
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 28 October 2021
Sec. Brain Disease Mechanisms
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.759729
This article is part of the Research Topic The Biochemistry of Amyloids in Neurodegenerative Diseases, Volume I View all 11 articles
 Somayra S. A. Mamsa1,2*
Somayra S. A. Mamsa1,2* Bruno P. Meloni2,3,4
Bruno P. Meloni2,3,4A substantial body of evidence indicates cationic, arginine-rich peptides (CARPs) are effective therapeutic compounds for a range of neurodegenerative pathologies, with beneficial effects including the reduction of excitotoxic cell death and mitochondrial dysfunction. CARPs, therefore, represent an emergent class of promising neurotherapeutics with multimodal mechanisms of action. Arginine itself is a known chaotrope, able to prevent misfolding and aggregation of proteins. The putative role of proteopathies in chronic neurodegenerative diseases such as Alzheimer’s disease (AD) warrants investigation into whether CARPs could also prevent the aggregation and cytotoxicity of amyloidogenic proteins, particularly amyloid-beta and tau. While monomeric arginine is well-established as an inhibitor of protein aggregation in solution, no studies have comprehensively discussed the anti-aggregatory properties of arginine and CARPs on proteins associated with neurodegenerative disease. Here, we review the structural, physicochemical, and self-associative properties of arginine and the guanidinium moiety, to explore the mechanisms underlying the modulation of protein aggregation by monomeric and multimeric arginine molecules. Arginine-rich peptide-based inhibitors of amyloid-beta and tau aggregation are discussed, as well as further modulatory roles which could reduce proteopathic cytotoxicity, in the context of therapeutic development for AD.
Alzheimer’s disease (AD) is a progressive, neurodegenerative disease estimated to affect over 44 million people worldwide, with a devastating impact on patients, their loved ones and caregivers, as well as vast social and economic consequences (Alzheimer’s Association, 2021). As the single biggest risk factor for AD is age, the prevalence of AD continues to increase as the average life expectancy rises; by 2050, it is predicted that over 100 million people worldwide will have the disease (GBD 2016 Dementia Collaborators, 2019). Clinically, AD is characterised by memory loss, impaired cognition, changes in mood, affect and behaviour, and a decline in the ability to carry out everyday tasks. These symptoms arise gradually, developing over the course of several years from mild cognitive impairment to dementia of increasing severity. Due to the progressive and insidious nature of AD, limited availability of tools for definitive diagnosis (Gofton and Weaver, 2006), and a lack of efficacious and disease-modifying therapeutics (Weller and Budson, 2018), AD remains a significant issue in global health.
AD is characterised by the development of proteopathies, specifically cytotoxic aggregates of amyloid-beta and tau. A range of therapeutic approaches for AD-associated proteopathies are therefore in various stages of development, including enzyme inhibitors targeting the production pathway of amyloid-beta (Kumar et al., 2018), gene silencing technologies to limit the expression of pro-aggregatory mutant tau proteins (Miller et al., 2004), kinase inhibitors aimed at preventing the pro-aggregatory hyperphosphorylation of tau, passive and active immunotherapies developed to drive protein clearance, and small molecule inhibitors of protein aggregation. These strategies are comprehensively reviewed in the literature (Hardy and De Strooper, 2017; Congdon and Sigurdsson, 2018; Pedrini et al., 2019). It remains unclear whether the proteopathies observed in AD are causative or consequential with regards to other aspects of AD pathogenesis, such as inflammation, neurovascular dysfunction, and metabolic disorders. Although the precise, mechanistic contributions of protein aggregation to the pathogenesis of AD are still contested, targeting protein aggregation remains a central priority in drug development; the recent approval of the amyloid-beta-lowering human monoclonal antibody Aducanumab by the United States Food and Drug Administration (FDA) represents the first drug to be approved by the FDA for the treatment of AD in 18 years.
Broadly, peptide-based therapeutics represent a particularly desirable class of candidates for pharmacotherapy, as peptides can be designed with high target specificity and bioactivity, and further optimised for increased safety, tolerability, efficacy and stability through sequence and structural modifications (Di, 2015; Räder et al., 2018; Evers et al., 2019). In the case of protein aggregation, the sequence and structural features of target proteins can be used to inform the rational design of peptides for binding and inhibition. Additionally, methods for screening peptide-based compounds are well-developed, including a variety of predictive in silico tools for assessing potential interactions between peptides and proteins (D’Annessa et al., 2020). As a general class, peptide-based inhibitors of protein aggregation developed for AD have been reviewed in detail by Goyal et al. (2017), Kumar and Sim (2014), and Funke and Willbold (2012). Despite their advantages, challenges in the development of anti-aggregatory peptides include lack of membrane permeability and difficulty in penetrating the blood-brain barrier. Cationic arginine-rich peptides (CARPs) demonstrate a particular ability to cross cellular membranes as well as the blood-brain barrier (Mitchell et al., 2000; Schmidt et al., 2010; Allolio et al., 2018), rendering them popular “carrier” molecules for a range of therapeutic “cargo” such as oligonucleotides, peptides and proteins (reviewed by Habault and Poyet, 2019). However, the aggregation-modulating properties of arginine itself may be favourable for peptide drugs targeting proteopathies. Here, we discuss the unique properties of arginine in modulating protein aggregation, as well as arginine-rich peptides, and peptides which have employed arginine as a key residue, in targeting the proteopathies associated with AD.
Arginine is one of two basic amino acids, alongside lysine, which is consistently protonated at physiological pH. The structure of arginine is highly unique; arginine monomers are comprised of a polar, hydrophilic head group conjugated to a hydrophobic body, and an aliphatic side chain capped with a guanidino group. At physiological pH, the carboxyl moiety of arginine is deprotonated, while protonation of both the amino group into an amide and the guanidino group into the cationic guanidinium moiety, confers the overall cationicity of the molecule to a net charge of +1. In proteins, the guanidinium moiety contributes extensively to the intra- and inter-molecular associations of arginine residues by imparting a strong capacity for electrostatic interactions such as hydrogen bonding. The structure of arginine in comparison to lysine is shown in Figure 1.
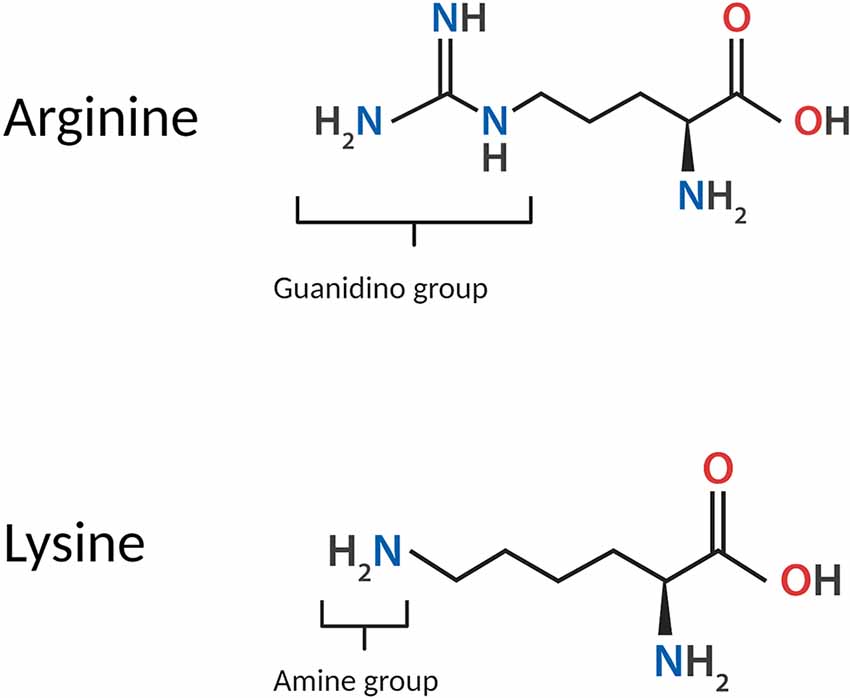
Figure 1. Structure of the arginine monomer in comparison to lysine.
Guanidinium itself is a planar molecule comprised of three amino groups conjugated to a central carbon atom; these three amino groups facilitate the formation of bidentate hydrogen bonds in three directions, enabling arginine to participate in a higher number of electrostatic interactions compared to lysine. Guanidinium also contributes to several distinctive properties of arginine and its behaviour in biological systems. The side chain of arginine is able to remain protonated under all physiological conditions and in even highly basic solutions (Xu et al., 2017), as the guanidinium moiety carries a highly stable, delocalised cationic charge with a pKa of 13.8 (Fitch et al., 2015) owing to resonance stabilisation. Due to its remarkable chemistry and emergent properties, guanidinium has been the subject of extensive study. The high stability of guanidinium is attributed to its Y-conjugated, quasi-aromatic structure with six delocalised pi electrons, which was regarded by Gund (1972) as a novel form of aromaticity. Additionally, the side chain of arginine is conferred partial hydrophobicity by the hydration structure of guanidinium; guanidinium is poorly hydrated above and below its plane, retaining only in-plane hydration (Mason et al., 2003, 2004).
Notably, the guanidinium moiety of arginine imparts a strong tendency for self-association. While it is a basic tenet of physical chemistry that ions with like charges repel each other, guanidinium ions in solution are able to overcome the Coulomb repulsion typically driving this effect and form thermodynamically stable pairs (Vazdar et al., 2018). This behaviour also extends to arginine; Shukla and Trout (2010) observed the tendency of arginine monomers to form clusters in solution, through three dominant modes of self-association: guanidinium-to-guanidinium stacking; bonding of the guanidinium moiety of an arginine monomer to the carboxylate group of another; and bonding of the arginine C-terminus to the N-terminus of an adjacent arginine monomer. The ability of arginine to form stable clusters has largely been attributed to the hydrogen-bonding ability of guanidinium (Li et al., 2010).
Computational simulations have also demonstrated like-charge pairing of di-arginine peptides through the association of their side chains, while the NH4+-containing side chains of di-lysine peptides do not exhibit any attraction to each other (Vazdar et al., 2018), evidencing the critical role of the guanidinium moiety in arginine-arginine interactions. Longer, poly-arginine peptides are also attracted to each other in solution. The self-association of polyarginine-10 (R10) peptides was demonstrated by Tesei et al. (2017) through computational modelling and experimentally confirmed by small-angle X-ray scattering experiments. The guanidinium cations of arginine side chains throughout R10 were found to associate with the corresponding guanidinium groups on pairing R10 molecules to form like-charge ion pairs, while the guanidinium cation of the ninth residue was also found to bind the negatively charged C-terminus of the adjacent R10 peptide through the formation of an intermolecular salt bridge (Tesei et al., 2017; Vazdar et al., 2018). The latter mechanism functions as an “adhesive patch” between two R10 peptides, a binding motif absent in the interactions between two molecules of the similarly cationic peptide polylysine-10 (K10), which only repels itself in solution (Tesei et al., 2017; Vazdar et al., 2018).
The formation of like-charge guanidinium pairing between arginine residues is also likely to impart stability in the tertiary structure of proteins; a number of biologically occurring protein structures are known to contain arginine residues oriented in close proximity to each other in three-dimensional space, with these steric arrangements enabled by the formation of guanidinium pairs within the structure (Tesei et al., 2017; Vazdar et al., 2018).
At physiological pH, where arginine is protonated, self-association of arginine also facilitates the formation of dynamic arginine clusters bound to protein surfaces. In silico, arginine ions bind to the surface of proteins such as lysozyme through interactions mediated by their guanidinium and carboxyl groups; while bound, the remaining guanidinium or carboxyl group of each arginine monomer interacts in a head-to-tail orientation with the carboxyl or guanidinium group of another arginine molecule in solution, which itself is able to interact with another arginine molecule through the same mechanism (Vagenende et al., 2013). Arginine clusters remain bound to proteins for a remarkably long period of time, and are thus considered to “extend” the surface of the protein they are bound to (Vagenende et al., 2013); indeed, experimental results confirm that binding of arginine ions to a protein in vitro results in a size increase of the protein detectable by chromatography (Vagenende et al., 2013). Corollary to this, it is expected that clusters of arginine can alter the physicochemical properties of the protein they are bound to, including charge and hydrophobicity, and could therefore alter their properties in vivo, such as solubility, stability and activity.
It is well-established that arginine modulates protein aggregation, acting as a molecular cosolvent and “chemical chaperone” in solution (Sharma et al., 2013). Arginine is commonly used as an additive to maintain protein stability in the biopharmaceutical industry and has conventionally been regarded as an aggregation suppressant (Golovanov et al., 2004; Arakawa et al., 2006; Ghosh et al., 2009). Shiraki et al. (2002) evaluated the effects of 15 amino acids on lysozyme under the conditions of thermal unfolding- and dilution-induced, aggregation; among them, arginine was found to be the most effective suppressor of aggregation, increasing protein solubility. Arginine has also proven particularly useful for solubilising membrane proteins otherwise prone to aggregation (Arakawa et al., 2011). Notably, the effects of arginine on proteins in solution are unique even in comparison to guanidine (Arakawa et al., 2007).
A number of experimental and computational studies have aimed to develop a mechanistic understanding of the molecular processes underlying the suppression of protein aggregation by arginine. It was previously reported by Das et al. (2007) that the self-association of arginine gives rise to hydrophobic surfaces through alignment of its methylene groups, which are then driven by the hydrophobic effect to bind the exposed hydrophobic residues of unfolded and intrinsically disordered proteins. Das et al. (2007) pertinently observed that the amyloid-beta peptide exhibits increased solubility and decreased fibril formation in the presence of arginine, and concluded, from their analyses, that these effects were due to hydrophobic surfaces of arginine clusters masking the pro-aggregatory hydrophobic residues of amyloid-beta monomers, preventing their self-association. However, while arginine does form hydrophobic columns along its crystallographic axis in crystal structures, molecular dynamics simulations subsequently performed by Shukla and Trout (2010) did not support the finding of methylene alignment in solution. While Shukla and Trout indeed confirmed the assembly of supramolecular arginine clusters, observing the formation of dimers and higher order n-mers at physiological pH, no hydrophobic surfaces were formed in these clusters through methylene alignment as proposed by Das et al. (2007).
Furthermore, Shukla and Trout (2010) also observed strong preferential interactions between arginine and the aromatic residues of their model proteins, mediated by cation-pi stacking; these observations were consistent with previous results by Arakawa et al. (2007), which demonstrated that while both arginine and guanidine displayed a strong affinity for most protein residues, their affinities were particularly strong for aromatic residues, which are generally hydrophobic. Tsumoto et al. (2004) had also previously observed that the aggregation-suppressive effects of arginine, which could not be explained through either surface tension effects or preferential interactions alone, were substantially mediated through the interactions of arginine guanidinium groups with the side chains of protein tryptophan residues. In the folded state of proteins, hydrophobic residues such as tryptophan are internalised or “buried” from the protein surface, while their externalisation in protein unfolding promotes aggregation; thus, arginine could indeed have a noticeable effect on the aggregation of unfolded or disordered proteins by binding hydrophobic surfaces and preventing their self-association, albeit through different molecular mechanisms than those proposed by Das et al. (2007).
Interestingly, however, Shukla and Trout (2010) reported that interactions between arginine and aromatic residues were insufficient to fully explain the effect of arginine on protein aggregation; in their study, the overall effect on aggregation was disproportionate when considering the relatively low number of aromatic residues comprising their model proteins. This led the authors to consider additional mechanisms, particularly the accumulation of arginine molecules on protein surfaces to form dynamic clusters; it was concluded that these clusters were able to “crowd out” the model proteins, preventing their self-association. Further research led to the classification of arginine as a “neutral crowder” in this context (Baynes et al., 2005). A comparative study by Schneider and Trout (2009) on the effects of arginine hydrochloride and guanidine hydrochloride on the aggregation of bovine serum albumin (BSA), lysozyme and α-chymotrypsinogen revealed that arginine had a unique effect: it was neither attracted to nor repelled from the protein surface. However, Schneider et al. (2011) later described a shift in the preferential interaction of arginine at high concentrations, from neutral to highly excluded, concluding that the “neutral crowder” effect was not able to completely describe the behaviour of salt forms of arginine such as arginine hydrochloride. Additionally, in silico analysis by Li et al. (2010) demonstrated the ability of arginine to solubilise hydrophobic and aromatic moieties by forming a “cage-like” solvation layer around the molecules. The mechanisms of aggregation suppression by arginine are therefore complex, involving not only interactions between arginine and proteins, but interactions between arginine and other arginine molecules; this tendency to form self-associative assemblies is an important factor in the aggregation-modulating effects of arginine.
Continued studies over the past decade, however, have demonstrated the effects of arginine on protein aggregation vary depending on its context and concentration, and are not always suppressive. Arginine potently inhibits the aggregation of lysozyme (Matsuoka et al., 2007; Ito et al., 2011), and porcine and mink growth hormones (Cirkovas and Sereikaite, 2011); monomeric arginine prevents the oligomerisation of insulin (Varughese and Newman, 2012; Březina et al., 2018; Haghighi-Poodeh et al., 2020), and di-arginine peptides exhibit even higher efficacy for suppressing insulin aggregation (Nuhu and Curtis, 2015). At physiological pH, arginine hydrochloride also suppresses the aggregation of immunoglobulin G1 (IgG1), with this effect attributed to the interactions between arginine and hydrophobic IgG1 residues (Fukuda et al., 2014). However, arginine and its derivatives arginine amide and arginine ethylester were found to alter the aggregation pathway of BSA, inducing the formation of larger BSA aggregates (Borzova et al., 2017) rather than inhibiting aggregation per se. Additionally, high concentrations of arginine suppress the aggregation of α-lactalbumin, while low concentrations are known to substantially alter its aggregation pathway, resulting in a distinctive aggregate morphology (Smirnova et al., 2013). These varied observations indicate the effects of arginine on protein aggregation are more complex and diverse than previously established. Thus, arginine is perhaps best described as a “complex molecular cosolvent”, and the molecular context of arginine greatly influences the manner in which it modulates the behaviour of proteins in solution.
Until the 1990s, the dominant theory of AD focused on the “cholinergic hypothesis” which posited the impairment of cholinergic neurotransmission as the primary cause of cognitive decline observed in patients with AD (reviewed by Francis et al., 1999). However, subsequent bodies of evidence have strongly implicated amyloid-beta deposition as a central event in the development of AD, regarded as the “amyloid cascade hypothesis” (first proposed by Hardy and Higgins, 1992; reviewed by Barage and Sonawane, 2015, and more recently by Ricciarelli and Fedele, 2017). The minimal correlation between cerebral amyloid-beta load and the severity of cognitive decline observed in patients with AD (Nelson et al., 2012), however, has led to an increased focus on the role of tauopathy in the pathogenesis of AD. Moreover, the presence of cerebral amyloid-beta plaques is not always concomitant with impaired cognition (Arboleda-Velasquez et al., 2019). The development of amyloid-beta pathology is therefore considered necessary, yet insufficient, for the progression of AD. Broad evidence from laboratory, preclinical and clinical studies suggests that amyloid-beta aggregation can also drive the progression of tauopathy (reviewed by Stancu et al., 2014; Hanseeuw et al., 2019), which is increasingly believed to play a key role in cognitive decline (Nelson et al., 2012; Di et al., 2016; Digma et al., 2019). The precise, mechanistic roles of these proteopathies in the aetiology and pathogenesis of AD are the subject of extensive research and debate; however, pathogenic protein aggregation remains strongly implicated in the disease (Lovestone and McLoughlin, 2002; Thal and Fändrich, 2015; Jouanne et al., 2017; Gandhi et al., 2019; Johnson et al., 2019). We will briefly outline the characteristics of amyloid-beta and tau and their aggregation pathways.
Amyloid-beta peptides are 38- to 43-residue peptides resulting from the sequential cleavage of amyloid precursor protein (APP) by the secretase family of enzymes (Crescenzi et al., 2002). APP itself is heterogeneous, ranging from 110 to 140 kDa, with three major isoforms (695, 751, and 770 residues in length) determined by the splicing pattern of its expression product, and is subjected to a range of post-translational modifications including sulfation, phosphorylation, and N- and O-linked glycosylation (Zheng and Koo, 2011). The generation of amyloid-beta results from a cleavage pathway of APP commonly referred to as the amyloidogenic pathway. This begins with the cleavage of APP by beta-secretase (BACE1), generating two fragments: a C-terminal fragment of APP referred to as C99, and a secreted, soluble N-terminal fragment termed sAPP-β. C99 is subsequently bound by the gamma-secretase complex, comprised of four protein subunits: presenilin (PSEN; PSEN1/PSEN2 isoforms), presenilin enhancer (PEN), Nicastrin, and APH-1. Processing of C99 by gamma-secretase results in a series of sequentially shorter cleavage products, until the amyloid-beta peptide is released (O’Brien and Wong, 2011). In the central nervous system, amyloid-beta is predominantly secreted by neurons and astrocytes into the extracellular space of the brain and physiologically cleared by the vascular system and cerebrospinal fluid. In AD, clearance mechanisms for amyloid-beta are impaired, leading to accumulation in the brain parenchyma (Ramanathan et al., 2015). Multiple amyloid-beta isoforms have been observed in the brain tissue of patients with AD, including amyloid-beta1–40 (Aβ40), amyloid-beta1–42 (Aβ42), and amyloid-beta1–43 (Aβ43; Welander et al., 2009); among them, Aβ42 is the primary constituent of neuritic plaques observed in end-stage AD (O’Brien and Wong, 2011).
While physiological roles of amyloid-beta peptides have been identified (Pearson and Peers, 2006; Morley et al., 2019), the aggregation of amyloid-beta monomers has causally been associated with neuronal toxicity (Yankner and Lu, 2009; Pauwels et al., 2012; Prasansuklab and Tencomnao, 2013; Carrillo-Mora et al., 2014). Amyloid-beta aggregates occur in a variety of assemblies, from low molecular weight oligomers (including dimers, trimers, tetramers, and pentamers) to higher molecular weight oligomers (hexamers, nonamers, dodecamers; Wolff et al., 2017), protofibrils, and fibrils, as well as amorphous aggregates (Jiang et al., 2012). in vitro, the formation of these aggregate species is affected by various factors including the presence and concentration of specific ions, such as metals, as well as pH and temperature (Valerio et al., 2008; Jiang et al., 2012; Bin et al., 2013; Faller et al., 2013; Bhowmik et al., 2014; Zhao and Ai, 2018). Amyloid-beta aggregation is illustrated in Figure 2.
Figure 2. Amyloid-beta aggregation in Alzheimer’s disease (AD).
Multiple regions of the amyloid-beta sequence are considered pro-aggregatory. A report by Liu et al. (2004) described six fragments of amyloid-beta1–40 (Aβ40), formed by residues Aβ401–28, Aβ4012–28, Aβ4017–28, Aβ4010–20, Aβ4025–35, and Aβ4017–40, which had a pro-aggregatory effect on the full-length peptide. Hsu et al. (2018) later identified several key residues involved in amyloid-beta aggregation: H14, E22, D23, G33, G37, and G38. Enache et al. (2018) later characterised the influence of key hydrophobic regions, particularly the well-characterised hydrophobic core domain KLVFF, as well as the C-terminal hydrophobic sequence IIGLMVGGVV and a histidine-containing tetrad, VHHQ, on amyloid-beta aggregation through atomic force microscopy and voltammetry. New insights into the aggregation pathway of amyloid-beta were also attained more recently by Nirmalraj et al. (2020) at nanometer resolution. Aβ42 was shown to aggregate faster than Aβ40 at all stages of assembly. Additionally, the study by Nirmalraj et al. (2020) confirmed that oligomers are not simply an intermediate aggregation species along a linear path to the formation of mature fibrils; oligomers were present even at timepoints where mature fibrils were detected. The pathological effects of different aggregate assemblies have been the subject of extensive research and debate (reviewed by Di Carlo, 2010), however, it is widely accepted that soluble oligomers are a highly cytotoxic species, preceding the development of end-stage neuritic plaques (Resende et al., 2008; Larson and Lesné, 2012; Sengupta et al., 2016; Chen et al., 2017).
Tau proteins are encoded by the MAPT gene on chromosome 7 (Neve et al., 1986); the pre-mRNA product of MAPT undergoes a range of alternative splicing events, resulting in transcript variants encoding six protein isoforms found in the central nervous system (D’Souza and Schellenberg, 2000). These isoforms range from 352 to 441 residues in length and are comprised of four distinct functional domains: the N-terminal projection domain (residues 1–165), proline-rich region (PRR; residues 166–242), microtubule-binding domain (MTBD; 243–367), and C-terminal domain (368–441). The N-terminal domain and MTBD are the primary sites affected by alternative splicing, and the resulting isoforms can be categorised by the number of repeat regions comprising the MTBD: 3R (three-repeat) and 4R (four-repeat) tau (D’Souza and Schellenberg, 2000). The absence of an insert, or the presence of one or two inserts, in the N-terminal projection domain also demarcates each of the isoforms as 0N, 1N or 2N, respectively; the six isoforms are thus referred to as 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, and 2N4R, depending on their number of N-terminal inserts and MTBD repeats. In the adult human brain, 3R and 4R isoforms of tau are present in equal abundance, and tau aggregates found in AD are comprised of both three- and four-repeat isoforms (Kolarova et al., 2012).
Physiologically, tau is involved in the assembly and stabilisation of microtubules (Weingarten et al., 1975), as well as in the regulation of intracellular trafficking (Vershinin et al., 2007; Dixit et al., 2008). These activities are regulated by post-translational modifications of the tau protein, which include phosphorylation, glycosylation, deamidation, oxidation, nitration, glycation, and ubiquitination. These modifications to tau and their physiological consequences have previously been reviewed in detail by Avila et al. (2004). Most pertinent to the development of tau aggregates found in AD is tau phosphorylation, which negatively regulates the binding of tau to microtubules and the cellular membrane (Brandt et al., 1995). Tau phosphorylation can occur at any of the multiple serine, threonine and tyrosine residues found on the protein (Williamson et al., 2002; Noble et al., 2013). Phosphorylation of tau induces a conformational change in the protein, reducing its ability to stimulate microtubule assembly (Jameson et al., 1980). In AD, phosphorylation of tau is observed to be over three times higher than in physiological conditions (Köpke et al., 1993). Hyperphosphorylated tau detaches from microtubules, aggregating in the cytosol, as illustrated in Figure 3. Tau acetylation, which similarly increases the negative charge of tau, has also been associated with AD (Irwin et al., 2012; Lucke-Wold et al., 2017).

Figure 3. Tauopathy in Alzheimer’s disease.
Tau aggregation primarily involves a shift in the conformation of two hexapeptide regions in the protein, VQIVYK and VQIINK (also referred to as PHF6 and PHF6* respectively), from a random coil to a beta sheet structure (von Bergen et al., 2005; Li and Lee, 2006; Eschmann et al., 2015). Oligomers of tau aggregate further to form paired helical filaments (PHFs) and, subsequently, the neurofibrillary tangles (NFTs) observed in end-stage AD. Tau multimers in a variety of conformations including straight filaments, twisted ribbons, and small oligomeric aggregates have also been observed in the brain tissue of patients with AD (Grundke-Iqbal et al., 1986; Meraz-Ríos et al., 2010). Recent, comprehensive reviews by Niewiadomska et al. (2021) and Shafiei et al. (2017) have established oligomeric assemblies of tau as cytotoxic species.
Through phage display, Kawasaki et al. (2010) identified libraries of three- and four-residue peptides capable of inhibiting (Aβ42) oligomerisation, and found arginine-containing peptides were enriched in both libraries; arginine comprised two of three, and two-to-three of four, residues in the majority of peptides identified, including RRRA, RRRL, RFRK, RRY, and RPR. Kawasaki et al. (2011) also found that while monomeric arginine and di-arginine were able to bind Aβ42, they did not have significant effects on the formation of Aβ42 oligomers, leading the authors to conclude that these molecules were too small to effectively inhibit Aβ42 oligomerisation. Together with the higher binding affinity and inhibitory effect of SRPGLRR in comparison to the three- and four-residue peptides, Kawasaki et al. (2011) concluded the size of the compound could be an important factor in the development of inhibitory peptides for amyloid-beta aggregation.
Notably, among the arginine-rich peptides screened by Kawasaki et al. (2011), RRRA and RRRL were found to be comparatively weaker, yet still effective, inhibitors of Aβ42 oligomerisation than RFRK. While the authors concluded that the higher efficacy of RFRK as an aggregation inhibitor was likely due to the phenylalanine residue of RFRK binding a phenylalanine residue of amyloid-beta, thereby strengthening the interaction between the inhibitor and amyloid-beta, an arginine residue in the second position of either inhibitory peptide could also bind a phenylalanine residue of Aβ42 through its aliphatic side chain, and ability to form cation-pi interactions with aromatic residues. It is, therefore, possible that the increased efficacy of RFRK was rather due to the cationicity of the lysine residue in RFRK imparting a stronger capacity for inhibition of Aβ42 aggregation than the terminal alanine and leucine residues of RRRA and RRRL, respectively. Regardless, the efficacy of RRRA, RRRL, RFRK, and SRPGLRR as inhibitors of Aβ42 oligomerisation led Kawasaki et al. (2011) to consider arginine an important residue for inhibiting Aβ42 aggregation.
Further work by Kawasaki and Kamijo (2012) demonstrated that two additional hybrid peptides combining arginine residues with organic moieties, Arg-Arg-7-amino-4-trifluoromethylcoumarin (RR-AFC) and Arg-Arg-thiobenzyl ester (ZRR-SBzl), were also able to inhibit amyloid-beta aggregation. Recent in silico characterisation of RR-AFC conducted by Barale et al. (2019) using molecular dynamics simulation indeed evidenced the critical role of arginine residues in the destabilisation of amyloid-beta protofibrils; in particular, the arginine residues of RR-AFC were found to bind amyloid-beta protofibrils through hydrogen bonding via their guanidinium moieties.
Many peptide-based candidates rationally designed to inhibit amyloid-beta aggregation have been derived from the sequence of amyloid-beta itself, exploiting its propensity for self-aggregation to facilitate targeted binding (Watanabe et al., 2002; Austen et al., 2008; Viet et al., 2011; Arai et al., 2014; Kino et al., 2015; Kumar et al., 2015; Cheng et al., 2017; Lu et al., 2019; Jokar et al., 2020). The majority of these peptides contain elements derived from amyloidogenic sequences involved in beta-sheet formation (Moss et al., 2003), such as the KLVFF (amyloid-beta16–20) and GGVVIA (amyloid-beta37–42) motifs, and are thus termed beta-sheet breakers (Soto et al., 1998). The design of these peptides generally includes an amyloid-derived sequence for target binding, conjugated to a flexible hydrophilic or cationic element to disrupt aggregation. One such inhibitor was a KLVFF-derived retro-inverso peptide, RI-OR2 (rGffvlkGr; lower case denoting D-amino acids), which effectively reduced Aβ42 aggregation in vitro (Parthsarathy et al., 2013). As RI-OR2 was not designed to penetrate the blood-brain barrier, it was subsequently fused to the arginine-rich cell-penetrating TAT peptide resulting in the hybrid peptide, RI-OR2-TAT (Ac-rGffvlkGrrrrqrrkkrGy-NH2; net charge +11.0). Intriguingly, experimental results from Parthsarathy et al. (2013) indicated that RI-OR2-TAT not only had beneficial effects on the aggregation and cytotoxicity of Aβ42 aggregation in cell models and in a transgenic mouse model of amyloidogenesis (APPSwe/PS1ΔE9) in vivo, but also demonstrated greater efficacy for inhibiting the aggregation of Aβ42 in vitro compared to RI-OR2. The inhibitory effects of RI-OR2-TAT on Aβ42 aggregation were evident at even the earliest detectable stages of Aβ42 oligomerisation. Surface plasmon resonance (SPR) experiments conducted by Parthsarathy et al. (2013) further indicated RI-OR2-TAT had a higher binding affinity for Aβ42 than RI-OR2 alone. It is likely that the increased efficacy of RI-OR2-TAT as an inhibitor of Aβ42 aggregation was mediated by the arginine content of TAT, specifically through: (a) greater capacity for hydrogen bonding imparted by the arginine residues of the TAT peptide, facilitating a higher number of electrostatic interactions between the inhibitory peptide and Aβ42; (b) increased cationic charge of the RI-OR2-TAT peptide conferred by the arginine-rich TAT component, driving further charge-based repulsion of bound Aβ42 monomers; and (c) the increased length of the inhibitory peptide likely causing greater steric and/or electrostatic interference between monomers of Aβ42, preventing their self-association. Importantly, these findings suggested that arginine-rich peptides could themselves have a mechanistic role in modulating the development of amyloid-beta aggregates beyond their use as “carrier” peptides alone.
Cheng et al. (2017) later developed a bipartite peptide comprised of polyarginine-8 (R8) conjugated to the sequence of amyloid-beta25–35 (RRRRRRRRGSNKGAIIGLM; net charge +9.0) which effectively reduced cerebral accumulation of amyloid-beta and ameliorated cognitive deficits in the APP/PS1 double transgenic mouse model of AD. The design of the peptide was comprised of the amyloid-beta25–35 self-recognition element to facilitate binding to amyloid-beta monomers, with the polyarginine component intended to drive charge-based repulsion of the bound monomers from each other, preventing their ability to aggregate.
More recently, Henning-Knechtel et al. (2020) designed two cell-penetrating peptides comprised of a hydrophobic signal sequence (MLRTKDLIWTLFFLGTAVS; NCAM1) conjugated to a polycationic sequence (KKRPKP; PrP23–28) or an amyloid-derived self-recognition motif sequence (KLVFF; Aβ16–20) via a lysine residue to increase the cationic charge of the peptide. The NCAM1-PrP peptide carried a higher overall charge of +6 compared to the +4 charge of NCAM1-Aβ, resulting from a higher proportion of cationic residues. While both NCAM1-PrP and NCAM1-Aβ were found to inhibit the aggregation of Aβ42 in vitro, NCAM1-PrP achieved effective inhibition at substoichiometric concentrations, while an equimolar concentration of NCAM1-Aβ to amyloid-beta was required. As amyloid-beta is negatively charged at physiological pH (net charge −2.7), the increased efficacy of NCAM1-PrP as an aggregation inhibitor compared to the KLVFF-containing NCAM1-Aβ peptide was attributed to its higher proportion of cationic residues, reportedly essential in stabilising the dimers formed between NCAM1-PrP and amyloid-beta. Additionally, although NCAM1 itself was able to bind amyloid-beta, the addition of a polycationic sequence was required to exert a modulatory effect on aggregation, suggesting the overall charge of an inhibitory peptide may be as important for preventing the aggregation of amyloid-beta as structural specificity. Indeed, Henning-Knechtel et al. suggested that the NCAM1-Aβ peptide could be optimised through further addition of cationic residues, thus it is conceivable that higher arginine content could increase its efficacy. These results also support the idea that electrostatic interactions between an inhibitory peptide and amyloid-beta are sufficient to drive target binding in the absence of amyloid-beta-derived sequence motifs, as was demonstrated by Parthsarathy et al. (2013).
The effects of a short peptide comprised entirely of arginine, polyarginine-9 (R9), were investigated by Fonar et al. (2018) in the triple transgenic (3xTg) mouse model of AD which harbours FAD mutations inducing the development of both amyloid-beta and tau pathology. 3xTg mice treated with R9 displayed a trend toward lower levels of cerebral amyloid-beta compared to untreated controls, as well as a trend toward reduced levels of oligomeric amyloid-beta in hippocampal lysates, however, these findings did not reach statistical significance. The effects of longer polyarginine peptides on amyloid-beta pathology may be an interesting area for further research; it is possible that increased cationic charge imparted by longer stretches of arginine could impart a greater capacity for electrostatic repulsion of amyloid-beta monomers, thereby inhibiting aggregation. Additionally, D-enantiomeric polyarginine peptides could potentially exert a stronger effect due to the increased proteolytic stability of D-amino acids (Feng and Xu, 2016) in comparison to L-isoforms.
Among the most promising aggregation inhibitors targeting amyloid-beta is the cationic arginine-rich RD2 peptide (ptlhthnrrrrr; +6.2 net charge), demonstrated to eliminate amyloid-beta oligomers in vivo, and rescue amyloid-beta pathology and cognitive deficits in a transgenic mouse model of AD (Schemmert et al., 2019; Zhang et al., 2019). RD2 is a rationally-optimised derivative of an arginine-rich precursor peptide, D3 (rprtrlhhrnr; +5.2 net charge), which was initially identified through mirror- image phage display (Wiesehan et al., 2003) and also shown to be effective in targeting amyloid-beta (van Groen et al., 2008). A series of C-terminally amidated D3 analogues were developed, outlined in Table 1, to identify a candidate for further development (Klein et al., 2017). Notably, increasing the charge of the inhibitory peptide was associated with increased binding affinity to amyloid-beta (Ziehm et al., 2016).
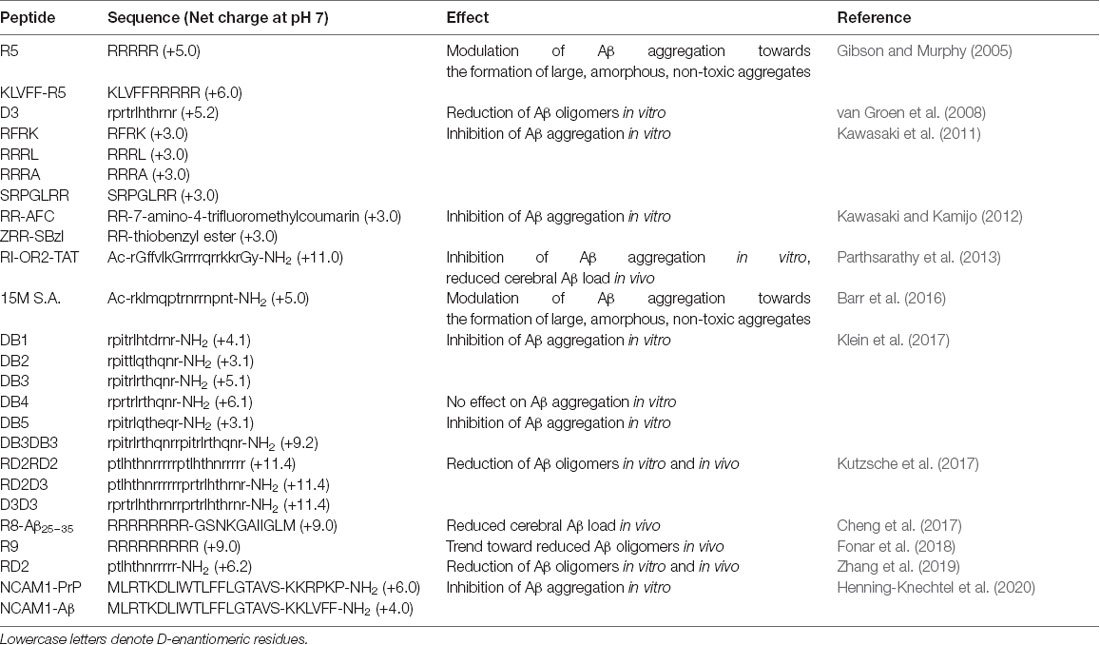
Table 1. Cationic arginine-rich peptides with modulatory effects on amyloid-beta aggregation.
Continued characterisation of D3 analogues indicated RD2 had the most favourable pharmacokinetic profile with regards to half-life and oral bioavailability (Leithold et al., 2016). Indeed, the initial success of RD2 in phase I clinical trials has led to its ongoing development as a potential therapeutic candidate for AD (Elfgen et al., 2021). The molecular mechanisms of both D3 and RD2 were investigated in detail through computational studies and experimental verification by Olubiyi et al. (2014), revealing that both D3 and RD2 bind amyloid-beta with high affinity, and reduce β-sheet formation, largely due to electrostatic interactions between the arginine residues of the inhibitory peptides and key anionic residues (E11, E22 and D23) of amyloid-beta.
Peptide-based inhibitors of tau aggregation have focused on the PHF6 and PHF6* regions of the MTBD, which are well-characterised as pro-aggregatory sequences (Li and Lee, 2006; Eschmann et al., 2015). Ralhan et al. (2017) reported inhibition of PHF6 aggregation by the poly-arginine-6 (R6) and R8 peptides, as well as the reversal of PHF6 fibril formation by R6 which acted as a beta-sheet breaker, inducing the disassembly of PH6 fibrils into sparser aggregates. While early peptides targeting the PHF6 region have shown effective inhibition of the isolated hexapeptide in vitro, these peptides were unable to prevent the aggregation of full-length tau (Sievers et al., 2011; Zheng et al., 2011; Seidler et al., 2018), which additionally contains the amyloidogenic PHF6* sequence considered to be a significant driver of tau aggregation. Seidler et al. (2018) subsequently developed new inhibitory peptides through combinatorial mutagenesis of the VQIINKKLD motif. These peptides are comprised of 10 residues, outlined in Table 2, and able to prevent aggregation of the full-length tau protein. Importantly, to achieve this, the inclusion of an arginine residue was required at the ninth position in order to prevent self-association of one of the two main interfaces involved in full-length tau aggregation: the KKL region of the tau KVQIINKKLD sequence, termed “interface B”.

Table 2. Polyarginine and arginine-rich peptides with modulatory effects on tau aggregation.
Longer polyarginine peptides (R32 to R96) were demonstrated by Nadimidla et al. (2017) to inhibit the aggregation of both PHF6 and the PHF6*-containing amyloidogenic tau fragment GKVQIINKLDL, as well as aggregation of the full-length mutant tau protein P301L, which is found in human tauopathies (Cosacak et al., 2017). The aggregation of PHF6 and PHF6* peptides was also inhibited, albeit to a lesser degree, by the cationic polymer polyethylenimine (PEI), implicating the positive charge of these compounds as a key factor in suppressing tau aggregation. Tau is aggregated in the presence of heparin in vitro to model the proaggregatory role of polyanionic molecules such as heparan sulfate proteoglycans found in NFTs in vivo (Fichou et al., 2018; Maïza et al., 2018); it is likely that, in addition to interacting electrostatically with negatively charged stretches of tau itself, strongly cationic compounds such as polyarginine could inhibit the seeding and nucleation of tau aggregation by complexing with essential polyanionic cofactors involved in the aggregation process. Furthermore, post-translational modifications which increase the negative charge of tau in vivo, such as phosphorylation, acetylation and nitration, could potentially increase the affinity of CARPs for tau, and their ability to modulate its aggregation.
Zhang et al. (2020) subsequently reported an arginine-rich, D-enantiomeric peptide termed p-NH (nitmnsrrrrnh; net charge +4.1), discovered through phage display, was able to inhibit tau aggregation in vitro and reduce tauP301S levels in transgenic mice. In vitro, p-NH was shown to inhibit PHF6 aggregation in a dose-dependent manner; this effect was optimal at an 8-fold molar excess of the peptide over PHF6, but remained effective at equimolar and substoichiometric concentrations. Remarkably, p-NH was also able to reverse aggregation when added to preformed PHF6 fibrils. Importantly, Zhang et al. (2020) reported p-NH was able to interact directly with PHF6 through hydrogen bond formation.
More recently, Aggidis et al. (2021) reported a D-enantiomeric, retro-inverso peptide (rrrrrrrrGpkyk(ac)iqvGr; net charge +11.0) based on the PHF6 sequence, termed RI-AG03, able to prevent tau aggregation. As a retro-inverso peptide, RI-AG03 was optimised for increased proteolytic stability from the AG03 peptide, itself selected from a family of peptides designed to inhibit tau aggregation (Aggidis, 2019) which are summarised in Table 2. The majority of the inhibitory peptides developed by Aggidis (2019) significantly reduced the aggregation of recombinant human tau Δ1–250, which exhibits faster aggregation kinetics than full-length tau protein, in a dose-dependent manner. Importantly, Aggidis (2019) reported that increasing arginine content of the inhibitory peptides, up to five residues, increased tau Δ1–250 solubility. An interesting observation is that R8 alone was unable to prevent tau Δ1–250 aggregation, in contrast to prior results indicating the effective inhibition of PHF6 aggregation by R6 and R8 (Ralhan et al., 2017); taken together, these effects are consistent with observations reported by Seidler et al. (2018) that PHF6* aggregation is a significant driver of aggregation in larger tau fragments.
As the precise, mechanistic roles of amyloid-beta and tau aggregates in the pathogenesis of AD remain unclear, CARPs could also be beneficial by mitigating the cellular effects of cytotoxic amyloids, rather than preventing the formation of aggregates per se. As comprehensively detailed in a review by Meloni et al. (2020), CARPs have favourable biological effects in models of neuronal injury and disease, such as stroke and traumatic brain injury, through multimodal mechanisms of action. Here, we will discuss how the bioactive properties of CARPs could be beneficial specifically in the context of proteopathic cytotoxicity associated with AD.
There are multiple potential mechanisms for CARPs to confer cytoprotection against toxic aggregates of amyloid-beta and tau. As described, the predominant approach has focused on inhibiting aggregation; CARPs which have demonstrated the ability to reduce amyloid-beta-induced cytotoxicity in vitro by inhibiting aggregation include those developed by Kawasaki et al. (2011), Parthsarathy et al. (2013), Cheng et al. (2017) and Henning-Knechtel et al. (2020). Additionally, Nadimidla et al. (2017), whose work reported inhibition of tau aggregation through high molecular-weight polyarginine peptides, demonstrated increased rates of cell survival in cultures exposed to cytotoxic concentrations of tau when treated with polyarginine peptides.
However, as the cytotoxicity of amyloid-beta aggregates varies depending on the aggregate conformation, inhibiting aggregation is not the only means of decreasing the concentration of cytotoxic aggregate species. CARPs, through their ability to electrostatically bind monomers of amyloid-beta, may be able to bind and stabilise monomers in conformations favouring non-oligomeric aggregation pathways. Gibson and Murphy (2005) previously reported that polyarginine-5 (R5) and KLVFF-R5 peptides broadly increased the aggregation of amyloid-beta1–42 overall, resulting in the formation of larger amyloid-beta1–42 aggregates with lower cytotoxicity. Barr et al. (2016) reported that a 15-residue cationic peptide, with 20% of its sequence comprised of arginine residues, reduced the formation of soluble, cytotoxic amyloid-beta1–42 oligomers by driving the aggregation pathway toward the formation of larger, amorphous aggregates; these amorphous aggregates were also observed to have lower cytotoxicity than the oligomers formed by amyloid-beta1–42 alone. A potential therapeutic mechanism for peptides targeting amyloid-beta could therefore involve the reduction of cytotoxic, soluble oligomeric amyloid-beta species by driving the aggregation pathway toward the formation of non-toxic insoluble aggregate species, rather than aiming to inhibit aggregation.
CARPs may be able to prevent the cellular conditions favouring the formation of cytotoxic aggregates. For example, a proteomic study of neuronal cultures treated with the polyarginine-18 peptide (R18) showed overall levels of tau were significantly decreased (MacDougall et al., 2019a, supplementary information), however the mechanisms underlying this effect are unclear. Arginine is also able to scavenge free radicals and mitigate oxidative stress (Wascher et al., 1997; Haklar et al., 1998), an ability that extends to polyarginine peptides (Marshall et al., 2015). Oxidative stress has a synergistic relationship with amyloid-beta pathology (reviewed in detail by Cheignon et al., 2018), as well as tau phosphorylation and polymerisation (reviewed by Zhao and Zhao, 2013). Reducing oxidative stress could potentially therefore aid, indirectly, in preventing the formation of aggregate species. Additionally, as oxidative stress is believed to be one of the main mechanisms through which amyloid-beta and tau aggregates induce toxic effects (Butterfield et al., 2013), there may be a potential role for arginine and CARPs in cytoprotection through the indirect, downstream effects of rescuing oxidative damage, an area which requires further investigation.
CARPs may also be able to prevent the development of tauopathy through inhibition of tau hyperphosphorylation, which is pro-aggregatory. The p-NH peptide developed by Zhang et al. (2020) was reported to significantly reduce tau phosphorylation at Thr181, Ser202, Thr231, Ser396, and Ser404 in the human neuroblastoma N2a cell line. More recently, a CARP developed by Kondo et al. (2021) termed TAT-7H (YGRKKRRQRRR-HHHHHHH; net charge +8.7) was shown to inhibit the phosphorylation of Ser202 and Thr205 in a neuronal cell line differentiated from human tauP301S double knock-in induced pluripotent stem cells.
Protein aggregates in AD also induce cytotoxicity through mitochondrial damage; tau oligomers are believed to induce mitochondrial damage via their detrimental effects on intracellular transport networks (Shafiei et al., 2017), while accumulation of amyloid-beta at the mitochondrial membranes induces mitochondrial damage through mechanisms including aberrant interactions with mitochondrial proteins, generation of reactive oxygen species, and disruption of the electron transport chain (reviewed in detail by Reddy et al., 2010, and Chen and Yan, 2007). The ability of CARPs to preserve mitochondrial function has been discussed at length in a review by MacDougall et al. (2019b); thus, another area for future research would be the potential for CARPs to preserve mitochondrial function and mitigate cytotoxicity in models of amyloid-beta and tau-induced neuronal injury. These mechanisms are summarized in Figure 4.
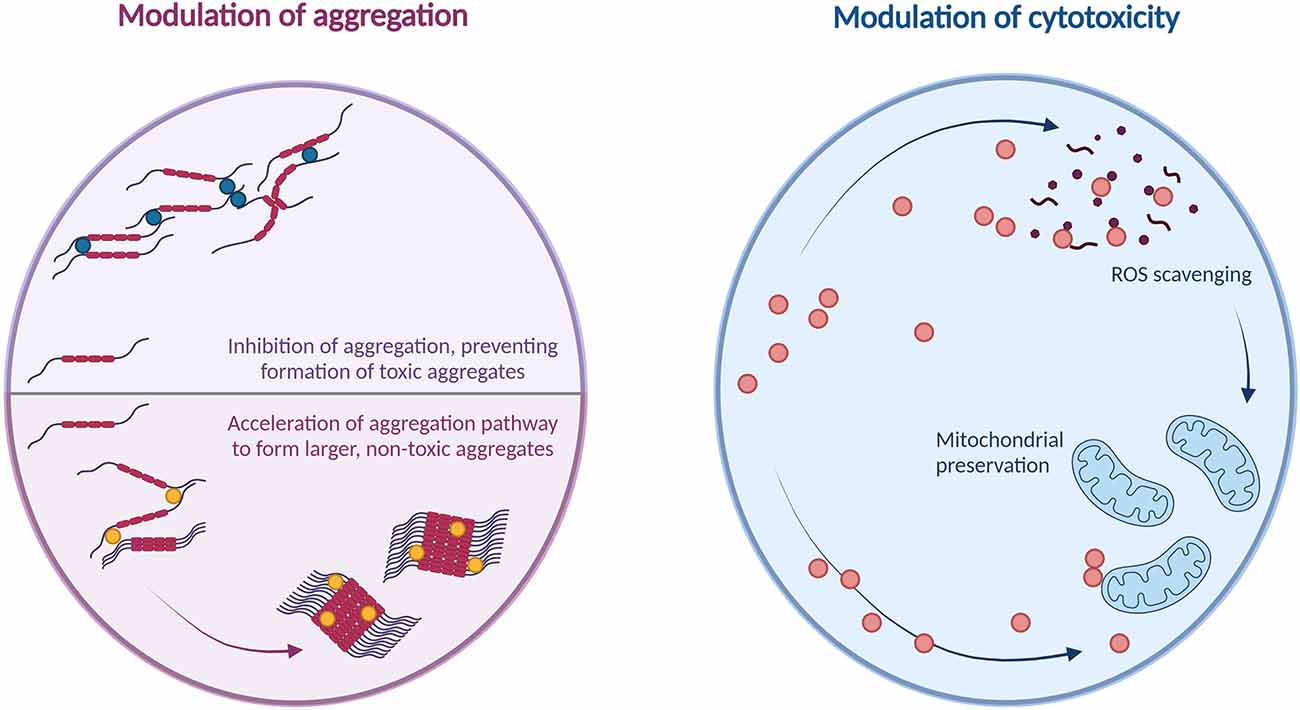
Figure 4. Summary of potential cytoprotective mechanisms of arginine and cationic arginine-rich peptides against amyloids.
It is also possible that arginine-rich peptides, through their extensive capacity for electrostatic interactions, may be able to bind pre-formed aggregates and prevent their deleterious cellular effects. It was recently discovered that fibrillar formations of tau exhibit an altered interactome than tau monomers and nano-aggregates. Ferrari et al. (2020) demonstrated that tau fibrils preferentially interact with a set of proteins containing disordered stretches significantly enriched for arginine residues, with these aberrant interactions mediated by pi-pi stacking interactions. Crucially, Ferrari et al. (2020) found that the replacement of arginine residues in these interacting proteins with lysine precluded their interactions with tau fibrils. The proteins comprising the altered interactome of tau fibrils were identified as predominantly belonging to three functional clusters: RNA-binding proteins, regulators of protein phosphorylation, and microtubule-associated proteins. It was inferred that these interactions may, in part, be responsible for the cytotoxicity of tau aggregates in AD (Ferrari et al., 2020). It is possible that exogenous arginine-rich compounds may be able to prevent cytotoxicity by competitively binding to tau fibrils through the same mechanism, preventing tau fibrils from associating with these aberrant interactors. Thus, the potential effects of CARPs on the cytotoxicity of preformed tau aggregates presents an interesting area for future research.
Monomeric arginine has demonstrated efficacy in preventing the aggregation of alpha-synuclein. While alpha-synuclein aggregation is predominantly associated with Parkinson’s disease (reviewed comprehensively by Fields et al., 2019), alpha-synuclein has also been implicated in AD (Kotzbauer et al., 2001; Crews et al., 2009; Twohig and Nielsen, 2019). Arginine was found to inhibit rotenone-induced aggregation of alpha-synuclein in vitro, even inhibiting further aggregation even when added past the stage of initial nucleation (Shristi, 2014). The inhibitory effects of arginine on alpha-synuclein aggregation were further confirmed in detail by Ghosh et al. (2018), who demonstrated arginine-based inhibition of alpha-synuclein aggregation both in vitro and in situ in live cells. Ghosh et al. (2018) also evidenced the cytoprotective effects of arginine on cultures treated with pre-aggregated alpha-synuclein. Interestingly, the use of lysine, a similarly cationic amino acid, was associated with increased alpha-synuclein aggregation in vitro, while guanidinium hydrochloride was associated with decreased viability of cells treated with aggregated alpha-synuclein. These results indicate that while the guanidinium moiety of arginine is essential to its activities, the overall properties of arginine rather than either its general cationicity or the guanidinium moiety in isolation, are key to its anti-aggregatory and cytoprotective effects in the context of alpha-synuclein aggregation.
Recently, the CARP RD2RD2 (ptlhthnrrrrrptlhthnrrrrr; +10.4 charge) was investigated as a potential therapeutic candidate for amyotrophic lateral sclerosis (ALS) in a mouse model of mutant superoxide dismutase 1 (SOD1) expression (Post et al., 2021). Administration of RD2RD2 was associated with reductions in motor deficits and neuroinflammation, although the precise molecular mechanisms underlying these effects remain to be elucidated.
Minakawa and Nagai (2021) recently discussed arginine as a modulator of aggregatory proteins containing polyglutamine (polyQ) stretches, a pathogenic feature of neurodegenerative diseases such as Huntington’s disease (HD) and spinocerebellar ataxias (SCAs). Oligomers of polyQ proteins, as well as polyQ protein monomers which are enriched for beta-sheet structure, have previously been associated with cytotoxicity (Kayed et al., 2003; Miller et al., 2011). When added to polyQ proteins in vitro, monomeric arginine was found to prevent the aggregation of polyQ proteins, including the initial transition from alpha-helical to beta-sheet-enriched monomers, subsequent oligomerisation, and the seeding of aggregation by pre-formed aggregates (Minakawa et al., 2020). Arginine, arginine methyl ester and arginine ethyl ester were also shown by Singh et al. (2019) to prevent aggregation of a polyQ-containing Huntington exon 1 protein (mHTTex1) in vitro, and rescue motor deficits in a model of HD in vivo. Minakawa et al. (2020) also investigated the effects of arginine administration in vivo and found motor deficits were rescued in multiple mouse and invertebrate models of SCA. Notably, arginine was able to rescue motor deficits even past the stage of initial symptom onset in a SCA1 mouse model (Minakawa et al., 2020).
Aggregation of an mHTT protein was also suppressed by a hybrid peptide, 8R10Q (RRRRRRRRQQQQQQQQQQ; net charge +8.0) designed by He et al. (2019) through a principle similar to the bipartite peptide designed for amyloid-beta aggregation by Cheng et al. (2017): inclusion of a self-recognition component (a ten-residue stretch of glutamine), conjugated to a polyarginine component to drive charge-based repulsion of bound monomers as well as increase solubility and membrane permeability.
The effects of arginine and its derivatives on polyQ aggregation and cytotoxicity are consistent with the advantages conferred by the structure and cationicity of arginine in amyloid-beta and tau-targeting peptides, as well as the effects of CARPs including polyarginine peptides on glutamic acid-induced excitotoxicity (reviewed comprehensively by Meloni et al., 2020). By evaluating a set of arginine derivatives with either N- or C- terminal substitutions, Singh et al. (2019) inferred the guanidino group of arginine was essential to its anti-aggregatory effect on a polyQ peptide (polyglutamine-35; Q35) in vitro. Citrulline, ornithine and lysine, which lack the guanidino moiety, were unable to prevent Q35 aggregation, while arginine and arginine ethyl ester prevented Q35 aggregation in a dose-dependent manner (Singh et al., 2019).
Arginine molecules can be converted to citrulline by arginine deiminase (ADI) through hydrolysis of their guanidinium groups, causing the strongly basic, cationic arginine side chain to be replaced by neutral urea. Whereas ADI converts free arginine molecules, peptidylarginine deiminases (PADs) catalyse the conversion of peptide-bound arginine residues to citrulline residues (Wang and Wang, 2013). Citrullination affects the ability of the residue to form hydrogen bonds, altering its intermolecular interactions.
Increased PAD activity has been associated with AD (Acharya et al., 2012; Wang et al., 2021); in particular, levels of PAD II detected in the hippocampus were found to be significantly higher in AD patients than in controls by Ishigami et al. (2005), concomitant with the presence of citrullinated proteins in AD-affected hippocampi. It is conceivable that higher levels of PAD activity in AD could affect the overall efficacy of CARP therapeutics through citrullination of their arginine residues, however, this remains to be observed. Moreover, understanding of PADs in AD more broadly remains limited. Further research on peptidylarginine citrullination in AD is required to hypothesise the potential effects of PADs on CARPs as therapeutic candidates in this context. Additionally, as current preclinical models of AD are unable to recapitulate all aspects of AD pathogenesis, it is difficult to predict how overexpression of PADs upregulated in human AD might affect CARPs shown to be highly efficacious in animal models, such as RD2.
It is worth noting, however, that PAD activity is tightly controlled by calcium ions (Lamensa and Moscarello, 1993; Sambandam et al., 2004; Mondal and Thompson, 2019), and PAD II is activated through elevated intracellular calcium levels (Slade et al., 2015). CARPs themselves have previously been shown to reduce intracellular calcium influx through multiple mechanisms (Meloni et al., 2015a,b; Edwards et al., 2017), including downregulation of the NMDAR subunit NR2B. Therefore, it is possible that CARPs may be able to prevent excessive activation of PADs in AD through upstream inhibition of calcium influx. In summary, although PADs could theoretically affect the ability of CARPs to exert beneficial effects in AD through citrullination of their arginine residues, it is difficult to predict whether this is likely to occur in vivo based on the paucity of evidence; additionally, the putative effects of CARPs on calcium signalling in states of neurological injury and disease render this a complex area. Regardless, the favourable safety profile of CARPs (Edwards et al., 2020) should allow CARPs to be evaluated at varying dose levels for efficacy in human AD.
Arginine has several distinctive properties, largely owing to the unique structure and chemistry of the guanidinium group. While monomeric arginine has long been regarded as a potent inhibitor of protein aggregation, polypeptides enriched for arginine residues also display interesting effects on the formation and cytotoxicity of protein aggregates. Arginine residues have substantial bioactive properties in the context of modulating protein aggregation, and should therefore be given particular consideration in the rational design of amyloid-targeting therapeutic peptides.
The lack of effective therapeutic options for AD presents a significant global health challenge. Peptide drugs are a rapidly growing class of therapeutic candidates, and while their design and optimisation carries a distinct set of challenges, they hold promise in the treatment of AD through their potential advantages of high target specificity and bioactivity. We have identified a number of CARPs which effectively modulate the aggregation pathway of amyloid-beta, as well as potential roles of arginine in peptide-based therapeutic design for targeting tau oligomerisation.
Interestingly, CARPs have also demonstrated significant neuroprotective effects in models of stroke and traumatic brain injury, particularly by targeting excitotoxicity, which is also a pathological feature of AD (reviewed by Wang and Reddy, 2017). The complex and multifactorial nature of AD pathogenesis particularly warrants therapeutic candidates with multiple mechanisms of action. The potential for CARPs to modulate proteopathies associated with AD, therefore, warrant further investigation. CARPs could potentially target the formation and/or deleterious cellular effects of amyloids in AD through diverse roles: decreasing the concentration of soluble, cytotoxic oligomers by either preventing aggregation or driving aggregation toward the formation of non-cytotoxic species; mitigating oxidative stress, which is known to drive the formation of cytotoxic amyloids; and preserving the function of mitochondria, which are a site of cellular damage from amyloids. These possibilities present several lines of inquiry for further research.
SM wrote the manuscript with input from BM. Both authors revised the manuscript and approved the final version.
BM is a named inventor of several patent applications regarding the use of CARPs as neuroprotective agents, and a shareholder in Argenica Therapeutics, a company developing R18 as a neurotherapeutic.
The remaining author declares the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Professor Frank Mastaglia for editorial suggestions. Figures throughout the manuscript were created using Biorender (https://biorender.com).
Acharya, N. K., Nagele, E. P., Han, M., Coretti, N. J., DeMarshall, C., Kosciuk, M. C., et al. (2012). Neuronal PAD4 expression and protein citrullination: possible role in production of autoantibodies associated with neurodegenerative disease. J. Autoimmun. 38, 369–380. doi: 10.1016/j.jaut.2012.03.004
Aggidis, A., Chatterjee, S., Townsend, D., Fullwood, N. J., Ortega, E. R., Tarutani, A., et al. (2021). Peptide-based inhibitors of tau aggregation as a potential therapeutic for Alzheimer’s disease and other tauopathies. bioRxiv [Preprint]. doi: 10.1101/2021.06.04.447069
Aggidis, A. (2019). The development of peptide-based inhibitors for Tau aggregation as a potential therapeutic for Alzheimer’s disease. Available online at: https://eprints.lancs.ac.uk/id/eprint/140396/1/2019AggidisPhD.pdf.
Allolio, C., Magarkar, A., Jurkiewicz, P., Baxová, K., Javanainen, M., Mason, P. E., et al. (2018). Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. 115, 11923–11928. doi: 10.1073/pnas.1811520115
Alzheimer’s Association. (2021). Alzheimer’s disease facts and figures. Alzheimers Dement. 17, 327–406. doi: 10.1002/alz.12328
Arai, T., Sasaki, D., Araya, T., Sato, T., Sohma, Y., and Kanai, M. A. (2014). A cyclic KLVFF-derived peptide aggregation inhibitor induces the formation of less-toxic off-pathway amyloid-β oligomers. Chembiochem. 15, 2577–2583. doi: 10.1002/cbic.201402430
Arakawa, J., Uegaki, M., and Ishimizu, T. (2011). Effects of L-arginine on solubilization and purification of plant membrane proteins. Protein Expr. Purif. 80, 91–96. doi: 10.1016/j.pep.2011.05.014
Arakawa, T., Ejima, D., Tsumoto, K., Obeyama, N., Tanaka, Y., Kita, Y., et al. (2007). Suppression of protein interactions by arginine: a proposed mechanism of the arginine effects. Biophys. Chem. 127, 1–8. doi: 10.1016/j.bpc.2006.12.007
Arakawa, T., Kita, Y., Ejima, D., Tsumoto, K., and Fukada, H. (2006). Aggregation suppression of proteins by arginine during thermal unfolding. Protein Pept. Lett. 13, 921–927. doi: 10.2174/092986606778256171
Arboleda-Velasquez, J. F., Lopera, F., O’Hare, M., Delgado-Tirado, S., Marino, C., Chmielewska, N., et al. (2019). Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. 25, 1680–1683. doi: 10.1038/s41591-019-0611-3
Austen, B. M., Paleologou, K. E., Ali, S. A., Qureshi, M. M., Allsop, D., and El-Agnaf, O. M. (2008). Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s beta-amyloid peptide. Biochemistry. 47, 1984–1992. doi: 10.1021/bi701415b
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Barage, S. H., and Sonawane, K. D. (2015). Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 52, 1–18. doi: 10.1016/j.npep.2015.06.008
Barale, S. S., Parulekar, R. S., Fandilolu, P. M., Dhanavade, M. J., and Sonawane, K. D. (2019). Molecular insights into destabilization of Alzheimer’s Aβ protofibril by arginine containing short peptides: a molecular modeling approach. ACS Omega 4, 892–903. doi: 10.1021/acsomega.8b02672
Barr, R. K., Verdile, G., Wijaya, L. K., Morici, M., Taddei, K., Gupta, V. B., et al. (2016). Validation and characterization of a novel peptide that binds monomeric and aggregated β-Amyloid and inhibits the formation of neurotoxic oligomers. J. Biol. Chem. 291, 547–559. doi: 10.1074/jbc.M115.679993
Baynes, B. M., Wang, D. I., and Trout, B. L. (2005). Role of arginine in the stabilization of proteins against aggregation. Biochemistry 44, 4919–4925. doi: 10.1021/bi047528r
Bhowmik, D., MacLaughlin, C. M., Chandrakesan, M., Ramesh, P., Venkatramani, R., Walker, G. C., et al. (2014). pH changes the aggregation propensity of amyloid-β without altering the monomer conformation. Phys. Chem. Chem. Phys. 16, 885–889. doi: 10.1039/c3cp54151g
Bin, Y., Li, X., He, Y., Chen, S., and Xiang, J. (2013). Amyloid-β peptide (1–42) aggregation induced by copper ions under acidic conditions. Acta Biochim. Biophys. Sin. (Shanghai) 45, 570–577. doi: 10.1093/abbs/gmt044
Borzova, V. A., Markossian, K. A., Kleymenov, S. Y., and Kurganov, B. I. (2017). A change in the aggregation pathway of bovine serum albumin in the presence of arginine and its derivatives. Sci. Rep. 7:3984. doi: 10.1038/s41598-017-04409-x
Brandt, R., Léger, J., and Lee, G. (1995). Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 131, 1327–1340. doi: 10.1083/jcb.131.5.1327
Březina, K., Duboué-Dijon, E., Palivec, V., Jiráček, J., Křížek, T., Viola, C. M., et al. (2018). Can arginine inhibit insulin aggregation? a combined protein crystallography, capillary electrophoresis and molecular simulation study. J. Phys. Chem. B. 122, 10069–10076. doi: 10.1021/acs.jpcb.8b06557
Butterfield, D. A., Swomley, A. M., and Sultana, R. (2013). Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox. Signal. 19, 823–835. doi: 10.1089/ars.2012.5027
Carrillo-Mora, P., Luna, R., and Colín-Barenque, L. (2014). Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell Longev. 2014:795375. doi: 10.1155/2014/795375
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chen, J. X., and Yan, S. D. (2007). Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimers Dis. 12, 177–184. doi: 10.3233/jad-2007-12208
Cheng, Y. S., Chen, Z. T., Liao, T. Y., Lin, C., Shen, H. C., Wang, Y. H., et al. (2017). An intranasally delivered peptide drug ameliorates cognitive decline in Alzheimer transgenic mice. EMBO Mol. Med. 9, 703–715. doi: 10.15252/emmm.201606666
Cirkovas, A., and Sereikaite, J. (2011). Different effects of (L)-arginine on the heat-induced unfolding and aggregation of proteins. Biologicals 39, 181–188. doi: 10.1016/j.biologicals.2011.04.003
Congdon, E. E., and Sigurdsson, E. M. (2018). Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415. doi: 10.1038/s41582-018-0013-z
Cosacak, M. I., Bhattarai, P., Bocova, L., Dzewas, T., Mashkaryan, V., Papadimitriou, C., et al. (2017). Human TAUP301L overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci. Rep. 7:12959. doi: 10.1038/s41598-017-13311-5
Crescenzi, O., Tomaselli, S., Guerrini, R., Salvadori, S., D’Ursi, A. M., Temussi, P. A., et al. (2002). Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 269, 5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x
Crews, L., Tsigelny, I., Hashimoto, M., and Masliah, E. (2009). Role of synucleins in Alzheimer’s disease. Neurotox Res. 16, 306–317. doi: 10.1007/s12640-009-9073-6
D’Annessa, I., Di Leva, F. S., La Teana, A., Novellino, E., Limongelli, V., and Di Marino, D. (2020). Bioinformatics and biosimulations as toolbox for peptides and peptidomimetics design: where are we? Front. Mol. Biosci. 7:66. doi: 10.3389/fmolb.2020.00066
Das, U., Hariprasad, G., Ethayathulla, A. S., Manral, P., Das, T. K., Pasha, S., et al. (2007). Inhibition of protein aggregation: supramolecular assemblies of arginine hold the key. PLoS One. 2:e1176. doi: 10.1371/journal.pone.0001176
Di Carlo, M. (2010). Beta amyloid peptide: from different aggregation forms to the activation of different biochemical pathways. Eur. Biophys. J. 39, 877–888. doi: 10.1007/s00249-009-0439-8
Di, J., Cohen, L. S., Corbo, C. P., Phillips, G. R., El Idrissi, A., and Alonso, A. D. (2016). Abnormal tau induces cognitive impairment through two different msechanisms: synaptic dysfunction and neuronal loss. Sci. Rep. 6:20833. doi: 10.1038/srep20833
Di, L. (2015). Strategic approaches to optimizing peptide ADME properties. AAPS J. 17, 134–143. doi: 10.1208/s12248-014-9687-3
Digma, L. A., Madsen, J. R., Reas, E. T., Dale, A. M., Brewer, J. B., Banks, S. J., et al. (2019). Tau and atrophy: domain-specific relationships with cognition. Alzheimers Res. Ther. 11:65. doi: 10.1186/s13195-019-0518-8
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
D’Souza, I., and Schellenberg, G. D. (2000). Determinants of 4-repeat tau expression. Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J. Biol. Chem. 275, 17700–17709. doi: 10.1074/jbc.M909470199
Edwards, A. B., Anderton, R. S., Knuckey, N. W., and Meloni, B. P. (2017). Characterisation of neuroprotective efficacy of modified poly-arginine-9 (R9) peptides using a neuronal glutamic acid excitotoxicity model. Mol. Cell Biochem. 426, 75–85. doi: 10.1007/s11010-016-2882-z
Edwards, A. B., Mastaglia, F. L., Knuckey, N. W., and Meloni, B. P. (2020). Neuroprotective cationic arginine-rich peptides (CARPs): an assessment of their clinical safety. Drug Saf. 43, 957–969. doi: 10.1007/s40264-020-00962-z
Elfgen, A., Santiago-Schübel, B., Hupert, M., Schemmert, S., Schartmann, E., Tusche, M., et al. (2021). Oral absorption enhancement of the amyloid-β oligomer eliminating compound RD2 by conjugation with folic acid. Eur. J. Pharm. Sci. 156:105581. doi: 10.1016/j.ejps.2020.105581
Enache, T. A., Chiorcea-Paquim, A. M., and Oliveira-Brett, A. M. (2018). Amyloid beta peptide VHHQ, KLVFF and IIGLMVGGVV domains involved in fibrilization: AFM and electrochemical characterization. Anal. Chem. 90, 2285–2292. doi: 10.1021/acs.analchem.7b04686
Eschmann, N. A., Do, T. D., LaPointe, N. E., Shea, J. E., Feinstein, S. C., Bowers, M. T., et al. (2015). Tau aggregation propensity engrained in its solution state. J. Phys. Chem. B. 119, 14421–14432. doi: 10.1021/acs.jpcb.5b08092
Evers, A., Pfeiffer-Marek, S., Bossart, M., Heubel, C., Stock, U., Tiwari, G., et al. (2019). Peptide optimization at the drug discovery-development interface: tailoring of physicochemical properties toward specific formulation requirements. J. Pharm. Sci. 108, 1404–1414. doi: 10.1016/j.xphs.2018.11.043
Faller, P., Hureau, C., and Berthoumieu, O. (2013). Role of metal ions in the self-assembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 52, 12193–12206. doi: 10.1021/ic4003059
Feng, Z., and Xu, B. (2016). Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 7, 179–187. doi: 10.1515/bmc-2015-0035
Ferrari, L., Stucchi, R., Konstantoulea, K., van de Kamp, G., Kos, R., Geerts, W. J. C., et al. (2020). Arginine π-stacking drives binding to fibrils of the Alzheimer protein Tau. Nat. Commun. 11:571. doi: 10.1038/s41467-019-13745-7
Fichou, Y., Lin, Y., Rauch, J. N., Vigers, M., Zeng, Z., Srivastava, M., et al. (2018). Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc. Natl. Acad. Sci. U S A 115, 13234–13239. doi: 10.1073/pnas.1810058115
Fields, C. R., Bengoa-Vergniory, N., and Wade-Martins, R. (2019). Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front. Mol. Neurosci. 12:299. doi: 10.3389/fnmol.2019.00299
Fitch, C. A., Platzer, G., Okon, M., Garcia-Moreno, B. E., and McIntosh, L. P. (2015). Arginine: its pKa value revisited. Protein Sci. 24, 752–761. doi: 10.1002/pro.2647
Fonar, G., Polis, B., Meirson, T., Maltsev, A., and Samson, A. O. (2018). Subcutaneous sustained-release of poly-arginine ameliorates cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Adv. Alzheimers Dis. 7, 153–182. doi: 10.4236/aad.2018.74011
Francis, P. T., Palmer, A. M., Snape, M., and Wilcock, G. K. (1999). The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry 66, 137–147. doi: 10.1136/jnnp.66.2.137
Fukuda, M., Kameoka, D., Torizawa, T., Saitoh, S., Yasutake, M., Imaeda, Y., et al. (2014). Thermodynamic and fluorescence analyses to determine mechanisms of IgG1 stabilization and destabilization by arginine. Pharm. Res. 31, 992–1001. doi: 10.1007/s11095-013-1221-2
Funke, S. A., and Willbold, D. (2012). Peptides for therapy and diagnosis of Alzheimer’s disease. Curr. Pharm. Des. 18, 755–767. doi: 10.2174/138161212799277752
Gandhi, J., Antonelli, A. C., Afridi, A., Vatsia, S., Joshi, G., Romanov, V., et al. (2019). Protein misfolding and aggregation in neurodegenerative diseases: a review of pathogeneses, novel detection strategies and potential therapeutics. Rev. Neurosci. 30, 339–358. doi: 10.1515/revneuro-2016-0035
GBD 2016 Dementia Collaborators. (2019). Global, regional and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet. Neurol. 18, 88–106. doi: 10.1016/S1474-4422(18)30403-4
Ghosh, R., Sharma, S., and Chattopadhyay, K. (2009). Effect of arginine on protein aggregation studied by fluorescence correlation spectroscopy and other biophysical methods. Biochemistry 48, 1135–1143. doi: 10.1021/bi802065j
Ghosh, S., Kundu, A., and Chattopadhyay, K. (2018). Small molecules attenuate the interplay between conformational fluctuations, early oligomerization and amyloidosis of alpha synuclein. Sci. Rep. 8:5481. doi: 10.1038/s41598-018-23718-3
Gibson, T. J., and Murphy, R. M. (2005). Design of peptidyl compounds that affect beta-amyloid aggregation: importance of surface tension and context. Biochemistry 44, 8898–8907. doi: 10.1021/bi050225s
Gofton, T., and Weaver, D. F. (2006). Challenges in the clinical diagnosis of Alzheimer’s disease: influence of "family coaching" on the mini-mental state examination. Am. J. Alzheimers Dis. Other Demen. 21, 109–112. doi: 10.1177/153331750602100210
Golovanov, A. P., Hautbergue, G. M., Wilson, S. A., and Lian, L. Y. (2004). A simple method for improving protein solubility and long-term stability. J. Am. Chem. Soc. 126, 8933–8939. doi: 10.1021/ja049297h
Goyal, D., Shuaib, S., Mann, S., and Goyal, B. (2017). Rationally designed peptides and peptidomimetics as inhibitors of Amyloid-β (Aβ) aggregation: potential therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 19, 55–80. doi: 10.1021/acscombsci.6b00116
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Nat. Acad. Sci. U S A 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Gund, P. (1972). Guanidine, trimethylenemethane and "Y-delocalization." Can acyclic compounds have "aromatic" stability? J. Chem. Educ. 49:100. doi: 10.1021/ed049p100
Habault, J., and Poyet, J. L. (2019). Recent advances in cell penetrating peptide-based anticancer therapies. Molecules 24:927. doi: 10.3390/molecules24050927
Haghighi-Poodeh, S., Kurganov, B., Navidpour, L., Yaghmaei, P., and Ebrahim-Habibi, A. (2020). Characterization of arginine preventive effect on heat-induced aggregation of insulin. Int. J. Biol. Macromol. 145, 1039–1048. doi: 10.1016/j.ijbiomac.2019.09.196
Haklar, G., Ulukaya-Durakba-a, C., Yüksel, M., Dağli, T., and Yalçin, A. S. (1998). Oxygen radicals and nitric oxide in rat mesenteric ischaemia-reperfusion: modulation by L-arginine and NG-nitro-L-arginine methyl ester. Clin. Exp. Pharmacol. Physiol. 25, 908–912. doi: 10.1111/j.1440-1681.1998.tb02342.x
Hanseeuw, B. J., Betensky, R. A., Jacobs, H. I. L., Schultz, A. P., Sepulcre, J., Becker, J. A., et al. (2019). Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924. doi: 10.1001/jamaneurol.2019.1424
Hardy, J., and De Strooper, B. (2017). Alzheimer’s disease: where next for anti-amyloid therapies?. Brain 140, 853–855. doi: 10.1093/brain/awx059
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
He, R. Y., Lai, X. M., Sun, C. S., Kung, T., Hong, J., Jheng, Y., et al. (2019). Nanoscopic insights of amphiphilic peptide against the oligomer assembly process to treat Huntington’s disease. Adv. Sci. (Weinh) 7:1901165. doi: 10.1002/advs.201901165
Henning-Knechtel, A., Kumar, S., Wallin, C., Krol, S., Warmlander, S. K. T. S., Jarvet, J., et al. (2020). Designed cell-penetrating peptide inhibitors of amyloid-beta aggregation and cytotoxicity. Cell Rep. Physic. Sci. 1:100014. doi: 10.1016/j.xcrp.2020.100014
Hsu, F., Park, G., and Guo, Z. (2018). Key residues for the formation of Aβ42 amyloid fibrils. ACS Omega 3, 8401–8407. doi: 10.1021/acsomega.8b00887
Irwin, D. J., Cohen, T. J., Grossman, M., Arnold, S. E., Xie, S. X., Lee, V. M., et al. (2012). Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135, 807–818. doi: 10.1093/brain/aws013
Ishigami, A., Ohsawa, T., Hiratsuka, M., Taguchi, H., Kobayashi, S., Saito, Y., et al. (2005). Abnormal accumulation of citrullinated proteins catalyzed by peptidylarginine deiminase in hippocampal extracts from patients with Alzheimer’s disease. J. Neurosci. Res. 80, 120–128. doi: 10.1002/jnr.20431
Ito, L., Shiraki, K., Matsuura, T., Okumura, M., Hasegawa, K., Baba, S., et al. (2011). High-resolution X-ray analysis reveals binding of arginine to aromatic residues of lysozyme surface: implication of suppression of protein aggregation by arginine. Protein Eng. Des. Sel. 24, 269–274. doi: 10.1093/protein/gzq101
Jameson, L., Frey, T., Zeeberg, B., Dalldorf, F., and Caplow, M. (1980). Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry 19, 2472–2479. doi: 10.1021/bi00552a027
Jiang, D., Rauda, I., Han, S., Chen, S., and Zhou, F. (2012). Aggregation pathways of the amyloid β(1–42) peptide depend on its colloidal stability and ordered β-sheet stacking. Langmuir 28, 12711–12721. doi: 10.1021/la3021436
Johnson, K., Hanseeuw, B. J., Betensky, R. A., Jacobs, H. L., Schltz, A. P., Sepulcre, J., et al. (2019). Association of amyloid and tau With cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924. doi: 10.1001/jamaneurol.2019.1424
Jokar, S., Erfani, M., Bavi, O., Khazaei, S., Sharifzadeh, M., Hajiramezanali, M., et al. (2020). Design of peptide-based inhibitor agent against amyloid-β aggregation: Molecular docking, synthesis and in vitro evaluation. Bioorg Chem. 102:104050. doi: 10.1016/j.bioorg.2020.104050
Jouanne, M., Rault, S., and Voisin-Chiret, A. S. (2017). Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 139, 153–167. doi: 10.1016/j.ejmech.2017.07.070
Kawasaki, T., and Kamijo, S. (2012). Inhibition of aggregation of amyloid β42 by arginine-containing small compounds. Biosci. Biotechnol. Biochem. 76, 762–766. doi: 10.1271/bbb.110879
Kawasaki, T., Onodera, K., and Kamijo, S. (2011). Identification of novel short peptide inhibitors of soluble 37/48 kDa oligomers of amyloid β42. Biosci. Biotechnol. Biochem. 75, 1496–1501. doi: 10.1271/bbb.110198
Kawasaki, T., Onodera, K., and Kamijo, S. (2010). Selection of peptide inhibitors of soluble Aβ(1–42) oligomer formation by phage display. Biosci. Biotechnol. Biochem. 74, 2214–2219. doi: 10.1271/bbb.100388
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kino, R., Araya, T., Arai, T., Sohma, Y., and Kanai, M. (2015). Covalent modifier-type aggregation inhibitor of amyloid-β based on a cyclo-KLVFF motif. Bioorg. Med. Chem. Lett. 25, 2972–2975. doi: 10.1016/j.bmcl.2015.05.027
Klein, A. N., Ziehm, T., van Groen, T., Kadish, I., Elfgen, A., Tusche, M., et al. (2017). Optimization of d-peptides for Aβ monomer binding specificity enhances their potential to eliminate toxic Aβ oligomers. ACS Chem. Neurosci. 8, 1889–1900. doi: 10.1021/acschemneuro.7b00045
Kolarova, M., García-Sierra, F., Bartos, A., Ricny, J., and Ripova, D. (2012). Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012:731526. doi: 10.1155/2012/731526
Kondo, K., Ikura, T., Tanaka, H., Fujita, K., Takayama, S., Yoshioka, Y., et al. (2021). Hepta-histidine inhibits tau aggregation. ACS Chem. Neurosci. 12, 3015–3027. doi: 10.1021/acschemneuro.1c00164
Köpke, E., Tung, Y. C., Shaikh, S., Alonso, A. C., Iqbal, K., and Grundke-Iqbal, I. (1993). Microtubule-associated protein tau. abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384.
Kotzbauer, P. T., Trojanowsk, J. Q., and Lee, V. M. (2001). Lewy body pathology in Alzheimer’s disease. J. Mol. Neurosci. 17, 225–232. doi: 10.1385/jmn:17:2:225
Kumar, D., Ganeshpurkar, A., Kumar, D., Modi, G., Gupta, S. K., and Singh, S. K. (2018). Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 148, 436–452. doi: 10.1016/j.ejmech.2018.02.035
Kumar, J., Namsechi, R., and Sim, V. L. (2015). Structure-based peptide design to modulate amyloid beta aggregation and reduce cytotoxicity. PLoS One 10:e0129087. doi: 10.1371/journal.pone.0129087
Kumar, J., and Sim, V. (2014). D-amino acid-based peptide inhibitors as early or preventative therapy in Alzheimer disease. Prion 8, 119–124. doi: 10.4161/pri.28220
Kutzsche, J., Schemmert, S., Tusche, M., Neddens, J., Rabl, R., Jürgens, D., et al. (2017). Large-scale oral treatment study with the four most promising D3-derivatives for the treatment of Alzheimer’s disease. Molecules 22:1693. doi: 10.3390/molecules22101693
Lamensa, J. W., and Moscarello, M. A. (1993). Deimination of human myelin basic protein by a peptidylarginine deiminase from bovine brain. J. Neurochem. 61, 987–996. doi: 10.1111/j.1471-4159.1993.tb03612.x
Larson, M. E., and Lesné, S. E. (2012). Soluble Aβ oligomer production and toxicity. J. Neurochem. 120, 125–139. doi: 10.1111/j.1471-4159.2011.07478.x
Leithold, L. H., Jiang, N., Post, J., Niemietz, N., Schartmann, E., Ziehm, T., et al. (2016). Pharmacokinetic properties of tandem d-peptides designed for treatment of Alzheimer’s disease. Eur. J. Pharm. Sci. 89, 31–38. doi: 10.1016/j.ejps.2016.04.016
Li, J., Garg, M., Shah, D., and Rajagopalan, R. (2010). Solubilization of aromatic and hydrophobic moieties by arginine in aqueous solutions. J. Chem. Phys. 133:054902. doi: 10.1063/1.3469790
Li, W., and Lee, V. M. (2006). Characterization of two VQIXXK motifs for tau fibrillization in vitro. Biochemistry 45, 15692–15701. doi: 10.1021/bi061422+
Liu, R., McAllister, C., Lyubchenko, Y., and Sierks, M. R. (2004). Residues 17–20 and 30–35 of beta-amyloid play critical roles in aggregation. J. Neurosci. Res. 75, 162–171. doi: 10.1002/jnr.10859
Lovestone, S., and McLoughlin, D. M. (2002). Protein aggregates and dementia: is there a common toxicity. J. Neurol. Neurosurg. Psychiatry 72, 152–161. doi: 10.1136/jnnp.72.2.152
Lu, J., Cao, Q., Wang, C., Zheng, J., Luo, F., Xie, J., et al. (2019). Structure-Based Peptide Inhibitor Design of Amyloid-β Aggregation. Front. Mol. Neurosci. 12:54. doi: 10.3389/fnmol.2019.00054
Lucke-Wold, B., Seidel, K., Udo, R., Omalu, B., Ornstein, M., Nolan, R., et al. (2017). Role of tau acetylation in Alzheimer’s disease and chronic traumatic encephalopathy: the way forward for successful treatment. J. Neurol. Neurosurg. 4:140.
MacDougall, G., Anderton, R. S., Mastaglia, F. L., Knuckey, N. W., and Meloni, B. P. (2019b). Mitochondria and neuroprotection in stroke: Cationic arginine-rich peptides (CARPs) as a novel class of mitochondria-targeted neuroprotective therapeutics. Neurobiol. Dis. 121, 17–33. doi: 10.1016/j.nbd.2018.09.010
MacDougall, G., Anderton, R. S., Mastaglia, F. L., Knuckey, N. W., and Meloni, B. P. (2019a). Proteomic analysis of cortical neuronal cultures treated with poly-arginine peptide-18 (R18) and exposed to glutamic acid excitotoxicity. Mol. Brain 12:66. doi: 10.1186/s13041-019-0486-8
Maïza, A., Chantepie, S., Vera, C., Fifre, A., Huynh, M. B., Stettler, O., et al. (2018). The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 592, 3806–3818. doi: 10.1002/1873-3468.13082
Marshall, J., Wong, K. Y., Rupasinghe, C. N., Tiwari, R., Zhao, X., Berberoglu, E. D., et al. (2015). Inhibition of N-Methyl-D-aspartate-induced retinal neuronal death by polyarginine peptides is linked to the attenuation of stress-induced hyperpolarization of the inner mitochondrial membrane potential. J. Biol. Chem. 290, 22030–22048. doi: 10.1074/jbc.M115.662791
Mason, P. E., Neilson, G. W., Dempsey, C. E., Barnes, A. C., and Cruickshank, J. M. (2003). The hydration structure of guanidinium and thiocyanate ions: implications for protein stability in aqueous solution. Proc. Natl. Acad. Sci. U S A 100, 4557–4561. doi: 10.1073/pnas.0735920100
Mason, P. E., Neilson, G. W., Enderby, J. E., Saboungi, M. L., Dempsey, C. E., MacKerell, A. D., Jr., et al. (2004). The structure of aqueous guanidinium chloride solutions. J. Am. Chem. Soc. 126, 11462–11470. doi: 10.1021/ja040034x
Matsuoka, T., Tomita, S., Hamada, H., and Shiraki, K. (2007). Amidated amino acids are prominent additives for preventing heat-induced aggregation of lysozyme. J. Biosci. Bioeng. 103, 440–443. doi: 10.1263/jbb.103.440
Meloni, B. P., Milani, D., Edwards, A. B., Anderton, R. S., O’Hare Doig, R. L., Fitzgerald, M., et al. (2015a). Neuroprotective peptides fused to arginine-rich cell penetrating peptides: Neuroprotective mechanism likely mediated by peptide endocytic properties. Pharmacol. Ther. 153, 36–54. doi: 10.1016/j.pharmthera.2015.06.002
Meloni, B. P., Brookes, L. M., Clark, V. W., Cross, J. L., Edwards, A. B., Anderton, R. S., et al. (2015b). Poly-arginine and arginine-rich peptides are neuroprotective in stroke models. J. Cereb. Blood Flow. Metab. 35, 993–1004. doi: 10.1038/jcbfm.2015.11
Meloni, B. P., Mastaglia, F. L., and Knuckey, N. W. (2020). Cationic arginine-rich peptides (CARPs): a novel class of neuroprotective agents with a multimodal mechanism of action. Front. Neurol. 11:108. doi: 10.3389/fneur.2020.00108
Meraz-Ríos, M. A., Lira-De León, K. I., Campos-Peña, V., De Anda-Hernández, M. A., and Mena-López, R. (2010). Tau oligomers and aggregation in Alzheimer’s disease. J. Neurochem. 112, 1353–1367. doi: 10.1097/JCN.0000000000000616
Miller, J., Arrasate, M., Brooks, E., Libeu, C. P., Legleiter, J., Hatters, D., et al. (2011). Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 7, 925–934. doi: 10.1038/nchembio.694
Miller, V. M., Gouvion, C. M., Davidson, B. L., and Paulson, H. L. (2004). Targeting Alzheimer’s disease genes with RNA interference: an efficient strategy for silencing mutant alleles. Nucleic Acids Res. 32, 661–668. doi: 10.1093/nar/gkh208
Minakawa, E. N., and Nagai, Y. (2021). Protein aggregation inhibitors as disease-modifying therapies for polyglutamine diseases. Front. Neurosci. 15:621996. doi: 10.3389/fnins.2021.621996
Minakawa, E. N., Popiel, H. A., Tada, M., Takahashi, T., Yamane, H., Saitoh, Y., et al. (2020). Arginine is a disease modifier for polyQ disease models that stabilizes polyQ protein conformation. Brain 143, 1811–1825. doi: 10.1093/brain/awaa115
Mitchell, D. J., Kim, D. T., Steinman, L., Fathman, C. G., and Rothbard, J. B. (2000). Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56, 318–325. doi: 10.1034/j.1399-3011.2000.00723.x
Mondal, S., and Thompson, P. R. (2019). Protein arginine deiminases (PADs): biochemistry and chemical biology of protein citrullination. Acc. Chem. Res. 52, 818–832. doi: 10.1021/acs.accounts.9b00024
Morley, J. E., Farr, S. A., Nguyen, A. D., and Xu, F. (2019). Editorial: what is the physiological function of amyloid-beta protein. J. Nutr. Health Aging 23, 225–226. doi: 10.1007/s12603-019-1162-5
Moss, M. A., Nichols, M. R., Reed, D. K., Hoh, J. H., and Rosenberry, T. L. (2003). The peptide KLVFF-K(6) promotes beta-amyloid(1–40) protofibril growth by association but does not alter protofibril effects on cellular reduction of 3- (4,5-dimethylthiazol-2-yl).-2,5-diphenyltetrazolium bromide (MTT). Mol. Pharmacol. 64, 1160–1168. doi: 10.1124/mol.64.5.1160
Nadimidla, K., Ismail, T., and Kanapathipillai, M. (2017). Tau peptides and tau mutant protein aggregation inhibition by cationic polyethyleneimine and polyarginine. Biopolymers 107:e23024. doi: 10.1002/bip.23024
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Neve, R. L., Harris, P., Kosik, K. S., Kurnit, D. M., and Donlon, T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280. doi: 10.1016/0169-328x(86)90033-1
Niewiadomska, G., Niewiadomski, W., Steczkowska, M., and Gasiorowska, A. (2021). Tau oligomers neurotoxicity. Life (Basel) 11:28. doi: 10.3390/life11010028
Nirmalraj, P. N., List, J., Battacharya, S., Howe, G., Xu, L., Thompson, D., et al. (2020). Complete aggregation pathway of amyloid β (1–40) and (1–42) resolved on an atomically clean interface. Sci. Adv. 6:eaaz6014. doi: 10.1126/sciadv.aaz6014
Noble, W., Hanger, D. P., Miller, C. C., and Lovestone, S. (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 4:83. doi: 10.3389/fneur.2013.00083
Nuhu, M. M., and Curtis, R. (2015). Arginine dipeptides affect insulin aggregation in a pH- and ionic strength-dependent manner. Biotechnol. J. 10, 404–416. doi: 10.1002/biot.201400190
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Olubiyi, O. O., Frenzel, D., Bartnik, D., Glück, J. M., Brener, O., Nagel-Steger, L., et al. (2014). Amyloid aggregation inhibitory mechanism of arginine-rich D-peptides. Curr. Med. Chem. 21, 1448–1457. doi: 10.2174/0929867321666131129122247
Parthsarathy, V., McClean, P. L., Hölscher, C., Taylor, M., Tinker, C., Jones, G., et al. (2013). A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS One 8:e54769. doi: 10.1371/journal.pone.0054769
Pauwels, K., Williams, T. L., Morris, K. L., Jonckheere, W., Vandersteen, A., Kelly, G., et al. (2012). Structural basis for increased toxicity of pathological aβ42:aβ40 ratios in Alzheimer disease. J. Biol. Chem. 287, 5650–5660. doi: 10.1074/jbc.M111.264473
Pearson, H. A., and Peers, C. (2006). Physiological roles for amyloid beta peptides. J. Physiol. 575, 5–10. doi: 10.1113/jphysiol.2006.111203
Pedrini, S., Morici, M., and Martins, R. N. (2019). “Chapter 14—Current and prospective treatments for Alzheimer’s disease (and other neurodegenerative diseases),” in Neurodegeneration and Alzheimer’s Disease: The Role of Diabetes, Genetics, Hormones, and Lifestyle, eds R. N. Martins, C. S. Brennan, W. M. A. D. Binosha Fernando, M. A. Brennan and S. J. Fuller (John Wiley & Sons), 391–442. doi: 10.1002/9781119356752.ch14
Post, J., Kogel, V., Schaffrath, A., Lohmann, P., Shah, N. J., Langen, K. J., et al. (2021). A novel anti-inflammatory d-peptide inhibits disease phenotype progression in an ALS mouse model. Molecules 26:1590. doi: 10.3390/molecules26061590
Prasansuklab, A., and Tencomnao, T. (2013). Amyloidosis in Alzheimer’s disease: the toxicity of amyloid beta (A β), mechanisms of its accumulation and implications of medicinal plants for therapy. Evid. Based Complement Alternat. Med. 2013:413808. doi: 10.1155/2013/413808
Räder, A. F. B., Reichart, F., Weinmüller, M., and Kessler, H. (2018). Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg. Med. Chem. 26, 2766–2773. doi: 10.1016/j.bmc.2017.08.031
Ralhan, K., Guru, K. V., and Gupta, S. (2017). “Arginine-rich β-sheet breaker peptides as potential tau protein aggregation inhibitors,” in SfN’s 47th Annual Meeting, Neuroscience 2017 (Washington, D.C., USA), 11–15. https://repository.iitgn.ac.in/handle/123456789/3328.
Ramanathan, A., Nelson, A. R., Sagare, A. P., and Zlokovic, B. V. (2015). Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Reddy, P. H., Manczak, M., Mao, P., Calkins, M. J., Reddy, A. P., and Shirendeb, U. (2010). Amyloid-beta and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 20, S499–S512. doi: 10.3233/JAD-2010-100504
Resende, R., Ferreiro, E., Pereira, C., and Resende de Oliveira, C. (2008). Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1–42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 155, 725–737. doi: 10.1016/j.neuroscience.2008.06.036
Ricciarelli, R., and Fedele, E. (2017). The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr. Neuropharmacol. 15, 926–935. doi: 10.2174/1570159X15666170116143743
Sambandam, T., Belousova, M., Accaviti-Loper, M. A., Blanquicett, C., Guercello, V., Raijmakers, R., et al. (2004). Increased peptidylarginine deiminase type II in hypoxic astrocytes. Biochem. Biophys. Res. Commun. 325, 1324–1329. doi: 10.1016/j.bbrc.2004.10.173
Schemmert, S., Schartmann, E., Zafiu, C., Kass, B., Hartwig, S., Lehr, S., et al. (2019). Aβ oligomer elimination restores cognition in transgenic Alzheimer’s mice with full-blown pathology. Mol. Neurobiol. 56, 2211–2223. doi: 10.1007/s12035-018-1209-3
Schmidt, N., Mishra, A., Lai, G. H., and Wong, G. C. (2010). Arginine-rich cell-penetrating peptides. FEBS Lett. 584, 1806–1813. doi: 10.1016/j.febslet.2009.11.046
Schneider, C., Shukla, D., and Trout, B. L. (2011). Arginine and the hofmeister series: the role of ion-ion interactions in protein aggregation suppression. J. Phys. Chem. B 115, 7447–7458. doi: 10.1021/jp111920y
Schneider, C. P., and Trout, B. L. (2009). Investigation of cosolute-protein preferential interaction coefficients: new insight into the mechanism by which arginine inhibits aggregation. J. Phys. Chem. B. 113, 2050–2058. doi: 10.1021/jp808042w
Seidler, P. M., Boyer, D. R., Rodriguez, J. A., Sawaya, M. R., Cascio, D., Murray, K., et al. (2018). Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176. doi: 10.1038/nchem.2889
Sengupta, U., Nilson, A. N., and Kayed, R. (2016). The role of amyloid-β oligomers in toxicity, propagation and immunotherapy. EBioMedicine 6, 42–49. doi: 10.1016/j.ebiom.2016.03.035
Shafiei, S. S., Guerrero-Muñoz, M. J., and Castillo-Carranza, D. L. (2017). Tau oligomers: cytotoxicity, propagation and mitochondrial damage. Front. Aging Neurosci. 9:83. doi: 10.3389/fnagi.2017.00083
Sharma, S., Sarkar, S., Paul, S. S., Roy, S., and Chattopadhyay, K. (2013). A small molecule chemical chaperone optimizes its unfolded state contraction and denaturant like properties. Sci. Rep. 3:3525. doi: 10.1038/srep03525
Shiraki, K., Kudou, M., Fujiwara, S., Imanaka, T., and Takagi, M. (2002). Biophysical effect of amino acids on the prevention of protein aggregation. J. Biochem. 132, 591–595. doi: 10.1093/oxfordjournals.jbchem.a003261
Shristi, A. (2014). Developing arginine as an inhibitor for α-Synuclein aggregation: an innovative therapy to combat Parkinson’s disease. Asian J. Sci. Technol. 5, 615–619.
Shukla, D., and Trout, B. L. (2010). Interaction of arginine with proteins and the mechanism by which it inhibits aggregation. J. Phys. Chem. B. 114, 13426–13438. doi: 10.1021/jp108399g
Sievers, S. A., Karanicolas, J., Chang, H. W., Zhao, A., Jiang, L., Zirafi, O., et al. (2011). Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475, 96–100. doi: 10.1038/nature10154
Singh, V., Patel, K. A., Sharma, R. K., Patil, P. R., Joshi, A. S., Parihar, R., et al. (2019). Discovery of arginine ethyl ester as polyglutamine aggregation inhibitor: conformational transitioning of huntingtin N-terminus augments aggregation suppression. ACS Chem. Neurosci. 10, 3969–3985. doi: 10.1021/acschemneuro.9b00167
Slade, D. J., Fang, P., Dreyton, C. J., Zhang, Y., Fuhrmann, J., Rempel, D., et al. (2015). Protein arginine deiminase 2 binds calcium in an ordered fashion: implications for inhibitor design. ACS Chem. Biol. 10, 1043–1053. doi: 10.1021/cb500933j
Smirnova, E., Safenkova, I., Stein-Margolina, B., Shubin, V., and Gurvits, B. (2013). L-arginine induces protein aggregation and transformation of supramolecular structures of the aggregates. Amino Acids 45, 845–855. doi: 10.1007/s00726-013-1528-7
Soto, C., Sigurdsson, E. M., Morelli, L., Kumar, R. A., Castaño, E. M., and Frangione, B. (1998). Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat. Med. 4, 822–826. doi: 10.1038/nm0798-822
Stancu, I. C., Vasconcelos, B., Terwel, D., and Dewachter, I. (2014). Models of β-amyloid induced Tau-pathology: the long and "folded" road to understand the mechanism. Mol. Neurodegener. 9:51. doi: 10.1186/1750-1326-9-51
Tesei, G., Vazdar, M., Jensen, M. R., Cragnell, C., Mason, P. E., Heyda, J., et al. (2017). Self-association of a highly charged arginine-rich cell-penetrating peptide. Proc. Natl. Acad. Sci. U S A 114, 11428–11433. doi: 10.1073/pnas.1712078114
Thal, D. R., and Fändrich, M. (2015). Protein aggregation in Alzheimer’s disease: Aβ and τ and their potential roles in the pathogenesis of AD. Acta Neuropathol. 129, 163–165. doi: 10.1007/s00401-015-1387-2
Tsumoto, K., Umetsu, M., Kumagai, I., Ejima, D., Philo, J. S., and Arakawa, T. (2004). Role of arginine in protein refolding, solubilization and purification. Biotechnol. Prog. 20, 1301–1308. doi: 10.1021/bp0498793
Twohig, D., and Nielsen, H. M. (2019). α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 14:23. doi: 10.1186/s13024-019-0320-x
Vagenende, V., Han, A. X., Mueller, M., and Trout, B. L. (2013). Protein-associated cation clusters in aqueous arginine solutions and their effects on protein stability and size. ACS Chem. Biol. 8, 416–422. doi: 10.1021/cb300440x
Valerio, M., Porcelli, F., Zbilut, J. P., Giuliani, A., Manetti, C., and Conti, F. (2008). pH effects on the conformational preferences of amyloid beta-peptide (1–40) in HFIP aqueous solution by NMR spectroscopy. Chem. Med. Chem. 3, 833–843. doi: 10.1002/cmdc.200700324
van Groen, T., Wiesehan, K., Funke, S. A., Kadish, I., Nagel-Steger, L., and Willbold, D. (2008). Reduction of Alzheimer’s disease amyloid plaque load in transgenic mice by D3, A D-enantiomeric peptide identified by mirror image phage display. Chem. Med. Chem. 3, 1848–1852. doi: 10.1002/cmdc.200800273
Varughese, M. M., and Newman, J. (2012). Inhibitory effects of arginine on the aggregation of bovine insulin. J. Biophys. 2012:434289. doi: 10.1155/2012/434289
Vazdar, M., Heyda, J., Mason, P. E., Tesei, G., Allolio, C., Lund, M., et al. (2018). Arginine "magic": guanidinium like-charge ion pairing from aqueous salts to cell penetrating peptides. Acc. Chem. Res. 51, 1455–1464. doi: 10.1021/acs.accounts.8b00098
Vershinin, M., Carter, B. C., Razafsky, D. S., King, S. J., and Gross, S. P. (2007). Multiple-motor based transport and its regulation by tau. Proc. Natl. Acad. Sci. U S A 104, 87–92. doi: 10.1073/pnas.0607919104
Viet, M. H., Ngo, S. T., Lam, N. S., and Li, M. S. (2011). Inhibition of aggregation of amyloid peptides by beta-sheet breaker peptides and their binding affinity. J. Phys. Chem. B. 115, 7433–7446. doi: 10.1021/jp1116728
von Bergen, M., Barghorn, S., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (2005). Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim. Biophys. Acta 1739, 158–166. doi: 10.1016/j.bbadis.2004.09.010
Wang, L. L., Song, Y. P., Mi, J. H., and Ding, M. L. (2021). Peptidyl arginine deiminase 4 and its potential role in Alzheimer’s disease. Med. Hypotheses. 146:110466. doi: 10.1016/j.mehy.2020.110466
Wang, R., and Reddy, P. H. (2017). Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 57, 1041–1048. doi: 10.3233/JAD-160763
Wang, S., and Wang, Y. (2013). Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta 1829, 1126–1135. doi: 10.1016/j.bbagrm.2013.07.003
Wascher, T. C., Posch, K., Wallner, S., Hermetter, A., Kostner, G. M., and Graier, W. F. (1997). Vascular effects of L-arginine: anything beyond a substrate for the NO-synthase. Biochem. Biophys. Res. Commun. 234, 35–38. doi: 10.1006/bbrc.1997.9994
Watanabe, K., Nakamura, K., Akikusa, S., Okada, T., Kodaka, M., Konakahara, T., et al. (2002). Inhibitors of fibril formation and cytotoxicity of beta-amyloid peptide composed of KLVFF recognition element and flexible hydrophilic disrupting element. Biochem. Biophys. Res. Commun. 290, 121–124. doi: 10.1006/bbrc.2001.6191
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U S A 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Welander, H., Frånberg, J., Graff, C., Sundström, E., Winblad, B., and Tjernberg, L. O. (2009). Abeta43 is more frequent than Abeta40 in amyloid plaque cores from Alzheimer disease brains. J. Neurochem. 110, 697–706. doi: 10.1111/j.1471-4159.2009.06170.x
Weller, J., and Budson, A. (2018). Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 7:F1000 Faculty Rev-1161. doi: 10.12688/f1000research.14506.1
Wiesehan, K., Buder, K., Linke, R. P., Patt, S., Stoldt, M., Unger, E., et al. (2003). Selection of D-amino-acid peptides that bind to Alzheimer’s disease amyloid peptide abeta1–42 by mirror image phage display. Chembiochem 4, 748–753. doi: 10.1002/cbic.200300631
Williamson, R., Scales, T., Clark, B. R., Gibb, G., Reynolds, C. H., Kellie, S., et al. (2002). Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J. Neurosci. 22, 10–20. doi: 10.1523/JNEUROSCI.22-01-00010.2002
Wolff, M., Zhang-Haagen, B., Decker, C., Barz, B., Schneider, M., Biehl, R., et al. (2017). Aβ42 pentamers/hexamers are the smallest detectable oligomers in solution. Sci. Rep. 7:2493. doi: 10.1038/s41598-017-02370-3
Xu, B., Jacobs, M. I., Kostko, O., and Ahmed, M. (2017). Guanidinium group remains protonated in a strongly basic arginine solution. Chemphyschem 18, 1503–1506. doi: 10.1002/cphc.201700197
Yankner, B. A., and Lu, T. (2009). Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease. J. Biol. Chem. 284, 4755–4759. doi: 10.1074/jbc.R800018200
Zhang, T., Gering, I., Kutzsche, J., Nagel-Steger, L., and Willbold, D. (2019). Toward the mode of action of the clinical stage all-d-enantiomeric peptide RD2 on Aβ42 aggregation. ACS Chem. Neurosci. 10, 4800–4809. doi: 10.1021/acschemneuro.9b00458
Zhang, X., Zhang, X., Zhong, M., Zhao, P., Guo, C., Li, Y., et al. (2020). Selection of a d-enantiomeric peptide specifically binding to PHF6 for inhibiting tau aggregation in transgenic mice. ACS Chem. Neurosci. 11, 4240–4253. doi: 10.1021/acschemneuro.0c00518
Zhao, W., and Ai, H. (2018). Effect of pH on Aβ42 monomer and fibril-like oligomers-decoding in silico of the roles of pK values of charged residues. Chemphyschem 19, 1103–1116. doi: 10.1002/cphc.201701384
Zhao, Y., and Zhao, B. (2013). Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell Longev. 2013:316523. doi: 10.1155/2013/316523
Zheng, H., and Koo, E. H. (2011). Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 6:27. doi: 10.1186/1750-1326-6-27
Zheng, J., Liu, C., Sawaya, M. R., Vadla, B., Khan, S., Woods, R. J., et al. (2011). Macrocyclic β-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J. Am. Chem. Soc. 133, 3144–3157. doi: 10.1021/ja110545h
Ziehm, T., Brener, O., van Groen, T., Kadish, I., Frenzel, D., Tusche, M., et al. (2016). Increase of positive net charge and conformational rigidity enhances the efficacy of d-enantiomeric peptides designed to eliminate cytotoxic Aβ species. ACS Chem. Neurosci. 7, 1088–1096. doi: 10.1021/acschemneuro.6b00047
Keywords: arginine, aggregation, Alzheimer’s disease, peptides, amyloid-beta (Aβ), tau & phospho-tau protein
Citation: Mamsa SSA and Meloni BP (2021) Arginine and Arginine-Rich Peptides as Modulators of Protein Aggregation and Cytotoxicity Associated With Alzheimer’s Disease. Front. Mol. Neurosci. 14:759729. doi: 10.3389/fnmol.2021.759729
Received: 17 August 2021; Accepted: 29 September 2021;
Published: 28 October 2021.
Edited by:
Wolfgang Hoyer, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Homira Behbahani, Karolinska Institutet (KI), SwedenCopyright © 2021 Mamsa and Meloni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Somayra S. A. Mamsa, c29tYXlyYS5tYW1zYUByZXNlYXJjaC51d2EuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.