 Shaunik Sharma
Shaunik Sharma Grant Tiarks
Grant Tiarks Joseph Haight
Joseph Haight
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 23 February 2021
Sec. Brain Disease Mechanisms
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.612073
This article is part of the Research Topic The Molecular Mechanisms of Epilepsy and Potential Therapeutics View all 12 articles
Traumatic brain injury (TBI) is a leading cause of death in young adults and a risk factor for acquired epilepsy. Severe TBI, after a period of time, causes numerous neuropsychiatric and neurodegenerative problems with varying comorbidities; and brain homeostasis may never be restored. As a consequence of disrupted equilibrium, neuropathological changes such as circuit remodeling, reorganization of neural networks, changes in structural and functional plasticity, predisposition to synchronized activity, and post-translational modification of synaptic proteins may begin to dominate the brain. These pathological changes, over the course of time, contribute to conditions like Alzheimer disease, dementia, anxiety disorders, and post-traumatic epilepsy (PTE). PTE is one of the most common, devastating complications of TBI; and of those affected by a severe TBI, more than 50% develop PTE. The etiopathology and mechanisms of PTE are either unknown or poorly understood, which makes treatment challenging. Although anti-epileptic drugs (AEDs) are used as preventive strategies to manage TBI, control acute seizures and prevent development of PTE, their efficacy in PTE remains controversial. In this review, we discuss novel mechanisms and risk factors underlying PTE. We also discuss dysfunctions of neurovascular unit, cell-specific neuroinflammatory mediators and immune response factors that are vital for epileptogenesis after TBI. Finally, we describe current and novel treatments and management strategies for preventing PTE.
More than 3 million people in United States suffer a TBI each year. Among these cases, 80% are mild, 10% moderate, and about 10% severe, accounting for ∼300,000 hospitalizations and ∼50,000 fatalities, annually (Maas et al., 2017). Many traumatic brain injuries cause long-term disabilities, cognitive decline, psychiatric illness, and post-traumatic disorders. About 35% of TBI result from falls, 17% from motor vehicle accidents, and 10% from assaults, while in 21% of the cases, the cause was not recorded (Ding et al., 2016; Centers for Disease Control and Prevention, 2019). Incidence rates are higher in both males and females up to 9 years of age, during teen years, and towards the end of life (>74 years of age). Approximately, 2% of U.S. population live with long-lasting disabilities stemming from TBI; and is one of the single greatest causes of deaths and permanent disability in people under the age of 45 (Maas et al., 2017). The total estimated annual cost for TBI treatment is over $56.3 billion (Faul and Coronado, 2015; Maas et al., 2017). Currently, no available therapies can limit secondary injury or foster repair and regeneration.
Traumatic brain injury can trigger seizures and account for 4% of epilepsy cases (Gupta et al., 2014). New-onset symptomatic epilepsy in adolescents and young adults is most often caused by developmental disorders, infections, skull fracture, intracranial hemorrhage, and subarachnoid or subdural hemorrhage. In contrast, amongst older populations, intracranial hematoma, strokes and tumors are more common causes (Mahler et al., 2015). More than 50% of people develop PTE after severe TBI. According to the American Academy of Neurology, severe TBI is defined as the condition in which a person stays in coma for longer than 24 h post-injury or requires a neurosurgical intervention. Among those who develop epilepsy after severe TBI, nearly 40% experience their first seizure within 6 months of injury, 50–60% within a year and about 80% in the later years of life (Annegers et al., 1998; Agrawal et al., 2006; Pohlmann-Eden et al., 2006; Ding et al., 2016).
Traumatic brain injury is the third most common cause of all epilepsies and results from either direct (primary) or indirect (secondary) damage to brain parenchyma (Kaur and Sharma, 2018; Fordington and Manford, 2020). Trauma or brain injury results in both focal and diffuse injury to the central nervous system (CNS) that can trigger epileptogenesis (Shlosberg et al., 2010; Webster et al., 2017). Focal injuries usually cause contusion, hemorrhage, infarction, and necrosis, causing cortical scarring that effects synaptic plasticity and recovery. On the other hand, diffuse injury leads to axonal shearing, microvasculature damage, release of inflammatory mediators, and free radical overload (Greenfield et al., 2008; Mckee and Daneshvar, 2015). These injuries sabotage vulnerable neuronal populations and white matter tracts; and reactive gliosis that follow neuroinflammation (Wang et al., 2008; Lamar et al., 2014). Later, secondary injury mechanisms reorganize the neural circuits and disrupt brain homeostasis, with the degree of secondary damage largely depending on the severity of primary damage. A mild injury may deteriorate and remodel neural circuits to a lesser extent, whereas a severe insult not only reorganizes neural networks but also cause long-term degenerative changes that results in neuropsychiatric conditions, and cognitive and behavioral deficits (Burda et al., 2016; Ladak et al., 2019). For instance, the release of glutamate after severe head injury causes excitotoxic cell death via excessive calcium release, and generation of free radicals such as reactive oxygen and nitrogen species (ROS/RNS), which elicit an oxidative response against the mitochondria. Further, the recruitment of glial cells and peripheral immune cells (such as leucocytes and macrophages) aggravate the neuroinflammatory response by secreting cytokines. This enhanced proinflammatory response, combined with endothelial ROS, deteriorates the blood-brain barrier (BBB) integrity (Rosenfeld et al., 2012). These combined mechanisms of primary and secondary insults commence a vicious cycle of neurodegenerative events that persist for months to years, executing permanent degenerative changes in the brain (Figure 1). This review highlights cellular and molecular mechanisms that promote seizures, epileptogenesis and epilepsy after TBI. We also discuss the role of immune system, contribution of glial cells, long-term consequences of TBI and therapeutic strategies for managing PTE.

Figure 1. Pathophysiological basis of epileptogenesis following TBI. Complex and multifaceted events triggered during primary injury contribute to secondary injury. These primary and secondary events, which take place over months or years, represent a period of epileptogenesis. Changes such as post-translational modifications of synaptic proteins, reorganization of neural circuits and production and activation of neuroinflammatory molecules/pathways result in PTE. PTE is a progressive process which is a result of mossy fiber sprouting, hippocampal sclerosis, neuroinflammation, neurodegeneration and SRS. PTE, post-traumatic epilepsy; SRS, spontaneous recurrent seizures.
After TBI, excitotoxicity in the brain is generally caused by an increase in extracellular glutamate. Under physiological conditions, glutamate is taken up by astrocytes and converted into glutamine which is then shuttled back to neurons as an alternative energy source (Dienel, 2014). However, excess glutamate overloads astrocytes’ capacity to remove glutamate from the extracellular space (ECS), triggering an enormous Ca2+ and Na+ influx and K+ efflux (Tehse and Taghibiglou, 2019). This resulting ionic imbalance depolarizes the postsynaptic cell membrane, causing a long-lasting increase in excitatory post-synaptic potential. Altered calcium signaling after TBI activates nitric oxide synthase (NOS), proteases, and lipases that trigger cell signaling cascades linked to excitotoxicity and cell death (Weber, 2012; Jarrahi et al., 2020). Elevations in nitric oxide (NO) levels interferes with mitochondrial bioenergetics leading to energy depletion, further adding oxidative stress in neurons. The changes in mitochondrial bioenergetics initiate the release of cytochrome-c- activating caspases, that cause inflammation-induced apoptosis (Adam-Vizi and Starkov, 2010; Rowley and Patel, 2013; Puttachary et al., 2015). Compromise to mitochondrial integrity after TBI elicits the release of ROS/RNS, which deteriorates membrane lipids, proteins, and DNA, and downregulates the expression of glutamate transporters such as GLT-1 and GLAST promoting cellular excitotoxicity (Trotti et al., 1998; Abdul-Muneer et al., 2015; Chen et al., 2020). TBI-induced ATP depletion cause loss of Na+/K+ ion concentration gradient across the plasma membrane due to dysfunctional Na+/K+-ATPase, and leads to excitotoxicity-induced cell stress (Lima et al., 2008). In addition to dysfunctional Na+/K+-ATPase induced excitotoxicity, cell death via lysis or apoptosis also releases cytoplasmic glutamate in ECS after TBI (Zhang et al., 2005). These two forms of glutamate release cause a continual domino effect of cellular excitability that elevates extracellular glutamate concentration in the injured brain.
Numerous in vivo studies on rodent models of TBI have reported an increased glutamate levels in the brain of injured mice, 1–2 days post-injury (Hinzman et al., 2010; Guerriero et al., 2015). Ex vivo studies on brain slices using extracellular field potential recordings have reported elevations in excitatory inputs and evoked synaptic connections between dentate granule cells with mossy fibers, when stimulated with glutamate photostimulation in controlled cortical impact (CCI) model (Hunt et al., 2010). Similar studies, using FRET-based glutamate sensors on hippocampal slices, also reported enhanced cortical excitability and glutamatergic signaling, and increased spread of perforant-path stimulation evoked depolarization in brain slices of CCI and weight drop animals, 2–4 weeks post-injury (Golarai et al., 2001; Cantu et al., 2015). These and other studies confirmed that increase in glutamate response after injury modulate neuronal microcircuits that correlates with an increase in epileptiform activity adjacent to the site of injury.
Blood-brain barrier disruption has a well-recognized role in the pathophysiology of CNS diseases; and understanding the anatomy and physiology of the neurovascular unit in health and disease is critical for advancing translational research into the clinics. Many studies demonstrated that BBB integrity is lost in CNS diseases such as meningitis, encephalitis, Alzheimer disease, Parkinson’s disease, multiple sclerosis, and epilepsy. Damage to the components of neurovascular unit (NVU) such as endothelial cells, after TBI, can impair BBB. Dysfunctional endothelial cell signaling and activation of the immune cell response stimulates the release of proinflammatory mediators, such as ROS, matrix metalloproteinases (MMPs), bradykinins, prostaglandins, cytokines, tachykinins, and excitatory amino acids (Paudel et al., 2019). The formation of intercellular adhesion molecule 1 and vascular cell adhesion protein 1/ERM complex with integrin, via Rac1, releases NADPH oxidase (enzyme involved in oxidative stress) in the endothelial cells generating ROS (Cerutti and Ridley, 2017; Jarrahi et al., 2020). Elevations in ROS levels stimulate the release of MMP-2 and 9 causing damage to tight and gap junction proteins such as occludins, claudins and connexin-43. A further rise in oxidative stress activates focal adhesion kinase, a non-receptor tyrosine kinase, and heat-shock protein 27, that results in receptor endocytosis and stress fiber formation within the cell (Hemphill et al., 2011; Cerutti and Ridley, 2017; Jarrahi et al., 2020). In addition, vascular endothelial growth factor stimulated increase in Src increases phosphorylation of VE-cadherins via serine/threonine-protein kinase, which results in receptor endocytosis. Concurrently, an increase in intracellular calcium activates calcium/calmodulin complex that generates endothelial nitric oxide synthase (eNOS). Rise in eNOS levels inhibits the transcription of claudin-5 and occludin, further increasing BBB permeability (Badaut et al., 2015; Andrews et al., 2016; Cerutti and Ridley, 2017) (Figure 2). An activation of inflammation and the immune response triggers a heightened neuronal response, stimulating neurotransmitter release from the endothelial cells via activation of the central-mediated hypothalamic-pituitary-adrenal axis (Licinio and Frost, 2000; Silverman et al., 2005; Burfeind et al., 2016). These deleterious events initiate multiple signaling transduction pathways, causing changes in BBB permeability and activation of signaling enzymes, such as kinases, to regulate calcium mobilization and gene expression. This affects the transport characteristics of proteins located on endothelial cells, promoting excitotoxicity (Dalal et al., 2020). Therefore, changes in BBB permeability and enhanced endothelial paracellular leak (due to tight junction protein modifications) alter the volume regulators that control BBB homeostasis. This alters tight junction proteins, leading to reorganization and remodeling of the cytoskeletal proteins disrupting brain homeostasis (Stamatovic et al., 2008; Burda et al., 2016).

Figure 2. Vascular endothelial signaling after brain injury. In a brain inflicted with traumatic injury, inflammatory conditions stimulate signaling pathways in the cells of NVU. Cytokines TNFα and IL-1 released from blood cells activate receptors present on the vascular lumen near the sites of inflammation. These cytokines upregulate expression of selectins, chemokines, and integrin ligands like ICAM-1 and VCAM-1 on the surface of the endothelial cells, causing increased production of NADPH oxidase. Elevations in NADPH oxidase-mediated release of ROS degrades tight and gap junction proteins such as occludins, claudins and connexins, by generating excessive amounts of Akt and MMPs (MMP-2 and MMP-9). Oxidative stress enhances the production of FAK2, that damages adhesion molecules, cadherins. Alternatively, increased calcium inside the cell activates IP3 receptors resulting in increased PKC production, which in turn generates PAK via phosphorylation of Src kinase. PAK is also activated by Src phosphorylation via the activation of VEGF receptors. These events collectively damage cadherins compromising integrity of cellular junctions. Elevations in calcium levels also produces eNOS, which blocks claudin-5 and occluding transcription. These events cause BBB breakdown, allowing increased migration of peripheral immune cells into the central nervous system. NVU, neurovascular unit; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion protein 1; MMPs, matrix metallopeptidase; FAK2, focal adhesion kinase 2. Figure created with BioRender.com and R&D Systems.
After TBI, loss in BBB integrity is primarily due to the release of excitotoxic factors by injured neurons and activated glial cells. These factors drive blood cell chemotaxis and their transmigration into the brain. Enhanced leukocyte infiltration and invasion of CNS parenchyma generates a cytokine storm which induces neuronal injury. Infiltration of leukocytes also increases accumulation of intracellular fluid and capillary pressure causing turnover in the transendothelial volume. This can lead to traumatic brain edema exemplifying a transcytosis response to injury (Castejon, 1984; Scallan et al., 2010). Additionally, concurrent modifications take place in glial cells that drive morphological and molecular changes in order to attain reactive morphology. Increased proinflammatory secretions from neurons and reactive glial cells facilitate recruitment of additional immune cells, such as neutrophils and monocytes, from the periphery further modulating brain activity by increasing proinflammatory receptor expression on their surface. The binding of molecules released by neighboring glial cells and injured neurons cause activation of these receptors, exacerbating neuronal excitotoxicity (Medzhitov, 2008; Aronica et al., 2012; Burda and Sofroniew, 2014; Sanz and Garcia-Gimeno, 2020). Increased blood immune cell infiltration and dysfunctional neuro-glia crosstalk cause further rise in cytokine storm, therefore damaging BBB and its components (Figure 3).

Figure 3. Comprised BBB integrity and neuro-glia crosstalk in PTE. Proinflammatory factors released by damaged neurons and glial cells initiate a vicious cycle of inflammation in the brain. (1) Brain injury results in an early activation of microglia and stimulate production of cytokines and expression of DAMPs associated receptors on their surface, leading to neuronal excitability. (2) Increased cellular hyperexcitability due to molecules released by reactive glia and neurons promote, (3) blood cell chemotaxis and transmigration into the brain. Disruption in BBB leads to brain infiltration of immune cells from the blood releasing (4) proinflammatory factors and promoting excitotoxicity. (5) Later, activated microglia release signals that bind to the receptors on astrocytes. (6) Serum albumin induced activation of astrocytes impairs potassium channel and glutamate transporter. In addition, activated astrocytes release proinflammatory factors which exacerbates neuronal hyperexcitability. (7) These factors collectively or on their own cause neurodegeneration. Cytokine mediated crosstalk between astrocyte-microglia and neuro-glia induces hyperexcitability and neuronal cell death. In contrast, molecules derived from neurons, such as HSP60, HMGB1, CD14, CX3CL1, and ATP, cause microglia activation leading to excitotoxicity. Monocytes and leukocytes also release cytokines at this point. This creates an activation cycle which causes release of additional inflammatory molecules triggering cytokine storm in the brain. Discharge of molecules like MHCII, complementary proteins, proinflammatory interleukins, iNOS, transcription factors and microRNAs by macrophages, cause excitotoxicity in neurons. These molecules promote neurodegeneration and initiate neuroinflammatory pathways lowering the seizure threshold. HSP60, heat shock proteins; HMGB1, high mobility group box 1; CD14, cluster of differentiation 14; CX3CL1, chemokine (C-X3-C motif) ligand 1; ATP, adenosine 5′-triphosphate; iNOS, inducible nitric oxide synthase. Figure created with BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates.
Traumatic brain injury can have deleterious effects on the neurovascular unit (NVU). Pericytes, an important component of NVU found in capillaries around the brain and other regions, play an important role in the maintenance of BBB, angiogenesis, regulation of blood flow and immune cell movement in the brain (Brown et al., 2019). After brain injury, dysfunctions in pericyte signaling results in the loss of pericyte–endothelium interactions, allowing easy passage for neurotoxins from the blood to enter brain. In vivo studies on the mouse model of TBI have reported reduced expression of pericyte markers, platelet-derived growth factor-B (PDGF-B), NG2 and CD13, 24 h post lateral fluid percussion injury (LFPI). Reduction in these markers also corresponds to a decrease in tight and gap junction proteins (Bhowmick et al., 2019). Alterations in these proteins cause increased water permeability in the brain due to a substantial changes in aquaporin (AQP4) expression around the perivascular region. These studies also reported higher expression of calcium binding protein and a reactive astrocyte marker, S100β, and neuron-specific enolase in the blood samples of TBI animals (Bhowmick et al., 2019). Using two different adult viable pericyte deficiency mouse strains with variable degrees of pericyte loss, Bell et al. (2010) demonstrated that pericyte loss during neurodegenerative conditions can influence brain capillary density, resting cerebral blood flow, blood flow responses to brain activation and blood brain integrity to serum proteins, and blood derived cytotoxic and neurotoxic molecules. Using in vivo multiphoton microscopy on mouse lines expressing PDGFRβ+ exclusively on pericytes, the same group further reported a significant reduction in the length of perfused capillaries that corresponded to a reduction in cerebral blood flow volume (Bell et al., 2010). BBB disruption after TBI causes dysfunction in pericyte-endothelium interaction and increase in toxic accumulation of serum derived proteins in the brain. Using time-lapse imaging of a low-molecular weight 40 KDa TMR-Dextran, studies have reported diminished brain capillary perfusion and changes in vascular permeability in PDGFRβ–/–deficient mice. Golgi histological analysis of neuronal structure and function revealed progressive loss of dendritic spines and significant structural abnormalities in CA1 region of hippocampus in PDGFRβ heterozygous mice at 8 and 16 months of age, supporting a crucial role of pericytes in neurovasculature (Bell et al., 2010). Studies on mouse models of TBI have shown diminished pericyte-endothelium interactions showing reduced oxygenation in ipsilateral and contralateral areas of the somatosensory cortex, as well as other regions of the brain, during early stages of TBI (Johnstone et al., 2014; Zehendner et al., 2015; Ichkova et al., 2020). Using acute brain slices and vascular staining, studies reported changes in neurovascular reactivity and morphological variations in the blood vessels of mice, 1 and 30 days post-injury (dpi). These changes reversed during early and late stages, revealing time-dependent alterations in the neurovasculature and dysfunction in oxygenation and vascular coupling (Ichkova et al., 2020). The results from these and other studies suggest that neurodegenerative changes develop following a primary vascular insult which impairs pericyte–endothelium interactions. Disruption in pericyte signaling alters brain microcirculation causing diminished brain capillary perfusion. This leads to chronic perfusion stress and cellular and molecular alterations of BBB, which includes, changes in transport functions of endothelium, loss of pericytes, decrease in cerebral blood flow, loss of vascular reactivity, changes in vascular morphology, alterations in glial metabolic rate and oxygen deficiency in tissues. These post traumatic brain injury events lead to cellular excitotoxicity and chronic neurodegeneration (Bell et al., 2010; Wu et al., 2020).
Mitochondrial dysfunction has long been recognized as a key source of oxidative stress in epilepsy. Emerging evidence suggests that acute seizures induce oxidative stress, and as a result of initial insult, the process of epileptogenesis begins to dominate the brain (Patel, 2004; Liang and Patel, 2006). During oxidative stress, deleterious changes in mitochondria include altered mitochondrial membrane potential, enhanced nicotinamide adenine dinucleotide phosphate (NADPH) production, impairment of electron transport chain complex 1, 3, and 4, rise in mitochondrial ROS, and mitochondrial DNA damage (Dexter et al., 1989; Cini and Moretti, 1995; Chuang et al., 2004; Kann et al., 2005; Chuang, 2010). These changes in mitochondrial activity cause progressive dysfunction, aligning with a common theme of epileptogenesis as a series of degenerative events that triggers a vicious cycle of oxidative stress and neurodegeneration, ultimately leading to PTE (Vezzani et al., 2011).
Free radicals are generated by oxidation and reduction reactions of electrons during hemolytic cleavage, when the bond is broken in such a way that the pair of electrons is shared equally by both the separating fragments. These separating fragments may carry one or more unpaired electrons, which makes free radicals highly reactive in nature. Free radicals are chemically unstable molecules that cause cellular and mitochondrial DNA fragmentation (Lobo et al., 2010; Cardenas-Rodriguez et al., 2013; Ozcan and Ogun, 2015). As a result of oxidative damage, alterations in morphological and functional properties of proteins and lipids takes place. This further impacts cellular and mitochondrial DNA, and cross-link base pairs and cause genetic mutations (Emerit et al., 2004; Waldbaum and Patel, 2010; Ramalingam and Kim, 2012). Free radical species of oxygen and nitrogen include superoxide anion, hydroxyl radical (OH), peroxyl and alkoxyl radicals, hydrogen peroxide (H2O2), peroxynitrite, nitroxyl anion, nitrogen dioxide and nitrate/nitrite (Cheeseman and Slater, 1993; Ozcan and Ogun, 2015; Puttachary et al., 2015). An excessive generation of these radicals within the cell causes oxidative stress.
Oxidative stress is a biochemical state when an excessive production of ROS/RNS cause damage to the cell membranes and proteins, as well as to cellular and mitochondrial genomes (Cardenas-Rodriguez et al., 2013). Majority of oxygen and nitrogen-centered free radicals are generated from interactions between NO and molecular oxygen (O2). NO is produced from the substrate L-arginine, with the help of a co-factor NADPH and O2, and enzyme nitric oxide synthase (NOS). In the cytoplasmic membrane, superoxides are primarily generated by NADPH oxidase, after an electron transfer from NADPH to O2. These superoxides are also generated by the action of O2 on xanthine oxidase. Under normal physiological conditions, superoxides are converted into H2O2 by the action of superoxide dismutase, which is then broken down into water and oxygen (with the help of catalase and glutathione peroxidase). Degradation of superoxide dismutase promotes enhanced production of highly reactive peroxynitrites (ONOO–), a powerful oxidizing agent, which results in increased ROS production, DNA, proteins and lipids oxidation and loss of ion channel dysfunction (Halliwell, 1999; Puttachary et al., 2015). Increased superoxide in a cell cause oxidative burst promoting oxidative damage by exacerbating inflammation, enhancing redox signaling and proinflammatory gene regulation (Agledal et al., 2010). Moreover, impairment of catalase and peroxides promote formation of hypochlorous acid from H2O2 by reacting with Cl–, NO2– and phenols. This causes cell death by destabilizing calcium homeostasis. Alternatively, H2O2 can also undergo Fenton and Haber–Weiss reaction to form OH radicals (a harmful free radical of oxygen with high reactivity and a short half-life) which results in proteins and lipid peroxidation, mitochondrial DNA damage and depletion of antioxidant enzymes (Bae et al., 2011; Puttachary et al., 2015).
The metabolic regulation and signaling of redox enzymes, such as NADPH oxidase, lipoxygenase and endoperoxide synthase is exceedingly altered after TBI. At basal levels, NADPH oxidase (NOX-2) is expressed widely in the brain where it plays an important role in learning, memory consolidation, innate immunity, phagocytic activity and apoptosis (Infanger et al., 2006; Aguiar et al., 2012; Eastman et al., 2020). However, under pathological conditions, such as in PTE, NADPH oxidase generates greater amounts of superoxide ions, triggering neuroinflammation and neurodegeneration, as evidenced by various animal models of TBI and chemoconvulsant-induced TLE (Ferreira et al., 2013; Angeloni et al., 2015; Ma et al., 2017; Eastman et al., 2020). For instance, Li et al. (2019) on the chemical induced brain injury model of mice, reported the accumulation of oxidative stress factors such as lipid ROS and 4-hydroxy-2-nonenal (4-HNE) adducts in the somatosensory cortex and hippocampal HT22 cells, 12–36 h post-injury. The same group also discussed the involvement of oxidative enzyme 12/15 lipoxygenase (12/15-LOX) associated ferroptosis in a trauma induced neuronal damage, that corresponds to reduced cell viability and glutathione peroxidase 4 activity in the cortex of mice and in hippocampal cultures (Li et al., 2019). 12/15 LOX plays an important role in modulating oxidative stress and increase post-traumatic seizures by generating oxidized phospholipids (Chinnici et al., 2005). In a study on the rat model of LFPI, Saraiva et al. (2012) demonstrated that an increased levels of thiobarbituric acid and protein carbonylation contents in the brain increased seizure and spiking activity, within a week after injury (Saraiva et al., 2012). These and other studies provide a strong evidence and the significance of synergistic interactions between the redox enzymes in maintaining TBI-induced oxidative stress. In addition, detrimental role of prostaglandin-endoperoxide synthase, such as cyclooxygenase (COX-2) have also widely been reported in various clinical and experimental models of TBI and epilepsy. COX-2 upregulates ROS by producing prostaglandins (specifically, F2 and H), and stimulate astrocytes to produce proinflammatory cytokines which signals for oxidative stress-mediated neuronal death (Madrigal et al., 2006; Hickey et al., 2007; Rojas et al., 2014). COX-2 also initiates inflammatory response in immune cells such as neutrophils and alters tissue homeostasis (Ricciotti and FitzGerald, 2011). Interactions between NOS and COX-2, after brain injury, can affect neocortical development by creating pathological milieu (Kaufmann et al., 1997). Studies on immature rats have reported enhanced COX-2 expression after TBI, that corresponds to an increased NOS and prostaglandin synthesis. Studies have shown that increased lesion size after TBI, corresponds with an increased COX-2 expression, that leads to impaired cognitive deficits in rats (Hickey et al., 2007). These studies demonstrate that the accumulation of oxidative stress factors, after TBI, cause increased cytokine levels, NO metabolites, oxidative enzymes, protein carbonylation contents, SRS and memory deficits over time- which altogether may lead to PTE (Table 1). Inhibition of these enzymes have been shown to prevent cognitive deficits, motor dysfunctions, cerebral edema, cerebral perfusion rate, neurodegeneration and neuroinflammation, in many clinical and animal models (Madrigal et al., 2006; Zhang et al., 2012; Ferreira et al., 2013; Liu et al., 2016; Li et al., 2019). Therefore, targeting these molecules can provide neuroprotection against TBI-induced epileptogenesis.
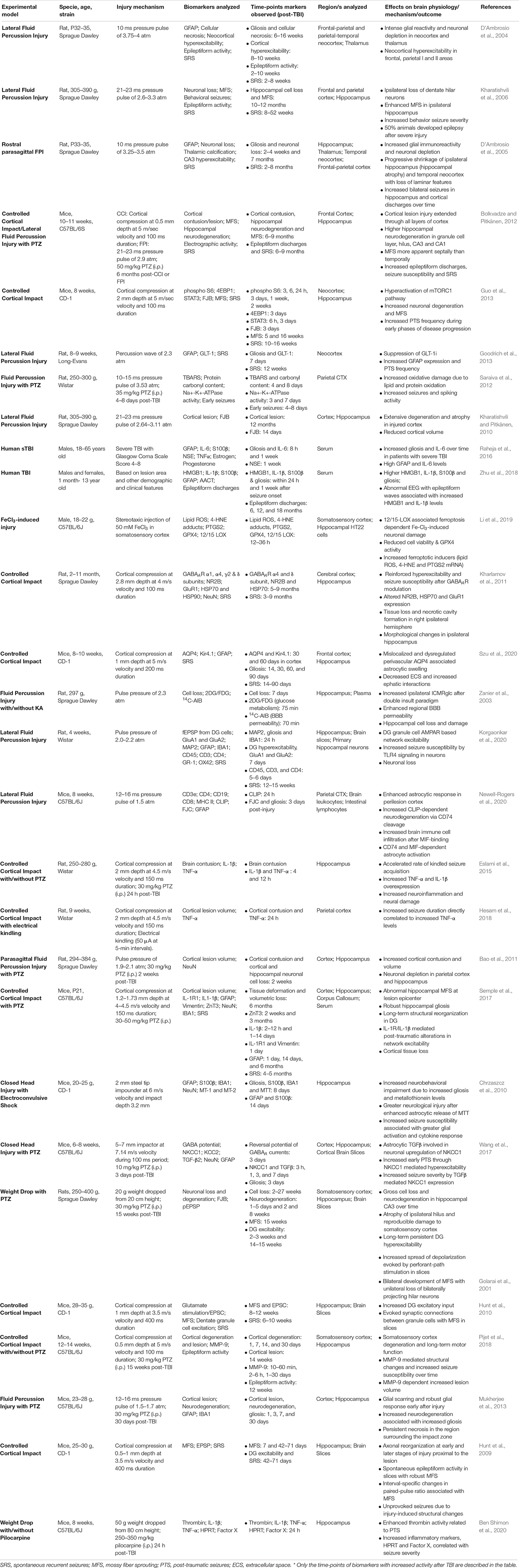
Table 1. Biomarkers of TBI-induced epileptogenesis.
Depending on the molecular mechanisms affected, neuronal cell death in TBI is classified as either physiologic or excitotoxic. Physiologic cell death is due to injuries that initiate cellular events such as mitochondrial swelling and nuclear membrane/cytoplasm rupture, whereas, excitotoxic cell death occurs a few hours after injury and causes chromatin agglutination and DNA fragmentation, but maintains an intact nuclear membrane (Stoica and Faden, 2010; Ladak et al., 2019). These intrinsic forms of cell death are primarily regulated by calcium release and enzyme-based regulators such as phospholipases, proteases, endonucleases, caspases, death-inducible complexes and pro-apoptotic proteins (Kögel and Prehn, 2000-2013; Broker et al., 2005; Raja et al., 2018). After TBI, the release of caspase-3 and caspase-12 disrupts the balance between pro-apoptotic and anti-apoptotic proteins, drawing the cell toward neurodegeneration and inflammation-induced apoptosis (Knoblach et al., 2002; Li and Yuan, 2008). Caspase-3 cleaves a specific serine-threonine protein kinase called PKCδ, causing its phosphorylation and activation. The activation of the NOX enzyme complex, either on its own or via TNFα, also increases PKCδ production. PKCδ trips the MAP kinase cascade, which allows NFκB to translocate into the nucleus, and transcriptionally activate proinflammatory genes (Sharma et al., 2018). PKCδ also regulates NOS expression and stimulates its release from reactive microglia and neurons, promoting lipid peroxidation by producing 4-HNE and malondialdehyde from hydroxyl radicals via a Fenton reaction (Puttachary et al., 2015; Sharma et al., 2018). 4-HNE impairs astrocytic proteins, such as glutamate transporter (GLT-1) which enhances free glutamate in the ECS. Free glutamate binds to NMDAR, causing NMDAR trafficking and calcium overload, free radical production, activation of gp91phox (heme-binding subunit of NADPH oxidase) and, ultimately, cell-membrane protein degradation and cell death (Reyes et al., 2012; Pecorelli et al., 2015; Sharma et al., 2018). These events are progressive in nature and drive long-term neurodegenerative changes in the brain over time (Figure 4).

Figure 4. Oxidative stress leads to neurodegeneration and neuroinflammation. ROS/RNS incites a multitude of different events that leads to oxidative stress and neurodegeneration. Binding of TNFα to its receptor triggers activation of transcription factors which stimulate the release of prostaglandins and cause neuronal injury via excessive calcium release. An inflammatory stimulus also activates NOX enzymes, either on its own or via TNFα, which produces exorbitant amounts of free radicals of oxygen and nitrogen. This results in peroxidation of proteins and lipids, DNA damage and depletion of anti-oxidant proteins, ultimately causing neurodegeneration. These events also impair mitochondrial bioenergetics leading to inflammation induced apoptosis. Increased calcium influx, due to NMDAR trafficking, also triggers NOX activation and blocks glutamate transporters causing excitotoxic cell death. Increased accumulation of free radicals activates MAP kinase which in turn activates PKCδ. PKCδ phosphorylation promotes transcription of proinflammatory proteins either directly or through NF-κB activation. All these events finally resolve into neuroinflammation and neurodegeneration. ROS/RNS, reactive oxygen/nitrogen species; NOX, nicotinamide adenine dinucleotide phosphate oxidase; MAP, mitogen-activated protein kinase; PKCδ, protein kinase C delta. Figure created with BioRender.com.
Physiological and structural evidence of dendritic loss, modulation of spine density and hippocampal sclerosis have widely been associated with increased seizure susceptibility after TBI (Golarai et al., 2001; Gao et al., 2011; Winston et al., 2013). There are numerous reports on the unilateral or bilateral loss of neurons in hilus and CA3 of hippocampus, progressive mossy fiber sprouting (MFS) in the inner molecular layer of DG and hyperexcitability in DG circuitry, several weeks after TBI (Lowenstein et al., 1992; Diaz-Arrastia et al., 2000; Golarai et al., 2001)-observations that were consistent with human PTE cases (Diaz-Arrastia et al., 2000). EEG and MRI studies on patients with intractable epilepsy, who suffered TBI, showed dysfunctions in temporal lobe as characterized by increased epileptiform spiking, dendritic spine remodeling, reactive gliosis and poor neuropsychologic response. These morphological changes in hippocampus were associated with MFS and hippocampal sclerosis (Diaz-Arrastia et al., 2000). Numerous studies on the rodent models of TBI have reported a strong association between dentate granule cell hyperexcitability and enhanced MFS with hippocampal sclerosis. These studies also demonstrated intense glial reactivity, DG hyperexcitability and neuronal loss in hilus of DG (Golarai et al., 2001; Kharatishvili et al., 2006; Hunt et al., 2009, 2010). Long-term persistent hyperexcitability in DG cause alterations in hippocampal pyramidal cell dendrites, that leads to reduction in spine density or spine loss (Jiang et al., 1998). Abnormalities in dendritic spines promote hyperexcitable circuits which directly influences neuronal excitability. The changes in number and morphology of spines are related to alterations in LTP and LTD, which can have a significant effect on the cognition (Wong and Guo, 2013). In PTE patients, the loss of dendritic spines has been observed in the pyramidal layers of hippocampus and in the granule cell layer of DG (Isokawa and Levesque, 1991; Wong, 2005). Dendritic atrophy, arborization, changes in dendritic length and even varicose swelling of dendrites were reported in some cases (Multani et al., 1994; Isokawa, 1998; Wong, 2005). Animal studies have not only provided a strong evidence for dendritic alterations after TBI, but also provided insights into the cellular and molecular mechanisms involved in such changes. It has been reported that changes in the neural circuits after TBI, and during early post-traumatic seizures can cause spine remodeling due to increased MMP-7 and 9 through NMDA-mediated receptor activation (Bilousova et al., 2006; Pijet et al., 2019). This alters neuromodulation resulting in excitotoxicity-induced neuronal death in the brain (Wong, 2005). For instance, experiments with GABA antagonist on hippocampal slice cultures revealed an increased spine loss in CA3 layer of hippocampus. This was partially reversed by an application of glutamate antagonist. Furthermore, application of glutamate agonist also caused loss of these spines due to NMDA-induced glutamate excitotoxicity due to activation of calcium-dependent enzymes, which degraded cytoskeletal structures (Müller et al., 1993; Jiang et al., 1998). Therefore, these studies strongly supports the role of NMDA receptors in dendritic spine loss and abnormalities that can be reversed using NMDAR modulators, suggesting the role of glutamate excitotoxicity in dendritic spine remodeling after TBI.
Numerous studies point to immunoregulatory molecules as master regulators of inflammation after injury. In cases of severe traumatic brain injuries, immunoregulators activate multiple signaling pathways that drives chronic microglial and immune response, and cause neurodegeneration (Loane et al., 2014). Interferons (IFNs) are among those pleiotropic signaling protein molecules that play a significant role in promoting neuroinflammation and neurodegeneration following PTE. IFNs are potent immune system activators and can act in an autocrine fashion to induce type I-IFN-driven inflammation and disease (Trinchieri, 2012; Uggenti and Crow, 2018). Type I IFNs play an important role in microglial activation and neurodegeneration, especially in the aging brain; and neutralization of such interferons alleviates cognitive deficits and slows down aging (Baruch et al., 2014). Recently, interest has been developed in identifying the DNA sensors responsible for IFN activation. An example of one such sensor is the cGAS-STING pathway. cGAS belongs to the nucleotidyltransferase family that activates STING by binding to DNA, which induces enormous amounts of type I IFN, driving neurodegeneration (Abdullah et al., 2018; Uggenti and Crow, 2018). In their CCI model, James Barret’s group recently reported increased cGAS and STING levels in the brain of juvenile mice, 3 days post-TBI. Using gene expression studies, they further reported enhanced mRNA expression of IFN-β and interferon regulatory factors such as IRF1, IRF4, and IRF7- factors that regulate amplification of type I IFNs in microglia. These studies show that microglia expressing high levels of IFNAR, following TBI, achieve reactive morphology and activation, and can prove to be a crucial target for IFNAR related diseases. Higher IFNAR expression subsequently increases production of TNF-α, NOX2, CCL5, and IL-1β mRNA, promoting cell death by driving synaptic and dendritic loss in neurons. In contrast, knocking out IFNAR and IFN-β has been shown to reverse these effects in the cortex and hippocampus of the mice (Karve et al., 2016; Barrett et al., 2020). Further, through behavioral studies, Barrett et al. (2020) demonstrated that knocking out IFN-β gene significantly improves motor and cognitive performance in the experimental subjects (Barrett et al., 2020). Moreover, several studies suggest that targeting IFN can prevent lesion volume, increase neuronal density, reduce the cytokine storm, decrease microglial activation and leucocyte infiltration, and limit neurodegeneration in the brain (Biliau, 2006; Mathur et al., 2017; Ta et al., 2019). Therefore, therapies targeting IFNAR can prove to be beneficial in treating TBI-associated neurological conditions. These therapeutic strategies can include using caspase inhibitors; cyclic dipeptides (to slow down cytochrome c release); use of pharmacological compounds (blocks cell cycle activators); progesterone and erythropoietin treatment (for edema and proinflammatory cytokine release); and statins (for governing Akt and slowing down microglial activation) (Ucciferri et al., 2007; Tayel et al., 2013; Zhu et al., 2013; Dejager et al., 2014).
Neuroinflammation in the brain is triggered by factors such as microbial infections, accumulation of toxic metabolites, traumatic brain and spinal cord injury, and tissue damage and malfunction. Acute inflammation, after TBI, activate molecules and signaling mechanisms that attempts to restore the body’s disrupted equilibrium by balancing inflammatory and resolution pathways. If these events are not controlled in time, they progress into a chronic stage, eliciting deleterious effects on the brain. The key molecules that regulate inflammation at this stage are granulocytes, platelets, prostaglandins, and cytokines released by lymphocytes, macrophages, microglia and stressed neurons. Their secretions cause intracellular modifications to recreate an unstable cellular microenvironment that disrupts cellular and molecular communications between cells (Herz et al., 2017; Davies et al., 2019; Scanlon, 2019).
Numerous studies on rodent models of epileptogenesis have reported on the post-TBI role of inflammatory mediators, prostaglandins and cytokines IL-1β and TNFα, in the hippocampus and other regions of the brain (Patel et al., 2017; Serhan, 2017). IL-1β, an immune cell mediator and IL-1RI ligand, has been associated with modulation of various neurological functions and in diseases. IL-1β increases NMDAR-mediated calcium release through the activation of Src family kinases (SFKs) (Viviani et al., 2003; Salter and Kalia, 2004). NMDAR are regulated by SFKs, especially by Fyn (Salter and Kalia, 2004). Substantial evidence suggests the link between increased IL-1β-NMDAR-SFK interactions in numerous neurological conditions influencing neuronal functions and enhancing neuronal excitability (Vezzani et al., 1999; Fogal and Hewett, 2008). Studies on hippocampal neurons have demonstrated that neurons exposed to IL-1β exhibit greater glutamatergic excitation and calcium release through NMDAR component, which induces excitotoxic cell death (Viviani et al., 2003). Interaction of IL-1β with IL-1R results in the recruitment of adaptor protein MYD88, which further recruits TRAF6 or IRAK I and II. The MYD88-TRAF6/IRAK I and II complex phosphorylates MAP kinase, causing NF-kB translocation into the nucleus, promoting transcription of proinflammatory genes (O’neill, 1995; Vezzani et al., 2011; Lalitha et al., 2018). These proinflammatory genes are primarily involved in cell death and survival, reorganization of molecular networks, plasticity, synaptogenesis and aberrant neurogenesis- events that takes place simultaneously with epileptogenesis (Vezzani et al., 2011). Activation of IL-1β/IL1R also promotes release of TNFα from astrocytes and glial cells. In contrast, toll-like receptor (TLR) activation stimulates TNFα expression as has been demonstrated in many experimental models of TBI (Yu and Zha, 2012; Shi et al., 2019). TNFα modulates neuronal excitability perhaps by internalizing inhibitory GABAA receptors (Stellwagen et al., 2005; Stück et al., 2012; Pribiag and Stellwagen, 2013). TNFα binding to its receptor activates the TRADD complex and PI3 kinase, resulting in NF-κB activation modulating apoptosis and inflammation (Ermolaeva et al., 2008; Ting and Bertrand, 2016; Holbrook et al., 2019). TNFα activation also increases COX-2 production in response to injury, which is followed by an increase in PGE2 synthesis. Activation of these events cause glutamate accumulation and increases calcium load in the cell exacerbating neuroinflammation (Figure 4).
IL-1β and TNFα are undoubtedly the most well studied and widely known mediators of inflammation following TBI. Exuberant amount of work is underway, in both animal and human models, to target these molecules and prevent neurological outcomes related to TBI. For instance, in a mouse model of blast-injury, IL-1β antagonist Anakinra, has been shown to reduce gliosis, retinal degeneration and neuronal dysfunction (Evans et al., 2020). Another IL-1β synthesis inhibitor, VX-765, delayed seizure onset, duration and the number of SRS in chemoconvulsant induced experimental model of epilepsy (Maroso et al., 2011). In a study on TNFα inhibitors, C7 and SGT11, on a mice model of midline FPI, Rowe et al. (2018) reported significant improvements in cognitive deficits and sensorimotor function tasks (Rowe et al., 2018). Therefore, these studies provide strong evidence on the roles of IL-1β and TNFα inhibitors in modulating TBI-induced inflammation, and improving neurocognitive deficits, linked to TBI.
The role of prostaglandins in the animal models of TBI and in epileptogenesis is well known. Prostaglandins are produced by the action of COX-2 on arachidonic acid, which can be converted into five different prostanoids by the action of specific enzymes, depending on cellular conditions and their requirements. Prostanoids activate 11 receptors that primarily play a role in smooth muscle relaxation and contraction. Depending on the type of receptors and ligands activated, prostaglandins can play a significant role in various physiological and pathological conditions (Jiang et al., 2013; Rojas et al., 2014; Du et al., 2016; Eastman et al., 2020). Numerous studies showed high concentrations of prostaglandins in the brains of human patients and animals with temporal lobe epilepsy (TLE) (Takemiya et al., 2006; Jiang et al., 2013; Rojas et al., 2014; Rana and Musto, 2018). Excess prostaglandins modulate calcium mobilization and cAMP activity, inducing neuronal injury and defects in neuronal plasticity (Hein and O’Banion, 2009; Figueiredo-Pereira et al., 2015; Kang et al., 2017). For example, during febrile seizures, inflammation in the hypothalamic neurons modulate systemic inflammatory response by recruiting prostaglandins from the system (Berg et al., 1998; Zetterström et al., 1998). This enhances EP1/EP2 receptor trafficking, stimulates COX-2 production, and increase prostaglandins within the brain, thereby reducing the threshold for seizures (Gatti et al., 2002). So far, multiple clinical trials of prostaglandin inhibitors for controlling febrile seizures have been largely contradictory: for e.g., patients treated with aspirin therapy had fewer seizures on day two of monitoring, whereas randomized placebo-controlled ibuprofen treatment, in children with febrile seizures, failed to prevent spontaneous recurrent seizures (SRS) (Godfred et al., 2013). An overproduction of prostaglandins and cytokines, along with the recruitment of other disease-causing molecules (such as platelet activating factors, MMPs and TLRs) trigger cellular damage, decrease long-term potentiation, elongate dendritic spines, increase production of forkhead transcription factor 3, modulate voltage-dependent ion channels, and impair BBB leukocyte-endothelium interactions (causing a leaky BBB) (Anderson and Delgado, 2008; Vezzani et al., 2012, 2015; Rana and Musto, 2018). Enhanced production of these molecules and the events they trigger lowers the seizure threshold post-injury and increases the brain’s susceptibility to PTE.
Immune cells play important roles in regulating normal functions of the brain, such as neurogenesis, cognition, aging, translation, formation of neural circuits, and stress responses. When this system stops functioning well, disease manifests. Therefore, it is essential to understand the functions of the immune system, to be able to evaluate its role as a repair mechanic that can be optimized, or a disease promoter that should be suppressed. The local inflammation surrounding an injured tissue is pivotal for its recovery. Although sometimes inflammation runs out of control, suppressing it may impact the normal functions of the system. Several studies report that circulating immune cells are vital for CNS protection and repair (Louveau et al., 2015; Morimoto and Nakajima, 2019; Norris and Kipnis, 2019). Blood macrophages are initially activated at the site of injury, and are generally anti-inflammatory and not proinflammatory in nature, challenging the notion of a strictly proinflammatory role for macrophages, post-injury (Popovich et al., 1996). These macrophages are reparative and alternate between an activated or M2 morphology (Rapalino et al., 1998). Rapalino et al. (1998) reported that animals injected with these macrophages, at the site of injury, recovered their locomotor activity and formed less scar tissue. Studies on a TBI chimeric mouse models of neurological diseases have addressed the need for the recruitment of monocytes/blood macrophages to fight progression of the disease, that follows post-injury. These studies proposed that blood macrophages degrade amyloid-β, elevate IL-10 levels, downregulate TNFα, and boost levels of growth factors, such as IGF-1, in the brain, which attenuates neuropathology (Shechter et al., 2009; Hu et al., 2012; Hsieh et al., 2013; Zyśk et al., 2019). Other studies argued that not only do macrophages have a reparative role, but so do circulating T-lymphocytes after injury (especially CD4+ lymphocytes) (Rapalino et al., 1998; Shechter et al., 2009). For instance, elevating the levels of myelin-recognizing T cells, after TBI, is protective and supports recovery, enhances neurogenesis, improves cognition and provides better protection and the ability to cope with stressful conditions (Mckee and Lukens, 2016; Krämer et al., 2019). It is notable that these protective T cells are different from those that cause autoimmune diseases, in terms of their antigen affinity and regulation. These studies validate an indirect role of T cells in maintaining brain homeostasis by regulating hippocampal neurogenesis, maintaining brain plasticity, enhancing cognition, and controlling the stress response.
Immune cells such as microglia (inflammatory microglia) initiate debris disposal after injury, whereas anti-inflammatory microglia initiate healing in response to sterile inflammation. During severe injury, if microglia cannot clear the debris, macrophages from the blood (or healing macrophages) enter the brain, and terminate the microglial response by releasing high amounts of IL-10. Resident microglia and blood macrophages have different functions in protecting the brain from neuroinflammation and behave differently, in a time-dependent manner. The infiltrating blood macrophages support cell survival and renewal after injury, whereas their depletion causes loss of cells (Shechter et al., 2009). Importantly, immune system activation does not always exacerbate the injury response and cause chronic inflammation. If the activated microglia can return to normal, then inflammation resolves itself; if not, however, they can trigger a systemic immune response. Therefore, it is crucial to understand whether the nature of the inflammation is local or systemic when employing anti-inflammatory therapies. In contrast to systemic inflammation, suppressing local inflammation may prove to be the more beneficial option.
The inflammation conundrum in neurodegenerative diseases occurs in the backdrop of ineffective recruitment or a dysfunctional immune system; it varies with model, strain, sex, region of the brain affected, severity and time of insult, age, etc. After an initial infection, the number of T cells remains steady for a long period of time and then declines; and when their number crosses a critical threshold, disease is manifested. The drop of T lymphocytes over time, as reported by Ho et al. (1995), is a very dynamic process, since immediately after infection T cells furiously regenerate themselves. The inflammatory response then kicks in to regenerate more T cells to fight the infection. When this process is exhausted, the disease is evinced (Ho et al., 1995). Researchers recently discovered that the brain is not as immune privileged as previously thought. In fact, CD4 T cells are present around leptomeninges, blood vessels and glia limitans, where they secrete immune signals into the CSF that bathes the brain; and these cells populate the brain meninges right around the time when all of the synaptic remodeling events are taking place, thereby exacerbating inflammation (Rauch, 2004; Koronyo et al., 2015; Pasciuto et al., 2020). In addition to this neuroinflammatory component, PTE also has a peripheral immune element, as the periphery too gets inflamed by the TBI-activated innate and adaptive responses. After traumatic injury, studies have reported a significant activation of immune cells, such as B cells, CD3+, CD4+, and CD8+ T cells, Tregs, and γδ-T cells in spleen as detected through flow cytometry. Evidence of innate and adaptive responses to injury were also observed in other tissues, such as the GI tract and liver (Tobin et al., 2014; Bai et al., 2017). For instance, PCR-arrays tracking cytokine expression showed increase in mRNA for chemokines, such as MCP-1, in the liver and gut, and proliferation of γδ-T cells (Tobin et al., 2014). Researchers are now beginning to realize that cytokines, for the most part, are not made by the neurons and astrocytes but by immune cells and microglia, which populate the entire body including the developing brain, and communicate with resident macrophages to promote tissue remodeling and cleanup (Röszer, 2015; Sridharan et al., 2015; Wynn and Vannella, 2016; Kumar, 2019). Interestingly, in a recent study on maternal immune activation, it was observed that maternal immune activation (from infection or autoimmune predisposition) induces T-cells to release IL-6 and IL-17. IL-17 can cross the placenta and cause cortical malformation and behavioral abnormalities in the baby (Choi et al., 2016; Wong and Hoeffer, 2018). Together, these studies conclusively support a crucial role for our immune system in health, and in the maintenance of inflammation after TBI.
Microglia are the resident immune cells that play an important role in immune surveillance of the CNS. Based on their morphology and activation, microglia have various subpopulation forms in the CNS. These subtypes include M0, M2a, M2b, M2c, M2d, and M1 (Franco and Fernandez-Suarez, 2015). M2 microglia have anti-inflammatory properties and play a significant role in maintenance of CNS homeostasis and plasticity, synaptic pruning, removal of pathogens through phagocytosis, neural development, regulating neurotransmitter release, neurogenesis, release of neurotrophic factors and tissue/synaptic remodeling. Microglia are acquisitively sensitive to changes in their local microenvironment. They dramatically change their phenotype and upregulate number of diverse cell-surface antigens. These microglia are typically referred to as M1 microglia. M1 microglia are in the hyper-activated state and can be amoeboid or rod-shaped. They promote immune cell recruitment into the CNS (such as Th1 and Th17), where they release proinflammatory cytokines, chemokines such as CCL2 and CCL20, and monocyte chemoattractant protein-1 and eotaxin. Under such circumstances, microglia stimulate iNOS production, trigger generation and release of ROS/RNS, activate the complementary proteins, and increase COX-2 production to produce prostaglandins (Streit et al., 2004; Dheen et al., 2007; Franco and Fernandez-Suarez, 2015).
During TBI, M1 microglia express several receptors on its surface as a result of either neural injury-derived damage-associated molecular patterns (DAMPs) or due to astrocytic secretions, that bind onto these receptors. Many of these receptors are a family of pathogen recognition receptors, such as TLRs, that recruit adapter proteins and initiate complex cascade of signaling events which regulate transcriptional events and inflammation. In response to DAMPs, and factors released by damaged neurons, astrocytes and immune cells, microglia drastically changes its morphology, proliferate, move along chemotactic gradience, express surface molecules for signaling, carry out cytotoxic attack on neurons and increase increase proinflammatory secretions. Activation of proinflammatory receptors and their downstream products, when released, either causes neural injury or modulate astrocytic activity, causing an A1 phenotype (Sharma and Naidu, 2016; Clark et al., 2019; Wofford et al., 2019). For instance, primary astrocyte-activating signals released by microglia include IL-1, TNFα, and C1q (Liddelow and Barres, 2017; Clark et al., 2019). These cytokines and complement proteins cause structural and functional changes in astrocytes. Reactive astrocytes disassemble synaptic connections between neurons and release neurotoxins that degenerates mature neurons and oligodendrocytes in CNS after TBI (Liddelow et al., 2017). Astrocytes, likewise microglia, also express high levels of proinflammatory receptors, which similarly alter microglial and neuronal activity. The crosstalk between astrocytes, microglia, and neurons causes degradation of the extracellular matrix (Hevin) and metabolic proteins (ADAMTs), triggers leukocyte mediated inflammatory responses (TREM2, complement proteins), promotes neutrophil chemotaxis (complement proteins), recruits immune cells (purinergic receptors), stimulates cell lysis, enhances production of miRNAs, disrupts lipid homeostasis and cell membranes (nuclear receptors), and impairs synaptogenesis (SPARC) (Figure 5). This cross-coupling between neuroglia induce changes in glial physiology causing long-term neurodegenerative changes after TBI, promoting epileptogenesis resulting in PTE (Smith, 2013; Izzy et al., 2019). Increased inflammasome binding onto TLRs, during the first few days post-injury is one of the major drivers of neuroinflammation that triggers epileptogenesis (O’Brien et al., 2020). In addition to the above events, microglial pruning of synapses is increased very early in the disease progression; as a result, the loss of synaptic density due to an increase in the phagocytic capacity of microglia could perhaps be an important factor that promotes epileptogenesis after TBI (Andoh et al., 2019).

Figure 5. Crosstalk between reactive microglia, reactive astrocytes and neurons during neuroinflammation. During inflammation, microglia and astrocytes achieve M1 and A1 like morphology. M1-like microglia, also known as reactive microglia, secrete multiple factors that modulate astrocytic functions by binding onto their receptors. Likewise, astrocytes too secrete molecules that bind onto microglia and modulate their activities. Molecules in purple are released by microglia and induce A1 phenotype in astrocytes and/or modulate astrocytic activity. Molecules in green are released by astrocytes and modulate microglia activity and/or forms reactive microglia. Molecules in orange are released by both astrocytes and microglia, co-regulating each other’s activities. Molecules released by these cells also effect neurons (in red). Beneath neurons are molecules (in purple) that microglia secrete which cause neuronal damage. In green are molecules secreted by astrocytes that cause neurodegeneration. Red dots next to purple and green text indicate molecules released by neurons that alter microglia and astrocytic activities. Some of the receptors on M1 microglia such as TLR4/6 complex, RAGE, CX3CR1, P2YR and P2XR cause microglia activation due to neural injury-derived DAMPs.
The signals and response modifiers in microglial activation can be triggered and modified by several factors based on the cellular origin, chemical structure and signaling. These include structures of infectious agents, immunoglobulins/immune complexes, complement system, cytokines, neurotrophic factors, proteins and peptides, and neurotransmission related compounds and ions such as ATP, purines and glutamate. Many of these molecules, and associated signaling events, are also released after TBI, which perhaps play a crucial role in the pathogenesis of PTE (Wofford et al., 2019). Signals emitted by neighboring resident cells or by immune cells from the periphery shape profiles of induced genes and functions in microglia. Studies have shown that activation of certain types of cytokines such as IFNγ, IL-1β, and TNFα after TBI, drives proinflammatory microglial response with increased expression of IL-12, supporting the role of immune cell (such as Th1) mediated reactions in regulating M1 state of microglia. These signals, in turn, influence the cocktail of chemoattractive factors to organize for a change in the composition of infiltrates to instruct the engagement of neutrophils, monocytes and distinct T-cell subpopulations (Hanisch and Kettenmann, 2007). Indeed, there are multiple reports of the phenotypic shifts in macrophages and microglia in vitro and in vivo, and cytokines produced by T-cell subtypes such as IFNγ are primarily known for this change. These series of events can orchestrate inflammatory reactions in response to traumatic insult (Hanisch and Kettenmann, 2007). Furthermore, time lapse images of microglia, after TBI, have revealed immediate microglial response to the focal injury. They undergo a rapid phenotypic change and form a bulbous body that extend towards the ablation site forming a spherical containment releasing ‘on’ signals (Davalos et al., 2005). On signals are inducible factors and includes a range of chemokines, but also neurotransmitters such as purines and glutamate. A prominent feature of reactive microglia is the high expression of receptors for purines and their wide range of responses to receptor activation, which have been reported in numerous experimental models TBI (Davalos et al., 2005; Jackson et al., 2016; Frenguelli, 2019). For instance, single focal injection of ATP in mice induced a localized response of activated microglia with higher P2Y6 and P2Y12 receptor expression, may support the role of purine receptors in TBI induced epileptogenesis (Davalos et al., 2005; Kumaria et al., 2008; Jackson et al., 2016).
The exciting development in microglia research in terms of origin and progenitors of microglia, their population and stability or turnover under normal and diseased conditions, their contribution to the maturation and support of neuronal development and glial functions, their protective and harmful actions in diseases and the options of therapeutic interventions by silencing or enhancing functions, will help to answer several key questions and help in understanding their role more clearly in health and disease.
Astrocytes, first identified by Virchow in 1846 as a glue filling the interstitial fluid, are star-shaped cells in the CNS that play an important role in maintaining brain homeostasis. Astrocytes use their “astrocytic end-feet” to support the metabolic demands of neurons by supplying nutrients from the blood vessels (Cui et al., 2012). They make ‘tripartite synapses’ with pre- and post-synaptic neurons, to integrate synaptic function by means of neurotransmitters and gliotransmitter release (Cui et al., 2012). Neuroactive molecules of astrocytes, such asD-serine, GABA, and adenosine triphosphate (ATP), regulate neuronal functions such as synaptic activity by inducing long-term depression and long-term potentiation, mediate tonic inhibition through Best1 ion channels, inhibit proinflammatory molecules (such as TNFα), assist GABA transporters in a calcium-independent manner and regulate sleep homeostasis, synaptic plasticity, and memory formation (Panatier et al., 2006; Haskó et al., 2008; Lee et al., 2010; Yoon et al., 2011; Fossat et al., 2012).
Astrocytes regulate neuronal functions under normal physiological conditions, but under pathological conditions, astrocytes phenotypically change in response to their microenvironment and become reactive during inflammation (Cui et al., 2012; Pekny and Pekna, 2016). After TBI, reactive astrocytes undergo morphological changes, that corresponds to changes in their functional and molecular properties. These alterations include dysfunctional potassium and glutamate buffering, modulation of aquaporins and adenosine activity, disturbances in gap junctions, disruption of glutamate-glutamine cycle, impairment of cysteine-glutamate antiporter system and mutations in potassium channel genes (Lewerenz et al., 2013; Burda et al., 2016; Zhou et al., 2020). Series of these events over time, results in the accumulation of neurotoxic molecules in the brain and cause BBB disruption. The damage to the BBB promotes extravasation of serum albumin into the brain (Puttachary et al., 2016). Serum albumin in the brain binds to TGFβ receptors on astrocytes, which phosphorylates Alk5 mediated SMAD2/3 complex and p-38 MAPK. This causes SMAD2/3 translocation into the nucleus activating transcription of proinflammatory genes promoting TGFβ and IL-6 production (Milikovsky et al., 2017). In numerous studies on rodent models of TBI, it has been reported that extravasation of serum albumin cause impairment of potassium buffering and glutamate reuptake by downregulating Kir4.1 potassium channels and glutamate transporters (Ranaivo et al., 2012; Weissberg et al., 2015; Zhou et al., 2020). This elevates extracellular K+ and glutamate concentration, and cause hyperexcitability (Puttachary et al., 2016; Steinhäuser et al., 2016). Infiltration of peripheral immune cells (such as T cells and monocytes) after BBB breakdown signals the release of complementary proteins. Up-regulation in complementary proteins promote leukocyte chemotaxis and migration at the lesion site (Cho, 2019). Many complement cascade genes are profoundly upregulated in the reactive astrocytes and neurons after TBI. They play an important roles in activating numerous pathological pathways involved in synaptic loss, increased synaptic pruning, impairment of neuromelanin clearance, increased stress in endoplasmic reticulum, decreased phagocytosis by dendritic cells, modulation in neurite outgrowth and regulating control of growth factors (Daglas and Adlard, 2018; Hammad et al., 2018; Cho, 2019). In response to proinflammatory insult, reactive astrocytes also produce an unknown factor called protein-X, which triggers the production and shedding of the complement components by neurons (Shi et al., 2010). Excessive tagging of neurons by activated complement proteins and their recognition by complement receptors or reactive microglia results in phagocytosis and removal of synapses, and eventually neuronal death.
The combination of aforementioned damaged signals and their relative concentrations most likely determine the type of astrogliosis experienced by astrocytes in different regions surrounding the initial insult zone. On a cellular level, insult to the brain such as TBI results in hypertrophy of astroglial processes and significant increase in astrocytic cytoskeleton (Sofroniew and Vinters, 2010; Burda et al., 2016; Steinhäuser et al., 2016; Chen et al., 2020). Brain damage very rapidly turns most of the astroglial cells into GFAP expressing reactive astrocytes. Both GFAP and vimentin are critically important for the development of reactive astrocytes. Severe stress in astroglia energetics leads to subsequent loss of ion homeostasis that triggers enormous amounts of glutamate in ECS. The astroglial involvement in controlling brain glutamate concentration is double edged. Upon severe injury, astrocytes may turn from being the sink for glutamate to being the main source of the latter. Astrocytes can release glutamate by several mechanisms which are triggered in PTE. First, the reversal of glutamate transporters can be caused by ATP depletion accompanied with an increase in intracellular Na+ concentration and cell depolarization. Second, elevation of cytoplasmic Ca2+ concentration in astrocytes, that follows traumatic injury, may trigger the release of glutamate stored in vesicles. Third, acidosis and lowering extracellular Ca2+ concentration may open glutamate-permeable hemichannels. Fourth, ATP released in higher concentrations by dying and disintegrating neurons can open astrocytic P2X7 purinoceptors which allows glutamate release. Fifth, brain oedema post-TBI can activate volume-sensitive channels which too allows the passage of glutamate.
Excess glutamate in the extracellular space disrupts the cysteine/glutamate antiporter system (CGS), a key anti-oxidant system in astrocytes that imports oxidized cysteine into the cell in exchange for glutamate. CGS regulates movement of amino acids in to and out of the cell, depending on the cellular requirements, and regulates the immune system, resistance against anti-cancer drugs, protection against carcinogenesis, cellular redox homeostasis, and modulates memory and behavior. In astrocytes, the intracellular concentration of cysteine (in its reduced form) is generally lower than glutamate (Lewerenz et al., 2013). Cysteine is an important substrate for the production of glutathione; and, inside the cell, oxidized cystine is reduced to form glutathione through the help of enzyme thioredoxin reductase 1 (Mandal et al., 2010; Lewerenz et al., 2013). Post-TBI, dysfunctional CGS upsets the balance between anti-oxidant and oxidants, causing oxidative stress as a result of glutamate excitotoxicity (Koza and Linseman, 2019). During inhibition of CGS, glutathione levels decline. Once glutathione depletion reaches a critical level, ROS production increases. This does not cause cell death immediately but instead facilitates the activation of signaling pathways and ultimately culminates in cell death. Therefore, neuroprotective compounds that generally are not beneficial during chronic stages of the disease can have favorable outcomes when administered at early time-points (when ROS concentration is gradually increasing, post injury) (Maher and Schubert, 2000; Lewerenz et al., 2013). Modulation in CGS levels have been reported in many neurodegenerative conditions. The increase in CGS in these conditions could primarily be due to an increased glutamate accumulation and release (Chung et al., 2005; Pampliega et al., 2011). This rise in extracellular glutamate is a result of downregulation of the excitatory amino acid transporter (EAAT) that balances CGS-mediated glutamate release. Therefore, specifically targeting glutamate by inhibiting CGS, can be an alternate approach for treating TBI related disorders as it aims to balance glutamate release into the ECS with glutamate uptake by EAATs. Drugs that protect from glutamate excitotoxicity act mainly through these mechanisms and inhibit excitotoxic effects of CGS by increasing glutathione synthesis modulating glutamate release (Lewerenz et al., 2013).
After TBI, initiation of secondary insult mechanisms can trigger epileptogenesis. If primary mechanisms are not controlled on time, they can cause long-term cellular and molecular alterations in astrocytes, leading to serious neurological consequences over time. For instance, astrocytic dysfunction can result in disruption of homeostatic regulation of brain volume and water content levels, causing edema. This can result in increased intracranial pressure, changes in extracellular osmotic pressure and compression damage to neural tissues (Dearden, 1992; Jha et al., 2019). After TBI, astrocytes are unable to remove excess water due to damage to their water channels, called aquaporins. Aquaporins have been widely studied as drivers of pathogenesis in epilepsy and other neurodegenerative conditions. Mutations in aquaporin 4 disrupt fluid osmolarity and potassium homeostasis (Heuser et al., 2010; Binder et al., 2012; Nagelhus and Ottersen, 2013). Although the role of gap junction dysregulation in epilepsy is still controversial, some studies have demonstrated an anti-epileptic role of gap junctions during astrocytic coupling. According to the spatial buffering concept, astrocytes pass excess K+ ions between their networks, reducing K+ concentration in the ECS. Dysfunctions in gap junction proteins, such as connexins, have been reported to increase cellular hyperexcitability and cause seizures. For instance, studies on Cx30–/– mice reported increased neuronal depolarization and lower seizure threshold with disturbances in potassium and glutamate clearance in astrocytes, causing astrocytic swelling (Wallraff et al., 2006; Steinhäuser et al., 2016). Damage to aquaporins and rapid swelling of astrocytes after injury is accompanied by a significant increase in astroglial surface area. Astroglial swelling can trigger numerous secondary effects that can exacerbate the brain damage. In particular, swelling of perivascular astrocytes and astrocyte endfeet may compress brain vessels and limit circulation. Swelling of astrocytes can result in the opening of volume regulated ion channels permeable to glutamate and other excitatory amino acids exacerbating excitotoxic cell death (Sun et al., 2003; Tran et al., 2010). Therefore, the functional and molecular changes in astrocytes, after TBI, promote epileptogenesis suggesting their role in the development of PTE.
The possibility of developing PTE, after post-traumatic seizures, is generally higher and so increases the risk of long-term consequences of TBI. These consequences depend on the severity of injury and the region of the brain affected. For instance, the odds of developing long-term implications diminish if the injury is a mild or moderate closed-head one, in contrast to a severe closed-head injury (Naalt et al., 1999; McCullagh and Feinstein, 2003). The closed-head TBI causes bleeding or intracranial hematoma which raises the risk of lasting impact on the brain. Recently, a 30 years followup study conducted in Sweden on patients with TBI, reported that all survivors of TBI, whether moderate or severe, developed dementia within 30 years of injury (Himanen et al., 2006). Moreover, studies on Vietnam War veterans, over the span of 40 years, revealed that treating with anti-convulsants during the acute phase of injury controlled severity and frequency of early seizures, whereas later treatments with anti-convulsants did not prevent the onset of PTE (Raymont et al., 2011). Over 40% of troops that suffer TBI develop PTE in their lifetime, with lasting effects including confusion, cognitive deficits, depression, and anxiety disorders. Long-term follow-up studies on veterans also revealed that about 18% of veterans experienced their first seizure after 15 years of injury (most had seizures after 1–5 years) and about two-thirds are on life-long medications (Raymont et al., 2010, 2011). Interestingly, having a family history of epilepsy or a genetic predisposition adds to the risk of developing PTE after brain injury. The genes involved mainly control plasticity, modulate levels of neurotransmitters, control ion channels, and regulate immune functions (Swartz et al., 2006; Raymont et al., 2010).
After TBI, the incidence of PTE increases and some patients are notoriously difficult to treat due to challenges in long-term follow up and therapeutics (Garga and Lowenstein, 2006; Schmidt et al., 2014; Szaflarski et al., 2014). Video-EEG monitoring and MRI studies on patients with PTE reported that approximately one quarter of the patients develop mesial temporal sclerosis and predicted the development of neocortical lesions on other half of the patients, at some stage in life; whereas, the vast majority of the cases develop focal epilepsy (Gupta et al., 2014). Swartz et al. reported that, of 200 consecutive temporal lobectomies performed on TBI survivors, 21 cases were of PTE, and about 50% of these cases had hippocampal sclerosis characterized by neuronal loss primarily in the hilar region of DG (Swartz et al., 2006). Moreover, a CEEG and PET scanning on 16 TBI patients revealed that ∼28% of these patients had non-convulsive seizures (NCS) over 7 days after injury and one had R temporal NCS during PET while comatosed (Vespa et al., 2010). Further, the same study reported the patients who had seizures several days after injury developed hippocampal atrophy, ipsilateral to the seizure, which was possibly why some develop PTE later in life (Vespa et al., 2010). High-resolution analysis of the brain (through diffusion tensor imaging of the perforant path) revealed that the white matter tracts that are either afferent and efferent to the hippocampus are particularly sensitive to shearing and stretching forces (Wang et al., 2008). This indicates that, at least in some cases, the mechanism of epileptogenesis results from a deafferentation or disconnection of the hippocampus from the long-term synaptic connections which develops over time.
Repeated TBI can alter neural circuits and lead to long-term degenerative changes in the brain and periphery. For instance, chronic traumatic encephalopathy (CTE; a neurological deterioration due to accumulation of hyperphosphorylated tau) causes release of TDP43 (transactive response DNA binding protein), which forms neurofibrillary tangles and increases oxidative stress. TDP43 is produced in high amounts, which affects the anti-oxidant enzyme SOD-1 and causes protein misfolding, damaging the BBB (Pokrishevsky et al., 2012). The breakdown of the BBB may persist for many months or years, gradually causing damage over time. The BBB disruption results in local inflammation which ultimately resolves into epileptogenesis (Tomkins et al., 2011; Vezzani et al., 2012). In the periphery, the cardiac complications of PTE cause morbidity and mortality due to enhanced cardiac contractility, high blood pressure, and production of myocardial ROS. The increased cardiac contractility results in sympathetic storm that causes arrhythmias, high blood pressure, reduced heart rate variability, and the manifestation of congenital heart problems. It also raises plasma catecholamine production, further damaging the myocardium (Shanlin et al., 1988). Elevated catecholamine enhances oxidative load in myocardial tissues, disrupting the balance between oxidants and anti-oxidants. This diminishes NO bioavailability in the heart, affecting general circulation and regulation of blood pressure (Larson et al., 2012).
Generally, after penetrating or severe closed TBI, altered homeostatic mechanisms generate the first seizure, usually a generalized seizure with focal onset, and a late seizure that is a partial complex seizure. A better understanding of the molecular mechanisms that cause these seizures and epilepsies is imperative for development of better drugs and treatments. Moreover, greater understanding of the brain’s immune system is also necessary to identify the causal mechanisms of long-term PTE-related consequences.
Management of brain injury focuses primarily on preventing signs of secondary injury. Currently, no therapies are available for permanently treating TBI-related injuries, although more than 20 drugs are available to treat epilepsy (however one-third of epilepsy patients are refractory to these drugs) (Dalic and Cook, 2016; Hogan, 2018). Moreover, over 40 failed drugs have been tested in the clinical trials against epilepsy in the past decade, most of which were ion channel targets. The failure of these compounds to treat PTE could perhaps be due to the complexity of PTE and the new unknown mechanisms that regulate epileptogenesis after TBI (Temkin, 2001; Varvel et al., 2014). Therefore, it is important to investigate novel non-neuronal targets/mechanisms other than ion channels, such as enzymes, glial cells, neurovascular components, oxidative stress molecules, and nuclear proteins (Table 2).
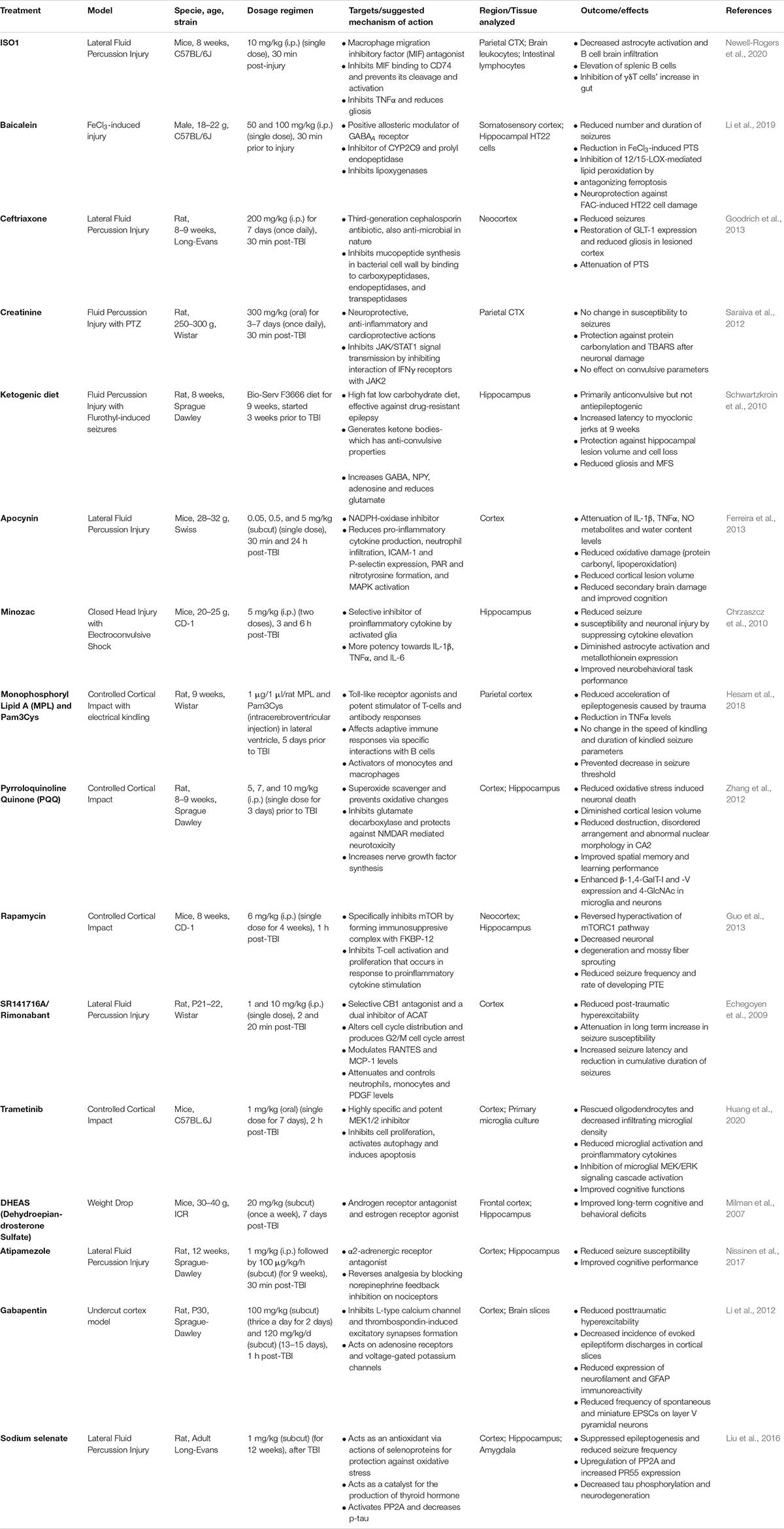
Table 2. Novel therapeutic interventions that may have potential to impact the outcome of TBI-induced epileptogenesis.
No existing treatments can prevent the long-lasting neurodegenerative changes in PTE, but targeting free radicals during the acute phase of inflammation might prove to be more effective. For example, increased levels of NADPH oxidase after TBI damages mitochondria and other organelles (Bordt and Polster, 2014; Angeloni et al., 2015; Ma et al., 2017). Pharmacological inhibitors of free radicals such as NOX, peroxides, peroxynitrites/nitrates, hypochlorites, phenols and prostanoid antagonists can modulate free radical production and suppress inflammation (Cheng et al., 2012; Korkina et al., 2016; Ma et al., 2017; Smith et al., 2019). Studies have shown that inhibition or genetic ablation of NADPH oxidase improves outcomes in terms of neurodegeneration, oxidative stress mediated mitochondrial dysfunction, gliosis, and increases neurogenesis (Cheng et al., 2012; Altenhöfer et al., 2015; Hirano et al., 2015; Maqbool et al., 2020). In contrast to reducing pro-oxidant levels, increasing anti-oxidants can be a useful, alternate therapy for preventing long-term changes in the brain post-TBI. Inhibitors of conventional targets, such as COX-2, IFN, and prostaglandin, can also help in combating poor outcomes. Interestingly, the chronic PTE signature is quite similar to the IFN signature, in terms of activation of neurodegenerative mechanisms; and inactivating type I IFN with αIFNAR-infusion therapy can block IFNα/IFNβ signaling, lowers expression of inflammatory mediators, diminishes neurodegeneration, and attenuates inflammation. Moreover, an ICV infusion of αIFNAR significantly improves cognitive deficits, motor functions, upregulates neuroprotective genes, and reduces lesion volume (Barrett et al., 2020).
Inhibiting hippocampal neurogenesis after TBI can be a viable therapeutic option for preventing mesial TLE. In most cases, the process of neurogenesis in the hippocampus after injury benefits the brain and allows for recovery of memory and normal behavior (Parent, 2002; Sun, 2014; Redell et al., 2020). Some of that repair may be imperfect and show synaptic reorganization of neural networks, which can create circuitry that is epileptogenic. These considerations show the need for caution and careful design of strategies that target aberrant neurogenesis and synaptogenesis, while leaving the neurogenesis and synaptogenesis that are important for recovery in place.
In terms of treatment and management, controlling swelling and preventing hypoxia or ischemia can prove to be another effective therapy. If, at a certain point, intracranial pressure (ICP) rises dramatically with an increase in intracranial volume, then the brain swells substantially. Preventing this is important because the degree of ICP directly correlates with cerebral perfusion pressure (CPP), the key pressure required by brain, in terms of delivery of oxygen and other nutrients. As ICP rises, CPP drops, and as with it the delivery of nutrients and oxygen decelerates having deleterious effects on the brain. This can further affect cerebral blood flow, reducing oxygen content in the blood (Bouzat et al., 2013; Kinoshita, 2016). Therefore, it is important to develop techniques that will sustain cerebral blood at an appropriate level—keeping CPP up without increasing ICP—to prevent permanent infarction from TBI (Zauner et al., 2002). Strategies for lowering ICP may include stepwise medical management or surgical options (like placing a ventricular drain for CSF) or employing strategies that would increase the intracranial vault size (like decompressive craniotomy). Medical management therapies can include normothermia, normoglycemia, targeting blood pressure, maintaining oxygen and carbon dioxide saturation levels, eliminating hyponatremia, decreasing cerebral metabolic demand/rate, increasing the mean arterial pressure to enhance CPP, and use of vasopressors such as norepinephrine and dopamine to prevent edema (Vespa et al., 1998; Hutchinson et al., 2002; Sookplung et al., 2011).
Prophylactic use of antiepileptic drugs (AEDs) can reduce the risk of early post-traumatic seizures but not the later ones. PTE perhaps represents the ideal model to study the mechanisms of epileptogenesis and develop therapies for epilepsy. Over the last several years it has become clear that, properly chosen, cases of PTE can successfully be treated with surgical interventions. Approximately 60% of the surgical cases of PTE end up being Engel class I, ∼20% Engel class II; and about 80% had favorable surgical outcomes. Gupta et al. (2014) reported that patients with mesial temporal sclerosis, as an epilepsy syndrome, have 92% class I and II outcomes. The lesional cases, from both frontal and temporal, and non-lesional cases were somewhat less favorable (Gupta et al., 2014). In contrast, the outcome of surgical intervention depended on the seizure-onset localization zone. The surgical intervention is not generally recommended in most cases of PTE, as seizure foci can be difficult to localize due to technical issues, such as craniotomies and breach rhythms. Patients with severe TBI may have undergone craniotomy, which may cause breath rhythms (special EEG rhythms that can be artifacts or misguide diagnosis). Moreover, patients with severe TBI may have diffuse cerebral or axonal injury evident on EEG recording as multiple epileptic foci that can overlap with localization of eloquent brain regions (Hakimian et al., 2012). Therefore, it is important to carefully select surgical candidates, as the patient with severe TBI may be at greater risk of surgical complications due to their structural damage or scar tissue and adhesion formation. For these reasons, other adjunctive treatment options such as stimulation of the vagus nerve, responsive nerve, and anterior nerve should be considered.
Medical treatment for PTE can be similar to that for other epilepsies, with the caveat that unnecessary treatment with AEDs may impair neurorehabilitation. There is no evidence that treatment with AEDs or anti-convulsants will be beneficial in cases of moderate to severe TBI; however, some evidence suggests AEDs could possibly reduce the incidence of post-traumatic seizure if administered during the acute phase of injury. One study conducted by Hernandez (1997) showed that treating patients with phenytoin during the acute phase lowers the incidence of early post-traumatic seizures from 14.2 to 3.6%. Yet continuing AED treatment beyond the acute phase has never been shown to change the prognosis for the ultimate development of epilepsy (Hernandez, 1997). Recently, levetiracetam (LEV) has gained popularity in the treatment of PTE, as LEV does not cause the same side effects and has lower cytotoxic effects than other AEDs. Even though no randomized controlled trials have been done, comparative studies show the efficacy of LEV seems slightly better than standard AEDs, and with fewer side-effects. On the downside, observational studies report that LEV is not effective in reducing PTE risk, so it is generally not recommended beyond 7 days post-injury (Szaflarski et al., 2010). As an alternate to LEV, sodium channel blockers are an ideal option, but some of these blockers do not appear to be effective against PTE. For example, Dilantin blocks early seizures but it is not regarded as an effective anti-epileptogenic agent (Szaflarski et al., 2010). Although not every sodium channel blocker has been tested in PTE models, additional replacement therapies are required to treat PTE. These alternate therapies must rely on things like blocking inflammation, promoting BBB repair, or perhaps promoting the integrity of damaged axonal pathways to prevent persistent brain inflammation. Therefore, the goal of developing anti-epileptogenic compounds should be tied to these strategies to promote integrity, resilience, and recovery of neural structures.
Four key elements—excitotoxicity, neuroinflammation, oxidative stress, and neurodegeneration—are the primary pathognomonic mechanisms responsible for PTE; and it is well known that TBI initiate cycles of neuroinflammatory events that elicit the oxidative stress response tripping a series of events and cycles that exacerbate the acute stage and lead to chronic conditions (Figure 6). The goal of this review is to understand the mechanisms of epileptogenesis after TBI and identify, develop, and validate therapeutic strategies to prevent PTE. In this regard, we can make several key conclusions: (1) the primary source of cellular excitotoxicity after TBI is elevation in extracellular glutamate, increased immune cell infiltration and crosstalk between glial cells and neurons governed mainly through cytokine and chemokine networks; (2) the initial immune response to injury is beneficial and, it works to counterbalance the disequilibrium in the system; (3) Impairment of mitochondria due to an excessive generation of ROS/RNS is a continuous process during epileptogenesis, and is associated with inflammation and neurodegeneration; (4) pro-inflammatory cytokines, and chemokines are the key players released by invading blood cells, microglia, astrocytes, and neurons; (5) over-production of cytokines, lipids, and chemokines over long periods of time triggers cell death; (6) invasion by leukocytes and activated microglia leads to tissue damage at later stages.

Figure 6. Pathophysiological cascades of events from TBI to PTE. A series of cellular and molecular events overwhelms the brain following TBI. TBI induces neurovascular damage and activates biomolecular mediators of injury. This neurovascular damage leads to vasogenic edema and necrotic cell death, while biomolecular mediators lead to necrotic cell death and brain damage. TBI also induces structural damage that leads to cerebral edema and decreases cerebral blood flow, causing acute neurological damage. In addition, cytokines and other molecules released in the extracellular space trigger free radical production in neurons and glial cells, promotes neuronal excitability, and neuronal death. Alterations in mitochondrial bioenergetics, fragmentation of DNA and structural proteins, and activation of neuroinflammatory pathways cause structural and functional changes in the brain that accompany recurrent post-epileptic seizures. All these events, after TBI, are interwoven and rely on other altered cellular and molecular events which exacerbates post-epileptic seizures. Asterisk represents changes that can also lead to secondary damage either directly or indirectly.
Post-traumatic epilepsy is phenotypically heterogenous in humans and it is important to understand this phenotypic heterogeneity to develop antiepileptogenic therapies. Both focal and diffuse mechanisms can result in PTE, and approximately 25–30% of PTE cases are associated with mesial temporal sclerosis. Although surgery is an alternate option, it is generally not recommended, so AEDs remain the first line of treatment. Yet, AEDs are not very effective in treating PTE, but are rather used to treat the symptoms without improving the underlying condition. Patients receiving AEDs often require lifelong AED treatment and some develop severe side effects over time. Furthermore, failure to control epileptic seizures can also lead to increased mortality, reduced quality of life, comorbidity, and depression. In spite of the many AEDs available, a little progress has been made in preventing the onset of new types of epilepsies. Moreover, repurposing anti-seizure drugs to prevent the onset of epilepsy has been entirely unsuccessful up to now.
Understanding epilepsy as a network disorder suggests early phases of epileptogenesis should be targeted before the imbalance spreads to other regions of the brain. This, however, is not as simple as it seems because many of the candidate compounds being investigated have multiple effects and target multiple pathways. These pathways can be different in humans and animal models, which devalues the translational significance of the latter and highlights the importance of designing experiments with the right timing, dosage, and targets, and with appropriate animal models (Smith, 2016; Saletti et al., 2019). Moreover, it is important to define targets with variable injury mechanics and to vary treatments at particular time-points. Ultimately, a clearer understanding of the molecular mechanisms of epilepsy will allow development of truly novel therapeutic targets for PTE.
SS reviewed the literature, wrote and edited manuscript, and prepared the tables and figures. GT and JH edited the manuscript. AB conceived the idea, and reviewed and edited manuscript. All the authors contributed to the manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Neha Pandya for editing and making figures.
Abdullah, A., Zhang, M., Frugier, T., Bedoui, S., Taylor, J. M., and Crack, P. J. (2018). STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J. Neuroinflamm. 15:323. doi: 10.1186/s12974-018-1354-7
Abdul-Muneer, P. M., Chandra, N., and Haorah, J. (2015). Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 51, 966–979. doi: 10.1007/s12035-014-8752-3
Adam-Vizi, V., and Starkov, A. A. (2010). Calcium and mitochondrial reactive oxygen species generation: how to read the facts. J. Alzheimer Dis. 20(Suppl. 2), S413–S426. doi: 10.3233/JAD-2010-100465
Agledal, L., Niere, M., and Ziegler, M. (2010). The phosphate makes a difference: cellular functions of NADP. Redox Rep. 15, 2–10. doi: 10.1179/174329210X12650506623122
Agrawal, A., Timothy, J., Pandit, L., and Manju, M. (2006). Post-traumatic epilepsy: an overview. Clin. Neurol. Neurosurg. 108, 433–439. doi: 10.1016/j.clineuro.2005.09.001
Aguiar, C. C. T., Almeida, A. B., Araújo, P. V. P., Abreu, R. N. D. C. D., Chaves, E. M. C., Vale, O. C. D., et al. (2012). Oxidative stress and epilepsy: literature review. Oxid. Med. Cell. Longev. 2012:795259. doi: 10.1155/2012/795259
Altenhöfer, S., Radermacher, K. A., Kleikers, P. W., Wingler, K., and Schmidt, H. H. (2015). Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 23, 406–427. doi: 10.1089/ars.2013.5814
Anderson, P., and Delgado, M. (2008). Endogenous anti-inflammatory neuropeptides and pro-resolving lipid mediators: a new therapeutic approach for immune disorders. J. Cell. Mol. Med. 12, 1830–1847. doi: 10.1111/j.1582-4934.2008.00387.x
Andoh, M., Ikegaya, Y., and Koyama, R. (2019). Synaptic pruning by microglia in epilepsy. J. Clin. Med. 8:2170. doi: 10.3390/jcm8122170
Andrews, A. M., Lutton, E. M., Merkel, S. F., Razmpour, R., and Ramirez, S. H. (2016). Mechanical injury induces brain endothelial-derived microvesicle release: implications for cerebral vascular injury during traumatic brain injury. Front. Cell. Neurosci. 10:43. doi: 10.3389/fncel.2016.00043
Angeloni, C., Prata, C., Vieceli Dalla Sega, F., Piperno, R., and Hrelia, S. (2015). Traumatic brain injury and NADPH oxidase: a deep relationship. Oxid. Med. Cell. Longev. 2015:370312. doi: 10.1155/2015/370312
Annegers, J. F., Hauser, W. A., Coan, S. P., and Rocca, W. A. (1998). A population-based study of seizures after traumatic brain injuries. New Engl. J. Med. 338, 20–24. doi: 10.1056/NEJM199801013380104
Aronica, E., Ravizza, T., Zurolo, E., and Vezzani, A. (2012). Astrocyte immune responses in epilepsy. Glia 60, 1258–1268. doi: 10.1002/glia.22312
Badaut, J., Ajao, D., Sorensen, D., Fukuda, A., and Pellerin, L. (2015). Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: Signs of blood-brain barrier healing? Neuroscience 285, 215–226. doi: 10.1016/j.neuroscience.2014.10.035
Bae, Y. S., Oh, H., Rhee, S. G., and Yoo, Y. D. (2011). Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 32, 491–509. doi: 10.1007/s10059-011-0276-3
Bai, R., Gao, H., Han, Z., Huang, S., Ge, X., Chen, F., et al. (2017). Flow cytometric characterization of T-cell subsets and microglia after repetitive mild traumatic brain injury in rats. Neurochem. Res. 42, 2892–2901. doi: 10.1007/s11064-017-2310-0
Bao, Y. H., Bramlett, H. M., Atkins, C. M., Lotocki, G., Alonso, O. F., Dietrich, W. D., et al. (2011). Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J. Neurotrauma 28, 35–42. doi: 10.1089/neu.2010.1383
Barrett, J. P., Henry, R. J., Shirey, K. A., Doran, S. J., Makarevich, O. D., Ritzel, R. M., et al. (2020). Interferon-β plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that drives chronic neurodegeneration. J. Neurosci. 40, 2357–2370. doi: 10.1523/JNEUROSCI.2516-19.2020
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., Larue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Ben Shimon, M., Shavit-Stein, E., Altman, K., Pick, C. G., and Maggio, N. (2020). Thrombin as key mediator of seizure development following traumatic brain injury. Front. Pharmacol. 10:1532. doi: 10.3389/fphar.2019.01532
Berg, A. T., Darefsky, A. S., Holford, T. R., and Shinnar, S. (1998). Seizures with fever after unprovoked seizures: an analysis in children followed from the time of a first febrile seizure. Epilepsia 39, 77–80. doi: 10.1111/j.1528-1157.1998.tb01277.x
Bhowmick, S., D’Mello, V., Caruso, D., Wallerstein, A., and Abdul-Muneer, P. M. (2019). Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 317, 260–270. doi: 10.1016/j.expneurol.2019.03.014
Biliau, A. (2006). Interferon: the pathways of discovery I. molecular and cellular aspects. Cytokine Growth Fact. Rev. 17, 381–409. doi: 10.1016/j.cytogfr.2006.07.001
Bilousova, T. V., Rusakov, D. A., Ethell, D. W., and Ethell, I. M. (2006). Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J. Neurochem. 97, 44–56. doi: 10.1111/j.1471-4159.2006.03701.x
Binder, D. K., Nagelhus, E. A., and Ottersen, O. P. (2012). Aquaporin-4 and epilepsy. Glia 60, 1203–1214. doi: 10.1002/glia.22317
Bolkvadze, T., and Pitkänen, A. (2012). Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J. Neurotrauma 29, 789–812. doi: 10.1089/neu.2011.1954
Bordt, E. A., and Polster, B. M. (2014). NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Rad. Biol. Med. 76, 34–46. doi: 10.1016/j.freeradbiomed.2014.07.033
Bouzat, P., Sala, N., Payen, J.-F., and Oddo, M. (2013). Beyond intracranial pressure: optimization of cerebral blood flow, oxygen, and substrate delivery after traumatic brain injury. Ann. Intens. Care 3:23. doi: 10.1186/2110-5820-3-23
Broker, L. E., Kruyt, F. A., and Giaccone, G. (2005). Cell death independent of caspases: a review. Clin. Cancer Res. 11, 3155–3162. doi: 10.1158/1078-0432.CCR-04-2223
Brown, L. S., Foster, C. G., Courtney, J., King, N. E., Howells, D. W., and Sutherland, B. A. (2019). Pericytes and neurovascular function in the healthy and diseased brain. Front. Cell. Neurosci. 13:282. doi: 10.3389/fncel.2019.00282
Burda, J. E., Bernstein, A. M., and Sofroniew, M. V. (2016). Astrocyte roles in traumatic brain injury. Exper. Neurol. 275, 305–315. doi: 10.1016/j.expneurol.2015.03.020
Burda, J. E., and Sofroniew, M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. doi: 10.1016/j.neuron.2013.12.034
Burfeind, K. G., Michaelis, K. A., and Marks, D. L. (2016). The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 54, 42–52. doi: 10.1016/j.semcdb.2015.10.038
Cantu, D., Walker, K., Andresen, L., Taylor-Weiner, A., Hampton, D., Tesco, G., et al. (2015). Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb. Cortex 25, 2306–2320. doi: 10.1093/cercor/bhu041
Cardenas-Rodriguez, N., Huerta-Gertrudis, B., Rivera-Espinosa, L., Montesinos-Correa, H., Bandala, C., Carmona-Aparicio, L., et al. (2013). Role of oxidative stress in refractory epilepsy: evidence in patients and experimental models. Intern. J. Mol. Sci. 14, 1455–1476. doi: 10.3390/ijms14011455
Castejon, O. J. (1984). Formation of transendothelial channels in traumatic human brain edema. Pathol. Res. Pract. 179, 7–12. doi: 10.1016/S0344-0338(84)80054-0
Centers for Disease Control and Prevention (2019). Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2014. Available online at: https://www.cdc.gov/traumaticbraininjury/pdf/TBI-Surveillance-Report-508.pdf (accessed August 15, 2020).
Cerutti, C., and Ridley, A. J. (2017). Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 358, 31–38. doi: 10.1016/j.yexcr.2017.06.003
Cheeseman, K. H., and Slater, T. F. (1993). An introduction to free radical biochemistry. Br. Med. Bull. 49, 481–493. doi: 10.1093/oxfordjournals.bmb.a072625
Chen, Y., Qin, C., Huang, J., Tang, X., Liu, C., Huang, K., et al. (2020). The role of astrocytes in oxidative stress of central nervous system: a mixed blessing. Cell Prolifer. 53:12781. doi: 10.1111/cpr.12781
Cheng, G., Kong, R. H., Zhang, L. M., and Zhang, J. N. (2012). Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 167, 699–719. doi: 10.1111/j.1476-5381.2012.02025.x
Chinnici, C. M., Yao, Y., Ding, T., Funk, C. D., and Praticò, D. (2005). Absence of 12/15 lipoxygenase reduces brain oxidative stress in apolipoprotein E-deficient mice. Am. J. Pathol. 167, 1371–1377. doi: 10.1016/S0002-9440(10)61224-2
Cho, K. (2019). Emerging Roles of Complement Protein C1q in Neurodegeneration. Aging Dis. 10:652. doi: 10.14336/ad.2019.0118
Choi, G. B., Yim, Y. S., Wong, H., Kim, S., Kim, H., Kim, S. V., et al. (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. doi: 10.1126/science.aad0314
Chrzaszcz, M., Venkatesan, C., Dragisic, T., Watterson, D. M., and Wainwright, M. S. (2010). Minozac treatment prevents increased seizure susceptibility in a mouse “two-hit” model of closed skull traumatic brain injury and electroconvulsive shock-induced seizures. J. Neurotrauma 27, 1283–1295. doi: 10.1089/neu.2009.1227
Chuang, T.-C., Chang, A. Y. W., Lin, J.-W., Hsu, S.-P., and Chan, S. H. H. (2004). Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia 45, 1202–1209. doi: 10.1111/j.0013-9580.2004.18204.x
Chuang, Y. C. (2010). Mitochondrial dysfunction and oxidative stress in seizure-induced neuronal cell death. Acta Neurol. Taiwa. 19, 3–15.
Chung, R. S., McCormack, G. H., King, A. E., West, A. K., and Vickers, J. C. (2005). Glutamate induces rapid loss of axonal neurofilament proteins from cortical neurons in vitro. Exper. Neurol. 193, 481–488. doi: 10.1016/j.expneurol.2005.01.005
Cini, M., and Moretti, A. (1995). Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiol. Aging 16, 53–57. doi: 10.1016/0197-4580(95)80007-e
Clark, D. P., Perreau, V. M., Shultz, S. R., Brady, R. D., Lei, E., Dixit, S., et al. (2019). Inflammation in traumatic brain injury: roles for toxic A1 astrocytes and microglial-astrocytic crosstalk. Neurochem. Res. 44, 1410–1424. doi: 10.1007/s11064-019-02721-8
Cui, H., Kong, Y., and Zhang, H. (2012). Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transd. 2012:646354.
Daglas, M., and Adlard, P. A. (2018). The involvement of iron in traumatic brain injury and neurodegenerative disease. Front. Neurosci. 12:981. doi: 10.3389/fnins.2018.00981
Dalal, P. J., Muller, W. A., and Sullivan, D. P. (2020). Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 190, 535–542. doi: 10.1016/j.ajpath.2019.11.004
Dalic, L., and Cook, M. J. (2016). Managing drug-resistant epilepsy: challenges and solutions. Neuropsychiatr. Dis. Treat. 12, 2605–2616. doi: 10.2147/NDT.S84852
D’Ambrosio, R., Fairbanks, J. P., Fender, J. S., Born, D. E., Doyle, D. L., and Miller, J. W. (2004). Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 127, 304–314. doi: 10.1093/brain/awh038
D’Ambrosio, R., Fender, J. S., Fairbanks, J. P., Born, D. E., Doyle, D. L., Miller, J. W., et al. (2005). Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain 128, 174–188. doi: 10.1093/brain/awh337
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Davies, C. L., Patir, A., and McColl, B. W. (2019). Myeloid cell and transcriptome signatures associated with inflammation resolution in a model of self-limiting acute brain inflammation. Front. Immunol. 10:1048. doi: 10.3389/fimmu.2019.01048
Dearden, N. M. (1992). “Brain edema and raised intracranial pressure after head injury,” in Yearbook of Intensive Care and Emergency Medicine Yearbook of Intensive Care and Emergency Medicine, ed. J. L. Vincent (Berlin: Springer), 537–552. doi: 10.1007/978-3-642-84734-9_52
Dejager, L., Vandevyver, S., Ballegeer, M., Van Wonterghem, E., An, L. L., Riggs, J., et al. (2014). Pharmacological inhibition of type I interferon signaling protects mice against lethal sepsis. J. Infect. Dis. 209, 960–970. doi: 10.1093/infdis/jit600
Dexter, D. T., Carter, C. J., Wells, F. R., Javoy-Agid, F., Agid, Y., Lees, A., et al. (1989). Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 52, 381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x
Dheen, S. T., Kaur, C., and Ling, E.-A. (2007). Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14, 1189–1197. doi: 10.2174/092986707780597961
Diaz-Arrastia, R., Agostini, A., Frol, A. B., Mickey, B., Fleckenstein, J., Bigio, E., et al. (2000). Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch. Neurol. 57, 1611–1616. doi: 10.1001/archneur.57.11.1611
Dienel, G. A. (2014). Lactate shuttling and lactate use as fuel after traumatic brain injury: metabolic considerations. J. Cereb. Blood Flow Metab. 34, 1736–1748. doi: 10.1038/jcbfm.2014.153
Ding, K., Gupta, P. K., and Diaz-Arrastia, R. (2016). “Ch. 14 Epilepsy after traumatic brain injury,” in Translational Research in Traumatic Brain Injury, eds D. Laskowitz and G. Grant (Boca Raton, FL: CRC Press).
Du, Y., Kemper, T., Qiu, J., and Jiang, J. (2016). Defining the therapeutic time window for suppressing the inflammatory prostaglandin E2 signaling after status epilepticus. Expert Rev. Neurotherapeut. 16, 123–130. doi: 10.1586/14737175.2016.1134322
Eastman, C. L., D’Ambrosio, R., and Ganesh, T. (2020). Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 172:107907. doi: 10.1016/j.neuropharm.2019.107907
Echegoyen, J., Armstrong, C., Morgan, R. J., and Soltesz, I. (2009). Single application of a CB1 receptor antagonist rapidly following head injury prevents long-term hyperexcitability in a rat model. Epilepsy Res. 85, 123–127. doi: 10.1016/j.eplepsyres.2009.02.019
Emerit, J., Edeas, M., and Bricaire, F. (2004). Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 58, 39–46. doi: 10.1016/j.biopha.2003.11.004
Ermolaeva, M. A., Michallet, M., Papadopoulou, N., Utermöhlen, O., Kranidioti, K., Kollias, G., et al. (2008). Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 9, 1037–1046. doi: 10.1038/ni.1638
Eslami, M., Sayyah, M., Soleimani, M., Alizadeh, L., and Hadjighassem, M. (2015). Lipopolysaccharide preconditioning prevents acceleration of kindling epileptogenesis induced by traumatic brain injury. J. Neuroimmunol. 289, 143–151. doi: 10.1016/j.jneuroim.2015.11.003
Evans, L. P., Woll, A. W., Wu, S., Todd, B. P., Hehr, N., Hedberg-Buenz, A., et al. (2020). Modulation of post-traumatic immune response using the IL-1 receptor antagonist anakinra for improved visual outcomes. J. Neurotrauma 37, 1463–1480. doi: 10.1089/neu.2019.6725
Faul, M., and Coronado, V. (2015). Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 127, 3–13. doi: 10.1016/B978-0-444-52892-6.00001-5
Ferreira, A. P., Rodrigues, F. S., Della-Pace, I. D., Mota, B. C., Oliveira, S. M., Velho Gewehr Cde, C., et al. (2013). The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: role of inflammatory and oxidative brain damage. Neurochem. Int. 63, 583–593. doi: 10.1016/j.neuint.2013.09.012
Figueiredo-Pereira, M. E., Rockwell, P., Schmidt-Glenewinkel, T., and Serrano, P. (2015). Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 7:104. doi: 10.3389/fnmol.2014.00104
Fogal, B., and Hewett, S. J. (2008). Interleukin-1β: a bridge between inflammation and excitotoxicity? J. Neurochem. 106, 1–23. doi: 10.1111/j.1471-4159.2008.05315.x
Fordington, S., and Manford, M. (2020). A review of seizures and epilepsy following traumatic brain injury. J. Neurol. 267, 3105–3111. doi: 10.1007/s00415-020-09926-w
Fossat, P., Turpin, F. R., Sacchi, S., Dulong, J., Shi, T., Rivet, J. M., et al. (2012). Glial D-serine gates NMDA receptors at excitatory synapses in prefrontal cortex. Cereb. Cortex 22, 595–606. doi: 10.1093/cercor/bhr130
Franco, R., and Fernandez-Suarez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Frenguelli, B. G. (2019). The purine salvage pathway and the restoration of cerebral ATP: implications for brain slice physiology and brain injury. Neurochem. Res. 44, 661–675. doi: 10.1007/s11064-017-2386-6
Gao, X., Deng, P., Xu, Z. C., and Chen, J. (2011). Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate Gyrus. PLoS One 6:e024566. doi: 10.1371/journal.pone.0024566
Garga, N., and Lowenstein, D. H. (2006). Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr. 6, 1–5. doi: 10.1111/j.1535-7511.2005.00083.x
Gatti, S., Vezzani, A., and Bartfai, T. (2002). “Mechanisms of fever and febrile seizures: putative role of the interleukin-1 system,” in Febrile Seizures, eds T. Z. Baram and S. Shinnar (Cambridge, MA: Academic Press), 169–188. doi: 10.1016/B978-012078141-6/50014-7
Godfred, R. M., Parikh, M. S., Haltiner, A. M., Caylor, L. M., Sepkuty, J. P., and Doherty, M. J. (2013). Does aspirin use make it harder to collect seizures during elective video-EEG telemetry? Epilepsy Behav. 27, 115–117. doi: 10.1016/j.yebeh.2012.12.031
Golarai, G., Greenwood, A. C., Feeney, D. M., and Connor, J. A. (2001). Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J. Neurosci. 21, 8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001
Goodrich, G. S., Kabakov, A. Y., Hameed, M. Q., Dhamne, S. C., Rosenberg, P. A., and Rotenberg, A. (2013). Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J. Neurotrauma 30, 1434–1441. doi: 10.1089/neu.2012.2712
Greenfield, J. G., Love, S., Louis, D. N., and Perry, A. (2008). Greenfield’s Neuropathology, 8th Edn. London: Hodder Arnold.
Guerriero, R. M., Giza, C. C., and Rotenberg, A. (2015). Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 15:27. doi: 10.1007/s11910-015-0545-1
Guo, D., Zeng, L., Brody, D. L., and Wong, M. (2013). Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One 8:e64078. doi: 10.1371/journal.pone.0064078
Gupta, P. K., Sayed, N., Ding, K., Agostini, M. A., Van Ness, P. C., Yablon, S., et al. (2014). Subtypes of post-traumatic epilepsy: clinical, electrophysiological, and imaging features. J. Neurotrauma 31, 1439–1443. doi: 10.1089/neu.2013.3221
Hakimian, S., Kershenovich, A., Miller, J. W., Ojemann, J. G., Hebb, A. O., D’Ambrosio, R., et al. (2012). Long-term outcome of extratemporal resection in posttraumatic epilepsy. Neurosurg. Focus 32:E10. doi: 10.3171/2012.1.FOCUS11329
Halliwell, B. (1999). Antioxidant defence mechanisms: from the beginning to the end (of the beginning). Free Rad. Res. 31, 261–272. doi: 10.1080/10715769900300841
Hammad, A., Westacott, L., and Zaben, M. (2018). The role of the complement system in traumatic brain injury: a review. J. Neuroinflamm. 15:24. doi: 10.1186/s12974-018-1066-z
Hanisch, U., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Haskó, G., Linden, J., Cronstein, B., and Pacher, P. (2008). Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 7, 759–770. doi: 10.1038/nrd2638
Hein, A. M., and O’Banion, M. K. (2009). Neuroinflammation and memory: the role of prostaglandins. Mol. Neurobiol. 40, 15–32. doi: 10.1007/s12035-009-8066-z
Hemphill, M. A., Dabiri, B. E., Gabriele, S., Kerscher, L., Franck, C., Goss, J. A., et al. (2011). A possible role for integrin signaling in diffuse axonal injury. PLoS One 6:e022899. doi: 10.1371/journal.pone.0022899
Hernandez, T. D. (1997). Preventing post-traumatic epilepsy after brain injury: weighing the costs and benefits of anticonvulsant prophylaxis. Trends Pharmacol. Sci. 18, 59–62. doi: 10.1016/S0165-6147(97)89801-X
Herz, J., Filiano, A. J., Smith, A., Yogev, N., and Kipnis, J. (2017). Myeloid cells in the central nervous system. Immunity 46, 943–956. doi: 10.1016/j.immuni.2017.06.007
Hesam, S., Khoshkholgh-Sima, B., Pourbadie, H. G., Babapour, V., Zendedel, M., and Sayyah, M. (2018). Monophosphoryl lipid A and Pam3Cys prevent the increase in seizure susceptibility and epileptogenesis in rats undergoing traumatic brain injury. Neurochem. Res. 43, 1978–1985. doi: 10.1007/s11064-018-2619-3
Heuser, K., Nagelhus, E. A., Taubøll, E., Indahl, U., Berg, P. R., Lien, S., et al. (2010). Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 88, 55–64. doi: 10.1016/j.eplepsyres.2009.09.023
Hickey, R. W., Adelson, P. D., Johnnides, M. J., Davis, D. S., and Yu, Z. (2007). Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr. Res. 62, 271–276. doi: 10.1203/pdr.0b013e3180db2902
Himanen, L., Portin, R., Isoniemi, H., Helenius, H., Kurki, T., and Tenovuo, O. (2006). Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology 66, 187–192. doi: 10.1212/01.wnl.0000194264.60150.d3
Hinzman, J. M., Thomas, T. C., Burmeister, J. J., Quintero, J. E., Huettl, P., Pomerleau, F., et al. (2010). Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: an enzyme-based microelectrode array study. J. Neurotrauma 27, 889–899. doi: 10.1089/neu.2009.1238
Hirano, K., Chen, W. S., Chueng, A. L., Dunne, A. A., Seredenina, T., Filippova, A., et al. (2015). Discovery of GSK2795039, a novel small molecule NADPH Oxidase 2 inhibitor. Antioxid. Redox Signal. 23, 358–374. doi: 10.1089/ars.2014.6202
Ho, D. D., Neumann, A. U., Perelson, A. S., Chen, W., Leonard, J. M., and Markowitz, M. (1995). Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126. doi: 10.1038/373123a0
Hogan, E. (2018). Drug resistant epilepsy and new AEDs: two perspectives. Epilepsy Curr. 18, 304–306. doi: 10.5698/1535-7597.18.5.304
Holbrook, J., Lara-Reyna, S., Jarosz-Griffiths, H., and McDermott, M. (2019). Tumour necrosis factor signalling in health and disease. F1000Research 8:F1000FacultyRev-111. doi: 10.12688/f1000research.17023.1
Hsieh, C. L., Kim, C. C., Ryba, B. E., Niemi, E. C., Bando, J. K., Locksley, R. M., et al. (2013). Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 43, 2010–2022. doi: 10.1002/eji.201243084
Hu, X., Li, P., Guo, Y., Wang, H., Leak, R. K., Chen, S., et al. (2012). Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. doi: 10.1161/strokeaha.112.659656
Huang, Y., Li, Q., Tian, H., Yao, X., Bakina, O., Zhang, H., et al. (2020). MEK inhibitor trametinib attenuates neuroinflammation and cognitive deficits following traumatic brain injury in mice. Am. J. Transl. Res. 12, 6351–6365.
Hunt, R. F., Scheff, S. W., and Smith, B. N. (2009). Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp. Neurol. 215, 243–252. doi: 10.1016/j.expneurol.2008.10.005
Hunt, R. F., Scheff, S. W., and Smith, B. N. (2010). Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J. Neurophysiol. 103, 1490–1500. doi: 10.1152/jn.00957.2009
Hutchinson, P. J., Gupta, A. K., Fryer, T. F., Al-Rawi, P. G., Chatfield, D. A., Coles, J. P., et al. (2002). Correlation between cerebral blood flow, substrate delivery, and metabolism in head injury: a combined microdialysis and triple oxygen positron emission tomography study. J. Cereb. Blood Flow Metab. 22, 735–745. doi: 10.1097/00004647-200206000-00012
Ichkova, A., Rodriguez-Grande, B., Zub, E., Saudi, A., Fournier, M. L., Aussudre, J., et al. (2020). Early cerebrovascular and long-term neurological modifications ensue following juvenile mild traumatic brain injury in male mice. Neurobiol. Dis. 2020:104952. doi: 10.1016/j.nbd.2020.104952
Infanger, D. W., Sharma, R. V., and Davisson, R. L. (2006). NADPH oxidases of the brain: distribution, regulation, and function. Antioxid. Redox Signal. 8, 1583–1596. doi: 10.1089/ars.2006.8.1583
Isokawa, M. (1998). Remodeling dendritic spines in the rat pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 258, 73–76. doi: 10.1016/s0304-3940(98)00848-9
Isokawa, M., and Levesque, M. F. (1991). Increased NMDA responses and dendritic degeneration in human epileptic hippocampal neurons in slices. Neurosci. Lett. 132, 212–216. doi: 10.1016/0304-3940(91)90304-c
Izzy, S., Liu, Q., Fang, Z., Lule, S., Wu, L., Chung, J. Y., et al. (2019). Time-dependent changes in microglia transcriptional networks following traumatic brain injury. Front. Cell. Neurosci. 13:307. doi: 10.3389/fncel.2019.00307
Jackson, E. K., Boison, D., Schwarzschild, M. A., and Kochanek, P. M. (2016). Purines: Forgotten mediators in traumatic brain injury. J. Neurochem. 137, 142–153. doi: 10.1111/jnc.13551
Jarrahi, A., Braun, M., Ahluwalia, M., Gupta, R. V., Wilson, M., Munie, S., et al. (2020). Revisiting traumatic brain injury: from molecular mechanisms to therapeutic interventions. Biomedicines 8:389. doi: 10.3390/biomedicines8100389
Jha, R. M., Kochanek, P. M., and Simard, J. M. (2019). Pathophysiology of cerebral edema in traumatic brain injury. Neuropharmacology 145, 230–246. doi: 10.1016/j.neuropharm.2018.08.004
Jiang, J., Quan, Y., Ganesh, T., Pouliot, W. A., Dudek, F. E., and Dingledine, R. (2013). Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc. Natl. Acad. Sci. U.S.A. 110, 3591–3596. doi: 10.1073/pnas.1218498110
Jiang, M., Lee, C. L., Smith, K. L., and Swann, J. W. (1998). Spine loss and other persistent alterations of hippocampal pyramidal cell dendrites in a model of early-onset epilepsy. J. Neurosci. 18, 8356–8368. doi: 10.1523/jneurosci.18-20-08356.1998
Johnstone, V. P., Shultz, S. R., Yan, E. B., O’Brien, T. J., and Rajan, R. (2014). The acute phase of mild traumatic brain injury is characterized by a distance-dependent neuronal hypoactivity. J. Neurotrauma 31, 1881–1895. doi: 10.1089/neu.2014.3343
Kang, X., Qiu, J., Li, Q., Bell, K. A., Du, Y., Jung, D. W., et al. (2017). Cyclooxygenase-2 contributes to oxidopamine-mediated neuronal inflammation and injury via the prostaglandin E2 receptor EP2 subtype. Sci. Rep. 7:9459. doi: 10.1038/s41598-017-09528-z
Kann, O., Kovács, R., Njunting, M., Behrens, C. J., Otáhal, J., Lehmann, T. N., et al. (2005). Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain 128, 2396–2407. doi: 10.1093/brain/awh568
Karve, I. P., Zhang, M., Habgood, M., Frugier, T., Brody, K. M., Sashindranath, M., et al. (2016). Ablation of Type-1 IFN signaling in hematopoietic cells confers protection following traumatic brain injury. eNeuro 3:ENEURO.0128-15.2016. doi: 10.1523/ENEURO.0128-15.2016
Kaufmann, W. E., Worley, P. F., Taylor, C. V., Bremer, M., and Isakson, P. C. (1997). Cyclooxygenase-2 expression during rat neocortical development and in Rett syndrome. Brain Dev. 19, 25–34. doi: 10.1016/s0387-7604(96)00047-2
Kaur, P., and Sharma, S. (2018). Recent advances in pathophysiology of traumatic brain injury. Curr. Neuropharmacol. 16, 1224–1238. doi: 10.2174/1871530318666180423121833
Kharatishvili, I., Nissinen, J. P., McIntosh, T. K., and Pitkänen, A. (2006). A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 140, 685–697. doi: 10.1016/j.neuroscience.2006.03.012
Kharatishvili, I., and Pitkänen, A. (2010). Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res. 90, 47–59. doi: 10.1016/j.eplepsyres.2010.03.007
Kharlamov, E. A., Lepsveridze, E., Meparishvili, M., Jiang, Q., Zhao, M., Hu, F., et al. (2011). Alterations of GABA(A) and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. 95, 20–34. doi: 10.1016/j.eplepsyres.2011.02.008
Kinoshita, K. (2016). Traumatic brain injury: pathophysiology for neurocritical care. J. Intens. Care 4:29. doi: 10.1186/s40560-016-0138-3
Knoblach, S. M., Nikolaeva, M., Huang, X., Fan, L., Krajewski, S., Reed, J. C., et al. (2002). Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J. Neurotrauma 19, 1155–1170. doi: 10.1089/08977150260337967
Kögel, D., and Prehn, J.H.M. (2000-2013). “Caspase-independent cell death mechanisms,” in Madame Curie Bioscience Database [Internet], (Austin, TX: Landes Bioscience). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK6197/
Korgaonkar, A. A., Li, Y., Sekhar, D., Wang, J., Zhao, K., Zhang, H., et al. (2020). Toll-like Receptor 4 signaling in neurons enhances calcium-permeable α-Amino-3-Hydroxy-5-Methyl-4-isoxazolepropionic acid receptor currents and drives post-traumatic epileptogenesis. Ann. Neurol. 87, 497–515. doi: 10.1002/ana.25698
Korkina, L., Ozben, T., and Saso, L. (2016). Modulation of oxidative stress: pharmaceutical and pharmacological aspects. Oxid. Med. Cell. Longev. 2016:6023417. doi: 10.1155/2016/6023417
Koronyo, Y., Salumbides, B. C., Sheyn, J., Pelissier, L., Li, S., Ljubimov, C., et al. (2015). Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain 138, 2399–2422. doi: 10.1093/brain/awv150
Koza, L., and Linseman, D. A. (2019). Glutathione precursors shield the brain from trauma. Neural Regen. Res. 14:1701. doi: 10.4103/1673-5374.257520
Krämer, T. J., Hack, N., Brühl, T. J., Menzel, L., Hummel, R., Griemert, E.-V., et al. (2019). Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-γ gene expression in acute experimental traumatic brain injury. J. Neuroinflamm. 16:163. doi: 10.1186/s12974-019-1550-0
Kumar, V. (2019). “Macrophages: the potent immunoregulatory innate immune cells,” in Macrophage at the Crossroads of Innate and Adaptive Immunity, ed. K. H. Bhat (London: IntechOpen), doi: 10.5772/intechopen.88013
Kumaria, A., Tolias, C. M., and Burnstock, G. (2008). ATP signalling in epilepsy. Purinerg. Signal. 4, 339–346. doi: 10.1007/s11302-008-9115-1
Ladak, A. A., Enam, S. A., and Ibrahim, M. T. (2019). A review of the molecular mechanisms of traumatic brain injury. World Neurosurg. 131, 126–132. doi: 10.1016/j.wneu.2019.07.039
Lalitha, S., Minz, R. W., and Medhi, B. (2018). Understanding the controversial drug targets in epilepsy and pharmacoresistant epilepsy. Rev. Neurosci. 29, 333–345. doi: 10.1515/revneuro-2017-0043
Lamar, C. D., Hurley, R. A., Rowland, J. A., and Taber, K. H. (2014). Post-traumatic epilepsy: review of risks, pathophysiology, and potential biomarkers. J. Neuropsychiatry Clin. Neurosci. 26, 4–113. doi: 10.1176/appi.neuropsych.260201
Larson, B. E., Stockwell, D. W., Boas, S., Andrews, T., Wellman, G. C., Lockette, W., et al. (2012). Cardiac reactive oxygen species after traumatic brain injury. J. Surg. Res. 173, e73–e81. doi: 10.1016/j.jss.2011.09.056
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., et al. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796. doi: 10.1126/science.1184334
Lewerenz, J., Hewett, S. J., Huang, Y., Lambros, M., Gout, P. W., Kalivas, P. W., et al. (2013). The cystine/glutamate antiporter system xc- in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 18, 522–555. doi: 10.1089/ars.2011.4391
Li, H., Graber, K. D., Jin, S., McDonald, W., Barres, B. A., and Prince, D. A. (2012). Gabapentin decreases epileptiform discharges in a chronic model of neocortical trauma. Neurobiol. Dis. 48, 429–438. doi: 10.1016/j.nbd.2012.06.019
Li, J., and Yuan, J. (2008). Caspases in apoptosis and beyond. Oncogene 27, 6194–6206. doi: 10.1038/onc.2008.297
Li, Q., Li, Q. Q., Jia, J. N., Sun, Q. Y., Zhou, H. H., Jin, W. L., et al. (2019). Baicalein exerts neuroprotective effects in FeCl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front. Pharmacol. 10:638. doi: 10.3389/fphar.2019.00638
Liang, L. P., and Patel, M. (2006). Seizure-induced changes in mitochondrial redox status. Free Rad. Biol. Med. 40, 316–322. doi: 10.1016/j.freeradbiomed.2005.08.026
Licinio, J., and Frost, P. (2000). The neuroimmune-endocrine axis: pathophysiological implications for the central nervous system cytokines and hypothalamus-pituitary-adrenal hormone dynamics. Braz. J. Med. Biol. Res. 33, 1141–1148. doi: 10.1590/s0100-879x2000001000003
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lima, F. D., Souza, M. A., Furian, A. F., Rambo, L. M., Ribeiro, L. R., Martignoni, F. V., et al. (2008). Na+, K+-ATPase activity impairment after experimental traumatic brain injury: relationship to spatial learning deficits and oxidative stress. Behav. Brain Res. 193, 306–310. doi: 10.1016/j.bbr.2008.05.013
Liu, S. J., Zheng, P., Wright, D. K., Zhou, H. H., and Mao, X. Y. (2016). Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139, 1919–1938. doi: 10.1093/brain/aww116
Loane, D. J., Kumar, A., Stoica, B. A., Cabatbat, R., and Faden, A. I. (2014). Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J. Neuropatho. Exper. Neurol. 73, 14–29. doi: 10.1097/NEN.0000000000000021
Lobo, V., Patil, A., Phatak, A., and Chandra, N. (2010). Free radicals, antioxidants and functional foods: impact on human health. Pharm. Rev. 4:118. doi: 10.4103/0973-7847.70902
Louveau, A., Harris, T. H., and Kipnis, J. (2015). Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577. doi: 10.1016/j.it.2015.08.006
Lowenstein, D. H., Thomas, M. J., Smith, D. H., and McIntosh, T. K. (1992). Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 12, 4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., et al. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegen. 12:7. doi: 10.1186/s13024-017-0150-7
Maas, A., Menon, D. K., Adelson, P. D., Andelic, N., Bell, M. J., Belli, A., et al. (2017). Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 16, 987–1048. doi: 10.1016/S1474-4422(17)30371-X
Madrigal, J. L., García-Bueno, B., Caso, J. R., Pérez-Nievas, B. G., and Leza, J. C. (2006). Stress-induced oxidative changes in brain. CNS Neurol. Disord. Drug Targets 5, 561–568. doi: 10.2174/187152706778559327
Maher, P., and Schubert, D. (2000). Signaling by reactive oxygen species in the nervous system. Cell. Mol. Life Sci. 57, 1287–1305. doi: 10.1007/pl00000766
Mahler, B., Carlsson, S., Andersson, T., Adelöw, C., Ahlbom, A., and Tomson, T. (2015). Unprovoked seizures after traumatic brain injury: a population-based case-control study. Epilepsia 56, 1438–1444. doi: 10.1111/epi.13096
Mandal, P. K., Seiler, A., Perisic, T., Kölle, P., Canak, A. B., Förster, H., et al. (2010). System xc- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 285, 22244–22253. doi: 10.1074/jbc.M110.121327
Maqbool, A., Watt, N. T., Haywood, N., Viswambharan, H., Skromna, A., Makava, N., et al. (2020). Divergent effects of genetic and pharmacological inhibition of Nox2 NADPH oxidase on insulin resistance-related vascular damage. Am. J. Physiol. Cell Physiol. 319, C64–C74. doi: 10.1152/ajpcell.00389.2019
Maroso, M., Balosso, S., Ravizza, T., Jiang, G. L., Wang, F., Xue, Y., et al. (2011). Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 8, 304–315. doi: 10.1007/s13311-011-0039-z
Mathur, V., Burai, R., Vest, R. T., Bonanno, L. N., Lehallier, B., Zardeneta, M. E., et al. (2017). Activation of the STING-dependent Type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 96, 1290–1302.e6. doi: 10.1016/j.neuron.2017.11.032
McCullagh, S., and Feinstein, A. (2003). Outcome after mild traumatic brain injury: an examination of recruitment bias. J. Neurol. Neurosurgery Psychiatry 74, 39–43. doi: 10.1136/jnnp.74.1.39
Mckee, A. C., and Daneshvar, D. H. (2015). The neuropathology of traumatic brain injury. Handb. Clini. Neurol. 127, 45–66. doi: 10.1016/B978-0-444-52892-6.00004-0
Mckee, C. A., and Lukens, J. R. (2016). Emerging roles for the immune system in traumatic brain injury. Front. Immunol. 7:556. doi: 10.3389/fimmu.2016.00556
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. doi: 10.1038/nature07201
Milikovsky, D. Z., Weissberg, I., Kamintsky, L., Lippmann, K., Schefenbauer, O., Frigerio, F., et al. (2017). Electrocorticographic dynamics as a novel biomarker in five models of epileptogenesis. J. Neurosci. 37, 4450–4461. doi: 10.1523/JNEUROSCI.2446-16.2017
Milman, A., Zohar, O., Maayan, R., Weizman, R., and Pick, C. G. (2007). DHEAS repeated treatment improves cognitive and behavioral deficits after mild traumatic brain injury. Eur. Neuropsychopharmacol. 18, 181–187. doi: 10.1016/j.euroneuro.2007.05.007
Morimoto, K., and Nakajima, K. (2019). Role of the immune system in the development of the central nervous system. Front. Neurosci. 13:916. doi: 10.3389/fnins.2019.00916
Mukherjee, S., Zeitouni, S., Cavarsan, C. F., and Shapiro, L. A. (2013). Increased seizure susceptibility in mice 30?days after fluid percussion injury. Front. Neurol. 4:28. doi: 10.3389/fneur.2013.00028
Müller, M., Gähwiler, B. H., Rietschin, L., and Thompson, S. M. (1993). Reversible loss of dendritic spines and altered excitability after chronic epilepsy in hippocampal slice cultures. Proc. Natl. Acad. Sci. U.S.A. 90, 257–261. doi: 10.1073/pnas.90.1.257
Multani, P., Myers, R. H., Blume, H. W., Schomer, D. L., and Sotrel, A. (1994). Neocortical dendritic pathology in human partial epilepsy: a quantitative Golgi study. Epilepsia 35, 728–736. doi: 10.1111/j.1528-1157.1994.tb02503.x
Naalt, J., vander Zomeren, A. H., van Sluiter, W. J., and Minderhoud, J. M. (1999). One year outcome in mild to moderate head injury: the predictive value of acute injury characteristics related to complaints and return to work. J. Neurol. Neurosurgery Psychiatry 66, 207–213. doi: 10.1136/jnnp.66.2.207
Nagelhus, E. A., and Ottersen, O. P. (2013). Physiological roles of aquaporin-4 in brain. Physiol. Rev. 93, 1543–1562. doi: 10.1152/physrev.00011.2013
Newell-Rogers, M. K., Rogers, S. K., Tobin, R. P., Mukherjee, S., and Shapiro, L. A. (2020). Antagonism of macrophage migration inhibitory factory (MIF) after traumatic brain injury ameliorates astrocytosis and peripheral lymphocyte activation and expansion. Int. J. Mol. Sci. 21:7448. doi: 10.3390/ijms21207448
Nissinen, J., Andrade, P., Natunen, T., Hiltunen, M., Malm, T., Kanninen, K., et al. (2017). Disease-modifying effect of atipamezole in a model of post-traumatic epilepsy. Epilepsy Res. 136, 18–34. doi: 10.1016/j.eplepsyres.2017.07.005
Norris, G. T., and Kipnis, J. (2019). Immune cells and CNS physiology: microglia and beyond. J. Exper. Med. 216, 60–70. doi: 10.1084/jem.20180199
O’Brien, W. T., Pham, L., Symons, G. F., Monif, M., Shultz, S. R., and Mcdonald, S. J. (2020). The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J. Neuroinflamm. 17:104. doi: 10.1186/s12974-020-01778-5
O’neill, L. A. (1995). Towards an understanding of the signal transduction pathways for interleukin 1. Biochim. Biophys. Acta Mol.Cell Res. 1266, 31–44. doi: 10.1016/0167-4889(94)00217-3
Ozcan, A., and Ogun, M. (2015). “Biochemistry of reactive oxygen and nitrogen species,” in Basic Principles and Clinical Significance of Oxidative Stress, ed. S. J. T. Gowder (London: IntechOpen), doi: 10.5772/61193
Pampliega, O., Domercq, M., Soria, F. N., Villoslada, P., Rodríguez-Antigüedad, A., and Matute, C. (2011). Increased expression of cystine/glutamate antiporter in multiple sclerosis. J. Neuroinflamm. 8:63. doi: 10.1186/1742-2094-8-63
Panatier, A., Theodosis, D. T., Mothet, J. P., Touquet, B., Pollegioni, L., Poulain, D. A., et al. (2006). Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784. doi: 10.1016/j.cell.2006.02.051
Parent, J. M. (2002). The role of seizure-induced neurogenesis in epileptogenesis and brain repair. Epilepsy Res. 50, 179–189. doi: 10.1016/s0920-1211(02)00078-5
Pasciuto, W., Burton, O. T., Roca, C. P., Lagou, V., Rajan, W. D., Theys, T., et al. (2020). Microglia require CD4 T cells to complete the fetal-to-adult transition. Cell 182, 625–640. doi: 10.1016/j.cell.2020.06.026
Patel, D. C., Wallis, G., Dahle, E. J., McElroy, P. B., Thomson, K. E., Tesi, R. J., et al. (2017). Hippocampal TNFα signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. eNeuro 4:ENEURO.0105-17.2017. doi: 10.1523/ENEURO.0105-17.2017
Patel, M. (2004). Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Rad. Biol. Med. 37, 1951–1962. doi: 10.1016/j.freeradbiomed.2004.08.021
Paudel, Y. N., Semple, B. D., Jones, N. C., Othman, I., and Shaikh, M. F. (2019). High mobility group box 1 (HMGB 1) as a novel frontier in epileptogenesis: from pathogenesis to therapeutic approaches. J. Neurochem. 151, 542–557. doi: 10.1111/jnc.14663
Pecorelli, A., Natrella, F., Belmonte, G., Miracco, C., Cervellati, F., Ciccoli, L., et al. (2015). NADPH oxidase activation and 4-hydroxy-2-nonenal/aquaporin-4 adducts as possible new players in oxidative neuronal damage presents in drug-resistant epilepsy. Biochim. Biophys. Acta 1852, 507–519. doi: 10.1016/j.bbadis.2014.11.016
Pekny, M., and Pekna, M. (2016). Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 1862, 483–491. doi: 10.1016/j.bbadis.2015.11.014
Pijet, B., Stefaniuk, M., and Kaczmarek, L. (2019). MMP-9 contributes to dendritic spine remodeling following traumatic brain injury. Neural Plast. 2019, 1–12. doi: 10.1155/2019/3259295
Pijet, B., Stefaniuk, M., Kostrzewska-Ksiezyk, A., Tsilibary, P. E., Tzinia, A., and Kaczmarek, L. (2018). Elevation of MMP-9 levels promotes epileptogenesis after traumatic brain injury. Mol. Neurobiol. 55, 9294–9306. doi: 10.1007/s12035-018-1061-5
Pohlmann-Eden, B., Beghi, E., Camfield, C., and Camfield, P. (2006). The first seizure and its management in adults and children. BMJ 332, 339–342. doi: 10.1136/bmj.332.7537.339
Pokrishevsky, E., Grad, L. I., Yousefi, M., Wang, J., Mackenzie, I. R., and Cashman, N. R. (2012). Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7:e35050. doi: 10.1371/journal.pone.0035050
Popovich, P. G., Stokes, B. T., and Whitacre, C. C. (1996). Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J. Neurosci. Res. 45, 349–363.
Pribiag, H., and Stellwagen, D. (2013). TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J. Neurosci. 33, 15879–15893. doi: 10.1523/JNEUROSCI.0530-13.2013
Puttachary, S., Sharma, S., Stark, S., and Thippeswamy, T. (2015). Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed. Res. Intern. 2015:745613. doi: 10.1155/2015/745613
Puttachary, S., Sharma, S., Verma, S., Yang, Y., Putra, M., Thippeswamy, A., et al. (2016). 1400W, a highly selective inducible nitric oxide synthase inhibitor is a potential disease modifier in the rat kainate model of temporal lobe epilepsy. Neurobiol. Dis. 93, 184–200. doi: 10.1016/j.nbd.2016.05.013
Raheja, A., Sinha, S., Samson, N., Windisch, M., Hutter-Paier, B., Jolkkonen, J., et al. (2016). Serum biomarkers as predictors of long-term outcome in severe traumatic brain injury: analysis from a randomized placebo-controlled Phase II clinical trial. J. Neurosurg. 125, 631–641. doi: 10.3171/2015.6.JNS15674
Raja, M., Chowdhury, J., and Radosevich, J. A. (2018). “Role of phospholipases in cell death,” in Apoptosis and Beyond: The Many Ways Cells Die, ed. J. Radosevich (Hoboken, NJ: Wiley), 395–410.
Ramalingam, M., and Kim, S. J. (2012). Reactive oxygen/nitrogen species and their functional correlations in neurodegenerative diseases. J. Neural Transm. 119, 891–910. doi: 10.1007/s00702-011-0758-7
Rana, A., and Musto, A. E. (2018). The role of inflammation in the development of epilepsy. J. Neuroinflamm. 15:144. doi: 10.1186/s12974-018-1192-7
Ranaivo, H. R., Hodge, J. N., Choi, N., and Wainwright, M. S. (2012). Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J. Neuroinflamm. 9:68. doi: 10.1186/1742-2094-9-68
Rapalino, O., Lazarov-Spiegler, O., Agranov, E., Velan, G. J., Yoles, E., Fraidakis, M., et al. (1998). Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat. Med. 4, 814–821. doi: 10.1038/nm0798-814
Rauch, U. (2004). Extracellular matrix components associated with remodelling processes in brain. Cell. Mol. Life Sci. 61, 2031–2045. doi: 10.1007/s00018-004-4043-x
Raymont, V., Salazar, A., Krueger, F., and Grafman, J. (2011). “Studying injured minds”-the Vietnam head injury study and 40 years of brain injury research. Front. Neurol. 2:15. doi: 10.3389/fneur.2011.00015
Raymont, V., Salazar, A. M., Lipsky, R., Goldman, D., Tasick, G., and Grafman, J. (2010). Correlates of posttraumatic epilepsy 35 years following combat brain injury. Neurology 75, 224–229. doi: 10.1212/WNL.0b013e3181e8e6d0
Redell, J. B., Maynard, M. E., Underwood, E. L., Vita, S. M., Dash, P. K., and Kobori, N. (2020). Traumatic brain injury and hippocampal neurogenesis: Functional implications. Exper. Neurol. 331:113372. doi: 10.1016/j.expneurol.2020.113372
Reyes, R. C., Brennan, A. M., Shen, Y., Baldwin, Y., and Swanson, R. A. (2012). Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 32, 12973–12978. doi: 10.1523/JNEUROSCI.1597-12.2012
Ricciotti, E., and FitzGerald, G. A. (2011). Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000. doi: 10.1161/ATVBAHA.110.207449
Rojas, A., Jiang, J., Ganesh, T., Yang, M. S., Lelutiu, N., Gueorguieva, P., et al. (2014). Cyclooxygenase-2 in epilepsy. Epilepsia 55, 17–25. doi: 10.1111/epi.12461
Rosenfeld, J. V., Maas, A. I., Bragge, P., Morganti-Kossmann, M. C., Manley, G. T., and Gruen, R. L. (2012). Early management of severe traumatic brain injury. Lancet 380, 1088–1098. doi: 10.1016/S0140-6736(12)60864-2
Röszer, T. (2015). Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Med. Inflamm. 2015:816460. doi: 10.1155/2015/816460
Rowe, R. K., Harrison, J. L., Zhang, H., Hesson, D. P., O’Hara, B. F., Greene, M. I., et al. (2018). Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J. Neuroinflamm. 15:154. doi: 10.1186/s12974-018-1200-y
Rowley, S., and Patel, M. (2013). Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Rad. Biol. Med. 62, 121–131. doi: 10.1016/j.freeradbiomed.2013.02.002
Saletti, P. G., Ali, I., Casillas-Espinosa, P. M., Semple, B. D., Lisgaras, C. P., Moshe, S. L., et al. (2019). In search of antiepileptogenic treatments for post-traumatic epilepsy. Neurobiol. Dis. 123, 86–99. doi: 10.1016/j.nbd.2018.06.017
Salter, M. W., and Kalia, L. V. (2004). Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328. doi: 10.1038/nrn1368
Sanz, P., and Garcia-Gimeno, M. A. (2020). Reactive Glia inflammatory signaling pathways and epilepsy. Intern. J. Mol. Sci. 21:4096. doi: 10.3390/ijms21114096
Saraiva, A. L., Ferreira, A. P., Silva, L. F., Wilcock, D. M., Goulding, D., Neltner, J. H., et al. (2012). Creatine reduces oxidative stress markers but does not protect against seizure susceptibility after severe traumatic brain injury. Brain Res. Bull. 87, 180–186. doi: 10.1016/j.brainresbull.2011.10.010
Scallan, J., Huxley, V. H., and Korthuis, R. J. (2010). “Capillary fluid exchange: Regulation functions and pathology,” in Colloquium Lectures on Integrated Systems Physiology: From Molecules to Function, eds D. N. Granger and J. P. Granger (San Rafael, CA: Morgan & Claypool Publishers).
Scanlon, S. T. (2019). A myeloid cell atlas of neuroinflammation. Science 363, 360–362. doi: 10.1126/science.363.6425.360-l
Schmidt, D., Friedman, D., and Dichter, M. A. (2014). Anti-epileptogenic clinical trial designs in epilepsy: issues and options. Neurotherapeutics 11, 401–411. doi: 10.1007/s13311-013-0252-z
Schwartzkroin, P. A., Wenzel, H. J., Lyeth, B. G., Watterson, D. M., and Van, E. L. J. (2010). Does ketogenic diet alter seizure sensitivity and cell loss following fluid percussion injury? Epilepsy Res. 92, 74–84. doi: 10.1016/j.eplepsyres.2010.08.009
Semple, B. D., O’Brien, T. J., Gimlin, K., Casillas-Espinosa, P. M., Webster, K. M., Petrou, S., et al. (2017). Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J. Neurosci. 37, 7864–7877. doi: 10.1523/JNEUROSCI.0982-17.2017
Serhan, C. N. (2017). Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. 31, 1273–1288. doi: 10.1096/fj.201601222R
Shanlin, R. J., Sole, M. J., Rahimifar, M., Tator, C. H., and Factor, S. M. (1988). Increased intracranial pressure elicils hypertension, increased sympathetic activity, electrocardiographic abnormalities and myocardial damage in rats. J. Am. Coll. Cardiol. 12, 727–736. doi: 10.1016/s0735-1097(88)80065-2
Sharma, S., Carlson, S., Puttachary, S., Sarkar, S., Showman, L., Putra, M., et al. (2018). Role of the Fyn-PKCδ signaling in SE-induced neuroinflammation and epileptogenesis in experimental models of temporal lobe epilepsy. Neurobiol. Dis. 110, 102–121. doi: 10.1016/j.nbd.2017.11.008
Sharma, S. K., and Naidu, G. (2016). The role of danger-associated molecular patterns (DAMPs) in trauma and infections. J. Thorac. Dis. 8, 1406–1409. doi: 10.21037/jtd.2016.05.22
Shechter, R., London, A., Varol, C., Raposo, C., Cusimano, M., Yovel, G., et al. (2009). Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 6:e1000113. doi: 10.1371/journal.pmed.1000113
Shi, H., Hua, X., Kong, D., Stein, D., and Hua, F. (2019). Role of toll-like receptor mediated signaling in traumatic brain injury. Neuropharmacology 145, 259–267. doi: 10.1016/j.neuropharm.2018.07.022
Shi, W., Zhao, W., Shen, A., Shao, B., Wu, X., Yang, J., et al. (2010). Traumatic brain injury induces an up-regulation of Hs1-associated protein X-1 (Hax-1) in rat brain cortex. Neurochem. Res. 36, 375–382. doi: 10.1007/s11064-010-0332-y
Shlosberg, D., Benifla, M., Kaufer, D., and Friedman, A. (2010). Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 6, 393–403. doi: 10.1038/nrneurol.2010.74
Silverman, M. N., Pearce, B. D., Biron, C. A., and Miller, A. H. (2005). Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 18, 41–78. doi: 10.1089/vim.2005.18.41
Smith, B. N. (2016). How and why study posttraumatic epileptogenesis in animal models? Epilepsy Curr. 16, 393–396. doi: 10.5698/1535-7511-16.6.393
Smith, C. (2013). Review: the long-term consequences of microglial activation following acute traumatic brain injury. Neuropathol. Appl. Neurobiol. 39, 35–44. doi: 10.1111/nan.12006
Smith, R. E., Ozben, T., and Saso, L. (2019). Modulation of oxidative stress: pharmaceutical and pharmacological aspects 2018. Oxid. Med. Cell. Longev. 2019:6380473. doi: 10.1155/2019/6380473
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sookplung, P., Siriussawakul, A., Malakouti, A., Sharma, D., Wang, J., Souter, M. J., et al. (2011). Vasopressor use and effect on blood pressure after severe adult traumatic brain injury. Neurocrit Care 15, 46–54. doi: 10.1007/s12028-010-9448-9
Sridharan, R., Cameron, A. R., Kelly, D. J., Kearney, C. J., and O’Brien, F. J. (2015). Biomaterial based modulation of macrophage polarization: a review and suggested design principles. Mater. Today 18, 313–325. doi: 10.1016/j.mattod.2015.01.019
Stamatovic, S. M., Keep, R. F., and Andjelkovic, A. V. (2008). Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr. Neuropharmacol. 6, 179–192. doi: 10.2174/157015908785777210
Steinhäuser, C., Grunnet, M., and Carmignoto, G. (2016). Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience 323, 157–169. doi: 10.1016/j.neuroscience.2014.12.047
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Stoica, B. A., and Faden, A. I. (2010). Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7, 3–12. doi: 10.1016/j.nurt.2009.10.023
Streit, W. J., Mrak, R. E., and Griffin, W. S. T. (2004). Microglia and neuroinflammation: a pathological perspective. J. Neuroinflamm. 1, 1–4. doi: 10.1186/1742-2094-1-14
Stück, E. D., Christensen, R. N., Huie, J. R., O’Brien, T. J., Rajan, R., Shultz, S. R., et al. (2012). Tumor necrosis factor alpha mediates GABA(A) receptor trafficking to the plasma membrane of spinal cord neurons in vivo. Neural Plast. 2012:261345. doi: 10.1155/2012/261345
Sun, D. (2014). The potential of endogenous neurogenesis for brain repair and regeneration following traumatic brain injury. Neural Regen. Res. 9, 688–692. doi: 10.4103/1673-5374.131567
Sun, M., Honey, C. R., Berk, C., Wong, N. L., and Tsui, J. K. (2003). Regulation of aquaporin-4 in a traumatic brain injury model in rats. J. Neurosurg. 98, 565–569. doi: 10.3171/jns.2003.98.3.0565
Swartz, B. E., Houser, C. R., Tomiyasu, U., Walsh, G. O., DeSalles, A., Rich, J. R., et al. (2006). Hippocampal cell loss in posttraumatic human epilepsy. Epilepsia 47, 1373–1382. doi: 10.1111/j.1528-1167.2006.00602.x
Szaflarski, J. P., Nazzal, Y., and Dreer, L. E. (2014). Post-traumatic epilepsy: current and emerging treatment options. Neuropsychiatr. Dis. Treat. 10, 1469–1477. doi: 10.2147/NDT.S50421
Szaflarski, J. P., Sangha, K. S., Lindsell, C. J., and Shutter, L. A. (2010). Prospective, randomized, single-blinded comparative trial of intravenous levetiracetam versus phenytoin for seizure prophylaxis. Neurocrit. Care 12, 165–172. doi: 10.1007/s12028-009-9304-y
Szu, J. I., Chaturvedi, S., Patel, D. D., and Binder, D. K. (2020). Aquaporin-4 dysregulation in a controlled cortical impact injury model of posttraumatic epilepsy. Neuroscience 428, 140–153. doi: 10.1016/j.neuroscience.2019.12.006
Ta, T., Dikmen, H. O., Schilling, S., Chausse, B., Lewen, A., Hollnagel, J., et al. (2019). Priming of microglia with IFN-γ slows neuronal gamma oscillations in situ. Proc. Natl. Acad. Sci. U.S.A. 116, 4637–4642. doi: 10.1073/pnas.1813562116
Takemiya, T., Maehara, M., Matsumura, K., Yasuda, S., Sugiura, H., and Yamagata, K. (2006). Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci. Res. 56, 103–110. doi: 10.1016/j.neures.2006.06.003
Tayel, S. S., Helmy, A. A., Ahmed, R., Esmat, G., Hamdi, N., and Abdelaziz, A. I. (2013). Progesterone suppresses interferon signaling by repressing TLR-7 and MxA expression in peripheral blood mononuclear cells of patients infected with hepatitis C virus. Archiv. Virol. 158, 1755–1764. doi: 10.1007/s00705-013-1673-z
Tehse, J., and Taghibiglou, C. (2019). The overlooked aspect of excitotoxicity: glutamate-independent excitotoxicity in traumatic brain injuries. Eur. J. Neurosci. 49, 1157–1170. doi: 10.1111/ejn.14307
Temkin, N. R. (2001). Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia 42, 515–524. doi: 10.1046/j.1528-1157.2001.28900.x
Ting, A. T., and Bertrand, M. (2016). More to life than NF-κB in TNFR1 Signaling. Trends Immunol. 37, 535–545. doi: 10.1016/j.it.2016.06.002
Tobin, R. P., Mukherjee, S., Kain, J. M., Rogers, S. K., Henderson, S. K., Motal, H. L., et al. (2014). Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol. Commun. 2, 1–10. doi: 10.1186/s40478-014-0143-5
Tomkins, O., Feintuch, A., Benifla, M., Cohen, A., Friedman, A., and Shelef, I. (2011). Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc. Psychiatry Neurol. 2011:765923. doi: 10.1155/2011/765923
Tran, N. D., Kim, S., Vincent, H. K., Rodriguez, A., Hinton, D. R., Bullock, M. R., et al. (2010). Aquaporin-1-mediated cerebral edema following traumatic brain injury: effects of acidosis and corticosteroid administration. J. Neurosurg. 112, 1095–1104. doi: 10.3171/2009.8.jns081704
Trinchieri, G. (2012). Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 30, 677–706. doi: 10.1146/annurev-immunol-020711-075008
Trotti, D., Danbolt, N. C., and Volterra, A. (1998). Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 19, 328–334. doi: 10.1016/s0165-6147(98)01230-9
Ucciferri, C., Falasca, K., Mancino, P., De Tullio, D., Pizzigallo, E., and Vecchiet, J. (2007). High dose of erythropoietin in management of interferon/ribavirin induced anemia. Hepatogastroenterology 54, 2181–2183.
Uggenti, C., and Crow, Y. J. (2018). Taking the STING out of inflammation. Nat. Rev. Rheumatol. 14, 508–509. doi: 10.1038/s41584-018-0071-z
Varvel, N. H., Jiang, J., and Dingledine, R. (2014). Candidate drug targets for prevention or modification of epilepsy. Annu. Rev. Pharmacol. Toxicol. 55, 229–247. doi: 10.1146/annurev-pharmtox-010814-124607
Vespa, P., Prins, M., Ronne-Engstrom, E., Caron, M., Shalmon, E., Hovda, D. A., et al. (1998). Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: a microdialysis study. J. Neurosurg. 89, 971–982. doi: 10.3171/jns.1998.89.6.0971
Vespa, P. M., McArthur, D. L., Xu, Y., Eliseo, M., Etchepare, M., Dinov, I., et al. (2010). Nonconvulsive seizures after traumatic brain injury are associated with hippocampal atrophy. Neurology 75, 792–798. doi: 10.1212/WNL.0b013e3181f07334
Vezzani, A., Conti, M., Luigi, A. D., Ravizza, T., Moneta, D., Marchesi, F., et al. (1999). Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J. Neurosci. 19, 5054–5065. doi: 10.1523/jneurosci.19-12-05054.1999
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Vezzani, A., Friedman, A., and Dingledine, R. J. (2012). The role of inflammation in epileptogenesis. Neuropharmacology 69, 16–24. doi: 10.1016/j.neuropharm.2012.04.004
Vezzani, A., Fujinami, R. S., White, H. S., Preux, P., Blümcke, I., Sander, J. W., et al. (2015). Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234. doi: 10.1007/s00401-015-1481-5
Viviani, B., Bartesaghi, S., Gardoni, F., Beynon, S., Schmidt, S. F., Kaye, A. H., et al. (2003). Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
Waldbaum, S., and Patel, M. (2010). Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 88, 23–45. doi: 10.1016/j.eplepsyres.2009.09.020
Wallraff, A., Köhling, R., Heinemann, U., Theis, M., Willecke, K., and Steinhäuser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006
Wang, F., Wang, X., Shapiro, L. A., Sun, M., Semple, B. D., Ozturk, E., et al. (2017). NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct. Funct. 222, 1543–1556. doi: 10.1007/s00429-016-1292-z
Wang, J. Y., Bakhadirov, K., Devous, M. D., Abdi, H., McColl, R., Moore, C., et al. (2008). Diffusion tensor tractography of traumatic diffuse axonal injury. Archiv. Neurol. 65, 619–626. doi: 10.1001/archneur.65.5.619
Weber, J. T. (2012). Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 3:60. doi: 10.3389/fphar.2012.00060
Webster, K. M., Sun, M., Crack, P., O’Brien, T. J., Shultz, S. R., and Semple, B. D. (2017). Inflammation in epileptogenesis after traumatic brain injury. J. Neuroinflamm. 14, 1–17. doi: 10.1186/s12974-016-0786-1
Weissberg, I., Wood, L., Kamintsky, L., Vazquez, O., Milikovsky, D. Z., Alexander, A., et al. (2015). Albumin induces excitatory synaptogenesis through astrocytic TGF-β/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol. Dis. 78, 115–125. doi: 10.1016/j.nbd.2015.02.029
Winston, C. N., Chellappa, D., Wilkins, T., Barton, D. J., Washington, P. M., Loane, D. J., et al. (2013). Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J. Neurotrauma 30, 1966–1972. doi: 10.1089/neu.2013.2960
Wofford, K. L., Loane, D. J., and Cullen, D. K. (2019). Acute drivers of neuroinflammation in traumatic brain injury. Neural. Regen. Res. 14, 1481–1489. doi: 10.4103/1673-5374.255958
Wong, H., and Hoeffer, C. (2018). Maternal IL-17A in autism. Exper. Neurol. 299, 228–240. doi: 10.1016/j.expneurol.2017.04.010
Wong, M. (2005). Modulation of dendritic spines in epilepsy: cellular mechanisms and functional implications. Epilepsy Behav. 7, 569–577. doi: 10.1016/j.yebeh.2005.08.007
Wong, M., and Guo, D. (2013). Dendritic spine pathology in epilepsy: cause or consequence? Neuroscience 251, 141–150. doi: 10.1016/j.neuroscience.2012.03.048
Wu, Y., Wu, H., Guo, X., Pluimer, B., and Zhao, Z. (2020). Blood-brain barrier dysfunction in mild traumatic brain injury: evidence from preclinical murine models. Front. Physiol. 11:1030. doi: 10.3389/fphys.2020.01030
Wynn, T. A., and Vannella, K. M. (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. doi: 10.1016/j.immuni.2016.02.015
Yoon, B. E., Jo, S., Woo, J., Lee, J. H., Kim, T., Kim, D., et al. (2011). The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol. Brain 4:42. doi: 10.1186/1756-6606-4-42
Yu, Z.-Q., and Zha, J.-H. (2012). Genetic ablation of toll-like receptor 2 reduces secondary brain injury caused by cortical contusion in mice. Ann. Clin. Lab. Sci. 42, 26–33.
Zanier, E. R., Lee, S. M., Vespa, P. M., Giza, C. C., and Hovda, D. A. (2003). Increased hippocampal CA3 vulnerability to low-level kainic acid following lateral fluid percussion injury. J. Neurotrauma 20, 409–420. doi: 10.1089/089771503765355496
Zauner, A., Daugherty, W. P., Bullock, M. R., and Warner, D. S. (2002). Brain oxygenation and energy metabolism: part I-biological function and pathophysiology. Neurosurgery 51, 289–302. doi: 10.1227/00006123-200208000-00003
Zehendner, C. M., Sebastiani, A., Hugonnet, A., Bischoff, F., Luhmann, H. J., and Thal, S. C. (2015). Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci. Rep. 5:13497. doi: 10.1038/srep13497
Zetterström, M., Lundkvist, J., Malinowski, D., Eriksson, G., and Bartfai, T. (1998). Interleukin-1-mediated febrile responses in mice and inter-leukin-1 beta activation of NFB in mouse primary astrocytes, involves the interleukin-1 receptor accessory protein. Eur. Cytokine Netw. 9, 131–138.
Zhang, L., Liu, J., Cheng, C., Yuan, Y., Yu, B., Shen, A., et al. (2012). The neuroprotective effect of pyrroloquinoline quinone on traumatic brain injury. J. Neurotrauma 29, 851–864. doi: 10.1089/neu.2011.1882
Zhang, X., Chen, Y., Jenkins, L. W., Kochanek, P. M., and Clark, R. S. B. (2005). Bench-to-bedside review: apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care 9, 66–75. doi: 10.1186/cc2950
Zhou, Y., Shao, A., Yao, Y., Tu, S., Deng, Y., and Zhang, J. (2020). Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 18:62. doi: 10.1186/s12964-020-00549-2
Zhu, M., Chen, J., Guo, H., Ding, L., Zhang, Y., and Xu, Y. (2018). High mobility group Protein B1 (HMGB1) and Interleukin-1β as prognostic biomarkers of epilepsy in children. J. Child Neurol. 33, 909–917. doi: 10.1177/0883073818801654
Zhu, Q., Li, N., Han, Q., Zhang, P., Yang, C., Zeng, X., et al. (2013). Statin therapy improves response to interferon alfa and ribavirin in chronic hepatitis C: a systematic review and meta-analysis. Antiv. Res. 98, 373–379. doi: 10.1016/j.antiviral.2013.04.009
Keywords: traumatic brain injury, post-traumatic epilepsy, excitotoxicity, neuroinflammation, oxidative stress, neurodegeneration, immune response, clinical management
Citation: Sharma S, Tiarks G, Haight J and Bassuk AG (2021) Neuropathophysiological Mechanisms and Treatment Strategies for Post-traumatic Epilepsy. Front. Mol. Neurosci. 14:612073. doi: 10.3389/fnmol.2021.612073
Received: 30 September 2020; Accepted: 26 January 2021;
Published: 23 February 2021.
Edited by:
Tobias Engel, Royal College of Surgeons in Ireland, IrelandReviewed by:
Edward Haig Beamer, Royal College of Surgeons in Ireland, IrelandCopyright © 2021 Sharma, Tiarks, Haight and Bassuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander G. Bassuk, YWxleGFuZGVyLWJhc3N1a0B1aW93YS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.