 Emmanouela Leandrou
Emmanouela Leandrou Evangelia Emmanouilidou
Evangelia Emmanouilidou Kostas Vekrellis
Kostas Vekrellis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci., 09 October 2019
Sec. Brain Disease Mechanisms
Volume 12 - 2019 | https://doi.org/10.3389/fnmol.2019.00237
This article is part of the Research TopicProtein Misfolding and Spreading Pathology in Neurodegenerative DiseasesView all 12 articles
Alpha-synuclein (α-syn) is biochemically and genetically linked to Parkinson’s disease (PD) and other synucleinopathies. It is now widely accepted that α-syn can be released in the extracellular space, even though the mechanism of its release is still unclear. In addition, pathology-related aggregated species of α-syn have been shown to propagate between neurons in synaptically connected areas of the brain thereby assisting the spreading of pathology in healthy neighboring neuronal cells. In neurons, calcium channels are key signaling elements that modulate the release of bioactive molecules (hormones, proteins, and neurotransmitters) through calcium sensing. Such calcium sensing activity is determined by the distinct biophysical and pharmacological properties and the ability of calcium channels to interact with other modulatory proteins. Although the function of extracellular α-syn is currently unknown, previous work suggested the presence of a calcium-dependent mechanism for α-syn secretion both in vitro, in neuronal cells in culture, and also in vivo, in the context of a trans-neuronal network in brain. Mechanisms regulating extracellular α-syn levels may be of particular importance as they could represent novel therapeutic targets. We discuss here how calcium channel activity may contribute to α-syn aggregation and secretion as a pathway to disease progression in synucleinopathies.
The affected neurons in Parkinson’s disease (PD) brains contain dense filamentous inclusions called Lewy bodies that primarily consist of the presynaptic protein α-synuclein (α-syn), a small neuronal protein that is abundant throughout the central nervous system under normal conditions (Goedert et al., 2017). The causal role of α-syn, a protein physiologically present in all neurons, in the pathogenesis of PD is further highlighted by human genetic studies that have linked multiplications and coding-region point mutations of the SNCA gene encoding for α-syn with familial PD (Houlden and Singleton, 2012). Increased levels of aggregated α-syn species have been strongly correlated with neuronal cell death partly due to a potent impairment of the degradation capacity of cellular proteolytic systems. Apart from acting in the cytoplasm, α-syn is normally secreted in the interstitial fluid (ISF) of the brain and can be taken up by neuronal cells (reviewed in Stefanis et al., 2019). High burden of extracellular α-syn has been shown to promote the production and transfer of pathology-linked, possibly hyper-aggregated, α-syn material. Under pathological conditions, such as PD, these pathologically relevant species of α-syn have the ability to propagate along interconnected neuronal networks leading to a progressive degeneration of neuronal function and ultimately cell death (Bieri et al., 2018).
It is now established that α-syn binds membranes with high affinity and this binding determines its functions especially at the synaptic sites. In particular, α-syn has been implicated in the process of neurotransmitter release where a number of potential roles of the protein in the maturation of synaptic vesicles, vesicle docking, priming and fusion have been proposed. The suggested mechanisms involve in part a direct or indirect interaction with the SNARE complex, but whether α-syn promotes or disrupts the formation of SNARE complex is still under debate (reviewed in Huang et al., 2019). How aggregated α-syn assemblies interfere with neurotransmitter release thereby affecting the functionality of neuronal synapses also remains elusive. In PD brains, deposits of small oligomers/aggregates of α-syn can be detected in the presynaptic sites prior to the formation of Lewy bodies and synaptic deficits seem to precede cell death in the course of the disease (Calo et al., 2016). The presence of abnormal α-syn assemblies extracellularly could modify neurotransmission and drastically affect the network dynamics of interconnected microcircuits and enhance the progression of disease pathology by facilitating the cell-to-cell transfer of pathological synuclein species.
Aggregation in the extracellular space may be favored by high local accumulation of α-syn. As such, the regulation of extracellular α-syn levels could be used therapeutically providing we understand the mechanism(s) of α-syn secretion and how this is triggered. Previous work indicated the presence of a Ca2+-dependent mechanism for α-syn secretion both in vitro, in neuronal cells in culture, and also in vivo, in the context of a trans-neuronal network in mouse brain, where α-syn release is mediated by presynaptic Ca2+ channels (Emmanouilidou et al., 2010, 2016). It is possible that abnormal function of the specific Ca2+ channel(s) regulating α-syn secretion could alter the levels of α-syn released to the extracellular space in a manner that favors the local aggregation of the protein at least in certain extra-synaptic sites. The aggregated assemblies may then be internalized by neighboring neurons acting as seeds to recruit the intracellular α-syn and facilitate the templating and subsequent release of pathological α-syn species by a cell-to-cell propagation mechanism.
The main presynaptic Ca2+ channels are the voltage-gated calcium channels (VGCCs) and the ligand-gated ion channels. The VGCCs consist of ten calcium channel isoforms, nine of which are widely expressed in the nervous system. VGCCs are responsible for calcium influx thereby controlling neuronal calcium homeostasis. As summarized in Table 1, dysregulation of VGCCs expression or activity has been linked with many neurological disorders such as Alzheimer’s disease, Parkinson’s disease, Multiple Sclerosis, iron, and zing neurotoxicity (Cataldi, 2013). In this review we focus on the role of VGCCs in PD and especially in neuronal degeneration that is apparent in PD. Up to date, there is no direct evidence that presynaptic calcium channels regulate α-syn propagation. In the paragraphs that follow, we try to analyze the role of Ca2+ signaling in α-syn aggregation and secretion, as they depict the major precursor mechanisms for α-syn propagation.
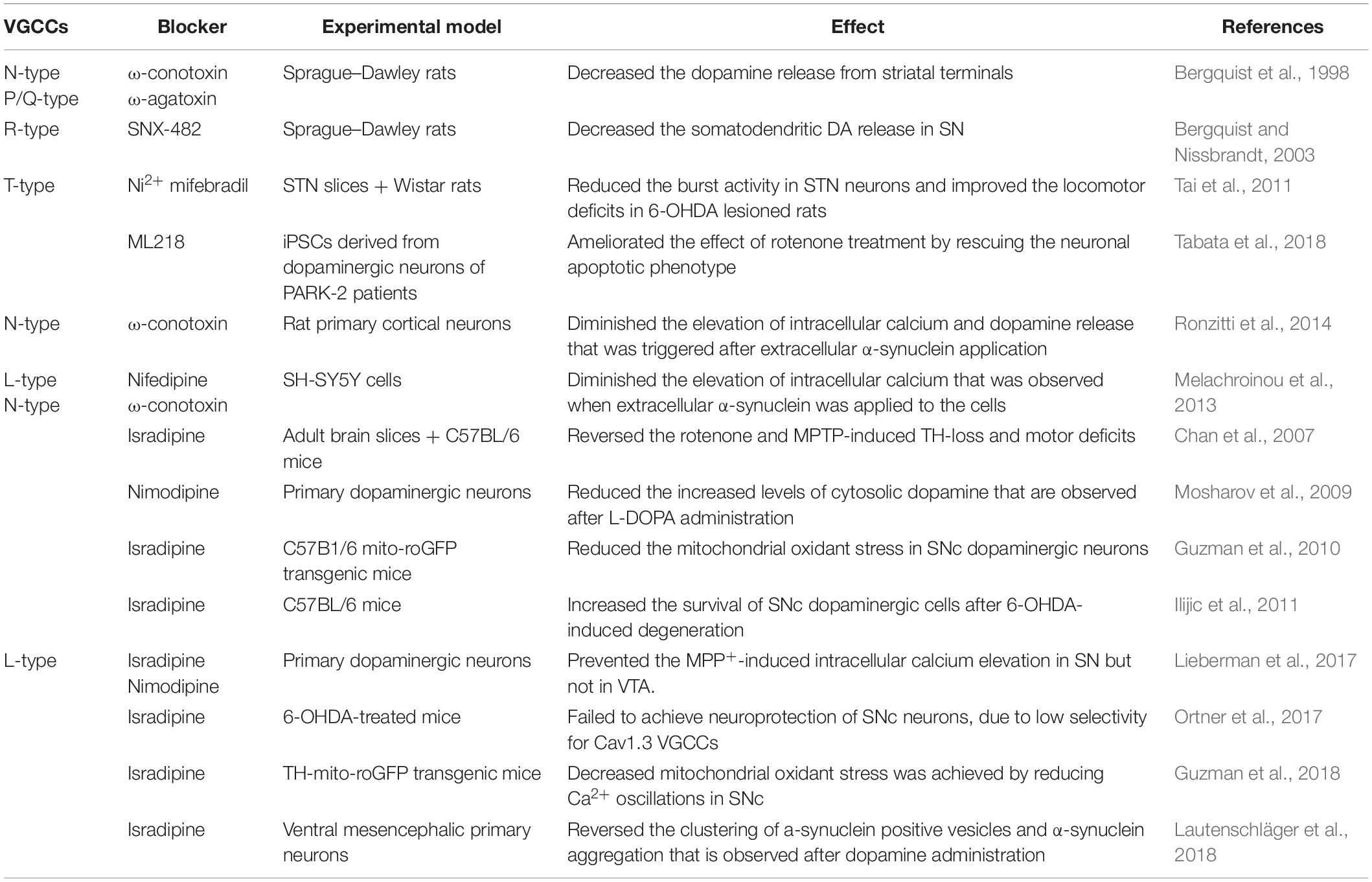
Table 1. Voltage-gated calcium channel blockers and their effect on PD pathophysiology.
Calcium homeostasis is important for the maintenance of neuronal integrity, since it is involved in synaptic transmission, neuronal plasticity, and cell survival. Activation of calcium signaling cascades is a result of intracellular calcium elevation, either via calcium influx or via calcium release from intracellular stores, such as the endoplasmic reticulum (ER) (reviewed in Yang et al., 2019). Among the different events that trigger α-syn pathology, the disruption of calcium homeostasis stands out as a possible mediator of aggregation and abnormal secretion of α-syn.
α-syn aggregation can be enhanced by alterations in intracellular calcium concentration in a direct or indirect manner. It has been shown that a transient increase of intracellular calcium leads to an increase in α-syn aggregates in human cell lines expressing α-syn (Nath et al., 2011; Follett et al., 2013). The mechanism through which calcium promotes α-syn aggregation is not clear. It has been shown that subsequent to calcium binding, the calcium-binding protein, calmodulin (CaM), changes its conformation and binds to α-syn inducing α-syn fibrillization (Lee et al., 2002; Martinez et al., 2003). CaM along with calbindin are calcium-binding proteins that are used by the cell as buffering proteins to maintain intracellular calcium homeostasis. The interplay between the presence of calbindin and the progression of PD was initially proposed after the discovery of a subgroup of calbindin-positive dopaminergic neurons that exhibited less pathologic features compared to calbindin-negative dopaminergic neurons in rat substantia nigra pars compacta (SNpc) and postmortem human brain material from PD patients (Gerfen et al., 1985; Yamada et al., 1990). Furthermore, a study in human brain tissues from dementia with Lewy bodies (DLB) patients showed that the Lewy bodies were exclusively formed in calbindin-negative neurons, a result that was confirmed using the mouse rotenone model, where α-syn aggregation was evident primarily in calbindin-negative neurons (Rcom-H’cheo-Gauthier et al., 2016). In the same line of evidence, striatal administration of a calbindin-expressing adenoviral vector in macaque monkeys protected the nigrostriatal dopamine system from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced degeneration (Inoue et al., 2019). These data suggest that α-syn aggregates are more prone to be formed in neurons with decreased calcium buffering capacity possibly due to irregular intracellular calcium levels.
Apart from calcium binding proteins, calcium-dependent proteases, such as calpain, seem to play a role in α-syn aggregation. In particular, increase in intracellular calcium concentrations lead to pathologically increased calpain activity. Mice deficient for calpastatin, a calpain-specific inhibitor, that were crossed with A30P α-syn expressing mice, exhibited higher calpain activity and increased α-syn aggregated species (Diepenbroek et al., 2014).
Finally, it has been proposed that calcium binds directly to the C-terminal of α-syn (Nielsen et al., 2001). Using time-resolved circular dichroism spectroscopy and infrared spectroscopy, it has been shown that calcium binding leads to exposure of the non-amyloid component (NAC) domain of the monomeric protein promoting the formation of β-sheet structures and thus accelerating the formation of α-syn aggregates (Han et al., 2018). In a different point of view, application of recombinant α-syn monomers or oligomers to primary neuronal cultures induced an increase in cytosolic transient [Ca2+] linking α-syn-induced neurotoxicity with increased intracellular calcium signaling (Angelova et al., 2016). By using the planar lipid bilayer approach, the authors concluded that the interaction of α-syn with the plasma membrane could facilitate calcium influx via affecting membrane permeability (Angelova et al., 2016). Alternatively, it has been proposed that α-syn aggregates can bind to the Ca2+ pump sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) on the endoplasmic reticulum modulating the intracellular calcium concentration. In this study, blockage of SERCA ameliorated the α-syn aggregated-induced cell death in neuronal cells in culture (Betzer et al., 2018).
Alpha-synuclein is considered to be secreted from neuronal cells though a stimulus-dependent mechanism that is regulated by the levels of intracellular Ca2+. It has been shown that an increase in intracellular calcium stimulate the secretion of α-syn in α-syn expressing SH-SY5Y cells (Emmanouilidou et al., 2010). Importantly, the secretion of α-syn in mouse striatum is thought to be regulated by the operation of presynaptic calcium channels as has been shown using a reverse microdialysis approach (Emmanouilidou et al., 2016). This is in accordance with a recent study that proposes, using a similar experimental setting, that the release of α-syn is dependent on neuronal activity in vivo (Yamada and Iwatsubo, 2018). The above studies support the idea that α-syn can be found in extracellular milieu not only as a monomer but also as an oligomeric conformer that may be internalized by neuronal cells.
The work described so far supports a multifactorial relationship of α-syn with calcium signaling where elevations in intracellular Ca2+ can result in the aggregation and release of α-syn or vice versa (Figure 1).
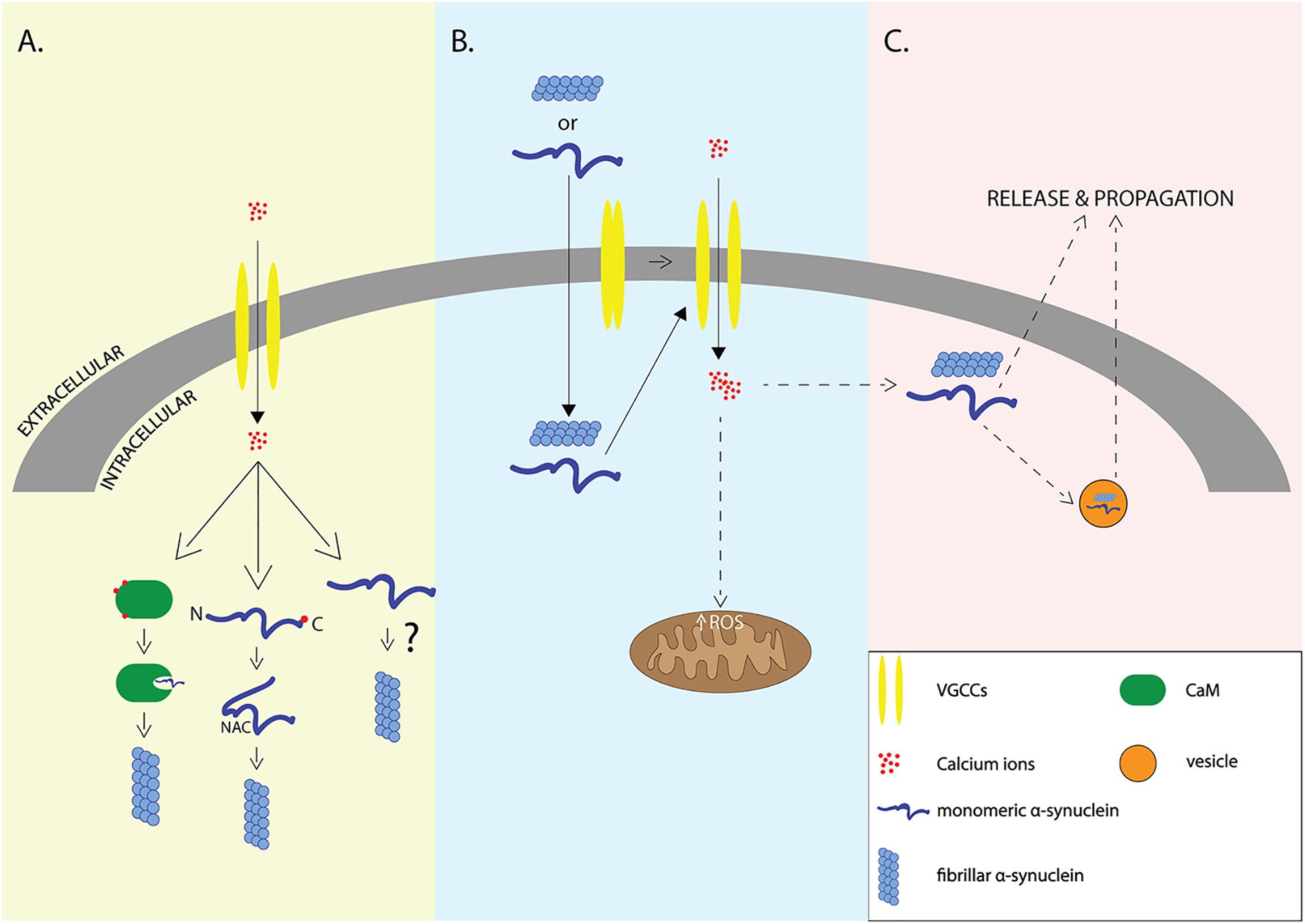
Figure 1. Interplay of α-syn and Ca2+. Elevation of intracellular Ca2+ levels through voltage-gated calcium channels (VGCCs) may lead to aggregation of α-syn via indirect interactions with calcium binding proteins such as Calmodulin, or via direct binding of Ca2+ to α-syn that leads to exposure of NAC domain (A), elevation of intracellular amounts of α-syn may lead to VGCCs opening and calcium influx that results in increased formation of reactive oxygen species (ROS) and neurodegeneration (B). It still remains unclear whether elevation of intracellular Ca2+ is the cause or the result of α-syn aggregation which promotes the release and propagation of α-syn -the release is accomplished either through exosomes or through other proposed secretory mechanisms (C).
In search of the mechanisms of disease progression in PD, previous work has focused on the connection between α-syn secretion, neurodegeneration, and alterations in calcium influx, mainly via the VGCCs. In neurons, VGCCs are key signaling elements that modulate the release of bioactive molecules (hormones, proteins, and neurotransmitters) through Ca2+ sensing. Following neuronal spiking and K+ channels opening, VGCCs modulate Ca2+ influx during repolarization back to the resting membrane potential. In most cases, VGCCs open at high voltages and close slowly during repolarization, leaving a time window where calcium influx occurs. The amount of calcium ions that enter the cell depend on the duration of the spike. Neurons regulate the amount of calcium influx by reducing the duration of the spikes (less than 1 ms) and by expressing calcium binding proteins (reviewed in James Surmeier et al., 2012). The VGCCs are divided to high-voltage activated (HVA) channels, which are activated at high membrane depolarization, or low-voltage activated (LVA) channels, which open at voltages near the resting membrane potential (Armstrong and Matteson, 1985; Bean, 1985). The HVA channels include L-type(Cav1 family), P-, Q-, and R-type (Cav2 family) VGCCs, whereas LVA channels include only T-type channels (Cav3 family) (reviewed in Zamponi, 2016).
It is noteworthy that in PD degeneration occurs preferentially in SNc dopaminergic neurons, while ventral tegmental area (VTA)-residing dopaminergic neurons remain mostly unaffected. It is proposed that differences in the regulation of calcium balance in these two neuronal populations could contribute to the onset and progression of neurodegeneration in PD. In particular, the adult SNc neurons mostly rely on L-type VGCCs for their basal activity and, more specifically, on the Cav1.3 subtype of L-type channels that open at relatively negative membrane potential (Mercuri et al., 1994). SNc dopaminergic neurons also express calcium binding proteins at lower level compared to VTA dopaminergic neurons (Foehring et al., 2009). These differences seem to be developmentally coordinated; it has been shown that during the embryonic stage and until postnatal week 3, the SNc dopaminergic neurons rely mostly on voltage-dependent Na+ channels, until a developmental switch renders Cav1.3-VGCCs the primary voltage-gated channels responsible for their pacemaking activities (Chan et al., 2007). In support for a critical role of VGCCs in the susceptibility of dopaminergic neurons, application of nimodipine, a Cav1.2/Cav1.3 blocker, in cultured L-DOPA-treated midbrain neurons decreased the levels of cytosolic dopamine suggesting a role of L-type channels in dopamine metabolism and neuronal survival (Mosharov et al., 2009). In addition, the L-type VGCCs were shown to be responsible for the elevation of intracellular calcium in primary dopaminergic neurons exposed to MPP+. Such calcium elevation was not observed in α-syn knock-out cultures after MPP+ exposure, suggesting that α-syn is implicated in intracellular calcium changes under stress conditions (Lieberman et al., 2017). Interestingly, increased intracellular calcium in SNc neurons overexpressing α-syn led to increased mitochondrial oxidation and neurotoxicity in these neurons, but not VTA neurons, probably due to the fact that VTA neurons do not depend on Cav1.3 VGCCs for neuronal firing (Lieberman et al., 2017). In support to these observations, a recent study depicted the importance of a fine balance between intracellular α-syn and intracellular calcium. In mesencephalic neurons, calcium binding in α-syn mediates the localization of the protein in synaptic vesicles and, under conditions of increased calcium or α-syn, this localization promotes synaptic vesicle clustering and α-syn aggregation. In this system, isradipine treatment reversed α-syn aggregation and improved neuronal survival (Lautenschläger et al., 2018). Several other studies using pharmacological inhibition emphasized on the importance of L-type channels in degeneration. Blockage of L-type calcium channels with the L-type inhibitor isradipine significantly reduced mitochondrial oxidation, indicating that calcium influx via the L-type VGCCs during pacemaking plays an important role in mitochondrial oxidant stress (Guzman et al., 2010). Furthermore, systemic administration of isradipine protected the striatal dopaminergic terminals, as well as the somata of dopaminergic neurons, after intrastriatal injection of 6-hydroxydopamine (6-OHDA) in mice (Ilijic et al., 2011).
Apart from the degeneration of dopaminergic neurons in SNpc of PD patients, the pathology is also evident in nuclei of the brainstem and olfactory bulbs (Jellinger, 2008). Cholinergic neurons in the dorsal motor nucleus of the vagus (DMV) in the caudal medulla have been shown to exhibit early Lewy body formation (Braak and Del Tredici, 2009). Interestingly, these neurons are autonomous slow pacemakers receiving an increased intracellular calcium load via the L-type VGCCs and they express low levels of calcium binding proteins. To induce mitochondrial stress, Goldberg et al. (2012) diminished DJ-1 from cholinergic DMV neurons and showed that pharmacological blockage of L-type VGCCs ameliorates the induced mitochondrial stress.
Further addressing a key role of L-type channels in PD, examination of post mortem material has revealed significant differences in the expression levels of these channels in PD patients compared with healthy subjects suggesting a possible role of the L-type family in the process of neurodegeneration, probably, via increased calcium influx that may lead to excitotoxicity. Immunostaining for Cav1 channels revealed that the expression ratio of Cav1.3/Cav1.2 of Cav1 was increased in early-stage PD brains compared to healthy controls (Hurley et al., 2013). Furthermore, two independent drug epidemiological studies targeting Cav1.3 channels concluded that administration of dihydropyridines lead to a reduced risk of developing PD (Ritz et al., 2010; Pasternak et al., 2012). However, since both Cav1.2 and Cav1.3 channels have very similar structural and pharmacological properties, the selectivity of 1,4-dihydropyridines is very low and the application of these L-type inhibitors could also block Cav1.2 channels (Xu and Lipscombe, 2001). In this context, isradipine-a well-known anti-hypertensive drug- is currently in phase III clinical trials to determine whether it can be effective against the progression of PD (Biglan et al., 2017). Isradipine has previously been used against neurodegeneration, but its selectivity is still under debate (Ortner et al., 2017; Guzman et al., 2018). In general, the variability in the blocking properties of dihydropyridines during different membrane depolarization states of dopaminergic neurons raises concerns about the usage of these drugs for PD treatment, since higher doses of dihydropyridines might not be tolerated during long-term treatment (Ortner et al., 2017).
Specific VGCCs have been implicated in α-syn-induced neurotoxicity. In SH-SY5Y cells, treatment with either nifedipine or ω-conotoxin, specific inhibitors for the L-type and N-type VGCCs, respectively, diminished the intracellular calcium raise induced by extracellular α-syn (Melachroinou et al., 2013). In rat cortical neurons, Ronzitti et al. (2014) showed that the calcium influx following application of extracellular α-syn was abolished by ω-conotoxin, but not by nifedipine or ω-agatoxin (specific inhibitors for L-type and P/Q type, respectively) indicating a possible role for the N-type VGCCs on α-syn-induced calcium increase.
Finally, targeting of T-type VGCCs has recently been considered a neuroprotective strategy for neurodegeneration and, more specifically, PD (Kopecky et al., 2014; Yang et al., 2014). Tabata et al. (2018) highlighted the importance of T-type VGCCs using induced pluripotent stem cell (iPSC) dopamine neurons derived from PARK2 patients by applying pharmacological or genetic silencing of T-type channels. In this study, the specific T-type antagonist ML218 ameliorated the effects of rotenone-induced mitochondrial stress by rescuing the apoptotic phenotype thereby leading to neuroprotection. Similar results were obtained after silencing of T-type VGCCs with specific siRNAs, while overexpression of T-type VGCCs led to the opposite effects. It has also been suggested that T-type VGCCs play a role in locomotor deficits accompanied after 6-OHDA lesion in rats. Specifically, in vitro as well as in vivo studies on subthalamic nuclei (STN) neurons revealed that blockage of T-type channels by the T-type specific inhibitors, Ni2+ and mifebradil, reduced the pathologically increased oscillations of STN that may be responsible for the tremor and other motor deficits present in PD. The above finding was also confirmed by behavioral experiments, where 6-OHDA lesioned rats showed a significant improvement in open field locomotor test after direct microinjection of either Ni2+ of mifebradil in STN (Tai et al., 2011).
We can conclude that all different types of VGCCs have been implicated in the progressive neurodegeneration present in PD. This is highlighted by a plethora of studies in which specific VGCCs are pharmacologically targeted in dopaminergic neurons to assess their role in preserving normal dopamine release and promoting cell survival under conditions of cellular stress. Several parameters could contribute to the discrepancies observed among the different studies. The different model systems used, ranging from in vitro cell models, such as cell lines and primary neurons, to the living rodent brain, could affect the magnitude and interpretation of the effects observed following the pharmacological manipulation of each VGCC. It is also possible that the different α-syn species (monomers, oligomers, and fibrils) have the ability to act through independent molecular mechanisms to trigger alterations in intracellular calcium. Finally, the pharmacological inhibition of certain VGCCs could stimulate compensatory mechanisms in which other calcium channels operate synergistically to regulate calcium levels adding further complexity to the interpretation of the results obtained so far.
Calcium influx can trigger α-syn aggregation thus providing an alternative pathway to PD neurodegeneration. There is also evidence that VGCCs can facilitate α-syn secretion under normal or pathological conditions, even though the mechanism for the stimulation of this process is still elusive. Abnormal function of these specific VGCCs may cause local accumulation of aggregated α-syn material into the extracellular space which could be taken up by recipient neurons thereby promoting the cell-to-cell spreading of disease pathology. As such, VGCCs that regulate α-syn properties could indicate specific molecular pathways to target as alternative therapeutic approaches for PD.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Part of this work has been financially supported by a grant to EE from the General Secretariat for Research and Technology (GSRT) and the Hellenic Foundation for Research and Innovation (HFRI) (Code: 1065) as well as by a grant to KV from the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (Project Code: T1EDK-03884).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
α-syn, alpha-synuclein; 6-OHDA, 6-hydroxydopamine; CaM, calmodulin; DLB, dementia with Lewy bodies; DMV, dorsal motor nucleus of the vagus; ER, endoplasmic reticulum; HVA, high-voltage activated; iPSC, induced pluripotent stem cell; ISF, interstitial fluid; LVA, low-voltage activated; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NAC domain, non-amyloid component domain; PD, Parkinson’s disease; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; SNpc, substantia nigra pars compacta; STN, subthalamic nuclei; VGCCs, voltage-gated calcium channels; VTA, ventral tegmental area.
Angelova, P. R., Ludtmann, M. H. R., Horrocks, M. H., Negoda, A., Cremades, N., Klenerman, D., et al. (2016). Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. doi: 10.1242/jcs.180737
Armstrong, C. M., and Matteson, D. R. (1985). Two distinct populations of calcium channels in a clonal line of pituitary cells. Science 227, 65–67. doi: 10.1126/science.2578071
Bean, B. P. (1985). Two kinds of calcium channels in canine atrial cells. Differences in kinetics, selectivity, and pharmacology. J. Gen. Physiol. 86, 1–30. doi: 10.1085/jgp.86.1.1
Bergquist, F., Jonason, J., Pileblad, E., and Nissbrandt, H. (1998). Effects of local administration of L-, N-, and P/Q-type calcium channel blockers on spontaneous dopamine release in the striatum and the substantia nigra: a microdialysis study in rat. J. Neurochem. 70, 1532–1540. doi: 10.1046/j.1471-4159.1998.70041532.x
Bergquist, F., and Nissbrandt, H. (2003). Influence of R-type (Cav2.3) and T-type (Cav3.1-3.3) antagonists on nigral somatodendritic dopamine release measured by microdialysis. Neuroscience 120, 757–764. doi: 10.1016/S0306-4522(03)00385-3
Betzer, C., Lassen, L. B., Olsen, A., Kofoed, R. H., Reimer, L., Gregersen, E., et al. (2018). Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 19, 1–21. doi: 10.15252/embr.201744617
Bieri, G., Gitler, A. D., and Brahic, M. (2018). Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 109, 219–225. doi: 10.1016/j.nbd.2017.03.007
Biglan, K. M., Oakes, D., Lang, A. E., Hauser, R. A., Hodgeman, K., Greco, B., et al. (2017). A novel design of a Phase III trial of isradipine in early Parkinson disease (STEADY-PD III). Ann. Clin. Transl. Neurol. 4, 360–368. doi: 10.1002/acn3.412
Braak, H., and Del Tredici, K. (2009). Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 201, 1–119.
Calo, L., Wegrzynowicz, M., Santivañez-Perez, J., and Grazia Spillantini, M. (2016). Synaptic failure and α-synuclein. Mov. Disord. 31, 169–177. doi: 10.1002/mds.26479
Cataldi, M. (2013). The changing landscape of voltage-gated calcium channels in neurovascular disorders and in neurodegenerative diseases. Curr. Neuropharmacol. 11, 276–297. doi: 10.2174/1570159x11311030004
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Diepenbroek, M., Casadei, N., Esmer, H., Saido, T. C., Takano, J., Kahle, P. J., et al. (2014). Over expression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum. Mol. Genet. 23, 3975–3989. doi: 10.1093/hmg/ddu112
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Emmanouilidou, E., Minakaki, G., Keramioti, M. V., Xylaki, M., Balafas, E., Chrysanthou-Piterou, M., et al. (2016). GABA transmission via ATP-dependent K+ channels regulates alpha-synuclein secretion in mouse striatum. Brain 139(Pt 3), 871–890. doi: 10.1093/brain/awv403
Foehring, R. C., Zhang, X. F., Lee, J. C. F., and Callaway, J. C. (2009). Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 102, 2326–2333. doi: 10.1152/jn.00038.2009
Follett, J., Darlow, B., Wong, M. B., Goodwin, J., and Pountney, D. L. (2013). Potassium depolarization and raised calcium induces α-synuclein aggregates. Neurotox. Res. 23, 378–392. doi: 10.1007/s12640-012-9366-z
Gerfen, C. R., Baimbridge, K. G., and Miller, J. J. (1985). The neostriatal mosaic: compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc. Natl. Acad. Sci. U.S.A. 82, 8780–8784. doi: 10.1073/pnas.82.24.8780
Goedert, M., Jakes, R., and Spillantini, M. G. (2017). The Synucleinopathies: twenty years on. J. Parkinson’s Dis. 7, S51–S69. doi: 10.3233/JPD-179005
Goldberg, J. A., Guzman, J. N., Estep, C. M., Ilijic, E., Kondapalli, J., Sanchez-Padilla, J., et al. (2012). Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat. Neurosci. 15, 1414–1421. doi: 10.1038/nn.3209
Guzman, J. N., Ilijic, E., Yang, B., Sanchez-Padilla, J., Wokosin, D., Galtieri, D., et al. (2018). Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Invest. 128, 2266–2280. doi: 10.1172/JCI95898
Guzman, J. N., Sanchez-Padilla, J., Wokosin, D., Kondapalli, J., Ilijic, E., Schumacker, P. T., et al. (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468, 696–700. doi: 10.1038/nature09536
Han, J. Y., Choi, T. S., and Kim, H. I. (2018). Molecular role of Ca2+ and hard divalent metal cations on accelerated fibrillation and interfibrillar aggregation of α-synuclein. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-20320-5
Houlden, H., and Singleton, A. B. (2012). The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 124, 325–338. doi: 10.1007/s00401-012-1013-5
Huang, X., Sun, S., Wang, X., Fan, F., Zhou, Q., Lu, S., et al. (2019). Mechanistic insights into the SNARE complex disassembly. Sci. Adv. 5:eaau8164. doi: 10.1126/sciadv.aau8164
Hurley, M. J., Brandon, B., Gentleman, S. M., and Dexter, D. T. (2013). Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 136, 2077–2097. doi: 10.1093/brain/awt134
Ilijic, E., Guzman, J. N., and Surmeier, D. J. (2011). The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 43, 364–371. doi: 10.1016/j.nbd.2011.04.007
Inoue, K.-I., Miyachi, S., Nishi, K., Okado, H., Nagai, Y., Minamimoto, T., et al. (2019). Recruitment of calbindin into nigral dopamine neurons protects against MPTP-Induced parkinsonism. Mov. Disord. 34, 200–209. doi: 10.1002/mds.107
James Surmeier, D., Guzman, J. N., Sanchez, J., and Schumacker, P. T. (2012). Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harbor Perspect. Med. 2, 1–27. doi: 10.1101/cshperspect.a009290
Jellinger, K. A. (2008). A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol. 116, 1–16. doi: 10.1007/s00401-008-0406-y
Kopecky, B. J., Liang, R., and Bao, J. (2014). T-type calcium channel blockers as neuroprotective agents. Pflugers Arch. Eur. J. Physiol. 466, 757–765. doi: 10.1007/s00424-014-1454-x
Lautenschläger, J., Stephens, A. D., Fusco, G., Ströhl, F., Curry, N., Zacharopoulou, M., et al. (2018). C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat. Commun. 9:712. doi: 10.1038/s41467-018-03111-4
Lee, D., Lee, S. Y., Lee, E. N., Chang, C. S., and Paik, S. R. (2002). A-synuclein exhibits competitive interaction between calmodulin and synthetic membranes. J. Neurochem. 82, 1007–1017. doi: 10.1046/j.1471-4159.2002.01024.x
Lieberman, O. J., Choi, S. J., Kanter, E., Saverchenko, A., Frier, M. D., Fiore, G. M., et al. (2017). alpha-synuclein-dependent calcium entry underlies differential sensitivity of cultured sn and vta dopaminergic neurons to a parkinsonian neurotoxin. ENeuro 4:ENEURO.0167-17.2017. doi: 10.1523/ENEURO.0167-17.2017
Martinez, J., Moeller, I., Erdjument-Bromage, H., Tempst, P., and Lauring, B. (2003). Parkinson’s disease-associated α-synuclein is a calmodulin substrate. J. Biol. Chem. 278, 17379–17387. doi: 10.1074/jbc.M209020200
Melachroinou, K., Xilouri, M., Emmanouilidou, E., Masgrau, R., Papazafiri, P., Stefanis, L., et al. (2013). Deregulation of calcium homeostasis mediates secreted alpha-synuclein-induced neurotoxicity. Neurobiol. Aging 34, 2853–2865. doi: 10.1016/j.neurobiolaging.2013.06.006
Mercuri, N. B., Bonci, A., Calabresi, P., Stratta, F., Stefani, A., and Bernardi, G. (1994). Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br. J. Pharmacol. 113, 831–838. doi: 10.1111/j.1476-5381.1994.tb17068.x
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Nath, S., Goodwin, J., Engelborghs, Y., and Pountney, D. L. (2011). Raised calcium promotes α-synuclein aggregate formation. Mol. Cell. Neurosci. 46, 516–526. doi: 10.1016/j.mcn.2010.12.004
Nielsen, M. S., Vorum, H., Lindersson, E., and Jensen, P. H. (2001). Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 276, 22680–22684. doi: 10.1074/jbc.M101181200
Ortner, N. J., Bock, G., Dougalis, A., Kharitonova, M., Duda, J., Hess, S., et al. (2017). Lower affinity of isradipine for L-Type Ca 2+ channels during substantia nigra dopamine neuron-like activity: implications for neuroprotection in parkinson’s disease. J. Neurosci. 37, 6761–6777. doi: 10.1523/jneurosci.2946-16.2017
Pasternak, B., Svanström, H., Nielsen, N. M., Fugger, L., Melbye, M., and Hviid, A. (2012). Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol. 175, 627–635. doi: 10.1093/aje/kwr362
Rcom-H’cheo-Gauthier, A. N., Davis, A., Meedeniya, A. C. B., and Pountney, D. L. (2016). Alpha-synuclein aggregates are excluded from calbindin-D28k-positive neurons in dementia with Lewy bodies and a unilateral rotenone mouse model. Mol. Cell. Neurosci. 77, 65–75. doi: 10.1016/j.mcn.2016.10.003
Ritz, B., Rhodes, S. L., Qian, L., Schernhammer, E., Olsen, J. H., and Friis, S. (2010). L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 67, 600–606. doi: 10.1002/ana.21937
Ronzitti, G., Bucci, G., Emanuele, M., Leo, D., Sotnikova, T. D., Mus, L. V., et al. (2014). Exogenous alpha-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 34, 10603–10615. doi: 10.1523/JNEUROSCI.0608-14.2014
Stefanis, L., Emmanouilidou, E., Pantazopoulou, M., Kirik, D., Vekrellis, K., and Tofaris, G. K. (2019). How is alpha-synuclein cleared from the cell? J. Neurochem. 150, 577–590. doi: 10.1111/jnc.14704
Tabata, Y., Imaizumi, Y., Sugawara, M., Andoh-Noda, T., Banno, S., Chai, M., et al. (2018). T-type calcium channels determine the vulnerability of dopaminergic neurons to mitochondrial stress in familial parkinson disease. Stem Cell Rep. 11, 1171–1184. doi: 10.1016/j.stemcr.2018.09.006
Tai, C.-H., Yang, Y.-C., Pan, M.-K., Huang, C.-S., and Kuo, C.-C. (2011). Modulation of subthalamic T-type Ca(2+) channels remedies locomotor deficits in a rat model of Parkinson disease. J. Clin. Invest. 121, 3289–3305. doi: 10.1172/JCI46482
Xu, W., and Lipscombe, D. (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944–5951. doi: 10.1523/jneurosci.21-16-05944.2001
Yamada, K., and Iwatsubo, T. (2018). Extracellular α-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 13, 4–11. doi: 10.1186/s13024-018-0241-0
Yamada, T., McGeer, P. L., Baimbridge, K. G., and McGeer, E. G. (1990). Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 526, 303–307. doi: 10.1016/0006-8993(90)91236-a
Yang, J., Zhao, Z., Gu, M., Feng, X., and Xu, H. (2019). Release and uptake mechanisms of vesicular Ca(2+) stores. Protein Cell 10, 8–19. doi: 10.1007/s13238-018-0523-x
Yang, Y.-C., Tai, C.-H., Pan, M.-K., and Kuo, C.-C. (2014). The T-type calcium channel as a new therapeutic target for Parkinson’s disease. Pflugers Arch. 466, 747–755. doi: 10.1007/s00424-014-1466-6
Keywords: alpha-synuclein, Parkinson’s and related diseases, protein aggregation, secretion, calcium, voltage gated Ca2+ channel, neurodegeneration
Citation: Leandrou E, Emmanouilidou E and Vekrellis K (2019) Voltage-Gated Calcium Channels and α-Synuclein: Implications in Parkinson’s Disease. Front. Mol. Neurosci. 12:237. doi: 10.3389/fnmol.2019.00237
Received: 15 July 2019; Accepted: 17 September 2019;
Published: 09 October 2019.
Edited by:
Tiago F. Outeiro, University Medical Center Göttingen, GermanyReviewed by:
Charles Robert Harrington, University of Aberdeen, United KingdomCopyright © 2019 Leandrou, Emmanouilidou and Vekrellis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kostas Vekrellis, dmVrcmVsbGlzQGJpb2FjYWRlbXkuZ3I=; Evangelia Emmanouilidou, ZWVtbWFuQGNoZW0udW9hLmdy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.