 Frédéric Ebstein
Frédéric Ebstein Sébastien Küry
Sébastien Küry Jonas Johannes Papendorf
Jonas Johannes Papendorf Elke Krüger
Elke Krüger- 1 Institute of Medical Biochemistry and Molecular Biology, University Medicine Greifswald, Greifswald, Germany
- 2CHU Nantes, Service de Génétique Médicale, Nantes, France
- 3l’Institut du Thorax, CNRS, INSERM, CHU Nantes, Université de Nantes, Nantes, France
Over thirty years have passed since the first description of ubiquitin-positive structures in the brain of patients suffering from Alzheimer’s disease. Meanwhile, the intracellular accumulation of ubiquitin-modified insoluble protein aggregates has become an indisputable hallmark of neurodegeneration. However, the role of ubiquitin and a fortiori the ubiquitin-proteasome system (UPS) in the pathogenesis of neurodevelopmental disorders (NDD) is much less described. In this article, we review all reported monogenic forms of NDD caused by lesions in genes coding for any component of the UPS including ubiquitin-activating (E1), -conjugating (E2) enzymes, ubiquitin ligases (E3), ubiquitin hydrolases, and ubiquitin-like modifiers as well as proteasome subunits. Strikingly, our analysis revealed that a vast majority of these proteins have a described function in the negative regulation of the innate immune response. In this work, we hypothesize a possible involvement of autoinflammation in NDD pathogenesis. Herein, we discuss the parallels between immune dysregulation and neurodevelopment with the aim at improving our understanding the biology of NDD and providing knowledge required for the design of novel therapeutic strategies.
Introduction
Neurodevelopmental disorders (NDD) are a broad spectrum of early onset syndromes affecting the development of the central nervous system (CNS) with a prevalence in children that exceeds 15% worldwide (Romero-Ayuso, 2021). Formerly referred to as “mental retardation” NDD are typically characterized by deficits in cognitive function and adaptive behavior (Micai et al., 2020; Hanly et al., 2021). They traditionally encompass a wide range of different neurologic diseases ranging from mild to severe that include intellectual disability (ID), developmental delay (DD), autism spectrum disorder (ASD), cerebral palsy (CP), attention deficit/hyperactivity disorder (ADHD), Down syndrome (DS), bipolar disorders (BP), and epilepsy and schizophrenia (Ismail and Shapiro, 2019). One usually discriminates between NDD and neurodegenerative diseases (ND), the latter being a heterogeneous group of late-onset disorders marked by the intracellular accumulation of insoluble protein aggregates perturbing CNS function (Johnson, 2000; Koziorowski et al., 2021). Prominent ND include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and Lewy body dementia (LBD) which all mostly affect elderly individuals (Popa-Wagner et al., 2020; Tittelmeier et al., 2020). The NDD/ND dichotomy is, however, not strict since neurodegeneration may in some cases accompany neurodevelopmental anomalies and vice versa (Thibaut, 2018).
Brain pathologies such as NDD and/or ND are complex disorders which are caused for the most part by genetic and/or environmental factors (Cardoso et al., 2019; Dunn et al., 2019). For instance, unquestionable risk factors for the development of NDD include prenatal asphyxia (Adhikari and Rao, 2017) as well as exposure to ethanol (Sokol et al., 2003), heavy metals (Ijomone et al., 2020), and/or organic pollutants (Mesnil et al., 2020). The genetic components of a large fraction of these psychiatric disorders are difficult to unravel since most of them are not necessarily Mendelian (dominant, recessive, or X-linked) and involve the participation of allelic variants in several genes (Au et al., 2020; Savatt and Myers, 2021). However, it is estimated that approximately 40% of NDD are monogenic conditions predominantly due to lesions of a single gene (Deciphering Developmental, and Disorders, 2017; Brunet et al., 2021), and this figure even rises to about 50% in the case of ID (Kaufman et al., 2010; Karam et al., 2015; Reichenberg et al., 2016; Vissers et al., 2016). Because many of these vulnerable genes do not necessarily encode proteins specifically expressed in the brain with documented functions in neurodevelopment, our current understanding of disease pathogenesis remains extremely limited.
Hence, since their initial descriptions, increasing efforts have been made to better understand how NDD/ND emerge from deteriorated genes. One major breakthrough in this field was made by identification of ubiquitin-positive inclusion bodies in the brain of patients with AD (Mori et al., 1987), which led to the assumption that dysfunctions of the ubiquitin-proteasome system (UPS) may contribute to neurodegeneration. This notion was confirmed 1 year later by a work from the same group showing that Lewy bodies in the brain of six cases with LBD and PD were enriched with ubiquitin (Kuzuhara et al., 1988). Shortly afterward, it became evident that the accumulation of ubiquitin aggregates was not necessarily a histological hallmark restricted to neurodegeneration, but could also be found in the brain of children suffering from various NDD (Del Bigio et al., 1997). Meanwhile, the constantly increasing number of genomic alterations in genes encoding components of the UPS identified in patients with neurological phenotypes unambiguously points to its participation in the pathogenesis of psychiatric disorders. Nevertheless, the extreme versatility of the UPS makes it difficult to fully pinpoint its precise implication in disease pathogenesis, as discussed below.
The Ubiquitin-Proteasome System (UPS)
The UPS is a highly conserved pathway across eukaryotic species which ensures the rapid elimination of ubiquitin-tagged proteins by the 26S proteasome (Cetin et al., 2021). The ability of the UPS to remove virtually any type of protein substrate makes it indispensable for almost –if not all– basic cellular processes such as cell division, gene expression and signal transduction (Ebstein et al., 2012). A prerequisite for protein breakdown by 26S proteasomes is the covalent modification of intracellular targets with ubiquitin molecules (Wilkinson et al., 1980; Pickart, 2001, 2004; Pickart and Eddins, 2004). In this process, also referred to as ubiquitination (or ubiquitylation), three enzymes (i.e., E1, E2, and E3) catalyze the coordinated transfer of ubiquitin moieties to acceptor residues of proteins destined for degradation (Haas and Siepmann, 1997). Ubiquitination requires the activation of ubiquitin by a E1 ubiquitin-activating enzyme in an ATP-dependent reaction prior to its subsequent transfer onto a E2 ubiquitin-conjugating enzyme. With the help of E3 ubiquitin ligases, the charged E2-ubiquitin transfer ensures the ubiquitination of protein substrates on lysine, cysteine, serine or threonine residues (Tait et al., 2007; McDowell et al., 2010; Golnik et al., 2016; Swatek and Komander, 2016). Depending on their mode of ubiquitin transfer, E3 ubiquitin ligases can be divided into RING-, HECT- and RBR-type E3 ubiquitin ligases (Petroski and Deshaies, 2005; Metzger et al., 2012; Metzger et al., 2014). In contrast to ligases containing the RING (Really Interesting New Gene) finger domain which catalyze the ubiquitin transfer directly from the E2 to substrate proteins, HECT (homologous to E6AP C-terminus)-type E3 ligases first receive ubiquitin from the E2 on a cysteine residue and then transfer it to substrate proteins (Metzger et al., 2012). Among the RING-type ligases, Cullin-RING-type ligases (CRL) are multi-subunit ligases whose major component is a specific cullin (CUL) molecule which itself binds simultaneously to a RING-box protein (Rbx1 or Rbx2) and a substrate receptor (via an adaptor subunit in some cases) at its C- and N-terminus, respectively, (Harper and Schulman, 2021). Because RING-box proteins recruit conjugated E2, CUL are widely regarded as scaffold molecules bridging E2 to substrate proteins. Typical substrate receptors include F-BOX proteins, BTB domain-containing proteins and DCAF proteins which are ligands for CUL1/7, CUL3, and CUL4, respectively, (Harper and Schulman, 2021). Finally, the RBR (RING-in -between RING)-type E3 ubiquitin ligase family includes members that transfer ubiquitin to substrates in a non-canonical manner via a RING/HECT combined process (Uchida and Kitagawa, 2016).
The tagging of intracellular proteins with one ubiquitin moiety is referred to as mono-ubiquitination and is widely viewed as a post-translational process regulating subcellular localization (Sigismund et al., 2004) and gene expression (Marsh et al., 2020). Multiple mono-ubiquitination, namely the addition of one ubiquitin molecule on multiple sites of the same substrate occurs as well and signals either endocytosis, protein trafficking and lysosomal degradation or proteasome-mediated degradation (Livneh et al., 2017).
Most importantly, the ubiquitin molecule itself may be subjected to ubiquitin modification on either one of its eight acceptor sites (K6, K11, K27, K29, K33, K48, K63, and Met-1), thereby generating poly ubiquitin chains carrying distinct ubiquitin linkages. The linkage type determines both the topology of the poly ubiquitin chain and the outcome of the modified substrate. It is well established that poly ubiquitin chains bearing K48-linkages typically deliver the modified protein for subsequent degradation by 26S proteasomes (Pickart and Fushman, 2004). The 26S proteasome is a multi-subunit complex consisting of a 19S regulatory particle and a barrel-shaped 20S core particle (Dahlmann, 2005; Tanaka et al., 2012). While the 19S regulatory particle is specialized in ubiquitin recognition and removing as well as substrate unfolding, the 20S core particle ensures protein breakdown into short peptides via its catalytic β-subunits (Finley et al., 2016; Bard et al., 2018). Substrates modified with K48-linked are rapidly recognized by the ubiquitin receptors PSMD4 and ADRM1 on the 19S regulatory particle which facilitate their translocation into the 20S core particle (Deveraux et al., 1995; Husnjak et al., 2008). The binding of ubiquitin-modified proteins with 26S proteasomes is usually strengthened with the help of protein shuttles which are capable of interacting with both ubiquitin chains and proteasomes via their UBA and UBL domains, respectively, (Chen et al., 2016).
The degradation signal exemplified by K48-linked ubiquitin chains represents just the tip of the iceberg of the ubiquitin code, as the other seven ubiquitination sites of ubiquitin may be used either singly or in combination to generate homotypic or mixed poly ubiquitin chains, respectively, that convey multiple cellular functions including lysosomal targeting and DNA repair to name a few (Akutsu et al., 2016; Grumati and Dikic, 2018). Complexity to the UPS pathway arises further with the existence of ubiquitin-like modifiers which, via a conjugation process similar to that of ubiquitin, are capable of modifying cellular targets in a covalent manner. Ubiquitin-like proteins encompass the ISG15, FAT10, NEDD8, URFM1, UFM1, and ATG12 modifiers as well as those of the ATG8 and SUMO families (Cappadocia and Lima, 2018). Thanks to their ability to tag intracellular substrates, ubiquitin-like modifiers generate an extreme variety of signals including proteolytic and non-proteolytic ones (Streich Jr., and Lima, 2014). Strikingly, SUMO, and to a lesser extent NEDD8 and ISG15, may themselves be subjected to ubiquitination at various lysine residues, thereby giving rise to hybrid chains whose biological functions, however, have not been fully elucidated (Perez Berrocal et al., 2019; Mulder et al., 2020).
Importantly, both ubiquitin and ubiquitin-like modifications are reversible processes which can be counteracted anytime by ubiquitin hydrolases (also called deubiquitinating enzymes, DUB). Up to now, an approximate number of 100 DUB have been identified, whereby the largest families are represented by the ubiquitin-specific proteases (USP), the ovarian tumor proteases (OTU), and ubiquitin C-terminal hydrolases (UCH) (Clague et al., 2019). Many DUB and E3 ubiquitin ligases regulate fundamental cellular pathways including cell division or death, genomic integrity, epigenetic control, developmental, and differentiation pathways as well as cellular homeostasis (Morgan and Crawford, 2021).
As alluded to earlier, the UPS pathway is frequently damaged in several forms of NDD by genomic alterations that may affect either one of the many genes encoding its various components. Because virtually any gene seems vulnerable, any stage of this process may be impaired from ubiquitin transfer to ubiquitin removal and/or proteasome-mediated breakdown of ubiquitin-modified proteins (Figure 1). These observations clearly point to a cause-and-effect relationship between perturbed UPS function and NDD onset, as discussed below.
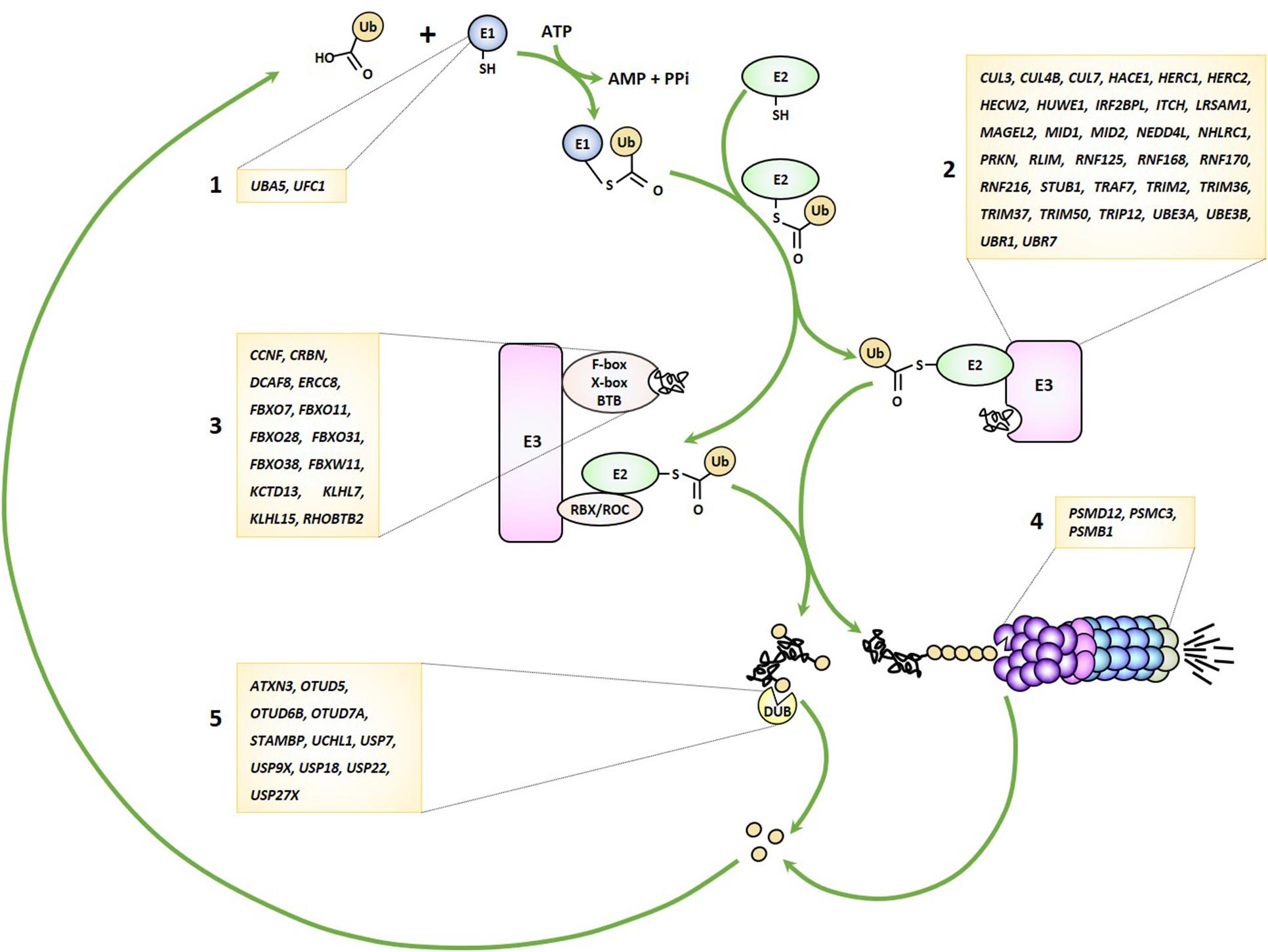
Figure 1. Genetic lesions identified in patients with NDD may affect any stage of the UPS pathway. UPS-related genes disrupted in NDD encode various components of the ubiquitin-conjugation system including E1 ubiquitin-activating enzymes (1), E3 ubiquitin ligases (2), Cullin-RING-type E3 ubiquitin ligases (3), proteasome subunits (4) and deubiquitinating enzymes (5), as indicated.
E3 Ubiquitin Ligases in NDD
Ubiquitin ligases are by far the largest group of UPS genes identified as NDD causing-genes. The first identified member of this constantly growing family is the UBE3A gene encoding the E6-AP HECT-type E3 ubiquitin ligase and whose loss-of-function has been shown to cause Angelman syndrome more than twenty years ago (Kishino et al., 1997; Matsuura et al., 1997; Sutcliffe et al., 1997). Because UBE3A is exclusively expressed from the maternal allele in neurons, any deletion or point mutations affecting the maternal chromosome leads to loss of E6-AP expression in these cells and result in the acquisition of a neuronal phenotype mostly characterized by absent speech, intellectual disability and happy demeanor with unusually frequent laughing smiling (Maranga et al., 2020).
Since the original reports associating UBE3A with Angelman syndrome in 1997, approximately forty-five further genes coding for E3 ubiquitin ligases or CRL substrate receptors have been identified as causative genes for forty-eight different forms of NDD (Tables 1, 2). Clinical features commonly observed in all these syndromes include developmental delay, cognitive deficits, dysmorphic facial features, hypotonia and seizures. However, given the large variety of signals generated by E3 ubiquitin ligases, the phenotypic spectrum of NDD subjects with loss-of-function in E3 genes may vary to a large degree. Herein, limb anomalies such as brachydactyly, polydactyly, or camptodactyly are frequently detected in patients carrying genomic alterations in the HUWE1, TRAF7, UBE3B, ITCH, or FBXW11 genes (Buntinx and Majewski, 1990; Lohr et al., 2010; Moortgat et al., 2018; Tokita et al., 2018; Holt et al., 2019), while gonadal dysfunction seems to be restricted to a subset of NDD cases carrying variants of the RNF216, STUB1, TRIM37, or KLHL15 genes (Seminara et al., 2002; Jagiello et al., 2003; Heimdal et al., 2014; Mignon-Ravix et al., 2014).
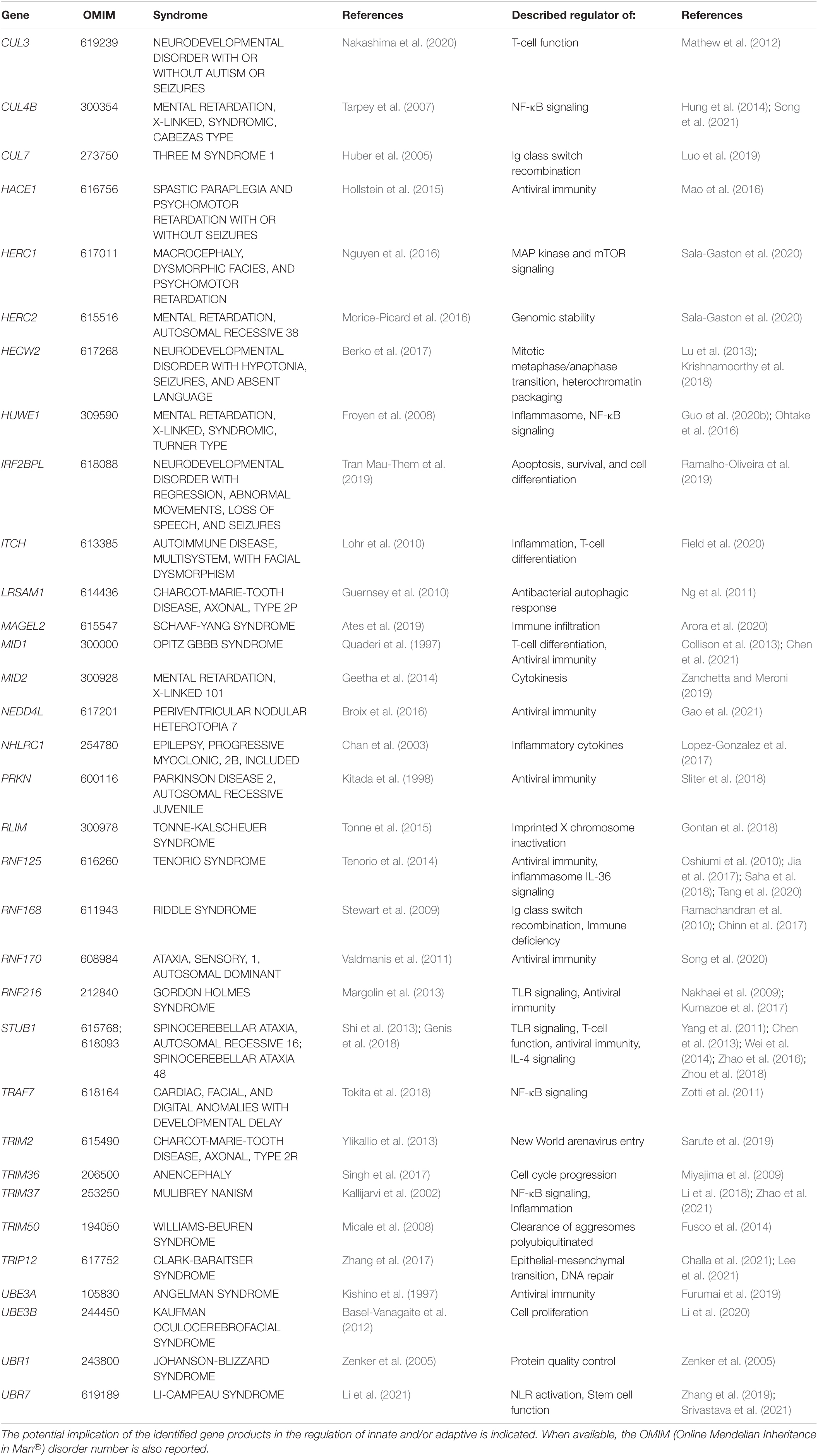
Table 1. NDD-causing genes encoding E3 ubiquitin ligases and associated syndromes.
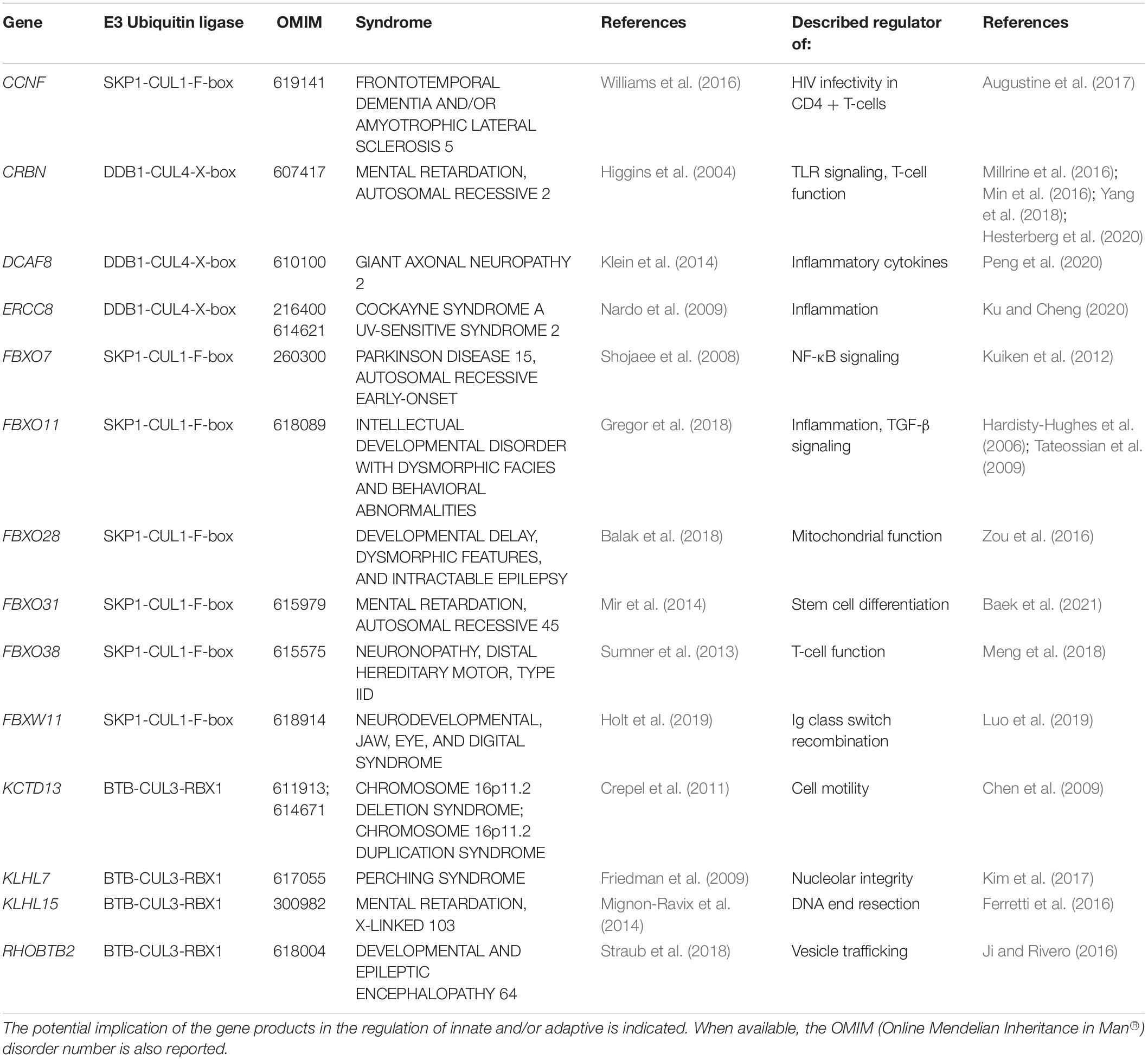
Table 2. NDD-causing genes encoding CUL substrate receptors and associated syndromes.
E3 Ubiquitin Ligases and Protein Aggregation in NDD
Over the last two decades, many attempts have been made to unravel the molecular pathogenesis of NDD caused by ubiquitin ligase dysfunction. One straightforward route to address this point consists of identifying downstream target substrates of the affected ligases by proteomic-based methods (Rayner et al., 2019). This strategy is nevertheless hampered by the fact that E3 ubiquitin ligases may have multiple substrates which themselves may fulfill many different functions. One prime example of such ligases is CHIP encoded by the STUB1 gene and whose genomic alterations cause spinocerebellar ataxia (Shi et al., 2013). Thanks to its ability to bind to cellular chaperones such as HSP70 and HSP90, CHIP mediates the ubiquitination of misfolded proteins, thereby targeting them for subsequent degradation by 26S proteasomes (Edkins, 2015; Wang et al., 2020). Such misfolded proteins typically encompass defective ribosomal products (DRIPS) which are generated during protein biosynthesis as a consequence of ribosomal mistranslation (Yewdell et al., 1996). Pioneering work of J. Yewdell and colleagues has estimated that DRIPS may account for 25% of the total pool of newly synthetized proteins in eukaryotic cells (Schubert et al., 2000; Princiotta et al., 2003; Qian et al., 2006). This implies that virtually any intracellular protein may become a target of CHIP, making it impossible to associate CHIP defects with one particular cellular pathway and/or function. The extremely broad substrate specificity of CHIP also presupposes that STUB1 loss-of-function results in the accumulation of various misfolded and/or damaged proteins that fail to undergo ubiquitination. Whether these protein aggregates are toxic as a whole and contribute to the pathogenesis of spinocerebellar ataxia is unclear. A fortiori, these inclusions would be devoid of ubiquitin molecules and, as such, not reminiscent of those typically accumulating during neurodegeneration. Herein, this assumption would underline a major distinction between NDD and ND, as it would preclude that the perturbations of protein homeostasis associated with NDD are not due to proteolytic dysfunction. Besides DRIPS, one cannot exclude that NDD due to STUB1 loss-of-function mutations may occur as a consequence of the inability of the cells to remove specific full-length mature proteins, which would then drive the disease by perturbing specific cellular pathways.
Like CHIP, UBR1, and UBR7, whose deficiencies reportedly cause the Johanson-Blizzard (Zenker et al., 2005) and Li-Campeau syndromes (Li et al., 2021), respectively, are E3 ubiquitin ligases with multiple potential substrates. As members of the N-end rule pathway, UBR1 and UBR7 target any protein carrying N-terminal destabilizing motifs (also referred to as “N-degrons”) such as arginine residues for degradation (Varshavsky, 2019). Substrates of the N-end rule pathway physiologically arise from limited proteolysis and encompass a wide variety of intracellular proteins fulfilling various functions in cell signaling, cellular homeostasis and apoptosis (Varshavsky, 2019). Herein, the multitude of pathways potentially affected by UBR1 and/or UBR7 loss-of-function mutations substantially challenges our understanding of NDD pathophysiology. In addition, one cannot exclude that the diseases may be triggered by the unspecific accumulation of N-end rule substrates that would affect cell function and/or integrity. In any case, CHIP, UBR1 and UBR7 exemplify the difficulty of deciphering the molecular pathogenesis of syndromes due to E3 ubiquitin ligase which have multiple substrates.
NDD-Associated E3 Ubiquitin Ligases and Their Roles in the Immune Response
Another E3 ubiquitin ligase potentially causing NDD with a wide range of substrates is ITCH, whose genetic disruption has been shown to cause a syndromic multisystem autoimmune disease referred to as autoimmune disease, multisystem, with facial dysmorphism (ADMFD) (Lohr et al., 2010). Interestingly, ADMFD is also a neurological disease with affected children exhibiting typical NDD features while developing autoimmune systemic responses at the same time. The immunological component of ADMFD is not surprising in view of the substantial number of ITCH cellular targets which play critical roles in T- and B-cell function. These notably include the T-cell receptor (TCR) chain-ζ as well as the RAR-related orphan receptor (ROR)-γt transcription factor, which control T-cell signaling and differentiation, respectively (Huang et al., 2010; Kathania et al., 2016).
The observation that NDD may be accompanied by immune manifestations is somehow intriguing and raises the question as to whether an unrestrained innate and/or adaptive immune response (i.e., autoinflammation and/or autoimmunity) might underlie the pathogenesis of NDD. Strikingly, besides ITCH, more than two-thirds of the E3 ubiquitin ligases reported to cause NDD have critical functions in the innate and adaptive immune systems. As listed in Tables 1, 2, the E3 ubiquitin ligases CUL4B (Hung et al., 2014; Song et al., 2021), HUWE1 (Ohtake et al., 2016; Guo et al., 2020b), RNF216 (Kumazoe et al., 2017), STUB1 (Yang et al., 2011), TRAF7 (Zotti et al., 2011), TRIM37 (Li et al., 2018; Zhao et al., 2021), CRBN (Min et al., 2016; Yang et al., 2018), and the substrate recognition component FBXO7 (Kuiken et al., 2012) have been shown to regulate the expression of inflammatory cytokines mostly thanks to their capacity of modulating NF-κB signaling and/or the inflammasome. It is worth noting that, except HUWE1, all these ligases are described as inflammation negative regulators of these pathways (Figure 2), implying that any loss-of-function of any one of these genes would result in the sustained production of pro-inflammatory cytokines.
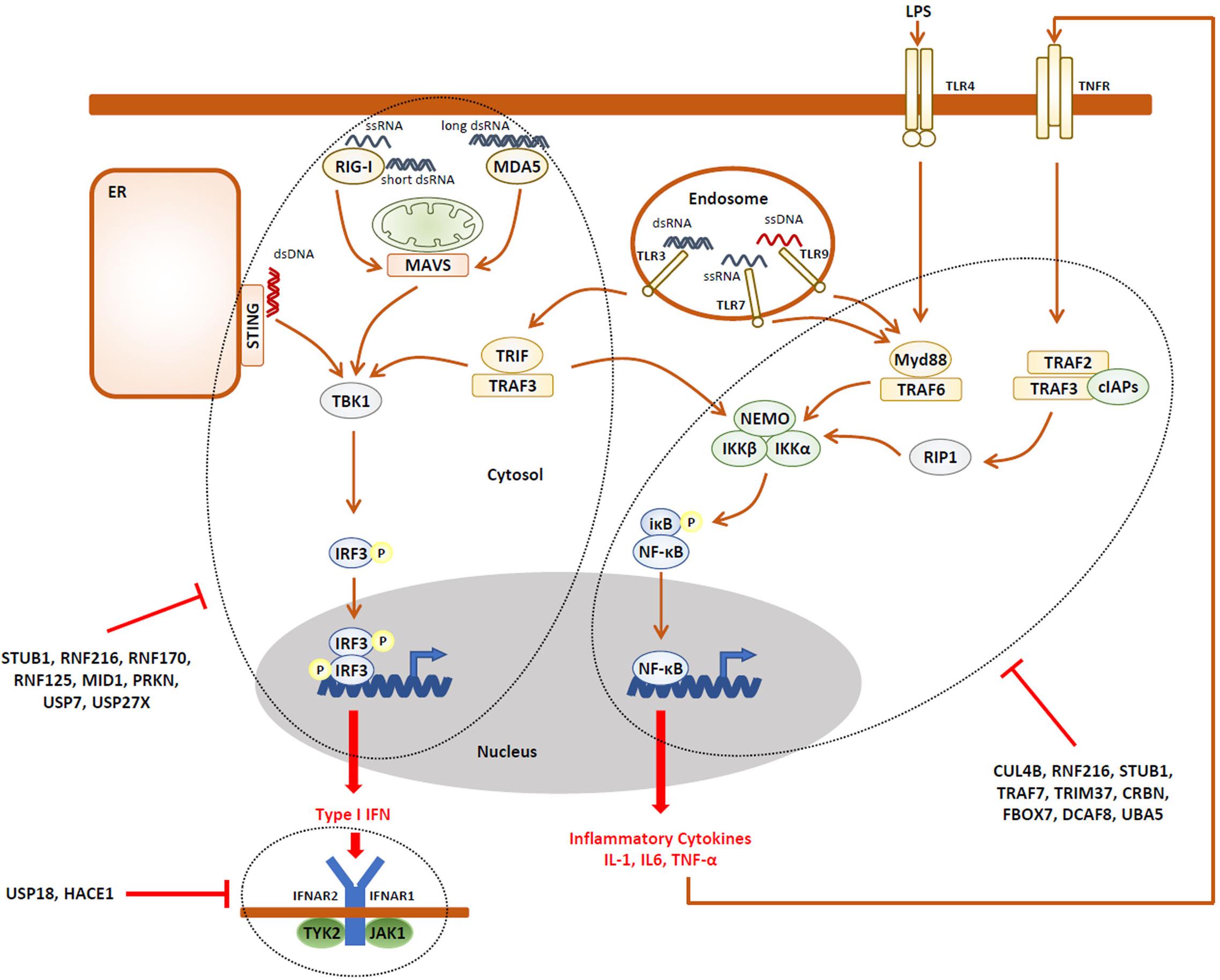
Figure 2. A large fraction of the UPS gene products associated with NDD are negative regulators of the innate immune response. Activation of pathogen recognition receptors (PRR, i.e., TLR3, TLR4, TLR7, TLR9, RIG-1, MDA5, and STING) by pathogen-associated molecular patterns (PAMP, i.e., LPS, ssRNA, dsRNA, and CpG) results in the activation of intracellular signaling cascades which ultimately promote the nuclear translocation of the IRF3 and NF-κB transcriptions factors, as indicated. These, in turn, directly induce the expression of pro-inflammatory cytokines (i.e., TNF-α) and type I IFN which engage the innate arm of the immune system through autocrine and/or paracrine loops. Both the IRF3 and NF-κB activation pathways are down-regulated by many components of the UPS which have been associated with NDD, as indicated. In addition, the autocrine/paracrine action of type I IFN is subjected to negative regulation by the UPS genes USP18 and HACE1, as indicated.
It is also understood that the E3 ubiquitin ligases HACE1 (Mao et al., 2016), MID1 (Chen et al., 2021), NEDD4L (Gao et al., 2021), PRKN (Sliter et al., 2018), RNF125 (Arimoto et al., 2007), RNF170 (Song et al., 2020), RNF216 (Nakhaei et al., 2009), STUB1 (Zhou et al., 2018), as well as UBE3A (Furumai et al., 2019) are involved in antiviral innate defense and the generation of type I interferon (IFN) responses (Figure 2). Again, besides NEDD4L, all these genes encode ligases involved in type I IFN negative feedback loops and, as such any dysfunction, would lead to uncontrolled type I IFN responses.
Some other NDD ubiquitin and/or CRL ligases seem to exert their activity predominantly during the adaptive immune response. These include CUL7 together with the substrate receptor FBXW11 (Luo et al., 2019) as well as RNF168 (Ramachandran et al., 2010) which regulate immunoglobulin switch recombination or CUL3 (Mathew et al., 2012), ITCH (Field et al., 2020), MID1 (Collison et al., 2013), STUB1 (Chen et al., 2013), CRBN (Hesterberg et al., 2020), and FBXO38 (Meng et al., 2018) that are involved in T-cell function and/or differentiation.
Despite the prominent implication of ligases in multiples levels of the innate immune system, only a very small number of NDD cases are associated with symptoms of autoinflammation. These include the Tenorio syndrome caused by RNF125 deficiency which, beside syndromic intellectual disability, leads to a severe inflammatory phenotype with recurrent episodes of conjunctivitis and stomatitis (Tenorio et al., 2014). The immune manifestations of the Tenorio syndrome clearly corroborate the fact that RNF125 acts as a negative regulator of innate immune signaling by targeting key pattern recognition receptors (i.e., RIG-1, MDA5) for degradation (Arimoto et al., 2007, 2018). Also found in this category of NDD are the Williams-Beuren and Cockayne syndrome which are characterized by intracranial calcifications (Neill and Dingwall, 1950; Knudtzon et al., 1987; Wilson et al., 2016), a typical trait of neuroinflammation (Saade et al., 2019). Ironically, and in contrast to the Tenorio syndrome, both Williams-Beuren and Cockayne syndromes are caused by genomic alterations in genes encoding E3 ubiquitin ligases (i.e., TRIM50 and ERCC8) with no described function in the immune system. Non-etheless, a closer look at their cellular targets reveals that both of these ligases may intersect with host innate immune defenses. In effect, one major substrate of TRIM50 includes BECN1 (Fusco et al., 2018), an important component of the autophagy lysosomal degradation pathway that recruits autophagy proteins to the phagophore assembly site (Kang et al., 2011). Interestingly, it has been shown that BECN1 is capable of activating NF-κB (Leonard et al., 2019), suggesting that any perturbations of its turnover due to TRIM50 deficiency might result in sustained inflammatory responses. In a similar manner, because of its implication in transcription-coupled nucleotide excision repair (TC-NER) in response to ultraviolet (UV) irradiation (Nardo et al., 2009), ERCC8 may render the cells susceptible for autoinflammation. In fact, it is conceivable that ERCC8 loss-of-function might result in abnormal cytosolic accumulation of damaged transcripts, which in turn may be sensed as non-self RNA by immune cells.
The lack of peripheral immune manifestations in NDD caused by the disruption of other E3 ubiquitin ligases may seem surprising at first sight, but it does not necessarily preclude the absence of ongoing autoinflammation and/or autoimmunity in these patients. Indeed, immune-inflammatory parameters have been frequently detected in NDD subjects seemingly devoid of clinical inflammatory symptoms. For instance, an elevated pro-inflammatory cytokine blood profile has been reported in patients with ASD (Eftekharian et al., 2018; Matta et al., 2019), epilepsy (Riazi et al., 2010), schizophrenia (Potvin et al., 2008; Miller et al., 2011; Tourjman et al., 2013), BP (Benedetti et al., 2020), and ADHD (Zhou et al., 2017). This also holds true for both Aicardi-Goutières and Down syndromes, two NDD which fail to exhibit clinical features of systemic inflammation but fall into the category of interferonopathies because of their sustained production of type I IFN (Crow and Manel, 2015; Livingston and Crow, 2016; Sullivan et al., 2016; Sullivan et al., 2017; Waugh et al., 2019).
Deubiquitinating Enzymes (DUB) in NDD
As illustrated in Table 3, a total of ten DUB have been reported as diseases-causing genes for various forms of NDD. Strikingly, eight of them have described roles in the immune system. These include STAMBP (Bednash et al., 2017, 2021), UCHL1 (Karim et al., 2013), USP7 (Daubeuf et al., 2009; Colleran et al., 2013; Palazon-Riquelme et al., 2018), and USP9X (Xiang et al., 2019) which have all been shown to modulate the expression of pro-inflammatory cytokines thanks to their capacity of interfering with the NF-κB signaling and/or inflammasome pathways. Other DUB exert a more specific action on type I IFN responses, rendering them essential at the very first line of innate antiviral defense. One prominent member of this family is undoubtedly USP18 which negatively regulates type I IFN signaling by competing with Janus kinase 1 (JAK1) for binding to IFN α/β receptor 2 (IFNAR2) (Malakhova et al., 2006). The observation that USP18 loss-of-function mutations give rise to brain malformations in patients with pseudo-Torch syndrome (Meuwissen et al., 2016) strongly suggests a cause-and-effect relationship between type I IFN and neurodevelopmental disabilities. Further support for this notion comes from the identification of USP7 and USP27X as disease-causing genes for the Hao-Fountain syndrome and X-linked mental retardation, respectively, (Hao et al., 2015; Hu et al., 2016). Both of these genes encode DUB that stimulate a type I IFN negative feedback mechanism by removing K63-linked poly ubiquitin chains on critical components of the antiviral signaling pathway such as RIG-I (Tao et al., 2020) and TBK1 (Cai et al., 2018).
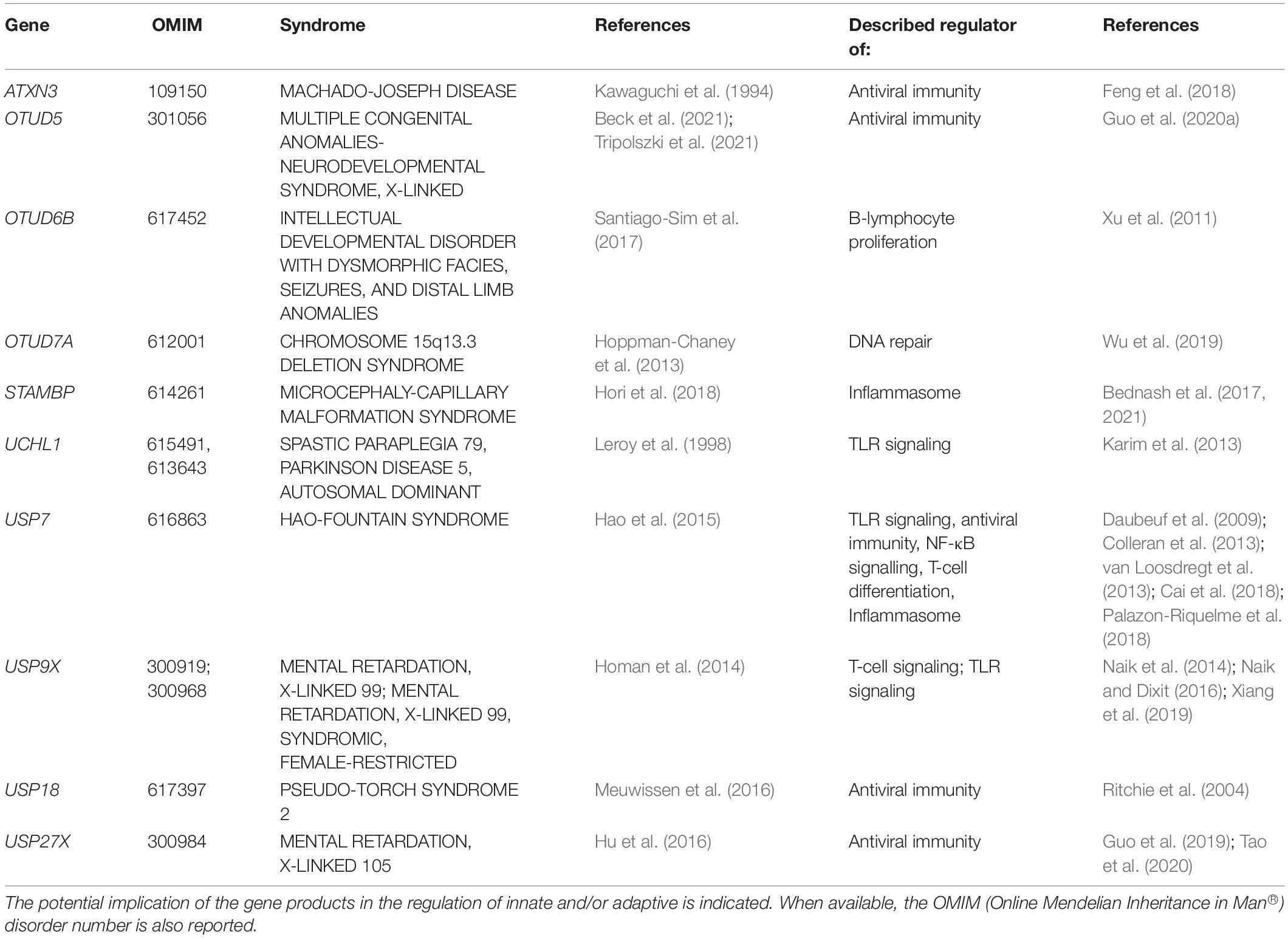
Table 3. NDD-causing genes encoding DUB and associated syndromes.
The association between type I IFN in NDD pathogenesis is, however, challenged by the fact that two DUB identified as NDD-causing have been also described as potent inducers of type I IFN. These include ATXN3 and OTUD5, respectively, causing the Machado-Joseph disease and a X-linked multiple congenital anomalies-neurodevelopmental syndrome (Kawaguchi et al., 1994; Beck et al., 2021; Tripolszki et al., 2021). Indeed, by removing proteolytic poly ubiquitin chains from the cytosolic DNA sensor STING, OTUD5 facilitates antiviral innate signaling and the subsequent transcription of type I IFN genes (Guo et al., 2020a). Likewise, ATXN3 has been shown to exacerbate type I antiviral response via the deubiquitination and stabilization of histone deacetylase 3 (HDAC3) (Feng et al., 2018). As such, any loss-of-function of any of these DUB would mitigate type I IFN responses and contradicts the view that NDD is associated with increased inflammation. However, it is highly likely that ATX3 deficiency exert its pathogenic effect independent of its deubiquitination function, since Machado-Joseph disease is a triplet (CAG encoding glutamine, Q) repeat expansion disorder whereby ATXN3 mutant proteins accumulate as toxic insoluble protein aggregates (Chai et al., 1999). Quite on the contrary, it has been shown that cell lines expressing expanded ATXN3 were characterized by increased transcription of pro-inflammatory cytokines (Evert et al., 2001; Evert et al., 2006). As for OTUD5, its recently described implication in DNA damage-induced transcriptional repression (de Vivo et al., 2019) opens the possibility that its genomic disruption would generate a danger signal leading to inflammation via excessive cytosolic mRNA accumulation.
Very little is known about the biological function and/or cellular targets of OTUD7A and OTUD6B, whose genetic lesions cause the chromosome 15q13.3 deletion syndrome and an intellectual developmental disorder with dysmorphic facies, seizures, and distal limb anomalies (Santiago-Sim et al., 2017; Uddin et al., 2018), respectively. Interestingly, common to both disorders is a decreased proteasome function ultimately resulting in the aggregation of ubiquitin-modified protein (Santiago-Sim et al., 2017; Garret et al., 2020). These studies suggest that OTUD7A and OTUD6B are directly or indirectly involved in the regulation of proteasome-mediated proteolysis and that their deficiencies would result in cellular situations very similar neurodegeneration ones. A distant and indirect member of the DUB family associated with NDD phenotypes is USP22 which participates in the pathogenesis of spinocerebellar ataxia 7 caused by poly Q repeats in the ATX7 gene (David et al., 1997). Both UPS22 and ATX7 are parts of the Spt-Ada-Gcn5 Acetyl transferase (SAGA) complex which promotes gene transcription via histone acetylation and deubiquitination activities (Melo-Cardenas et al., 2016). It is argued that the generation of poly Q Ataxin-7 protein aggregates substantially affects the access of USP22 to its cellular substrates notably histone H2B (Henry et al., 2003), a dysfunction likely contributing to disease onset. Most interestingly, it has been shown that USP22 gene silencing is accompanied by activation of the JAK-STAT1 signaling pathway (Han et al., 2020), thereby raising the possibility that type I IFN might be a component of spinocerebellar ataxia 7.
Proteasomes in NDD
As shown in Table 4, the most recent identified group of UPS genes associated with NDD include those encoding proteasome subunits. In this short list are found the PSMD12, PSMC3 and PSMB1 genes that cause the Stankiewicz-Isidor syndrome (STISS), a neurosensory syndrome combining deafness and cataract as well as a disorder characterized by microcephaly, intellectual disability, developmental delay and short stature, respectively, (Kury et al., 2017; Ansar et al., 2020; Kroll-Hermi et al., 2020). Like most of the NDD due to genomic alterations of UPS genes, these syndromes are seemingly devoid of systemic signs of immune dysregulation. This observation is even more surprising considering the fact that proteasome loss-of-function mutations have been described to cause autoinflammatory diseases referred to as proteasome-associated autoinflammatory syndromes (PRAAS) or chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) (Agarwal et al., 2010; Arima et al., 2011; Kitamura et al., 2011; Liu et al., 2012; Brehm et al., 2015; Poli et al., 2018; de Jesus et al., 2019; Sarrabay et al., 2019; Kataoka et al., 2021). In contrast to the NDD alterations that may affect 20S or 19S proteasome complexes, the CANDLE/PRAAS mutations are exclusively located in the 20S core particle and/or proteasome assembly chaperones (Ebstein et al., 2019). Common to all CANDLE/PRAAS subject is a type I IFN gene signature characterized by increased amounts of transcripts encoding canonical IFN-stimulated genes such as ISG15, SIGLEC-1, IFI44L, IFIT1, IFI27, and RSAD2 (Brehm and Krüger, 2015). Interestingly, resetting the immune system of patients with mutation in the proteasome assembly maturation protein (POMP) via hematopoietic stem cell transplantation (HSCT) could successfully reverse the clinical and molecular features of CANDLE/PRAAS (Martinez et al., 2021), indicating that the signature is mostly generated by immune cells. Although some patients may exhibit signs of cognitive impairment, CANDLE/PRAAS are usually not dominated by typical neuropsychological and biological features of a neurodevelopmental disorder, thereby making them distinct from classical NDD. Conversely, and unlike CANDLE/PRAAS, NDD due to lesions in PSMD12, PSMC3 and PSMB1 genes fail to develop systemic autoinflammation, which prevent them from falling into autoinflammatory disease categories. The reasons why proteasomes loss-of-function mutations lead to two clinically distinct phenotypes are unclear and warrant further investigations. Clearly, the divergence between the two diseases is not dictated by the location of the affected subunit within the 26S proteasome, as initially assumed (Ebstein et al., 2019). The notion that CANDLE/PRAAS develop peripheral autoimmunity is not unexpected given the pleiotropic role of proteasomes in multiple inflammatory signal cascades (Cetin et al., 2021; Goetzke et al., 2021). On the contrary, the lack of systemic manifestations in NDD due to proteasome loss-of-function mutations is intriguing, but again does not necessarily imply the absence of autoinflammation in some tissues and/or the generation of atypical inflammatory signatures that may have been overlooked.

Table 4. NDD-causing genes encoding proteasome subunits and associated syndromes.
UFMylation in NDD
As shown in Table 5, the last and smallest group of UPS-related genes responsible for NDD comprises the two E1 ubiquitin-activating enzymes UBA5 (Duan et al., 2016; Muona et al., 2016) and UFC1 (Nahorski et al., 2018). Both of these proteins belong to the recently described Ubiquitin-fold modifier 1 (Ufm1)-conjugation system whose biological relevance remains to be fully understood. Because the only E3 Ufm1 ligase identified so far (i.e., UFL1) is recruited at the cytosolic side of the endoplasmic reticulum (ER) membrane (Wu et al., 2010), it is thought that Ufm1 modification is involved in ER protein quality control and/or homeostasis (Adamson et al., 2016). This notion is in line with recent studies showing that proteins involved in these processes such as RPL26 and RPN1 are cellular targets of the (Ufm1)-conjugation pathway (Walczak et al., 2019; Liang et al., 2020). Most importantly, it seems that Ufm1 modification at the ER represses the unfolded protein response (UPR) (Liang et al., 2020), a pathway known to cause sterile inflammation (Ebstein et al., 2019). It is therefore conceivable that Ufm1 loss-of-function might result in sustained overactivation of the UPR and expression of inflammatory markers. This assumption is in agreement with recent reports showing that Ufm1 attenuates inflammation induced by LPS (Li et al., 2017, 2019). Although patients with UBA5 loss-of-function mutations fail to show noticeable symptoms of inflammation, one can again not exclude that these syndromes are devoid of inflammatory process.
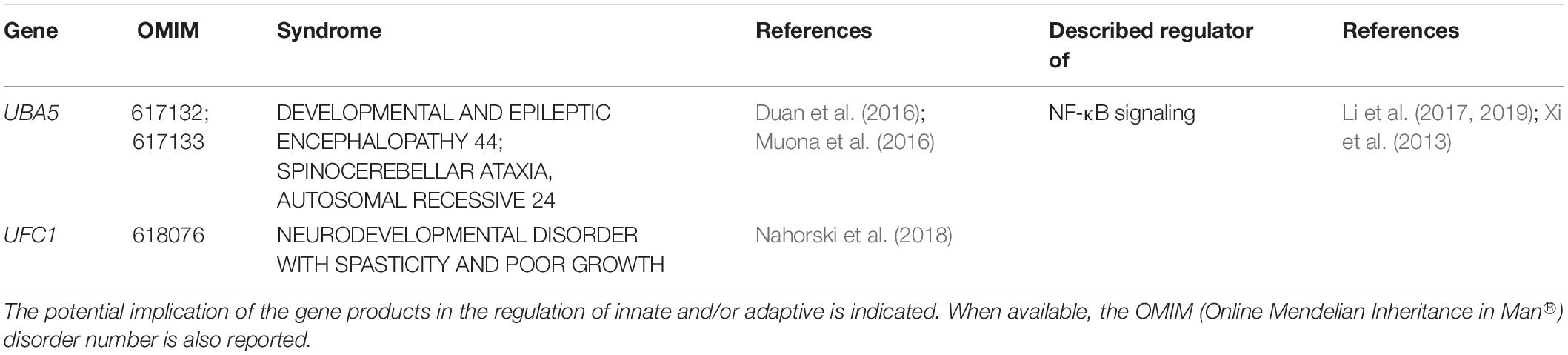
Table 5. NDD-causing genes encoding components of the Umf1-conjugation pathway and associated syndromes.
Conclusion and Future Directions
In this review, we have identified 62 reported monogenic NDD directly caused by lesions in genes encoding components of the UPS (Tables 1–5). To our surprise, 37 of these genes encode products that have been shown to regulate the immune system at various levels (Tables 1–5). Specifically, 20 of them are negative regulators of the two major (i.e., NF-κB and IRF) pathways in inflammation and antiviral response as well as type I IFN signaling (Figure 2). We believe that this number is likely underestimated, as many cellular targets of the identified ubiquitin ligases and/or DUB encompass proteins involved in DNA/RNA processing which may alert the immune system upon dysfunction through the generation of dangers signals. Altogether, this analysis strengthens the straightforward assumption that uncontrolled inflammation contributes to the pathogenesis of psychiatric disorders including NDD, although this remains to be formally demonstrated. One general contradiction stemming from our work is the fact that a great majority of NDD patients does not exhibit typical symptoms of chronic inflammation. However, suspecting subtle and stealthy levels of inflammation remains a challenge for pediatricians and it is likely that children apparently devoid of clinical signs of inflammation are not tested for immune disorders. In this regard, the absence of standardized diagnostic assays for a number of pro-inflammatory cytokines, particularly type I IFN, makes also the detection of specific and atypical inflammatory signatures difficult. One further possible explanation for this discrepancy may be that the UPS components affected in NDD exhibit a tissue-specific distribution, thereby promoting a more localized inflammation rather than a systemic one. In view of the neuronal phenotype of these disorders, it is highly likely that most of these genes are expressed in the CNS including microglia cells and astrocytes which might represent a potential source of inflammation in response to UPS dysfunction. As such, it is conceivable that inflammation might be restricted to the cerebrospinal fluid (CSF) in these patients. Future work aiming to address the role of pro-inflammatory mediators on neuron differentiation and/or function will help improve our understanding of disease pathogenesis and identify therapeutic targets for NDD.
Author Contributions
FE conceived, wrote, and edited the manuscript. SK, JJP, and EK participated in data analysis and provided intellectual input into the manuscript. FE and SK have designed the figures and tables.
Funding
This work was supported by grants of the German Research foundation (DFG SFBTR 167 TP A4) to EK. We acknowledge support for the Article Processing Charge from the DFG (German Research Foundation, 393148499) and the Open Access Publication Fund of the University of Greifswald.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adamson, B., Norman, T. M., Jost, M., Cho, M. Y., Nunez, J. K., Chen, Y., et al. (2016). A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21.
Adhikari, S., and Rao, K. S. (2017). Neurodevelopmental outcome of term infants with perinatal asphyxia with hypoxic ischemic encephalopathy stage II. Brain Dev. 39, 107–111. doi: 10.1016/j.braindev.2016.09.005
Agarwal, A. K., Xing, C., Demartino, G. N., Mizrachi, D., Hernandez, M. D., Sousa, A. B., et al. (2010). PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet. 87, 866–872. doi: 10.1016/j.ajhg.2010.10.031
Akutsu, M., Dikic, I., and Bremm, A. (2016). Ubiquitin chain diversity at a glance. J. Cell Sci. 129, 875–880.
Ansar, M., Ebstein, F., Ozkoc, H., Paracha, S. A., Iwaszkiewicz, J., Gesemann, M., et al. (2020). Biallelic variants in PSMB1 encoding the proteasome subunit beta6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 29, 1132–1143. doi: 10.1093/hmg/ddaa032
Arima, K., Kinoshita, A., Mishima, H., Kanazawa, N., Kaneko, T., Mizushima, T., et al. (2011). Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder. nakajo-nishimura syndrome. Proc. Natl. Acad. Sci. U S A. 108, 14914–14919. doi: 10.1073/pnas.1106015108
Arimoto, K., Takahashi, H., Hishiki, T., Konishi, H., Fujita, T., and Shimotohno, K. (2007). Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. U S A. 104, 7500–7505. doi: 10.1073/pnas.0611551104
Arimoto, K. I., Miyauchi, S., Stoner, S. A., Fan, J. B., and Zhang, D. E. (2018). Negative regulation of type I IFN signaling. J. Leukoc Biol. Online ahead of print.
Arora, M., Kumari, S., Singh, J., Chopra, A., and Chauhan, S. S. (2020). Downregulation of brain enriched Type 2 MAGEs is associated with immune infiltration and poor prognosis in glioma. Front. Oncol. 10:573378. doi: 10.3389/fonc.2020.573378
Ates, T., Oncul, M., Dilsiz, P., Topcu, I. C., Civas, C. C., Alp, M. I., et al. (2019). Inactivation of Magel2 suppresses oxytocin neurons through synaptic excitation-inhibition imbalance. Neurobiol. Dis. 121, 58–64. doi: 10.1016/j.nbd.2018.09.017
Au, P. Y. B., Eaton, A., and Dyment, D. A. (2020). Genetic mechanisms of neurodevelopmental disorders. Handb. Clin. Neurol. 173, 307–326.
Augustine, T., Chaudhary, P., Gupta, K., Islam, S., Ghosh, P., Santra, M. K., et al. (2017). Cyclin F/FBXO1 interacts with HIV-1 Viral Infectivity Factor (Vif) and restricts progeny virion infectivity by ubiquitination and proteasomal degradation of Vif Protein through SCF(cyclin F) E3 ligase machinery. J. Biol. Chem. 292, 5349–5363. doi: 10.1074/jbc.m116.765842
Baek, D., Park, K. H., Lee, K. M., Jung, S., Joung, S., Kim, J., et al. (2021). Ubiquitin-specific protease 53 promotes osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Cell Death Dis. 12:238.
Balak, C., Belnap, N., Ramsey, K., Joss, S., Devriendt, K., Naymik, M., et al. (2018). A novel FBXO28 frameshift mutation in a child with developmental delay, dysmorphic features, and intractable epilepsy: a second gene that may contribute to the 1q41-q42 deletion phenotype. Am. J. Med. Genet. A 176, 1549–1558. doi: 10.1002/ajmg.a.38712
Bard, J. A. M., Goodall, E. A., Greene, E. R., Jonsson, E., Dong, K. C., and Martin, A. (2018). Structure and function of the 26S proteasome. Annu. Rev. Biochem. 87, 697–724.
Basel-Vanagaite, L., Dallapiccola, B., Ramirez-Solis, R., Segref, A., Thiele, H., Edwards, A., et al. (2012). Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am. J. Hum. Genet. 91, 998–1010. doi: 10.1016/j.ajhg.2012.10.011
Beck, D. B., Basar, M. A., Asmar, A. J., Thompson, J. J., Oda, H., Uehara, D. T., et al. (2021). Linkage-specific deubiquitylation by OTUD5 defines an embryonic pathway intolerant to genomic variation. Sci. Adv. 7:eabe2116. doi: 10.1126/sciadv.abe2116
Bednash, J. S., Johns, F., Patel, N., Smail, T. R., Londino, J. D., and Mallampalli, R. K. (2021). The deubiquitinase STAMBP modulates cytokine secretion through the NLRP3 inflammasome. Cell. Signal. 79:109859. doi: 10.1016/j.cellsig.2020.109859
Bednash, J. S., Weathington, N., Londino, J., Rojas, M., Gulick, D. L., Fort, R., et al. (2017). Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat. Commun. 8:15203.
Benedetti, F., Aggio, V., Pratesi, M. L., Greco, G., and Furlan, R. (2020). Neuroinflammation in bipolar depression. Front. Psychiatry 11:71. doi: 10.3389/fpsyt.2020.00071
Berko, E. R., Cho, M. T., Eng, C., Shao, Y., Sweetser, D. A., Waxler, J., et al. (2017). De novo missense variants in HECW2 are associated with neurodevelopmental delay and hypotonia. J. Med. Genet. 54, 84–86. doi: 10.1136/jmedgenet-2016-103943
Brehm, A., and Krüger, E. (2015). Dysfunction in protein clearance by the proteasome: impact on autoinflammatory diseases. Semin. Immunopathol. 37, 323–333. doi: 10.1007/s00281-015-0486-4
Brehm, A., Liu, Y., Sheikh, A., Marrero, B., Omoyinmi, E., Zhou, Q., et al. (2015). Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest. 125, 4196–4211. doi: 10.1172/jci81260
Broix, L., Jagline, H., Ivanova, E., Schmucker, S., Drouot, N., Clayton-Smith, J., et al. (2016). Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia. Nat. Genet. 48, 1349–1358. doi: 10.1038/ng.3676
Brunet, T., Jech, R., Brugger, M., Kovacs, R., Alhaddad, B., Leszinski, G., et al. (2021). De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin. Genet. 100, 14–28.
Buntinx, I., and Majewski, F. (1990). Blepharophimosis, iris coloboma, microgenia, hearing loss, postaxial polydactyly, aplasia of corpus callosum, hydroureter, and developmental delay. Am. J. Med. Genet. 36, 273–274. doi: 10.1002/ajmg.1320360304
Cai, J., Chen, H. Y., Peng, S. J., Meng, J. L., Wang, Y., Zhou, Y., et al. (2018). USP7-TRIM27 axis negatively modulates antiviral type I IFN signaling. FASEB J. 32, 5238–5249. doi: 10.1096/fj.201700473rr
Cappadocia, L., and Lima, C. D. (2018). Ubiquitin-like protein conjugation: structures. Chem. Mechan. Chem. Rev. 118, 889–918. doi: 10.1021/acs.chemrev.6b00737
Cardoso, A. R., Lopes-Marques, M., Silva, R. M., Serrano, C., Amorim, A., Prata, M. J., et al. (2019). Essential genetic findings in neurodevelopmental disorders. Hum. Genomics 13:31.
Cetin, G., Klafack, S., Studencka-Turski, M., Krüger, E., and Ebstein, F. (2021). The ubiquitin-proteasome system in immune cells. Biomolecules 11:60. doi: 10.3390/biom11010060
Chai, Y., Koppenhafer, S. L., Shoesmith, S. J., Perez, M. K., and Paulson, H. L. (1999). Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Genet. 8, 673–682. doi: 10.1093/hmg/8.4.673
Challa, K., Schmid, C. D., Kitagawa, S., Cheblal, A., Iesmantavicius, V., Seeber, A., et al. (2021). Damage-induced chromatome dynamics link Ubiquitin ligase and proteasome recruitment to histone loss and efficient DNA repair. Mol. Cell. 81, 811–829.e16.
Chan, E. M., Young, E. J., Ianzano, L., Munteanu, I., Zhao, X., Christopoulos, C. C., et al. (2003). Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 35, 125–127. doi: 10.1038/ng1238
Chen, X., Randles, L., Shi, K., Tarasov, S. G., Aihara, H., and Walters, K. J. (2016). Structures of Rpn1 T1:Rad23 and hRpn13:hPLIC2 Reveal Distinct Binding Mechanisms between Substrate Receptors and Shuttle Factors of the Proteasome. Structure 24, 1257–1270. doi: 10.1016/j.str.2016.05.018
Chen, X., Xu, Y., Tu, W., Huang, F., Zuo, Y., Zhang, H. G., et al. (2021). Ubiquitin E3 ligase MID1 inhibits the innate immune response by ubiquitinating IRF3. Immunology 163, 278–292. doi: 10.1111/imm.13315
Chen, Y., Yang, Z., Meng, M., Zhao, Y., Dong, N., Yan, H., et al. (2009). Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 35, 841–855. doi: 10.1016/j.molcel.2009.09.004
Chen, Z., Barbi, J., Bu, S., Yang, H. Y., Li, Z., Gao, Y., et al. (2013). The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 39, 272–285. doi: 10.1016/j.immuni.2013.08.006
Chinn, I. K., Sanders, R. P., Stray-Pedersen, A., Coban-Akdemir, Z. H., Kim, V. H., Dadi, H., et al. (2017). Novel combined immune deficiency and radiation sensitivity blended phenotype in an adult with biallelic variations in ZAP70 and RNF168. Front. Immunol. 8:576. doi: 10.3389/fimmu.2017.00576
Clague, M. J., Urbe, S., and Komander, D. (2019). Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 20, 338–352. doi: 10.1038/s41580-019-0099-1
Colleran, A., Collins, P. E., O’carroll, C., Ahmed, A., Mao, X., Mcmanus, B., et al. (2013). Deubiquitination of NF-kappaB by ubiquitin-Specific Protease-7 promotes transcription. Proc. Natl. Acad. Sci. U S A. 110, 618–623. doi: 10.1073/pnas.1208446110
Collison, A., Hatchwell, L., Verrills, N., Wark, P. A., De Siqueira, A. P., Tooze, M., et al. (2013). The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat. Med. 19, 232–237. doi: 10.1038/nm.3049
Crepel, A., Steyaert, J., De La Marche, W., De Wolf, V., Fryns, J. P., Noens, I., et al. (2011). Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 243–245. doi: 10.1002/ajmg.b.31163
Crow, Y. J., and Manel, N. (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440. doi: 10.1038/nri3850
Daubeuf, S., Singh, D., Tan, Y., Liu, H., Federoff, H. J., Bowers, W. J., et al. (2009). HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113, 3264–3275. doi: 10.1182/blood-2008-07-168203
David, G., Abbas, N., Stevanin, G., Durr, A., Yvert, G., Cancel, G., et al. (1997). Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70. doi: 10.1038/ng0997-65
de Jesus, A. A., Brehm, A., Vantries, R., Pillet, P., Parentelli, A. S., Montealegre Sanchez, G. A., et al. (2019). Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 143, 1939–1943.e38.
de Vivo, A., Sanchez, A., Yegres, J., Kim, J., Emly, S., and Kee, Y. (2019). The OTUD5-UBR5 complex regulates FACT-mediated transcription at damaged chromatin. Nucleic Acids Res. 47, 729–746. doi: 10.1093/nar/gky1219
Deciphering Developmental, and Disorders, S. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Del Bigio, M. R., Greenberg, C. R., Rorke, L. B., Schnur, R., Mcdonald-Mcginn, D. M., and Zackai, E. H. (1997). Neuropathological findings in eight children with cerebro-oculo-facio-skeletal (COFS) syndrome. J. Neuropathol. Exp. Neurol. 56, 1147–1157. doi: 10.1097/00005072-199710000-00009
Deveraux, Q., Van Nocker, S., Mahaffey, D., Vierstra, R., and Rechsteiner, M. (1995). Inhibition of ubiquitin-mediated proteolysis by the Arabidopsis 26 S protease subunit S5a. J. Biol. Chem. 270, 29660–29663. doi: 10.1074/jbc.270.50.29660
Duan, R., Shi, Y., Yu, L., Zhang, G., Li, J., Lin, Y., et al. (2016). UBA5 mutations cause a new form of autosomal recessive cerebellar ataxia. PLoS One 11:e0149039. doi: 10.1371/journal.pone.0149039
Dunn, A. R., O’connell, K. M. S., and Kaczorowski, C. C. (2019). Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci. Biobehav. Rev. 103, 73–80.
Ebstein, F., Kloetzel, P. M., Krüger, E., and Seifert, U. (2012). Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol. Life. Sci. 69, 2543–2558. doi: 10.1007/s00018-012-0938-0
Ebstein, F., Poli Harlowe, M. C., Studencka-Turski, M., and Krüger, E. (2019). Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front. Immunol. 10:2756. doi: 10.3389/fimmu.2019.02756
Edkins, A. L. (2015). CHIP: a co-chaperone for degradation by the proteasome. Subcell Biochem. 78, 219–242. doi: 10.1007/978-3-319-11731-7_11
Eftekharian, M. M., Ghafouri-Fard, S., Noroozi, R., Omrani, M. D., Arsang-Jang, S., Ganji, M., et al. (2018). Cytokine profile in autistic patients. Cytokine 108, 120–126. doi: 10.1016/j.cyto.2018.03.034
Evert, B. O., Schelhaas, J., Fleischer, H., De Vos, R. A., Brunt, E. R., Stenzel, W., et al. (2006). Neuronal intranuclear inclusions, dysregulation of cytokine expression and cell death in spinocerebellar ataxia type 3. Clin. Neuropathol. 25, 272–281.
Evert, B. O., Vogt, I. R., Kindermann, C., Ozimek, L., De Vos, R. A., Brunt, E. R., et al. (2001). Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains. J. Neurosci. 21, 5389–5396. doi: 10.1523/jneurosci.21-15-05389.2001
Feng, Q., Miao, Y., Ge, J., Yuan, Y., Zuo, Y., Qian, L., et al. (2018). ATXN3 positively regulates Type I IFN antiviral response by deubiquitinating and stabilizing HDAC3. J. Immunol. 201, 675–687. doi: 10.4049/jimmunol.1800285
Ferretti, L. P., Himmels, S. F., Trenner, A., Walker, C., Von Aesch, C., Eggenschwiler, A., et al. (2016). Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat. Commun. 7:12628.
Field, N. S., Moser, E. K., and Oliver, P. M. (2020). Itch regulation of innate and adaptive immune responses in mice and humans. J. Leukoc. Biol. 108, 353–362. doi: 10.1002/jlb.3mir0320-272r
Finley, D., Chen, X., and Walters, K. J. (2016). Gates, channels, and switches: elements of the proteasome machine. Trends Biochem. Sci. 41, 77–93. doi: 10.1016/j.tibs.2015.10.009
Friedman, J. S., Ray, J. W., Waseem, N., Johnson, K., Brooks, M. J., Hugosson, T., et al. (2009). Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 84, 792–800.
Froyen, G., Corbett, M., Vandewalle, J., Jarvela, I., Lawrence, O., Meldrum, C., et al. (2008). Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 82, 432–443. doi: 10.1016/j.ajhg.2007.11.002
Furumai, R., Tamada, K., Liu, X., and Takumi, T. (2019). UBE3A regulates the transcription of IRF, an antiviral immunity. Hum. Mol. Genet. 28, 1947–1958. doi: 10.1093/hmg/ddz019
Fusco, C., Mandriani, B., Di Rienzo, M., Micale, L., Malerba, N., Cocciadiferro, D., et al. (2018). TRIM50 regulates Beclin 1 proautophagic activity. Biochim. Biophys. Acta Mol. Cell Res. 1865, 908–919. doi: 10.1016/j.bbamcr.2018.03.011
Fusco, C., Micale, L., Augello, B., Mandriani, B., Pellico, M. T., De Nittis, P., et al. (2014). HDAC6 mediates the acetylation of TRIM50. Cell. Signal. 26, 363–369. doi: 10.1016/j.cellsig.2013.11.036
Gao, P., Ma, X., Yuan, M., Yi, Y., Liu, G., Wen, M., et al. (2021). E3 ligase Nedd4l promotes antiviral innate immunity by catalyzing K29-linked cysteine ubiquitination of TRAF3. Nat. Commun. 12:1194.
Garret, P., Ebstein, F., Delplancq, G., Dozieres-Puyravel, B., Boughalem, A., Auvin, S., et al. (2020). Report of the first patient with a homozygous OTUD7A variant responsible for epileptic encephalopathy and related proteasome dysfunction. Clin. Genet. 97, 567–575.
Geetha, T. S., Michealraj, K. A., Kabra, M., Kaur, G., Juyal, R. C., and Thelma, B. K. (2014). Targeted deep resequencing identifies MID2 mutation for X-linked intellectual disability with varied disease severity in a large kindred from India. Hum. Mutat. 35, 41–44. doi: 10.1002/humu.22453
Genis, D., Ortega-Cubero, S., San Nicolas, H., Corral, J., Gardenyes, J., De Jorge, L., et al. (2018). Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91:e1988. doi: 10.1212/wnl.0000000000006550
Goetzke, C. C., Ebstein, F., and Kallinich, T. (2021). Role of proteasomes in inflammation. J. Clin. Med. 10:1783. doi: 10.3390/jcm10081783
Golnik, R., Lehmann, A., Kloetzel, P. M., and Ebstein, F. (2016). Major Histocompatibility Complex (MHC) Class I processing of the NY-ESO-1 antigen is regulated by Rpn10 and Rpn13 proteins and immunoproteasomes following non-lysine ubiquitination. J. Biol. Chem. 291, 8805–8815. doi: 10.1074/jbc.m115.705178
Gontan, C., Mira-Bontenbal, H., Magaraki, A., Dupont, C., Barakat, T. S., Rentmeester, E., et al. (2018). REX1 is the critical target of RNF12 in imprinted X chromosome inactivation in mice. Nat. Commun. 9:4752.
Gregor, A., Sadleir, L. G., Asadollahi, R., Azzarello-Burri, S., Battaglia, A., Ousager, L. B., et al. (2018). De novo variants in the F-Box Protein FBXO11 in 20 individuals with a variable neurodevelopmental disorder. Am. J. Hum. Genet. 103, 305–316. doi: 10.1016/j.ajhg.2018.07.003
Grumati, P., and Dikic, I. (2018). Ubiquitin signaling and autophagy. J. Biol. Chem. 293, 5404–5413. doi: 10.1074/jbc.tm117.000117
Guernsey, D. L., Jiang, H., Bedard, K., Evans, S. C., Ferguson, M., Matsuoka, M., et al. (2010). Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 6:e1001081. doi: 10.1371/journal.pgen.1001081
Guo, Y., Jiang, F., Kong, L., Li, B., Yang, Y., Zhang, L., et al. (2019). Cutting edge: USP27X deubiquitinates and stabilizes the DNA sensor cGAS to regulate cytosolic DNA-Mediated signaling. J. Immunol. 203, 2049–2054. doi: 10.4049/jimmunol.1900514
Guo, Y., Jiang, F., Kong, L., Wu, H., Zhang, H., Chen, X., et al. (2020a). OTUD5 promotes innate antiviral and antitumor immunity through deubiquitinating and stabilizing STING. Cell Mol Immunol. 18, 1945–1955. doi: 10.1038/s41423-020-00531-5
Guo, Y., Li, L., Xu, T., Guo, X., Wang, C., Li, Y., et al. (2020b). HUWE1 mediates inflammasome activation and promotes host defense against bacterial infection. J. Clin. Invest. 130, 6301–6316. doi: 10.1172/jci138234
Haas, A. L., and Siepmann, T. J. (1997). Pathways of ubiquitin conjugation. FASEB J. 11, 1257–1268. doi: 10.1096/fasebj.11.14.9409544
Han, B., Sun, Y., Yang, D., Zhang, H., Mo, S., Chen, X., et al. (2020). USP22 promotes development of lung adenocarcinoma through ubiquitination and immunosuppression. Aging (Albany NY) 12, 6990–7005. doi: 10.18632/aging.103056
Hanly, C., Shah, H., Au, P. Y. B., and Murias, K. (2021). Description of neurodevelopmental phenotypes associated with 10 genetic neurodevelopmental disorders: a scoping review. Clin. Genet. 99, 335–346. doi: 10.1111/cge.13882
Hao, Y. H., Fountain, M. D. Jr., Fon Tacer, K., Xia, F., Bi, W., et al. (2015). USP7 acts as a molecular rheostat to promote WASH-Dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol. Cell 59, 956–969. doi: 10.1016/j.molcel.2015.07.033
Hardisty-Hughes, R. E., Tateossian, H., Morse, S. A., Romero, M. R., Middleton, A., Tymowska-Lalanne, Z., et al. (2006). A mutation in the F-box gene, Fbxo11, causes otitis media in the Jeff mouse. Hum. Mol. Genet. 15, 3273–3279. doi: 10.1093/hmg/ddl403
Harper, J. W., and Schulman, B. A. (2021). Cullin-RING ubiquitin ligase regulatory circuits: a quarter century beyond the F-box hypothesis. Annu. Rev. Biochem. 90, 403–429. doi: 10.1146/annurev-biochem-090120-013613
Heimdal, K., Sanchez-Guixe, M., Aukrust, I., Bollerslev, J., Bruland, O., Jablonski, G. E., et al. (2014). STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet. J. Rare Dis. 9:146.
Henry, K. W., Wyce, A., Lo, W. S., Duggan, L. J., Emre, N. C., Kao, C. F., et al. (2003). Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 17, 2648–2663. doi: 10.1101/gad.1144003
Hesterberg, R. S., Beatty, M. S., Han, Y., Fernandez, M. R., Akuffo, A. A., Goodheart, W. E., et al. (2020). Cereblon harnesses Myc-dependent bioenergetics and activity of CD8+ T lymphocytes. Blood 136, 857–870. doi: 10.1182/blood.2019003257
Higgins, J. J., Pucilowska, J., Lombardi, R. Q., and Rooney, J. P. (2004). A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931. doi: 10.1212/01.wnl.0000146196.01316.a2
Hollstein, R., Parry, D. A., Nalbach, L., Logan, C. V., Strom, T. M., Hartill, V. L., et al. (2015). HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J. Med. Genet. 52, 797–803.
Holt, R. J., Young, R. M., Crespo, B., Ceroni, F., Curry, C. J., Bellacchio, E., et al. (2019). De novo missense variants in FBXW11 cause diverse developmental phenotypes including brain, eye, and digit anomalies. Am. J. Hum. Genet. 105, 640–657. doi: 10.1016/j.ajhg.2019.07.005
Homan, C. C., Kumar, R., Nguyen, L. S., Haan, E., Raymond, F. L., Abidi, F., et al. (2014). Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. Am. J. Hum. Genet. 94, 470–478. doi: 10.1016/j.ajhg.2014.02.004
Hoppman-Chaney, N., Wain, K., Seger, P. R., Superneau, D. W., and Hodge, J. C. (2013). Identification of single gene deletions at 15q13.3: further evidence that CHRNA7 causes the 15q13.3 microdeletion syndrome phenotype. Clin. Genet. 83, 345–351. doi: 10.1111/j.1399-0004.2012.01925.x
Hori, I., Miya, F., Negishi, Y., Hattori, A., Ando, N., Boroevich, K. A., et al. (2018). A novel homozygous missense mutation in the SH3-binding motif of STAMBP causing microcephaly-capillary malformation syndrome. J. Hum. Genet. 63, 957–963. doi: 10.1038/s10038-018-0482-3
Hu, H., Haas, S. A., Chelly, J., Van Esch, H., Raynaud, M., De Brouwer, A. P., et al. (2016). X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 21, 133–148.
Huang, H., Jeon, M. S., Liao, L., Yang, C., Elly, C., Yates, J. R. III, et al. (2010). K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity 33, 60–70. doi: 10.1016/j.immuni.2010.07.002
Huber, C., Dias-Santagata, D., Glaser, A., O’sullivan, J., Brauner, R., Wu, K., et al. (2005). Identification of mutations in CUL7 in 3-M syndrome. Nat. Genet. 37, 1119–1124.
Hung, M. H., Jian, Y. R., Tsao, C. C., Lin, S. W., and Chuang, Y. H. (2014). Enhanced LPS-induced peritonitis in mice deficiency of cullin 4B in macrophages. Genes Immun. 15, 404–412. doi: 10.1038/gene.2014.32
Husnjak, K., Elsasser, S., Zhang, N., Chen, X., Randles, L., Shi, Y., et al. (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. doi: 10.1038/nature06926
Ijomone, O. M., Olung, N. F., Akingbade, G. T., Okoh, C. O. A., and Aschner, M. (2020). Environmental influence on neurodevelopmental disorders: potential association of heavy metal exposure and autism. J. Trace Elem. Med. Biol. 62:126638. doi: 10.1016/j.jtemb.2020.126638
Ismail, F. Y., and Shapiro, B. K. (2019). What are neurodevelopmental disorders? Curr. Opin. Neurol. 32, 611–616.
Jagiello, P., Hammans, C., Wieczorek, S., Arning, L., Stefanski, A., Strehl, H., et al. (2003). A novel splice site mutation in the TRIM37 gene causes mulibrey nanism in a Turkish family with phenotypic heterogeneity. Hum. Mutat. 21, 630–635. doi: 10.1002/humu.10220
Ji, W., and Rivero, F. (2016). Atypical Rho GTPases of the RhoBTB subfamily: roles in vesicle trafficking and tumorigenesis. Cells 5:28. doi: 10.3390/cells5020028
Jia, X., Zhou, H., Wu, C., Wu, Q., Ma, S., Wei, C., et al. (2017). The ubiquitin ligase RNF125 targets innate immune adaptor protein TRIM14 for ubiquitination and degradation. J. Immunol. 198, 4652–4658. doi: 10.4049/jimmunol.1601322
Johnson, W. G. (2000). Late-onset neurodegenerative diseases–the role of protein insolubility. J. Anat. 196(Pt 4), 609–616. doi: 10.1046/j.1469-7580.2000.19640609.x
Kallijarvi, J., Avela, K., Lipsanen-Nyman, M., Ulmanen, I., and Lehesjoki, A. E. (2002). The TRIM37 gene encodes a peroxisomal RING-B-box-coiled-coil protein: classification of mulibrey nanism as a new peroxisomal disorder. Am. J. Hum. Genet. 70, 1215–1228. doi: 10.1086/340256
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death. Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
Karam, S. M., Riegel, M., Segal, S. L., Felix, T. M., Barros, A. J., Santos, I. S., et al. (2015). Genetic causes of intellectual disability in a birth cohort: a population-based study. Am. J. Med. Genet. A 167, 1204–1214. doi: 10.1002/ajmg.a.37011
Karim, R., Tummers, B., Meyers, C., Biryukov, J. L., Alam, S., Backendorf, C., et al. (2013). Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog 9:e1003384. doi: 10.1371/journal.ppat.1003384
Kataoka, S., Kawashima, N., Okuno, Y., Muramatsu, H., Miwata, S., Narita, K., et al. (2021). Successful treatment of a novel type I interferonopathy due to a de novo PSMB9 gene mutation with a Janus kinase inhibitor. J. Allergy Clin. Immunol. 148, 639–644. doi: 10.1016/j.jaci.2021.03.010
Kathania, M., Khare, P., Zeng, M., Cantarel, B., Zhang, H., Ueno, H., et al. (2016). Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-gammat ubiquitination. Nat. Immunol. 17, 997–1004. doi: 10.1038/ni.3488
Kaufman, L., Ayub, M., and Vincent, J. B. (2010). The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord. 2, 182–209. doi: 10.1007/s11689-010-9055-2
Kawaguchi, Y., Okamoto, T., Taniwaki, M., Aizawa, M., Inoue, M., Katayama, S., et al. (1994). CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8, 221–228. doi: 10.1038/ng1194-221
Kim, J., Tsuruta, F., Okajima, T., Yano, S., Sato, B., and Chiba, T. (2017). KLHL7 promotes TUT1 ubiquitination associated with nucleolar integrity: implications for retinitis pigmentosa. Biochem. Biophys. Res. Commun. 494, 220–226. doi: 10.1016/j.bbrc.2017.10.049
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kitamura, A., Maekawa, Y., Uehara, H., Izumi, K., Kawachi, I., Nishizawa, M., et al. (2011). A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Invest. 121, 4150–4160. doi: 10.1172/jci58414
Klein, C. J., Wu, Y., Vogel, P., Goebel, H. H., Bonnemann, C., Zukosky, K., et al. (2014). Ubiquitin ligase defect by DCAF8 mutation causes HMSN2 with giant axons. Neurology 82, 873–878. doi: 10.1212/wnl.0000000000000206
Knudtzon, J., Aksnes, L., Akslen, L. A., and Aarskog, D. (1987). Elevated 1,25-dihydroxyvitamin D and normocalcaemia in presumed familial Williams syndrome. Clin. Genet. 32, 369–374. doi: 10.1111/j.1399-0004.1987.tb03151.x
Koziorowski, D., Figura, M., Milanowski, L. M., Szlufik, S., Alster, P., Madetko, N., et al. (2021). Mechanisms of neurodegeneration in various forms of parkinsonism-similarities and differences. Cells 10:656. doi: 10.3390/cells10030656
Krishnamoorthy, V., Khanna, R., and Parnaik, V. K. (2018). E3 ubiquitin ligase HECW2 mediates the proteasomal degradation of HP1 isoforms. Biochem. Biophys. Res. Commun. 503, 2478–2484. doi: 10.1016/j.bbrc.2018.07.003
Kroll-Hermi, A., Ebstein, F., Stoetzel, C., Geoffroy, V., Schaefer, E., Scheidecker, S., et al. (2020). Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 12, e11861.
Ku, H. C., and Cheng, C. F. (2020). Master regulator activating transcription Factor 3 (ATF3) in metabolic homeostasis and cancer. Front. Endocrinol. (Lausanne) 11:556. doi: 10.3389/fendo.2020.00556
Kuiken, H. J., Egan, D. A., Laman, H., Bernards, R., Beijersbergen, R. L., and Dirac, A. M. (2012). Identification of F-box only protein 7 as a negative regulator of NF-kappaB signalling. J. Cell Mol. Med. 16, 2140–2149. doi: 10.1111/j.1582-4934.2012.01524.x
Kumazoe, M., Nakamura, Y., Yamashita, M., Suzuki, T., Takamatsu, K., Huang, Y., et al. (2017). Green tea polyphenol Epigallocatechin-3-gallate suppresses toll-like receptor 4 expression via up-regulation of E3 Ubiquitin-protein ligase RNF216. J. Biol. Chem. 292, 4077–4088. doi: 10.1074/jbc.m116.755959
Kury, S., Besnard, T., Ebstein, F., Khan, T. N., Gambin, T., Douglas, J., et al. (2017). De Novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am. J. Hum. Genet. 100, 352–363.
Kuzuhara, S., Mori, H., Izumiyama, N., Yoshimura, M., and Ihara, Y. (1988). Lewy bodies are ubiquitinated. a light and electron microscopic immunocytochemical study. Acta Neuropathol. 75, 345–353. doi: 10.1007/bf00687787
Lee, K. K., Rajagopalan, D., Bhatia, S. S., Tirado-Magallanes, R., Chng, W. J., and Jha, S. (2021). The oncogenic E3 ligase TRIP12 suppresses epithelial-mesenchymal transition (EMT) and mesenchymal traits through ZEB1/2. Cell Death Discov. 7:95.
Leonard, A., Millar, M. W., Slavin, S. A., Bijli, K. M., Dionisio Santos, D. A., Dean, D. A., et al. (2019). Critical role of autophagy regulator Beclin1 in endothelial cell inflammation and barrier disruption. Cell. Signal. 61, 120–129. doi: 10.1016/j.cellsig.2019.04.013
Leroy, E., Boyer, R., Auburger, G., Leube, B., Ulm, G., Mezey, E., et al. (1998). The ubiquitin pathway in Parkinson’s disease. Nature 395, 451–452.
Li, C., Beauregard-Lacroix, E., Kondratev, C., Rousseau, J., Heo, A. J., Neas, K., et al. (2021). UBR7 functions with UBR5 in the Notch signaling pathway and is involved in a neurodevelopmental syndrome with epilepsy, ptosis, and hypothyroidism. Am. J. Hum. Genet. 108, 134–147. doi: 10.1016/j.ajhg.2020.11.018
Li, C., Li, L., Chen, K., Wang, Y., Yang, F., and Wang, G. (2019). UFL1 alleviates lipopolysaccharide-induced cell damage and inflammation via regulation of the TLR4/NF-kappaB pathway in bovine mammary epithelial cells. Oxid. Med. Cell Longev. 2019:6505373.
Li, K., Wang, F., Yang, Z. N., Zhang, T. T., Yuan, Y. F., Zhao, C. X., et al. (2020). TRIB3 promotes MYC-associated lymphoma development through suppression of UBE3B-mediated MYC degradation. Nat. Commun. 11:6316.
Li, Y., Deng, L., Zhao, X., Li, B., Ren, D., Yu, L., et al. (2018). Tripartite motif-containing 37 (TRIM37) promotes the aggressiveness of non-small-cell lung cancer cells by activating the NF-kappaB pathway. J. Pathol. 246, 366–378. doi: 10.1002/path.5144
Li, Y. Y., Zhang, G. Y., He, J. P., Zhang, D. D., Kong, X. X., Yuan, H. M., et al. (2017). Ufm1 inhibits LPS-induced endothelial cell inflammatory responses through the NF-kappaB signaling pathway. Int. J. Mol. Med. 39, 1119–1126. doi: 10.3892/ijmm.2017.2947
Liang, J. R., Lingeman, E., Luong, T., Ahmed, S., Muhar, M., Nguyen, T., et al. (2020). A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-Resident UFMylation. Cell 180, 1160–1177.e20.
Liu, Y., Ramot, Y., Torrelo, A., Paller, A. S., Si, N., Babay, S., et al. (2012). Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 64, 895–907. doi: 10.1002/art.33368
Livingston, J. H., and Crow, Y. J. (2016). Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: aicardi-goutieres syndrome and beyond. Neuropediatrics 47, 355–360. doi: 10.1055/s-0036-1592307
Livneh, I., Kravtsova-Ivantsiv, Y., Braten, O., Kwon, Y. T., and Ciechanover, A. (2017). Monoubiquitination joins polyubiquitination as an esteemed proteasomal targeting signal. Bioessays 39:e201700027.
Lohr, N. J., Molleston, J. P., Strauss, K. A., Torres-Martinez, W., Sherman, E. A., Squires, R. H., et al. (2010). Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 86, 447–453. doi: 10.1016/j.ajhg.2010.01.028
Lopez-Gonzalez, I., Viana, R., Sanz, P., and Ferrer, I. (2017). Inflammation in lafora disease: evolution with disease progression in laforin and malin knock-out mouse models. Mol. Neurobiol. 54, 3119–3130. doi: 10.1007/s12035-016-9884-4
Lu, L., Hu, S., Wei, R., Qiu, X., Lu, K., Fu, Y., et al. (2013). The HECT type ubiquitin ligase NEDL2 is degraded by anaphase-promoting complex/cyclosome (APC/C)-Cdh1, and its tight regulation maintains the metaphase to anaphase transition. J. Biol. Chem. 288, 35637–35650. doi: 10.1074/jbc.m113.472076
Luo, Y., Liu, Y., Wu, L., Ma, X., Liu, Q., Huang, F., et al. (2019). CUL7 E3 ubiquitin ligase mediates the degradation of activation-induced cytidine deaminase and regulates the ig class switch recombination in b lymphocytes. J. Immunol. 203, 269–281. doi: 10.4049/jimmunol.1900125
Malakhova, O. A., Kim, K. I., Luo, J. K., Zou, W., Kumar, K. G., Fuchs, S. Y., et al. (2006). UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 25, 2358–2367. doi: 10.1038/sj.emboj.7601149
Mao, H. T., Wang, Y., Cai, J., Meng, J. L., Zhou, Y., Pan, Y., et al. (2016). HACE1 negatively regulates virus-triggered type I IFN signaling by impeding the formation of the MAVS-TRAF3 complex. Viruses 8:146. doi: 10.3390/v8050146
Maranga, C., Fernandes, T. G., Bekman, E., and Da Rocha, S. T. (2020). Angelman syndrome: a journey through the brain. FEBS J. 287, 2154–2175. doi: 10.1111/febs.15258
Margolin, D. H., Kousi, M., Chan, Y. M., Lim, E. T., Schmahmann, J. D., Hadjivassiliou, M., et al. (2013). Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 368, 1992–2003. doi: 10.1056/nejmoa1215993
Marsh, D. J., Ma, Y., and Dickson, K. A. (2020). Histone monoubiquitination in chromatin remodelling: focus on the histone H2B interactome and cancer. Cancers (Basel) 12:3462. doi: 10.3390/cancers12113462
Martinez, C. A., Ebstein, F., Nicholas, S. K., De Guzman, M., Forbes, L. R., Delmonte, O. M., et al. (2021). HSCT corrects primary immunodeficiency and immune dysregulation in patients with POMP-related auto-inflammatory disease. Blood. Online ahead of print.
Mathew, R., Seiler, M. P., Scanlon, S. T., Mao, A. P., Constantinides, M. G., Bertozzi-Villa, C., et al. (2012). BTB-ZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature 491, 618–621. doi: 10.1038/nature11548
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S., et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77.
Matta, S. M., Hill-Yardin, E. L., and Crack, P. J. (2019). The influence of neuroinflammation in Autism Spectrum disorder. Brain Behav. Immun. 79, 75–90. doi: 10.1016/j.bbi.2019.04.037
McDowell, G. S., Kucerova, R., and Philpott, A. (2010). Non-canonical ubiquitylation of the proneural protein Ngn2 occurs in both Xenopus embryos and mammalian cells. Biochem. Biophys. Res. Commun. 400, 655–660. doi: 10.1016/j.bbrc.2010.08.122
Melo-Cardenas, J., Zhang, Y., Zhang, D. D., and Fang, D. (2016). Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 7, 44848–44856. doi: 10.18632/oncotarget.8602
Meng, X., Liu, X., Guo, X., Jiang, S., Chen, T., Hu, Z., et al. (2018). FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 564, 130–135. doi: 10.1038/s41586-018-0756-0
Mesnil, M., Defamie, N., Naus, C., and Sarrouilhe, D. (2020). Brain disorders and chemical pollutants: a gap junction link? Biomolecules 11:51. doi: 10.3390/biom11010051
Metzger, M. B., Hristova, V. A., and Weissman, A. M. (2012). HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 125, 531–537. doi: 10.1242/jcs.091777
Metzger, M. B., Pruneda, J. N., Klevit, R. E., and Weissman, A. M. (2014). RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60. doi: 10.1016/j.bbamcr.2013.05.026
Meuwissen, M. E., Schot, R., Buta, S., Oudesluijs, G., Tinschert, S., Speer, S. D., et al. (2016). Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 213, 1163–1174. doi: 10.1084/jem.20151529
Micai, M., Fulceri, F., Caruso, A., Guzzetta, A., Gila, L., and Scattoni, M. L. (2020). Early behavioral markers for neurodevelopmental disorders in the first 3 years of life: an overview of systematic reviews. Neurosci. Biobehav. Rev. 116, 183–201. doi: 10.1016/j.neubiorev.2020.06.027
Micale, L., Fusco, C., Augello, B., Napolitano, L. M., Dermitzakis, E. T., Meroni, G., et al. (2008). Williams-Beuren syndrome TRIM50 encodes an E3 ubiquitin ligase. Eur. J. Hum. Genet. 16, 1038–1049. doi: 10.1038/ejhg.2008.68
Mignon-Ravix, C., Cacciagli, P., Choucair, N., Popovici, C., Missirian, C., Milh, M., et al. (2014). Intragenic rearrangements in X-linked intellectual deficiency: results of a-CGH in a series of 54 patients and identification of TRPC5 and KLHL15 as potential XLID genes. Am. J. Med. Genet. A 164A, 1991–1997. doi: 10.1002/ajmg.a.36602
Miller, B. J., Buckley, P., Seabolt, W., Mellor, A., and Kirkpatrick, B. (2011). Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol. Psychiatry 70, 663–671. doi: 10.1016/j.biopsych.2011.04.013
Millrine, D., Miyata, H., Tei, M., Dubey, P., Nyati, K., Nakahama, T., et al. (2016). Immunomodulatory drugs inhibit TLR4-induced type-1 interferon production independently of Cereblon via suppression of the TRIF/IRF3 pathway. Int. Immunol. 28, 307–315. doi: 10.1093/intimm/dxw005
Min, Y., Wi, S. M., Kang, J. A., Yang, T., Park, C. S., Park, S. G., et al. (2016). Cereblon negatively regulates TLR4 signaling through the attenuation of ubiquitination of TRAF6. Cell Death Dis. 7:e2313. doi: 10.1038/cddis.2016.226
Mir, A., Sritharan, K., Mittal, K., Vasli, N., Araujo, C., Jamil, T., et al. (2014). Truncation of the E3 ubiquitin ligase component FBXO31 causes non-syndromic autosomal recessive intellectual disability in a Pakistani family. Hum. Genet. 133, 975–984. doi: 10.1007/s00439-014-1438-0
Miyajima, N., Maruyama, S., Nonomura, K., and Hatakeyama, S. (2009). TRIM36 interacts with the kinetochore protein CENP-H and delays cell cycle progression. Biochem. Biophys. Res. Commun. 381, 383–387. doi: 10.1016/j.bbrc.2009.02.059
Moortgat, S., Berland, S., Aukrust, I., Maystadt, I., Baker, L., Benoit, V., et al. (2018). HUWE1 variants cause dominant X-linked intellectual disability: a clinical study of 21 patients. Eur. J. Hum. Genet. 26, 64–74.
Morgan, J. J., and Crawford, L. J. (2021). The ubiquitin proteasome system in genome stability and cancer. Cancers (Basel) 13, 2235. doi: 10.3390/cancers13092235
Mori, H., Kondo, J., and Ihara, Y. (1987). Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 235, 1641–1644.
Morice-Picard, F., Benard, G., Rezvani, H. R., Lasseaux, E., Simon, D., Moutton, S., et al. (2016). Complete loss of function of the ubiquitin ligase HERC2 causes a severe neurodevelopmental phenotype. Eur. J. Hum. Genet. 25, 52–58.
Mulder, M. P. C., Witting, K. F., and Ovaa, H. (2020). Cracking the ubiquitin code: the ubiquitin toolbox. Curr. Issues Mol. Biol. 37, 1–20. doi: 10.21775/cimb.037.001
Muona, M., Ishimura, R., Laari, A., Ichimura, Y., Linnankivi, T., Keski-Filppula, R., et al. (2016). Biallelic variants in UBA5 link dysfunctional UFM1 ubiquitin-like modifier pathway to severe infantile-onset encephalopathy. Am. J. Hum. Genet. 99, 683–694. doi: 10.1016/j.ajhg.2016.06.020
Nahorski, M. S., Maddirevula, S., Ishimura, R., Alsahli, S., Brady, A. F., Begemann, A., et al. (2018). Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 141, 1934–1945. doi: 10.1093/brain/awy135
Naik, E., and Dixit, V. M. (2016). Usp9X is required for lymphocyte activation and homeostasis through its control of ZAP70 ubiquitination and PKCbeta kinase activity. J. Immunol. 196, 3438–3451. doi: 10.4049/jimmunol.1403165
Naik, E., Webster, J. D., Devoss, J., Liu, J., Suriben, R., and Dixit, V. M. (2014). Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J. Exp. Med. 211, 1947–1955. doi: 10.1084/jem.20140860
Nakashima, M., Kato, M., Matsukura, M., Kira, R., Ngu, L. H., Lichtenbelt, K. D., et al. (2020). De novo variants in CUL3 are associated with global developmental delays with or without infantile spasms. J. Hum. Genet. 65, 727–734. doi: 10.1038/s10038-020-0758-2
Nakhaei, P., Mesplede, T., Solis, M., Sun, Q., Zhao, T., Yang, L., et al. (2009). The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog 5:e1000650. doi: 10.1371/journal.ppat.1000650
Nardo, T., Oneda, R., Spivak, G., Vaz, B., Mortier, L., Thomas, P., et al. (2009). A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. U S A. 106, 6209–6214. doi: 10.1073/pnas.0902113106
Neill, C. A., and Dingwall, M. M. (1950). A syndrome resembling progeria: a review of two cases. Arch. Dis. Child. 25, 213–223. doi: 10.1136/adc.25.123.213
Ng, A. C., Eisenberg, J. M., Heath, R. J., Huett, A., Robinson, C. M., Nau, G. J., et al. (2011). Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl. Acad. Sci. U S A. 108(Suppl. 1), 4631–4638. doi: 10.1073/pnas.1000093107
Nguyen, L. S., Schneider, T., Rio, M., Moutton, S., Siquier-Pernet, K., Verny, F., et al. (2016). A nonsense variant in HERC1 is associated with intellectual disability, megalencephaly, thick corpus callosum and cerebellar atrophy. Eur. J. Hum. Genet. 24, 455–458. doi: 10.1038/ejhg.2015.140
Ohtake, F., Saeki, Y., Ishido, S., Kanno, J., and Tanaka, K. (2016). The K48-K63 branched ubiquitin chain regulates NF-kappaB signaling. Mol. Cell. 64, 251–266. doi: 10.1016/j.molcel.2016.09.014
Oshiumi, H., Miyashita, M., Inoue, N., Okabe, M., Matsumoto, M., and Seya, T. (2010). The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8, 496–509. doi: 10.1016/j.chom.2010.11.008
Palazon-Riquelme, P., Worboys, J. D., Green, J., Valera, A., Martin-Sanchez, F., Pellegrini, C., et al. (2018). USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 19:e44766.
Peng, Q., Xu, H., Xiao, M., and Wang, L. (2020). The small molecule PSSM0332 disassociates the CRL4A(DCAF8) E3 ligase complex to decrease the ubiquitination of NcoR1 and inhibit the inflammatory response in a mouse sepsis-induced myocardial dysfunction model. Int. J. Biol. Sci. 16, 2974–2988. doi: 10.7150/ijbs.50186
Perez Berrocal, D. A., Witting, K. F., Ovaa, H., and Mulder, M. P. C. (2019). Hybrid chains: a collaboration of ubiquitin and ubiquitin-like modifiers introducing cross-functionality to the ubiquitin code. Front. Chem. 7:931. doi: 10.3389/fchem.2019.00931
Petroski, M. D., and Deshaies, R. J. (2005). Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20.
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Pickart, C. M. (2004). Back to the future with ubiquitin. Cell 116, 181–190. doi: 10.1016/s0092-8674(03)01074-2
Pickart, C. M., and Eddins, M. J. (2004). Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta 1695, 55–72. doi: 10.1016/j.bbamcr.2004.09.019
Pickart, C. M., and Fushman, D. (2004). Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 8, 610–616. doi: 10.1016/j.cbpa.2004.09.009
Poli, M. C., Ebstein, F., Nicholas, S. K., De Guzman, M. M., Forbes, L. R., Chinn, I. K., et al. (2018). Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am. J. Hum. Genet. 102, 1126–1142. doi: 10.1016/j.ajhg.2018.04.010
Popa-Wagner, A., Dumitrascu, D. I., Capitanescu, B., Petcu, E. B., Surugiu, R., Fang, W. H., et al. (2020). Dietary habits, lifestyle factors and neurodegenerative diseases. Neural Regen. Res. 15, 394–400. doi: 10.4103/1673-5374.266045
Potvin, S., Stip, E., Sepehry, A. A., Gendron, A., Bah, R., and Kouassi, E. (2008). Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol. Psychiatry 63, 801–808. doi: 10.1016/j.biopsych.2007.09.024
Princiotta, M. F., Finzi, D., Qian, S. B., Gibbs, J., Schuchmann, S., Buttgereit, F., et al. (2003). Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18, 343–354. doi: 10.1016/s1074-7613(03)00051-7
Qian, S. B., Princiotta, M. F., Bennink, J. R., and Yewdell, J. W. (2006). Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J. Biol. Chem. 281, 392–400. doi: 10.1074/jbc.m509126200
Quaderi, N. A., Schweiger, S., Gaudenz, K., Franco, B., Rugarli, E. I., Berger, W., et al. (1997). Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 17, 285–291. doi: 10.1038/ng1197-285
Ramachandran, S., Chahwan, R., Nepal, R. M., Frieder, D., Panier, S., Roa, S., et al. (2010). The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. U S A. 107, 809–814. doi: 10.1073/pnas.0913790107
Ramalho-Oliveira, R., Oliveira-Vieira, B., and Viola, J. P. B. (2019). IRF2BP2: a new player in the regulation of cell homeostasis. J. Leukoc. Biol. 106, 717–723. doi: 10.1002/jlb.mr1218-507r
Rayner, S. L., Morsch, M., Molloy, M. P., Shi, B., Chung, R., and Lee, A. (2019). Using proteomics to identify ubiquitin ligase-substrate pairs: how novel methods may unveil therapeutic targets for neurodegenerative diseases. Cell Mol. Life. Sci. 76, 2499–2510. doi: 10.1007/s00018-019-03082-9
Reichenberg, A., Cederlof, M., Mcmillan, A., Trzaskowski, M., Kapra, O., Fruchter, E., et al. (2016). Discontinuity in the genetic and environmental causes of the intellectual disability spectrum. Proc. Natl. Acad. Sci. U S A. 113, 1098–1103. doi: 10.1073/pnas.1508093112
Riazi, K., Galic, M. A., and Pittman, Q. J. (2010). Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 89, 34–42. doi: 10.1016/j.eplepsyres.2009.09.004
Ritchie, K. J., Hahn, C. S., Kim, K. I., Yan, M., Rosario, D., Li, L., et al. (2004). Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 10, 1374–1378. doi: 10.1038/nm1133
Romero-Ayuso, D. (2021). Future challenges in research in children with neurodevelopmental disorders. Children (Basel) 8:328. doi: 10.3390/children8050328
Saade, C., Najem, E., Asmar, K., Salman, R., El Achkar, B., and Naffaa, L. (2019). Intracranial calcifications on CT: an updated review. J. Radiol. Case Rep. 13, 1–18.
Saha, S. S., Caviness, G., Yi, G., Raymond, E. L., Mbow, M. L., and Kao, C. C. (2018). E3 ubiquitin ligase RNF125 activates interleukin-36 receptor signaling and contributes to its turnover. J. Innate Immun. 10, 56–69. doi: 10.1159/000481210
Sala-Gaston, J., Martinez-Martinez, A., Pedrazza, L., Lorenzo-Martin, L. F., Caloto, R., Bustelo, X. R., et al. (2020). HERC ubiquitin ligases in cancer. Cancers (Basel) 12, 1653. doi: 10.3390/cancers12061653
Santiago-Sim, T., Burrage, L. C., Ebstein, F., Tokita, M. J., Miller, M., Bi, W., et al. (2017). Biallelic variants in OTUD6B cause an intellectual disability syndrome associated with seizures and dysmorphic features. Am. J. Hum. Genet. 100, 676–688.
Sarrabay, G., Mechin, D., Salhi, A., Boursier, G., Rittore, C., Crow, Y., et al. (2019). PSMB10, the last immunoproteasome gene missing for PRAAS. J Allergy Clin Immunol. 143, 1015–1017.e6.
Sarute, N., Ibrahim, N., Medegan Fagla, B., Lavanya, M., Cuevas, C., Stavrou, S., et al. (2019). TRIM2, a novel member of the antiviral family, limits new world arenavirus entry. PLoS Biol. 17:e3000137. doi: 10.1371/journal.pbio.3000137
Savatt, J. M., and Myers, S. M. (2021). Genetic testing in neurodevelopmental disorders. Front. Pediatr. 9:526779. doi: 10.3389/fped.2021.526779
Schubert, U., Anton, L. C., Gibbs, J., Norbury, C. C., Yewdell, J. W., and Bennink, J. R. (2000). Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. doi: 10.1038/35008096
Seminara, S. B., Acierno, J. S. Jr., Abdulwahid, N. A., Crowley, W. F. Jr., et al. (2002). Hypogonadotropic hypogonadism and cerebellar ataxia: detailed phenotypic characterization of a large, extended kindred. J. Clin. Endocrinol. Metab. 87, 1607–1612. doi: 10.1210/jcem.87.4.8384
Shi, Y., Wang, J., Li, J. D., Ren, H., Guan, W., He, M., et al. (2013). Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One 8:e81884. doi: 10.1371/journal.pone.0081884
Shojaee, S., Sina, F., Banihosseini, S. S., Kazemi, M. H., Kalhor, R., Shahidi, G. A., et al. (2008). Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am. J. Hum. Genet. 82, 1375–1384. doi: 10.1016/j.ajhg.2008.05.005
Sigismund, S., Polo, S., and Di Fiore, P. P. (2004). Signaling through monoubiquitination. Curr. Top. Microbiol. Immunol. 286, 149–185. doi: 10.1007/978-3-540-69494-6_6
Singh, N., Kumble Bhat, V., Tiwari, A., Kodaganur, S. G., Tontanahal, S. J., Sarda, A., et al. (2017). A homozygous mutation in TRIM36 causes autosomal recessive anencephaly in an Indian family. Hum. Mol. Genet. 26, 1104–1114.
Sliter, D. A., Martinez, J., Hao, L., Chen, X., Sun, N., Fischer, T. D., et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. doi: 10.1038/s41586-018-0448-9
Sokol, R. J., Delaney-Black, V., and Nordstrom, B. (2003). Fetal alcohol spectrum disorder. JAMA 290, 2996–2999.
Song, X., Liu, S., Wang, W., Ma, Z., Cao, X., and Jiang, M. (2020). E3 ubiquitin ligase RNF170 inhibits innate immune responses by targeting and degrading TLR3 in murine cells. Cell Mol. Immunol. 17, 865–874. doi: 10.1038/s41423-019-0236-y
Song, Y., Li, P., Qin, L., Xu, Z., Jiang, B., Ma, C., et al. (2021). CUL4B negatively regulates Toll-like receptor-triggered proinflammatory responses by repressing Pten transcription. Cell Mol. Immunol. 18, 339–349. doi: 10.1038/s41423-019-0323-0
Srivastava, S., Sahu, U., Zhou, Y., Hogan, A. K., Sathyan, K. M., Bodner, J., et al. (2021). NOTCH1-driven UBR7 stimulates nucleotide biosynthesis to promote T cell acute lymphoblastic leukemia. Sci. Adv. 7:eabc9781. doi: 10.1126/sciadv.abc9781
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434. doi: 10.1016/j.cell.2008.12.042
Straub, J., Konrad, E. D. H., Gruner, J., Toutain, A., Bok, L. A., Cho, M. T., et al. (2018). Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in Drosophila. Am. J. Hum. Genet. 102, 44–57. doi: 10.1016/j.ajhg.2017.11.008
Streich, F. C. Jr., and Lima, C. D. (2014). Structural and functional insights to ubiquitin-like protein conjugation. Annu. Rev. Biophys. 43, 357–379. doi: 10.1146/annurev-biophys-051013-022958
Sullivan, K. D., Evans, D., Pandey, A., Hraha, T. H., Smith, K. P., Markham, N., et al. (2017). Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 7:14818.
Sullivan, K. D., Lewis, H. C., Hill, A. A., Pandey, A., Jackson, L. P., Cabral, J. M., et al. (2016). Trisomy 21 consistently activates the interferon response. eLife 5:e16220. doi: 10.7554/eLife.16220
Sumner, C. J., D’ydewalle, C., Wooley, J., Fawcett, K. A., Hernandez, D., Gardiner, A. R., et al. (2013). A dominant mutation in FBXO38 causes distal spinal muscular atrophy with calf predominance. Am. J. Hum. Genet. 93, 976–983. doi: 10.1016/j.ajhg.2013.10.006
Sutcliffe, J. S., Jiang, Y. H., Galijaard, R. J., Matsuura, T., Fang, P., Kubota, T., et al. (1997). The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 7, 368–377. doi: 10.1101/gr.7.4.368
Tait, S. W., De Vries, E., Maas, C., Keller, A. M., D’santos, C. S., and Borst, J. (2007). Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J. Cell Biol. 179, 1453–1466. doi: 10.1083/jcb.200707063
Tanaka, K., Mizushima, T., and Saeki, Y. (2012). The proteasome: molecular machinery and pathophysiological roles. Biol. Chem. 393, 217–234. doi: 10.1515/hsz-2011-0285
Tang, J., Tu, S., Lin, G., Guo, H., Yan, C., Liu, Q., et al. (2020). Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 217:e20182091.
Tao, X., Chu, B., Xin, D., Li, L., and Sun, Q. (2020). USP27X negatively regulates antiviral signaling by deubiquitinating RIG-I. PLoS Pathog 16:e1008293. doi: 10.1371/journal.ppat.1008293
Tarpey, P. S., Raymond, F. L., O’meara, S., Edkins, S., Teague, J., Butler, A., et al. (2007). Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 80, 345–352. doi: 10.1086/511134
Tateossian, H., Hardisty-Hughes, R. E., Morse, S., Romero, M. R., Hilton, H., Dean, C., et al. (2009). Regulation of TGF-beta signalling by Fbxo11, the gene mutated in the Jeff otitis media mouse mutant. Pathogenetics 2:5. doi: 10.1186/1755-8417-2-5
Tenorio, J., Mansilla, A., Valencia, M., Martinez-Glez, V., Romanelli, V., Arias, P., et al. (2014). A new overgrowth syndrome is due to mutations in RNF125. Hum. Mutat. 35, 1436–1441.
Thibaut, F. (2018). Psychiatric disorders: neurodevelopmental disorders, neurodegenerative disorders, or both? Dial. Clin. Neurosci. 20, 251–252. doi: 10.31887/dcns.2018.20.4/fthibaut
Tittelmeier, J., Nachman, E., and Nussbaum-Krammer, C. (2020). Molecular chaperones: a double-edged sword in neurodegenerative diseases. Front. Aging Neurosci. 12:581374. doi: 10.3389/fnagi.2020.581374
Tokita, M. J., Chen, C. A., Chitayat, D., Macnamara, E., Rosenfeld, J. A., Hanchard, N., et al. (2018). De novo missense variants in TRAF7 cause developmental delay, congenital anomalies, and dysmorphic features. Am. J. Hum. Genet. 103, 154–162. doi: 10.1016/j.ajhg.2018.06.005
Tonne, E., Holdhus, R., Stansberg, C., Stray-Pedersen, A., Petersen, K., Brunner, H. G., et al. (2015). Syndromic X-linked intellectual disability segregating with a missense variant in RLIM. Eur. J. Hum. Genet. 23, 1652–1656. doi: 10.1038/ejhg.2015.30
Tourjman, V., Kouassi, E., Koue, M. E., Rocchetti, M., Fortin-Fournier, S., Fusar-Poli, P., et al. (2013). Antipsychotics’ effects on blood levels of cytokines in schizophrenia: a meta-analysis. Schizophr. Res. 151, 43–47. doi: 10.1016/j.schres.2013.10.011
Tran Mau-Them, F., Guibaud, L., Duplomb, L., Keren, B., Lindstrom, K., Marey, I., et al. (2019). De novo truncating variants in the intronless IRF2BPL are responsible for developmental epileptic encephalopathy. Genet. Med. 21, 1008–1014. doi: 10.1038/s41436-018-0143-0
Tripolszki, K., Sasaki, E., Hotakainen, R., Kassim, A. H., Pereira, C., Rolfs, A., et al. (2021). An X-linked syndrome with severe neurodevelopmental delay, hydrocephalus, and early lethality caused by a missense variation in the OTUD5 gene. Clin. Genet. 99, 303–308. doi: 10.1111/cge.13873
Uchida, C., and Kitagawa, M. (2016). RING-, HECT-, and RBR-type E3 ubiquitin ligases: involvement in human cancer. Curr. Cancer Drug. Targets 16, 157–174. doi: 10.2174/1568009616666151112122801
Uddin, M., Unda, B. K., Kwan, V., Holzapfel, N. T., White, S. H., Chalil, L., et al. (2018). OTUD7A regulates neurodevelopmental phenotypes in the 15q13.3 Microdeletion Syndrome. Am. J. Hum. Genet. 102, 278–295. doi: 10.1016/j.ajhg.2018.01.006
Valdmanis, P. N., Dupre, N., Lachance, M., Stochmanski, S. J., Belzil, V. V., Dion, P. A., et al. (2011). A mutation in the RNF170 gene causes autosomal dominant sensory ataxia. Brain 134, 602–607. doi: 10.1093/brain/awq329
van Loosdregt, J., Fleskens, V., Fu, J., Brenkman, A. B., Bekker, C. P., Pals, C. E., et al. (2013). Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases treg-cell-suppressive capacity. Immunity 39, 259–271. doi: 10.1016/j.immuni.2013.05.018
Varshavsky, A. (2019). N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. U S A. 116, 358–366. doi: 10.1073/pnas.1816596116
Vissers, L. E., Gilissen, C., and Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18. doi: 10.1038/nrg3999
Walczak, C. P., Leto, D. E., Zhang, L., Riepe, C., Muller, R. Y., Darosa, P. A., et al. (2019). Ribosomal protein RPL26 is the principal target of UFMylation. Proc. Natl. Acad. Sci. U S A. 116, 1299–1308. doi: 10.1073/pnas.1816202116
Wang, T., Wang, W., Wang, Q., Xie, R., Landay, A., and Chen, D. (2020). The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Ann. N. Y. Acad. Sci. 1460, 3–10. doi: 10.1111/nyas.14206
Waugh, K. A., Araya, P., Pandey, A., Jordan, K. R., Smith, K. P., Granrath, R. E., et al. (2019). Mass cytometry reveals global immune remodeling with multi-lineage hypersensitivity to Type I interferon in down syndrome. Cell Rep. 29, 1893–1908.e4.
Wei, Q., Sha, Y., Bhattacharya, A., Abdel Fattah, E., Bonilla, D., Jyothula, S. S., et al. (2014). Regulation of IL-4 receptor signaling by STUB1 in lung inflammation. Am. J. Respir. Crit. Care Med. 189, 16–29.
Wilkinson, K. D., Urban, M. K., and Haas, A. L. (1980). Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J. Biol. Chem. 255, 7529–7532. doi: 10.1016/s0021-9258(19)43857-x
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253.
Wilson, B. T., Stark, Z., Sutton, R. E., Danda, S., Ekbote, A. V., Elsayed, S. M., et al. (2016). The Cockayne Syndrome Natural History (CoSyNH) study: clinical findings in 102 individuals and recommendations for care. Genet. Med. 18, 483–493. doi: 10.1038/gim.2015.110
Wu, J., Lei, G., Mei, M., Tang, Y., and Li, H. (2010). A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. J. Biol. Chem. 285, 15126–15136. doi: 10.1074/jbc.m110.110619
Wu, X., Liu, S., Sagum, C., Chen, J., Singh, R., Chaturvedi, A., et al. (2019). Crosstalk between Lys63- and Lys11-polyubiquitin signaling at DNA damage sites is driven by Cezanne. Genes Dev. 33, 1702–1717. doi: 10.1101/gad.332395.119
Xi, P., Ding, D., Zhou, J., Wang, M., and Cong, Y. S. (2013). DDRGK1 regulates NF-kappaB activity by modulating IkappaBalpha stability. PLoS One 8:e64231. doi: 10.1371/journal.pone.0064231
Xiang, Y., Zhang, S., Lu, J., Zhang, W., Cai, M., Qiu, D., et al. (2019). USP9X promotes LPS-induced pulmonary epithelial barrier breakdown and hyperpermeability by activating an NF-kappaBp65 feedback loop. Am. J. Physiol. Cell Physiol. 317, C534–C543.
Xu, Z., Zheng, Y., Zhu, Y., Kong, X., and Hu, L. (2011). Evidence for OTUD-6B participation in B lymphocytes cell cycle after cytokine stimulation. PLoS One 6:e14514. doi: 10.1371/journal.pone.0014514
Yang, J., Huang, M., Zhou, L., He, X., Jiang, X., Zhang, Y., et al. (2018). Cereblon suppresses the lipopolysaccharide-induced inflammatory response by promoting the ubiquitination and degradation of c-Jun. J. Biol. Chem. 293, 10141–10157. doi: 10.1074/jbc.ra118.002246
Yang, M., Wang, C., Zhu, X., Tang, S., Shi, L., Cao, X., et al. (2011). E3 ubiquitin ligase CHIP facilitates Toll-like receptor signaling by recruiting and polyubiquitinating Src and atypical PKC{zeta}. J. Exp. Med. 208, 2099–2112. doi: 10.1084/jem.20102667
Yewdell, J. W., Anton, L. C., and Bennink, J. R. (1996). Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 157, 1823–1826.
Ylikallio, E., Poyhonen, R., Zimon, M., De Vriendt, E., Hilander, T., Paetau, A., et al. (2013). Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum. Mol. Genet. 22, 2975–2983. doi: 10.1093/hmg/ddt149
Zanchetta, M. E., and Meroni, G. (2019). Emerging roles of the TRIM E3 ubiquitin ligases MID1 and MID2 in cytokinesis. Front. Physiol. 10:274. doi: 10.3389/fphys.2019.00274
Zenker, M., Mayerle, J., Lerch, M. M., Tagariello, A., Zerres, K., Durie, P. R., et al. (2005). Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat. Genet. 37, 1345–1350. doi: 10.1038/ng1681
Zhang, J., Gambin, T., Yuan, B., Szafranski, P., Rosenfeld, J. A., Balwi, M. A., et al. (2017). Haploinsufficiency of the E3 ubiquitin-protein ligase gene TRIP12 causes intellectual disability with or without autism spectrum disorders, speech delay, and dysmorphic features. Hum. Genet. 136, 377–386. doi: 10.1007/s00439-017-1763-1
Zhang, Y., Song, G., Lal, N. K., Nagalakshmi, U., Li, Y., Zheng, W., et al. (2019). TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat. Commun. 10:3252.
Zhao, K., Zhang, Q., Li, X., Zhao, D., Liu, Y., Shen, Q., et al. (2016). Cytoplasmic STAT4 promotes antiviral Type I IFN production by blocking CHIP-Mediated degradation of RIG-I. J. Immunol. 196, 1209–1217. doi: 10.4049/jimmunol.1501224
Zhao, L., Hao, Y., Song, Z., Fan, Y., and Li, S. (2021). TRIM37 negatively regulates inflammatory responses induced by virus infection via controlling TRAF6 ubiquitination. Biochem. Biophys. Res. Commun. 556, 87–92. doi: 10.1016/j.bbrc.2021.03.147
Zhou, P., Ding, X., Wan, X., Liu, L., Yuan, X., Zhang, W., et al. (2018). MLL5 suppresses antiviral innate immune response by facilitating STUB1-mediated RIG-I degradation. Nat. Commun. 9:1243.
Zhou, R. Y., Wang, J. J., Sun, J. C., You, Y., Ying, J. N., and Han, X. M. (2017). Attention deficit hyperactivity disorder may be a highly inflammation and immune-associated disease (Review). Mol. Med. Rep. 16, 5071–5077. doi: 10.3892/mmr.2017.7228
Zotti, T., Uva, A., Ferravante, A., Vessichelli, M., Scudiero, I., Ceccarelli, M., et al. (2011). TRAF7 protein promotes Lys-29-linked polyubiquitination of IkappaB kinase (IKKgamma)/NF-kappaB essential modulator (NEMO) and p65/RelA protein and represses NF-kappaB activation. J. Biol. Chem. 286, 22924–22933. doi: 10.1074/jbc.m110.215426
Keywords: ubiquitin, proteasome, autoinflammation, neurodevelopmental disorders, protein aggregation
Citation: Ebstein F, Küry S, Papendorf JJ and Krüger E (2021) Neurodevelopmental Disorders (NDD) Caused by Genomic Alterations of the Ubiquitin-Proteasome System (UPS): the Possible Contribution of Immune Dysregulation to Disease Pathogenesis. Front. Mol. Neurosci. 14:733012. doi: 10.3389/fnmol.2021.733012
Received: 29 June 2021; Accepted: 10 August 2021;
Published: 08 September 2021.
Edited by:
Miguel Diaz-Hernandez, Complutense University of Madrid, SpainReviewed by:
Nico P. Dantuma, Karolinska Institutet (KI), SwedenAlessio Cardinale, Bambino Gesù Children’s Hospital (IRCCS), Italy
Copyright © 2021 Ebstein, Küry, Papendorf and Krüger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frédéric Ebstein, ZWJzdGVpbmZAdW5pLWdyZWlmc3dhbGQuZGU=; Elke Krüger, ZWxrZS5rcnVlZ2VyQHVuaS1ncmVpZnN3YWxkLmRl