
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 15 January 2019
Sec. Neuroplasticity and Development
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00494
This article is part of the Research Topic Neuronal Calcium Sensors in Health and Disease View all 26 articles
 Grit Bornschein
Grit Bornschein Hartmut Schmidt*
Hartmut Schmidt*Ca2+ concentrations drop rapidly over a distance of a few tens of nanometers from an open voltage-gated Ca2+ channel (Cav), thereby, generating a spatially steep and temporally short-lived Ca2+ gradient that triggers exocytosis of a neurotransmitter filled synaptic vesicle. These non-steady state conditions make the Ca2+-binding kinetics of the Ca2+ sensors for release and their spatial coupling to the Cavs important parameters of synaptic efficacy. In the mammalian central nervous system, the main release sensors linking action potential mediated Ca2+ influx to synchronous release are Synaptotagmin (Syt) 1 and 2. We review here quantitative work focusing on the Ca2+ kinetics of Syt2-mediated release. At present similar quantitative detail is lacking for Syt1-mediated release. In addition to triggering release, Ca2+ remaining bound to Syt after the first of two successive high-frequency activations was found to be capable of facilitating release during the second activation. More recently, the Ca2+ sensor Syt7 was identified as additional facilitation sensor. We further review how several recent functional studies provided quantitative insights into the spatial topographical relationships between Syts and Cavs and identified mechanisms regulating the sensor-to-channel coupling distances at presynaptic active zones. Most synapses analyzed in matured cortical structures were found to operate at tight, nanodomain coupling. For fast signaling synapses a developmental switch from loose, microdomain to tight, nanodomain coupling was found. The protein Septin5 has been known for some time as a developmentally down-regulated “inhibitor” of tight coupling, while Munc13-3 was found only recently to function as a developmentally up-regulated mediator of tight coupling. On the other hand, a highly plastic synapse was found to operate at loose coupling in the matured hippocampus. Together these findings suggest that the coupling topography and its regulation is a specificity of the type of synapse. However, to definitely draw such conclusion our knowledge of functional active zone topographies of different types of synapses in different areas of the mammalian brain is too incomplete.
The release of neurotransmitter from presynaptic terminals and its modulation via synaptic plasticity are the bedrocks of directed information flow within neuronal circuits of the central nervous system (CNS). An action potential (AP) triggers the release of a neurotransmitter filled synaptic vesicle (SV) by opening voltage-gated Ca2+ channels (Cavs) in the presynaptic active zone (AZ). The inflowing Ca2+ diffuses toward the SV, which bears the primary Ca2+ sensor proteins Synaptotagmin (Syt) 1 or 2 on its surface that are required for triggering its fusion with the presynaptic plasma membrane. Ca2+ binding to Syt changes its conformation and the resulting interaction with proteins of the core release machinery, the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, and other proteins at the AZ ultimately triggers the fusion of the SV with the presynaptic plasma membrane (Südhof, 2013; Kaeser and Regehr, 2014). Thus, although aspects of this process are still not understood, it can be noted that Syts link Ca2+ influx to SNARE mediated SV fusion.
The process of transmitter release is probabilistic, i.e., not every AP leads to exocytosis; rather it triggers the release of a SV only with a certain probability. The average vesicular release probability (pr) can be quantified by way of analyzing fluctuations in postsynaptic current amplitudes (PSCs) under conditions of different pr, e.g., at different concentrations of extracellular Ca2+ ([Ca2+]e; Clements and Silver, 2000). Instead of recording PSCs, recently it became also feasible to more directly monitor glutamate release from individual boutons by imaging the fluorescence of a genetically encoded, intensity-based glutamate-sensing fluorescent reporter (iGlusnFr; Jensen et al., 2017; Helassa et al., 2018; Marvin et al., 2018).
The initial pr (pr1) is an important factor not only in determining the release fidelity for a single AP but also in setting the properties of short-term plasticity of a synapse (Zucker and Regehr, 2002; Abbott and Regehr, 2004). This can be illustrated by a simple example of paired-pulse ratio (PPR) in the absence of replenishment of SVs between the two APs of a paired-pulse experiment. In this case PPR = pr2/pr1 (1-pr1), where pr2 is the release probability of the second release process, which may differ from pr1. In general, it can be noted that if pr1 > 0.5 the synapse will depress, i.e., PPR < 1, and only the magnitude of the depression will depend on pr2. However, if pr1 < 0.5 the synapse will show facilitation or depression depending on pr2.
The pr depends on the Ca2+-binding kinetics of the release machinery, i.e., the Ca2+-binding kinetics of Syt in the context of the SNARE and other proteins at the AZ, and on the amplitude and time course of the Ca2+ signal “seen” by Syt. The latter depends on different factors, including the number and types of Cavs, their diffusional distance to Syt, and the characteristics of other Ca2+-binding proteins present in the terminal. Ca2+ entering the presynaptic terminal builds a steep, short-lasting concentration gradient around the mouth of the open Cavs that rapidly diminishes with increasing distance from the channel. Due to the steepness and short duration of this Ca2+ gradient a chemical equilibrium is never established in this process. This makes the intracellular Ca2+-binding kinetics of the release sensor, rather than its affinity alone, as well as its diffusional distance to the Cavs crucial to the control of speed and reliability of transmitter release (Bollmann et al., 2000; Schneggenburger and Neher, 2000; Eggermann and Jonas, 2012). In this review we will focus on these two prominent factors in the regulation of pr, the synaptic Ca2+-binding kinetics of Syt proteins and their topographical relationships to Cavs. We will put an emphasis to more recent findings at small synapses in cortical structures of the mammalian brain.
Synaptotagmin–1,−2, and −9 (Syt1, 2, 9) are the known Ca2+ sensors for fast, synchronous transmitter release in the millisecond time window following an AP (Südhof, 2014). Syt1 and Syt2 are the dominating Syt isoforms for synchronous release in the mammalian brain while Syt9 expression appears to be restricted to the limbic system and the striatum (Berton et al., 1997; Fox and Sanes, 2007; Xu et al., 2007). In addition to fast synchronous release, a second, slow and asynchronous component of transmitter release has been described (Geppert et al., 1994; Goda and Stevens, 1994). Asynchronous release is primarily activated during and following repetitive stimulation and operates via sensors different from those for synchronous release (Sun et al., 2007; Kochubey et al., 2011). Due to their dominating role for rapid neuronal communication, we will focus here on Syt1 and Syt2 triggered release processes. Molecular and structural aspects of Syt1, 2 proteins and their interactions with SNARE- and scaffold proteins were covered by several comprehensive recent reviews (Südhof and Rothman, 2009; Südhof, 2012, 2013; Kaeser and Regehr, 2014; Brunger et al., 2018; Park and Ryu, 2018).
Briefly, a synaptic vesicle bears approximately 15 copies of Syt on its surface (Takamori et al., 2006). Each Syt has two C2 domains that constitute Ca2+-binding and in addition might mediate protein-protein interactions with SNAREs and other Syt proteins or interactions with the membrane. One of the C2 domains is a C2A domain that binds three Ca2+ ions, while the other one is a C2B domain that binds two Ca2+ ions (Südhof, 2013). Upon Ca2+ binding Syt triggers rapid synchronous vesicle fusion but the detailed molecular mechanisms are complex and still controversial (Brunger et al., 2018; Park and Ryu, 2018). Some of the proposed models discuss the role of Syt in at least two processes: First, prior to Ca2+ influx spontaneous fusion of synaptic vesicles has to be prevented by inhibiting the constititively active SNARE complex from full zippering (SNARE clamping). Second, upon Ca2+ influx fusion is triggered by relieving SNAREs from the clamp (SNARE unclamping). Among the proposed models it is under debate if SNARE clamping is mediated directly by Syt or if and in as much it involves a second protein called Complexin (Cpx), which is discussed to also have a SNARE clamping function (Südhof, 2013; Trimbuch and Rosenmund, 2016), and can form a protein complex with SNAREs and Syt (Zhou et al., 2017). Hence, according to these models, Syt either has a dual function by first clamping SNARE zippering and an uncalmping function by relieving the clamp upon Ca2+ binding or only by relieving a Cpx-mediated SNARE clamp upon Ca2+ binding. Other models emphasize the membrane binding properties of Syt and suggest that membrane insertion of Ca2+-bound Syt could cross-link vesicle and plasma membrane or lower the energy barrier for fusion by either regulating the vesicle to plasma membrane distance or by locally curving the plasma membrane. In addition, there is evidence that Syt also directly binds to Ca2+ channels (Sheng et al., 1997). Since a detailed discussion of the molecular mechanisms of the fusion process is beyond the scope of this review, we refer the reader to most recent reviews (Trimbuch and Rosenmund, 2016; Brunger et al., 2018; Park and Ryu, 2018). We will focus here on the kinetic aspects of the interaction between Ca2+ ions and Syt1, 2.
It has been known for half a century that transmitter release has a non-linear, approximately power of 4 dependency on [Ca2+]e (Dodge and Rahamimoff, 1967). However, a quantification of the intracellular presynaptic Ca2+-binding kinetics of a CNS release process became available only more recently (Bollmann et al., 2000; Schneggenburger and Neher, 2000), resulting in a detailed kinetic model of Ca2+-binding and release for the young calyx of Held synapse in the auditory brainstem (Figure 1), which expresses the Syt2 isoform as prime release sensor (Kochubey et al., 2016). The model was established based on an elegant combination of presynaptic Ca2+ uncaging and Ca2+ imaging with pre- and postsynaptic patch-clamp recordings (Box 1). The established model covers five cooperative, low-affinity Ca2+-binding sites with fast kinetic rate constants for Ca2+-binding and -unbinding (kon ~108 M−1s−1, koff ~5000 s−1, respectively) and accounted for the experimental, cooperative power of 4 dependency of the release rate onto the intracellular Ca2+ concentration ([Ca2+]i) as well as brief synaptic delays (Figure 1, Table 1). It should be noted that this model does not reflect the Ca2+-binding kinetics of Syt2 alone but rather the kinetics of Syt2 embedded in its natural synaptic environment. For simplicity we will refer to it as the Syt2 model.
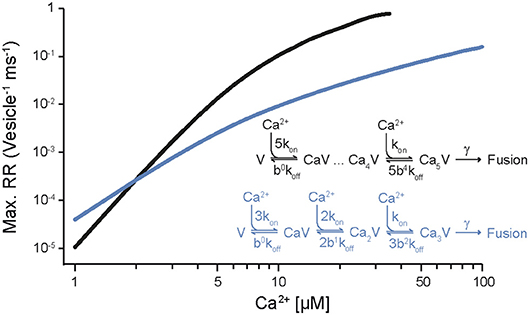
Figure 1. Dependency of release rates on the intracellular Ca2+ concentration. Release rates were calculated for different [Ca2+]i using the sensor models developed for Syt2 at the calyx of Held (black; Schneggenburger and Neher, 2000) and for Syt1 at chromaffin cells (blue; Voets, 2000). Peak release rates per vesicle were plotted against the corresponding [Ca2+]i. Insets show the kinetic binding schemes for the reaction between vesicular sensor (V) and Ca2+.
Box 1. Quantification of the Ca2+-binding kinetics of Syt in presynaptic terminals via Ca2+ uncaging.
Syt is integrated in the supra-molecular protein complex of the release machinery, which will influence its Ca2+-binding kinetics in a non-predictable manner similar to other Ca2+ sensor proteins (Xia and Storm, 2005). Consequently, Syt2 has been analyzed in Syt2 expressing synapses (cf. above). Toward this end, it was required to first define the local [Ca2+]i at the release sensor and second, to quantify corresponding release rates.
At present it is difficult or even impossible to directly quantify the local [Ca2+]i at the release sensor. Even if it were possible to measure [Ca2+]i at areas as small as ~0.5 μm2 as performed at squid giant synapses (Llinás et al., 1992), the local [Ca2+]i at the sensor would remain rather ill-defined due to the steep spatial gradient of synaptic [Ca2+]i elevations, the unknown distance to the sensor and uncertainties about endogenous Ca2+ buffers (Neher, 1998a; Bucurenciu et al., 2008, 2010; Bornschein et al., 2013; Schmidt et al., 2013).
Ca2+ uncaging has been shown to be a useful method to resolve this problem (Heidelberger et al., 1994). Ca2+ uncaging elevates [Ca2+]i uniformly in a presynaptic terminal. Due to this uniform [Ca2+]i elevation throughout the terminal, local [Ca2+]i will be identical to global [Ca2+]i, which in turn is quantified by concomitant Ca2+ imaging. Uniform elevations of synaptic [Ca2+]i to different levels by flash photolysis of caged Ca2+ have been employed for establishing the relationship between [Ca2+]i and release and permitted the construction of the above described Syt2-based release models. This method was applied at the giant calyx of Held (Bollmann et al., 2000; Schneggenburger and Neher, 2000; Lou et al., 2005; Sun et al., 2007; Kochubey and Schneggenburger, 2011), which permits direct whole-cell patch-clamp equilibration with caged Ca2+ compounds and Ca2+ indicator dyes, Ca2+ uncaging at the presynaptic terminal and concomitant patch-clamp recordings from the postsynaptic site. Thus, differences in PSC amplitudes and synaptic delays recorded at the postsynaptic site can be directly correlated to differences in [Ca2+]i at the presynaptic release sensor. Finally, recording of quantal PSCs (“minis”) allows for calculating the release rates by deconvolution analysis (Van der Kloot, 1988; Diamond and Jahr, 1995; Bollmann et al., 2000; Schneggenburger and Neher, 2000; Neher and Sakaba, 2001). Deconvolution decomposes the PSC into the times of release of individual quanta, thereby, giving the release rate in quanta/s during the PSC. The recorded mini serves as elementary quantal event for the deconvolution. Deconvolution assumes that there are no quantal interactions at the synapse, i.e., the PSC arises from linearly summing minis.
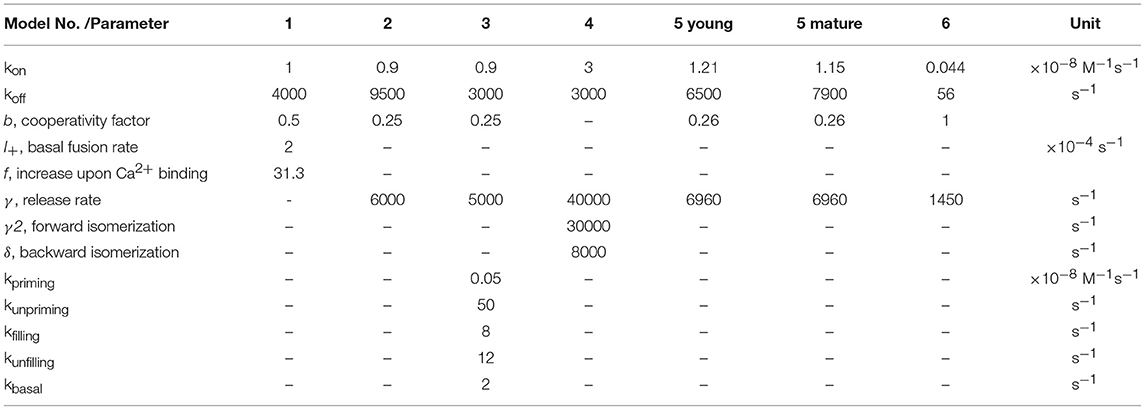
Table 1. Parameters of release sensors.
In following work the Syt2 model has been extended (Scheme 1) to also account for release at low [Ca2+]i (Lou et al., 2005), for phasic and tonic release (Millar et al., 2005; Pan and Zucker, 2009), for asynchronous release (Sun et al., 2007), and to address mechanisms of synaptic plasticity (Felmy et al., 2003; Sakaba, 2008; Pan and Zucker, 2009; Bornschein et al., 2013; Brachtendorf et al., 2015). In addition, it has been shown that the intracellular Ca2+ sensitivity of Syt2-driven release is slightly reduced between postnatal day (P) 8-9 and P12-15 at the calyx of Held (Wang et al., 2008; Kochubey et al., 2009). Currently, the established Syt2 models are widely used in quantitative descriptions of transmitter release (Eggermann et al., 2012; Stanley, 2016).
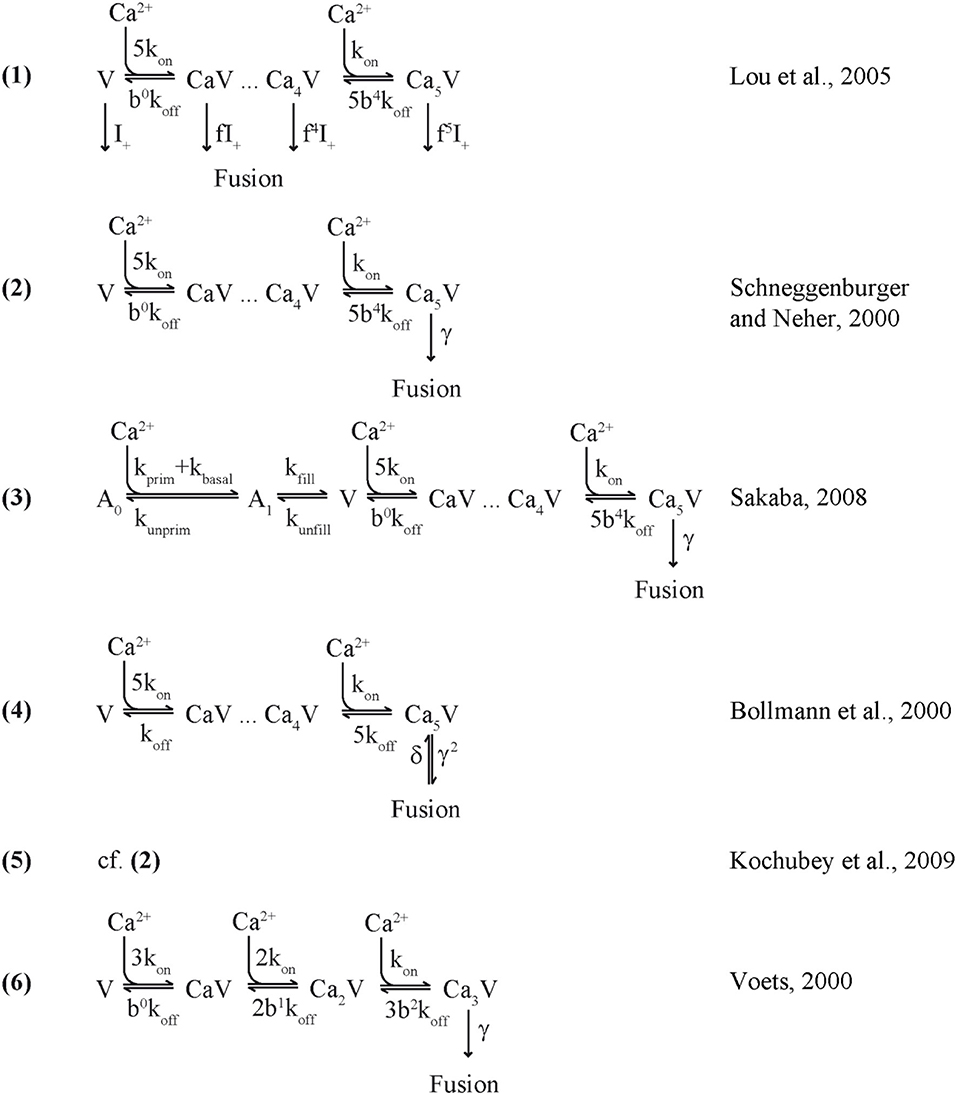
Scheme 1. Release sensor models.
For mammalian CNS synapses, the Syt2-based models were originally constructed at the young (1–2 weeks old) calyx of Held but subsequently also at a small CNS synapse, the immature cerebellar basket cell to Purkinje cell (PC) synapse (Sakaba, 2008), at which Syt2 also represents the dominant Syt isoform (Chen et al., 2017). Notably, Syt2 is the dominating fast release sensor in hindbrain structures, while in most forebrain structures, including the neocortex, Syt1 is the sensor mediating fast synchronous release (Berton et al., 1997; Fox and Sanes, 2007; Xu et al., 2007). Importantly, a detailed kinetic model similar to that of Syt2 is at present not available for Ca2+-binding to Syt1 in mammalian CNS synapses. At very young, P5-6 pre-calyx synapses in the brainstem a fast release component has been reported to be mediated via Syt1 but no kinetic model has been constructed (Kochubey et al., 2016). This Syt1 triggered release process had a less than power of 2 dependency on [Ca2+]i, i.e., its [Ca2+]i dependency was substantially shallower than that of Syt2 triggered release at the young calyx of Held. A kinetic model for Syt1-mediated release has been constructed for fusion of dense core vesicles at chromaffin cells of the adrenal gland (Voets, 2000; Sørensen et al., 2003). In this model three to four Ca2+-binding sites with rate constants of about two orders of magnitude smaller than those for the synaptic Syt2 model were found to be suitable to describe secretion from chromaffin cells, which is much slower than rapid synaptic release (Table 1). Consequently, the resulting dependency of the release rate onto [Ca2+]i was again much shallower than for synaptic Syt2 (Figure 1). Also, a quantitative comparison of the dependency on [Ca2+]e of release processes mediated by Sy1 and Syt2 in cultured neurons revealed differences between the two proteins. Finally, differences were found for the kinetics of Syt1 and Syt2 mediated postsynaptic currents (PSCs), indicating differences in the kinetics of Syt1 vs. Syt2 triggered release. Consequently, it has been suggested that the expression of a particular Syt isoform dictates the properties of release at its synapse (Xu et al., 2007). Thus, it will be interesting to see, whether Syt1-triggered release at mature synapses in the mammalian brain indeed has a dependency on [Ca2+]i different from Syt2-triggered release.
Paired pulse facilitation (PPF) is a form of short-term synaptic plasticity important for synaptic computation (Abbott and Regehr, 2004). PPF is a use-dependent enhancement of transmitter release following the second of two successive APs separated by a millisecond time interval. Although PPF was discovered more than 70 years ago, its mechanisms remain controversial and may differ between synapses (Zucker and Regehr, 2002). Originally, it has been suggested that “Ca2+ remaining attached to specific sites on the inner axon membrane” causes facilitation. For this Ca2+ the term “active Ca2+” was coined (Katz and Miledi, 1968). In a simpler form of the “residual Ca2+ hypothesis” a residue of free Ca2+ ([Ca2+]res) from the first AP summates with free Ca2+ ([Ca2+]i) from the second AP, thereby, causing amplified release. However, it has been recognized early that the decay of [Ca2+]res deviates from the time course of facilitation, such that [Ca2+]res cannot fully account for facilitation (Blundon et al., 1993). Additionally, due to the large amplitude difference between [Ca2+]res (~100 nM) and nano- or microdomain [Ca2+]i at the release site during the second AP (~20–100 μM) simple Ca2+ summation is unlikely to be the exclusive source of facilitation (Zucker and Regehr, 2002). Consequently, at different synapses different conceptions were developed to account for facilitation. These include slow Ca2+ relaxation of the bound sensor (Yamada and Zucker, 1992; Bertram et al., 1996; Matveev et al., 2002), separate sites for release and facilitation (Atluri and Regehr, 1996), elevated release site [Ca2+]i during the second pulse (Geiger and Jonas, 2000; Felmy et al., 2003; Bollmann and Sakmann, 2005), buffer effects (Neher, 1998a; Rozov et al., 2001), or activity dependent recruitment of additional release sites (Valera et al., 2012; Miki et al., 2016; Doussau et al., 2017). For a recent comprehensive review on mechanisms of PPF (see Jackman and Regehr, 2017).
Here, we focus on Syt-related mechanisms of PPF. Ca2+-unbinding from the release sensor has been suggested as one mechanism of PPF (Yamada and Zucker, 1992; Bertram et al., 1996; Matveev et al., 2002). Young cerebellar PCs are connected among each other via recurrent GABAergic synapses that show PPF during high-frequency activation. Although PCs strongly express the “slow” and “fast” native Ca2+ buffers Parvalbumin (PV) and Calbindin-D28k (CB), respectively, PPF was not affected by loss of either of the buffers (Bornschein et al., 2013). Rather the results indicated that a residue of Ca2+ remaining bound to the release sensor Syt2 (Schneggenburger and Neher, 2000; Sakaba, 2008) after the first AP is the probable main cause of PPF at PC to PC synapses, a mechanism highly reminiscent of the original “active Ca2+” mechanism (Katz and Miledi, 1968).
Another suggested mechanism reminding on the original “active Ca2+” mechanism was that a facilitation sensor separate from the release sensor could exist (Atluri and Regehr, 1996). The molecular identity of the facilitation sensor, however, remained elusive until recently Syt7 has been identified to operate as a facilitation sensor (Jackman et al., 2016). Syt7 is abundantly found in presynaptic plasma membranes (Li et al., 2017), while Syt1 and Syt2 rather locate to SV membranes. The intrinsic Ca2+ affinities of Syt1 and Syt7 are comparably low in solution (KD ≥ 100 μM; Radhakrishnan et al., 2009; Voleti et al., 2017). In the presence of lipids the apparent Ca2+ affinity of both proteins increases, albeit for Syt7 stronger than for Syt1, such that the apparent Ca2+ affinity of Syt7 is ~10fold higher than that of Syt1 (Sugita et al., 2002). The apparent Ca2+-sensing properties of Syt1 and Syt7 correlate with their specific functions, such that Syt1 is activated only by high Ca2+ concentrations (~10–100 μM) typical for AP-evoked [Ca2+]i elevations in the vicinity of Ca2+ channels, while Syt7 can also operate during longer lasting increases in residual Ca2+ in the low micromolar range (Volynski and Krishnakumar, 2018). These characteristics made Syt7 a promising candidate for the proposed facilitation sensor. Indeed, it was found that Syt7 contributes to PPF at different types of facilitating synapses in the hippocampus and at cortico-thalamic synapses. Mechanistically, Ca2+-binding to the C2A domain of Syt7 was required for facilitation (Jackman et al., 2016; Jackman and Regehr, 2017; Turecek et al., 2017).
For Syt7 at least two other functions were reported (Volynski and Krishnakumar, 2018): It was found to act as a Ca2+ sensor for SV replenishment (Liu et al., 2014) and to mediate slow, asynchronous transmitter release (Bacaj et al., 2013; Luo and Südhof, 2017). Interestingly, the different proposed functions of Syt7 need not be mutually exclusive (Chen and Jonas, 2017). Consistently, it was found at cerebellar PF to PC synapses that Syt7 is involved in mediating both, PPF and asynchronous release (Turecek and Regehr, 2018). PPF at PF synapses has further been reported to depend on rapid SV replenishment with recruitment of additional release sites that resulted in an activity dependent, transient increase in the RRP (Valera et al., 2012; Brachtendorf et al., 2015; Miki et al., 2016; Doussau et al., 2017). It is tempting to speculate that the “overfilling” of an RRP by additional release sites could involve Syt7.
Besides the Ca2+-binding kinetics of Syt, its spatial relationship to the presynaptic Cavs is crucial for setting fundamental synaptic properties, including pr, synchronicity of release and synaptic delays (Bucurenciu et al., 2008). The distance between Syt and the Cavs is frequently referred to as the coupling distance. In general it may be asserted that for AP evoked release a tight coupling favors high pr (Bucurenciu et al., 2008; Baur et al., 2015; Kusch et al., 2018), short synaptic delays (Bucurenciu et al., 2008), energy efficacy (Eggermann et al., 2012; Lu et al., 2016) and renders the release process less modifiable by Ca2+ buffers (Adler et al., 1991; Eggermann and Jonas, 2012; Bornschein et al., 2013; Schmidt et al., 2013; Brachtendorf et al., 2015). Loose coupling, on the other hand, offers more options for regulation and plasticity (Nadkarni et al., 2012; Vyleta and Jonas, 2014). There has been a comprehensive review on influx-release coupling at mammalian synapses of the peripheral NS (PNS) and the CNS (Eggermann et al., 2012). However, since then, a large body of work at AZs focused directly or indirectly on coupling distances and greatly advanced our knowledge about coupling and its regulation at different synapses. Some of these insights stem from classical model synapses, like the calyx of Held in the auditory brainstem, the frog neuromuscular junction, the squid giant synapse, or chick ciliary ganglion cells, which offer favorable conditions for electrophysiological analysis, in particular due to their large size. Insights from these synapses were covered by two recent comprehensive reviews (Wang and Augustine, 2015; Stanley, 2016) to which we refer the reader here. We will review advances in understanding AP-mediated Ca2+ influx-evoked transmitter release coupling at mammalian cortical AZs as well as their regulation during postnatal development and emerging roles of specific proteins in this regulatory process.
We will start this chapter with a brief note on nomenclature. The border between “tight” and “loose” coupling is not clearly defined (Box 2). A border line in the range of 70–100 nm has been suggested previously to distinguish between the two coupling regimes (Eggermann et al., 2012; Vyleta and Jonas, 2014). In light of the most recent quantitative estimates of coupling distances and domain topographies at mammalian CNS synapses (Table 2), we suggest an even narrower line of demarcation of no larger than 50 nm. At this coupling distance a given open Cav will be essentially ineffective in triggering release of a SV (Figure 3). Throughout this review we will use “loose coupling” if the coupling distance is ≥ 50 nm and “tight coupling” otherwise. In addition, we will use “single domain topography” (SDT) if only a single open Cav triggers release, and “domain overlap topography” (DOT) if a cluster of open Cavs with overlapping Ca2+ signaling domains controls release. Finally, we will use “nanodomain” as synonym for tight coupling plus SDT and “microdomain” as synonym for loose coupling plus DOT (Fedchyshyn and Wang, 2005; Table 3; Box 3).
Box 2. Deriving quantitative estimates of coupling distances.
We are not aware of any report of a direct quantification of the coupling distance between Syt and Cavs at AZs by microscopic techniques. In particular this appears to be due to the non-availability of appropriately sized labels. Hence, information about the average coupling distance is classically obtained by dialyzing a presynaptic terminus with exogenous Ca2+ chelators of similar affinity (KD) but different Ca2+-binding kinetics, i.e., different on-rates (kon; Adler et al., 1991; Augustine et al., 1991; Neher, 1998b; Eggermann et al., 2012). Typically the Ca2+ chelators EGTA (ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid; KD = 70 nM, kon = 107 M−1s−1; Nägerl et al., 2000) and BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid; KD = 220 nM, kon = 4∗108 M−1s−1; Naraghi and Neher, 1997) are used for this approach since they have similar KD values but BAPTA is ~40 times faster than EGTA. Ca2+ chelators suppress synaptic transmission by reducing the amount of Ca2+ that binds to Syt (Figure 2). The exact amount of interference depends on four factors: the average coupling distance, the mobility of the chelator, its kon, and its concentration. If influx-release coupling is tight, only a chelator with a rapid kon like BAPTA is able to capture Ca2+ in the nanodomain in the immediate vicinity of the channel before it reaches Syt, while chelators with slow kon like EGTA fail to influence the nanodomain Ca2+. Thus, at moderate concentrations only the fast BAPTA will reduce the amount of transmitter released in a tight coupling regime. In a loose coupling regime, on the other hand, both, BAPTA and EGTA will interfere with transmitter release since Ca2+ has to diffuse a larger distance from the Cavs to reach the sensor. This allows also the slower EGTA to capture Ca2+ in the microdomain before the ions reach the release sensor. Using this exogenous chelator dialysis approach, most of the estimates of coupling distances reviewed here were derived. It should be noted that the degree of interference actually not only depends on the kon but also on the concentration of the buffer, i.e., a large concentration of EGTA interferes with release similar to a much smaller concentration of BAPTA (Figure 3). In order to obtain quantitative values of the coupling distance, additional information about the magnitude and duration of the Ca2+ influx and potential Ca2+ sensor saturation is required (e.g., Bucurenciu et al., 2008; Schmidt et al., 2013; Nakamura et al., 2015, 2018; Kusch et al., 2018). Finally, by combining all results in experimentally constrained computer simulations quantitative estimates of the average coupling distance can be obtained (Bucurenciu et al., 2008; Bornschein et al., 2013; Schmidt et al., 2013; Vyleta and Jonas, 2014; Nakamura et al., 2015; Kusch et al., 2018).
The exogenous chelator dialysis approach was applied to large synapses that can be directly infused with chelator containing solution (Adler et al., 1991; Borst and Sakmann, 1996) and to large neurons that permit dialyzing the distant presynaptic sites by prolonged somatic whole-cell patch-clamp recordings (Ohana and Sakmann, 1998; Bucurenciu et al., 2008; Bornschein et al., 2013). The advantage of this approach is that the intracellular concentrations of the Ca2+ chelators are well-defined.
Another way of loading neurons with exogenous chelators is by application of membrane permeant acetoxymethyl ester variants of the Ca2+ chelators (EGTA-AM or BAPTA-AM) to the extracellular bath solution (Atluri and Regehr, 1996; Matsui and Jahr, 2003; Hefft and Jonas, 2005). The AM-chelator compound passes the lipophilic plasma membrane and enters the presynaptic cytosol. There, the ester group is cleaved by enzymes, which makes the chelator membrane-impermeable. Depending on loading time, its intracellular concentration can substantially exceed its bath concentration due to continuous intracellular accumulation of the chelator as long as its AM-form is present in the bath. The advantage of this approach is its relative experimental ease and that it is well-tolerated also by small neurons. It has the disadvantage that the intracellular chelator concentration remains rather ill defined. Thus, it permits a rapid initial assessment of relative differences in coupling e.g., between age groups, if differently aged synapses are compared under otherwise identical experimental conditions (Matsui and Jahr, 2003; Hefft and Jonas, 2005; Baur et al., 2015).
Neurons express endogenous Ca2+ buffers with quantified Ca2+-binding kinetics (Lee et al., 2000; Faas et al., 2007). Knowledge about the expression of specific native Ca2+ buffers and there Ca2+-binding kinetics offers an alternative route to deriving quantitative estimates of coupling distances by comparing transmitter-release from wild-type terminals to release from mutant terminals lacking a specific native buffer (Bornschein et al., 2013; Schmidt et al., 2013).
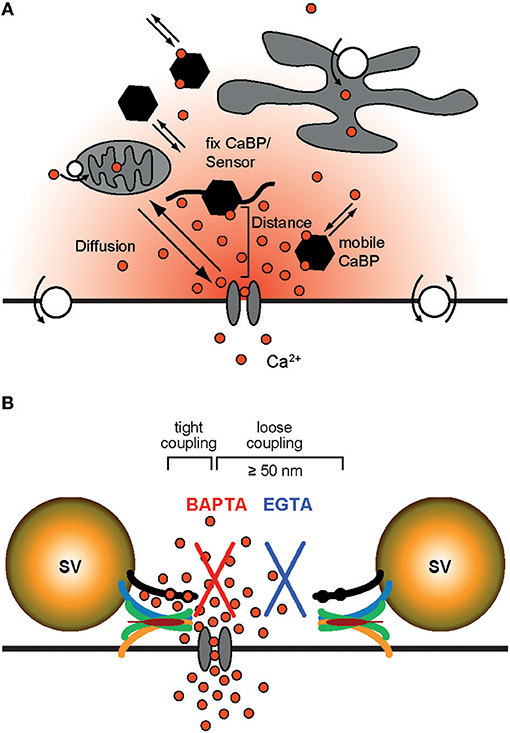
Figure 2. Ca2+ signaling domains. (A) General Ca2+ dynamics: Ca2+ enters a presynaptic terminal through a voltage-gated Ca2+ channel. Due to rapid diffusion (indicated by red gradient and the arrows) Ca2+ forms a steep, short-lived spatio-temporal gradient around the mouth of the open channel. It binds to mobile or fix Ca2+-binding proteins (CaBPs); some CaBP are pure buffers, others have an additional Ca2+ sensor function. Ultimately Ca2+ is cleared from the cytosol via Ca2+-ATPases (white circles with arrows) that either pump Ca2+ into the extracellular space or sequester it into organelles. (B) In a tight coupling regime a Syt bearing SV is located very close to the site of Ca2+ entry (<50 nm). If coupling is tight, at moderate concentrations only a buffer with rapid Ca2+ binding kinetics like BAPTA (red) can interfere with Ca2+ binding to Syt and prevent release. In a loose coupling regime, on the other hand, the SV is further away from the site of Ca2+ entry and also a slow buffer like EGTA (blue) can bind Ca2+ before it reaches the release sensor.
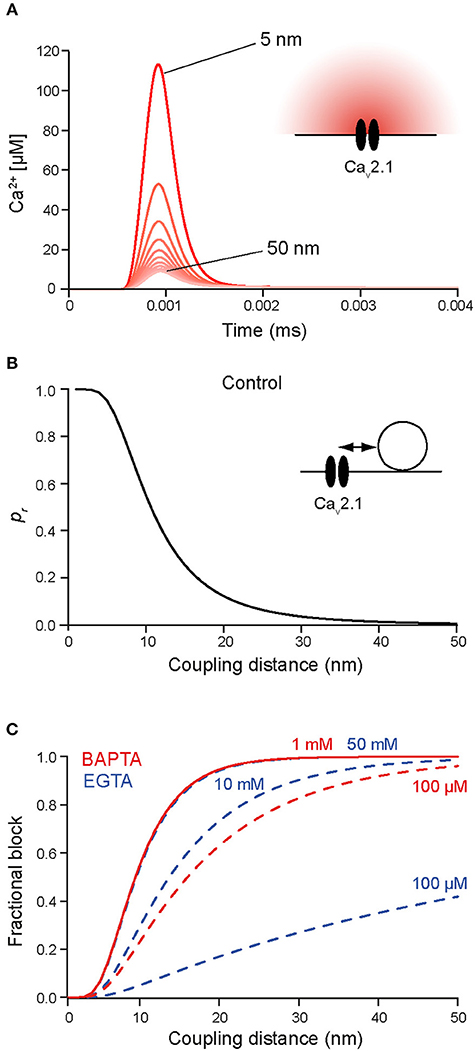
Figure 3. (A) Simulated [Ca2+]i transients at increasing distances between 5 and 50 nm (5 nm increments) from a single Cav2.1 channel (inset; Li et al., 2007) opening during an AP in the absence of Ca2+ buffers. (B) Release rates were simulated at increasing distances from the Cav2.1 channel (1 nm increments) using the Syt2 sensor model from Figure 1. Release probabilities were calculated by integrating the release rates over time and plotted against the corresponding sensor-to-Cav coupling distances. Note the steep decline in pr between 6 and 20 nm coupling distance and that pr at 50 nm is almost 0. (C) Simulated relative reduction of pr for different concentrations of EGTA (blue) and BAPTA (red). Moderate concentrations of EGTA are not very effective in blocking release close to a channel, while moderate concentrations of BAPTA are highly effective already at coupling distances of 10–20 nm. Higher concentration of EGTA mimic the effects of lower concentrations of BAPTA (concentrations are indicated). Note that in native boutons the concentrations of EGTA and BAPTA that yield corresponding effects on pr will be different due to the presence of native Ca2+-binding proteins, which were not included in the simulations.
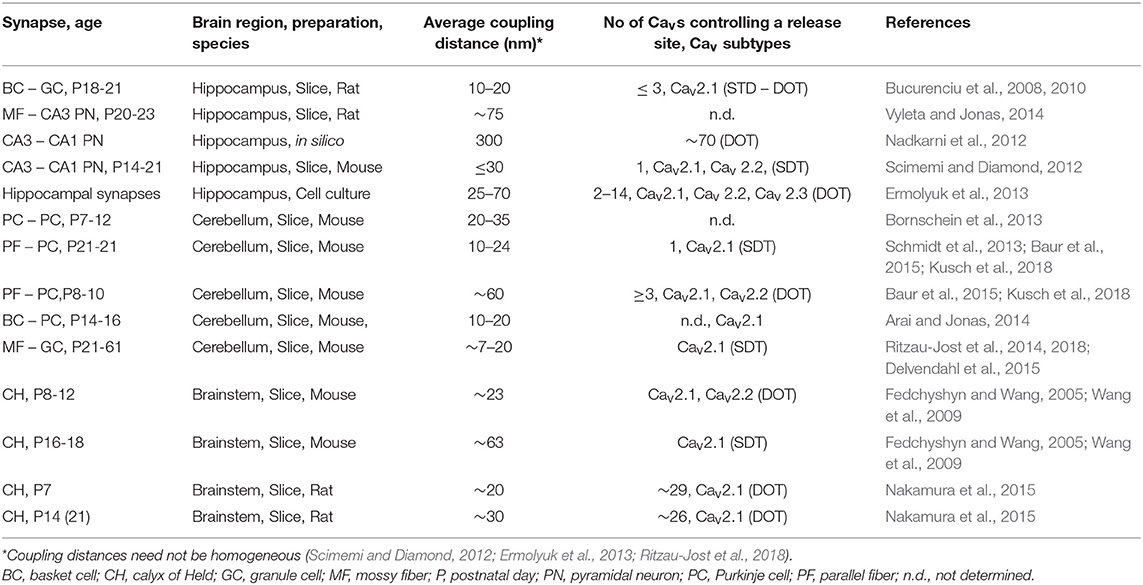
Table 2. Quantitative estimates of coupling topographies at mammalian CNS synapses.
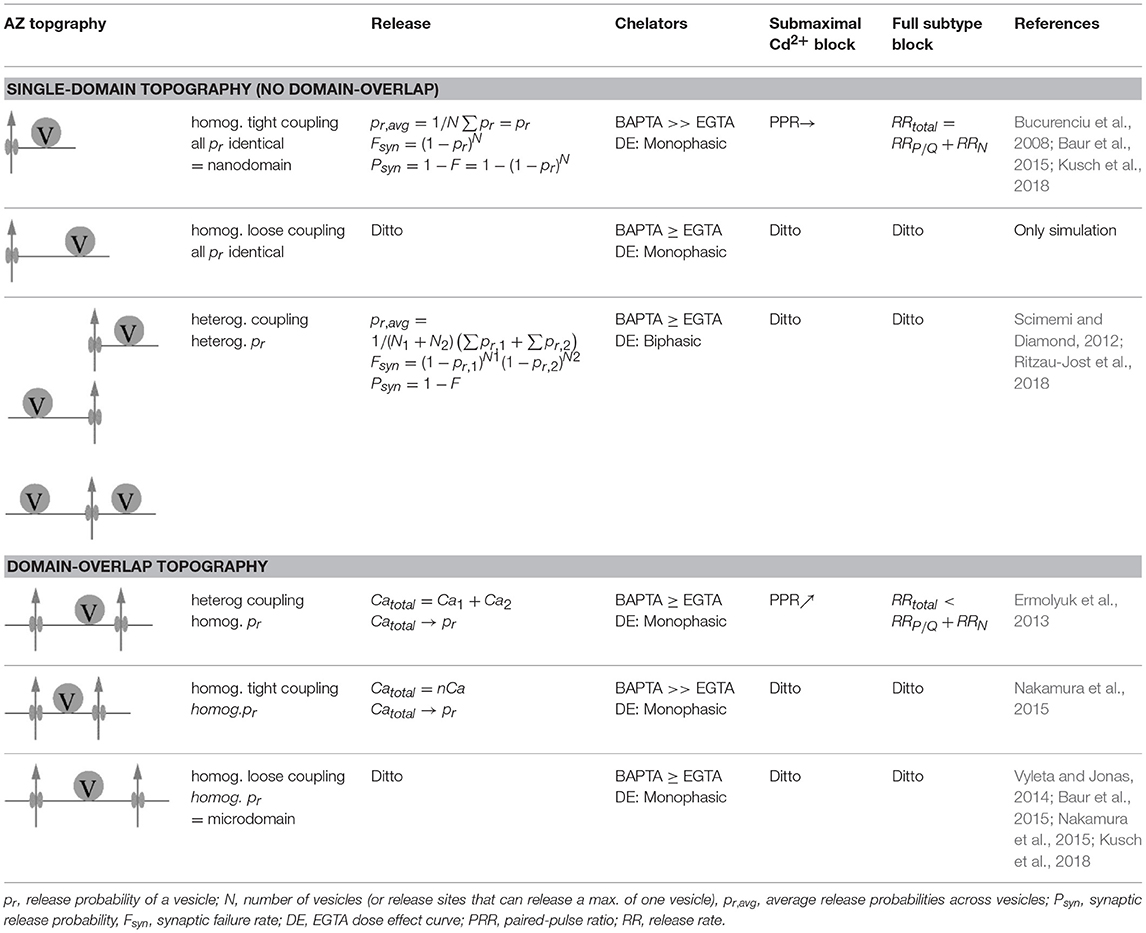
Table 3. Active zone topographies.
Box 3. Estimating the functional domain topography.
How many Cavs open during an AP and do their Ca2+ signaling domains overlap? Immunolabelling techniques combined with electron microscopy provide highly valuable insights into the structural organization of Cav clusters and Cav subtypes at the AZ. Yet, in order to answer the above question for functional SDT or DOT they need to be combined with physiological studies at the synapse (Holderith et al., 2012; Baur et al., 2015; Nakamura et al., 2015; Kusch et al., 2018). Specifically, the use of the unspecific Cav blocker Cd2+ and/or a combination of Cav subtype specific blockers were shown to yield valuable insights into the functional domain topography (Table 3; Augustine et al., 1991; Mintz et al., 1995; Scimemi and Diamond, 2012).
At physiological temperature Cd2+ dissociates slowly from a Cav (Chow, 1991), thus, blocking a channel in an all-or-none fashion on the time-scale of an AP. The shape of Cd2+ dose-effect curves onto EPSC amplitudes as read-out for release will depend on the domain topography. If a presynaptic terminal harbors release sites with DOT, the curve will be non-linear whereas it will be linear if the release sites operate with SDT (Augustine et al., 1991). The construction of full dose-effect curves may be circumvented by analyzing the effects of a subsaturating concentration of Cd2+ onto the PPR (Scimemi and Diamond, 2012).
Application of a subsaturating concentration of Cd2+ reduces the amplitude of the first postsynaptic current (PSC) of a paired pulse experiment irrespective of the domain topography. However, its impact on the PPR markedly depends on whether the release sites operate with SDT or DOT. At a subsaturating concentration of Cd2+ some but not all Cavs will be blocked during an AP. In a DOT, blocking some of the Cavs controlling a synaptic vesicle will have effects similar to reducing [Ca2+]e, i.e., the initial pr will be reduced while the PPR will increase. On the other hand, if a release site is controlled by a single Cav (SDT), release of synaptic vesicles encountering no Ca2+ would be blocked while release or facilitation of vesicles encountering Ca2+ would be the same as in the absence of Cd2+. In consequence, application of Cd2+ will increase PPR in a DOT but leave it unaltered in a SDT (Scimemi and Diamond, 2012).
These results can further be substantiated by using Cav subtype specific blockers, if more than one channel subtype contributes to release. For a SDT in which a given vesicle is linked to either of the Cav subtypes, the sum of the toxin-sensitive release fractions will not exceed release measured in the absence of toxin, i.e., the toxin sensitive release fractions will sum linearly. Contrariwise, for an AZ at which release of a vesicle is controlled in a DOT composed of different Cav subtypes, the sum of the blocked release fractions can exceed the control value (“supralinear” summation) because of the non-linear dependency of release on Ca2+ (Mintz et al., 1995; Scimemi and Diamond, 2012).
Tight influx-release coupling has been reported for giant synapses specialized for escape reflexes in the squid (Adler et al., 1991), bipolar cells in the goldfish retina (von Gersdorff and Matthews, 1994; Burrone et al., 2002) and for the frog neuromuscular junction (Harlow et al., 2001). The first descriptions of nanodomain coupling came from the squid giant synapse and chick ciliary ganglia cells (Adler et al., 1991; Stanley, 1993). In the mammalian CNS, inhibitory synapses in the hippocampus and cerebellum were found to operate at tight coupling and at least in part with SDT, i.e., with nanodomains (Bucurenciu et al., 2008, 2010; Eggermann and Jonas, 2012; Bornschein et al., 2013). Surprisingly, cortical glutamatergic synapses seemed to forgo the benefits of tight coupling. Experimental studies performed on young pyramidal neurons (PNs; Ohana and Sakmann, 1998; Rozov et al., 2001) and in hippocampal cell cultures (Ermolyuk et al., 2013) as well as computational models of hippocampal CA3 – CA1 PN synapses (Nadkarni et al., 2012) showed loose coupling and established the view that small glutamatergic synapses in the brain, in particular excitatory cortical synapses, operate at microdomain coupling (Eggermann et al., 2012).
Initial experimental evidence against the generality of this view was available from the CA3 – CA1 PN synapse (Scimemi and Diamond, 2012), showing SDT and results that were more consistent with a tighter coupling at this synapse rather than with DOT and a very large number of Cavs loosely coupled to the release sensor as suggested by the above mentioned study of Nadkarni et al. (2012). In the cerebellar cortex of 3 to 4 weeks old mice, subsequent work quantified the coupling distance at the parallel fiber (PF) to PC synapse, an excitatory, glutamatergic synapse in the cerebellar cortex and probably the most abundant synapse in the mammalian brain. It was found that this synapse operates at tight coupling of ~24 nm (Schmidt et al., 2013). In successional work it was found that at this age coupling is not only tight but that these synapses operate with a nanodomain topography (Baur et al., 2015; Kusch et al., 2018) and that also further excitatory synapses in the cerebellar cortex operate with tight coupling (Ritzau-Jost et al., 2014; Delvendahl et al., 2015). Together these studies clearly contradicted the generality of microdomain coupling at excitatory synapses in mammalian cortical structures.
An interesting difference between the experiments suggesting microdomain coupling in glutamatergic cortical synapses (Ohana and Sakmann, 1998; Rozov et al., 2001) and the experiments showing nanodomain coupling (Schmidt et al., 2013; Ritzau-Jost et al., 2014) was the age of the experimental animals: While the former studies were performed in young rats (~2 weeks old), in the latter studies, the coupling distance was assessed in more matured mice (>3 weeks old). Considering that the postnatal development of rats likely proceeds slower than that of mice, the age difference most likely corresponds to an even larger difference in brain maturation. This raised the possibility that the coupling distance could be regulated developmentally.
Support for the idea of a developmental regulation of coupling came from experiments performed at the calyx of Held. Experiments performed on young (~10 days old) and matured (~3 weeks old) calyces indicated a substantial developmental tightening of the coupling distance during postnatal development (Taschenberger et al., 2002; Fedchyshyn and Wang, 2005; Wang et al., 2008; Kochubey et al., 2009). Simulations quantified that the experimental results are explained by a developmental tightening of the spatial coupling between Ca2+ channels and Syt from ~60 to ~20 nm at the calyx of Held (Wang et al., 2009).
Coupling distances and domain topographies were quantified more recently in a developmental context (Table 2), again at the calyx of Held (Nakamura et al., 2015) and at the PF to PC synapse (Baur et al., 2015). At the calyx of Held, a moderate developmental tightening of the coupling distance between Syt and the closest Cav of a cluster from ~30 nm to ~20 nm between P7 and P14 was found, while the number of Cavs within a cluster controlling a given release site remained relatively constant with an average in the range of 25 to 30 (Nakamura et al., 2015). Thus, although a developmental shortening of the coupling distance was found at the calyx of Held, it operated at fairly tight coupling with DOT, independent of age in the range of P7 to P14. By contrast, at the PF to PC synapse a switch from DOT to SDT was found. At ~P9, PF terminals operated with a DOT with a distance of ~60 nm between the closest Cav within a cluster and Syt, while at ~P23 a coupling distance of ~20 nm and SDT were found (Baur et al., 2015; Kusch et al., 2018). The DOT at young PF terminals was composed of Cav2.1 (P/Q-type) and Cav2.2 (N-type) concomitantly controlling a release site, with likely 2 Cav2.1 and 1 Cav2.2 triggering release during an AP. The nanodomain at more matured PF terminals comprised only a Cav2.1 (Kusch et al., 2018).
These results may suggest that developmental tightening of the coupling distance is a common phenomenon in the mammalian brain, which could be accompanied by a switch from DOT to SDT in small, but not in large synapses. However, an alternative is that coupling distances, domain topographies and their regulation are synapse specific properties. At the glutamatergic MF to CA3 PN synapse a loose coupling distance of ~75 nm has been quantified in the matured hippocampus (Table 2; Vyleta and Jonas, 2014). However, this finding does not necessarily exclude a developmental tightening of the coupling distance at the MF to CA3 synapse. It remains possible that the young synapse operates at an even larger coupling distance. Thus, while developmental tightening and loose coupling in mature brain are not mutually exclusive, the finding of loose coupling at the mature MF – CA3 synapse clearly suggests that the coupling distance is a synapse specific property in the context of its developmental stage.
Are there other forms of regulation of the coupling distance besides developmental regulation? An intriguing possibility would be a regulation of the coupling distance depending on the activity of a synapse, i.e., as a mechanism of synaptic plasticity. Evidence for such use-dependent regulation of the coupling distance came from a recent study at hippocampal mossy fiber boutons (Midorikawa and Sakaba, 2017). It was found that increasing the level of cAMP in the boutons, which is a crucial step in the induction of long term potentiation, results in increased release from the bouton, while not increasing the number of synaptic vesicles in the RRP nor altering the Ca2+ influx. Based on the differential action of EGTA prior and following the induction of cAMP-mediated plasticity the study provides evidence for a tightening of the coupling distance following cAMP application (Midorikawa and Sakaba, 2017).
The MF – CA3 PN synapse, which was found to operate at loose coupling in the matured hippocampus (Vyleta and Jonas, 2014), is highly plastic and expresses several forms of presynaptic plasticity (Salin et al., 1996). It has been suggested that loose coupling provides a molecular framework for high plasticity (Vyleta and Jonas, 2014). Consistent with this idea, synapses with tight coupling are mostly fast-signaling synapses in neuronal circuits specialized for high-frequency coding of sensory information or in motor control (Table 2). However, some of these synapses also show pronounced presynaptic plasticity. For example, the PF to PC synapse exhibits low-frequency depression and high-frequency facilitation (Doussau et al., 2017).
Loose coupling offers more possibilities for regulating transmitter release and plasticity, e.g., via the action of Ca2+ buffers, since in loose coupling also slow Ca2+ buffers can intercept sizable amounts of Ca2+ before it reaches Syt (Adler et al., 1991). In tight coupling regimes, on the other hand, only rapid buffers like BAPTA (Adler et al., 1991), Calretinin (Schmidt et al., 2013; Brachtendorf et al., 2015), or Calbindin (Bornschein et al., 2013) were found to be regulators of pr, while the “slow” buffer Parvalbumin (PV) did not affect pr (Bornschein et al., 2013). At high concentrations, however, even PV becomes effective in affecting [Ca2+]i and release in tight coupling regimes (Eggermann and Jonas, 2012). This is because PV actually is a rapid, high-affinity Ca2+ buffer but its Ca2+-binding sites also have a medium affinity for Mg2+ such that most binding sites are occupied by Mg2+ under physiological resting conditions and only a small amount of binding sites (~5%) are metal free (Lee et al., 2000). Thus, Ca2+-binding has to be preceded by Mg2+-unbinding, which proceeds with slow kinetics, i.e., the slow Mg2+-unbinding kinetics makes PV a slow Ca2+ buffer (Lee et al., 2000). However, if PV is expressed strongly in a synapse the small relative fraction of Mg2+-free binding sites can constitute a sufficiently large absolute concentration of rapidly Ca2+-binding PV to significantly affect [Ca2+]i even in the nanodomain around a Cav channel. Metal free binding sites are then continuously replenished efficiently from the large pool of Mg2+-bound sites (Eggermann and Jonas, 2012). It should be noted that this action of PV is different from the effects of large concentrations of EGTA in tight coupling regimes. PV was already effective at concentrations ~500 μM due to rapid Ca2+-binding and replenishment via Mg2+-unbinding, while slow buffering by EGTA requires concentrations >10 mM to intercept [Ca2+]i in the nanodomain.
Tight coupling increases speed and efficacy of synaptic transmission (Eggermann et al., 2012). In addition, it can provide an energy efficient design compared to loose coupling. To obtain a certain [Ca2+]i level at the release sensor less Cavs have to open in a tight than in a loose coupling regime (Eggermann et al., 2012). As the ATP cost of Ca2+ removal is a significant post of the presynaptic energy consumption (Kim et al., 2005), tight coupling can save energy. This, however, requires that Ca2+ influx would indeed be different between terminals with tight or loose coupling. Indeed results from the calyx of Held conform to this requirement, showing that concomitant with developmental coupling distance tightening the amplitudes of presynaptic Ca2+ transients decreased (Nakamura et al., 2015). On the other hand, at the PF – PC synapse presynaptic Ca2+ transients did not change developmentally despite the developmental switch from loose to tight coupling (Baur et al., 2015). Several Cavs opening during the presynaptic AP no longer contributed to driving release at later developmental stages (Kusch et al., 2018). Their primary function remains speculative but could be in Ca2+-driven replenishment of synaptic vesicles into the readily releasable pool (Brachtendorf et al., 2015; Miki et al., 2016; Doussau et al., 2017).
Ca2+ influx-transmitter release coupling is mediated via proteins of the AZ scaffold, albeit, this process is still not well-understood at present and a detailed review of the AZ scaffold is far beyond the scope of this paper. We briefly focus on some recent advances directly related to establishing influx—release coupling. RIMs (Rab3-interacting molecules) are known as central organizer of the AZ (Südhof, 2012). Specifically, they are required for recruiting Cav2.1 and Cav2.2 channels to the AZ (Kaeser et al., 2011), which can be considered as a first step in coupling Ca2+ influx to transmitter release, in particular since these channel subtypes are the most important ones for AP-mediated fusion (Table 2). The protein Septin5 was identified as a negative regulator of tight coupling during development, i.e., its downregulation was permissive to the establishment of tight coupling (Yang et al., 2010). Proteins involved in mediating tight coupling were identified more recently, suggesting RIM-BPs (RIM-binding proteins; Acuna et al., 2015; Grauel et al., 2016) and Munc13-3 (Ishiyama et al., 2014; Kusch et al., 2018), as positive regulators of the coupling distance. Thereby, Munc13-3 was found to be a developmental mediator of tight coupling (Kusch et al., 2018). However, details of the interplay between identified regulators of the coupling distance, their relationships to other regulatory proteins at the AZ, and details of their interaction with the exocytotic core complex remain essentially unclear.
More than 30 years after the steep non-linear dependency of transmitter release onto [Ca2+]e has been established (Dodge and Rahamimoff, 1967), detailed kinetic five-site models of the [Ca2+]i dependency of Syt2-triggered transmitter release were developed (Bollmann et al., 2000; Schneggenburger and Neher, 2000) and subsequently elaborated to cover sub-modes and subtleties of release (Lou et al., 2005; Sun et al., 2007; Pan and Zucker, 2009) and to capture developmental aspects (Kochubey et al., 2009). These models are widely applied in functional quantitative studies of transmitter release and AZ topography.
During the past decade several functional studies focused directly or indirectly on the coupling distance between Syts and Cavs at mammalian cortical synapses. Initially it was thought that only GABAergic synapses in cortical structures make use of tight coupling, while cortical glutamatergic synapses seemed to operate with loose coupling (Ohana and Sakmann, 1998; Rozov et al., 2001; Eggermann et al., 2012; Nadkarni et al., 2012; Stanley, 2016). However, results from a glutamatergic synapse in the mature cerebellar cortex falsified the generality of this hypothesis (Schmidt et al., 2013). From subsequent work (Table 2) it became evident that synapses in the matured mammalian brain, including synapses in cortical structures of hippocampus and cerebellum, indeed make widespread use of tight coupling and, furthermore, that release of a SV was frequently triggered by opening of only a few or even a single Cav. At synapses investigated in a developmental context, it was found that tight coupling at matured synapses develops from an initially loose coupling at younger synapses. This latter result provides an explanation why previous studies predominantly found microdomain coupling at glutamatergic cortical synapses. These earlier studies were performed at synapses of very young animals (Eggermann et al., 2012; Stanley, 2016). The concept of nanodomain coupling was developed 20 years ago at the squid giant synapse (Adler et al., 1991) and calyx-type synapses in the chick ciliary ganglion (Stanley, 1993) and now experiences a revival at matured mammalian central synapses.
However, tight coupling is not universal for synapses of the mature mammalian brain (Vyleta and Jonas, 2014). As suggested by Vyleta and Jonas, the present state of knowledge indicates that coupling distances are specific adaptations to the function of a synapse. GABAergic central synapses appear to operate at tight coupling, most probably irrespective of age (Table 2). For glutamatergic synapses the situation is more complex. While excitatory synapses specialized for rapid signaling develop a tight, nanodomain coupling topography, synapses highly adaptive via plasticity make use of loose coupling even in matured brain. To learn more about the rules that regulate coupling distances will require to investigate further types of synapses in different brain regions. For example, a particularly striking lack of quantitative data on coupling distances and AZ topographies exists for neocortical synapses (Eggermann et al., 2012; Stanley, 2016; Table 2). To our knowledge, a coupling distance has never been quantified at a neocortical synapse.
For understanding the rules regulating coupling, it will be also required to identify the proteins that link Syt bearing SVs to Cavs. Recent studies indicated RIM-BPs (Acuna et al., 2015; Grauel et al., 2016) and Munc13-3 (Kusch et al., 2018) to be involved in organizing Cav clusters at the AZ and in narrowing the coupling distance. Munc13-3 was identified as a specific developmental mediator of nanodomain coupling at a glutamatergic synapse in cerebellar cortex (Kusch et al., 2018). Interestingly, Munc13-3 protein is expressed strongly in the cerebellar cortex, more weekly in the brainstem and is essentially absent from the hippocampus and cerebral cortex (Augustin et al., 1999). Does this indicate that developmental tightening of the coupling distance is a specificity of glutamatergic synapses in the cerebellum and brainstem? To answer this question, it will be required to quantify coupling distances in a developmental context also at neocortical synapses. Since coupling distances are key parameters of synaptic function, understanding the rules regulating this distance will advance our general understanding of the rules regulating synaptic transmission, which is the basic substrate of information flow in neuronal networks.
HS wrote the first draft of the manuscript. GB and HS prepared figures and tables. GB and HS contributed to manuscript revision, read and approved the submitted version.
This work was supported by the German Research Foundation (DFG SCHM1838/2) and DFG and University of Leipzig within the program of open access publishing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Stefan Hallermann for critical discussion of the manuscript.
Abbott, L. F., and Regehr, W. G. (2004). Synaptic computation. Nature 431, 796–803. doi: 10.1038/nature03010
Acuna, C., Liu, X., Gonzalez, A., and Südhof, T. C. (2015). RIM-BPs mediate tight coupling of action potentials to Ca2+-triggered neurotransmitter release. Neuron 87, 1234–1247. doi: 10.1016/j.neuron.2015.08.027
Adler, E. M., Augustine, G. J., Duffy, S. N., and Charlton, M. P. (1991). Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J. Neurosci. 11, 1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991
Arai, I., and Jonas, P. (2014). Nanodomain coupling explains Ca2+ independence of transmitter release time course at a fast central synapse. Elife 3:e04057. doi: 10.7554/eLife.04057
Atluri, P. P., and Regehr, W. G. (1996). Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J. Neurosci. 16, 5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996
Augustin, I., Betz, A., Herrmann, C., Jo, T., and Brose, N. (1999). Differential expression of two novel Munc13 proteins in rat brain. Biochem. J. 337, 363–371. doi: 10.1042/bj3370363
Augustine, G. J., Adler, E. M., and Charlton, M. P. (1991). The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann. N. Y. Acad. Sci. 635, 365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x
Bacaj, T., Wu, D., Yang, X., Morishita, W., Zhou, P., Xu, W., et al. (2013). Synaptotagmin-1 and Synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. doi: 10.1016/j.neuron.2013.10.026
Baur, D., Bornschein, G., Althof, D., Watanabe, M., Kulik, A., Eilers, J., et al. (2015). Developmental tightening of cerebellar cortical synaptic influx-release coupling. J. Neurosci. 35, 1858–1871. doi: 10.1523/JNEUROSCI.2900-14.2015
Berton, F., Iborra, C., Boudier, J. A., Seagar, M. J., and Marqueze, B. (1997). Developmental regulation of synaptotagmin I, II, III, and IV mRNAs in the rat CNS. J. Neurosci. 17, 1206–1216. doi: 10.1523/JNEUROSCI.17-04-01206.1997
Bertram, R., Sherman, A., and Stanley, E. F. (1996). Single-domain/bound calcium hypothesis of transmitter release and facilitation. J. Neurophysiol. 75, 1919–1931. doi: 10.1152/jn.1996.75.5.1919
Blundon, J. A., Wright, S. N., Brodwick, M. S., and Bittner, G. D. (1993). Residual free calcium is not responsible for facilitation of neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 90, 9388–9392. doi: 10.1073/pnas.90.20.9388
Bollmann, J. H., and Sakmann, B. (2005). Control of synaptic strength and timing by the release-site Ca2+ signal. Nat. Neurosci. 8, 426–434. doi: 10.1038/nn1417
Bollmann, J. H., Sakmann, B., and Borst, J. G. (2000). Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289, 953–957. doi: 10.1126/science.289.5481.953
Bornschein, G., Arendt, O., Hallermann, S., Brachtendorf, S., Eilers, J., and Schmidt, H. (2013). Paired-pulse facilitation at recurrent Purkinje neuron synapses is independent of calbindin and parvalbumin during high-frequency activation. J. Physiol. 591, 3355–3370. doi: 10.1113/jphysiol.2013.254128
Borst, J. G., and Sakmann, B. (1996). Calcium influx and transmitter release in a fast CNS synapse. Nature 383, 431–434. doi: 10.1038/383431a0
Brachtendorf, S., Eilers, J.-K., and Schmidt, H. (2015). A use-dependent increase in release sites drives facilitation at calretinin-deficient cerebellar parallel-fiber synapses. Front. Cell Neurosci. 9:27. doi: 10.3389/fncel.2015.00027
Brunger, A. T., Choi, U. B., Lai, Y., Leitz, J., and Zhou, Q. (2018). Molecular mechanisms of fast neurotransmitter release. Annu. Rev. Biophys. 47, 469–497. doi: 10.1146/annurev-biophys-070816-034117
Bucurenciu, I., Bischofberger, J., and Jonas, P. (2010). A small number of open Ca2+ channels trigger transmitter release at a central GABAergic synapse. Nat. Neurosci. 13, 19–21. doi: 10.1038/nn.2461
Bucurenciu, I., Kulik, A., Schwaller, B., Frotscher, M., and Jonas, P. (2008). Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 57, 536–545. doi: 10.1016/j.neuron.2007.12.026
Burrone, J., Neves, G., Gomis, A., Cooke, A., and Lagnado, L. (2002). Endogenous calcium buffers regulate fast exocytosis in the synaptic terminal of retinal bipolar cells. Neuron 33, 101–112. doi: 10.1016/S0896-6273(01)00565-7
Chen, C., Arai, I., Satterfield, R., Young, S. M. Jr., and Jonas, P. (2017). Synaptotagmin 2 is the fast Ca2+ sensor at a central inhibitory synapse. Cell Rep. 18, 723–736. doi: 10.1016/j.celrep.2016.12.067
Chen, C., and Jonas, P. (2017). Synaptotagmins: that's why so many. Neuron 94, 694–696. doi: 10.1016/j.neuron.2017.05.011
Chow, R. H. (1991). Cadmium block of squid calcium currents. Macroscopic data and a kinetic model. J. Gen. Physiol. 98, 751–770. doi: 10.1085/jgp.98.4.751
Clements, J. D., and Silver, R. A. (2000). Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 23, 105–113. doi: 10.1016/S0166-2236(99)01520-9
Delvendahl, I., Jablonski, L., Baade, C., Matveev, V., Neher, E., and Hallermann, S. (2015). Reduced endogenous Ca2+ buffering speeds active zone Ca2+ signaling. Proc. Natl. Acad. Sci. U.S.A. 112, E3075–E3084. doi: 10.1073/pnas.1508419112
Diamond, J. S., and Jahr, C. E. (1995). Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15, 1097–1107. doi: 10.1016/0896-6273(95)90098-5
Dodge, F. A., and Rahamimoff, R. (1967). Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193, 419–432. doi: 10.1113/jphysiol.1967.sp008367
Doussau, F., Schmidt, H., Dorgans, K., Valera, A. M., Poulain, B., and Isope, P. (2017). Frequency-dependent mobilization of heterogeneous pools of synaptic vesicles shapes presynaptic plasticity. Elife 6:e28935. doi: 10.7554/eLife.28935
Eggermann, E., Bucurenciu, I., Goswami, S. P., and Jonas, P. (2012). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 13, 7–21. doi: 10.1038/nrn3125
Eggermann, E., and Jonas, P. (2012). How the 'slow' Ca2+ buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat. Neurosci. 15, 20–22. doi: 10.1038/nn.3002
Ermolyuk, Y. S., Alder, F. G., Surges, R., Pavlov, I. Y., Timofeeva, Y., Kullmann, D. M., et al. (2013). Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2+ channels. Nat. Neurosci. 16, 1754–1763. doi: 10.1038/nn.3563
Faas, G. C., Schwaller, B., Vergara, J. L., and Mody, I. (2007). Resolving the fast kinetics of cooperative binding: Ca2+ buffering by calretinin. PLoS Biol. 5:e311. doi: 10.1371/journal.pbio.0050311
Fedchyshyn, M. J., and Wang, L. Y. (2005). Developmental transformation of the release modality at the calyx of Held synapse. J. Neurosci. 25, 4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005
Felmy, F., Neher, E., and Schneggenburger, R. (2003). Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron 37, 801–811. doi: 10.1016/S0896-6273(03)00085-0
Fox, M. A., and Sanes, J. R. (2007). Synaptotagmin I and II are present in distinct subsets of central synapses. J. Comp. Neurol. 503, 280–296. doi: 10.1002/cne.21381
Geiger, J. R., and Jonas, P. (2000). Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron 29, 927–939. doi: 10.1016/S0896-6273(00)00164-1
Geppert, M., Goda, Y., Hammer, R. E., Li, C., Rosahl, T. W., Stevens, C. F., et al. (1994). Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. doi: 10.1016/0092-8674(94)90556-8
Goda, Y., and Stevens, C. F. (1994). Two components of transmitter release at a central synapse. Proc. Natl. Acad. Sci. U.S.A. 91, 12942–12946. doi: 10.1073/pnas.91.26.12942
Grauel, M. K., Maglione, M., Reddy-Alla, S., Willmes, C. G., Brockmann, M. M., Trimbuch, T., et al. (2016). RIM-binding protein 2 regulates release probability by fine-tuning calcium channel localization at murine hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 113, 11615–11620. doi: 10.1073/pnas.1605256113
Harlow, M. L., Ress, D., Stoschek, A., Marshall, R. M., and McMahan, U. J. (2001). The architecture of active zone material at the frog's neuromuscular junction. Nature 409, 479–484. doi: 10.1038/35054000
Hefft, S., and Jonas, P. (2005). Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat. Neurosci. 8, 1319–1328. doi: 10.1038/nn1542
Heidelberger, R., Heinemann, C., Neher, E., and Matthews, G. (1994). Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371, 513–515. doi: 10.1038/371513a0
Helassa, N., Dürst, C. D., Coates, C., Kerruth, S., Arif, U., Schulze, C., et al. (2018). Ultrafast glutamate sensors resolve high-frequency release at Schaffer collateral synapses. Proc. Natl. Acad. Sci. U.S.A. 115, 5594–5599. doi: 10.1073/pnas.1720648115
Holderith, N., Lorincz, A., Katona, G., Rozsa, B., Kulik, A., Watanabe, M., et al. (2012). Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci. 15, 988–997. doi: 10.1038/nn.3137
Ishiyama, S., Schmidt, H., Cooper, B. H., Brose, N., and Eilers, J. (2014). Munc13-3 superprimes synaptic vesicles at granule cell-to-basket cell synapses in the mouse cerebellum. J. Neurosci. 34, 14687–14696. doi: 10.1523/JNEUROSCI.2060-14.2014
Jackman, S. L., and Regehr, W. G. (2017). The mechanisms and functions of synaptic facilitation. Neuron 94, 447–464. doi: 10.1016/j.neuron.2017.02.047
Jackman, S. L., Turecek, J., Belinsky, J. E., and Regehr, W. G. (2016). The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529, 88–91. doi: 10.1038/nature16507
Jensen, T. P., Zheng, K., Tyurikova, O., Reynolds, J. P., and Rusakov, D. A. (2017). Monitoring single-synapse glutamate release and presynaptic calcium concentration in organised brain tissue. Cell Calcium 64, 102–108. doi: 10.1016/j.ceca.2017.03.007
Kaeser, P. S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., et al. (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295. doi: 10.1016/j.cell.2010.12.029
Kaeser, P. S., and Regehr, W. G. (2014). Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu. Rev. Physiol. 76, 333–363. doi: 10.1146/annurev-physiol-021113-170338
Katz, B., and Miledi, R. (1968). The role of calcium in neuromuscular facilitation. J. Physiol. 195, 481–492. doi: 10.1113/jphysiol.1968.sp008469
Kim, M.-H., Korogod, N., Schneggenburger, R., Ho, W.-K., and Lee, S.-H. (2005). Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J. Neurosci. 25, 6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005
Kochubey, O., Babai, N., and Schneggenburger, R. (2016). A synaptotagmin isoform switch during the development of an identified CNS synapse. Neuron 90:1183. doi: 10.1016/j.neuron.2016.08.024
Kochubey, O., Han, Y., and Schneggenburger, R. (2009). Developmental regulation of the intracellular Ca2+ sensitivity of vesicle fusion and Ca2+-secretion coupling at the rat calyx of Held. J. Physiol. 587, 3009–3023. doi: 10.1113/jphysiol.2009.172387
Kochubey, O., Lou, X., and Schneggenburger, R. (2011). Regulation of transmitter release by Ca2+ and synaptotagmin: insights from a large CNS synapse. Trends Neurosci. 34, 237–246. doi: 10.1016/j.tins.2011.02.006
Kochubey, O., and Schneggenburger, R. (2011). Synaptotagmin Increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69, 736–748. doi: 10.1016/j.neuron.2011.01.013
Kusch, V., Bornschein, G., Loreth, D., Bank, J., Jordan, J., Baur, D., et al. (2018). Munc13-3 is required for the developmental localization of Ca2+ channels to active zones and the nanopositioning of Cav2.1 near release sensors. Cell Rep. 22, 1965–1973. doi: 10.1016/j.celrep.2018.02.010
Lee, S.-H., Schwaller, B., and Neher, E. (2000). Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J. Physiol. 525, 419–432. doi: 10.1111/j.1469-7793.2000.t01-2-00419.x
Li, L., Bischofberger, J., and Jonas, P. (2007). Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J. Neurosci. 27, 13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007
Li, Y. C., Chanaday, N. L., Xu, W., and Kavalali, E. T. (2017). Synaptotagmin-1- and Synaptotagmin-7-dependent fusion mechanisms target synaptic vesicles to kinetically distinct endocytic pathways. Neuron 93, 616-631.e3. doi: 10.1016/j.neuron.2016.12.010
Liu, H., Bai, H., Hui, E., Yang, L., Evans, C. S., Wang, Z., et al. (2014). Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. Elife 3:e01524. doi: 10.7554/eLife.01524
Llinás, R., Sugimori, M., and Silver, R. B. (1992). Microdomains of high calcium concentration in a presynaptic terminal. Science 256, 677–679. doi: 10.1126/science.1350109
Lou, X., Scheuss, V., and Schneggenburger, R. (2005). Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature 435, 497–501. doi: 10.1038/nature03568
Lu, Z., Chouhan, A. K., Borycz, J. A., Lu, Z., Rossano, A. J., Brain, K. L., et al. (2016). High-probability neurotransmitter release sites represent an energy-efficient design. Curr. Biol. 26, 1–10. doi: 10.1016/j.cub.2016.07.032
Luo, F., and Südhof, T. C. (2017). Synaptotagmin-7-mediated asynchronous release boosts high-fidelity synchronous transmission at a central synapse. Neuron 94, 826-839 e3. doi: 10.1016/j.neuron.2017.04.020
Marvin, J. S., Scholl, B., Wilson, D. E., Podgorski, K., Kazemipour, A., Müller, J. A., et al. (2018). Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat. Meth. 15, 936–939. doi: 10.1038/s41592-018-0171-3
Matsui, K., and Jahr, C. E. (2003). Ectopic release of synaptic vesicles. Neuron 40, 1173–1183. doi: 10.1016/S0896-6273(03)00788-8
Matveev, V., Sherman, A., and Zucker, R. S. (2002). New and corrected simulations of synaptic facilitation. Biophys. J. 83, 1368–1373. doi: 10.1016/S0006-3495(02)73907-6
Midorikawa, M., and Sakaba, T. (2017). Kinetics of releasable synaptic vesicles and their plastic changes at hippocampal mossy fiber synapses. Neuron 96, 1033–1040 e3. doi: 10.1016/j.neuron.2017.10.016
Miki, T., Malagon, G., Pulido, C., Llano, I., Neher, E., and Marty, A. (2016). Actin- and myosin-dependent vesicle loading of presynaptic docking sites prior to exocytosis. Neuron 91, 808–823. doi: 10.1016/j.neuron.2016.07.033
Millar, A. G., Zucker, R. S., Ellis-Davies, G. C., Charlton, M. P., and Atwood, H. L. (2005). Calcium sensitivity of neurotransmitter release differs at phasic and tonic synapses. J. Neurosci. 25, 3113–3125. doi: 10.1523/JNEUROSCI.4717-04.2005
Mintz, I. M., Sabatini, B. L., and Regehr, W. G. (1995). Calcium control of transmitter release at a cerebellar synapse. Neuron 15, 675–688. doi: 10.1016/0896-6273(95)90155-8
Nadkarni, S., Bartol, T. M., Stevens, C. F., Sejnowski, T. J., and Levine, H. (2012). Short-term plasticity constrains spatial organization of a hippocampal presynaptic terminal. Proc. Natl. Acad. Sci. U.S.A. 109, 14657–14662. doi: 10.1073/pnas.1211971109
Nägerl, U. V., Novo, D., Mody, I., and Vergara, J. L. (2000). Binding kinetics of calbindin-D28k determined by flash photolysis of caged Ca2+. Biophys. J. 79, 3009–3018. doi: 10.1016/S0006-3495(00)76537-4
Nakamura, Y., Harada, H., Kamasawa, N., Matsui, K., Rothman, J. S., Shigemoto, R., et al. (2015). Nanoscale distribution of presynaptic Ca2+ channels and its impact on vesicular release during development. Neuron 85, 145–158. doi: 10.1016/j.neuron.2014.11.019
Nakamura, Y., Reva, M., and DiGregorio, D. A. (2018). Variations in Ca2+ influx can alter chelator-based estimates of Ca2+ channel-synaptic vesicle coupling distance. J. Neurosci. 38, 3971–3987. doi: 10.1523/JNEUROSCI.2061-17.2018
Naraghi, M., and Neher, E. (1997). Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J. Neurosci. 17, 6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997
Neher, E. (1998a). Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium 24, 345–357. doi: 10.1016/S0143-4160(98)90058-6
Neher, E. (1998b). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399. doi: 10.1016/S0896-6273(00)80983-6
Neher, E., and Sakaba, T. (2001). Combining deconvolution and noise analysis for the estimation of transmitter release rates at the calyx of held. J. Neurosci. 21, 444–461. doi: 10.1523/JNEUROSCI.21-02-00444.2001
Ohana, O., and Sakmann, B. (1998). Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. J. Physiol. 513, 135–148. doi: 10.1111/j.1469-7793.1998.135by.x
Pan, B., and Zucker, R. S. (2009). A general model of synaptic transmission and short-term plasticity. Neuron 62, 539–554. doi: 10.1016/j.neuron.2009.03.025
Park, Y., and Ryu, J. K. (2018). Models of synaptotagmin-1 to trigger Ca2+ -dependent vesicle fusion. FEBS Lett. 592, 3480–3492. doi: 10.1002/1873-3468.13193
Radhakrishnan, A., Stein, A., Jahn, R., and Fasshauer, D. (2009). The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 284, 25749–25760. doi: 10.1074/jbc.M109.042499
Ritzau-Jost, A., Delvendahl, I., Rings, A., Byczkowicz, N., Harada, H., Shigemoto, R., et al. (2014). Ultrafast action potentials mediate kilohertz signaling at a central synapse. Neuron 84, 152–163. doi: 10.1016/j.neuron.2014.08.036
Ritzau-Jost, A., Jablonski, L., Viotti, J., Lipstein, N., Eilers, J.-K., and Hallermann, S. (2018). Apparent calcium dependence of vesicle recruitment. J. Physiol. 596, 4693–4707. doi: 10.1113/JP275911
Rozov, A., Burnashev, N., Sakmann, B., and Neher, E. (2001). Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol. 531, 807–826. doi: 10.1111/j.1469-7793.2001.0807h.x
Sakaba, T. (2008). Two Ca2+-dependent steps controlling synaptic vesicle fusion and replenishment at the cerebellar basket cell terminal. Neuron 57, 406–419. doi: 10.1016/j.neuron.2007.11.029
Salin, P. A., Malenka, R. C., and Nicoll, R. A. (1996). Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16, 797–803. doi: 10.1016/S0896-6273(00)80099-9
Schmidt, H., Brachtendorf, S., Arendt, O., Hallermann, S., Ishiyama, S., Bornschein, G., et al. (2013). Nanodomain coupling at an excitatory cortical synapse. Curr. Biol. 244, 244–249. doi: 10.1016/j.cub.2012.12.007
Schneggenburger, R., and Neher, E. (2000). Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406, 889–893. doi: 10.1038/35022702
Scimemi, A., and Diamond, J. S. (2012). The number and organization of Ca2+ channels in the active zone shapes neurotransmitter release from Schaffer collateral synapses. J. Neurosci. 32, 18157–18176. doi: 10.1523/JNEUROSCI.3827-12.2012
Sheng, Z. H., Yokoyama, C. T., and Catterall, W. A. (1997). Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc. Natl. Acad. Sci. U.S.A. 94, 5405–5410. doi: 10.1073/pnas.94.10.5405
Sørensen, J. B., Fernandez-Chacon, R., Sudhof, T. C., and Neher, E. (2003). Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+. J. Gen. Physiol. 122, 265–276. doi: 10.1085/jgp.200308855
Stanley, E. F. (1993). Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron 11, 1007–1011. doi: 10.1016/0896-6273(93)90214-C
Stanley, E. F. (2016). The nanophysiology of fast tansmitter release. Trends Neurosci. 39, 183–197. doi: 10.1016/j.tins.2016.01.005
Südhof, T. C. (2012). The presynaptic active zone. Neuron 75, 11–25. doi: 10.1016/j.neuron.2012.06.012
Südhof, T. C. (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. doi: 10.1016/j.neuron.2013.10.022
Südhof, T. C. (2014). The molecular machinery of neurotransmitter release (Nobel lecture). Angew. Chem. Int. Ed Engl. 53, 12696–12717. doi: 10.1002/anie.201406359
Südhof, T. C., and Rothman, J. E. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477. doi: 10.1126/science.1161748
Sugita, S., Shin, O.-H., Han, W., Lao, Y., and Südhof, T. C. (2002). Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 21, 270–280. doi: 10.1093/emboj/21.3.270
Sun, J., Pang, Z. P., Qin, D., Fahim, A. T., Adachi, R., and Sudhof, T. C. (2007). A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450, 676–682. doi: 10.1038/nature06308
Takamori, S., Holt, M., Stenius, K., Lemke, E. A., Grønborg, M., Riedel, D., et al. (2006). Molecular anatomy of a trafficking organelle. Cell 127, 831–846. doi: 10.1016/j.cell.2006.10.030
Taschenberger, H., Leao, R. M., Rowland, K. C., Spirou, G. A., and von Gersdorff, H. (2002). Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 36, 1127–1143. doi: 10.1016/S0896-6273(02)01137-6
Trimbuch, T., and Rosenmund, C. (2016). Should I stop or should I go? The role of complexin in neurotransmitter release. Nat. Rev. Neurosci. 17, 118–125. doi: 10.1038/nrn.2015.16
Turecek, J., Jackman, S. L., and Regehr, W. G. (2017). Synaptotagmin 7 confers frequency invariance onto specialized depressing synapses. Nature 551, 503–506. doi: 10.1038/nature24474
Turecek, J., and Regehr, W. G. (2018). Synaptotagmin 7 mediates both facilitation and asynchronous release at granule cell synapses. J. Neurosci. 38, 3240–3251. doi: 10.1523/JNEUROSCI.3207-17.2018
Valera, A. M., Doussau, F., Poulain, B., Barbour, B., and Isope, P. (2012). Adaptation of granule cell to Purkinje cell synapses to high-frequency transmission. J. Neurosci. 32, 3267–3280. doi: 10.1523/JNEUROSCI.3175-11.2012
Van der Kloot, W. (1988). Estimating the timing of quantal releases during end-plate currents at the frog neuromuscular junction. J. Physiol. 402, 595–603. doi: 10.1113/jphysiol.1988.sp017224
Voets, T. (2000). Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 537–545. doi: 10.1016/S0896-6273(00)00131-8
Voleti, R., Tomchick, D. R., Südhof, T. C., and Rizo, J. (2017). Exceptionally tight membrane-binding may explain the key role of the synaptotagmin-7 C2A domain in asynchronous neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 114, E8518–E8527. doi: 10.1073/pnas.1710708114
Volynski, K. E., and Krishnakumar, S. S. (2018). Synergistic control of neurotransmitter release by different members of the synaptotagmin family. Curr. Opin. Neurobiol. 51, 154–162. doi: 10.1016/j.conb.2018.05.006
von Gersdorff, H., and Matthews, G. (1994). Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature 370, 652–655. doi: 10.1038/370652a0
Vyleta, N. P., and Jonas, P. (2014). Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science 343, 665–670. doi: 10.1126/science.1244811
Wang, L.-Y., and Augustine, G. J. (2015). Presynaptic nanodomains: a tale of two synapses. Front. Cell. Neurosci. 8:455. doi: 10.3389/fncel.2014.00455
Wang, L.-Y., Fedchyshyn, M. J., and Yang, Y. M. (2009). Action potential evoked transmitter release in central synapses: insights from the developing calyx of Held. Mol. Brain 2:36. doi: 10.1186/1756-6606-2-36
Wang, L.-Y., Neher, E., and Taschenberger, H. (2008). Synaptic vesicles in mature calyx of Held synapses sense higher nanodomain calcium concentrations during action potential-evoked glutamate release. J. Neurosci. 28, 14450–14458. doi: 10.1523/JNEUROSCI.4245-08.2008
Xia, Z., and Storm, D. R. (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 6, 267–276. doi: 10.1038/nrn1647
Xu, J., Mashimo, T., and Sudhof, T. C. (2007). Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54, 567–581. doi: 10.1016/j.neuron.2007.05.004
Yamada, W. M., and Zucker, R. S. (1992). Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys. J. 61, 671–682. doi: 10.1016/S0006-3495(92)81872-6
Yang, Y. M., Fedchyshyn, M. J., Grande, G., Aitoubah, J., Tsang, C. W., Xie, H., et al. (2010). Septins regulate developmental switching from microdomain to nanodomain coupling of Ca2+ influx to neurotransmitter release at a central synapse. Neuron 67, 100–115. doi: 10.1016/j.neuron.2010.06.003
Zhou, Q., Zhou, P., Wang, A. L., Wu, D., Zhao, M., Südhof, T. C., et al. (2017). The primed SNARE–complexin–synaptotagmin complex for neuronal exocytosis. Nature 548, 420–425. doi: 10.1038/nature23484
Keywords: Synaptotagmin, release sensor, Ca2+ channel, coupling distance, nanodomain, microdomain
Citation: Bornschein G and Schmidt H (2019) Synaptotagmin Ca2+ Sensors and Their Spatial Coupling to Presynaptic Cav Channels in Central Cortical Synapses. Front. Mol. Neurosci. 11:494. doi: 10.3389/fnmol.2018.00494
Received: 11 October 2018; Accepted: 21 December 2018;
Published: 15 January 2019.
Edited by:
Beat Schwaller, Université de Fribourg, SwitzerlandReviewed by:
Kirill Volynski, University College London, United KingdomCopyright © 2019 Bornschein and Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hartmut Schmidt, aGFydG11dC5zY2htaWR0QG1lZGl6aW4udW5pLWxlaXB6aWcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.