 Yining Hao
Yining Hao Min Guo
Min Guo Yiwei Feng1
Yiwei Feng1 Mei Cui
Mei Cui- 1Department of Neurology, Huashan Hospital, Fudan University, Shanghai, China
- 2Department of Neurology, Huashan Hospital, State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, Fudan University, Shanghai, China
Lysophospholipids (LPLs) are bioactive signaling lipids that are generated from phospholipase-mediated hydrolyzation of membrane phospholipids (PLs) and sphingolipids (SLs). Lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) are two of the best-characterized LPLs which mediate a variety of cellular physiological responses via specific G-protein coupled receptor (GPCR) mediated signaling pathways. Considerable evidence now demonstrates the crucial role of LPA and S1P in neurodegenerative diseases, especially in Alzheimer’s disease (AD). Dysfunction of LPA and S1P metabolism can lead to aberrant accumulation of amyloid-β (Aβ) peptides, the formation of neurofibrillary tangles (NFTs), neuroinflammation and ultimately neuronal death. Summarizing LPA and S1P signaling profile may aid in profound health and pathological processes. In the current review, we will introduce the metabolism as well as the physiological roles of LPA and S1P in maintaining the normal functions of the nervous system. Given these pivotal functions, we will further discuss the role of dysregulation of LPA and S1P in promoting AD pathogenesis.
Introduction
The human brain is highly rich in lipids, which account for approximately 60%–70% of its dry weight (Yung et al., 2015). These lipids can be generally divided into two major clusters: lipids which act as structural components and lipids which function as signaling molecules, according to their functional heterogeneity (Yung et al., 2015). Structural lipids are the primary components of cellular membranes. They form dynamic functional rafts and serve as platforms for the membrane proteins (Simons and Ikonen, 1997). Meanwhile, the signaling lipids, which are considered to be the bioactive lipids, exert their effects by binding to specific receptors acting in a signal transduction mechanism (Choi and Chun, 2013). Lysophospholipids (LPLs) are metabolic intermediates generated via the active hydrolyzation of phospholipase on membrane phospholipids (PLs) and sphingolipids (SLs). Two major bioactive lipid derivatives are playing a crucial role in various cellular physiological processes as well as pathological events, the well-characterized lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P; Li et al., 2016). LPA and S1P function mainly as extracellular mediators by activating cognate cell surface G-protein coupled receptors (GPCRs) and stimulate intracellular responses through different signaling transduction pathways. Sophisticated and well-balanced modulation of LPA and S1P metabolism has been suggested to be critical in the developing and mature nervous system (Choi and Chun, 2013).
Most neurodegenerative diseases are accompanied by changes in both the composition and metabolism of LPLs (Wang and Bieberich, 2018). Reoccurring evidence has indicated that loss homeostatic LPA and S1P metabolism may act as a co-participator in the pathogenesis of multiple neurodegenerative disorders, particularly in Alzheimer’s disease (AD; Ghasemi et al., 2016; Wang and Bieberich, 2018). Indeed, both LPA and S1P have been demonstrated to participate in the generation of neuropathological hallmarks that characterize AD by binding to their G protein-coupled LPLs receptors (LPL-GPCRs). Dysfunction of LPA and S1P metabolism results in aberrant amyloid-β peptide (Aβ) aggregation (Shi et al., 2013), neurofibrillary tangle (NFT) formation (Sayas et al., 2002b), neuroinflammation (Awada et al., 2014; Kwon et al., 2018) and ultimately neuronal apoptosis (Robinson, 2015). In this review, we summarized the metabolism of LPA and S1P as well as their GPCRs cell signaling, with emphasis on the physiological role of LPA and S1P in the nervous system, and their underlying crosstalk with AD pathogenesis.
Metabolism of Cellular LPA and S1P
LPA Metabolism
LPA, also known as 1-acyl 2-hydroxylglycerol 3-phosphate, is an autocoid PL that is formed on-demand and functions near to the location of its production (Li et al., 2016). The generation of bioactive LPA requires phospholipase A2 (PLA2) mediated cleavage of a membrane PL, for example, the phosphatidylcholine. In this instance, arachidonic acid (AA) is generated in addition to a LPL, such as lysophosphatidylcholine (LPC). The latter then acts as the substrate for producing LPA by a dual-function ectoenzyme named autotaxin (ATX), while AA is further converted to pro-inflammatory mediators. ATX, also known as the ectonucleotide pyrophosphatase/phosphodiesterase-2 (ENPP2), is a soluble enzyme mainly found in plasma and cerebrospinal fluid (CSF; Herr et al., 2020). Aberrant ATX expression and malfunction in the autotaxin–LPA (ATX–LPA) axis have been suggested to promote the initiation and progression of AD pathology (Ramesh et al., 2018; Herr et al., 2020).
S1P Metabolism
S1P levels in human tissues are under sophisticated regulation with two bioactive enzymes; sphingosine kinase (SphK), which is related to S1P biosynthesis, and sphingosine-1-phosphate lyase (SPL), which governs S1P degradation. Physiologically, membrane sphingomyelin is degraded to ceramide, which is subsequently converted to sphingosine by the enzyme ceramidase. Sphingosine is then phosphorylated to S1P by highly regulated SphKs in various cellular compartments (Spiegel and Milstien, 2003; Santos and Lynch, 2015). Sphingosine kinases (SphKs) are a cluster of evolutionarily conserved lipid kinases, which modulate S1P production. There are two isoforms of SphK, known as SphK1 and SphK2, of which the subcellular localizations are consistent with the compartmentalization and biological effects of S1P (Chan and Pitson, 2013). SphK1 is found localized in the cytoplasm and is activated only when recruited to the cell membrane, while SphK2, as a membrane related lipid kinase, mainly concentrates on cellular organelles, such as the nucleus, mitochondria and endoplasmic reticulum (ER; Neubauer and Pitson, 2013). The discrepancy of the subcellular localization between SphK1 and SphK2 indicates the different biological functions they mediate (Spiegel and Milstien, 2003; Alvarez et al., 2010). SphK1 generated S1P signaling requires the re-localization of SphK1 from the cytosol to the plasma membrane, thus stimulating cell migration, proliferation, and survival (Zhu et al., 2010; Chan and Pitson, 2013; Gassowska et al., 2014). By contrast, the biological effects of SphK2 are more complicated. SphK2 has been implicated in the inhibition of DNA synthesis in the nuclei (Hait et al., 2009). It has also been proved to promote apoptosis by interacting with Bax and Bak in mitochondria (Chipuk et al., 2012). Furthermore, in serum deprivation conditions, localization of SphK2 in the ER has been suggested to activate a “salvage” pathway named “sphingolipid rheostat” by promoting the generation of pro-apoptotic ceramide instead of protective S1P (Neubauer and Pitson, 2013).
G-Coupled Protein Receptors (GPCRs) of LPA and S1P in The Cell Signaling
Both LPA and S1P are cell membrane-derived bioactive LPLs. They function via the 7-transmembrane GPCRs on the cell surface to activate multiple downstream signaling cascades (Choi and Chun, 2013; Li et al., 2016). There are four classes of heterotrimeric G proteins involved in the LPL mediated GPCR signals: Gs, Gq/11, Gi, and G12/13. By binding to specific GPCRs, LPA and S1P mediate physiological processes as well as pathological events within the nervous system (Choi and Chun, 2013).
Six LPA receptors (LPARs) have been identified : LPA1-LPA6, with genes named LPAR1-LPAR6 (human) and Lpar1-Lpar6 (non-human; Kihara et al., 2014; Yung et al., 2014, 2015). LPARs have been found to function in neurogenesis and brain development (Castilla-Ortega et al., 2011), neurodifferentiation (Fukushima et al., 2002b; Spohr et al., 2008), neural network formation and morphogenesis (Furuta et al., 2012; Roza et al., 2019), neuroplasticity (Fujiwara et al., 2003; Rhee et al., 2006) and glia cell modulation (Shano et al., 2008; Awada et al., 2014).
Similarly, five S1P receptors (S1PR1-5) belonging to the Edg family engage in the regulation of cellular biological events through Gq, Gi, G12/13, and Rho proteins (Li et al., 2016; Czubowicz et al., 2019). S1P with specific high-affinity S1PRs, have multiple ancillary roles in the regulation of oxidative stress (Sinha et al., 2013; Pyszko and Strosznajder, 2014), apoptosis (Edsall et al., 1997; Ghasemi et al., 2016), mitochondrial dysfunction (Chan and Pitson, 2013; Santos and Lynch, 2015; Ghasemi et al., 2016), autophagy (Moruno Manchon et al., 2015) and neuronal development (Mizugishi et al., 2005; Hait et al., 2014; Ghasemi et al., 2016). It should also be noted that S1P employs several mechanisms to act as a bioactive participant in subcellular compartmentalization, thus indicating a complicated biological effect of S1P and S1P mediated intracellular transduction (Maceyka and Spiegel, 2014). Identification of LPA and S1P associated G-protein-coupled LPL receptors, signaling mechanisms, and biological and pathological functions in the central nervous system (CNS) are summarized in Table 1.
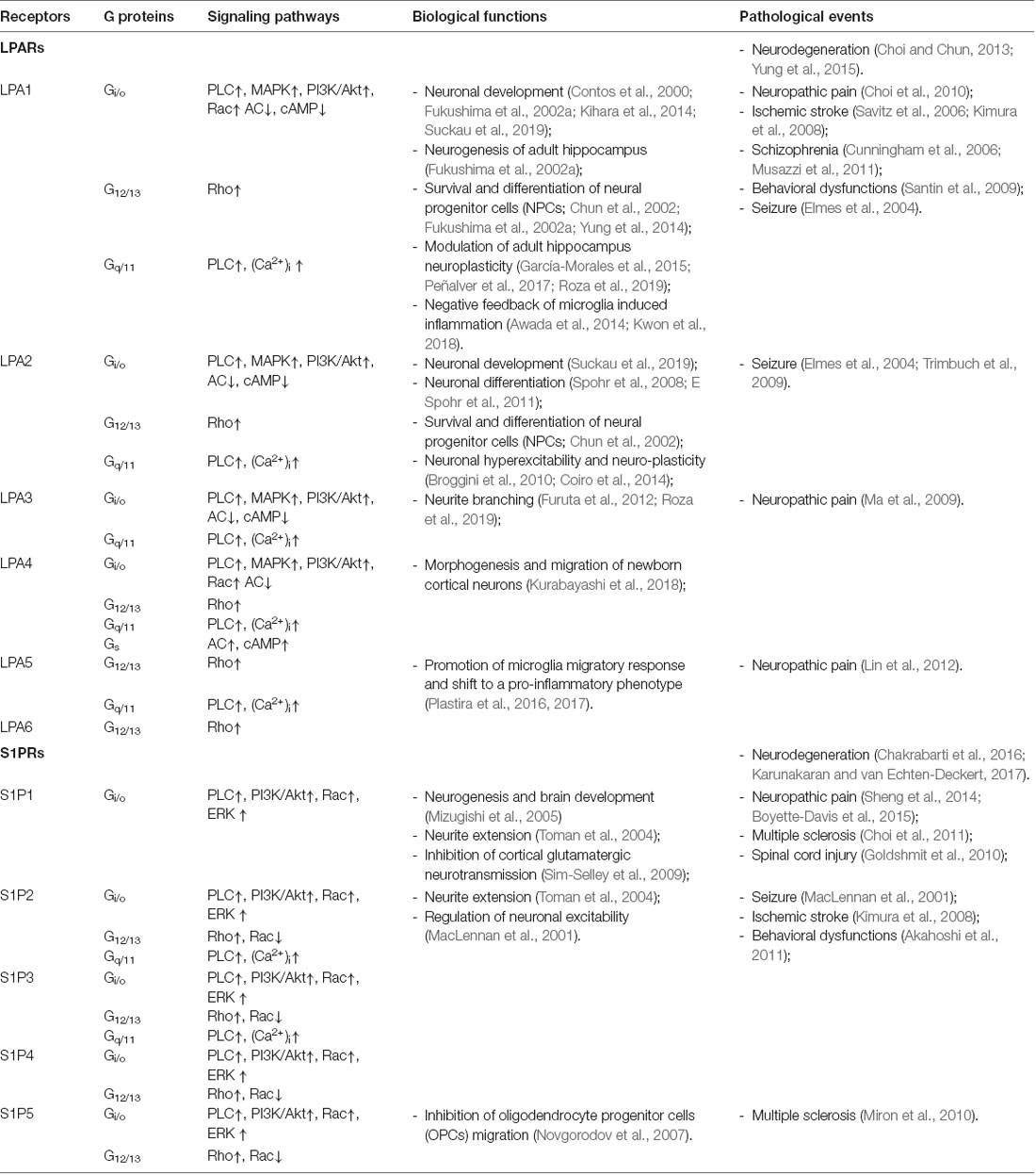
Table 1. Signaling mechanism, G-protein-coupled lysophospholipids receptors, and their identified physiological and pathological functions.
Physiological Roles of LPA in The Nervous System
LPA acts as a functional bioactive lipid, exerting its effects by binding to six cognate cell surface GPCRs termed LPARs (LPA1-LPA6). The importance of LPA signaling has been stressed by accumulating evidence about their involvement in the regulation of cell proliferation, survival, migration, cytoskeletal change, and cell to cell interaction in the nervous system (Choi and Chun, 2013; Yung et al., 2014). In this section, the LPA-mediated biological functions in the CNS will be discussed with detail.
LPA and Brain Development
It has been proven that LPA acts as a crucial regulator of cortical development and neurogenesis. Embryos with mutations of ATX, the key ectoenzyme that modulates LPA biosynthesis, are unable to achieve cranial neural tube closure. In this regard, using expression analysis of neural marker genes, Koike et al. (2011) have demonstrated reduced lateral expansion of the rostral forebrain and impaired midbrain-hindbrain boundary in ATX mutant embryos. Also, animal experiments suggested defects in neural tube development in Enpp−/− mutant mice (Moolenaar et al., 2013). In the same way, LPARs are also required in the modulation of cortical development since the LPAR genes are dynamically regulated in time and space-dependent manner at various developmental stages in the mouse brain and hippocampal primary neurons (Suckau et al., 2019).
Deficiency of LPA1 has been shown to reduce cortical width as well as cerebral wall thickness, thus leading to smaller brain size, and more severely, 50% perinatal lethality (Contos et al., 2000). Consistently, similar work also indicated disabled migration, morphological changes, proliferation, and differentiation in LPA1-null neuronal cells (Contos et al., 2000; Kihara et al., 2014), suggesting a crucial role of LPA1 in modulating neurogenesis-related events. Moreover, it is also well evidenced that LPA4 remodels the actin cytoskeleton and promotes microtubule formation in neurons, thus leading to bipolar morphogenesis and radial migration of newborn cortical neurons (Fukushima et al., 2002b; Kihara et al., 2014; Kurabayashi et al., 2018).
Several lines of evidence are available which indicate that the LPA mediated cortical development and neurogenesis are realized by modulating neural progenitor cells (NPCs). LPA has been shown to mediate NPC ionic conductance alterations (Yung et al., 2014). It must be emphasized that, among all of the subtypes of LPAR, LPA1 and LPA2 are two of the best-described receptors that mediate the LPA-induced survival and differentiation of NPCs (Chun et al., 2002). Besides the mentioned roles, LPA1 and LPA2, along with LPA6, have been characterized as key players in regulating astrocyte and oligodendrocyte development, synapse formation, maturation and stabilization (Suckau et al., 2019). Collectively, this evidence illustrates the regulatory role of LPA and its receptors in brain development.
LPA and Neuronal Differentiation
The first step of neuronal differentiation requires the formation of neurites, through a key process known as neuritogenesis. An array of evidence indicates that LPA has a controlling role in neurite growth within the embryonic brain (Hecht et al., 1996), and collective observations have suggested LPA-elicited neurite retraction, cell rounding, and growth cone collapse in various neuronal cell lines (Tigyi and Miledi, 1992). Satoh et al have reported that LPA-induced neurite retraction can be prevented by inhibiting the Rho/Rho-kinase pathway (Satoh et al., 2011), thus suggesting an underlying signaling pathway. Consistently, fasudil, a Rho-kinase inhibitor that acts as an efficacious treatment of ischemic brain infarction by improving hemodynamic function as well as preventing inflammatory response (Satoh et al., 2008), has recently been demonstrated to prohibit neurite retraction (Satoh et al., 2008). Moreover, research has also shown that LPA can activate the small GTPase Rho, which will in turn contract the cytoskeleton in neurite retraction (Tigyi and Miledi, 1992; Savitz et al., 2006; Kimura et al., 2008). These results are in line with the study conducted by Yamazaki et al. (2008) which showed that LPA induces growth cone collapse and neurite retraction via the G12/13-RhoA-GSK3β pathway. Indeed, LPA has been identified to primarily influence GSK3β-mediated rearrangement of microfilaments (MFs) and polymerization-dependent microtubules (MTs). Such interactions between MF-MT have been indicated to regulate LPA-induced neurite outgrowth (Fukushima and Morita, 2006; Fukushima et al., 2011). In this regard, conclusions can be drawn that LPA-mediated G12/13 activation increases RhoA GTPase activity, thus leading to the upregulation of GSK-3, downstream disorganization of MTs, growth cone collapse as well as the retraction of neurites (Sayas et al., 1999).
In addition to the LPA induced Rho/Rho-kinase pathway activation of cytoskeletal rearrangement, it has also been suggested that LPA could also stimulate another form of actin rearrangement by modulating intracellular calcium. It has been well evidenced that activation of LPARs can stimulate phospholipase C (PLC), thus leading to a temporary release of calcium from the ER. Furthermore, it has also been validated that LPA can directly induce Ca2+ influx from the extracellular fluid (Jang et al., 2014). Fukushima et al. (2002a) have demonstrated two distinct types of actin remodeling induced by LPA within neuroblasts (Fukushima et al., 2002a). By binding to the same LPAR, activation of the Rho/Rho-kinase pathway induced actin polymerization and the subsequent neurite retraction, whereas elevation of the intracellular calcium concentration led to α-actinin depolymerization and resulted in the loss of membrane ruffling (Fukushima et al., 2002a). This LPA-PLC-Ca2+ mediated cytoskeletal rearrangement could be significantly reversed by decreasing intracellular calcium (Fukushima et al., 2002a).
Besides the mentioned role of LPA in modulating cytoskeletal rearrangement, Spohr et al. (2008) have also indicated that LPA could regulate neuronal differentiation by affecting other cellular events such as cell fate determination and maturation in direct and indirect ways. Similar results are also demonstrated by Dottori et al. (2008) that high concentrations of LPA can inhibit neuronal differentiation by either affecting cell cycle or increasing cell death.
LPA and Neural Morphogenesis
Another important biological function of LPA lies in the regulation of neural morphogenesis, which involves the modulation of axons and neurites branching. There are two ways in which LPA mediates axon and neurite branch formation: direct and indirect. The former requires a novel signaling transduction pathway involving LPA3, Gq, and Rnd2, while the indirect mechanism involves the small GTPase family. Furuta et al. (2012) have detected inhibition of neurite branching when both Gα i and Gα q were restrained. Besides the mentioned roles of Rho GTPase in regulating GSK3β-mediated cytoskeleton rearrangement, it has also been shown to modulate the axon’s response to neurotrophins. There has been validated evidence showing that the small GTPase family members such as Rho, Rac and Cdc42 are involved in the regulation of neurite outgrowth as well as neurite morphology (Govek et al., 2005). Govek et al. (2005) have observed that in the embryonic trigeminal cells, the Rho GTPases acted in a crucial role in neurotrophin-mediated axonal differentiation. They went on to further show that in the differentiation of axons and dendrites, Rac, another member of the small GTPase family, activates the response of trigeminal cells to neurotrophins (Yamazaki et al., 2008). In addition to these effects, the small GTPases, Rac and Rho have also been implicated in altering microglial morphology by regulating Kir2.1 channels (Muessel et al., 2013).
LPA and Neuroplasticity
The LPAR LPA1 is widely expressed in human hippocampal progenitor cells (Castilla-Ortega et al., 2011), and several lines of evidence have indicated a regulatory role of LPA1 in promoting synaptic modifications in adult hippocampal neurons (Fujiwara et al., 2003; Rhee et al., 2006; Castilla-Ortega et al., 2011). Further evidence identified that the LPAR genes are under dynamic regulation throughout mouse brain development, modulating synaptic plasticity in a temporal- and spatial-dependent manner (Suckau et al., 2019). Roza et al. (2019) have proposed the underlying mechanism by which LPAR modulates synaptic plasticity. They detected presynaptic glutamate release when stimulating LPAR localized in the presynaptic terminals, whereas excessive LPA stimulation was found to cause seizures (Roza et al., 2019). These results are in line with the observation that LPA modulates the excitatory as well as the inhibitory synapses to regulate synaptic strength and neuronal activity (García-Morales et al., 2015). It has been demonstrated that LPA functions as a dual regulatory factor in modulating synaptic excitability. High levels of LPA have been shown to lessen the size of available vesicle pools at glutamatergic pre-synaptic neurons, thus providing negative feed-back to prohibit the transmission at excitatory synaptic terminals. Nevertheless, in the presence of low concentrations of LPA, inhibitory postsynaptic receptors, namely gamma-aminobutyric acid receptor type A (GABAAR), were internalized to restrain transmission at inhibitory synaptic terminals, thereby briefly increasing the synaptic excitability (García-Morales et al., 2015; Roza et al., 2019). In line with these findings, it is believed that LPA plays a crucial role in modulating glutamatergic transmission in the nervous system. Accordingly, LPA is also demonstrated to induce the phosphorylation of NMDA receptors, thus regulating the synaptic plasticity that is associated with learning and memory (Roza et al., 2019). Direct evidence is published showing that LPA triggers the phosphorylation of NMDA receptors and mediates the Ca2+ influx in the postsynaptic terminals, leading to either long-term potentiation or long-term depression at the synapses of both excitatory and inhibitory neurons (Roza et al., 2019).
In addition to the functional plasticity regulated by LPA, it has also been shown that the LPARs play a vital role in modulating the structural plasticity of neurons. Roza et al. (2019) have demonstrated that inhibition of LPA1 mediates the downregulation of both glutaminase isoform (GLS) and active matrix-metalloproteinase 9 (MMP-9) in the cerebral cortex and hippocampus. This downregulation thereby results in an alteration in the morphology of glutamatergic pyramidal cells to an immature phenotype, as well as cognitive and memory deficiency (Roza et al., 2019). Moreover, this phenomenon has also been well illustrated by Peñalver et al. (2017) who have shown that silencing of LPA signaling affected the expression of the biosynthesis of glutamate isoenzyme and induced the dendritic spines to differentiate towards a less mature phenotype. In line with these results, it is plausible to assume that LPA signaling contributes to the regulation of cognitive and memory-related neuroplasticity at a molecular level.
Plasticity-related gene 1 (PRG-1) is a member of the integrin family, which acts as a co-factor with LPA in modulating glutamate neurotransmission. It has been shown that the post-synaptic deficiency of PGR-1 prohibits LPA from entering the post-synaptic membrane, thereby leading to the accumulation of LPA in the synaptic cleft (Vogt et al., 2016). Thus, an increased level of LPA in the synaptic gap has been shown to have two complementary mechanisms which result in increased glutamate concentration in the synaptic cleft. First, the increased LPA level has been shown to stimulate pre-synaptical LPA2 and induce the release of glutamate (Trimbuch et al., 2009; Roza et al., 2019). Second, the accumulated LPA promotes the release of autotaxin (ATX) from astrocytes, further enhancing the glutamate concentration in the synaptic cleft (Thalman et al., 2018). Other evidence is also available showing that PRG-1 modulates LPA release in a non-cell-autonomous pattern, thus leading to an elevation of pre-synaptical glutamate vesicle release, as well as a series of alterations in structural plasticity such as filopodia formation, neurite extension, and brain reorganization after lesion (Broggini et al., 2010; Coiro et al., 2014).
LPA and Modulation of Glial Cell Activity
LPA modulates the activity of neural glial cells through three actions: (1) LPA mediates the development of oligodendrocytes; (2) LPA regulates the activation of microglia and plays a crucial role in modulating neuroinflammation; and (3) LPA interacts with astrocytes and induces neuronal differentiation.
Accumulating evidence has proposed the involvement of LPA in oligodendrocyte development. Studies on neonatal Lpar1−/− mice have revealed that a deficiency of the LPA1 gene in adult mice could lead to a decrease in myelination and oligodendrocyte survival, suggesting a pro-differentiation role LPA1 on developing and mature oligodendrocytes (Weiner et al., 1998; Choi and Chun, 2013; García-Díaz et al., 2015). Furthermore, ATX has been shown to reduce the adhesion between oligodendrocytes and extracellular matrix molecules, such as fibronectin, laminin 2, and vitronectin through the ATX-LPA-Gα i pathway in CNS development (Fox et al., 2003; Nogaroli et al., 2009). Besides, ATX has also been demonstrated to modulate oligodendrocyte development indirectly. Clair et al. (2003) observed that ATX was capable of catalyzing S1P formation, which has been considered as a crucial bioactive factor, which regulates oligodendrocyte maturation and function (Jung et al., 2007; Cui et al., 2014).
Several years of hard work have expanded our knowledge of microglia and their role as a resident immune regulator in the brain. Microglia have been found to produce inflammatory factors in the CNS as well as modulate inflammation and repair. It has been demonstrated that ATX acts as a negative regulator during oxidative stress by inhibiting microglia activation and subsequent release of pro-inflammatory mediators, TNF-α and IL-6, through the LPA-LPA1-ERK1/2 pathway (Kwon et al., 2018). Meanwhile, another LPA pathway has been found to drive microglia towards a pro-inflammatory phenotype. Plastira et al. (2017) demonstrated that LPA could enhance the migratory capacity of microglia, increase the secretion of pro-inflammatory cytokines, as well as produce reactive oxygen species (ROS) and NO to exert cytotoxicity towards neurons. These effects were proven to be mediated through the LPA-LPA5-PKD pathway. Interfering with this axis leads to a reduction in microglial migration and pro-inflammatory cytokine secretion, thus abrogating the LPA-induced neuroinflammation (Plastira et al., 2017). Other research has indicated that LPA can promote BV-2 (expressing LPARs 2, 3, 5, and 6) and primary murine microglia (PMM, expressing LPARs 1, 2, 4, 5, and 6) to transform towards a pro-inflammatory M1-like phenotype. Furthermore, inhibition of microglia with pharmacological LPA5 antagonist TCLPA5 disrupted most of the pro-inflammatory effects, indicating the potential regulatory role of the LPA-LPA5 signaling axis in activating microglia-mediated neuroinflammation (Plastira et al., 2016). Besides the mentioned effects, ATX has also been suggested to function in both acute and chronic inflammatory conditions, while more involvement of ATX-mediated pro-inflammatory response was detected in chronic inflammation (Swaney et al., 2010).
In addition to the above-mentioned roles of LPA in regulating oligodendrocyte development and microglia-mediated inflammation, LPA has also been suggested to stimulate astrocyte-induced neuronal communication (Shano et al., 2008; García-Díaz et al., 2015). Spohr et al. (2008) have proposed an underlying mechanism by which neurons and astrocytes communicate. They have indicated that LPA plays a role in affecting cellular events such as neurite outgrowth, cell fate commitment, and maturation. These cellular responses are mediated by LPA induced rearrangement of fibronectin and laminin through the LPA1/2-EGF-MAPK-fibronectin/laminin pathway as well as the LPA4-PKA-fibronectin/laminin pathway (Spohr et al., 2008; E Spohr et al., 2011). Also, other cellular responses such as neuronal differentiation require the participants of LPA signaling, as it has been shown that the generation of nerve growth factor (NGF) by astrocytes can be increased by LPA (Furukawa et al., 2007).
Crosstalk Between LPA and The Pathogenesis of Alzheimer’s Disease
For several years, AD has been clinically characterized by progressive dementia with aberrant pathological deposits of intracellular NFTs and aggregation of extracellular Aβ peptides. Recent etiological studies of AD have focused on the vascular factors which participate in neuronal degeneration (Greenberg et al., 2020). In particular, oxidized low-density lipoprotein (oxLDL) has been demonstrated to contribute to AD pathology by regulating Aβ generation (Sun et al., 2003). As the primary component of oxLDL, LPA has been suggested to disrupt the normal functions of the blood-brain barrier and lead to AD-related cellular dysfunction (Frisardi et al., 2011).
Decades before the occurrence of dementia symptoms, senile plaque (SP) deposits containing Aβ peptides are a pathological hallmark in the brains of AD patients. Excessive aggregation of Aβ peptides has been ascribed to the imbalance between Aβ production and clearance. Sequential cleavage of amyloid precursor protein (APP) by its processing enzymes, β-secretase (β-site of APP-cleaving enzyme, BACE1) and γ-secretase (presenilin subunits PS1 or PS2), has been suggested as the main contributor to Aβ-induced AD pathology (Tiwari et al., 2019). Several lines of evidence have suggested that LPA plays a role in Aβ production. Shi et al observed that LPA could significantly upregulate the binding activity of CREB (cAMP response element-binding protein) by utilizing the BACE1 promoter at the CRE site. This work thus increased the CREB-mediated β-secretase (BACE1) expression without altering the expression level of APP or γ-secretase complex (Shi et al., 2013). This alleged role of LPA in promoting Aβ production was later shown to be mediated through the PKCδ–MEK–MAPK–p90RSK–CREB signaling cascade (Shi et al., 2013; Figure 1).
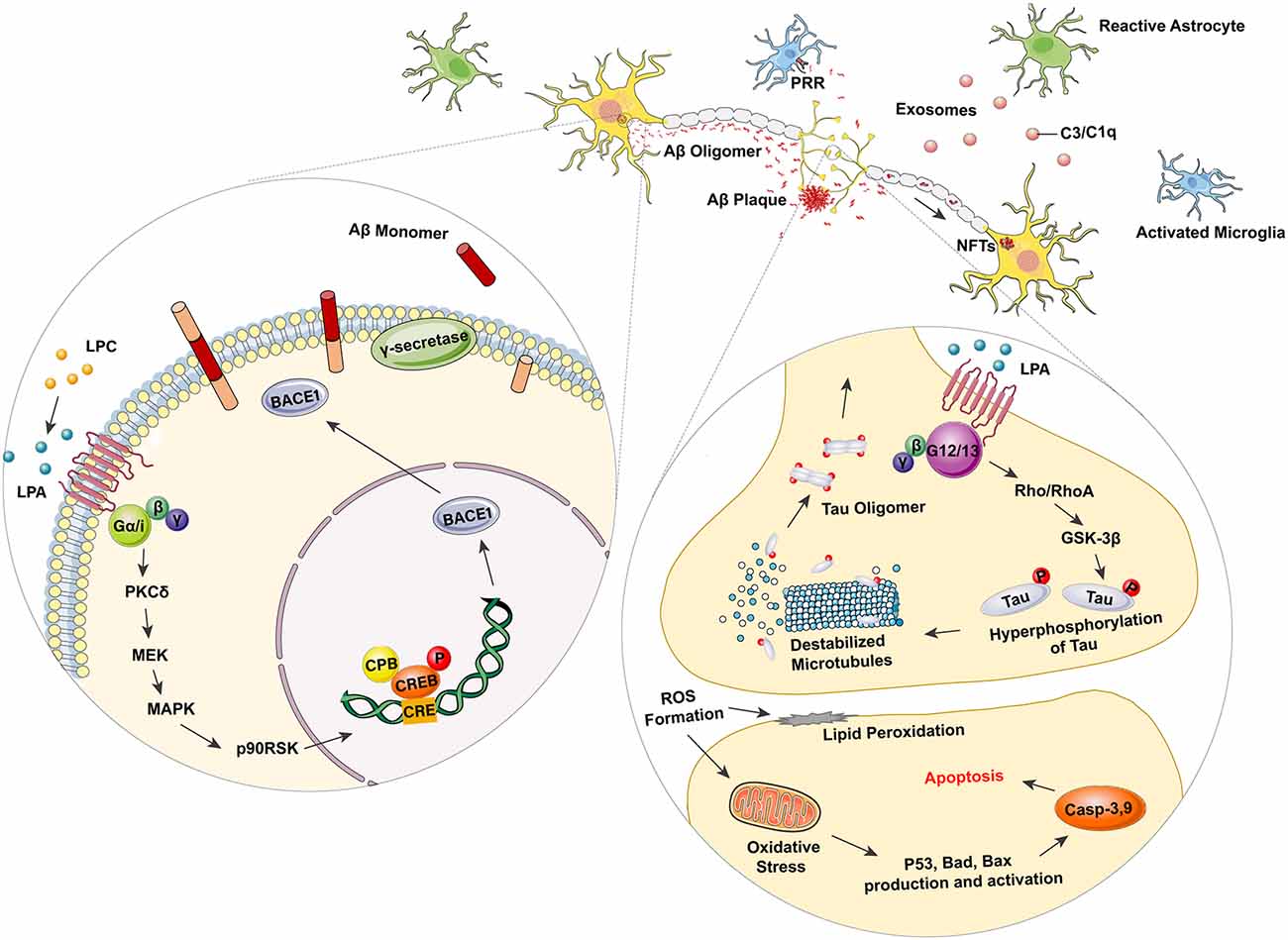
Figure 1. Lysophosphatidic acid (LPA) as a promoter in Alzheimer’s disease (AD) related neurodegeneration. LPA can elevate the binding activity of cAMP response element-binding protein (CREB) with β-site of amyloid precursor protein (APP)-cleaving enzyme (β-secretase, BACE 1) promoter at the CRE site through the Gα/i-PKCδ–MEK–MAPK–p90RSK–CREB signaling pathway. Overexpressed BACE 1 with γ-secretase then acts as the processing enzymes to cleave APP and produce Aβ monomers, thus leading to the dynamic polymerization and depolymerization of Aβ, as well as the AD-related pathology. Excessive activation of LPA mediated Gα12/13-RhoA/Rock-GSK3β pathway also results in the hyperphosphorylation of Tau, as well as the subsequent disorganization of microtubule (MT). The hyperphosphorylated Tau protein can be further polymerized to form intracellular NFT deposits, which are considered as another hallmark of AD. Moreover, microglia are also activated in AD-related pathogenesis. Astrocytes become stimulated and transform into their A1 phase. The active A1 astrocytes promote the secretion of complement-contained exosomes to mediate neurological damage via the complement pathway. Microglia combines with toxic amyloid-β (Aβ) oligomer and fibrils through pattern recognition receptors (PRRs) and initiates the release of pro-inflammatory mediators, such as iNOS, TNF-α, IL-1β, and IL-6.
In addition to the Aβ-associated pathogenesis, the function of phosphorylated tau in forming intracellular NFTs, has been considered as another key promoter in AD pathology. As the key element in forming microtubule-associated proteins (MAPs), tau has been validated to play a crucial role in microtube assembly as well as maintaining axonal homeostasis (Evans et al., 2000). As mentioned in earlier sections, LPA has been demonstrated to affect the GSK3β-mediated interactions between microfilaments (MF) and polymerization-dependent microtubule (MT), thus promoting microtubule depolymerization, neurite extension and axon differentiation (Fukushima et al., 2002a,b, 2011; Fukushima and Morita, 2006). Under pathological circumstances, however, the finely tuned MF-MT dynamics, which require sophisticated regulation through the LPA-mediated Gα12/13-RhoA/Rock-GSK3β pathway, is altered in AD (Sayas et al., 1999, 2002b). Additional studies have shown redundant neurite retraction in neurodegeneration (Sayas et al., 2002a), suggesting excessive activation of GSK3β induced by LPA. By using the GSK-3 inhibitor lithium, Ramesh et al. (2018) have also identified the involvement of GSK-3β induced tau hyperphosphorylation. These results have demonstrated a dual modulatory role of LPA in regulating MT dynamics. In physiological conditions, the equilibrium of MT depolymerization is shown to promote neurite outgrowth, while pathological MT depolymerization and tau hyperphosphorylation leads to excessive NFT deposits (Figure 1).
Another important pathological feature in AD is the activation of glial cells, particularly the activation of astrocytes and microglia in the CNS. Astrocytes are a type of glial cell that acts as key regulators in neurotransmission, calcium homeostasis, and the formation, maturation, and elimination of synapses. Astrocytes have also been demonstrated to provide nutritional and trophic support for neurons. Several lines of evidence have identified morphological changes of astrocytes in a range of CNS disorders. These alterations, including astroglial atrophy and reactive astrogliosis, are closely related to the pathological response of astrocytes in AD pathology. Collective evidence has indicated that reactive astrocytes can promote the inflammatory processes observed in AD. Reactive astrocytes have been found near to activated microglia, surrounding SPs in the neurodegenerative regions of post-mortem AD brains (Olabarria et al., 2010; Liddelow et al., 2017). Astroglial atrophy was also identified in AD models, as progressive cognitive deterioration caused by the decrease in astrocytic arborization and related loss of synaptic connectivity was also detected (Kulijewicz-Nawrot et al., 2012). Microglia are a cluster of resident immune cells in the CNS which function in maintaining brain homeostasis as well as the plasticity of neural circuits (Ji et al., 2013). There are numerous highly conserved pattern recognition receptor (PRR) expressing in microglia, such as the toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NOD-like receptors, NLRs), scavenger receptors (SRs) and receptors for advanced glycation end products (RAGEs; Parkhurst et al., 2013; Dansokho and Heneka, 2018). These PRRs then recognize exogenous signals (such as pathogen-associated molecular patterns, PAMPs) as well as self-derived signals (such as danger-associated molecular patterns, DAMPs) to activate microglia and generate inflammatory mediators (Pereira et al., 2019). Evidence is available showing that microglia function as promoting factors rather than a relevant feature in AD pathogenesis (Hansen et al., 2018). Zhang et al. (2016) found that most of the AD-related risk genes were preferentially expressed in microglia rather than any other cell types in the brain. This is in line with the results demonstrated by several emission tomography (PET) studies, which identified that the activation of microglia was positively correlated to Aβ deposits, tau aggregation and cognitive deficiency in MCI and AD patients (Hamelin et al., 2016; Dani et al., 2018). Similar histopathological research also suggested the co-localization of activated microglia with Aβ aggregation as well as tau oligomers in AD brains (Zotova et al., 2013; Nilson et al., 2017). The activation of microglia has been widely recognized as a major contributor to neuroinflammation (Heneka et al., 2015a), it has also been verified that Aβ plaques and NFTs were both invariably correlated with microglia-induced neuroinflammation (Pereira et al., 2019). Several studies have proposed a plausible mechanism of microglial activation, which is initiated by interaction with toxic Aβ oligomers and fibrils through PRRs, such as NLRP3, NLRP1, CD36, CD14, TLR2, RAGE, thus inducing the release of pro-inflammatory mediators like iNOS, TNF-α, IL-1β and, IL-6, as well as the elevated expression of cell adhesion markers CD11b and CD68 (Fassbender et al., 2004; Halle et al., 2008; Jana et al., 2008; Stewart et al., 2010; Venegas and Heneka, 2017). Similar results have also demonstrated that Aβ peptides and fibrils can act as disease-associated molecular patterns (DAMPs). This, in turn, leads to activation of Toll-like receptors (TRLs) and the NRLP3 inflammasome, leading to the generation of TNFα, IL-1β, and other pro-inflammatory mediators by microglia (Heneka et al., 2013; Heneka et al., 2015b). Evidence is also available showing that tau oligomers, as well as NFTs, can induce the activation of microglia and generation of NO and IL-6 (Morales et al., 2013). In addition to the outlined evidence, it is also well-illustrated that microglia and astrocytes can act as co-contributors to promote neuroinflammation. It has been shown that activated microglia release a great range of pro-inflammatory mediators, such as IL-1α, TNF-α, and C1q, and trigger the transformation of astrocytes into a neurotoxic “A1” reactive state, in which astrocytes lose the function of maintaining the survival of neurons and oligodendrocytes (Liddelow et al., 2017). Moreover, Goetzl et al. (2018) have also demonstrated an elevated level of complement factors, such as C3 and C1q in astrocyte-derived exosomes from AD patients, supporting the finding that “A1” reactive astrocytes induce AD pathogenesis by promoting the secretion of complement proteins (Goetzl et al., 2018; Figure 1).
Physiological Roles of S1P in The Nervous System
As mentioned in earlier sections, the generation of S1P is under precise modulation by two evolutionarily conserved lipid kinases known as the sphingosine kinase-1 (SphK1) and sphingosine kinase-2 (SphK2). The subcellular localization, as well as the biological effect of S1P, have been suggested to be determined by the two isoenzymes (Spiegel and Milstien, 2003; Chan and Pitson, 2013; Santos and Lynch, 2015). It has been demonstrated that SphK1 activation requires the re-localization from the cytoplasm to the plasma membrane (Neubauer and Pitson, 2013). Furthermore, SphK1 has been shown to mediate S1P induced effects such as cell proliferation, migration, and survival (Zhu et al., 2010; Chan and Pitson, 2013; Gassowska et al., 2014). However, another lipid kinase SphK2 has been proved to localize mainly in cellular organelles, such as the nucleus and ER, and the downstream intracellular functions mediated by SphK2 induced S1P are yet more complicated (Neubauer and Pitson, 2013). It has been shown that the SphK2 mediated S1P generation can also function as a pro-survival factor as well as a cellular apoptosis contributor without activating S1PRs (Pyne and Pyne, 2010). In this part, the S1P-mediated biological functions in the CNS will be discussed in detail.
S1P and Brain Development
In the decades following the discovery of S1P, it was regarded as an inactive end product of sphingosine metabolism. Recently immense amounts of research have now indicated the crucial role of S1P in regulating intracellular signaling transduction as an active lipid metabolite (Ghasemi et al., 2016). Both SphK1 and SphK2 have been identified as being expressed in the CNS. Studies have shown that the deficiency of either enzyme would not cause obvious CNS phenotype changes, while the deletion of both Sphk1 and Sphk2 in animals can lead to severe developmental brain defects (Mizugishi et al., 2005; Ghasemi et al., 2016). As demonstrated by Mizugishi et al. (2005), mice lacking both Sphk1 and Sphk2 can result in a deficiency of S1P production, thus leading to severe abnormalities in neurogenesis as well as angiogenesis, and ultimately impair brain development. There is also evidence showing that the single deletion of Sphk2, which leads to a reduction of nuclear S1P level, can induce abnormalities in spatial memory and fear extinction (Hait et al., 2014). These authors believed that the downregulation of Sphk2 could impair S1P-mediated endogenous inhibition of histone deacetylase (HDAC), thus decreasing histone acetylation as well as depressing the expression of cognitive related genes (Hait et al., 2014).
S1P functions in modulating brain development by binding to the widely expressed S1P receptors in a range of CNS cells, including oligodendrocytes, neurons, astrocytes and neurogenic microglia (Anelli et al., 2005; Jaillard et al., 2005; Satoh et al., 2008). Evidence demonstrates that the expression profile of S1P receptors is dynamic, changing depending on the developmental stage and the certain physiological function they exert (Jaillard et al., 2005). Moreover, growth factors have also been validated to act as modulators in determining the expression of receptors and the activation of cellular responses. For example Jung et al. (2007) have shown that platelet-derived growth factor (PDGF) can trigger the down-regulation of oligodendrocyte progenitor cells (OPCs)-mediated S1P5 expression as well as the up-regulation of S1P1, S1P2 and S1P3 expression in these cells. Although it has been demonstrated that the widely expressed S1P receptors are associated with the signaling pathways required for maintaining brain functions, the cells that express S1P receptors are mainly glial rather than neuronal. Several studies have suggested a high expression of S1P receptors (S1P3 > S1P1 > S1P2) in astrocytes, while the expression of S1P5 is relatively low (Rao et al., 2003; Anelli et al., 2005). In oligodendrocytes, however, the expression of S1P receptors presented a distinct profile among different subtypes, as S1P5 demonstrated a relatively high expression compared to the other S1P receptors (S1P1 = S1P2 > S1P3; Yu et al., 2004). Accordingly, various cellular responses have been validated to be induced by the activation of different S1P receptor subtypes (Jung et al., 2007). Novgorodov et al. (2007) have verified that S1P can inhibit the migration of OPCs by binding to S1P5, while it has also been demonstrated to promote the differentiation of oligodendrocytes through other S1P receptor subtypes (Novgorodov et al., 2007; Wang and Bieberich, 2018). Similar results have also demonstrated that S1P can interact with S1P receptors and induce the migration of neural stem progenitor cells (NSPCs) to the sites of injury (Kimura et al., 2007). Other evidence has also indicated that S1P-induced cellular responses are cell types specific, as S1P was shown to mediate neurite contraction and soma rounding in N1E-115 neuronal cells, which can not be observed in PC12 cells (Postma et al., 1996; Edsall et al., 1997).
S1P as a Neuroprotective Factor
Several lines of evidence have identified that oxidative stress leads to a range of cytopathic events, and therefore, leads to cell apoptosis in both mitochondrial-dependent as well as mitochondrial-independent ways (Sinha et al., 2013). Evidence shows that the impact of oxidative stress on cell fate is bilateral. It has been suggested that high levels of ROS can induce the activation of sphingomyelinase (SMase) and shift the “sphingolipid rheostat” towards ceramide accumulation, thus triggering cell apoptosis (Petrache and Berdyshev, 2016). Meanwhile, temperate oxidative stress can activate SphK1 and lead to the upregulation of S1P, thereby promoting cell survival (Martín-Montañez et al., 2019). The protective role of S1P against oxidative stress-induced cellular damage has been validated, as proven by Pyszko and Strosznajder that S1P treatment can reverse MPP+ mediated free radical production and cellular apoptosis (Pyszko and Strosznajder, 2014). Similar results have also demonstrated that S1P can activate the Akt pathway and inhibit the JNK pathway to alleviate H2O2-mediated growth arrest (Lee et al., 2012). Furthermore, it has been suggested that S1P could act as an antioxidant factor by enhancing the activity of superoxide dismutase and catalase (Chawla et al., 2014).
In line with the above-mentioned roles of S1P in protecting cells from oxidative stress, there are also several studies showing that S1P can act as an inhibitor of neuronal apoptosis (Edsall et al., 1997). S1P has been shown to exert anti-apoptotic effects by inhibiting oxidative stress (Ghasemi et al., 2016). S1P has also been demonstrated to prevent apoptosis by blocking Aβ toxicity (Edsall et al., 1997; Gomez-Brouchet et al., 2007). Studies have suggested that S1P may also play a two-sided role in modulating cell apoptosis since transient S1P exposure is neuroprotective, while critically high levels of S1P are neurotoxic and can trigger neuronal apoptosis (Hagen et al., 2009; van Echten-Deckert et al., 2014; Ghasemi et al., 2016). However, Hagen et al. (2009) have demonstrated that only SphK2-mediated production of S1P can lead to apoptosis, indicating the role of subcellular localization of S1P in determining its biological function. Moreover, the key molecules in S1P-related signalings, such as the AP-1, ERK, or NF-κB, also play an ambiguous role in regulating cell apoptosis, thus exerting a dual effect on cell fate (Oh et al., 2017; Park et al., 2018). One of the most important signaling pathways related to S1P-mediated anti-apoptosis is the S1PR- Jnk/p38/ERK-AP-1 cascade (O’Neill et al., 2011; Jazvinšćak Jembrek et al., 2015). S1P exerts its anti-apoptotic role mainly by activating p38 and ERK, as well as inhibiting Jnk, thus triggering the downstream inhibition of transcription activator AP-1 in the nucleus, thus preventing cell death (Hasegawa et al., 2010; Van Brocklyn and Williams, 2012). Another crucial anti-apoptotic signaling pathway that is activated by S1P is the S1PR-PI3K-Akt-Bad/GSK-3β/FOXOs pathway, the disruption of which can lead to severe AD pathogenesis (Moloney et al., 2010; Safarian et al., 2015). S1P has been shown to suppress the activation of tau kinase GSK-3β as well as inhibit the pro-apoptotic proteins Bad, FOXO1a, 3a, 4, and 6 via PI3K-Akt, functioning to protect cells from apoptosis (Santos and Lynch, 2015; Maiese, 2017). Other mediators including transcription factor NF-κB and HDACs HDAC1 and HDAC2 are also under the regulation of S1P related SL signaling (Dai et al., 2005; Hait et al., 2009). Also, S1P has been shown to inhibit acid sphingomyelinase (aSMase) mediated ceramide production, thus suppressing ceramide-induced cell apoptosis (Jazvinšćak Jembrek et al., 2015; Figure 2).
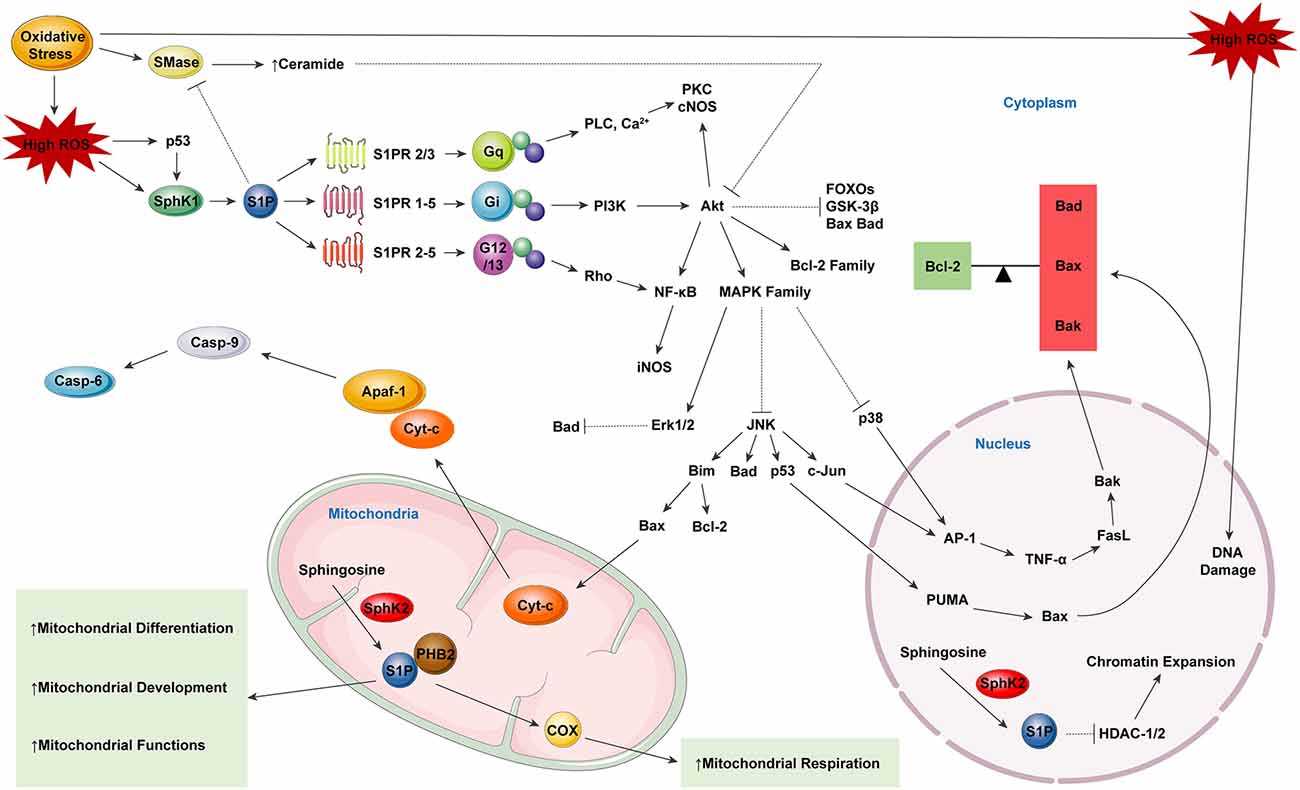
Figure 2. Sphingosine-1-phosphate (S1P) as a neuroprotector, modulating reactive oxygen species (ROS) induced cell apoptosis. S1P serves in an indirect way of protecting neural function in neurodegeneration. By binding to specific S1PRs, S1P in the cytoplasm can modulate the balance between the pro-apoptotic factors Bad, Bax, and Bak as well as the anti-apoptotic mediator Bcl-2. The equilibrium between the factors of the two major categories determine the cell fate. Besides, S1P produced in mitochondria can specifically bind to prohibitin 2 (PHB2), which in combination with cytochrome-c (Cyt-c), helps to stabilize mitochondrial assembly. S1P can also promote the differentiation, development, respiration and biological functions of mitochondria.
Mitochondria have been considered to play a crucial role in maintaining metabolic homeostasis, regulating apoptotic processes and oxidative energy metabolism, as well as modulating ROS production (Lou et al., 2020). SphK2 has also been found to be expressed in mitochondria in addition to being expressed in the nucleus, and evidence has suggested that SphK2 may function in the production of S1P in mitochondria (Chan and Pitson, 2013; Santos and Lynch, 2015; Ghasemi et al., 2016). Several lines of evidence have indicated a protective function of SphK2-induced S1P in mitochondrial homeostasis. As shown by Strub et al. (2011), S1P that is produced within mitochondria can specifically bind to prohibitin 2 (PHB2), which functions in conjunction with cytochrome c oxidase (complex V) in mitochondria and stabilizes its assembly. It has been demonstrated that SphK2 deficiency can lead to mitochondrial respiratory dysfunction and subsequent energy metabolism disorders, which can be restored by exogenous S1P treatment (Strub et al., 2011). Furthermore, S1P has been proved to promote mitochondrial biogenesis by increasing mitochondrial DNA replication, transcription and mitochondrial mass (Shen et al., 2014). There is also evidence showing that S1P treatment can alleviate oxygen-glucose deprivation (OGD) induced inner membrane depolarization, which can induce the formation of MMP (mitochondrial membrane permeability) and directly affect mitochondrial function (Agudo-López et al., 2010). S1P has been shown to play an indirect role in protecting mitochondrial function by modulating the SL rheostat. Accordingly, S1P can inhibit sphingomyelinase (SMase) enzyme activity as well as the subsequent production of ceramide, thus altering the ceramide-induced mobilization of ER Ca2+, the activity of mitochondrial respiratory complexes I and III, and also forming channels in the outer mitochondrial membrane (OMM; Kim and Sieburth, 2018; Oleinik et al., 2019).
Autophagy plays an important physiological role in the survival of cells with high energy demands such as neurons (Menzies et al., 2015). While normal levels of autophagy act as a cellular survival and adaptation program that is essential for maintaining cell homeostasis under different stress conditions, excessive autophagy can lead to autophagic cell death (Mariño et al., 2014). S1P, as one of the important bioactive SL metabolites, has been shown to regulate cellular apoptosis as well as autophagy (Wang and Bieberich, 2018). Recent studies have focused on the underlying mechanisms by which S1P-associated autophagic pathways might influence neurodegeneration (Wang and Bieberich, 2018). Several lines of evidence have indicated that the dynamic equilibrium between the formation and degradation of S1P by three enzymes (SphKs, which catalyze S1P formation, S1P phosphatases (SGPPs) and sphingosine phosphate lyase 1 (SGPL1), which catalyze S1P degradation) are of vital importance for S1P-related autophagic processes (Spiegel and Milstien, 2003; Lépine et al., 2011; Moruno Manchon et al., 2015). It has been demonstrated that SphK-1 and S1P can induce autophagy in nutrient deprivation conditions to protect cells from apoptosis (Lavieu et al., 2006). Also, similar results have been shown in primary neurons, as cytosolic S1P has been identified to modulate neuronal autophagy (Lépine et al., 2011; Moruno Manchon et al., 2015). Sheng et al. (2014) have also demonstrated that SphK2 can mediate hypoxic preconditioning-induced autophagy, and found that pretreatment with SphK2 inhibitors prevented S1P-induced apoptosis. It has been proposed that S1P may induce autophagy by inhibiting the mTOR pathway which could be considered as a potential underlying mechanism for autophagy inhibition (Orsini et al., 2019). In line with the previous studies, it has also been suggested that S1P could promote cell autophagy, as inhibition of SphK-1 with SK1-I increased autophagic flux and cell apoptosis (Lima et al., 2018). However, recent studies have demonstrated that SPHK1 deletion leads to upregulated autophagic flux in primary embryonic fibroblasts of mice, indicating an inhibitory role of SphKs in autophagy (Young et al., 2016). Moreover, several lines of evidence have suggested an indirect pathway by which S1P could modulate cell autophagy, as the S1P degradation product, phosphatidylethanolamine (PE), has been observed to result in deficient autophagy (Fyrst and Saba, 2008; Mitroi et al., 2017).
S1P and Neurotrophic Factors
It has been widely demonstrated that S1P interacts with various NGFsto promote cell development and survival (Ghasemi et al., 2016). Yamagata et al. (2003) have observed that S1P mediates the expression and release of glial-derived neurotrophic factor (GDNF) in cortical astrocytes, which can subsequently induce the growth and proliferation of these cells. Similar results have indicated that the S1P receptor agonist fingolimod phosphate (FTY720-P) can elevate the expression level of brain-derived neurotrophic factor (BDNF) in neurons, thus mediating a protective effect against oligomeric amyloid β-induced neurotoxicity (Doi et al., 2013). In line with these findings, FTY720-P has also been proved to induce the production of neurotrophic mediators, such as LIF, HBEGF, and IL-11 in astrocytes, as well as inhibit the expression of TNF-induced inflammatory genes (Brinkmann et al., 2002; Hoffmann et al., 2015). Furthermore, S1P is shown to mediate NGF-induced neurofilament expression and cell survival, while inhibition of Sphk activity suppressed NGF mediated cell differentiation (Edsall et al., 1997). Moreover, Hall et al. (1988) have identified the S1P decrease, that resulted from the shifting of the “sphingolipid rheostat” towards ceramide, could inhibit NGF-induced neurite outgrowth. In addition to the mentioned roles of S1P in modulating NGF-mediated biological response, it has also been demonstrated that NGF can, in turn, stimulate Sphk1 through TrkA signaling to elevate S1P expression, thus activating S1P1 and S1P2 to promote neurite extension (Toman et al., 2004). Similar results have also demonstrated that the GDNF can increase the expression of SphK1, thereby promoting the production and secretion of S1P in a neuroblastoma cell line (TGW; Murakami et al., 2011).
S1P and Neurotransmitter
Several lines of evidence have indicated the role of S1P in regulating neurotransmitter release as well as neuronal excitability in neural tissues, among which regulation mainly involves modulation of epinephrine and glutamate synaptic transmission. Recent studies have demonstrated that S1P is involved in bortezomib-induced neuropathic pain by increasing presynaptic glutamate release; this effect was later shown to be mediated by S1PR signaling (Sheng et al., 2014; Boyette-Davis et al., 2015; shown in Table 1). In addition to this, there is a wealth of evidence indicating that exogenous, as well as endogenous, S1P can stimulate glutamate release from the excitatory synaptic terminals (Kajimoto et al., 2007). It has also been shown that the S1P-induced hippocampal glutamate release is exclusively mediated by S1P3 (Kajimoto et al., 2007; Kanno and Nishizaki, 2011). This is in line with the evidence showing that in vitro treatment of Sphk and S1P in hippocampal slices or cultures can demonstrate increased glutamate release from synaptosomes as well as an elevated rate of spontaneous glutamate transmission (Darios et al., 2009). Furthermore, S1P is also validated to mediate glutamate release through an intrinsic pathway, as deletion of SphK-1 can lead to a decrease in activated synaptic glutamate transmitters, thereby impairing the long term-potentiation as well as causing cognitive dysfunction (Kajimoto et al., 2007; Chan and Sieburth, 2012). Schenk et al. (2005) have proposed that S1P induced endogenous glutamate release may function in activating presynaptic AMPA receptors to mediate the dispersion of SynI, which plays a crucial role in modulating exocytosis. In contrast, it has also been suggested that S1P may inhibit glutamate transmission through S1P1 signaling in cortical regions (Sim-Selley et al., 2009), which suggests that S1P may function in a region-specific way to modulate glutamatergic neurotransmission (Welch et al., 2012). In addition to this, a role for S1P in depolarization mediated release of noradrenaline has also been indicated (Alemany et al., 2001).
Crosstalk Between S1P and The Pathogenesis of Alzheimer’s Disease
Alterations in SL metabolism, as well as composition, have been considered as crucial contributors to neurodegeneration (Wang and Bieberich, 2018). AD-related lesions including synaptic loss, aberrant Aβ aggregations, NFTs, gliosis, and neuroinflammation have all been demonstrated to promote cerebral atrophy and cognitive impairment (van der Kant et al., 2020). It has been shown that S1P participates in a vast range of pathological conditions such as cancer, autoimmunity, cardiovascular diseases and diabetes (Proia and Hla, 2015), and recent studies have also indicated a key role of S1P in neurodegeneration (Chakrabarti et al., 2016; Karunakaran and van Echten-Deckert, 2017). Abnormal SL metabolism was initially observed in brain samples from AD patients when compared to age-matched normal individuals, with the S1P level being found to decrease with elevated ceramide expression (He et al., 2010). This is in line with cellular studies suggesting that Aβ treatment could drive the “sphingolipid rheostat” towards ceramide and lead to a reduction in S1P level of neuronal and glial cells (Lee et al., 2004; Gomez-Brouchet et al., 2007). Recent studies have identified a decreased level of SphK1 and S1P coupled with elevated SPL in the entorhinal cortex of AD patients, further illustrating the deregulation of SL metabolism in AD pathogenesis (Ceccom et al., 2014). SphK1 overexpression has been identified to switch the “sphingolipid rheostat” toward S1P production, thus protecting the neurons against Aβ-induced neurotoxicity (Yang et al., 2014). This effect was later shown to be mediated by the S1P-induced inhibition of acid sphingomyelinase (aSMase) activation as well as the subsequent ceramide production (Claus et al., 2009; Justice et al., 2018). In line with these observations, He et al. (2010) have identified decreased S1P expression with elevated Aβ peptides and phosphorylated tau protein in AD brains. Besides, the authors showed that in vitro treatment of neuronal cells with Aβ induced S1P inhibition and neuronal apoptosis (He et al., 2010).
In contrast to the identified reduction of SphK1 expression, the role of SphK2 in AD brains remains controversial. Several lines of evidence have suggested that there is an increase in SphK2 activity in the frontal cortex while SphK2 expression inhibition was observed in the temporal cortex and hippocampus of AD brains (Takasugi et al., 2011; Maceyka et al., 2012; Asle-Rousta et al., 2013). Dominguez et al. (2018) have demonstrated the re-localization of SphK2 from the cytosol to the nucleus relative to Aβ pathology, leading to the upregulation of intranuclear S1P expression as well as a series of deleterious responses. This is following the evidence that S1P functions by indirectly modulating β-site APP cleaving enzyme-1 (β-secretase, BACE1), which is considered as the rate-limiting enzyme for amyloid-β peptide (Aβ) production (Takasugi et al., 2011; Maceyka et al., 2012). Since SphK2 and S1P have been indicated to regulate the expression of HDAC1/2, there is also evidence suggesting that S1P acts as an underlying epigenetic regulator in AD-related cognitive dysfunction. Graff et al. (2012) have demonstrated in their study that elevated levels of HDAC2 could epigenetically repress the synaptic genes that related to cognitive deficits, while Panikker et al. (2018) suggested that decreasing HDAC2 levels in AD-related APP brain could reverse the neuroepigenetic changes in activating synaptic plasticity genes, as well as restoring brain morphology and cognition (Graff et al., 2012; Panikker et al., 2018).
Conclusion
LPLs are a cluster of bioactive intermediates, among which the well-characterized LPA and S1P have been identified as playing a crucial role in a series of cellular responses such as neurogenesis, proliferation, survival, cytoskeleton remodeling, morphological changes, migration and differentiation through the G protein-coupled receptor signaling. Several years of research have identified the importance of well-regulated LPA and S1P metabolism in maintaining neuronal health as an integrative process. Dysfunction of the regulating circuits of the two best-characterized signaling lipids has been verified to be a participant in a range of pathological processes, especially the neurodegeneration. Herein, we have demonstrated plausible mechanisms by which LPA and S1P modulate AD-related pathologies. Meanwhile more specific studies are still needed to explore the remaining questions such as as: (1) what are the exact functional roles of LPA and S1P in modulating physiological responses in the CNS? (2) Is LPL disruption the primary initiator in promoting AD pathogenesis? (3) How do LPLs work in an integrative mechanism to promote AD development? and (4) Is there any way to prevent LPL-related neurodegeneration? Due to the wide distributions of LPLs and their signaling pathways in CNS, a wide range of work is required in the future to illustrate their physiological activities.
Author Contributions
YH and MC conceptualized the manuscript. YH wrote the initial draft. YH, MG, YF, QD and MC contributed substantially to discussions of the article content and review and/or editing of the manuscript before submission.
Funding
This study was supported by the National Natural Science Foundation of China (81870915, 81971013) and grants from the Science and Technology Commission of Shanghai Municipality (16411970200).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agudo-López, A., Miguel, B. G., Fernández, I., and Martínez, A. M. (2010). Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia. Neurosci. Lett. 470, 130–133. doi: 10.1016/j.neulet.2009.12.070
Akahoshi, N., Ishizaki, Y., Yasuda, H., Murashima, Y. L., Shinba, T., Goto, K., et al. (2011). Frequent spontaneous seizures followed by spatial working memory/anxiety deficits in mice lacking sphingosine 1-phosphate receptor 2. Epilepsy Behav. 22, 659–665. doi: 10.1016/j.yebeh.2011.09.002
Alemany, R., Kleuser, B., Ruwisch, L., Danneberg, K., Lass, H., Hashemi, R., et al. (2001). Depolarisation induces rapid and transient formation of intracellular sphingosine-1-phosphate. FEBS Lett. 509, 239–244. doi: 10.1016/s0014-5793(01)03168-4
Alvarez, S. E., Harikumar, K. B., Hait, N. C., Allegood, J., Strub, G. M., Kim, E. Y., et al. (2010). Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088. doi: 10.1038/nature09128
Anelli, V., Bassi, R., Tettamanti, G., Viani, P., and Riboni, L. (2005). Extracellular release of newly synthesized sphingosine-1-phosphate by cerebellar granule cells and astrocytes. J. Neurochem. 92, 1204–1215. doi: 10.1111/j.1471-4159.2004.02955.x
Asle-Rousta, M., Oryan, S., Ahmadiani, A., and Rahnema, M. (2013). Activation of sphingosine 1-phosphate receptor-1 by SEW2871 improves cognitive function in Alzheimer’s disease model rats. EXCLI J. 12, 449–461. doi: 10.17877/DE290R-7354
Awada, R., Saulnier-Blache, J. S., Grès, S., Bourdon, E., Rondeau, P., Parimisetty, A., et al. (2014). Autotaxin downregulates LPS-induced microglia activation and pro-inflammatory cytokines production. J. Cell Biochem. 115, 2123–2132. doi: 10.1002/jcb.24889
Boyette-Davis, J. A., Walters, E. T., and Dougherty, P. M. (2015). Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain Manag. 5, 285–296. doi: 10.2217/pmt.15.19
Brinkmann, V., Davis, M. D., Heise, C. E., Albert, R., Cottens, S., Hof, R., et al. (2002). The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 277, 21453–21457. doi: 10.1074/jbc.c200176200
Broggini, T., Nitsch, R., and Savaskan, N. E. (2010). Plasticity-related gene 5 (PRG5) induces filopodia and neurite growth and impedes lysophosphatidic acid- and nogo-A-mediated axonal retraction. Mol. Biol. Cell 21, 521–537. doi: 10.1091/mbc.e09-06-0506
Castilla-Ortega, E., Hoyo-Becerra, C., Pedraza, C., Chun, J., Rodríguez De Fonseca, F., Estivill-Torrús, G., et al. (2011). Aggravation of chronic stress effects on hippocampal neurogenesis and spatial memory in LPA1 receptor knockout mice. PLoS One 6:e25522. doi: 10.1371/journal.pone.0025522
Ceccom, J., Loukh, N., Lauwers-Cances, V., Touriol, C., Nicaise, Y., Gentil, C., et al. (2014). Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol. Commun. 2:12. doi: 10.1186/2051-5960-2-12
Chakrabarti, S. S., Bir, A., Poddar, J., Sinha, M., Ganguly, A., and Chakrabarti, S. (2016). Ceramide and sphingosine-1-phosphate in cell death pathways: relevance to the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 13, 1232–1248. doi: 10.2174/1567205013666160603004239
Chan, H., and Pitson, S. M. (2013). Post-translational regulation of sphingosine kinases. Biochim. Biophys. Acta 1831, 147–156. doi: 10.1016/j.bbalip.2012.07.005
Chan, J. P., and Sieburth, D. (2012). Localized sphingolipid signaling at presynaptic terminals is regulated by calcium influx and promotes recruitment of priming factors. J. Neurosci. 32, 17909–17920. doi: 10.1523/JNEUROSCI.2808-12.2012
Chawla, S., Sahni, C., Tulsawani, R., Singh, M., Saraswat, D., Bansal, A., et al. (2014). Exogenous sphingosine 1-phosphate protects murine splenocytes against hypoxia-induced injury. Lipids 49, 191–202. doi: 10.1007/s11745-013-3860-9
Chipuk, J. E., McStay, G. P., Bharti, A., Kuwana, T., Clarke, C. J., Siskind, L. M., et al. (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. doi: 10.1016/j.cell.2012.01.038
Choi, J. W., and Chun, J. (2013). Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 1831, 20–32. doi: 10.1016/j.bbalip.2012.07.015
Choi, J. W., Gardell, S. E., Herr, D. R., Rivera, R., Lee, C. W., Noguchi, K., et al. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U S A 108, 751–756. doi: 10.1073/pnas.1014154108
Choi, J. W., Herr, D. R., Noguchi, K., Yung, Y. C., Lee, C. W., Mutoh, T., et al. (2010). LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 50, 157–186. doi: 10.1146/annurev.pharmtox.010909.105753
Chun, J., Goetzl, E. J., Hla, T., Igarashi, Y., Lynch, K. R., Moolenaar, W., et al. (2002). International union of pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 54, 265–269. doi: 10.1124/pr.54.2.265
Clair, T., Aoki, J., Koh, E., Bandle, R. W., Nam, S. W., Ptaszynska, M. M., et al. (2003). Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res. 63, 5446–5453.
Claus, R. A., Dorer, M. J., Bunck, A. C., and Deigner, H. P. (2009). Inhibition of sphingomyelin hydrolysis: targeting the lipid mediator ceramide as a key regulator of cellular fate. Curr. Med. Chem. 16, 1978–2000. doi: 10.2174/092986709788682182
Coiro, P., Stoenica, L., Strauss, U., and Bräuer, A. U. (2014). Plasticity-related gene 5 promotes spine formation in murine hippocampal neurons. J. Biol. Chem. 289, 24956–24970. doi: 10.1074/jbc.m114.597880
Contos, J. J., Fukushima, N., Weiner, J. A., Kaushal, D., and Chun, J. (2000). Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. U S A 97, 13384–13389. doi: 10.1073/pnas.97.24.13384
Cui, Q. L., Fang, J., Kennedy, T. E., Almazan, G., and Antel, J. P. (2014). Role of p38MAPK in S1P receptor-mediated differentiation of human oligodendrocyte progenitors. Glia 62, 1361–1375. doi: 10.1002/glia.22688
Cunningham, M. O., Hunt, J., Middleton, S., LeBeau, F. E., Gillies, M. J., Davies, C. H., et al. (2006). Region-specific reduction in entorhinal γ oscillations and parvalbumin-immunoreactive neurons in animal models of psychiatric illness. J. Neurosci. 26, 2767–2776. doi: 10.1523/JNEUROSCI.5054-05.2006
Czubowicz, K., Jśko, H., Wencel, P., Lukiw, W. J., and Strosznajder, R. P. (2019). The role of ceramide and sphingosine-1-phosphate in Alzheimer’s disease and other neurodegenerative disorders. Mol. Neurobiol. 56, 5436–5455. doi: 10.1007/s12035-018-1448-3
Dai, Y., Rahmani, M., Dent, P., and Grant, S. (2005). Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell. Biol. 25, 5429–5444. doi: 10.1128/mcb.25.13.5429-5444.2005
Dani, M., Wood, M., Mizoguchi, R., Fan, Z., Walker, Z., Morgan, R., et al. (2018). Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754. doi: 10.1093/brain/awy188
Dansokho, C., and Heneka, M. T. (2018). Neuroinflammatory responses in Alzheimer’s disease. J. Neural Transm. 125, 771–779. doi: 10.1007/s00702-017-1831-7
Darios, F., Wasser, C., Shakirzyanova, A., Giniatullin, A., Goodman, K., Munoz-Bravo, J. L., et al. (2009). Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron 62, 683–694. doi: 10.1016/j.neuron.2009.04.024
Doi, Y., Takeuchi, H., Horiuchi, H., Hanyu, T., Kawanokuchi, J., Jin, S., et al. (2013). Fingolimod phosphate attenuates oligomeric amyloid β-induced neurotoxicity via increased brain-derived neurotrophic factor expression in neurons. PLoS One 8:e61988. doi: 10.1371/journal.pone.0061988
Dominguez, G., Maddelein, M. L., Pucelle, M., Nicaise, Y., Maurage, C. A., Duyckaerts, C., et al. (2018). Neuronal sphingosine kinase 2 subcellular localization is altered in Alzheimer’s disease brain. Acta Neuropathol. Commun. 6:25. doi: 10.1186/s40478-018-0527-z
Dottori, M., Leung, J., Turnley, A. M., and Pébay, A. (2008). Lysophosphatidic acid inhibits neuronal differentiation of neural stem/progenitor cells derived from human embryonic stem cells. Stem Cells 26, 1146–1154. doi: 10.1634/stemcells.2007-1118
E Spohr, T. C., Dezonne, R. S., Rehen, S. K., and Gomes, F. C. (2011). Astrocytes treated by lysophosphatidic acid induce axonal outgrowth of cortical progenitors through extracellular matrix protein and epidermal growth factor signaling pathway. J. Neurochem. 119, 113–123. doi: 10.1111/j.1471-4159.2011.07421.x
Edsall, L. C., Pirianov, G. G., and Spiegel, S. (1997). Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J. Neurosci. 17, 6952–6960. doi: 10.1523/JNEUROSCI.17-18-06952.1997
Elmes, S. J., Millns, P. J., Smart, D., Kendall, D. A., and Chapman, V. (2004). Evidence for biological effects of exogenous LPA on rat primary afferent and spinal cord neurons. Brain Res. 1022, 205–213. doi: 10.1016/j.brainres.2004.07.005
Evans, D. B., Rank, K. B., Bhattacharya, K., Thomsen, D. R., Gurney, M. E., and Sharma, S. K. (2000). Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. 275, 24977–24983. doi: 10.1074/jbc.m000808200
Fassbender, K., Walter, S., Kühl, S., Landmann, R., Ishii, K., Bertsch, T., et al. (2004). The LPS receptor (CD14) links innate immunity with Alzheimers disease. FASEB J. 18, 203–205. doi: 10.1096/fj.03-0364fje
Fox, M. A., Colello, R. J., Macklin, W. B., and Fuss, B. (2003). Phosphodiesterase-Iα/autotaxin: a counteradhesive protein expressed by oligodendrocytes during onset of myelination. Mol. Cell. Neurosci. 23, 507–519. doi: 10.1016/s1044-7431(03)00073-3
Frisardi, V., Panza, F., Seripa, D., Farooqui, T., and Farooqui, A. A. (2011). Glycerophospholipids and glycerophospholipid-derived lipid mediators: a complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 50, 313–330. doi: 10.1016/j.plipres.2011.06.001
Fujiwara, Y., Sebök, A., Meakin, S., Kobayashi, T., Murakami-Murofushi, K., and Tigyi, G. (2003). Cyclic phosphatidic acid elicits neurotrophin-like actions in embryonic hippocampal neurons. J. Neurochem. 87, 1272–1283. doi: 10.1046/j.1471-4159.2003.02106.x
Fukushima, N., Furuta, D., and Tsujiuchi, T. (2011). Coordinated interactions between actin and microtubules through crosslinkers in neurite retraction induced by lysophosphatidic acid. Neurochem. Int. 59, 109–113. doi: 10.1016/j.neuint.2011.04.020
Fukushima, N., Ishii, I., Habara, Y., Allen, C. B., and Chun, J. (2002a). Dual regulation of actin rearrangement through lysophosphatidic acid receptor in neuroblast cell lines: actin depolymerization by Ca2+-α-actinin and polymerization by rho. Mol. Biol. Cell 13, 2692–2705. doi: 10.1091/mbc.01-09-0465
Fukushima, N., Weiner, J. A., Kaushal, D., Contos, J. J., Rehen, S. K., Kingsbury, M. A., et al. (2002b). Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol. Cell. Neurosci. 20, 271–282. doi: 10.1006/mcne.2002.1123
Fukushima, N., and Morita, Y. (2006). Actomyosin-dependent microtubule rearrangement in lysophosphatidic acid-induced neurite remodeling of young cortical neurons. Brain Res. 1094, 65–75. doi: 10.1016/j.brainres.2006.04.007
Furukawa, A., Kita, K., Toyomoto, M., Fujii, S., Inoue, S., Hayashi, K., et al. (2007). Production of nerve growth factor enhanced in cultured mouse astrocytes by glycerophospholipids, sphingolipids, and their related compounds. Mol. Cell. Biochem. 305, 27–34. doi: 10.1007/s11010-007-9524-4
Furuta, D., Yamane, M., Tsujiuchi, T., Moriyama, R., and Fukushima, N. (2012). Lysophosphatidic acid induces neurite branch formation through LPA3. Mol. Cell. Neurosci. 50, 21–34. doi: 10.1016/j.mcn.2012.03.006
Fyrst, H., and Saba, J. D. (2008). Sphingosine-1-phosphate lyase in development and disease: sphingolipid metabolism takes flight. Biochim. Biophys. Acta 1781, 448–458. doi: 10.1016/j.bbalip.2008.05.005
García-Díaz, B., Riquelme, R., Varela-Nieto, I., Jiménez, A. J., de Diego, I., Gómez-Conde, A. I., et al. (2015). Loss of lysophosphatidic acid receptor LPA1 alters oligodendrocyte differentiation and myelination in the mouse cerebral cortex. Brain Struct. Funct. 220, 3701–3720. doi: 10.1007/s00429-014-0885-7
García-Morales, V., Montero, F., González-Forero, D., Rodríguez-Bey, G., Gómez-Pérez, L., Medialdea-Wandossell, M. J., et al. (2015). Membrane-derived phospholipids control synaptic neurotransmission and plasticity. PLoS Biol. 13:e1002153. doi: 10.1371/journal.pbio.1002153
Gassowska, M., Cieslik, M., Wilkaniec, A., and Strosznajder, J. B. (2014). Sphingosine kinases/sphingosine-1-phosphate and death Signalling in APP-transfected cells. Neurochem. Res. 39, 645–652. doi: 10.1007/s11064-014-1240-3
Ghasemi, R., Dargahi, L., and Ahmadiani, A. (2016). Integrated sphingosine-1 phosphate signaling in the central nervous system: from physiological equilibrium to pathological damage. Pharmacol. Res. 104, 156–164. doi: 10.1016/j.phrs.2015.11.006
Goetzl, E. J., Schwartz, J. B., Abner, E. L., Jicha, G. A., and Kapogiannis, D. (2018). High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 83, 544–552. doi: 10.1002/ana.25172
Goldshmit, Y., Munro, K., Leong, S. Y., Pébay, A., and Turnley, A. M. (2010). LPA receptor expression in the central nervous system in health and following injury. Cell Tissue Res. 341, 23–32. doi: 10.1007/s00441-010-0977-5
Gomez-Brouchet, A., Pchejetski, D., Brizuela, L., Garcia, V., Altié, M. F., Maddelein, M. L., et al. (2007). Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-β peptide. Mol. Pharmacol. 72, 341–349. doi: 10.1124/mol.106.033738
Govek, E. E., Newey, S. E., and Aelst, L. (2005). The role of the Rho GTPases in neuronal development. Genes Dev. 19, 1–49. doi: 10.1101/gad.1256405
Graff, J., Rei, D., Guan, J. S., Wang, W. Y., Seo, J., Hennig, K. M., et al. (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. doi: 10.1038/nature10849
Greenberg, S. M., Bacskai, B. J., Hernandez-Guillamon, M., Pruzin, J., Sperling, R., and Veluw, S. J. (2020). Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat Rev Neurol 16, 30–42. doi: 10.1038/s41582-019-0281-2
Hagen, N., Van Veldhoven, P. P., Proia, R. L., Park, H., Merrill, A. H. Jr., and van Echten-Deckert, G. (2009). Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 284, 11346–11353. doi: 10.1074/jbc.m807336200
Hait, N. C., Allegood, J., Maceyka, M., Strub, G. M., Harikumar, K. B., Singh, S. K., et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. doi: 10.1126/science.1176709
Hait, N. C., Wise, L. E., Allegood, J. C., O’Brien, M., Avni, D., Reeves, T. M., et al. (2014). Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci. 17, 971–980. doi: 10.1038/nn.3728
Hall, F. L., Fernyhough, P., Ishii, D. N., and Vulliet, P. R. (1988). Suppression of nerve growth factor-directed neurite outgrowth in PC12 cells by sphingosine, an inhibitor of protein kinase C. J. Biol. Chem. 263, 4460–4466.
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. J., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hamelin, L., Lagarde, J., Dorothée, G., Leroy, C., Labit, M., Comley, R. A., et al. (2016). Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264. doi: 10.1093/brain/aww017
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Hasegawa, Y., Suzuki, H., Sozen, T., Rolland, W., and Zhang, J. H. (2010). Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 41, 368–374. doi: 10.1161/strokeaha.109.568899
He, X., Huang, Y., Li, B., Gong, C. X., and Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
Hecht, J. H., Weiner, J. A., Post, S. R., and Chun, J. (1996). Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 135, 1071–1083. doi: 10.1083/jcb.135.4.1071
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015a). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, M. T., Golenbock, D. T., and Latz, E. (2015b). Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236. doi: 10.1038/ni.3102
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Herr, D. R., Chew, W. S., Satish, R. L., and Ong, W. Y. (2020). Pleotropic roles of autotaxin in the nervous system present opportunities for the development of novel therapeutics for neurological diseases. Mol. Neurobiol. 57, 372–392. doi: 10.1007/s12035-019-01719-1
Hoffmann, F. S., Hofereiter, J., Rübsamen, H., Melms, J., Schwarz, S., Faber, H., et al. (2015). Fingolimod induces neuroprotective factors in human astrocytes. J. Neuroinflammation 12:184. doi: 10.1186/s12974-015-0393-6
Jaillard, C., Harrison, S., Stankoff, B., Aigrot, M. S., Calver, A. R., Duddy, G., et al. (2005). Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 25, 1459–1469. doi: 10.1523/JNEUROSCI.4645-04.2005
Jana, M., Palencia, C. A., and Pahan, K. (2008). Fibrillar amyloid-β peptides activate microglia via TLR2: implications for Alzheimer’s disease. J. Immunol. 181, 7254–7262. doi: 10.4049/jimmunol.181.10.7254
Jang, Y., Lee, M. H., Lee, J., Jung, J., Lee, S. H., Yang, D. J., et al. (2014). TRPM2 mediates the lysophosphatidic acid-induced neurite retraction in the developing brain. Pflugers Arch. 466, 1987–1998. doi: 10.1007/s00424-013-1436-4
Jazvinšćak Jembrek, M., Hof, P. R., and Šimić, G. (2015). Ceramides in Alzheimer’s disease: key mediators of neuronal apoptosis induced by oxidative stress and Aβ accumulation. Oxid. Med. Cell. Longev. 2015:346783. doi: 10.1155/2015/346783
Ji, K., Akgul, G., Wollmuth, L. P., and Tsirka, S. E. (2013). Microglia actively regulate the number of functional synapses. PLoS One 8:e56293. doi: 10.1371/journal.pone.0056293
Jung, C. G., Kim, H. J., Miron, V. E., Cook, S., Kennedy, T. E., Foster, C. A., et al. (2007). Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 55, 1656–1667. doi: 10.1002/glia.20576
Justice, M. J., Bronova, I., Schweitzer, K. S., Poirier, C., Blum, J. S., Berdyshev, E. V., et al. (2018). Inhibition of acid sphingomyelinase disrupts LYNUS signaling and triggers autophagy. J. Lipid Res. 59, 596–606. doi: 10.1194/jlr.m080242
Kajimoto, T., Okada, T., Yu, H., Goparaju, S. K., Jahangeer, S., and Nakamura, S. (2007). Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol. Cell. Biol. 27, 3429–3440. doi: 10.1128/mcb.01465-06
Kanno, T., and Nishizaki, T. (2011). Endogenous sphingosine 1-phosphate regulates spontaneous glutamate release from mossy fiber terminals via S1P3 receptors. Life Sci. 89, 137–140. doi: 10.1016/j.lfs.2011.05.021
Karunakaran, I., and van Echten-Deckert, G. (2017). Sphingosine 1-phosphate—a double edged sword in the brain. Biochim. Biophys. Acta Biomembr. 1859, 1573–1582. doi: 10.1016/j.bbamem.2017.03.008
Kihara, Y., Maceyka, M., Spiegel, S., and Chun, J. (2014). Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 171, 3575–3594. doi: 10.1111/bph.12678
Kim, S., and Sieburth, D. (2018). Sphingosine kinase activates the mitochondrial unfolded protein response and is targeted to mitochondria by stress. Cell Rep. 24, 2932.e4–2945.e4. doi: 10.1016/j.celrep.2018.08.037
Kimura, A., Ohmori, T., Kashiwakura, Y., Ohkawa, R., Madoiwa, S., Mimuro, J., et al. (2008). Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain. Stroke 39, 3411–3417. doi: 10.1161/strokeaha.108.514612
Kimura, A., Ohmori, T., Ohkawa, R., Madoiwa, S., Mimuro, J., Murakami, T., et al. (2007). Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem Cells 25, 115–124. doi: 10.1634/stemcells.2006-0223
Koike, S., Yutoh, Y., Keino-Masu, K., Noji, S., Masu, M., and Ohuchi, H. (2011). Autotaxin is required for the cranial neural tube closure and establishment of the midbrain-hindbrain boundary during mouse development. Dev. Dyn. 240, 413–421. doi: 10.1002/dvdy.22543
Kulijewicz-Nawrot, M., Verkhratsky, A., Chvátal, A., Syková, E., and Rodríguez, J. J. (2012). Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J. Anat. 221, 252–262. doi: 10.1111/j.1469-7580.2012.01536.x
Kurabayashi, N., Tanaka, A., Nguyen, M. D., and Sanada, K. (2018). The LPA-LPA4 axis is required for establishment of bipolar morphology and radial migration of newborn cortical neurons. Development 145:dev162529. doi: 10.1242/dev.162529
Kwon, J. H., Gaire, B. P., Park, S. J., Shin, D. Y., and Choi, J. W. (2018). Identifying lysophosphatidic acid receptor subtype 1 (LPA1) as a novel factor to modulate microglial activation and their TNF-α production by activating ERK1/2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 1237–1245. doi: 10.1016/j.bbalip.2018.07.015
Lavieu, G., Scarlatti, F., Sala, G., Carpentier, S., Levade, T., Ghidoni, R., et al. (2006). Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 281, 8518–8527. doi: 10.1074/jbc.m506182200
Lee, J. P., Cha, H. J., Lee, K. S., Lee, K. K., Son, J. H., Kim, K. N., et al. (2012). Phytosphingosine-1-phosphate represses the hydrogen peroxide-induced activation of c-Jun N-terminal kinase in human dermal fibroblasts through the phosphatidylinositol 3-kinase/Akt pathway. Arch. Dermatol. Res. 304, 673–678. doi: 10.1007/s00403-012-1241-5
Lee, J. T., Xu, J., Lee, J. M., Ku, G., Han, X., Yang, D. I., et al. (2004). Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 164, 123–131. doi: 10.1083/jcb.200307017
Lépine, S., Allegood, J. C., Park, M., Dent, P., Milstien, S., and Spiegel, S. (2011). Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 18, 350–361. doi: 10.1038/cdd.2010.104
Li, Y. F., Li, R. S., Samuel, S. B., Cueto, R., Li, X. Y., Wang, H., et al. (2016). Lysophospholipids and their G protein-coupled receptors in atherosclerosis. Front. Biosci. 21, 70–88. doi: 10.2741/4377
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lima, S., Takabe, K., Newton, J., Saurabh, K., Young, M. M., Leopoldino, A. M., et al. (2018). TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 14, 942–957. doi: 10.1080/15548627.2018.1429875
Lin, M. E., Rivera, R. R., and Chun, J. (2012). Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J. Biol. Chem. 287, 17608–17617. doi: 10.1074/jbc.m111.330183
Lou, G., Palikaras, K., Lautrup, S., Scheibye-Knudsen, M., Tavernarakis, N., and Fang, E. F. (2020). Mitophagy and neuroprotection. Trends Mol. Med. 26, 8–20. doi: 10.1016/j.molmed.2019.07.002
Ma, L., Uchida, H., Nagai, J., Inoue, M., Chun, J., Aoki, J., et al. (2009). Lysophosphatidic acid-3 receptor-mediated feed-forward production of lysophosphatidic acid: an initiator of nerve injury-induced neuropathic pain. Mol. Pain 5:64. doi: 10.1186/1744-8069-5-64
Maceyka, M., Harikumar, K. B., Milstien, S., and Spiegel, S. (2012). Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 22, 50–60. doi: 10.1016/j.tcb.2011.09.003
Maceyka, M., and Spiegel, S. (2014). Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67. doi: 10.1038/nature13475
MacLennan, A. J., Carney, P. R., Zhu, W. J., Chaves, A. H., Garcia, J., Grimes, J. R., et al. (2001). An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur. J. Neurosci. 14, 203–209. doi: 10.1046/j.0953-816x.2001.01634.x
Maiese, K. (2017). Forkhead transcription factors: formulating a FOXO target for cognitive loss. Curr. Neurovasc. Res. 14, 415–420. doi: 10.2174/1567202614666171116102911
Mariño, G., Niso-Santano, M., Baehrecke, E. H., and Kroemer, G. (2014). Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94. doi: 10.1038/nrm3735
Martín-Montañez, E., Pavia, J., Valverde, N., Boraldi, F., Lara, E., Oliver, B., et al. (2019). The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 137, 116–130. doi: 10.1016/j.freeradbiomed.2019.04.022
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Miron, V. E., Ludwin, S. K., Darlington, P. J., Jarjour, A. A., Soliven, B., Kennedy, T. E., et al. (2010). Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol. 176, 2682–2694. doi: 10.2353/ajpath.2010.091234
Mitroi, D. N., Karunakaran, I., Gräler, M., Saba, J. D., Ehninger, D., Ledesma, M. D., et al. (2017). SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 13, 885–899. doi: 10.1080/15548627.2017.1291471
Mizugishi, K., Yamashita, T., Olivera, A., Miller, G. F., Spiegel, S., and Proia, R. L. (2005). Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 25, 11113–11121. doi: 10.1128/mcb.25.24.11113-11121.2005
Moloney, A. M., Griffin, R. J., Timmons, S., O’Connor, R., Ravid, R., and O’Neill, C. (2010). Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243. doi: 10.1016/j.neurobiolaging.2008.04.002
Moolenaar, W. H., Houben, A. J., Lee, S. J., and van Meeteren, L. A. (2013). Autotaxin in embryonic development. Biochim. Biophys. Acta 1831, 13–19. doi: 10.1016/j.bbalip.2012.09.013
Morales, I., Jiménez, J. M., Mancilla, M., and Maccioni, R. B. (2013). Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 37, 849–856. doi: 10.3233/jad-131843
Moruno Manchon, J. F., Uzor, N. E., Dabaghian, Y., Furr-Stimming, E. E., Finkbeiner, S., and Tsvetkov, A. S. (2015). Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci. Rep. 5:15213. doi: 10.1038/srep15213
Muessel, M. J., Harry, G. J., Armstrong, D. L., and Storey, N. M. (2013). SDF-1α and LPA modulate microglia potassium channels through rho gtpases to regulate cell morphology. Glia 61, 1620–1628. doi: 10.1002/glia.22543
Murakami, M., Ito, H., Hagiwara, K., Kobayashi, M., Hoshikawa, A., Takagi, A., et al. (2011). Sphingosine kinase 1/S1P pathway involvement in the GDNF-induced GAP43 transcription. J. Cell. Biochem. 112, 3449–3458. doi: 10.1002/jcb.23275
Musazzi, L., Di Daniel, E., Maycox, P., Racagni, G., and Popoli, M. (2011). Abnormalities in α/β-CaMKII and related mechanisms suggest synaptic dysfunction in hippocampus of LPA1 receptor knockout mice. Int. J. Neuropsychopharmacol. 14, 941–953. doi: 10.1017/s1461145710001240
Neubauer, H. A., and Pitson, S. M. (2013). Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J. 280, 5317–5336. doi: 10.1111/febs.12314
Nilson, A. N., English, K. C., Gerson, J. E., Barton Whittle, T., Nicolas Crain, C., Xue, J., et al. (2017). Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J. Alzheimers Dis. 55, 1083–1099. doi: 10.3233/jad-160912
Nogaroli, L., Yuelling, L. M., Dennis, J., Gorse, K., Payne, S. G., and Fuss, B. (2009). Lysophosphatidic acid can support the formation of membranous structures and an increase in MBP mRNA levels in differentiating oligodendrocytes. Neurochem. Res. 34, 182–193. doi: 10.1007/s11064-008-9772-z
Novgorodov, A. S., El-Alwani, M., Bielawski, J., Obeid, L. M., and Gudz, T. I. (2007). Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 21, 1503–1514. doi: 10.1096/fj.06-7420com
Oh, Y. T., Yue, P., and Sun, S. Y. (2017). DR5 suppression induces sphingosine-1-phosphate-dependent TRAF2 polyubiquitination, leading to activation of JNK/AP-1 and promotion of cancer cell invasion. Cell Commun. Signal. 15:18. doi: 10.1186/s12964-017-0174-1
Olabarria, M., Noristani, H. N., Verkhratsky, A., and Rodríguez, J. J. (2010). Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838. doi: 10.1002/glia.20967
Oleinik, N., Kim, J., Roth, B. M., Selvam, S. P., Gooz, M., Johnson, R. H., et al. (2019). Mitochondrial protein import is regulated by p17/PERMIT to mediate lipid metabolism and cellular stress. Sci. Adv. 5:eaax1978. doi: 10.1126/sciadv.aax1978
O’Neill, S. M., Houck, K. L., Yun, J. K., Fox, T. E., and Kester, M. (2011). AP-1 binding transcriptionally regulates human neutral ceramidase. Arch Biochem. Biophys. 511, 31–39. doi: 10.1016/j.abb.2011.04.009
Orsini, M., Chateauvieux, S., Rhim, J., Gaigneaux, A., Cheillan, D., Christov, C., et al. (2019). Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells. Cell Death Differ. 26, 1796–1812. doi: 10.1038/s41418-018-0245-x
Panikker, P., Xu, S. J., Zhang, H., Sarthi, J., Beaver, M., Sheth, A., et al. (2018). Restoring Tip60 HAT/HDAC2 balance in the neurodegenerative brain relieves epigenetic transcriptional repression and reinstates cognition. J. Neurosci. 38, 4569–4583. doi: 10.1523/JNEUROSCI.2840-17.2018
Park, S. J., Kim, J. M., Kim, J., Hur, J., Park, S., Kim, K., et al. (2018). Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. U S A 115, E11721–E11730. doi: 10.1073/pnas.1811432115
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Peñalver, A., Campos-Sandoval, J. A., Blanco, E., Cardona, C., Castilla, L., Martín-Rufián, M., et al. (2017). Glutaminase and MMP-9 downregulation in cortex and hippocampus of LPA1 receptor null mice correlate with altered dendritic spine plasticity. Front. Mol. Neurosci. 10:278. doi: 10.3389/fnmol.2017.00278
Pereira, C. F., Santos, A. E., Moreira, P. I., Pereira, A. C., Sousa, F. J., Cardoso, S. M., et al. (2019). Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 56:100966. doi: 10.1016/j.arr.2019.100966
Petrache, I., and Berdyshev, E. V. (2016). Ceramide signaling and metabolism in pathophysiological states of the lung. Annu. Rev. Physiol. 78, 463–480. doi: 10.1146/annurev-physiol-021115-105221
Plastira, I., Bernhart, E., Goeritzer, M., DeVaney, T., Reicher, H., Hammer, A., et al. (2017). Lysophosphatidic acid via LPA-receptor 5/protein kinase D-dependent pathways induces a motile and pro-inflammatory microglial phenotype. J. Neuroinflammation 14:253. doi: 10.1186/s12974-017-1024-1
Plastira, I., Bernhart, E., Goeritzer, M., Reicher, H., Kumble, V. B., Kogelnik, N., et al. (2016). 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J. Neuroinflammation 13:205. doi: 10.1186/s12974-016-0701-9
Postma, F. R., Jalink, K., Hengeveld, T., and Moolenaar, W. H. (1996). Sphingosine-1-phosphate rapidly induces Rho-dependent neurite retraction: action through a specific cell surface receptor. EMBO J. 15, 2388–2392. doi: 10.1002/j.1460-2075.1996.tb00595.x
Proia, R. L., and Hla, T. (2015). Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Invest. 125, 1379–1387. doi: 10.1172/jci76369
Pyne, N. J., and Pyne, S. (2010). Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 10, 489–503. doi: 10.1038/nrc2875
Pyszko, J., and Strosznajder, J. B. (2014). Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 50, 38–48. doi: 10.1007/s12035-013-8622-4
Ramesh, S., Govindarajulu, M., Suppiramaniam, V., Moore, T., and Dhanasekaran, M. (2018). Autotaxin lysophosphatidic acid signaling in Alzheimer’s disease. Int. J. Mol. Sci. 19:E1827. doi: 10.3390/ijms19071827
Rao, T. S., Lariosa-Willingham, K. D., Lin, F. F., Palfreyman, E. L., Yu, N., Chun, J., et al. (2003). Pharmacological characterization of lysophospholipid receptor signal transduction pathways in rat cerebrocortical astrocytes. Brain Res. 990, 182–194. doi: 10.1016/s0006-8993(03)03527-3
Rhee, H. J., Nam, J. S., Sun, Y., Kim, M. J., Choi, H. K., Han, D. H., et al. (2006). Lysophosphatidic acid stimulates cAMP accumulation and cAMP response element-binding protein phosphorylation in immortalized hippocampal progenitor cells. Neuroreport 17, 523–526. doi: 10.1097/01.wnr.0000209011.16718.68
Robinson, R. (2015). One lipid, two synaptic plasticity pathways. PLoS Biol. 13:e1002154. doi: 10.1371/journal.pbio.1002154
Roza, C., Campos-Sandoval, J. A., Gómez-García, M. C., Peñalver, A., and Márquez, J. (2019). Lysophosphatidic acid and glutamatergic transmission. Front. Mol. Neurosci. 12:138. doi: 10.3389/fnmol.2019.00138
Safarian, F., Khallaghi, B., Ahmadiani, A., and Dargahi, L. (2015). Activation of S1P1 receptor regulates PI3K/Akt/FoxO3a pathway in response to oxidative stress in PC12 cells. J. Mol. Neurosci. 56, 177–187. doi: 10.1007/s12031-014-0478-1
Santin, L. J., Bilbao, A., Pedraza, C., Matas-Rico, E., López-Barroso, D., Castilla-Ortega, E., et al. (2009). Behavioral phenotype of maLPA1-null mice: increased anxiety-like behavior and spatial memory deficits. Genes Brain Behav. 8, 772–784. doi: 10.1111/j.1601-183x.2009.00524.x
Santos, W. L., and Lynch, K. R. (2015). Drugging sphingosine kinases. ACS Chem. Biol. 10, 225–233. doi: 10.1021/cb5008426
Satoh, S., Kawasaki, K., Hitomi, A., Ikegaki, I., and Asano, T. (2011). Fasudil protects cultured N1E-115 cells against lysophosphatidic acid-induced neurite retraction through inhibition of Rho-kinase. Brain Res. Bull. 84, 174–177. doi: 10.1016/j.brainresbull.2010.11.013
Satoh, S., Toshima, Y., Hitomi, A., Ikegaki, I., Seto, M., and Asano, T. (2008). Wide therapeutic time window for Rho-kinase inhibition therapy in ischemic brain damage in a rat cerebral thrombosis model. Brain Res. 1193, 102–108. doi: 10.1016/j.brainres.2007.11.050
Savitz, S. I., Dhallu, M. S., Malhotra, S., Mammis, A., Ocava, L. C., Rosenbaum, P. S., et al. (2006). EDG receptors as a potential therapeutic target in retinal ischemia-reperfusion injury. Brain Res. 1118, 168–175. doi: 10.1016/j.brainres.2006.05.060
Sayas, C. L., Avila, J., and Wandosell, F. (2002a). Glycogen synthase kinase-3 is activated in neuronal cells by Gα12 and Gα13 by Rho-independent and Rho-dependent mechanisms. J. Neurosci. 22, 6863–6875. doi: 10.1523/jneurosci.22-16-06863.2002
Sayas, C. L., Avila, J., and Wandosell, F. (2002b). Regulation of neuronal cytoskeleton by lysophosphatidic acid: role of GSK-3. Biochim. Biophys. Acta 1582, 144–153. doi: 10.1016/s1388-1981(02)00149-x
Sayas, C. L., Moreno-Flores, M. T., Avila, J., and Wandosell, F. (1999). The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like tau phosphorylation. J. Biol. Chem. 274, 37046–37052. doi: 10.1074/jbc.274.52.37046
Schenk, U., Menna, E., Kim, T., Passafaro, M., Chang, S., De Camilli, P., et al. (2005). A novel pathway for presynaptic mitogen-activated kinase activation via AMPA receptors. J. Neurosci. 25, 1654–1663. doi: 10.1523/jneurosci.3074-04.2005
Shano, S., Moriyama, R., Chun, J., and Fukushima, N. (2008). Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochem. Int. 52, 216–220. doi: 10.1016/j.neuint.2007.07.004
Shen, Z., Liu, C., Liu, P., Zhao, J., and Xu, W. (2014). Sphingosine 1-phosphate (S1P) promotes mitochondrial biogenesis in Hep G2 cells by activating Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Cell Stress Chaperones 19, 541–548. doi: 10.1007/s12192-013-0480-5
Sheng, R., Zhang, T. T., Felice, V. D., Qin, T., Qin, Z. H., Smith, C. D., et al. (2014). Preconditioning stimuli induce autophagy via sphingosine kinase 2 in mouse cortical neurons. J. Biol. Chem. 289, 20845–20857. doi: 10.1074/jbc.m114.578120
Shi, J., Dong, Y., Cui, M. Z., and Xu, X. (2013). Lysophosphatidic acid induces increased BACE1 expression and Aβ formation. Biochim. Biophys. Acta 1832, 29–38. doi: 10.1016/j.bbadis.2012.09.010
Simons, K., and Ikonen, E. (1997). Functional rafts in cell membranes. Nature 387, 569–572. doi: 10.1038/42408
Sim-Selley, L. J., Goforth, P. B., Mba, M. U., Macdonald, T. L., Lynch, K. R., Milstien, S., et al. (2009). Sphingosine-1-phosphate receptors mediate neuromodulatory functions in the CNS. J. Neurochem. 110, 1191–1202. doi: 10.1111/j.1471-4159.2009.06202.x
Sinha, K., Das, J., Pal, P. B., and Sil, P. C. (2013). Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 87, 1157–1180. doi: 10.1007/s00204-013-1034-4
Spiegel, S., and Milstien, S. (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407. doi: 10.1038/nrm1103
Spohr, T. C., Choi, J. W., Gardell, S. E., Herr, D. R., Rehen, S. K., Gomes, F. C., et al. (2008). Lysophosphatidic acid receptor-dependent secondary effects via astrocytes promote neuronal differentiation. J. Biol. Chem. 283, 7470–7479. doi: 10.1074/jbc.M707758200
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A., et al. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161. doi: 10.1038/ni.1836
Strub, G. M., Paillard, M., Liang, J., Gomez, L., Allegood, J. C., Hait, N. C., et al. (2011). Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612. doi: 10.1096/fj.10-167502
Suckau, O., Gross, I., Schrötter, S., Yang, F., Luo, J., Wree, A., et al. (2019). LPA1, LPA2, LPA4, and LPA6 receptor expression during mouse brain development. Dev. Dyn. 248, 375–395. doi: 10.1002/dvdy.23
Sun, Y. X., Minthon, L., Wallmark, A., Warkentin, S., Blennow, K., and Janciauskiene, S. (2003). Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 16, 136–144. doi: 10.1159/000071001
Swaney, J. S., Chapman, C., Correa, L. D., Stebbins, K. J., Bundey, R. A., Prodanovich, P. C., et al. (2010). A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 160, 1699–1713. doi: 10.1111/j.1476-5381.2010.00828.x
Takasugi, N., Sasaki, T., Suzuki, K., Osawa, S., Isshiki, H., Hori, Y., et al. (2011). BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 31, 6850–6857. doi: 10.1523/jneurosci.6467-10.2011
Thalman, C., Horta, G., Qiao, L., Endle, H., Tegeder, I., Cheng, H., et al. (2018). Synaptic phospholipids as a new target for cortical hyperexcitability and E/I balance in psychiatric disorders. Mol. Psychiatry 23, 1699–1710. doi: 10.1038/s41380-018-0053-1
Tigyi, G., and Miledi, R. (1992). Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. 267, 21360–21367.
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int. J. Nanomedicine 14, 5541–5554. doi: 10.2147/IJN.S200490
Toman, R. E., Payne, S. G., Watterson, K. R., Maceyka, M., Lee, N. H., Milstien, S., et al. (2004). Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J. Cell Biol. 166, 381–392. doi: 10.1083/jcb.200402016
Trimbuch, T., Beed, P., Vogt, J., Schuchmann, S., Maier, N., Kintscher, M., et al. (2009). Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell 138, 1222–1235. doi: 10.1016/j.cell.2009.06.050
Van Brocklyn, J. R., and Williams, J. B. (2012). The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 163, 26–36. doi: 10.1016/j.cbpb.2012.05.006
van der Kant, R., Goldstein, L. S. B., and Ossenkoppele, R. (2020). Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35. doi: 10.1038/s41583-019-0240-3
van Echten-Deckert, G., Hagen-Euteneuer, N., Karaca, I., and Walter, J. (2014). Sphingosine-1-phosphate: boon and bane for the brain. Cell. Physiol. Biochem. 34, 148–157. doi: 10.1159/000362991
Venegas, C., and Heneka, M. T. (2017). Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 101, 87–98. doi: 10.1189/jlb.3MR0416-204R
Vogt, J., Yang, J. W., Mobascher, A., Cheng, J., Li, Y., Liu, X., et al. (2016). Molecular cause and functional impact of altered synaptic lipid signaling due to a prg-1 gene SNP. EMBO Mol. Med. 8, 25–38. doi: 10.15252/emmm.201505677
Wang, G., and Bieberich, E. (2018). Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv. Biol. Regul. 70, 51–64. doi: 10.1016/j.jbior.2018.09.013
Weiner, J. A., Hecht, J. H., and Chun, J. (1998). Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J. Comp. Neurol. 398, 587–598. doi: 10.1002/(sici)1096-9861(19980907)398:4<587::aid-cne10>3.0.co;2-5
Welch, S. P., Sim-Selley, L. J., and Selley, D. E. (2012). Sphingosine-1-phosphate receptors as emerging targets for treatment of pain. Biochem. Pharmacol. 84, 1551–1562. doi: 10.1016/j.bcp.2012.08.010
Yamagata, K., Tagami, M., Torii, Y., Takenaga, F., Tsumagari, S., Itoh, S., et al. (2003). Sphingosine 1-phosphate induces the production of glial cell line-derived neurotrophic factor and cellular proliferation in astrocytes. Glia 41, 199–206. doi: 10.1002/glia.10180
Yamazaki, J., Katoh, H., and Negishi, M. (2008). Lysophosphatidic acid and thrombin receptors require both G α12 and G α13 to regulate axonal morphology in hippocampal neurons. Biol. Pharm. Bull. 31, 2216–2222. doi: 10.1248/bpb.31.2216
Yang, Y., Wang, M., Lv, B., Ma, R., Hu, J., Dun, Y., et al. (2014). Sphingosine kinase-1 protects differentiated N2a cells against β-amyloid25–35-induced neurotoxicity via the mitochondrial pathway. Neurochem. Res. 39, 932–940. doi: 10.1007/s11064-014-1290-6
Young, M. M., Takahashi, Y., Fox, T. E., Yun, J. K., Kester, M., and Wang, H. G. (2016). Sphingosine kinase 1 cooperates with autophagy to maintain endocytic membrane trafficking. Cell Rep. 17, 1532–1545. doi: 10.1016/j.celrep.2016.10.019
Yu, N., Lariosa-Willingham, K. D., Lin, F. F., Webb, M., and Rao, T. S. (2004). Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 45, 17–27. doi: 10.1002/glia.10297
Yung, Y. C., Stoddard, N. C., and Chun, J. (2014). LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 55, 1192–1214. doi: 10.1194/jlr.r046458
Yung, Y. C., Stoddard, N. C., Mirendil, H., and Chun, J. (2015). Lysophosphatidic acid signaling in the nervous system. Neuron 85, 669–682. doi: 10.1016/j.neuron.2015.01.009
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. doi: 10.1016/j.neuron.2015.11.013
Zhu, D., Sreekumar, P. G., Hinton, D. R., and Kannan, R. (2010). Expression and regulation of enzymes in the ceramide metabolic pathway in human retinal pigment epithelial cells and their relevance to retinal degeneration. Vision Res. 50, 643–651. doi: 10.1016/j.visres.2009.09.002
Keywords: lysophospholipids, lysophosphatidic acid (LPA), sphingosine-1-phosphate (S1P), G-protein coupled receptor (GPCR), Alzheimer’s disease (AD)
Citation: Hao Y, Guo M, Feng Y, Dong Q and Cui M (2020) Lysophospholipids and Their G-Coupled Protein Signaling in Alzheimer’s Disease: From Physiological Performance to Pathological Impairment. Front. Mol. Neurosci. 13:58. doi: 10.3389/fnmol.2020.00058
Received: 24 January 2020; Accepted: 24 March 2020;
Published: 15 April 2020.
Edited by:
Aurelio Galli, University of Alabama at Birmingham, United StatesReviewed by:
Roberta Squecco, University of Florence, ItalyPeter Hamilton, Virginia Commonwealth University, United States
Copyright © 2020 Hao, Guo, Feng, Dong and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Dong, ZG9uZ19xaWFuZ0BmdWRhbi5lZHUuY24=; Mei Cui, Y3VpbWVpQGZ1ZGFuLmVkdS5jbg==
† These authors have contributed equally to this work