 Richard Seist1,2,3†
Richard Seist1,2,3† Lukas D. Landegger1,2,4†Nahid G. Robertson5†
Lukas D. Landegger1,2,4†Nahid G. Robertson5† Sasa Vasilijic1,2Cynthia C. Morton5,6,7,8
Sasa Vasilijic1,2Cynthia C. Morton5,6,7,8 Konstantina M. Stankovic1,2,8,9*
Konstantina M. Stankovic1,2,8,9*- 1Eaton-Peabody Laboratories and Department of Otolaryngology – Head and Neck Surgery, Massachusetts Eye and Ear, Boston, MA, United States
- 2Department of Otolaryngology – Head and Neck Surgery, Harvard Medical School, Boston, MA, United States
- 3Department of Otorhinolaryngology – Head and Neck Surgery, Paracelsus Medical University, Salzburg, Austria
- 4Department of Otorhinolaryngology – Head and Neck Surgery, Medical University of Vienna, Vienna, Austria
- 5Department of Obstetrics and Gynecology and of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
- 6Broad Institute of MIT and Harvard, Cambridge, MA, United States
- 7Manchester Centre for Audiology and Deafness, School of Health Sciences, University of Manchester, Manchester, United Kingdom
- 8Program in Speech and Hearing Bioscience and Technology, Harvard Medical School, Boston, MA, United States
- 9Harvard Program in Therapeutic Science, Harvard Medical School, Boston, MA, United States
Cochlin is the most abundant protein in the inner ear. To study its function in response to noise trauma, we exposed adolescent wild-type (Coch+/+) and cochlin knock-out (Coch–/–) mice to noise (8–16 kHz, 103 dB SPL, 2 h) that causes a permanent threshold shift and hair cell loss. Two weeks after noise exposure, Coch–/– mice had substantially less elevation in noise-induced auditory thresholds and hair cell loss than Coch+/+ mice, consistent with cochlin deficiency providing protection from noise trauma. Comparison of pre-noise exposure thresholds of auditory brain stem responses (ABRs) and distortion product otoacoustic emissions (DPOAEs) in Coch–/– mice and Coch+/+ littermates revealed a small and significant elevation in thresholds of Coch–/– mice, overall consistent with a small conductive hearing loss in Coch–/– mice. We show quantitatively that the pro-inflammatory component of cochlin, LCCL, is upregulated after noise exposure in perilymph of wild-type mice compared to unexposed mice, as is the enzyme catalyzing LCCL release, aggrecanase1, encoded by Adamts4. We further show that upregulation of pro-inflammatory cytokines in perilymph and cochlear soft-tissue after noise exposure is lower in cochlin knock-out than wild-type mice. Taken together, our data demonstrate for the first time that cochlin deficiency results in conductive hearing loss that protects against physiologic and molecular effects of noise trauma.
Introduction
Coagulation factor C Homolog (COCH) is the most highly expressed gene in the cochlea (Robertson et al., 1994, 1997; Shen et al., 2015), and its encoded protein, cochlin, is the most abundant protein detected in the inner ear (Ikezono et al., 2001; Robertson et al., 2006). Cochlin consists of an N-terminal signal peptide (SP) for secretion, an LCCL (Limulus factor C, Cochlin, and late gestation Lung protein) domain, followed by two vWFA (von Willebrand-factor A-like) domains, separated by intervening domains, ivd1 and ivd2. Mutations in COCH cause the adult-onset progressive sensorineural hearing loss (HL) and vestibular disorder, DFNA9 (Khetarpal et al., 1991). To date, there are 29 known missense and 2 in-frame deletions causative of autosomal dominant hearing loss, DFNA9. These mutations result in deleterious function of cochlin with a distinct aggregative histopathology in inner ear structures, pathognomonic for DFNA9 (Khetarpal et al., 1991; Robertson et al., 1998, 2006; Khetarpal, 2000), as well as dimerization and high-molecular-weight multimerization of mutant cochlins in vitro (Yao et al., 2010; Bae et al., 2014), indicative of the dominant negative properties of mutant cochlin.
To study cochlin function, we previously utilized Coch “knock-out” (Coch–/–) mice that lack cochlin and Coch “knock-in” (CochG88E/G88E) mice that incorporate the human p.G88E missense mutation (Robertson et al., 2008). Both heterozygous and homozygous CochG88E/G88E mice recapitulate DFNA9 hearing loss and vestibular phenotype (Robertson et al., 2008; Jones et al., 2011). However, the heterozygous knock-out mice (Coch+/–) have hearing function comparable to wild-type littermates (Jones et al., 2011). This observation in a murine model is consistent with evidence that COCH haploinsufficiency is not the cause of hearing loss in humans (Masuda et al., 2016). However, mice that lack cochlin (homozygous Coch–/– mice) show a trend for mild HL limited to the highest frequency tested, and no apparent histological defects (Robertson et al., 2008; Jones et al., 2011). Relevantly, mice lacking cochlin have been utilized to demonstrate a fundamental role for cochlin in protecting the cochlea from bacteria and in systemic innate immunity (Py et al., 2013; Nyström et al., 2018; Choe et al., 2019).
Based on the mild hearing loss phenotype in Coch–/– mice, we hypothesized that a more pronounced phenotype would become apparent after noise exposure. We therefore utilized the cochlin-knockout mouse model compared to wild-type (Coch+/+) and heterozygous (Coch+/–) littermates to investigate cochlin’s function and biology in noise-induced hearing loss.
Materials and Methods
Animals and Experimental Design
The Coch–/– mouse model backcrossed into the CBA/CaJ mouse strain (Jones et al., 2011) was bred with wild-type CBA/CaJ mice purchased from Jackson Laboratory (Bar Harbor, ME, United States). Coch+/– mice were bred to generate Coch+/+, Coch+/–, and Coch–/– littermates. Genotypes were confirmed by Transnetyx company (Cordova, TN, United States) after completion of experimental procedures by researchers blinded to the genotype. Four litters of either sex were used. Cochlear function was tested by measuring ABRs and DPOAEs 2–4 days before noise exposure at 6 weeks of age (N = 10 Coch+/+, N = 13 Coch+/–, N = 12 Coch–/– mice). Animals were then exposed to noise causing permanent threshold shift (PTS), as described below. Fourteen days after noise exposure, cochlear function was reassessed with ABRs and DPOAEs. Cochlear tissue and perilymph were collected from Coch+/+ mice bred homozygous (N = 32 mice) and Coch–/– mice (N = 28).
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Massachusetts Eye and Ear and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Noise Exposure
Mice were exposed to PTS-causing octave-band noise (8–16 kHz) for 2 h at 103 dB sound pressure level (SPL) in a reverberant, acoustically transparent wire box on a rotating platform, as routine in our laboratory (Jensen et al., 2015; Landegger et al., 2019). Animals were awake and unrestrained during noise exposure. The digitally created noise using a fifth-order Butterworth filter was amplified through a power amplifier (D75A, Crown, Elkhart, IN, United States) and delivered by a loudspeaker (2446H, JBL, Los Angeles, CA, United States) coupled to an exponential horn in the roof of the box. Exposure levels were measured in each cage with a 0.25-inch Brüel & Kjaer (Naerum, Denmark) condenser microphone.
Cochlear Function Testing
ABRs and DPOAEs were recorded as detailed previously (Seist et al., 2020). Briefly, mice were anesthetized with intraperitoneally administered ketamine (100 mg/kg) and xylazine (10 mg/kg). A small incision was made in the external ear canal to provide a clear view and access to the tympanic membrane. Two miniature earphones serving as sound sources (CDMG15008-03A, CUI, Tualatin, OR, United States) and a microphone (FG-23329-P07, Knowles, Itasca, IL, United States) coupled to a probe tube within a custom acoustic system were used to measure sound pressure near the eardrum. DPOAEs were measured as ear canal pressure in response to two tones presented into the ear canal (f1 and f2, with f2/f1 = 1.2 and f1 being 10 dB above f2) at half octave steps from f2 = 5.66 to 45.25 kHz, and in 5 dB intensity increments from 10 to 80 dB SPL. DPOAE thresholds were defined as the f2 intensity required to generate a DP response 10 dB SPL over noise floor. Five ms tone pips were used to elicit ABR, which was measured between subdermal electrodes (adjacent to the ipsilateral incision, at the vertex, and near the tail), amplified 10,000 times and filtered (0.3–3.0 kHz). A total of 512 responses were recorded for each frequency and sound level and averaged using custom LabVIEW data-acquisition software run on a PXI chassis (National Instruments Corp., Austin, TX, United States). ABR waveforms stacked from lowest to highest SPL were visually inspected to define threshold as the first level at which a repeatable waveform was visually detected.
Cochlear Whole Mounts and Quantitative Confocal Fluorescence Immunohistochemistry
After intracardial perfusion of deeply anesthetized animals with 4% paraformaldehyde (PFA, #P6148, Sigma-Aldrich, St. Louis, MO, United States), both cochleae were extracted. The round and oval window membranes were punctured and gentle intracochlear perfusion with PFA was performed. Cochleae were post-fixed for 2 h in 4% PFA and decalcified in 0.12M EDTA (#17892, Thermo Fisher Scientific, Waltham, MA, United States) for 48 h. The spiraling cochleae were microdissected into six pieces to prepare whole mounts of the organ of Corti. These pieces were blocked with 5% normal horse serum (NHS, #16050130, Thermo Fisher Scientific, Waltham, MA, United States) and 1% Triton X-100 (#NC9903183, Integra Chemical, Kent, WA, United States) in PBS (#10010023, Thermo Fisher Scientific, Waltham, MA, United States) for 1/2 h at room temperature, then immunostained overnight at room temperature with rabbit anti-myosin 7A (1:200, #25-6790, Proteus Biosciences, Ramona, CA, United States) diluted in 1% NHS (#16050130, Thermo Fisher Scientific, Waltham, MA, United States) with 0.4% Triton X-100 to label hair cells. After washing in PBS three times, cochlear pieces were incubated in Alexa Fluor 647-conjugated chicken anti-rabbit antibody at 1:200 (#A21443, Thermo Fisher Scientific, Waltham, MA, United States) and phalloidin at 1:200 (#A22283, Thermo Fisher Scientific, Waltham, MA, United States). A cochlear frequency map was created by imaging specimens at low magnification (10X objective) using a fluorescence microscope (E800, Nikon, Tokyo, Japan), and then applying a custom ImageJ plug-in developed at Massachusetts Eye and Ear1. Cochlear whole mounts were subsequently imaged with a confocal microscope (SP5, Leica, Wetzlar, Germany) and a glycerol-immersion 63X objective (1.3 N.A.) at log-spaced cochlear frequency regions from 4 to 64 kHz.
RNA Isolation, cDNA Synthesis and Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Six hours after noise exposure, cochleae were extracted after decapitating deeply anesthetized mice and cochlear soft tissue was microdissected away from the bony otic capsule with fine forceps in RNAlater (Ambion, Austin, TX, United States). One specimen consisted of both cochleae from one animal. Total RNA was extracted using RNeasy Micro Kit (Qiagen GmbH, Hilden, Germany) and reverse transcribed with TaqMan Reverse Transcription Reagents kit (Applied Biosystems, Foster City, CA, United States) according to the manufacturers’ protocols. Real-time quantitative RT-PCR was performed using an Applied Biosystems 7700 Sequence Detection System and TaqMan Primers for Coch (Mm00483360_m1), Adamts4 (Mm00556068_m1), Adamts5 (Mm00478620_m1), Il6 (Mm00446190_m1), Tnf (Mm00443258_m1), Il1b (Mm008409 04_m1), Pycard (Mm00445747_g1), Cxcl1 (Mm04207460_m1), Casp1 (Mm00438023_m1), Nlrp3 (Mm00840904_m1), and reference gene 18S ribosomal RNA (Hs99999901_s1). Expression changes were analyzed by relative quantification and plotted as fold-change using 2–ΔΔCT method (Livak and Schmittgen, 2001; Stankovic and Corfas, 2003).
Perilymph Collection
Perilymph was sampled 6 h after noise exposure using the approach adapted by our laboratory in a terminal procedure via the posterior semicircular canal (PSCC) (Landegger et al., 2019). In brief, mice anesthetized with ketamine (100 mg/kg i.p.) and xylazine (10 mg/kg i.p.) were given a booster as needed at half of the initial dose. A retroauricular stereotaxic approach permitted the exposure of the PSCC, which was opened after appropriate preparation. Sampling was achieved with the help of calibrated disposable micropipettes (VWR, Radnor, PA, United States) and three specimens per ear were collected (0.5 μL for vestibular perilymph = vPLF; 0.5 μL for cochlear perilymph = cPLF; up to 5 μL of cerebrospinal fluid = CSF). We observed minimal amounts of cochlin (data not shown) in CSF, suggesting some perilymph residue in the CSF sample. Only clear specimens were quantified and stored in an Eppendorf tube (Eppendorf, Hamburg, Germany) in 10 μL of double-filtered PBS (Millipore Sigma, Burlington, MA, United States). Tubes were sealed and placed into a −80°C freezer until further use.
Perilymph Analysis
As described earlier (Landegger et al., 2019), the V-PLEX Pro-inflammatory Panel 1 Mouse Kit (#K15048D, Meso Scale Discovery = MSD, Rockville, MD, United States), an immunoassay based on electrochemiluminescence, was used to analyze the murine perilymph. Manufacturer’s instructions were followed, and the levels of the following cytokines were measured on a matching MSD SI2400 instrument by fitting electrochemiluminescence signals to the calibration curve: IFN-γ, IL-1β, IL-2, IL-4, IL-5, IL-6, CXCL1, IL-10, IL-12p70, and TNF-α.
Western Blot
Western blot of cochlear perilymph was performed on perilymph samples collected through the PSCC as described above. Proteins were separated by SDS gel electrophoresis using reducing 4–12% polyacrylamide gradient gels and transferred to Immobilon-P (Millipore, Bedford, MA, United States) membranes using a wet transfer apparatus (Invitrogen, Carlsbad, CA, United States). Membranes were incubated with primary antibody monoclonal anti-cochlin (EMD Millipore, clone 9A10D2, Cat# MABF267) at 1:1,000 dilution. Blots were subjected to radiography after secondary antibody and PBST (Tween) washes.
The relative protein quantification of LCCL fragments was performed with ImageJ software (National Institutes of Health, MD, United States) (Davarinejad, 2018). Briefly, gel and western blotting PVDF membrane were imaged and analyzed using ImageJ after exporting images at 600 dpi in TIFF file format. Raw images were converted into a JPEG file format using Adobe Photoshop CS2 software (Adobe Systems Inc., San Jose, CA, United States) and changed to the grayscale picture mode. Using ImageJ tools, a single region of interest was created covering the minimum area containing the whole content of the largest band. This process was applied for each row of protein bands for each lane. The same frame was used for all of the protein bands across other lanes. Using ImageJ tools, density of the protein bands, including the background of empty fields, was converted into the pixel density, and exported into Excel (Microsoft Office, Microsoft Corporation, Redmond, WA, United States). Inversion of pixel density was performed using subtraction of the recorded values from 255. The net value for all protein bands was determined by deducting the inverted background from the inverted band values. Relative quantification values were expressed as a ratio of the net band values of LCCL fragments in each sample to the sum of the net bands of all proteins in the sample (the total protein value).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 9.0.0 (GraphPad Software, Inc., San Diego, CA, United States). Statistical significance in ABR and DPOAE data (thresholds, I/O-functions) (Figures 1A,B, 2) as well as cytokine expression in perilymph (Figure 3C) was determined using ordinary two-way ANOVA and subsequent Tukey’s multiple comparisons test. For outer hair cell-loss, ordinary two-way ANOVA with subsequent Šidak test for multiple comparisons was employed (Figure 1D). qPCR data were analyzed by unpaired t-test with Holm-Šidak method for multiple comparisons (Figure 3B and Supplementary Figures 2A,B). Western blot of perilymph was analyzed by unpaired t-test (Figure 3A). A probability value of P < 0.05 was considered statistically significant. All data are presented as means ± standard errors of the mean (SEMs).
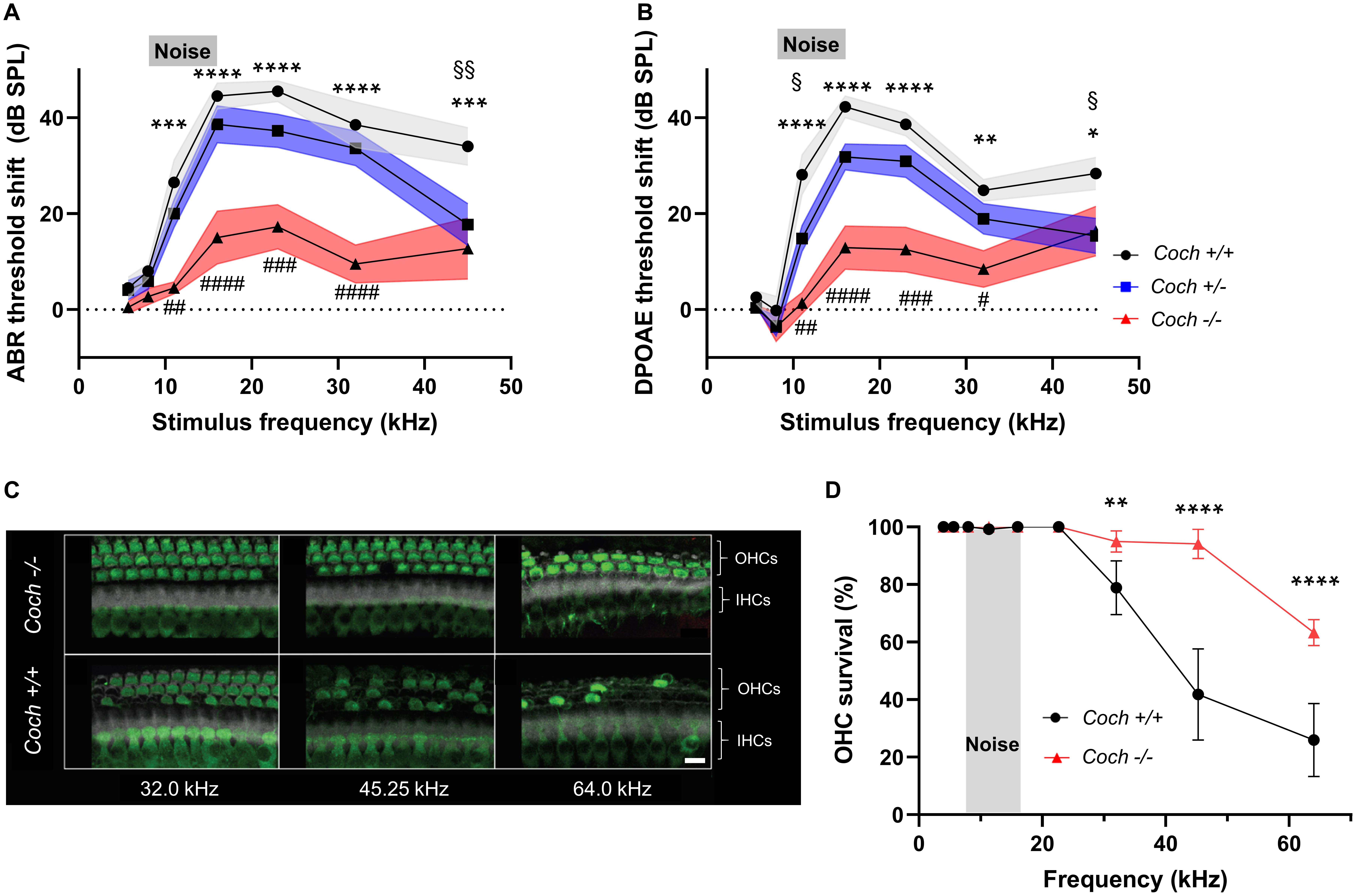
Figure 1. Absence of cochlin reduces the level of cochlear dysfunction and sensory cell damage after acoustic injury. Six-week-old mice of each genotype were exposed to 8–16 kHz noise for 2 h at 103 dB SPL. (A) ABR and (B) DPOAE threshold shifts 2 weeks after noise trauma demonstrate profound mid-to-high frequency hearing loss in wild-type Coch+/+ mice, and statistically significant lower threshold shifts in Coch–/– mice. A trend toward lower threshold shifts is observed in heterozygous mice, which reaches statistical significance at 11.33 kHz (DPOAE) and 45.25 kHz (ABR and DPOAE). The gray rectangle indicates frequency of noise band. Data are shown as group means ± standard error of the mean; N = 10 Coch+/+, N = 11 Coch+/–, and N = 11 Coch–/– animals. *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001; asterisks: Coch+/+ vs. Coch–/–, #: Coch+/–. Coch–/–, §: Coch+/– vs. Coch+/+. (C) Representative cochlear whole mounts from Coch+/+ and Coch–/– mice 2 weeks after acoustic trauma. IHC, inner hair cells. OHC, outer hair cells. Green = myosin 7A, white = phalloidin. Scale bar: 10 μm. (D) Cochleogram showing fewer missing outer hair cells in Coch–/– mice 2 weeks after acoustic trauma. Data are shown as group means ± standard error of the mean; N = 4 Coch+/+ and N = 4 Coch–/– mice. **P < 0.01 or ****P < 0.0001.
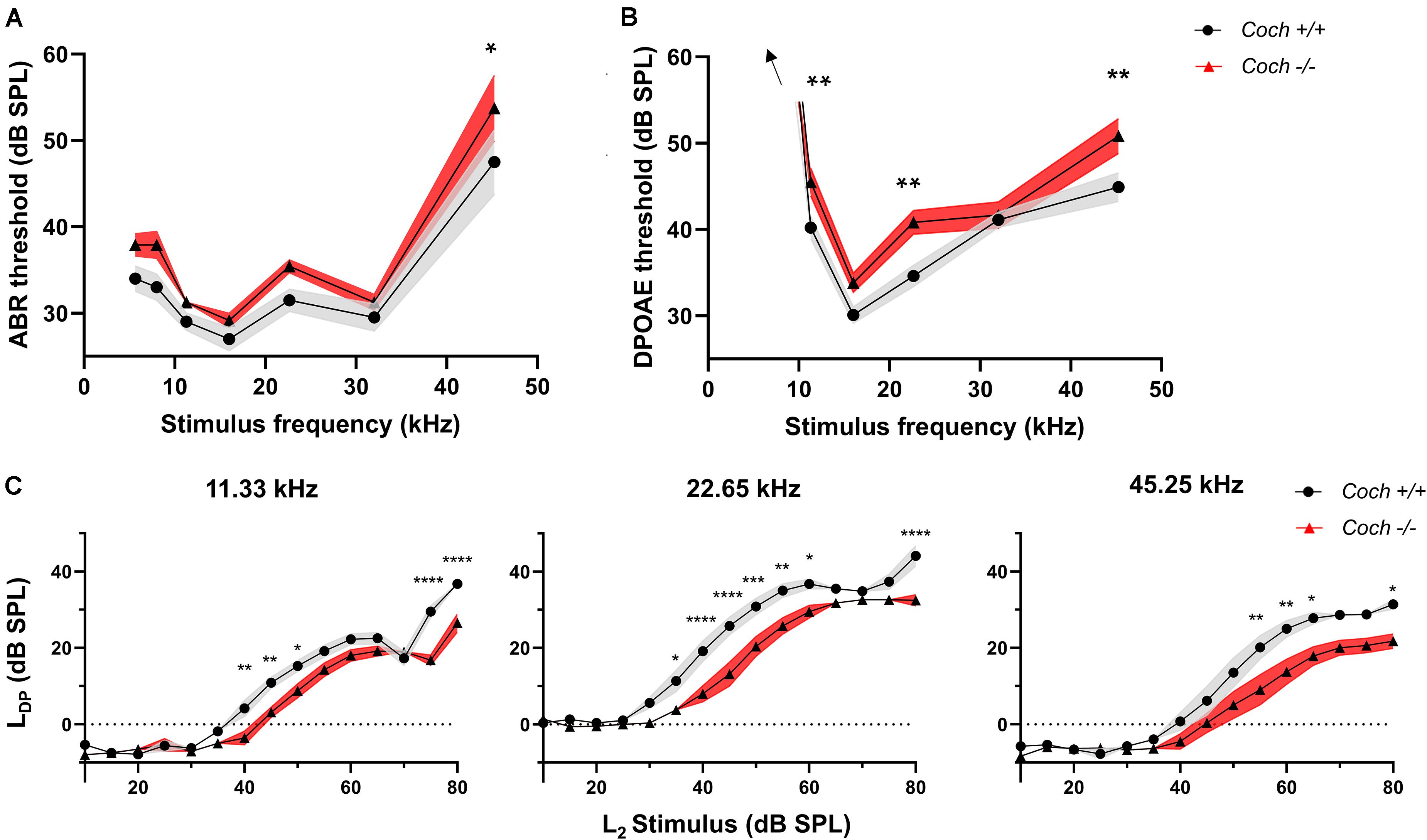
Figure 2. Cochlin knock-out mice have small but significant elevations in audiometric thresholds compared to wild-type mice. (A) ABR and (B) DPOAE thresholds are higher in Coch–/– compared to Coch+/+ mice. The ABR threshold difference reaches statistical significance at 45.25 kHz. DPOAE threshold difference reaches significance at 11.33, 22.65, and 45.25 kHz. Thresholds at 5.66 and 8 kHz >60 dB SPL. (C) Input/output functions of DPOAEs at 11.33 kHz, 22.65 kHz, and 45.25 kHz show a right shift and largely unaltered growth function in Coch–/– animals compared to Coch+/+. Data are shown as group means ± standard error of the mean; N = 10 Coch+/+ and N = 12 Coch–/– animals. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. LDP = DPOAE level, L2 = primary tone level.
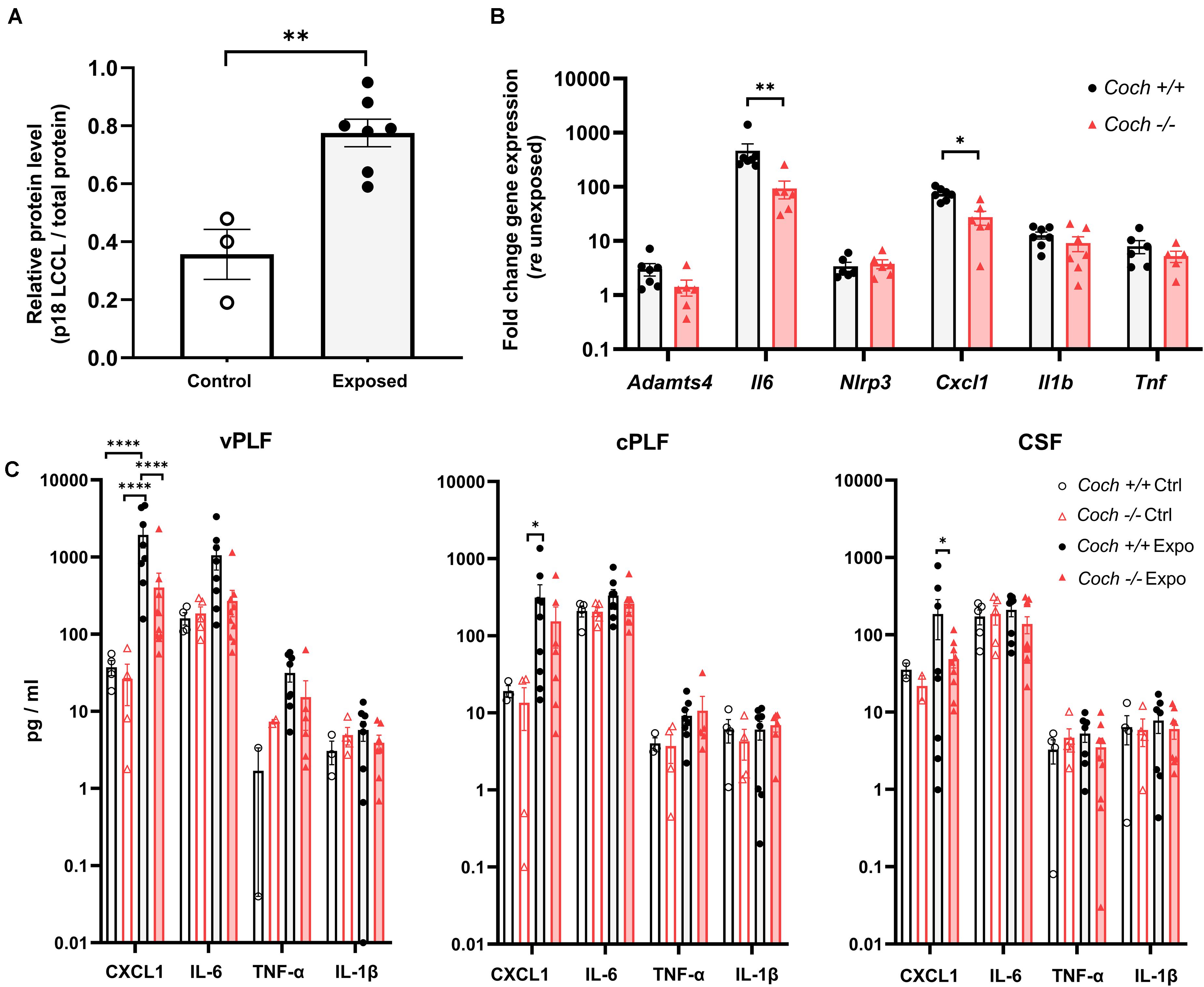
Figure 3. Cochlin-deficient mice demonstrate reduced pro-inflammatory response to acoustic trauma in cochlear soft tissue and perilymph. Six-week-old Coch–/– and Coch+/+ mice were exposed to 8–16 kHz noise for 2 h at 103 dB SPL. Unexposed mice served as controls. (A) Six hours post exposure, cochlear perilymph of wild-type animals demonstrated significantly increased levels of the cleaved LCCL domain (18 kDa fragment) compared to unexposed animals. Data are shown as group means of relative protein level of 18 kDa LCCL fragment (p18) ± standard error of the mean. ∗∗P < 0.01 (unpaired t-test). Cochlear perilymph was collected through the posterior semicircular canal. Expression of LCCL fragment was determined by Western blotting using an anti-cochlin monoclonal antibody. Protein level of LCCL fragment was calculated as a ratio of LCCL p18 protein bands relative to the total protein bands. N = 3 ears from unexposed mice, 7 ears from exposed mice. (B) Cochleae collected 6 h post exposure had statistically significant elevation of Il6 and Cxcl1 gene expression in Coch+/+ compared to Coch–/– mice, and demonstrated a similar trend that did not meet our criterion for significance for Adamts4, Il1b, and Tnf. N = 7 animals per group. Data are shown as group means ± standard error of the mean. ∗P < 0.05, ∗∗P < 0.01. (C) Six hours post exposure, perilymph demonstrated significantly lower CXCL1 levels in Coch–/– compared to Coch+/+ mice and trends toward lower IL-6, TNF-α, and IL-1β levels, that did not meet our criterion of statistical significance. Data are shown as group means ± standard error of the mean. ∗P < 0.05, ****P < 0.0001. Vestibular perilymph (vPLF), cochlear perilymph (cPLF), and cerebrospinal fluid (CSF) were collected through the posterior semicircular canal. N = 4 Coch+/+ unexposed ears, 5 Coch+/+ exposed ears, 9 Coch–/– unexposed ears, and 10 Coch–/– exposed ears.
Results
Protection From Permanent Noise Trauma in Coch–/– Mice
Sound entering the inner ear causes vibration of the basilar membrane, which is amplified by OHCs, leading to stimulation of inner hair cells, neurotransmitter release, and transmission of electrical signals via the auditory nerve to the brain. DPOAEs are produced by OHCs not only because cochleae amplify vibrations, but also because they exhibit non-linearity (Hudspeth, 2014). The ABR captivates the neural response to sound, initiated at the inner hair cell-spiral ganglion neuron synapse. While a previous study showed mild physiological changes (slightly elevated ABR threshold only in the highest frequency tested, in older mice) but no morphological cochlear deficits in Coch–/– mice (Jones et al., 2011), here we investigated how Coch–/– mice would respond to an external challenge, in this case high noise exposure. For these measurements, we exposed 6-week-old littermates of three genotypes – wild-type Coch+/+ (N = 10) mice, heterozygous Coch+/– (N = 10) mice and homozygous Coch–/– (N = 11) mice – to 8–16 kHz octave-band noise at 103 dB SPL for 2 h. We have previously shown that these noise parameters cause permanent hearing loss accompanied by outer hair cell loss in the cochlear basal turn and substantial intracochlear inflammation (Landegger et al., 2019). Hearing thresholds were measured by ABR (Figure 1A) and DPOAE (Figure 1B) 2 weeks after noise exposure and demonstrated threshold elevations in all genotypes. To our surprise, compared to the wild-type genotype, Coch–/– mice had significantly lower ABR threshold shifts (Figure 1A) at 11.33 kHz (mean difference = 21.95 dB SPL, P = 0.0001), 16.00 kHz (mean difference = 29.50 dB SPL, P < 0.0001), 22.65 kHz (mean difference = 28.23 dB SPL, P < 0.0001), 32.00 kHz (mean difference = 28.9 dB SPL, P < 0.0001), and 45.25 kHz (mean difference = 21.27 dB SPL, P = 0.0001), showing a protective effect to noise as compared to wild-type mice. Coch+/– mice demonstrated a trend toward lower threshold shifts than wild-type mice, which reached statistical significance at 45.25 kHz (mean difference = 16.27 dB SPL, P = 0.0049). Compared to wild-type mice, DPOAE threshold shifts (Figure 1B) in Coch–/– mice were significantly lower at 11.33 kHz (mean difference = 26.74 dB SPL, P < 0.0001), 16.00 kHz (mean difference = 29.39 dB SPL, P < 0.0001), 22.65 kHz (mean difference = 26.05 dB SPL, P < 0.0001), 32.00 kHz (mean difference = 16.45 dB SPL, P = 0.0010), and 45.25 kHz (mean difference = 12.04 dB SPL, P = 0.0227). Coch+/– mice showed a trend toward lower DPOAE threshold shifts than wild-type mice, which reached significance at 11.33 kHZ (mean difference = 13.28 dB SPL, P = 0.0103) and 45.25 kHz (mean difference = 13.04 dB SPL, P = 0.0121). Sex-specific analysis did not show any major differences between female and male animals (Supplementary Figure 1).
Next, we assessed morphological changes in cochlear whole mounts from noise-exposed wild-type and Coch–/– mice to identify structural evidence for the observed functional outcome. Hair cells were identified using phalloidin stain and antibodies to Myo7A as hair cell marker. The number of surviving outer hair cells (OHCs) in Coch–/– mice was higher than in wild-type animals across the frequencies of maximal noise susceptibility, as illustrated in representative images in Figure 1C. Acoustic injury typically causes maximal damage at frequencies higher than the stimulus frequency (Cody and Johnstone, 1981). This upward spread of cochlear damage arises because of level-dependent non-linearities in cochlear mechanics (Robles and Ruggero, 2001). The qualitative observation illustrated in Figure 1C was supported by quantitative measurements (Figure 1D) demonstrating that Coch–/– animals had significantly higher OHC counts at 32.00 kHz (P = 0.0013), 45.25 kHz (P < 0.0001), and 64.00 kHz (P < 0.0001). Taken together, cochlin-lacking mice exhibited a greater survival of OHCs than their wild-type littermates after noise exposure, which led to reduced ABR and DPOAE thresholds.
Coch–/– Mice Exhibit Signs of Mild Conductive Hearing Loss
To determine whether cochlin deficiency confers protection from noise-induced hearing loss via a conductive or sensorineural mechanism, we measured audiometric thresholds in littermates of Coch–/– and Coch+/+ mice. To maximize robustness and reproducibility of the data, all measurements were performed by a researcher blinded to the genotype. Six-week-old unexposed Coch–/– mice (N = 12) demonstrated two to six dB higher ABR (Figure 2A) and DPOAE (Figure 2B) thresholds compared to Coch+/+ mice (N = 10). This difference was statistically significant in ABR thresholds at 45.25 kHz (P = 0.0252) and DPOAE thresholds at 11.33 kHz (P = 0.0187), 22.65 kHz (P = 0.0043), and 45.25 kHz (P = 0.0071). Coch+/+ and Coch+/– animals (N = 13) were statistically indistinguishable (data not shown).
To assess a possible mechanical contribution to the elevated thresholds in Coch–/– mice, we further evaluated DPOAE input/output functions. Cochlear dysfunction is known to lead to a change in slope of the growth function, whereas middle ear pathologies cause a right shift of the curve with unaltered slope of the growth function (Gehr et al., 2004). Input/output functions of DPOAEs at 11.33 kHz, 22.65 kHz, and 45.25 kHz show a statistically significant right shift and largely unaltered slope of the growth function in Coch–/– animals compared to Coch+/+ animals (Figure 2C), with the exception of a slight reduction in slope at 45.25 kHz. Taken together, these data are consistent with a small conductive hearing loss in Coch–/– mice compared to Coch+/+ and a possible additional sensorineural component at the highest tested frequency.
Noise Trauma Causes Weaker Pro-inflammatory Response in Coch–/– Mice
To further study the mechanism behind the protection from noise exposure in cochlin-deficient animals, we collected perilymph from the posterior semicircular canal 6 h after noise exposure, because we previously reported a robust immune reaction at that time point (Landegger et al., 2019). Six hours after exposure of Coch+/+ mice to 103 dB noise, we detected an increase in the level of cleaved LCCL domain p18 (18 kDa fragment) compared to unexposed Coch+/+ mice (P = 0.0018), as assessed by western blot of cochlear perilymph (Figure 3A and Supplementary Figure 3). We also found the expression of the gene Adamts4 encoding the metalloprotease enzyme, aggrecanase1 responsible for cleaving the cochlin LCCL, and initiating immune response (Py et al., 2013), to be elevated after noise exposure. Adamts4 expression as measured by quantitative real-time PCR, was upregulated in cochlear soft tissue 6 h after 103 dB noise in Coch+/+ and Coch–/– mice; Coch–/– mice showed a trend toward lower Adamts4 expression than Coch+/+ mice after noise exposure (Figure 3B). The expression of genes encoding the pro-inflammatory cytokines IL-6 (P = 0.005881) and CXCL1 (P = 0.032899) were significantly elevated in Coch+/+ mice compared to Coch–/– mice. A similar trend was demonstrated for Il1b and Tnf genes (Figure 3B) although it did not meet our criterion for significance. Inflammasome-associated genes were significantly upregulated in exposed wild-type cochleae: Il1b (P = 0.002001), and Nlrp3 (P = 0.008397) (Supplementary Figure 2A). A trend toward reduced activation of inflammasome-associated genes in Coch–/– compared to Coch+/+ cochleae was measured 6 h after noise exposure (Supplementary Figure 2B). In vestibular perilymph, we observed significantly lower CXCL1 levels in Coch–/– compared to Coch+/+ mice (P < 0.0001) and trends for lower levels of IL-6, IL-1β, and TNF-α that did not meet our criterion for statistical significance (Figure 3C).
Discussion
Using a combination of physiological tests that assess cochlear function, here we demonstrate for the first time that Coch–/– mice have a small conductive hearing loss compared to Coch+/+ littermates, and possibly additional sensorineural component at the highest tested frequency. Together with the previously reported expression of cochlin in the murine and human middle ear, precisely on the articular surfaces of the incudomalleal and incudostapedial joints as well as pars tensa of the tympanic membrane (Robertson et al., 2014), we highlight cochlin’s importance in sound conduction across the middle ear. In previous studies, a trend for increased ABR thresholds was present in young Coch–/– mice compared to Coch+/+ mice, although this trend did not reach the authors’ criterion for significance (Choe et al., 2019). The small number of mice tested in that study, four per group, may have prevented detection of a statistically significant change. In contrast, by more than doubling the number of mice in our cohorts, we were able to detect a statistically significant elevation in ABR and DPOAE thresholds due to cochlin deficiency.
Precisely quantifying the degree of conductive hearing loss due to cochlin deficiency is important in this era of emerging inner ear gene therapy, as correction of COCH mutations via localized delivery to the inner ear will leave the middle ear unaddressed. Long-term conductive hearing loss is known to lead to speech intelligibility deficits in patients and loss of synapses in an animal model of conductive hearing loss (Liberman et al., 2015; Okada et al., 2020). Future murine studies should therefore quantify air- and bone-conduction evoked ABRs (Qin et al., 2010; Chhan et al., 2017) to further delineate conductive vs. sensorineural contribution to HL in Coch–/– mice. In addition, future clinical studies should evaluate for potential presence of conductive hearing loss in patients with COCH pathogenic variants.
In the meantime, our finding that noise exposure in wild-type mice causes an increase of the pro-inflammatory LCCL component of cochlin in perilymph compared to unexposed controls is significant because it is consistent with noise-causing sterile inflammation. It is known that inflammatory stimuli such as lipopolysaccharide (LPS) and TNF-α, and pathogens including Pseudomonas aeruginosa and Staphylococcus aureus lead to post-translational cleavage of full-length cochlin (p60, 60 kD) between the LCCL and vWFA1 domains by aggrecanase1. This, in turn, releases the cochlin-LCCL self-folding modular protein isoform (p8-p16, 8 kD–16 kD, including glycosylated forms) into the bloodstream or perilymph. This process results in local downstream effects on inflammatory cytokine production, macrophage activation, and immune cell recruitment in the infected organ (Moussion and Sixt, 2013; Py et al., 2013; Nyström et al., 2018; Choe et al., 2019; Komatsu and Li, 2019; Rhyu et al., 2020).
The first and typically irreversible cellular changes after noise exposure are degeneration of spiral ligament fibrocytes (Kujawa and Liberman, 2006) and cochlear afferent neurites (Kujawa and Liberman, 2009). Whereas there is abundant cochlin expression in the spiral ligament, there is no immunohistochemically detectable cochlin in spiral ganglion cell bodies and neurites (Robertson et al., 2006). However, cochlin is immunohistochemically detectable in cells lining Rosenthal’s canal and the osseus spiral lamina channels that surround the neural compartments of the cochlea (Robertson et al., 2006). Cochlin’s location surrounding neural tissue warrants future studies to determine whether cochlin plays a role in cochlear neuropathy and synaptopathy. The importance of cochlin for spiral ligament function is demonstrated by histopathological analysis of human DFNA9 temporal bones that show cell loss and replacement with eosinophilic acellular material in the spiral ligament, as well as the spiral limbus, and stroma of the cristae and maculae (Robertson et al., 1998, 2006; Merchant et al., 2000). We show for the first time that cochlin’s LCCL component is cleaved after intense noise exposure through aggrecanase1 and released into the perilymph. It is likely that this LCCL component is cleaved from the spiral ligament extracellular matrix given the abundance of cochlin in the spiral ligament and the spiral ligament’s central role in inflammatory responses (Adams, 2002). Precise mechanisms of LCCL entry into the perilymph and how this affects patients suffering from DFNA9, noise, and age-related hearing loss remains to be determined in the future.
Due to the apparent and surprising conductive hearing loss in Coch knock-out mice, the observed protection from noise in these Coch–/– mice cannot be entirely attributed to the lack of LCCL cleaving in the inner ear and its downstream reduction of inflammatory cytokines. It is, however, possible that the mild conductive loss attenuates the amount of noise carried into the cochlea, thus preventing some of the noise-induced damage. Strategically designed additional experiments are needed to dissect out contributions from cochlin deficiency in the inner ear and cochlin deficiency in the middle ear as the latter causes conductive hearing loss and effectively reduces noise input to the inner ear. These future experiments could involve mouse models lacking cochlin in the inner ear only or middle ear only, as well as pharmacologic (Durham et al., 2017) or genetic inhibition of aggrecanases.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Massachusetts Eye and Ear.
Author Contributions
KS, LL, and RS conceived and planned the experiments. LL, RS, NR, and SV performed the experiments and analyzed the data. KS, RS, and NR wrote the manuscript with inputs from all authors. KS supervised all aspects of research. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health grants R01 DC015824 (to KS) and R01 DC03402 (to CM), as well as Nancy Sayles Day Foundation, Lauer Tinnitus Research Center, Zwanziger Foundation, Barnes Foundation, and Sheldon and Dorothea Buckler (all to KS) and CM is also supported by the NIHR Manchester Biomedical Research Center.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2021.670013/full#supplementary-material
Supplementary Figure 1 | Protection from noise-induced hearing loss in Coch–/– compared to Coch+/+ mice is similar in both sexes. Replotting of results depicted in Figure 1. Six-week-old mice of each genotype were exposed to 8–16 kHz noise for 2 h at 103 dB SPL; ABR threshold shifts were measured 2 weeks later. The gray rectangle indicates frequency of noise band. There was no significant difference between the sexes of each genotype. N = 3 Coch+/+ males, N = 7 Coch+/+ females, N = 5 Coch–/– males, and N = 6 Coch–/– females.
Supplementary Figure 2 | Acoustic trauma leads to upregulation of inflammasome-associated genes in WT cochleae and a trend toward reduced activation of inflammasome-associated genes in Coch–/– compared to Coch+/+ cochleae. Six-week-old Coch+/+ and Coch–/– mice were exposed to 8–16 kHz noise for 2 h at 103 dB SPL. Unexposed mice served as controls. (A) Wild-type cochleae collected 6 h post exposure (gray bars) had statistically significant elevation in expression of Nlrp3 and Il1b genes compared to unexposed (white bars) cochleae and demonstrated a similar trend for Casp1. Data are shown as group means ± standard error of the mean. ∗∗P < 0.01. (B) Six hours post exposure, Coch–/– cochleae showed a trend toward reduced expression of inflammasome-associated genes; this trend did not meet our criterion for significance. N = 7 mice per group.
Supplementary Figure 3 | Raw western blot data used for quantification presented in Figure 3A. Six-week-old Coch+/+ mice were exposed to 8–16 kHz noise for 2 h at 103 dB SPL. Unexposed mice served as controls. Six hours post exposure, perilymph was collected through the posterior semicircular canal. Left, representative Ponceau stained gel. Right, corresponding PVDF membrane stained with anti-cochlin monoclonal antibody. The anti-cochlin antibody identified strong protein bands of 18 kDa LCCL fragment in perilymph of noise-exposed animals (lanes 4–10) (N = 7 ears) and weaker bands of the same fragment in perilymph of control animals (lanes 1–3) (N = 3 ears). The arrows point to 18 kDa (p18), 16 kDa (p16), and 8 kDa (p8) LCCL-specific bands.
Footnotes
References
Adams, J. C. (2002). Clinical implications of inflammatory cytokines in the cochlea: a technical note. Otol. Neurotol. 23, 316–322. doi: 10.1097/00129492-200205000-00015
Bae, S. H., Robertson, N. G., Cho, H. J., Morton, C. C., Jung, D. J., Baek, J. I., et al. (2014). Identification of pathogenic mechanisms of COCH mutations, abolished cochlin secretion, and intracellular aggregate formation: genotype-phenotype correlations in DFNA9 deafness and vestibular disorder. Hum. Mutat. 35, 1506–1513. doi: 10.1002/humu.22701
Chhan, D., McKinnon, M. L., and Rosowski, J. J. (2017). Identification of induced and naturally occurring conductive hearing loss in mice using bone conduction. Hear. Res. 346, 45–54. doi: 10.1016/j.heares.2017.02.001
Choe, Y. H., Yoon, J., Jung, J., Yoo, J. E., Choe, Y. H., and Park, S. C. (2019). Cleaved cochlin sequesters Pseudomonas aeruginosa and activates innate immunity in the inner ear. Cell Host Microbe 25, 513.e6–525.e6. doi: 10.1016/j.chom.2019.02.001
Cody, A. R., and Johnstone, B. M. (1981). Acoustic trauma: single neuron basis for the “half-octave shift”. J. Acoust. Soc. Am. 70, 707–711. doi: 10.1121/1.386906
Davarinejad, H. (2018). Quantifications of Western Blots With ImageJ. Avaliable at: www.yorku.ca/yisheng/Internal/Protocols/ImageJ.pdf (accessed 10.1.10).
Durham, T. B., Marimuthu, J., Toth, J. L., Liu, C., Adams, L., Mudra, D. R., et al. (2017). A highly selective hydantoin inhibitor of aggrecanase-1 and aggrecanase-2 with a low projected human dose. J. Med. Chem. 60, 5933–5939. doi: 10.1021/acs.jmedchem.7b00650
Gehr, D. D., Janssen, T., Michaelis, C. E., Deingruber, K., and Lamm, K. (2004). Middle ear and cochlear disorders result in different DPOAE growth behaviour: implications for the differentiation of sound conductive and cochlear hearing loss. Hear. Res. 193, 9–19. doi: 10.1016/j.heares.2004.03.018
Hudspeth, A. J. (2014). Integrating the active process of hair cells with cochlear function. Nat. Rev. Neurosci. 15, 600–614. doi: 10.1038/nrn3786
Ikezono, T., Omori, A., Ichinose, S., Pawankar, R., Watanabe, A., and Yagi, T. (2001). Identification of the protein product of the Coch gene (hereditary deafness gene) as the major component of bovine inner ear protein. Biochim. Biophys. Acta Mol. Basis Dis. 1535, 258–265. doi: 10.1016/S0925-4439(00)00101-0
Jensen, J. B., Lysaght, A. C., Liberman, M. C., Qvortrup, K., and Stankovic, K. M. (2015). Immediate and delayed cochlear neuropathy after noise exposure in pubescent mice. PLoS ONE 10:e0125160. doi: 10.1371/journal.pone.0125160
Jones, S. M., Robertson, N. G., Given, S., Giersch, A. B. S., Liberman, M. C., and Morton, C. C. (2011). Hearing and vestibular deficits in the Coch-/- null mouse model: comparison to the CochG88E/G88E mouse and to DFNA9 hearing and balance disorder. Hear. Res. 272, 42–48. doi: 10.1016/j.heares.2010.11.002
Khetarpal, U. (2000). DFNA9 is a progressive audiovestibular dysfunction with a microfibrillar deposit in the inner ear. Laryngoscope 110, 1379–1384. doi: 10.1097/00005537-200008000-00030
Khetarpal, U., Schuknecht, H. F., Gacek, R. R., and Holmes, L. B. (1991). Autosomal dominant sensorineural hearing loss. Pedigrees, audiologic findings, and temporal bone findings in two kindreds. Arch. Otolaryngol. Head. Neck Surg. 117, 1032–1042.
Komatsu, K., and Li, J.-D. (2019). Cleaved cochlin: one guard with two roles in surveillance. Cell Host Microbe 25, 475–476. doi: 10.1016/j.chom.2019.03.011
Kujawa, S. G., and Liberman, M. C. (2006). Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J. Neurosci. 26, 2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006
Kujawa, S. G., and Liberman, M. C. (2009). Adding insult to injury: cochlear nerve degeneration after “Temporary” noise-induced hearing loss. J. Neurosci. 29, 14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009
Landegger, L. D., Vasilijic, S., Fujita, T., Soares, V. Y., Seist, R., Xu, L., et al. (2019). Cytokine levels in inner ear fluid of young and aged mice as molecular biomarkers of noise-induced hearing loss. Front. Neurol. 10, 1–14. doi: 10.3389/fneur.2019.00977
Liberman, M. C., Liberman, L. D., and Maison, S. F. (2015). Chronic conductive hearing loss leads to cochlear degeneration. PLoS One 10:e0142341. doi: 10.1371/journal.pone.0142341
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Masuda, M., Mutai, H., Arimoto, Y., Nakano, A., and Matsunaga, T. (2016). A novel frameshift variant of COCH supports the hypothesis that haploinsufficiency is not a cause of autosomal dominant nonsyndromic deafness 9. Biochem. Biophys. Res. Commun. 469, 270–274. doi: 10.1016/j.bbrc.2015.11.106
Merchant, S. N., Linthicum, F. H., and Nadol, J. B. (2000). Histopathology of the inner ear in DFNA9. Adv. Otorhinolaryngol. 56, 212–217. doi: 10.1159/000059105
Moussion, C., and Sixt, M. (2013). A conduit to amplify innate immunity. Immunity 38, 853–854. doi: 10.1016/j.immuni.2013.05.005
Nyström, A., Bornert, O., Kühl, T., Gretzmeier, C., Thriene, K., Dengjel, J., et al. (2018). Impaired lymphoid extracellular matrix impedes antibacterial immunity in epidermolysis bullosa. Proc. Natl. Acad. Sci. U.S.A. 115, E705–E714. doi: 10.1073/pnas.1709111115
Okada, M., Welling, D. B., Liberman, M. C., and Maison, S. F. (2020). Chronic conductive hearing loss is associated with speech intelligibility deficits in patients with normal bone conduction thresholds. Ear Hear 41, 500–507. doi: 10.1097/AUD.0000000000000787
Py, B. F., Gonzalez, S. F., Long, K., Kim, M. S., Kim, Y. A., Zhu, H., et al. (2013). Cochlin produced by follicular dendritic cells promotes antibacterial innate immunity. Immunity 38, 1063–1072. doi: 10.1016/j.immuni.2013.01.015
Qin, Z., Wood, M., and Rosowski, J. J. (2010). Measurement of conductive hearing loss in mice. Hear. Res. 263, 93–103. doi: 10.1016/j.heares.2009.10.002
Rhyu, H.-J., Bae, S. H., Jung, J., and Hyun, Y.-M. (2020). Cochlin-cleaved LCCL is a dual-armed regulator of the innate immune response in the cochlea during inflammation. BMB Rep. 53, 449–452. doi: 10.5483/BMBRep.2020.53.9.104
Robertson, N. G., Cremers, C. W. R. J., Huygen, P. L. M., Ikezono, T., Krastins, B., Kremer, H., et al. (2006). Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum. Mol. Genet. 15, 1071–1085. doi: 10.1093/hmg/ddl022
Robertson, N. G., Jones, S. M., Sivakumaran, T. A., Giersch, A. B. S., Jurado, S. A., Call, L. M., et al. (2008). A targeted Coch missense mutation: a knock-in mouse model for DFNA9 late-onset hearing loss and vestibular dysfunction. Hum. Mol. Genet. 17, 3426–3434. doi: 10.1093/hmg/ddn236
Robertson, N. G., Khetarpal, U., Gutiérrez-Espeleta, G. A., Bieber, F. R., and Morton, C. C. (1994). Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics 23, 42–50. doi: 10.1006/geno.1994.1457
Robertson, N. G., Lu, L., Heller, S., Merchant, S. N., Eavey, R. D., McKenna, M., et al. (1998). Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat. Genet. 20, 299–303. doi: 10.1038/3118
Robertson, N. G., O’Malley, J. T., Ong, C. A., Giersch, A. B. S., Shen, J., Stankovic, K. M., et al. (2014). Cochlin in normal middle ear and abnormal middle ear deposits in DFNA9 and CochG88E/G88EMice. JARO J. Assoc. Res. Otolaryngol. 15, 961–974. doi: 10.1007/s10162-014-0481-9
Robertson, N. G., Skvorak, A. B., Yin, Y., Weremowicz, S., Johnson, K. R., Kovatch, K. A., et al. (1997). Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder. DFNA9. Genomics 46, 345–354. doi: 10.1006/geno.1997.5067
Robles, L., and Ruggero, M. A. (2001). Mechanics of the mammalian cochlea. Physiol. Rev. 81, 1305–1352. doi: 10.1152/physrev.2001.81.3.1305
Seist, R., Tong, M., Landegger, L. D., Vasilijic, S., Hyakusoku, H., Katsumi, S., et al. (2020). Regeneration of cochlear synapses by systemic administration of a bisphosphonate. Front. Mol. Neurosci. 13:87. doi: 10.3389/fnmol.2020.00087
Shen, J., Scheffer, D. I., Kwan, K. Y., and Corey, D. P. (2015). SHIELD: an integrative gene expression database for inner ear research. Database 2015:bav071. doi: 10.1093/database/bav071
Stankovic, K. M., and Corfas, G. (2003). Real-time quantitative RT-PCR for low-abundance transcripts in the inner ear: analysis of neurotrophic factor expression. Hear. Res. 185, 97–108. doi: 10.1016/S0378-5955(03)00298-3
Keywords: cochlin, noise-induced hearing loss, conductive hearing loss, cytokines, LCCL, sterile inflammation
Citation: Seist R, Landegger LD, Robertson NG, Vasilijic S, Morton CC and Stankovic KM (2021) Cochlin Deficiency Protects Against Noise-Induced Hearing Loss. Front. Mol. Neurosci. 14:670013. doi: 10.3389/fnmol.2021.670013
Received: 19 February 2021; Accepted: 16 April 2021;
Published: 24 May 2021.
Edited by:
Maria Eulalia Rubio, University of Pittsburgh, United StatesReviewed by:
Jian Zuo, Creighton University, United StatesEsperanza Bas Infante, University of Miami, United States
Copyright © 2021 Seist, Landegger, Robertson, Vasilijic, Morton and Stankovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantina M. Stankovic, a29uc3RhbnRpbmFfc3RhbmtvdmljQG1lZWkuaGFydmFyZC5lZHU=
†These authors have contributed equally to this work