). PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 139, 503–526. doi: 10.1007/s00401-019-02114-9
Crozet, C., Beranger, F., and Lehmann, S. (2008). Cellular pathogenesis in prion diseases. Vet. Res. 39:44. doi: 10.1051/vetres:2008021
De Cecco, E., Celauro, L., Vanni, S., Grandolfo, M., Bistaffa, E., Moda, F., et al. (2020). The uptake of tau amyloid fibrils is facilitated by the cellular prion protein and hampers prion propagation in cultured cells. J. Neurochem. 155, 577–591. doi: 10.1111/jnc.15040
De Cecco, E., and Legname, G. (2018). The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion 12, 23–27. doi: 10.1080/19336896.2017.1423186
Dohler, F., Sepulveda-Falla, D., Krasemann, S., Altmeppen, H., Schluter, H., Hildebrand, D., et al. (2014). High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain. 137, 873–886. doi: 10.1093/brain/awt375
Dron, M., Moudjou, M., Chapuis, J., Salamat, M. K., Bernard, J., Cronier, S., et al. (2010). Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J. Biol. Chem. 285, 10252–10264. doi: 10.1074/jbc.M109.083857
Engelke, A. D., Gonsberg, A., Thapa, S., Jung, S., Ulbrich, S., Seidel, R., et al. (2018). Dimerization of the cellular prion protein inhibits propagation of scrapie prions. J. Biol. Chem. 293, 8020–8031. doi: 10.1074/jbc.RA117.000990
Fabiani, C., and Antollini, S. S. (2019). Alzheimer’s disease as a membrane disorder: spatial cross-talk among beta-amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front. Cell Neurosci. 13:309. doi: 10.3389/fncel.2019.00309
Ferreira, D. G., Temido-Ferreira, M., Vicente Miranda, H., Batalha, V. L., Coelho, J. E., Szego, E. M. I., et al. (2017). alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 20, 1569–1579. doi: 10.1038/nn.4648
Fluharty, B. R., Biasini, E., Stravalaci, M., Sclip, A., Diomede, L., Balducci, C., et al. (2013). An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866. doi: 10.1074/jbc.M112.423954
Gasperini, L., and Legname, G. (2014). Prion protein and aging. Front. Cell Dev. Biol. 2:44. doi: 10.3389/fcell.2014.00044
Gasperini, L., Meneghetti, E., Pastore, B., Benetti, F., and Legname, G. (2015). Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 22, 772–784. doi: 10.1089/ars.2014.6032
Gavin, R., Lidon, L., Ferrer, I., and Del Rio, J. A. (2020). The quest for cellular prion protein functions in the aged and neurodegenerating brain. Cells 9:591. doi: 10.3390/cells9030591
Ghirardini, E., Restelli, E., Morini, R., Bertani, I., Ortolan, D., Perrucci, F., et al. (2020). Mutant prion proteins increase calcium permeability of AMPA receptors, exacerbating excitotoxicity. PLoS Pathog. 16:e1008654. doi: 10.1371/journal.ppat.1008654
Giachin, G., Mai, P. T., Tran, T. H., Salzano, G., Benetti, F., Migliorati, V., et al. (2015). The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci. Rep. 5:15253. doi: 10.1038/srep15253
Gimbel, D. A., Nygaard, H. B., Coffey, E. E., Gunther, E. C., Lauren, J., Gimbel, Z. A., et al. (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010
Hedden, T., and Gabrieli, J. D. (2004). Insights into the ageing mind: a view from cognitive neuroscience. Nat. Rev. Neurosci. 5, 87–96. doi: 10.1038/nrn1323
Hegde, R. S., Mastrianni, J. A., Scott, M. R., DeFea, K. A., Tremblay, P., Torchia, M., et al. (1998). A transmembrane form of the prion protein in neurodegenerative disease. Science 279, 827–834. doi: 10.1126/science.279.5352.827
Heller, U., Winklhofer, K. F., Heske, J., Reintjes, A., and Tatzelt, J. (2003). Post-translational import of the prion protein into the endoplasmic reticulum interferes with cell viability: a critical role for the putative transmembrane domain. J. Biol. Chem. 278, 36139–36147. doi: 10.1074/jbc.M304002200
Herms, J. W., Korte, S., Gall, S., Schneider, I., Dunker, S., and Kretzschmar, H. A. (2000). Altered intracellular calcium homeostasis in cerebellar granule cells of prion protein-deficient mice. J. Neurochem. 75, 1487–1492. doi: 10.1046/j.1471-4159.2000.0751487.x
Herms, J. W., Tings, T., Dunker, S., and Kretzschmar, H. A. (2001). Prion protein affects Ca2+-activated K+ currents in cerebellar purkinje cells. Neurobiol. Dis. 8, 324–330. doi: 10.1006/nbdi.2000.0369
Heske, J., Heller, U., Winklhofer, K. F., and Tatzelt, J. (2004). The C-terminal globular domain of the prion protein is necessary and sufficient for import into the endoplasmic reticulum. J. Biol. Chem. 279, 5435–5443. doi: 10.1074/jbc.M309570200
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581.
Huang, S., Chen, L., Bladen, C., Stys, P. K., and Zamponi, G. W. (2018). Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol. Brain 11:62. doi: 10.1186/s13041-018-0406-3
Isaacs, J. D., Jackson, G. S., and Altmann, D. M. (2006). The role of the cellular prion protein in the immune system. Clin. Exp. Immunol. 146, 1–8.
Jeon, J. W., Park, B. C., Jung, J. G., Jang, Y. S., Shin, E. C., and Park, Y. W. (2013). The Soluble form of the cellular prion protein enhances phagocytic activity and cytokine production by human monocytes via activation of ERK and NF-kappaB. Immune Netw. 13, 148–156. doi: 10.4110/in.2013.13.4.148
Jeong, J. K., and Park, S. Y. (2015). Neuroprotective effect of cellular prion protein (PrPC) is related with activation of alpha7 nicotinic acetylcholine receptor (alpha7nAchR)-mediated autophagy flux. Oncotarget 6, 24660–24674. doi: 10.18632/oncotarget.4953
Kang, M., Kim, S. Y., An, S. S., and Ju, Y. R. (2013). Characterizing affinity epitopes between prion protein and beta-amyloid using an epitope mapping immunoassay. Exp. Mol. Med. 45:e34. doi: 10.1038/emm.2013.63
Kellett, K. A., and Hooper, N. M. (2009). Prion protein and Alzheimer disease. Prion 3, 190–194. doi: 10.4161/pri.3.4.9980
Khosravani, H., Zhang, Y., and Zamponi, G. W. (2008b). Cellular prion protein null mice display normal AMPA receptor mediated long term depression. Prion 2, 48–50. doi: 10.4161/pri.2.2.6628
Khosravani, H., Zhang, Y., Tsutsui, S., Hameed, S., Altier, C., Hamid, J., et al. (2008a). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 181, 551—65.
Kim, J., Nadal, M. S., Clemens, A. M., Baron, M., Jung, S. C., Misumi, Y., et al. (2008). Kv4 accessory protein DPPX (DPP6) is a critical regulator of membrane excitability in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 100, 1835–1847. doi: 10.1152/jn.90261.2008
Kim, S. J., Rahbar, R., and Hegde, R. S. (2001). Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J. Biol. Chem. 276, 26132–26140. doi: 10.1074/jbc.M101638200
Kleene, R., Loers, G., Langer, J., Frobert, Y., Buck, F., and Schachner, M. (2007). Prion protein regulates glutamate-dependent lactate transport of astrocytes. J. Neurosci. 27, 12331–12340. doi: 10.1523/JNEUROSCI.1358-07.2007
Kostylev, M. A., Kaufman, A. C., Nygaard, H. B., Patel, P., Haas, L. T., Gunther, E. C., et al. (2015). Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple alzheimer mouse models. J. Biol. Chem. 290, 17415–17438. doi: 10.1074/jbc.M115.643577
Kovacs, G. G. (2015). Invited review: neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol. 41, 3–23. doi: 10.1111/nan.12208
Kuffer, A., Lakkaraju, A. K., Mogha, A., Petersen, S. C., Airich, K., Doucerain, C., et al. (2016). The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 536, 464–468. doi: 10.1038/nature19312
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U. S. A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lansbury, P. T. (1994). Mechanism of scrapie replication. Science 265:1510. doi: 10.1126/science.8079159
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871. doi: 10.1523/JNEUROSCI.1858-12.2012
Lau, A., McDonald, A., Daude, N., Mays, C. E., Walter, E. D., Aglietti, R., et al. (2015). Octarepeat region flexibility impacts prion function, endoproteolysis and disease manifestation. EMBO Mol. Med. 7, 339–356. doi: 10.15252/emmm.201404588
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Legname, G. (2017). Elucidating the function of the prion protein. PLoS Pathog. 13:e1006458. doi: 10.1371/journal.ppat.1006458
Legname, G., and Scialo, C. (2020). On the role of the cellular prion protein in the uptake and signaling of pathological aggregates in neurodegenerative diseases. Prion 14, 257–270.
Lidon, L., Vergara, C., Ferrer, I., Hernandez, F., Avila, J., Del Rio, J.A., et al. (2020). Tau protein as a new regulator of cellular prion protein transcription. Mol. Neurobiol. 57, 4170–4186.
Lima, F. R., Arantes, C. P., Muras, A. G., Nomizo, R., Brentani, R. R., and Martins, V. R. (2007). Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J. Neurochem. 103, 2164–2176.
Linden, R. (2017). The biological function of the prion protein: a cell surface scaffold of signaling modules. Front. Mol. Neurosci. 10:77. doi: 10.3389/fnmol.2017.00077
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728.
Llinas, R., Steinberg, I. Z., and Walton, K. (1976). Presynaptic calcium currents and their relation to synaptic transmission: voltage clamp study in squid giant synapse and theoretical model for the calcium gate. Proc. Natl. Acad. Sci. U. S. A. 73, 2918–2922. doi: 10.1073/pnas.73.8.2918
Llorens, F., Carulla, P., Villa, A., Torres, J. M., Fortes, P., Ferrer, I., et al. (2013). PrP(C) regulates epidermal growth factor receptor function and cell shape dynamics in Neuro2a cells. J. Neurochem. 127, 124–138. doi: 10.1111/jnc.12283
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi: 10.1007/s00401-011-0845-8
Maglio, L. E., Perez, M. F., Martins, V. R., Brentani, R. R., and Ramirez, O. A. (2004). Hippocampal synaptic plasticity in mice devoid of cellular prion protein. Brain Res. Mol. Brain Res. 131, 58–64.
Malaga-Trillo, E., and Ochs, K. (2016). Uncontrolled SFK-mediated protein trafficking in prion and Alzheimer’s disease. Prion 10, 352–361. doi: 10.1080/19336896.2016.1221873
Mallucci, G. R., Ratte, S., Asante, E. A., Linehan, J., Gowland, I., Jefferys, J. G., et al. (2002). Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 21, 202–210. doi: 10.1093/emboj/21.3.202
Mange, A., Beranger, F., Peoc’h, K., Onodera, T., Frobert, Y., and Lehmann, S. (2004). Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 96, 125–132. doi: 10.1016/j.biolcel.2003.11.007
Manson, J., West, J. D., Thomson, V., McBride, P., Kaufman, M. H., and Hope, J. (1992). The prion protein gene: a role in mouse embryogenesis? Development 115, 117–122.
Manson, J. C., Clarke, A. R., Hooper, M. L., Aitchison, L., McConnell, I., and Hope, J. (1994). 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 8, 121–127. doi: 10.1007/BF02780662
Marijanovic, Z., Caputo, A., Campana, V., and Zurzolo, C. (2009). Identification of an intracellular site of prion conversion. PLoS Pathog. 5:e1000426. doi: 10.1371/journal.ppat.1000426
Martellucci, S., Santacroce, C., Santilli, F., Manganelli, V., Sorice, M., and Mattei, V. (2020). Prion protein in stem cells: a lipid raft component involved in the cellular differentiation process. Int. J. Mol. Sci. 21:4168. doi: 10.3390/ijms21114168
Martinez, J., Sanchez, R., Castellanos, M., Makarava, N., Aguzzi, A., Baskakov, I. V., et al. (2015). PrP charge structure encodes interdomain interactions. Sci. Rep. 5:13623. doi: 10.1038/srep13623
Mercer, R. C., Ma, L., Watts, J. C., Strome, R., Wohlgemuth, S., Yang, J., et al. (2013). The prion protein modulates A-type K+ currents mediated by Kv4.2 complexes through dipeptidyl aminopeptidase-like protein 6. J. Biol. Chem. 288, 37241–37255. doi: 10.1074/jbc.M113.488650
Miranzadeh Mahabadi, H., and Taghibiglou, C. (2020). Cellular Prion Protein (PrPc): putative interacting partners and consequences of the interaction. Int. J. Mol. Sci. 21:7058.
Munoz-Montesino, C., Larkem, D., Barbereau, C., Igel-Egalon, A., Truchet, S., Jacquet, E., et al. (2020). A seven-residue deletion in PrP leads to generation of a spontaneous prion formed from C-terminal C1 fragment of PrP. J. Biol. Chem. 295, 14025–14039. doi: 10.1074/jbc.RA120.014738
Munoz-Montesino, C., Sizun, C., Moudjou, M., Herzog, L., Reine, F., Chapuis, J., et al. (2016). Generating bona fide mammalian prions with internal deletions. J. Virol. 90, 6963–6975. doi: 10.1128/JVI.00555-16
Munoz-Montesino, C., Sizun, C., Moudjou, M., Herzog, L., Reine, F., Igel-Egalon, A., et al. (2017). A stretch of residues within the protease-resistant core is not necessary for prion structure and infectivity. Prion 11, 25–30. doi: 10.1080/19336896.2016.1274851
Musiek, E. S., and Schindler, S. E. (2013). Alzheimer disease: current concepts & future directions. Mo. Med. 110, 395–400.
Nicoll, A. J., Panico, S., Freir, D. B., Wright, D., Terry, C., Risse, E., et al. (2013). Amyloid-β nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 4:2416. doi: 10.1038/ncomms3416
Nguyen, X. T. A., Tran, T. H., Cojoc, D., and Legname, G. (2019). Copper binding regulates cellular prion protein function. Mol. Neurobiol. 56, 6121–6133. doi: 10.1007/s12035-019-1510-9
Ondrejcak, T., Klyubin, I., Corbett, G. T., Fraser, G., Hong, W., Mably, A. J., et al. (2018). Cellular prion protein mediates the disruption of hippocampal synaptic plasticity by soluble tau in vivo. J. Neurosci. 38, 10595–10606. doi: 10.1523/JNEUROSCI.1700-18.2018
Pantera, B., Bini, C., Cirri, P., Paoli, P., Camici, G., Manao, G., et al. (2009). PrPc activation induces neurite outgrowth and differentiation in PC12 cells: role for caveolin-1 in the signal transduction pathway. J. Neurochem. 110, 194–207. doi: 10.1111/j.1471-4159.2009.06123.x
Park, J. Y., Jeong, J. K., Lee, J. H., Moon, J. H., Kim, S. W., Lee, Y. J., et al. (2015). Induction of cellular prion protein (PrPc) under hypoxia inhibits apoptosis caused by TRAIL treatment. Oncotarget 6, 5342–5353. doi: 10.18632/oncotarget.3028
Peters, C., Espinoza, M. P., Gallegos, S., Opazo, C., and Aguayo, L. G. (2015). Alzheimer’s Abeta interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity. Neurobiol. Aging 36, 1369–1377. doi: 10.1016/j.neurobiolaging.2014.11.019
Petit, C. S., Besnier, L., Morel, E., Rousset, M., and Thenet, S. (2013). Roles of the cellular prion protein in the regulation of cell-cell junctions and barrier function. Tissue Barriers 1:e24377. doi: 10.4161/tisb.24377
Prado, M. B., Melo Escobar, M. I., Alves, R. N., Coelho, B. P., Fernandes, C. F. L., Boccacino, J. M., et al. (2020). Prion protein at the leading edge: its role in cell motility. Int. J. Mol. Sci. 21:6677. doi: 10.3390/ijms21186677
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 Misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Premzl, M., and Gamulin, V. (2007). Comparative genomic analysis of prion genes. BMC Genomics 8:1. doi: 10.1186/1471-2164-8-1
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B. (2012). Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336, 1511–1513. doi: 10.1126/science.1222951
Puzzo, D., Piacentini, R., Fa, M., Gulisano, W., Li Puma, D. D., Staniszewski, A., et al. (2017). LTP and memory impairment caused by extracellular Abeta and Tau oligomers is APP-dependent. Elife 6:e26991. doi: 10.7554/eLife.26991
Rabinovici, G. D., and Miller, B. L. (2010). Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24, 375–398. doi: 10.2165/11533100-000000000-00000
Rammes, G., Seeser, F., Mattusch, K., Zhu, K., Haas, L., Kummer, M., et al. (2018). The NMDA receptor antagonist Radiprodil reverses the synaptotoxic effects of different amyloid-beta (Abeta) species on long-term potentiation (LTP). Neuropharmacology 140, 184–192. doi: 10.1016/j.neuropharm.2018.07.021
Rangel, A., Burgaya, F., Gavin, R., Soriano, E., Aguzzi, A., and Del Rio, J. A. (2007). Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: role of AMPA/kainate receptors. J. Neurosci. Res. 85, 2741–2755. doi: 10.1002/jnr.21215
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wuthrich, K. (1996). NMR structure of the mouse prion protein domain PrP(121-231). Nature 382, 180–182.
Roffe, M., Beraldo, F. H., Bester, R., Nunziante, M., Bach, C., Mancini, G., et al. (2010). Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. U. S. A. 107, 13147–13152. doi: 10.1073/pnas.1000784107
Rutishauser, D., Mertz, K. D., Moos, R., Brunner, E., Rulicke, T., Calella, A. M., et al. (2009). The comprehensive native interactome of a fully functional tagged prion protein. PLoS One 4:e4446. doi: 10.1371/journal.pone.0004446
Sakaguchi, S., Katamine, S., Nishida, N., Moriuchi, R., Shigematsu, K., Sugimoto, T., et al. (1996). Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 380, 528–531. doi: 10.1038/380528a0
Sakudo, A., Lee, D. C., Nakamura, I., Taniuchi, Y., Saeki, K., Matsumoto, Y., et al. (2005). Cell-autonomous PrP-Doppel interaction regulates apoptosis in PrP gene-deficient neuronal cells. Biochem. Biophys. Res. Commun. 333, 448–454. doi: 10.1016/j.bbrc.2005.05.128
Salamat, K., Moudjou, M., Chapuis, J., Herzog, L., Jaumain, E., Beringue, V., et al. (2012). Integrity of helix 2-helix 3 domain of the PrP protein is not mandatory for prion replication. J. Biol. Chem. 287, 18953–18964. doi: 10.1074/jbc.M112.341677
Salamat, M. K., Munoz-Montesino, C., Moudjou, M., Rezaei, H., Laude, H., Beringue, V., et al. (2013). Mammalian prions: tolerance to sequence changes-how far? Prion 7, 131–135. doi: 10.4161/pri.23110
Salzano, G., Giachin, G., and Legname, G. (2019). Structural Consequences of Copper Binding to the Prion Protein. Cells 8:770. doi: 10.3390/cells8080770
Santuccione, A., Sytnyk, V., Leshchyns’ka, I., and Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169, 341–354. doi: 10.1083/jcb.200409127
Sarnataro, D., Pepe, A., and Zurzolo, C. (2017). Cell Biology of Prion Protein. Prog. Mol. Biol. Transl. Sci. 150, 57–82. doi: 10.1016/bs.pmbts.2017.06.018
Schilling, K. M., Tao, L., Wu, B., Kiblen, J. T. M., Ubilla-Rodriguez, N. C., Pushie, M. J., et al. (2020). Both N-Terminal and C-Terminal histidine residues of the prion protein are essential for copper coordination and neuroprotective self-regulation. J. Mol. Biol. 432, 4408–4425. doi: 10.1016/j.jmb.2020.05.020
Schmitt-Ulms, G., Hansen, K., Liu, J., Cowdrey, C., Yang, J., DeArmond, S. J., et al. (2004). Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat. Biotechnol. 22, 724–731. doi: 10.1038/nbt969
Schmitt-Ulms, G., Legname, G., Baldwin, M. A., Ball, H. L., Bradon, N., Bosque, P. J., et al. (2001). Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314, 1209–1225. doi: 10.1006/jmbi.2000.5183
Schmitz, M., Greis, C., Ottis, P., Silva, C. J., Schulz-Schaeffer, W. J., Wrede, A., et al. (2014). Loss of prion protein leads to age-dependent behavioral abnormalities and changes in cytoskeletal protein expression. Mol. Neurobiol. 50, 923–936. doi: 10.1007/s12035-014-8655-3
Scialo, C., Celauro, L., Zattoni, M., Tran, T. H., Bistaffa, E., Moda, F., et al. (2021). The cellular prion protein increases the uptake and toxicity of TDP-43 Fibrils. Viruses 13:1625. doi: 10.3390/v13081625
Senatore, A., Colleoni, S., Verderio, C., Restelli, E., Morini, R., Condliffe, S. B., et al. (2012). Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC alpha(2)delta-1 Subunit. Neuron 74, 300–313. doi: 10.1016/j.neuron.2012.02.027
Simons, K., and Gerl, M. J. (2010). Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 11, 688–699. doi: 10.1038/nrm2977
Smith, L. M., Kostylev, M. A., Lee, S., and Strittmatter, S. M. (2019). Systematic and standardized comparison of reported amyloid-beta receptors for sufficiency, affinity, and Alzheimer’s disease relevance. J. Biol. Chem. 294, 6042–6053. doi: 10.1074/jbc.RA118.006252
Smith, L. M., and Strittmatter, S. M. (2017). Binding sites for amyloid-beta oligomers and synaptic toxicity. Cold Spring Harb. Perspect Med. 7:a024075. doi: 10.1101/cshperspect.a024075
Sparkes, R. S., Simon, M., Cohn, V. H., Fournier, R. E., Lem, J., Klisak, I., et al. (1986). Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc. Natl. Acad. Sci. U. S. A. 83, 7358–7362. doi: 10.1073/pnas.83.19.7358
Stahl, N., Borchelt, D. R., Hsiao, K., and Prusiner, S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229–240. doi: 10.1016/0092-8674(87)90150-4
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., et al. (2004). A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U. S. A. 101, 6062–6067. doi: 10.1073/pnas.0400782101
Turnbaugh, J. A., Unterberger, U., Saa, P., Massignan, T., Fluharty, B. R., Bowman, F. P., et al. (2012). The N-terminal, polybasic region of PrP(C) dictates the efficiency of prion propagation by binding to PrP(Sc). J. Neurosci. 32, 8817–8830. doi: 10.1523/JNEUROSCI.1103-12.2012
Urrea, L., Ferrer, I., Gavin, R., and Del Rio, J. A. (2017). The cellular prion protein (PrP(C)) as neuronal receptor for alpha-synuclein. Prion 11, 226–233. doi: 10.1080/19336896.2017.1334748
Walsh, D. M., and Selkoe, D. J. (2007). A beta oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x
Wang, F., Yin, S., Wang, X., Zha, L., Sy, M. S., and Ma, J. (2010). Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 49, 8169–8176. doi: 10.1021/bi101146v
Warwicker, J. (2000). Modeling a prion protein dimer: predictions for fibril formation. Biochem. Biophys. Res. Commun. 278, 646–652. doi: 10.1006/bbrc.2000.3829
Watt, N. T., Taylor, D. R., Kerrigan, T. L., Griffiths, H. H., Rushworth, J. V., Whitehouse, I. J., et al. (2012). Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 3:1134.
Westergard, L., Christensen, H. M., and Harris, D. A. (2007). The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim. Biophys. Acta 1772, 629–644. doi: 10.1016/j.bbadis.2007.02.011
Westergard, L., Turnbaugh, J. A., and Harris, D. A. (2011). A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J. Biol. Chem. 286, 44234–44242. doi: 10.1074/jbc.M111.286195
Wiatrak, B., Piasny, J., Kuzniarski, A., and Gasiorowski, K. (2021). Interactions of Amyloid-beta with Membrane Proteins. Int. J. Mol. Sci. 22:6075. doi: 10.3390/ijms22116075
Williams, W. M., Stadtman, E. R., and Moskovitz, J. (2004). Ageing and exposure to oxidative stress in vivo differentially affect cellular levels of PrP in mouse cerebral microvessels and brain parenchyma. Neuropathol. Appl. Neurobiol. 30, 161–168. doi: 10.1111/j.1365-2990.2003.00523.x
Wu, B., McDonald, A. J., Markham, K., Rich, C. B., McHugh, K. P., Tatzelt, J., et al. (2017). The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife 6:e23473. doi: 10.7554/eLife.23473
Wulf, M. A., Senatore, A., and Aguzzi, A. (2017). The biological function of the cellular prion protein: an update. BMC Biol. 15:34. doi: 10.1186/s12915-017-0375-5
Wyss-Coray, T. (2016). Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186. doi: 10.1038/nature20411
Yim, Y. I., Park, B. C., Yadavalli, R., Zhao, X., Eisenberg, E., and Greene, L. E. (2015). The multivesicular body is the major internal site of prion conversion. J. Cell Sci. 128, 1434–1443. doi: 10.1242/jcs.165472
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T. J., Chen, L., Xia, P., et al. (2012). Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U. S. A. 109, 1737–1742. doi: 10.1073/pnas.1110789109
Zanata, S. M., Lopes, M. H., Mercadante, A. F., Hajj, G. N., Chiarini, L. B., Nomizo, R., et al. (2002). Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316. doi: 10.1093/emboj/cdf325
Zeng, F., Watt, N. T., Walmsley, A. R., and Hooper, N. M. (2003). Tethering the N-terminus of the prion protein compromises the cellular response to oxidative stress. J. Neurochem. 84, 480–490. doi: 10.1046/j.1471-4159.2003.01529.x
Zhang, Y., Zhao, Y., Zhang, L., Yu, W., Wang, Y., and Chang, W. (2019). Cellular prion protein as a receptor of Toxic Amyloid-beta42 oligomers is important for Alzheimer’s disease. Front. Cell Neurosci. 13:339. doi: 10.3389/fncel.2019.00339
Keywords: PrP, Aβ, PrPC signaling, PrPC role, PrPC in CNS, Alzheimer’s disease
Citation: Panes JD, Saavedra P, Pineda B, Escobar K, Cuevas ME, Moraga-Cid G, Fuentealba J, Rivas CI, Rezaei H and Muñoz-Montesino C (2021) PrPC as a Transducer of Physiological and Pathological Signals. Front. Mol. Neurosci. 14:762918. doi: 10.3389/fnmol.2021.762918
Received: 23 August 2021; Accepted: 18 October 2021;
Published: 22 November 2021.
Edited by:
Célia Duarte Cruz, University of Porto, PortugalReviewed by:
Giuseppe Legname, International School for Advanced Studies (SISSA), ItalyCopyright © 2021 Panes, Saavedra, Pineda, Escobar, Cuevas, Moraga-Cid, Fuentealba, Rivas, Rezaei and Muñoz-Montesino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carola Muñoz-Montesino, Y2FybXVub3ptQHVkZWMuY2w=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.