 Cristina Catoni1
Cristina Catoni1 Tito Calì
Tito Calì Marisa Brini
Marisa Brini- 1Department of Biology, University of Padova, Padua, Italy
- 2Department of Biomedical Sciences, University of Padova, Padua, Italy
Parkinson’s disease (PD) is a debilitating neurodegenerative disorder characterized by loss of dopaminergic neurons in the substantia nigra pars compacta. The causes of PD in humans are still unknown, although metabolic characteristics of the neurons affected by the disease have been implicated in their selective susceptibility. Mitochondrial dysfunction and proteostatic stress are recognized to be important in the pathogenesis of both familial and sporadic PD, and they both culminate in bioenergetic deficits. Exposure to calcium overload has recently emerged as a key determinant, and pharmacological treatment that inhibits Ca2+ entry diminishes neuronal damage in chemical models of PD. In this review, we first introduce general concepts on neuronal Ca2+ signaling and then summarize the current knowledge on fundamental properties of substantia nigra pars compacta dopaminergic neurons, on the role of the interplay between Ca2+ and dopamine signaling in neuronal activity and susceptibility to cell death. We also discuss the possible involvement of a “neglected” player, the Neuronal Calcium Sensor-1 (NCS-1), which has been shown to participate to dopaminergic signaling by regulating dopamine dependent receptor desensitization in normal brain but, data supporting a direct role in PD pathogenesis are still missing. However, it is intriguing to speculate that the Ca2+-dependent modulation of NCS-1 activity could eventually counteract dopaminergic neurons degeneration.
Neuronal Calcium Signaling
Calcium (Ca2+) homeostasis is essential for neuronal function and survival. Intracellular Ca2+ signaling in neurons is extremely fine-tuned, because it controls gene transcription, membrane excitability, neurotransmitters secretion and many other cellular processes, including synaptic plasticity (Berridge, 1998; Brini et al., 2014). Like other cells, neurons use both extracellular and intracellular sources of Ca2+ and, as a consequence of their excitability, they are exposed to large Ca2+ fluctuations and thus to a major risk of Ca2+ overload.
The coordinated action of the different systems that handle Ca2+ fluxes guarantees the generation of high Ca2+ concentration microdomains with precise spatiotemporal features that are crucial to specifically activate different cellular processes (La Rovere et al., 2016; Filadi et al., 2017a; Samanta and Parekh, 2017). For instance, those generated upon the opening of the endoplasmic reticulum Ca2+ channels are sensed by mitochondria that use them to drive bioenergetic metabolism for the production of ATP and mitochondrial substrates for anabolic process (Tarasov et al., 2012).
However, exaggerated mitochondrial Ca2+ accumulation may be dangerous, since can lead to mitochondrial permeability transition pore (mPTP) opening, cytochrome c release and can activate apoptotic cell death (Bernardi et al., 2015). Thus, once Ca2+-regulated processes have been engaged, Ca2+ ions must be rapidly extruded (and/or buffered) to avoid that their excessive accumulation could trigger mitochondrial dysfunction (Calì et al., 2012a; Muller et al., 2018). The “Ca2+ machinery” that is in place to tune Ca2+ concentration includes transport proteins such as channels, exchangers and pumps that move the ion across the membranes (i.e., the plasma membrane and the membranes of organelles), and Ca2+ binding proteins that act as Ca2+ buffer and/or transducer (Figure 1).
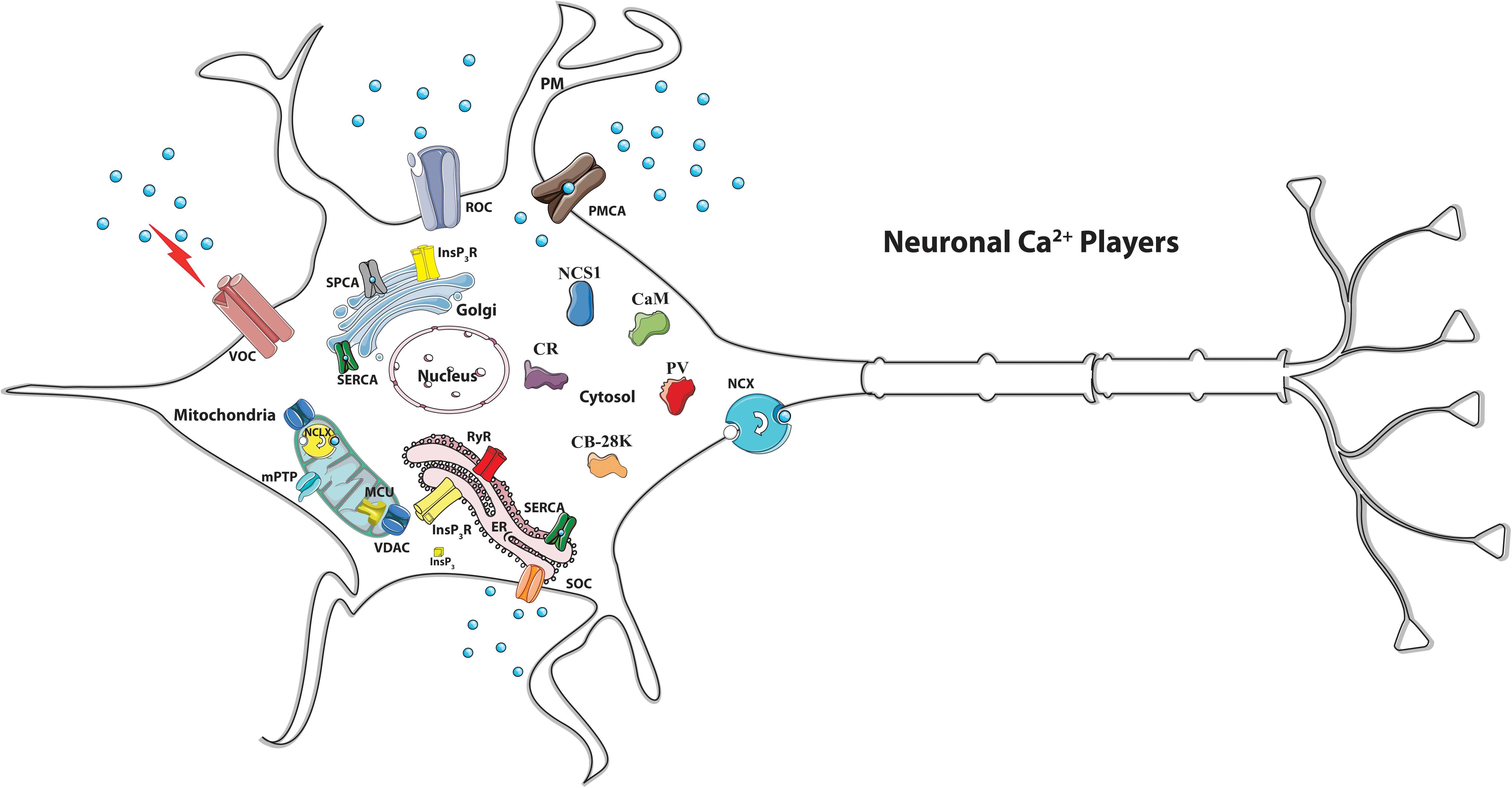
Figure 1. Schematic view of the main neuronal Ca2+ players. Many Ca2+ transport proteins contribute to Ca2+ handling: the inositol 1,4,5-trisphosphate receptor (InsP3R), the ryanodine receptor (RyR) at the sarco/endoplasmic reticulum (SR/ER) membranes, and the voltage (VOC), the receptor (ROC)-activated and the store-operated (SOC) Ca2+ channels of the plasma membrane. Ca2+ extrusion depends on the activity of the plasma membrane Ca2+ ATPase (PMCA) and the plasma membrane Na+/Ca2+ exchanger (NCX). Ca2+ reuptake in the intracellular stores is operated by the ER/SR Ca2+ ATPase (SERCA) and the secretory pathway Ca2+ ATPase (SPCA) of the Golgi apparatus. The mitochondrial Ca2+ handling systems and the cytosolic Ca2+-binding/buffering proteins are indicated. MCU, mitochondrial Ca2+ uniporter. NCLX, mitochondrial Na+/Ca2+ exchanger. VDAC, voltage-dependent anion channels. MPTP, mitochondrial permeability transition pore. CaM, calmodulin. CR, calretinin. CB-28K, calbindin D-28K. NCS-1, Neuronal Calcium Sensor 1.
Increasing evidence suggests that defective Ca2+ handling plays an important role in aging and neurodegeneration (Berridge, 1998; Calì et al., 2014; Pchitskaya et al., 2018). Despite of neurodegenerative diseases are a large group of heterogeneous disorders characterized by relative selectivity in the death of neuronal subtypes, they share some common tracts that include disturbance in cellular quality mechanisms (i.e., ER stress, autophagy, accumulation of aggregated proteins), oxidative stress, neuroinflammation and defective Ca2+ signaling (Brini et al., 2014; Hetz and Saxena, 2017; Kurtishi et al., 2018; Muller et al., 2018). Furthermore, recent studies have indicated that defective ER-mitochondria communication, by impinging on energetic metabolism, lipid synthesis, autophagy, could have detrimental consequences for cell function and survival (Filadi et al., 2017b). Many regulators of ER-mitochondria interface are proteins whose mutations are linked to familial forms of Alzheimer’s disease (AD) and PD, suggesting that defects at the ER-mitochondria contact sites could have a role in the onset and/or the progression of these neurodegenerative diseases (Calì et al., 2013a; Filadi et al., 2016; Area-Gomez and Schon, 2017).
As mentioned above, in addition to the Ca2+ transport across the membranes, another important mechanism that contributes to the regulation of Ca2+ homeostasis is the processes of Ca2+ buffering that is managed by mitochondrial Ca2+ uptake but largely relies on the existence of several cytosolic Ca2+ binding proteins. Among them, the ubiquitous EF-hand Ca2+ protein calmodulin (CaM) is mainly responsible for translating the increases of the cytosolic Ca2+ concentration into a biochemical signal through conformational changes of its targets (Sharma and Parameswaran, 2018). It is present at high concentration in the brain, where it plays a key role in the regulation of ions channels activity and synaptic plasticity (Xia and Storm, 2005). Other Ca2+-binding proteins such as Calbindin D-28K (CB-28K), calretinin (CR), and parvalbumin (PV) are also present in the nervous system. By buffering Ca2+ levels with different capacity, affinity and kinetics (Schwaller, 2012; Paillusson et al., 2017) and thanks to their cell-specific abundance, they guarantee the selective activation of different biological processes. Cell-type-specific distribution of Ca2+ binding proteins could also account for the selective susceptibility to cell death of the specific neuronal populations affected in different neurodegenerative diseases. Indeed, it has been observed that CB-28K containing cells are spared from cell death in pharmacological-induced parkinsonism in mice and that CB-28K immunoreactivity in cholinergic neurons of the basal forebrain (the same neurons that are preferentially loss in AD) was reduced in an age-related manner in humans, suggesting a role for CB-28K also in the selective neuronal vulnerability in AD (Yamada et al., 1990; German et al., 1992; Mouatt-Prigent et al., 1994; Damier et al., 1999; Geula et al., 2003; Zallo et al., 2018).
Parkinson’s Disease, Calcium and Selective Vulnerability of Substantia Nigra Par Compacta
PD is the second most common neurodegenerative disorder in humans after AD. PD patients present motor symptoms such as resting tremor, bradykinesia and postural rigidity. However, the appearance of other disturbances such as constipation, sleep disorders, olfactory deficit, apathy, pain, sexual difficulties, and in some case cognitive decline is currently observed to anticipate motor deficits in many patients (de Lau and Breteler, 2006) and indicates that regions of the brain that are not involved in motor symptoms are also compromised. At histological levels, the hallmarks for PD are the selective loss of the dopamine (DA)-containing neurons of the substantia nigra pars compacta (SNc) and the presence of proteinaceous cytosolic inclusions known as Lewy bodies, mainly constituted by alpha-synuclein (Goedert et al., 2013). The progressive SNc DA cells death leads to decreased DA levels and the worsening of the symptoms. SNc DA cells release DA from their axonal terminals and from their cell bodies and dendrites within both the dorsal striatum and the midbrain, respectively. DA release is crucial for voluntary movement and it is strictly Ca2+- and electrical activity-dependent. Indeed, the continuous supply of DA to the connected brain areas is guaranteed by autonomous pacemaking, which occurs in the absence of conventional synaptic input and thank to the orchestrated action of different ion channels. In particular, the presence of voltage-dependent L-type Ca2+ channels containing a distinctive Cav1.3 pore forming subunit, which opens at relatively hyperpolarized potentials, allows Ca2+ entry with an oscillatory pathway that contributes to the membrane potential threshold, underlying autonomous pacemaking (Chan et al., 2007; Puopolo et al., 2007; Guzman et al., 2010). Continuous Ca2+ influx is necessary to modulate physiological DA release by SNc DA neurons, but, its long-lasting presence may synergize with the exposure to risk factors (i.e., aging, mitochondrial toxins, mutations) and generate metabolic stress and mitochondrial damage (Surmeier et al., 2011; Guzman et al., 2018).
It is widely recognized that in PD, the major risk of Ca2+-induced toxicity is represented by Ca2+ entry through the voltage gated Ca2+ channels during the normal activity of the dopaminergic neurons (Ilijic et al., 2011; Liss and Striessnig, 2019), that, in this way, become more vulnerable to death than other neuronal populations. Cell damage could be further exacerbated by environmental factors such as exposure to mitochondrial toxins [i.e., MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), rotenone, 6-hydroxy dopamine (6-OHDA), paraquat (1,1′-dimethyl-4,4′-bipyridine)] or upon loss of function of specific proteins such as alpha-synuclein, Parkin, PINK1 and DJ-1, whose mutations are linked to genetic forms of PD. Interestingly, all these proteins, despite their different intracellular localization and function, are able to interfere with Ca2+ signaling (Calì et al., 2014). Indeed extracellular and intracellular deposition of alpha-synuclein aggregates has been proposed to enhance Ca2+ influx through the plasma membrane by forming pore-like structures (Danzer et al., 2007; Surguchev and Surguchov, 2015; Angelova et al., 2016) or by interfering with Ca2+ channels (Liu et al., 2013; Ronzitti et al., 2014), as well as PINK1 has been proposed to participate to the regulation of both influx or efflux of Ca2+ ions from mitochondria (Gandhi et al., 2009; Marongiu et al., 2009). We have found that the overexpression of PD-linked alpha-synuclein, parkin and DJ-1 proteins enhanced ER-mitochondria Ca2+ transfer by favoring ER-mitochondria juxtaposition, and provided evidence that through this action, physiological amounts of these proteins are able to tune ATP production (Calì et al., 2012b, 2013b; Ottolini et al., 2013). The loss of this function is likely to be particularly damaging to neurons that are heavily dependent on proper Ca2+signaling and ATP production. Accordingly, Paillusson et al. (2017) have documented loss of ER-mitochondria association in induced pluripotent stem cells derived neurons from PD patients harboring alpha-synuclein gene triplication.
In summary, if by one side Ca2+ entry through Cav1.3 pore subunit is essential to sustain pacemaking activity of SNc DA neurons, by the other it exposes these neurons to metabolic burden and mitochondrial stress. Differently, DA neurons from the ventral tegumental area (VTA), which are also autonomous pacemakers, are significantly less vulnerable than SNc DA neurons from which they differ in respect with two main features: they have smaller Ca2+ currents (Khaliq and Bean, 2010) and strong intrinsic Ca2+ buffering capacity due to higher calbindin levels (Dopeso-Reyes et al., 2014).
The most convincing argument in favor of the “Ca2+ hypothesis” in PD onset is that epidemiologic studies on patients under clinical trial with L-type channel antagonists for the treatment of hypertension have shown a reduced risk of developing PD (Becker et al., 2008; Ritz et al., 2010; Pasternak et al., 2012). The voltage gated L-type Ca2+ plasma membrane channels inhibitor isradipine has been demonstrated to be neuroprotective in a mouse model of PD (Ilijic et al., 2011) and phase III of clinical trial is currently under evaluation to establish whether treatment with isradipine is able to slow the progression of PD in humans (Liss and Striessnig, 2019).
Despite general consensus agrees with the fact that the anatomical, physiological, and biochemical phenotype of the SNc DA neurons predisposes them to mitochondrial dysfunction, the molecular bases of the subtype-selective neuronal vulnerability are still obscure and of big interest.
Interestingly, computer imaging and immunohistochemical staining techniques have revealed a strict correlation between the distribution of the Ca2+-binding proteins CB-28K and CR and cell survival in midbrain dopaminergic regions: cells that are spared from death in animals treated with the DA neurotoxin MPTP (German et al., 1992; Mouatt-Prigent et al., 1994) are those that display higher expression levels of CB-28K and CR in control untreated animals. Interestingly, this observation has been reinforced by a comparative study performed on post-mortem brain from neurologically normal individuals and PD patients in which the distribution of calbindin, calmodulin and calretinin did not associate with the regions prone to neurodegeneration. It has also been observed that the expression of Cav1.3 subtypes increased in the brain of patients at early stage of the disease, even before the appearance of recognized pathological signs (Hurley et al., 2013), suggesting that Ca2+ dysregulation could be an early event in PD pathogenesis.
Low expression levels of Ca2+-binding proteins in the brain area more susceptible to cell death in PD suggest that those neuronal populations are characterized by low Ca2+ buffering capacity. This parameter has been directly evaluated in neurons from the ventral and medial SNc by applying a protocol originally developed by E. Neher (Neher and Augustine, 1992; Zhou and Neher, 1993; Neher, 1998). Foehring and colleagues (Foehring et al., 2009) have loaded the cells with an exogenous Ca2+-indicator/buffer and calculated the Ca2+ binding ratio (KS) by measuring changes in Ca2+-bound buffer and dividing by the free Ca2+ increase. Interestingly, despite the intrinsic Ca2+ buffering in DA cells increases with postnatal age (KS ≃ 110 at postnatal day 13–18 and ≃179 at postnatal day 25–32), it remains low at both age ranges. Other neuronal populations (e.g., neocortical pyramidal cells or cortical GABAergic interneurons), that are not endowed with pacemaking activity, display similar or higher values and Purkinje cells have the highest KS values (∼2,000) (Fierro and Llano, 1996).
Considering that, in addition to Ca2+ binding proteins, also mitochondria play a role in buffering cytosolic Ca2+, a reduction of mitochondria amount or/and the presence of dysfunctional mitochondria could account for differences in Ca2+ buffering capacity among different neuronal midbrain populations. In line with these considerations, a study has found that the mitochondrial mass in SNc DA neurons is reduced in respect with that of other neurons from the midbrain (Liang et al., 2007). Thus, also this peculiarity may account for selective vulnerability of DA SNc neurons.
At the end of this discussion, it is worth to mention that other observations suggest that additional sources of Ca2+ (other than Ca2+ entry from the extracellular ambient) could contribute to SNc DA neurons vulnerability. In this respect, defects in intracellular Ca2+ stores handling and ER stress have been frequently documented in PD cellular models (Wang and Takahashi, 2007; Mercado et al., 2013).
All together it is clear that the equilibrium between Ca2+ signaling and SNc DA neurons activity is extremely critical: upon conditions of increased metabolic demand, i.e., when continuous dopamine release into the dorsal striatum is required for movement, elevated metabolic burden could originate a vicious cycle that further impairs mitochondrial function, resulting in increased metabolic stress. Interestingly, it has been proposed that Ca2+ load may further contribute to exacerbate neurodegeneration by promoting an increase of the neurotoxic catecholamine intracellular levels (Mosharov et al., 2009).
Dopamine Release and Neuronal Calcium Sensor 1: Possible Implications in Parkinson Disease?
Among the Ca2+-binding proteins, the components of the subfamily of Neuronal Ca2+ Sensors (NCS) are particularly abundant in neurons and photoreceptors and deserve special attention since their properties distinguish them from CaM or CB-28K, CR and PV and allow them to play non-reduntant roles. Differences in Ca2+ affinities, in cellular expression and distribution and in target proteins are at the basis of the specialization of NCS function (McCue et al., 2010). Neuronal Ca2+ Sensor-1 (NCS-1) is the most ancient member of the family (Pongs et al., 1993), and it is implicated in the regulation of cell-surface receptors and ion channels, and in neurotransmitter release, gene transcription, cell growth and survival (Burgoyne, 2007).
NCS-1 has been linked to a large spectrum of diseases possibly because its differential interaction with partners. Changes in the abundance of NCS-1 result in altered relationship with target proteins and determine cell dysfunction. An up-regulation of NCS-1 mRNA was found in a variety of non-neurological and neurological diseases. NCS-1 has been proposed to be a biomarker in aggressive breast cancer (Moore et al., 2017). In the heart, altered Ca2+ signaling mediated by NCS-1 and inositol 1,4,5 trisphosphate receptor interaction was linked to cardiac arrhythmias (Zhang et al., 2010). Schizophrenia, bipolar disorder (BD) (Koh et al., 2003) and autism (Piton et al., 2008; Handley et al., 2010) have been associated with upregulation or mutations in NCS-1 protein.
Increased levels of NCS-1 mRNA were measured in neurons from SNc of PD patients (Dragicevic et al., 2014) and NCS-1 was shown to co-localize with the D2 type-dopamine receptors in dendrites, spines, and occasionally in axonal buttons of rat and monkey striatal neurons (Kabbani et al., 2002), thus supporting the involvement of NCS-1 in the process of dopaminergic signaling, but also suggesting its possible link with PD.
As mentioned above, numerous convincing biophysical and pharmacological findings support the hypothesis that Cav1.3 channels by sustaining pacemaker-activity exposes SNc DA neurons to continuous Ca2+ load and mitochondrial stress (Surmeier et al., 2011). However, other studies investigating dopamine receptor mediated autoinhibition of neuronal activity have shown that Ca2+ entry through Cav1.3 channels, in addition to sustain pacemaker activity, regulates dopamine autoreceptors (Dragicevic et al., 2014). Considering that current therapies to alleviate PD symptoms and progression are based on the administration of dopamine precursor L-Dopa and/or dopamine D2 receptor agonists (Oertel and Schulz, 2016), the understanding of Cav1.3 physiology becomes crucial to better define the pathways involved in PD onset and develop therapeutic strategies.
Dopaminergic transmission is dependent on two main families of DA receptors, namely D1- and D2-type (Beaulieu and Gainetdinov, 2011) that are both members of the G protein-coupled receptor (GPCR) superfamily. The D1-like receptors activate Gαs/olf and stimulate cAMP production, whereas the D2-like receptors activate Gαi/o and inhibit adenylate cyclase activity and cAMP production. The two DA receptor types differ in their localization: the D1-like receptors are predominately localized post-synaptically (Levey et al., 1993), whereas the D2-like receptors are present post-synaptically on dopaminergic target neurons (Levey et al., 1993; Sesack et al., 1994), but pre-synaptically and as autoreceptors (D2-AR) on DA neurons (Mercuri et al., 1997; L’hirondel et al., 1998). The response of SNc neurons to DA is highly regulated and chronic loss of dopamine leads to receptor sensitization (Schultz and Ungerstedt, 1978). In particular, DA binding to the D2-AR leads to activation of G-protein-coupled, inwardly rectifying potassium channels (GIRK2) (Luscher and Slesinger, 2010; Beaulieu and Gainetdinov, 2011) that promotes K+ efflux and hyperpolarization, and consequently reduces SNc DA activity (Beckstead et al., 2004). At the same time, however, D2-AR internalization occurring in response to DA stimulation reduces the DA-induced inhibitory effect on SNc DA neurons firing and tonic Ca2+ entry through L-type voltage channels promotes desensitization of D2 receptor-dependent activation of GIRK channels (Gantz et al., 2015).
In other words, DA itself, upon release, acts in a negative feedback loop: by binding to D2-subtype receptors, it inhibits SNc DA neurons electrical activity and further DA release, but both Ca2+ influx and receptor desensitization limit this action.
Dragicevic et al. (2014) have observed that, in contrast to juvenile SNc neurons, mature neurons have lost D2-autoreceptors desensitization, and, accordingly, upon in vivo exposure to high DA level also juvenile neurons present the same D2-autoreceptors desensitizing response. According to their results, Cav 1.3 mediated Ca2+ influx is essential for age-dependent modulation of somatodendritic D2-autoreceptors responses and D2 autoreceptor sensitization requires both Cav1.3 and NCS-1 activation.
NCS-1 and D2 receptors co-localize both in primate and rodent brain (Kabbani et al., 2002) and NCS-1 attenuates agonist-induced receptor internalization via a mechanism that involves a reduction in D2 receptor phosphorylation. Interestingly, amino acid substitutions that affect NCS-1 Ca2+ binding ability abolished its modulation on D2 receptor signaling (Kabbani et al., 2012) and NCS-1 deletion in mouse has been reported to decrease DA secretion (Ng et al., 2016), thus implying important contribution of NCS-1 impairment in defective dopaminergic signaling.
The finding that, in juvenile mice, Cav1.3 can adapt SNc DA neurons activity in response to high extracellular DA-levels by providing the Ca2+ source for neuronal Ca2+ sensor NCS-1 (Dragicevic et al., 2014) strongly indicates the existence of an adaptive signaling network (Cav1.3/NCS-1/D2/GIRK2) that may have protective role by preventing D2 autoreceptors desensitization. A simplified model that summarizes this concept is shown in Figure 2. According to it, increases in the intracellular Ca2+ concentration activate NCS-1 that opposes somatodendritic D2-autoreceptors internalization and blocks their desensitization counteracting in this way the inhibitory effect mediated by GIRK2 channels on Cav1.3 and finally promotes dopamine release also through this mechanism. Apparently, this could result in a sort of vicious circle that exacerbate Ca2+ entry. However, no desensitization was found during development in KO mice for Cav1.3 and no evidence for exacerbated excitotoxicity upon treatment with the dihydropyridine L-type Ca2+ channel blocker isradipine has been reported so far, thus suggesting that other compensatory mechanisms intervene.
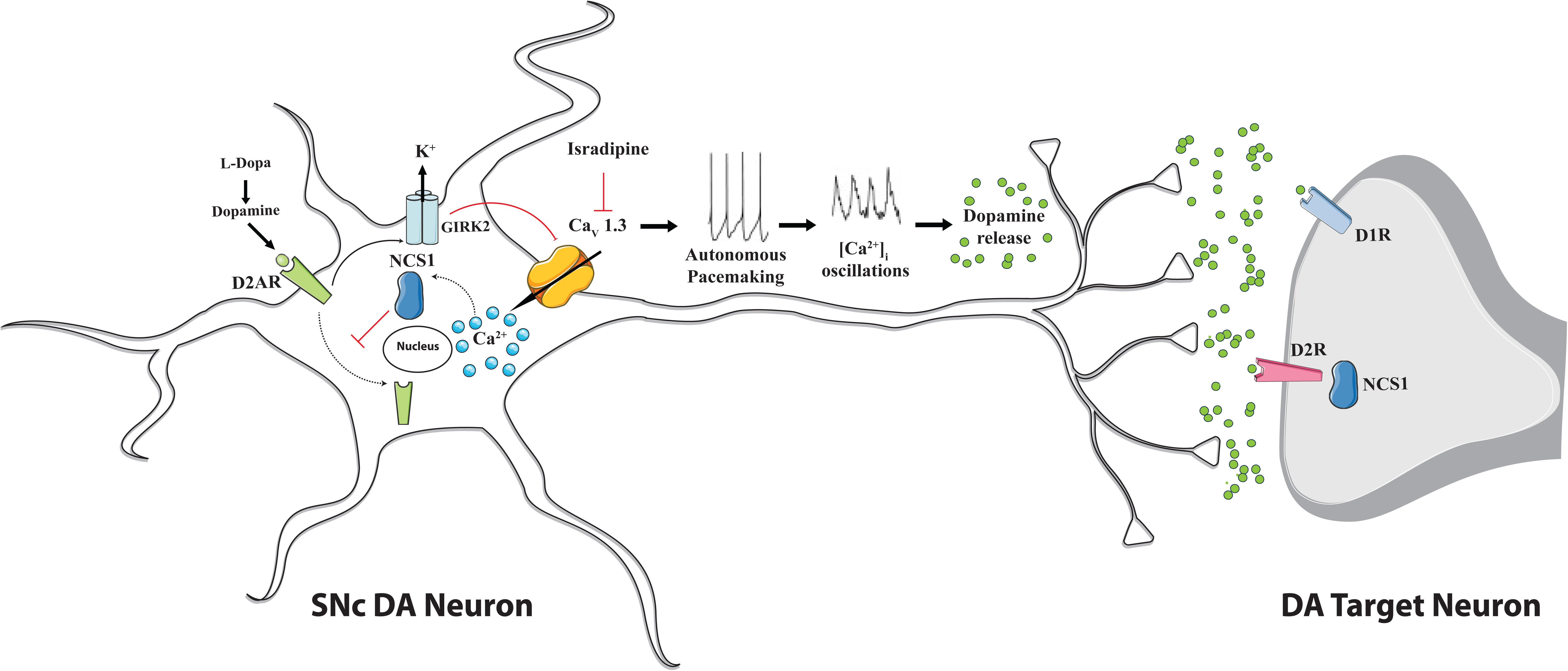
Figure 2. Proposed mechanism for Cav1.3 L-type Ca2+ channel action and NCS-1 contribute during autonomous firing of substantia nigra pars compacta dopaminergic neurons. Cav 1.3 channels activity sustains dopamine release. Dopamine binding to D1 and D2 receptors on dopaminergic target neurons promotes motor function. Dopamine binding to D2-autoreceptors (D2-AR) on SNc DA neurons controls their firing rate by promoting the inhibitory effect of GIRK2 K+ channels and, at the same time, the D2-AR internalization thus contributing to desensitization process. NCS-1 participates in the regulation of dopaminergic signaling since upon Cav 1.3 channels-mediated Ca2+ influx it becomes active and, by blocking D2-AR internalization, prevents receptors desensitization. Upon pharmacological treatment with L-DOPA and/or Cav.1.3 antagonist isradipine a vicious loop may be activated: D2-AR desensitization could be facilitated, since reduced Ca2+ influx may affect NCS-1 inhibition of receptor internalization and the ability of dopamine to inhibit neuronal activity may be compromised, altogether leading to excitotoxicity. This possibility deserves further investigation, even if no evidence in this direction has been provided so far.
In line with this suggestion, loss of Cav1.3 (or its pharmacological inhibition) does not severely compromise pacemaking activity both in juvenile and adult SNc DA neurons, but rather altered its precision and regular occurrence (Poetschke et al., 2015). The appearance of compensatory response due both to NCS-1 upregulation and to the existence of alternative Ca2+ source in SNc DA cells that are able to mediate NCS-1/D2-AR interactions could explain the findings. Indeed, an upregulation of both T-type Ca2+ Cav 1.2 channels and NCS-1 protein has been found in Cav1.3 KO mice (Poetschke et al., 2015).
All together these observations strongly support the idea that Ca2+ and DA are critical components in the disease and underline the complexity of their interplay in the modulation of dopaminergic response.
Conclusion
The distinctive physiology of the DA midbrain neurons within the SNc has attracted attention as possible reason for their selective vulnerability. Slow rhythmic activity (accompanied by oscillations in intracellular Ca2+ concentration) and high dendritic arborization distinguishes these neurons from the other neurons in the brain. Cav1.3 mediated Ca2+ influx is essential to sustain DA release, to guarantee high energy demands that are required for this function and to provide necessary amount of ATP at axonal and dendrites sites. But if continuous Ca2+ entry sustains DA secretion and mitochondrial metabolism, at the same time it exposes cells to “Ca2+ stress,” that may synergize with intrinsic low Ca2+ buffering capacity, aging, mutations or mitochondria damage and culminate in cell degeneration. In vitro and in vivo studies strongly implicated Cav1.3 activity in the high vulnerability of SNc DA neurons, however the complexity of DA metabolism that includes an autoregulatory nature of DA secretion underlines that selective vulnerability of SNc neurons is still an obscure issue. The characterization of the Cav1.3 Ca2+ channels physiology and of the alternative pathways that are engaged to compensate pharmacological inhibition of Cav.1.3 channels upon isradipine treatment certainly deserves more investigations. The outcome of isradipine phase III clinical trial will shed light on these aspects.
At this point we can conclude that the deciphering of the molecular mechanisms involved in dopaminergic signaling is the best we can do to develop therapeutic strategy, but we have to be aware that the complexity of the system is increased by interactive pathways that are engaged in compensatory mechanisms and this makes the investigations very challenging.
Data Availability
All datasets generated for this study are included in the manuscript and/or the supplementary files.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the Università degli Studi di Padova (Progetto di Ateneo 2015 n. CPDA 153402 to MB, Progetto Giovani 2012 n.GRIC128SP0 to TC and Progetto di Ateneo 2016 n. CALI_SID16_01 to TC) and from the Ministry of University and Research (Bando SIR 2014 n. RBSI14C65Z to TC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Angelova, P. R., Ludtmann, M. H., Horrocks, M. H., Negoda, A., Cremades, N., Klenerman, D., et al. (2016). Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. doi: 10.1242/jcs.180737
Area-Gomez, E., and Schon, E. A. (2017). On the pathogenesis of alzheimer’s disease: the mam hypothesis. FASEB J. 31, 864–867. doi: 10.1096/fj.201601309
Beaulieu, J. M., and Gainetdinov, R. R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217. doi: 10.1124/pr.110.002642
Becker, C., Jick, S. S., and Meier, C. R. (2008). Use of antihypertensives and the risk of parkinson disease. Neurology 70, 1438–1444. doi: 10.1212/01.wnl.0000303818.38960.44
Beckstead, M. J., Grandy, D. K., Wickman, K., and Williams, J. T. (2004). Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42, 939–946. doi: 10.1016/j.neuron.2004.05.019
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by f-atp synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Berridge, M. J. (1998). Neuronal calcium signaling. Neuron 21, 13–26. doi: 10.1016/S0896-6273(00)80510-3
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: function and dysfunction. Cell Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193. doi: 10.1038/nrn2093
Calì, T., Ottolini, D., and Brini, M. (2012a). Mitochondrial Ca(2+) and neurodegeneration. Cell Calcium 52, 73–85. doi: 10.1016/j.ceca.2012.04.015
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2012b). alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Calì, T., Ottolini, D., and Brini, M. (2013a). Calcium and endoplasmic reticulum-mitochondria tethering in neurodegeneration. DNA Cell. Biol. 32, 140–146. doi: 10.1089/dna.2013.2011
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2013b). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi: 10.1016/j.bbadis.2013.01.004
Calì, T., Ottolini, D., and Brini, M. (2014). Calcium signaling in parkinson’s disease. Cell Tissue Res. 357, 439–454. doi: 10.1007/s00441-014-1866-0
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuvenation’ protects neurons in mouse models of parkinson’s disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Damier, P., Hirsch, E. C., Agid, Y., and Graybiel, A. M. (1999). The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain 122(Pt 8), 1421–1436. doi: 10.1093/brain/122.8.1421
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
de Lau, L. M., and Breteler, M. M. (2006). Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
Dopeso-Reyes, I. G., Rico, A. J., Roda, E., Sierra, S., Pignataro, D., Lanz, M., et al. (2014). Calbindin content and differential vulnerability of midbrain efferent dopaminergic neurons in macaques. Front. Neuroanat. 8:146. doi: 10.3389/fnana.2014.00146
Dragicevic, E., Poetschke, C., Duda, J., Schlaudraff, F., Lammel, S., Schiemann, J., et al. (2014). Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain 137, 2287–2302. doi: 10.1093/brain/awu131
Fierro, L., and Llano, I. (1996). High endogenous calcium buffering in purkinje cells from rat cerebellar slices. J. Physiol. 496(Pt 3), 617–625. doi: 10.1113/jphysiol.1996.sp021713
Filadi, R., Basso, E., Lefkimmiatis, K., and Pozzan, T. (2017a). Beyond intracellular signaling: the ins and outs of second messengers microdomains. Adv. Exp. Med. Biol. 981, 279–322. doi: 10.1007/978-3-319-55858-5_12
Filadi, R., Theurey, P., and Pizzo, P. (2017b). The endoplasmic reticulum-mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium 62, 1–15. doi: 10.1016/j.ceca.2017.01.003
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2016). Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 15, 2226–2238. doi: 10.1016/j.celrep.2016.05.013
Foehring, R. C., Zhang, X. F., Lee, J. C., and Callaway, J. C. (2009). Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 102, 2326–2333. doi: 10.1152/jn.00038.2009
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Gantz, S. C., Robinson, B. G., Buck, D. C., Bunzow, J. R., Neve, R. L., Williams, J. T., et al. (2015). Distinct regulation of dopamine D2S and D2L autoreceptor signaling by calcium. eLife 4:e09358. doi: 10.7554/eLife.09358
German, D. C., Manaye, K. F., Sonsalla, P. K., and Brooks, B. A. (1992). Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann. N. Y. Acad. Sci. 648, 42–62. doi: 10.1111/j.1749-6632.1992.tb24523.x
Geula, C., Bu, J., Nagykery, N., Scinto, L. F., Chan, J., Joseph, J., et al. (2003). Loss of calbindin-D28k from aging human cholinergic basal forebrain: relation to neuronal loss. J. Comp. Neurol. 455, 249–259. doi: 10.1002/cne.10475
Goedert, M., Spillantini, M. G., Del Tredici, K., and Braak, H. (2013). 100 years of lewy pathology. Nat. Rev. Neurol. 9, 13–24. doi: 10.1038/nrneurol.2012.242
Guzman, J. N., Ilijic, E., Yang, B., Sanchez-Padilla, J., Wokosin, D., Galtieri, D., et al. (2018). Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Invest. 128, 2266–2280. doi: 10.1172/JCI95898
Guzman, J. N., Sanchez-Padilla, J., Wokosin, D., Kondapalli, J., Ilijic, E., Schumacker, P. T., et al. (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468, 696–700. doi: 10.1038/nature09536
Handley, M. T., Lian, L. Y., Haynes, L. P., and Burgoyne, R. D. (2010). Structural and functional deficits in a neuronal calcium sensor-1 mutant identified in a case of autistic spectrum disorder. PLoS One 5:e10534. doi: 10.1371/journal.pone.0010534
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hurley, M. J., Brandon, B., Gentleman, S. M., and Dexter, D. T. (2013). Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 136, 2077–2097. doi: 10.1093/brain/awt134
Ilijic, E., Guzman, J. N., and Surmeier, D. J. (2011). The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 43, 364–371. doi: 10.1016/j.nbd.2011.04.007
Kabbani, N., Negyessy, L., Lin, R., Goldman-Rakic, P., and Levenson, R. (2002). Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J. Neurosci. 22, 8476–8486. doi: 10.1523/JNEUROSCI.22-19-08476.2002
Kabbani, N., Woll, M. P., Nordman, J. C., and Levenson, R. (2012). Dopamine receptor interacting proteins: targeting neuronal calcium sensor-1/D2 dopamine receptor interaction for antipsychotic drug development. Curr. Drug Targets 13, 72–79. doi: 10.2174/138945012798868515
Khaliq, Z. M., and Bean, B. P. (2010). Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 30, 7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010
Koh, P. O., Undie, A. S., Kabbani, N., Levenson, R., Goldman-Rakic, P. S., and Lidow, M. S. (2003). Up-regulation of neuronal calcium sensor-1 (NCS-1) in the prefrontal cortex of schizophrenic and bipolar patients. Proc. Natl. Acad. Sci. U.S. A. 100, 313–317. doi: 10.1073/pnas.232693499
Kurtishi, A., Rosen, B., Patil, K. S., Alves, G. W., and Moller, S. G. (2018). Cellular proteostasis in neurodegeneration. Mol. Neurobiol. doi: 10.1007/s12035-018-1334-z [Epub ahead of print].
L’hirondel, M., Cheramy, A., Godeheu, G., Artaud, F., Saiardi, A., Borrelli, E., et al. (1998). Lack of autoreceptor-mediated inhibitory control of dopamine release in striatal synaptosomes of D2 receptor-deficient mice. Brain Res. 792, 253–262. doi: 10.1016/S0006-8993(98)00146-2
La Rovere, R. M., Roest, G., Bultynck, G., and Parys, J. B. (2016). Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 60, 74–87. doi: 10.1016/j.ceca.2016.04.005
Levey, A. I., Hersch, S. M., Rye, D. B., Sunahara, R. K., Niznik, H. B., Kitt, C. A., et al. (1993). Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc. Natl. Acad. Sci. U.S.A. 90, 8861–8865. doi: 10.1073/pnas.90.19.8861
Liang, C. L., Wang, T. T., Luby-Phelps, K., and German, D. C. (2007). Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson’s disease. Exp. Neurol. 203, 370–380. doi: 10.1016/j.expneurol.2006.08.015
Liss, B., and Striessnig, J. (2019). The potential of l-type calcium channels as a drug target for neuroprotective therapy in Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 59, 263–289. doi: 10.1146/annurev-pharmtox-010818-021214
Liu, Q., Emadi, S., Shen, J. X., Sierks, M. R., and Wu, J. (2013). Human alpha4beta2 nicotinic acetylcholine receptor as a novel target of oligomeric alpha-synuclein. PLoS One 8:e55886. doi: 10.1371/journal.pone.0055886
Luscher, C., and Slesinger, P. A. (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315. doi: 10.1038/nrn2834
Marongiu, R., Spencer, B., Crews, L., Adame, A., Patrick, C., Trejo, M., et al. (2009). Mutant pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 108, 1561–1574. doi: 10.1111/j.1471-4159.2009.05932.x
McCue, H. V., Haynes, L. P., and Burgoyne, R. D. (2010). The diversity of calcium sensor proteins in the regulation of neuronal function. Cold Spring Harb. Perspect. Biol. 2:a004085. doi: 10.1101/cshperspect.a004085
Mercado, G., Valdes, P., and Hetz, C. (2013). An ERcentric view of Parkinson’s disease. Trends Mol. Med. 19, 165–175. doi: 10.1016/j.molmed.2012.12.005
Mercuri, N. B., Saiardi, A., Bonci, A., Picetti, R., Calabresi, P., Bernardi, G., et al. (1997). Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience 79, 323–327.
Moore, L. M., England, A., Ehrlich, B. E., and Rimm, D. L. (2017). Calcium sensor, ncs-1, promotes tumor aggressiveness and predicts patient survival. Mol. Cancer Res. 15, 942–952. doi: 10.1158/1541-7786.MCR-16-0408
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Mouatt-Prigent, A., Agid, Y., and Hirsch, E. C. (1994). Does the calcium binding protein calretinin protect dopaminergic neurons against degeneration in Parkinson’s disease? Brain Res. 668, 62–70. doi: 10.1016/0006-8993(94)90511-8
Muller, M., Ahumada-Castro, U., Sanhueza, M., Gonzalez-Billault, C., Court, F. A., and Cardenas, C. (2018). Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 12:470. doi: 10.3389/fnins.2018.00470
Neher, E. (1998). Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium 24, 345–357. doi: 10.1016/S0143-4160(98)90058-6
Neher, E., and Augustine, G. J. (1992). Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 450, 273–301. doi: 10.1113/jphysiol.1992.sp019127
Ng, E., Varaschin, R. K., Su, P., Browne, C. J., Hermainski, J., Le Foll, B., et al. (2016). Neuronal calcium sensor-1 deletion in the mouse decreases motivation and dopamine release in the nucleus accumbens. Behav. Brain Res. 301, 213–225. doi: 10.1016/j.bbr.2015.12.037
Oertel, W., and Schulz, J. B. (2016). Current and experimental treatments of parkinson disease: a guide for neuroscientists. J. Neurochem. 139(Suppl. 1), 325–337. doi: 10.1111/jnc.13750
Ottolini, D., Calì, T., Negro, A., and Brini, M. (2013). The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 22, 2152–2168. doi: 10.1093/hmg/ddt068
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149. doi: 10.1007/s00401-017-1704-z
Pasternak, B., Svanstrom, H., Nielsen, N. M., Fugger, L., Melbye, M., and Hviid, A. (2012). Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol. 175, 627–635. doi: 10.1093/aje/kwr362
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Piton, A., Michaud, J. L., Peng, H., Aradhya, S., Gauthier, J., Mottron, L., et al. (2008). Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 17, 3965–3974. doi: 10.1093/hmg/ddn300
Poetschke, C., Dragicevic, E., Duda, J., Benkert, J., Dougalis, A., Dezio, R., et al. (2015). Compensatory T-type Ca2+ channel activity alters D2-autoreceptor responses of Substantia nigra dopamine neurons from Cav1.3 L-type Ca2+ channel KO mice. Sci. Rep. 5:13688. doi: 10.1038/srep13688
Pongs, O., Lindemeier, J., Zhu, X. R., Theil, T., Engelkamp, D., Krah-Jentgens, I., et al. (1993). Frequenin–a novel calcium-binding protein that modulates synaptic efficacy in the Drosophila nervous system. Neuron 11, 15–28. doi: 10.1016/0896-6273(93)90267-U
Puopolo, M., Raviola, E., and Bean, B. P. (2007). Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J. Neurosci. 27, 645–656. doi: 10.1523/JNEUROSCI.4341-06.2007
Ritz, B., Rhodes, S. L., Qian, L., Schernhammer, E., Olsen, J. H., and Friis, S. (2010). L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 67, 600–606.
Ronzitti, G., Bucci, G., Emanuele, M., Leo, D., Sotnikova, T. D., Mus, L. V., et al. (2014). Exogenous alpha-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 34, 10603–10615. doi: 10.1523/JNEUROSCI.0608-14.2014
Samanta, K., and Parekh, A. B. (2017). Spatial Ca(2+) profiling: decrypting the universal cytosolic Ca(2+) oscillation. J. Physiol. 595, 3053–3062. doi: 10.1113/JP272860
Schultz, W., and Ungerstedt, U. (1978). Striatal cell supersensitivity to apomorphine in dopamine-lesioned rats correlated to behaviour. Neuropharmacology 17, 349–353. doi: 10.1016/0028-3908(78)90005-9
Schwaller, B. (2012). The use of transgenic mouse models to reveal the functions of Ca2+ buffer proteins in excitable cells. Biochim. Biophys. Acta 1820, 1294–1303. doi: 10.1016/j.bbagen.2011.11.008
Sesack, S. R., Aoki, C., and Pickel, V. M. (1994). Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J. Neurosci. 14, 88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994
Sharma, R. K., and Parameswaran, S. (2018). Calmodulin-binding proteins: a journey of 40 years. Cell Calcium 75, 89–100. doi: 10.1016/j.ceca.2018.09.002
Surguchev, A., and Surguchov, A. (2015). Effect of alpha-synuclein on membrane permeability and synaptic transmission: a clue to neurodegeneration? J. Neurochem. 132, 619–621. doi: 10.1111/jnc.13045
Surmeier, D. J., Guzman, J. N., Sanchez-Padilla, J., and Schumacker, P. T. (2011). The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience 198, 221–231. doi: 10.1016/j.neuroscience.2011.08.045
Tarasov, A. I., Griffiths, E. J., and Rutter, G. A. (2012). Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 52, 28–35. doi: 10.1016/j.ceca.2012.03.003
Wang, H. Q., and Takahashi, R. (2007). Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid. Redox Signal. 9, 553–561. doi: 10.1089/ars.2006.1524
Xia, Z., and Storm, D. R. (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 6, 267–276. doi: 10.1038/nrn1647
Yamada, T., Mcgeer, P. L., Baimbridge, K. G., and Mcgeer, E. G. (1990). Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 526, 303–307. doi: 10.1016/0006-8993(90)91236-A
Zallo, F., Gardenal, E., Verkhratsky, A., and Rodriguez, J. J. (2018). Loss of calretinin and parvalbumin positive interneurones in the hippocampal CA1 of aged Alzheimer’s disease mice. Neurosci. Lett. 681, 19–25. doi: 10.1016/j.neulet.2018.05.027
Zhang, K., Heidrich, F. M., Degray, B., Boehmerle, W., and Ehrlich, B. E. (2010). Paclitaxel accelerates spontaneous calcium oscillations in cardiomyocytes by interacting with NCS-1 and the InsP3R. J. Mol. Cell. Cardiol. 49, 829–835. doi: 10.1016/j.yjmcc.2010.08.018
Keywords: calcium signaling, Cav1.3 calcium channel, ncs-1, dopamine, Parkinson’s disease
Citation: Catoni C, Calì T and Brini M (2019) Calcium, Dopamine and Neuronal Calcium Sensor 1: Their Contribution to Parkinson’s Disease. Front. Mol. Neurosci. 12:55. doi: 10.3389/fnmol.2019.00055
Received: 25 October 2018; Accepted: 14 February 2019;
Published: 22 March 2019.
Edited by:
Daniele Dell’Orco, University of Verona, ItalyReviewed by:
Rosanna Parlato, University of Ulm, GermanyTeresa Duda, Salus University, United States
Copyright © 2019 Catoni, Calì and Brini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marisa Brini, bWFyaXNhLmJyaW5pQHVuaXBkLml0