 Lučka Bibič
Lučka Bibič Leanne Stokes
Leanne Stokes- School of Pharmacy, University of East Anglia, Norwich, United Kingdom
The P2X7 receptor (P2X7) is a cell surface ligand-gated ion channel, activated by its physiological nucleotide agonist ATP and a synthetic analog (BzATP). However, it has also been suggested that there may be structurally unrelated, non-nucleotide agonists such as the amyloidogenic β peptide. Here we aimed to reassess the effect of amyloid β peptides in various in vitro cell models, namely HEK293 overexpressing human P2X7, the microglial BV-2 cell line, and BV-2 cells lacking P2X7. We measured YO-PRO-1 dye uptake in response to full-length amyloid β peptide (1–42) or the shorter amyloid β peptide (25–35) and there was a concentration-dependent increase in YO-PRO-1 dye uptake in HEK-hP2X7 cells. However, these amyloid β peptide-induced increases in YO-PRO-1 dye uptake were also identical in non-transfected HEK-293 cells. We could observe small transient increases in [Ca2+]i induced by amyloid β peptides in BV-2 cells, however these were identical in BV-2 cells lacking P2X7. Furthermore, our metabolic viability and LDH release experiments suggest no significant change in viability or cell membrane damage in HEK-hP2X7 cells. In the BV-2 cells we found that high concentrations of amyloid β peptides (1–42) and (25–35) could reduce cell viability by up to 35% but this was also seen in BV-2 cells lacking P2X7. We found no evidence of LDH release by amyloid β peptides. In summary, we found no evidence that amyloid β peptides act as agonists of P2X7 in our in vitro models. Our study raises the possibility that amyloid β peptides simply mimic features of P2X7 activation.
Introduction
The most prevalent cause of dementia is Alzheimer’s disease, a fatal neurodegenerative disorder that is characterized by a progressive cognitive and functional impairment and memory loss (Heppner et al., 2015). In the field of Alzheimer’s disease research, the amyloid cascade hypothesis has been the major hallmark of pathogenesis. This states that the generation of amyloid plaques, primarily composed of the amyloid-β peptide (Aβ), represents the initial event triggering neurobiological dysfunction (Hardy and Selkoe, 2002). Over two decades of research (Hardy and Higgins, 1992) have recently revealed many layers of complexity (Lee et al., 2018), however, the bulk of data still supports the role of the Aβ peptide as the primary initiator of Alzheimer’s disease pathogenesis (Musiek and Holtzman, 2015; Cheignon et al., 2018).
Previous data have indicated that the immune system may have a role in Alzheimer’s disease and that activated microglia have been observed in patients (Sarlus and Heneka, 2017). Microglia, the resident macrophages of the CNS, alter their morphology and phenotype to adopt a so-called activated state in response to neurophysiological brain insults (Heneka et al., 2015; Sarlus and Heneka, 2017). Morphologically activated microglia are believed to contribute to the progression of Alzheimer’s disease via receptors such as the scavenger receptor CD36 (El Khoury et al., 1996), α6β1 integrins (Koenigsknecht and Landreth, 2004), the formyl peptide receptor-like protein (Le et al., 2001), TLR2 (Chen et al., 2006), TLR4 (Michaud et al., 2013) and the TLR-interacting molecule CD14 (Fassbender et al., 2004). As a result, this leads to inflammatory mediator secretion and microglial responsiveness to injury as comprehensively reviewed by Sarlus and Heneka (2017). However, another receptor that is also highly expressed by microglial cells is P2X7, an ATP-gated ion channel (Chessell et al., 1997; Ferrari et al., 1997; Bartlett et al., 2013). In recent years, several studies suggested the participation of P2X7 in Aβ-mediated brain damage (Haughey and Mattson, 2003; Parvathenani et al., 2003; Rampe et al., 2004; McLarnon et al., 2006). Furthermore, Sanz et al. (2009) suggested that P2X7 may participate in microglia activation by Aβ peptides and it has since been proposed that ATP might not be the only endogenous agonist for P2X7 receptors (Di Virgilio et al., 2018). This may, in turn, open new avenues for the development of novel therapies for Alzheimer’s disease. Intrigued by this proposal of alternative agonists, we set out to determine if Aβ could act as a P2X7 agonist and investigate the effect of positive allosteric modulators of P2X7 that we have previously characterized (Helliwell et al., 2015). However, we have been unable to validate the results that suggest that Aβ-induced responses require the expression of P2X7. Based on our data, we conclude that Aβ peptides may not directly cause P2X7-dependent signaling in microglial cells.
Materials and Methods
Cell Culture
Microglial BV-2 and BV-2 P2X7-deficient cells were maintained in DMEM/F12 with L-glutamine (Gibco 11320-074, Fisher Scientific, United Kingdom), containing 10% (v/v) FBS (Gibco, US origin), penicillin and streptomycin (Fisher Scientific, United Kingdom). HEK-293 cells were maintained under the same media conditions. HEK-293 cells stably expressing human P2X7 were generated previously (Bhaskaracharya et al., 2014) and maintained under similar conditions with the addition of 400 μg/ml geneticin (Fisher Scientific, United Kingdom). All cells were maintained at 37°C with 5% CO2 in a humidified incubator.
Materials
Aβ peptides corresponding to human Aβ amino acids Aβ25–35, Aβ35–25 (inactive scrambled peptide) and Aβ1–42 were purchased from GenScript, United States and prepared as 10 mM stock solutions in either water or DMSO. Apyrase (an ATP-hydrolyzing enzyme), ATP and BzATP were purchased from Sigma Aldrich, United Kingdom. Stock solutions of P2X7 antagonists AZ10606120 and JNJ47965567 (Tocris Bioscience, Bio-Techne, United Kingdom) were prepared in DMSO (10 mM) and stocks were kept frozen at −20°C.
Measurements of Intracellular Ca2+
The cells were seeded at 2 × 105 cells/well (100 μl) and plated on poly-D-Lysine coated 96-well plates (Nunc 167008, Fisher Scientific, United Kingdom) and used for experiments 24 h after plating. Cells were loaded with the indicator dye Fura-2-AM using a concentration of 2 μM (HelloBio, United Kingdom) in HBSS for 45 min at 37°C. Loading buffer was removed and replaced with 180 μl of assay buffer containing 147 mM NaCl, 2 mM KCl, 0.1 mM CaCl2, 13 mM Glucose, 10 mM HEPES; pH 7.35. The plate was warmed in the FlexStation 3 (Molecular Devices, United Kingdom) for 10 min before recording using excitation wavelengths 340 and 380 nm and emission at 520 nm (Bibic et al., 2019).
Agonists (10x concentration) were automatically injected at 30 s using the Flex function. Data was converted to Fura-2 ratio (340/380) and normalized using a zero baseline correction. Area under the curve was calculated and plotted. All data was collected in triplicate.
Measurements of YO-PRO-1 Dye Uptake
Impermeant dye uptake was measured with the extracellular fluorescent tracer YO-PRO-1, a probe that enters the cells through P2X7 activation-induced pores and emits fluorescence when it binds DNA (Idziorek et al., 1995). A solution of 2 μM YO-PRO-1 in assay buffer (see above) was added to wells, and the 96-well plate was placed at 37°C for 10 min. The fluorescence signal in response to drug injection was then measured using a Flexstation 3 (Molecular Devices) as described previously (Bidula et al., 2019; Dhuna et al., 2019). Excitation wavelength was 490 nm and emission was measured at 520 nm. Machine settings include bottom read fluorescence, PMT medium, 6 reads/well with a sample interval of 3.5 s. RFU data was normalized using a zero baseline correction and area under the curve was calculated and plotted. All data was collected from triplicate wells in each independent experiment.
Cell Viability Measurements
Cells were plated at 2 × 104/well (in a volume of 100 μl) and plated on non-coated 96-well plates (Fisher Scientific, United Kingdom) in culture medium containing 1% FBS 24 h before stimulation. Stimuli included concentrations of Aβ peptides (10–60 μM), ATP at 500 μM and 3 mM concentrations, as well as staurosporine (5 μM), and the vehicle control (DMSO). Following incubation with different stimuli, resazurin (0.1 mg/ml in PBS, Sigma Aldrich) was added to cells for final 2 h at 37°C. The plate was then read on a Flexstation 3 plate reader using an excitation wavelength of 570 nm and emission wavelength of 600 nm (Bidula et al., 2019).
LDH Release Assay
Lactate dehydrogenase (LDH) release into cell culture supernatants was measured using an LDH assay kit (Pierce, Fisher Scientific, United Kingdom) following the manufacturer’s instructions. Control cells were lysed with the lysis buffer provided to harvest total intracellular LDH. For measuring LDH release, cells were cultured in 96-well plates, stimuli applied for 24 h, and supernatants were collected. Absorbance of duplicate 50 μl aliquots of supernatants were measured on a Flexstation 3 plate reader at 490 nm.
Data Analysis and Statistics
All results are expressed as mean ± SD using data from collated experiments. All experimental data was collected from triplicate wells. All data for cell viability and cytotoxicity were obtained as relative fluorescence units (RFU) and are expressed as a percentage of the negative control (culture medium). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison post-hoc test (GraphPad Prism v8). Statistically significant differences from controls are indicated by ∗ using p < 0.05 as a threshold.
Results
A study using N13 microglial cells indicated that Aβ peptide, both the full-length 1–42 and the shorter 25–35, may induce responses similar to the activation of P2X7 (Sanz et al., 2009). We show that stimulation of BV-2 microglial cells with ATP and BzATP, both known agonists for P2X7, led to [Ca2+]i increases (Figures 1A,C). Using a P2X7-deficient BV-2 cell line generated by CRISPR/Cas9 gene editing (Dhuna et al., 2019), the [Ca2+]i responses were different to the parental BV-2 line, with the response to BzATP almost completely abolished (Figures 1B,D). We first tested Aβ25–35 and found that this peptide did induce a transient increase in [Ca2+]i increase in BV-2 cells (Figures 1A,E) that displayed a concentration-dependent effect. We observed a similar Aβ25–35 -induced [Ca2+]i increase in the P2X7-deficient BV-2 cells (Figures 1B,F). Furthermore, we show that the Aβ25–35 induced [Ca2+]i increases were not affected by the P2X7-selective antagonist AZ10606120 whereas the ATP-induced [Ca2+]i increase was reduced 30% by AZ10606120 (Figure 1A) in BV-2 cells. As expected, there was no significant effect of AZ10606120 in P2X7-deficient BV-2 cells (Figure 1B). The inactive scrambled Aβ35–25 peptide did not increase [Ca2+]i to the same level as Aβ25–35 in BV-2 microglial cells, whether expressing P2X7 (Figure 1G) or not (Figure 1H). Some evidence suggests that the solvent, such as DMSO, acetonitrile, and water, may influence the self-assembly and thus the biological activity of Aβ peptides (Busciglio et al., 1992; Shen and Murphy, 1995; Zagorski et al., 1999). In our experiments Aβ25–35 was prepared in either DMSO or water (Figure 1) and peptide dissolved in DMSO displayed higher Aβ25–35 -induced [Ca2+]i increases (Figures 1E,F and Supplementary Figure S1).
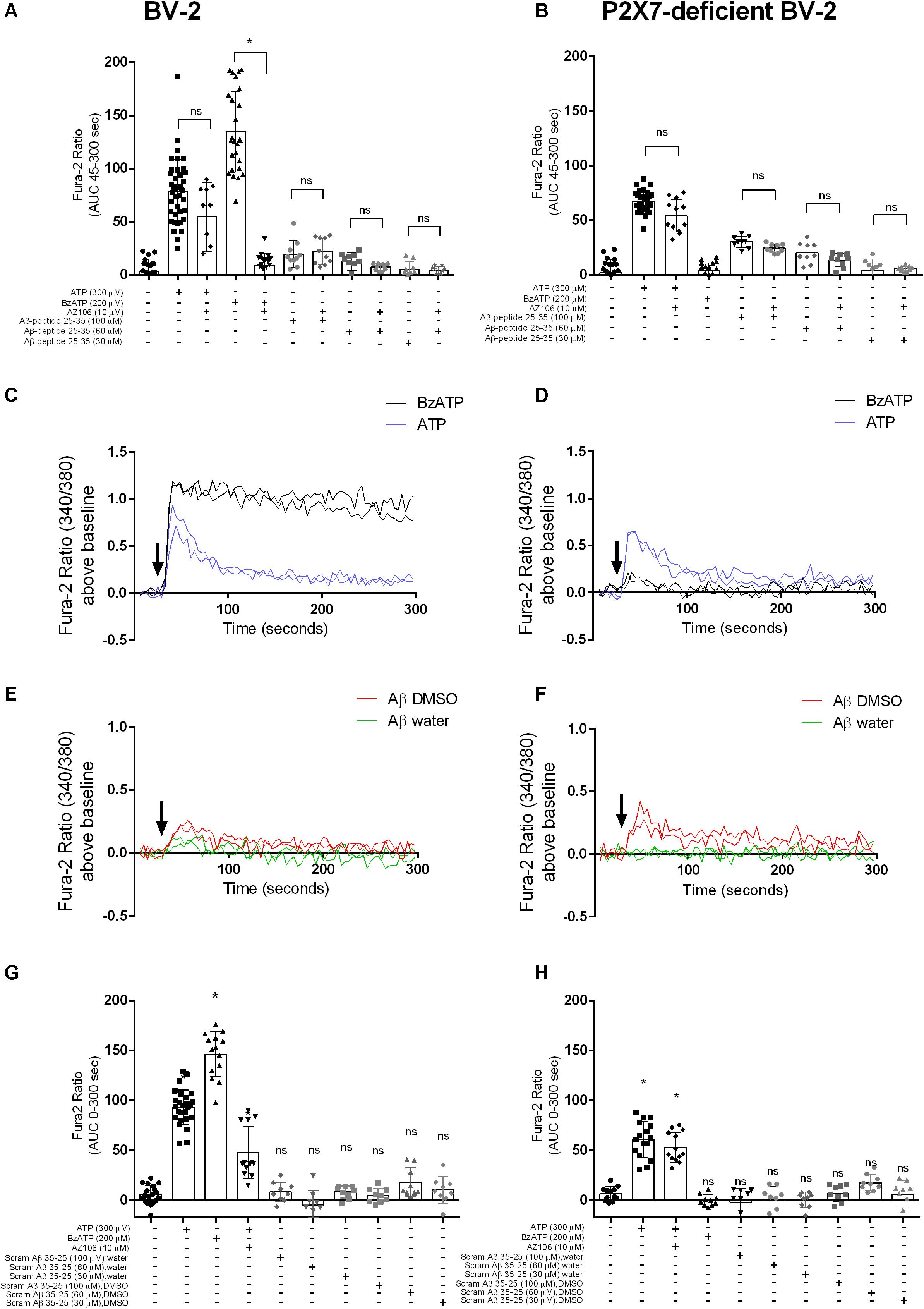
Figure 1. Aβ25–35 induced similar [Ca2+]i responses in microglial BV-2 cells in the presence/absence of P2X7. BV-2 cells were loaded with Fura-2AM (2 μM) and challenged with the Aβ25–35 peptides (DMSO) in the concentration range 30–100 μM in either BV-2 (A) or P2X7-deficient BV-2 (B). One-way ANOVA was performed with Tukey’s multiple comparison test where * indicates a significant difference to the paired control (with AZ1060610) and ns denotes no significant difference. Each symbol represents a single well and data has been collated from all independent experiments. Kinetics of the [Ca2+]i response in either BV-2 (C,E) or P2X7-deficient BV-2 (D,F) are plotted from a representative experiment. Similarly, BV-2 cells were loaded with Fura-2AM (2 μM) and challenged with the inactive scrambled Aβ35–25 peptide in the concentration range 30 μM -100 μM in either BV-2 (G) or P2X7-deficient BV-2 (H). AZ10606120 is a selective antagonist of P2X7, and BzATP is a synthetic agonist for P2X7 receptors. Experiments were repeated five times with triplicates on each plate, and the results are presented as area under curve (mean ± SD). One-way ANOVA was performed with Tukey’s multiple comparison test where * indicates a significant difference (P < 0.05) to the control (buffer alone) and ns denotes no significant difference.
We next tested whether Aβ25–35 would induce responses in a stable HEK-293 cell line over-expressing human P2X7 (HEK-hP2X7). We used a standard YO-PRO-1 dye uptake assay to assess the P2X7 large pore formation (Bibic et al., 2019). We observed that Aβ25–35 (Figures 2A,B), but not the inactive scrambled Aβ35–25 (Figure 2C), induced a significant YO-PRO-1 uptake over the concentration range 30–100 μM. Notably, this Aβ25–35-induced dye uptake was not abrogated by the P2X7-specific antagonist AZ10606120 despite complete blockade of ATP- and BzATP-induced dye uptake in this cell line (Figure 2A). Furthermore, Aβ25–35-induced dye uptake was not affected by the ATP-degrading enzyme apyrase (Supplementary Figure S2). In addition, we observed that Aβ25–35 induced YO-PRO-1 dye uptake into non-transfected HEK-293 cells which fail to display ATP-induced dye uptake (Figure 2D) supporting the hypothesis that the observed effects of Aβ25–35 are not dependent on the expression of P2X7. The inactive scrambled Aβ35–25-peptide did not induce YO-PRO-1 dye uptake responses (Figures 2B,C). The Aβ25–35-induced dye uptake was affected by the solvent used with the DMSO-peptide having a greater effect than the water-solubilized peptide (Figure 2B and Supplementary Figure S2).
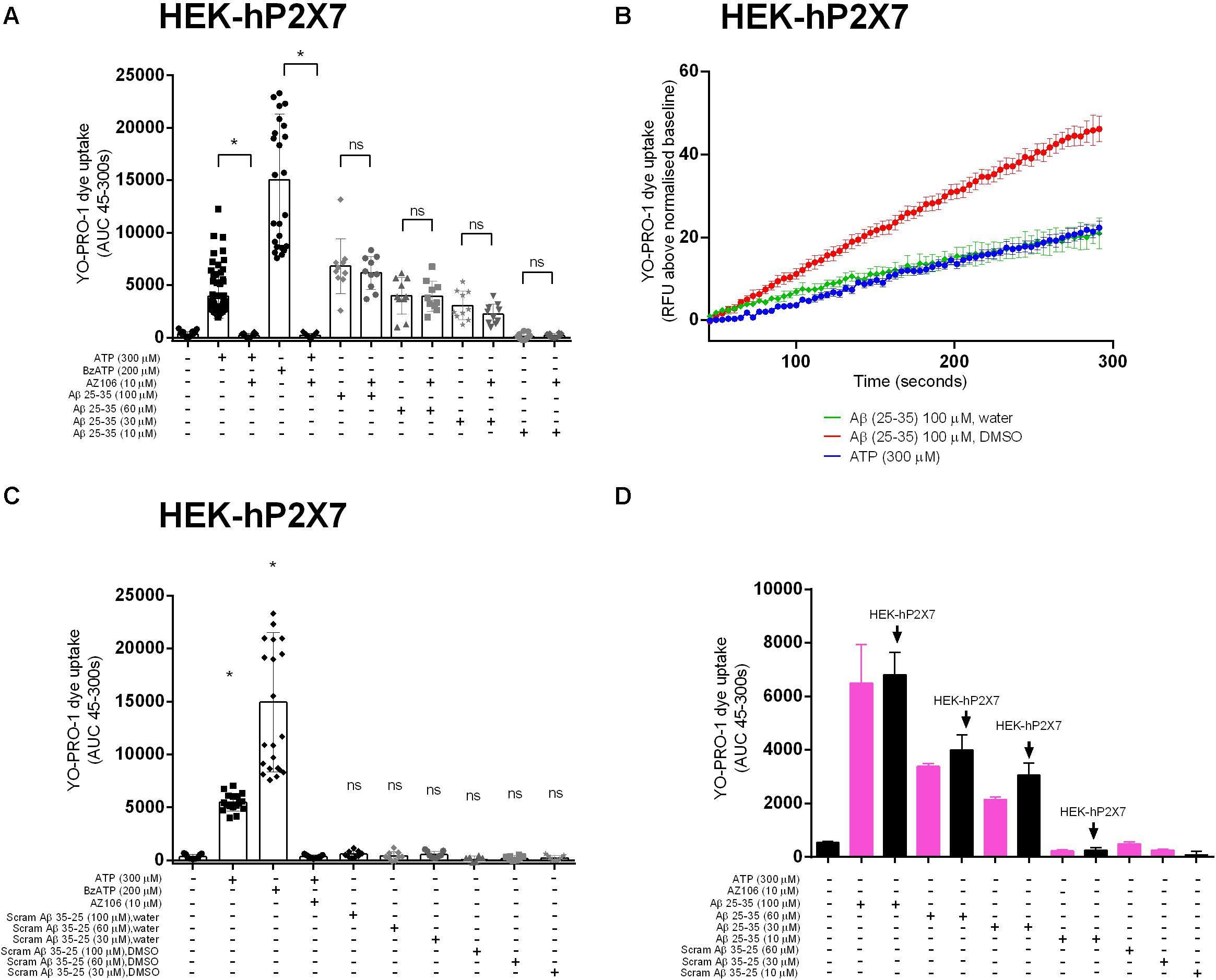
Figure 2. Aβ peptides triggered a non-specific YO-PRO-1 uptake in HEK-hP2X7 and HEK-293 cells. HEK-hP2X7 and plain HEK-293 cells were incubated with YO-PRO-1 dye in low-divalent assay buffer (2 μM) and challenged with either Aβ25–35 (A,B) or the inactive scrambled version Aβ35–25 (C). Kinetics of the Aβ25–35 dye uptake response in HEK-hP2X7 are plotted together with the appropriate controls (B). AZ10606120 is a selective antagonist of hP2X7, and BzATP is a synthetic agonist for hP2X7 receptors. (D) shows summary data from HEK-293 cells comparing responses to HEK-hP2X7 cells. Experiments were repeated five times with triplicates on each plate, and the results are presented as mean ± SD. One-way ANOVA was performed with Tukey’s multiple comparison test where * indicates a significant difference (P < 0.05) to the paired control [with AZ1060610 in (A) or control in (C)] and ns denotes no significant difference.
We investigated the full-length Aβ peptide, 1–42, which was also reported by Sanz et al. (2009) to have an effect on microglial IL-1β secretion. We did not observe [Ca2+]i increases in BV-2 (Figure 3A) or YO-PRO-1 dye uptake in HEK-hP2X7 (Figure 3B) in response to Aβ1–42 peptide at concentrations of 10, 60 or 100 μM. Both ATP and BzATP induced robust responses in both cell lines and the P2X7-selective antagonists AZ10606120 and JNJ47965567 could abolish these responses (Figure 3). Even at the highest concentration of Aβ1–42, this peptide did not cause a significant increase in [Ca2+]i release relative to the buffer control.
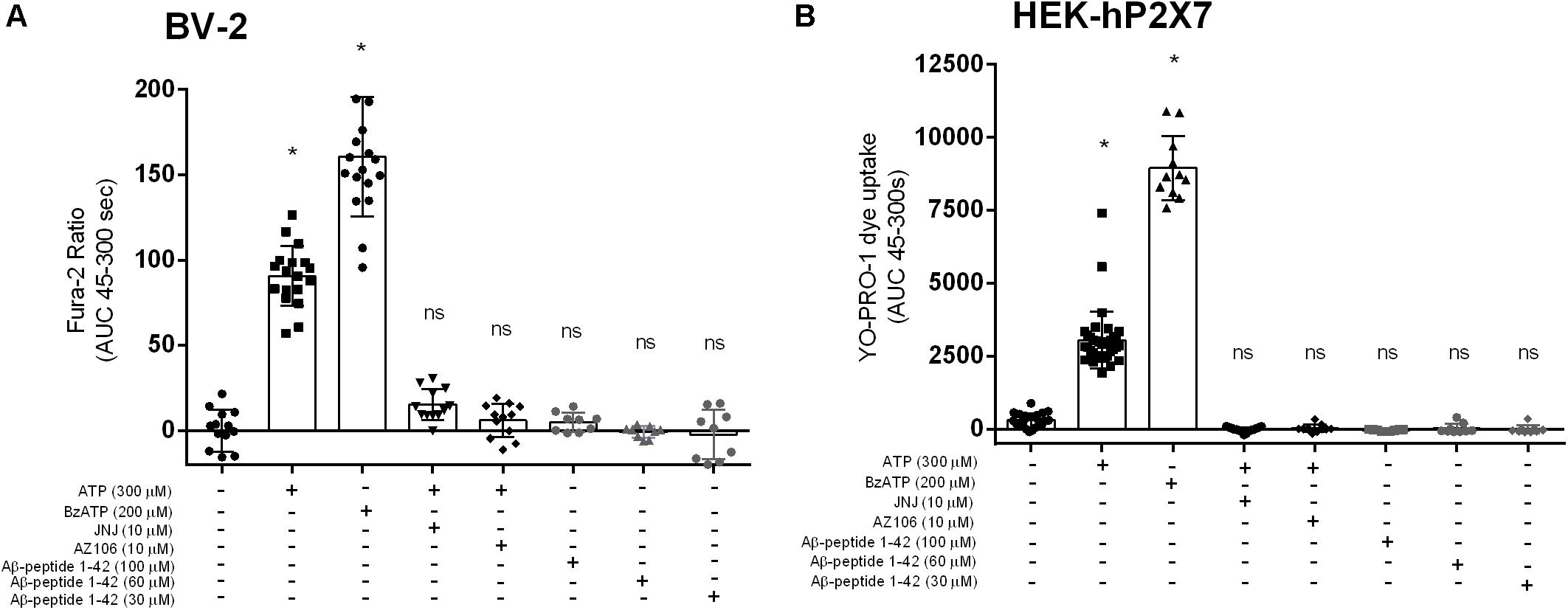
Figure 3. Aβ1–42 do not trigger responses in BV-2 cells or HEK-hP2X7 cells. BV-2 cells were loaded with Fura-2AM (2 μM) and challenged with the Aβ–1–42 (A) peptides in the concentration range 30–100 μM. HEK-hP2X7 cells were exposed to YO-PRO-1 dye in low-divalent assay buffer (2 μM) and challenged similarly as BV-2 cells (B). AZ10606120 is a commercially available antagonist of hP2X7. Experiments were repeated five times with triplicates on each plates, and the results are presented as mean ± SD. One-way ANOVA was performed with Tukey’s multiple comparison test where * indicates a significant difference (P < 0.05) to the control (buffer alone) and ns denotes no significant difference.
Once activated, P2X7 is endowed with the ability to kill microglia, either by necrosis or apoptosis, as well as to trigger many responses such as inflammation and oxidative stress (Surprenant et al., 1996; Ferrari et al., 1997; Brough et al., 2002; Volonte et al., 2012). Thus, we examined whether Aβ25–35 or Aβ1–42 could trigger cell death in BV-2, P2X7-deficient BV-2 cells, or HEK-hP2X7 cells at either 10, 30 or 60 μM, using a cell viability assay (Figure 4). In BV-2 (Figure 4A) and P2X7-deficient BV-2 cells (Figure 4B), Aβ1–42 or Aβ25–35 (60 μM) reduced cell viability similarly up to 35 and 30% of control, respectively, suggesting that this effect was not P2X7 dependent. Lower concentrations of Aβ1–42 or Aβ25–35 (10, 30 μM) had lesser effects and no significant decrease in cell viability was found with Aβ1–42 or Aβ25–35 on HEK-hP2X7 cells (Figure 4C). The inactive scrambled Aβ35–25 peptide did not have any effect on the cell viability (Figures 4A–C). Cell supernatants were also measured for the presence of LDH, which is released upon cell lysis. Figures 4D–F shows LDH levels from BV-2 cells, P2X7-deficient BV-2 cells, and HEK-hP2X7 cells. None of the Aβ-treated HEK-hP2X7 cell supernatants contained significant levels of released LDH as compared with control samples of cells stimulated with staurosporine or 3 mM ATP. In the BV-2 cells and P2X7-deficient BV-2 cells, we found a relatively high spontaneous LDH release and none of the Aβ peptide treatments were higher than this (Figures 4D,E).
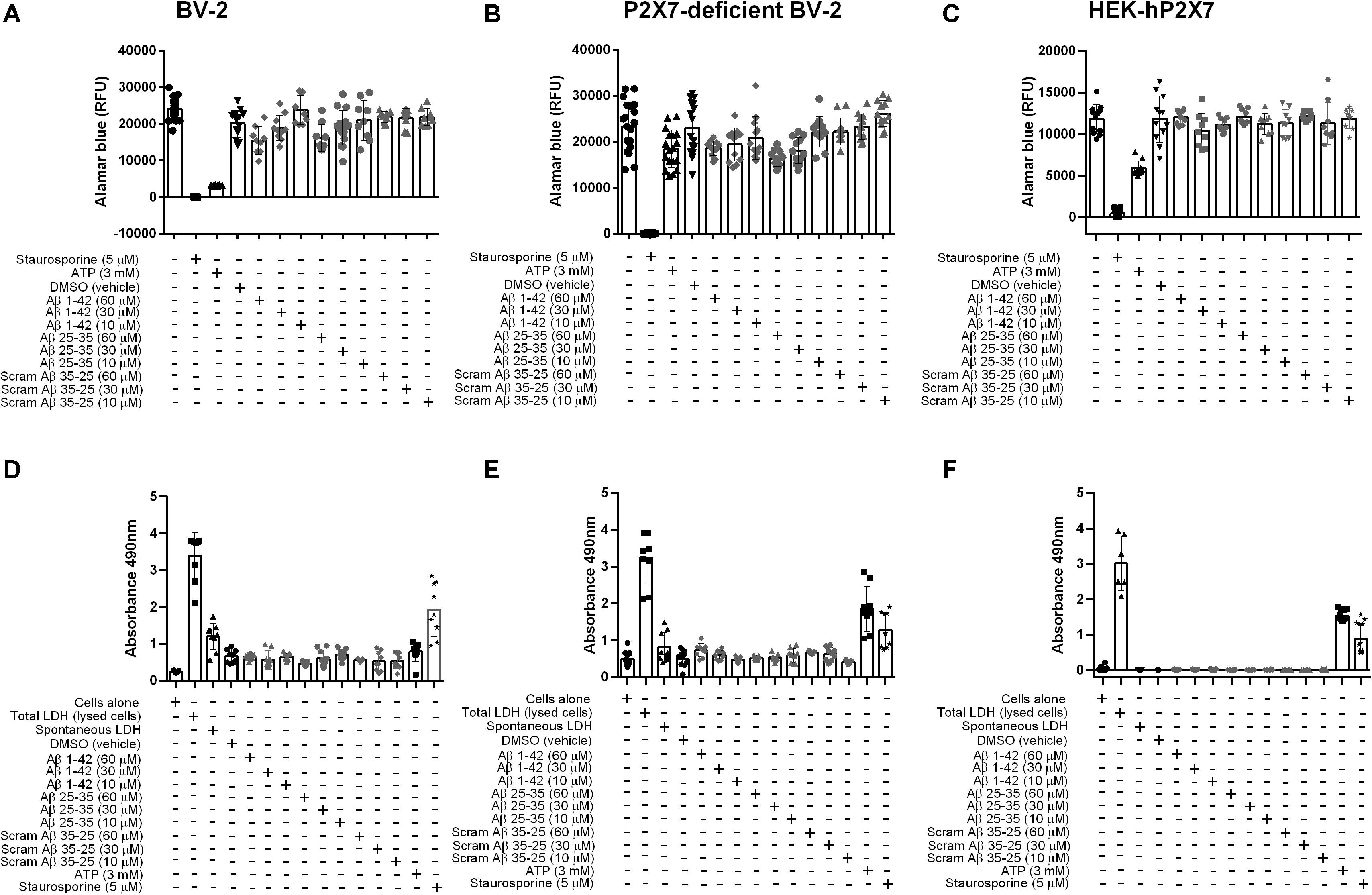
Figure 4. Aβ peptides do not trigger P2X7-specific cell death nor compromise the cell membrane in microglial BV-2 cells or HEK-hP2X7 cells. (A–C) An AlamarBlue assay and (D–F) an LDH assay were performed to assess cell viability and cell death respectively after 24 h administration of Aβ peptides. At indicated times (see “Materials and Methods”), the extracellular medium was collected and assayed for lactate dehydrogenase (LDH) activity. Control cells were lysed with the lysis buffer to induce maximal LDH release for normalization of the LDH release (% of max). Staurosporine and 3 mM ATP were used as a positive control. Data points represent the mean ± SD of 5 replicated experiments with triplicates on each plate. One-way ANOVA was performed with Dunnett’s multiple comparison test where * indicates a significant difference (P < 0.05) to the control (media alone).
Discussion
ATP is a crucial extracellular messenger serving as the physiological agonist of both P2Y and P2X receptors (Wang et al., 2004; Davalos et al., 2005; Haynes et al., 2006; Koizumi et al., 2007). The identification of alternative agonists for P2X receptors is of particular interest when ascribing physiological roles to individual receptors in different cells and tissues. There has been a suggestion that ATP may not be the only agonist at P2X7 receptors (Di Virgilio et al., 2018). It is proposed that agents such as amyloid–β (Sanz et al., 2009), serum amyloid (Niemi et al., 2011) and the cathelicidin LL-37 peptide (Elssner et al., 2004), may function as non-nucleotide agonists at P2X7. In the current study, we revisited the role of the amyloid-β peptide as an agonist for the P2X7 receptor using a microglial cell line and HEK-293 cells over-expressing human P2X7. We have previously used the microglial BV-2 cells and a clonal P2X7-deficient BV-2 cell line, generated using Cas9 gene editing, in order to assess Ca2+ influx upon P2X7 activation with ATP and the effect of positive modulators (Dhuna et al., 2019).
The biological effect of synthetic amyloid-β peptides, including Aβ25–35, may vary due to differences in aggregation states (Pike et al., 1993; Wei and Shea, 2006), therefore we performed the experiments using two common solvents, DMSO (Mattson et al., 1993) and water (Whitson et al., 1994). Our data shows that Aβ25–35 directly induced intracellular Ca2+ responses in BV-2 microglial cells regardless of the solvent (Figure 1) although responses were noted to be higher for the DMSO-dissolved peptide. We saw no increase in [Ca2+]i to the full length human Aβ1–42 peptide. Furthermore, when these two amyloid-β peptides (Aβ25–35 and Aβ1–42) were studied in a HEK-hP2X7 stable cell line using a dye uptake assay (Helliwell et al., 2015; Bibic et al., 2019; Bidula et al., 2019; Dhuna et al., 2019), we observed some YO-PRO-1 dye uptake in response to Aβ25–35. However, this also occurred in non-transfected HEK-293 cells that do not express P2X7 receptors (Figure 2). We saw no increase in YO-PRO-1 dye uptake to the full length human Aβ1–42 peptide. Collectively this data suggests that amyloid-β peptides may act on other receptors that can induce similar responses to P2X7 or the peptide may act by insertion into the lipid bilayer forming similar pores. It is known that other agents such as maitotoxin and ionomycin can induce dye uptake responses to P2X7 stimulation (Schilling et al., 1999; Verhoef et al., 2004) likely through insertion into the membrane and subsequent pore formation. There is evidence that amyloid-β peptides may act via other routes such as on pannexin-1 (Orellana et al., 2011) to cause ATP release from cells rather than acting to directly activate P2X7. This was also suggested to be the likely (indirect) effect on microglial cells in the work by Sanz et al. (2009). We did not directly measure ATP release in our study but we hypothesized that any ATP released by the amyloid-β peptides would elicit responses at P2X receptors and therefore we would have seen an effect of the P2X7-selective antagonists, apyrase, or in the BV-2 cells with P2X7 deficiency.
Human and rodent (rat/mouse) full length amyloid-β peptide (1–42) are highly similar and differ only in three amino acid substitutions at the N-terminus. It is not clear which isoform of the Aβ1–42 was used in the study by Sanz et al. (2009) however, we believe this minor sequence difference is unlikely to contribute to our lack of effect at P2X7. Indeed, should there be a species difference, we would expect to see responses at human P2X7 (which we did not). The amino acid sequence for Aβ25–35 peptide is identical between human and rodent. In our study we focused on addressing whether the amyloid-β peptides could act as agonists at P2X7. We did not extend our experiments to investigate the effect of amyloid-β peptides on IL-1β secretion from microglial cells. Gustin et al. demonstrated that Aβ25–35 could induce IL-1β secretion from LPS-primed mouse microglia but did not detect any ATP secretion in response to Aβ25–35. Furthermore, this team of researchers used primary microglia from the P2X7–/– mouse and observed a similar IL- 1β secretion in response to Aβ25–35 (Gustin et al., 2015). This notion led them to conclude that Aβ25–35-induced IL-1β secretion was P2X7-independent (Gustin et al., 2015). This contradicts the earlier work from Sanz et al. who demonstrated that amyloid peptides could induce IL-1β release from primary microglia but only when P2X7 was present (Sanz et al., 2009). Furthermore, in vivo evidence showed that amyloid β-induced IL-1β secretion in the hippocampus was reduced in P2X7 knockout mice (Sanz et al., 2009). Other studies have shown that activation of microglia with Aβ1–42, followed by exposure to BzATP, may result in enhanced secretion of IL-1β (Rampe et al., 2004). This suggests that amyloid-β peptides may be working as positive allosteric modulators rather than as agonists. Furthermore, McLarnon et al. demonstrated that Ca2+ responses in adult microglial cells from Alzheimer’s disease patients were significantly increased following Aβ1–42 pre-treatment when activated by a P2X7 selective agonist BzATP (McLarnon et al., 2006). We have tested the idea that amyloid-β peptides could act as positive allosteric modulators since this was our entry point into this set of experiments. However, we saw no enhancement of ATP-induced [Ca2+]i responses by Aβ1–42 peptide and no potentiation of ATP-induced YO-PRO-1 dye uptake in HEK-hP2X7 cells (Supplementary Figure S3).
Microglial cell death (measured as a reduction in cellular viability) can be observed when cells were stimulated by 3 mM ATP (Nishida et al., 2012; Dhuna et al., 2019). When the BV-2 cells were stimulated by either Aβ1–42, Aβ25–35 or the inactive scrambled Aβ35–25 peptide (60 μM), microglial cell viability was reduced by 35% compared to control. There was no significant increase in LDH release above vehicle treatment or spontaneous LDH release induced by water. Our data indicate that Aβ peptides may not act as non-nucleotide agonists of the P2X7 receptor and that Aβ peptides are unable to induce cytotoxicity or decrease cell viability directly via P2X7. We are not discarding P2X7 as an emerging therapeutic target for Alzheimer’s disease. Others have shown that P2X7 is involved in amyloid protein precursor (APP) processing (Delarasse et al., 2011) and affects phagocytosis of Aβ peptides (Ni et al., 2013). More recently, the P2X7 knockout mouse was investigated in the APPPS1 mouse model of Alzheimer’s disease (Martin et al., 2019). P2X7–/–mice had a reduced amyloid-β load and were protected from cognitive defects. In this model the chemokines CCL3, CCL4, and CCL5 were elevated only when P2X7 was present and this affected CD8+ T cell recruitment to the choroid plexus and hippocampus (Martin et al., 2019). It appears that there is still more to be discovered about how P2X7 can influence the development of neurodegenerative disorders.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
LB conceived and designed the study together with LS. LB performed the experiments and analyzed the data. LS and LB wrote the manuscript and edited the final version. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the BBSRC NRPDTP (Grant No. 1794654). This funding body had no direct role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Matthew Felgate for help generating the BV-2 P2X7-deficient cell line. We thank Professor Ronald Sluyter (University of Wollongong, Australia) for critical evaluation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2020.00166/full#supplementary-material
References
Bartlett, R., Yerbury, J. J., and Sluyter, R. (2013). P2X7 receptor activation induces reactive oxygen species formation and cell death in murine EOC13 microglia. Mediators Inflamm. 2013:271813.
Bhaskaracharya, A., Dao-Ung, P., Jalilian, I., Spildrejorde, M., Skarratt, K. K., Fuller, S. J., et al. (2014). Probenecid blocks human P2X7 receptor-induced dye uptake via a pannexin-1 independent mechanism. PLoS One 9:e93058. doi: 10.1371/journal.pone.0093058
Bibic, L., Herzig, V., King, G. F., and Stokes, L. (2019). Development of high-throughput fluorescent-based screens to accelerate discovery of P2X inhibitors from animal venoms. J. Nat. Prod. 82, 2559–2567. doi: 10.1021/acs.jnatprod.9b00410
Bidula, S. M., Cromer, B. A., Walpole, S., Angulo, J., and Stokes, L. (2019). Mapping a novel positive allosteric modulator binding site in the central vestibule region of human P2X7. Sci. Rep. 9:3231.
Brough, D., Le Feuvre, R. A., Iwakura, Y., and Rothwell, N. J. (2002). Purinergic (P2X7) receptor activation of microglia induces cell death via an interleukin-1-independent mechanism. Mol. Cell. Neurosci. 19, 272–280. doi: 10.1006/mcne.2001.1054
Busciglio, J., Lorenzo, A., and Yankner, B. A. (1992). Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol. Aging 13, 609–612. doi: 10.1016/0197-4580(92)90065-6
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464.
Chen, K., Iribarren, P., Hu, J., Chen, J., Gong, W., Cho, E. H., et al. (2006). Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid β peptide. J. Biol. Chem. 281, 3651–3659. doi: 10.1074/jbc.m508125200
Chessell, I. P., Michel, A. D., and Humphrey, P. P. A. (1997). Properties of the pore-forming P2X7 purinoceptor in mouse NTW8 microglial cells. Br. J. Pharmacol. 121, 1429–1437. doi: 10.1038/sj.bjp.0701278
Davalos, D., Grutzendler, J., Yang, Y., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8:752. doi: 10.1038/nn1472
Delarasse, C., Auger, R., Gonnord, P., Fontaine, B., and Kanellopoulos, J. M. (2011). The purinergic receptor P2X7 triggers α-secretase-dependent processing of the amyloid precursor protein. J. Biol. Chem. 286, 2596–2606. doi: 10.1074/jbc.m110.200618
Dhuna, K., Felgate, M., Bidula, S. M., Walpole, S., Bibic, L., Cromer, B. A., et al. (2019). Ginsenosides act as positive modulators of P2X4 receptors. Mol. Pharmacol. 95, 210–221. doi: 10.1124/mol.118.113696
Di Virgilio, F., Giuliani, A. L., Vultaggio-Poma, V., Falzoni, S., and Sarti, A. C. (2018). Non-nucleotide agonists triggering P2X7 receptor activation and pore formation. Front. Pharmacol. 9:39. doi: 10.3389/fphar.2018.00039
El Khoury, J., Hickman, S. E., Thomas, C. A., Cao, L., Silverstein, S. C., and Loike, J. D. (1996). Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 382, 716–719. doi: 10.1038/382716a0
Elssner, A., Duncan, M., Gavrilin, M., and Wewers, M. D. (2004). A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1β processing and release. J. Immunol. 172, 4987–4994. doi: 10.4049/jimmunol.172.8.4987
Fassbender, K., Walter, S., Kuhl, S., Landmann, R., Ishii, K., Bertsch, T., et al. (2004). The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 18, 203–205. doi: 10.1096/fj.03-0364fje
Ferrari, D., Chiozzi, P., Falzoni, S., Hanau, S., and Di Virgilio, F. (1997). Purinergic modulation of interleukin-1β release from microglial cells stimulated with bacterial endotoxin. J. Exp. Med. 185, 579–582. doi: 10.1084/jem.185.3.579
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10:e0130624. doi: 10.1371/journal.pone.0130624
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–186.
Haughey, N. J., and Mattson, M. P. (2003). Alzheimer’s amyloid β-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes. Neuromol. Med. 3, 173–180. doi: 10.1385/nmm:3:3:173
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W.-B., et al. (2006). The P2Y 12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9:1512. doi: 10.1038/nn1805
Helliwell, R. M., ShioukHuey, C. O., Dhuna, K., Molero, J. C., Ye, J. M., Xue, C. C., et al. (2015). Selected ginsenosides of the protopanaxdiol series are novel positive allosteric modulators of P2X7 receptors. Br. J. Pharmacol. 172, 3326–3340. doi: 10.1111/bph.13123
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Idziorek, T., Estaquier, J., De Bels, F., and Ameisen, J.-C. (1995). YOPRO-1 permits cytofluorometric analysis of programmed cell death (apoptosis) without interfering with cell viability. J. Immunol. Methods 185, 249–258. doi: 10.1016/0022-1759(95)00172-7
Koenigsknecht, J., and Landreth, G. (2004). Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846. doi: 10.1523/jneurosci.2557-04.2004
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y 6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. doi: 10.1038/nature05704
Le, Y., Gong, W., Tiffany, H. L., Tumanov, A., Nedospasov, S., Shen, W., et al. (2001). Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 21, RC123–RC123.
Lee, M.-H., Siddoway, B., Kaeser, G. E., Segota, I., Rivera, R., Romanow, W. J., et al. (2018). Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563, 639–645. doi: 10.1038/s41586-018-0718-6
Martin, E., Amar, M., Dalle, C., Youssef, I., Boucher, C., Le Duigou, C., et al. (2019). New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 24, 108–125. doi: 10.1038/s41380-018-0108-3
Mattson, M. P., Cheng, B., Culwell, A. R., Esch, F. S., Lieberburg, I., and Rydel, R. E. (1993). Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 10, 243–254. doi: 10.1016/0896-6273(93)90315-i
McLarnon, J. G., Ryu, J. K., Walker, D. G., and Choi, H. B. (2006). Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 65, 1090–1097. doi: 10.1097/01.jnen.0000240470.97295.d3
Michaud, J.-P., Hallé, M., Lampron, A., Thériault, P., Préfontaine, P., Filali, M., et al. (2013). Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. U.S.A. 110, 1941–1946. doi: 10.1073/pnas.1215165110
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Ni, J., Wang, P., Zhang, J., Chen, W., and Gu, L. (2013). Silencing of the P2X7 receptor enhances amyloid-β phagocytosis by microglia. Biochem. Biophys. Res. Commun. 434, 363–369. doi: 10.1016/j.bbrc.2013.03.079
Niemi, K., Teirilä, L., Lappalainen, J., Rajamäki, K., Baumann, M. H., Öörni, K., et al. (2011). Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 186, 6119–6128. doi: 10.4049/jimmunol.1002843
Nishida, K., Nakatani, T., Ohishi, A., Okuda, H., Higashi, Y., Matsuo, T., et al. (2012). Mitochondrial dysfunction is involved in P2X7 receptor-mediated neuronal cell death. J. Neurochem. 122, 1118–1128. doi: 10.1111/j.1471-4159.2012.07868.x
Orellana, J. A., Froger, N., Ezan, P., Jiang, J. X., Bennett, M. V. L., Naus, C. C., et al. (2011). ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 118, 826–840. doi: 10.1111/j.1471-4159.2011.07210.x
Parvathenani, L. K., Tertyshnikova, S., Greco, C. R., Roberts, S. B., Robertson, B., and Posmantur, R. (2003). P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 278, 13309–13317. doi: 10.1074/jbc.m209478200
Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G., and Cotman, C. W. (1993). Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13, 1676–1687. doi: 10.1523/jneurosci.13-04-01676.1993
Rampe, D., Wang, L., and Ringheim, G. E. (2004). P2X7 receptor modulation of β-amyloid-and LPS-induced cytokine secretion from human macrophages and microglia. J. Neuroimmunol. 147, 56–61. doi: 10.1016/j.jneuroim.2003.10.014
Sanz, J. M., Chiozzi, P., Ferrari, D., Colaianna, M., Idzko, M., Falzoni, S., et al. (2009). Activation of microglia by amyloid β requires P2X7 receptor expression. J. Immunol. 182, 4378–4385. doi: 10.4049/jimmunol.0803612
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249.
Schilling, W. P., Sinkins, W. G., and Estacion, M. (1999). Maitotoxin activates a nonselective cation channel and a P2Z/P2X7-like cytolytic pore in human skin fibroblasts. Am. J. Physiol. Cell Physiol. 277, C755–C765.
Shen, C.-L., and Murphy, R. M. (1995). Solvent effects on self-assembly of beta-amyloid peptide. Biophys J. 69, 640–651. doi: 10.1016/s0006-3495(95)79940-4
Surprenant, A., Rassendren, F., Kawashima, E., North, R. A., and Buell, G. (1996). The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738. doi: 10.1126/science.272.5262.735
Verhoef, P. A., Kertesy, S. B., Estacion, M., Schilling, W. P., and Dubyak, G. R. (2004). Maitotoxin induces biphasic interleukin-1β secretion and membrane blebbing in murine macrophages. Mol. Pharmacol. 66, 909–920. doi: 10.1124/mol.66.4.909
Volonte, C., Apolloni, S., Skaper, S. D., and Burnstock, S. D. (2012). P2X7 receptors: channels, pores and more. CNS Neurol. Disord. Drug Targets 11, 705–721. doi: 10.2174/187152712803581137
Wang, X., Arcuino, G., Takano, T., Lin, J., Peng, W. G., Wan, P., et al. (2004). P2X7 receptor inhibition improves recovery after spinal cord injury. Nat. Med. 10, 821–827. doi: 10.1038/nm1082
Wei, G., and Shea, J.-E. (2006). Effects of solvent on the structure of the Alzheimer amyloid-β (25–35) peptide. Biophys. J. 91, 1638–1647. doi: 10.1529/biophysj.105.079186
Whitson, J. S., Mims, M. P., Strittmatter, W. J., Yamaki, T., Morrisett, J. D., and Appel, S. H. (1994). Attenuation of the neurotoxic effect of Aβ amyloid peptide by apolipoprotein E. Biochem. Biophys. Res. Commun. 199, 163–170. doi: 10.1006/bbrc.1994.1209
Keywords: Alzheimer disease, P2X7 receptor, microglia, reproducibility, amyloid beta peptide
Citation: Bibič L and Stokes L (2020) Revisiting the Idea That Amyloid-β Peptide Acts as an Agonist for P2X7. Front. Mol. Neurosci. 13:166. doi: 10.3389/fnmol.2020.00166
Received: 04 December 2019; Accepted: 11 August 2020;
Published: 17 September 2020.
Edited by:
Beata Sperlagh, Institute of Experimental Medicine (MTA), HungaryReviewed by:
Eric Boué-Grabot, Université de Bordeaux, FranceSimonetta Falzoni, University of Ferrara, Italy
Copyright © 2020 Bibič and Stokes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leanne Stokes, bC5zdG9rZXNAdWVhLmFjLnVr