 Silvia Landi1*
Silvia Landi1* Luigi Petrucco
Luigi Petrucco Federico Sicca
Federico Sicca Gian Michele Ratto
Gian Michele Ratto
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 07 January 2019
Sec. Neuroplasticity and Development
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00458
This article is part of the Research Topic Molecular Mechanisms Underlying Psychiatric Disorders View all 18 articles
Impairments of the dialog between excitation and inhibition (E/I) is commonly associated to neuropsychiatric disorders like autism, bipolar disorders and epilepsy. Moderate levels of hyperexcitability can lead to mild alterations of the EEG and are often associated with cognitive deficits even in the absence of overt seizures. Indeed, various testing paradigms have shown degraded performances in presence of acute or chronic non-ictal epileptiform activity. Evidences from both animal models and the clinics suggest that anomalous activity can cause cognitive deficits by transiently disrupting cortical processing, independently from the underlying etiology of the disease. Here, we will review our understanding of the influence of an abnormal EEG activity on brain computation in the context of the available clinical data and in genetic or pharmacological animal models.
Epilepsy is a heterogeneous disorder that includes a great variety of phenotypes and pathological manifestations, all characterized by enhanced neuronal excitability due to an impaired balance between excitation and inhibition (E/I). In this review article, we will deal mostly with subclinical interictal activity, that can impact on everyday life by causing transient alterations of cortical computation. First, we will attempt to delineate the difference between chronic cognitive decline and transient cognitive deficits due to brief episodes of altered network excitability in humans. These studies require to correlate the EEG recording with the outcome of the cognitive task, to verify whether the presence of interictal epileptic discharges (IEDs) causes a temporary deficit. Second, we will review the available animal models that can be used to analyze brain computation during chronic and transient hypersynchronous activity. Addressing these topics can foster a better understanding of the still ambiguous role of interictal epileptiform activity in modulating development and cognition. This knowledge is necessary to assess the utility of treating interictal EEG abnormalities to reduce cognitive impairment.
Epileptic seizures are transient episodes of excessive synchronous activity in the brain (Noachtar et al., 1999; Chang and Lowenstein, 2003; Shorvon, 2011; Fisher et al., 2014; Trevelyan, 2016; Wang et al., 2017). They encompass a diverse group of events ranging from minimal clinical manifestations, e.g., brief and nearly undetectable losses of consciousness, to vigorous episodes of muscular shaking that can result in physical injuries. Epileptic events can also be transiently induced by brain trauma, injuries, drugs, temperature, hypoxia and other deviations from normal brain homeostasis (Goldberg and Coulter, 2013). Even if epilepsy is a worldwide pathology affecting more than 65 million people (Ngugi et al., 2010), we are still far from a clear understanding of the common mechanisms underlying its very diverse spectrum of presentations (Dichter, 2009; Pitkänen and Lukasiuk, 2009; Vezzani, 2014). The analysis of the EEG of patients with recurrent seizures demonstrates the existence of abnormal electrophysiological events in between seizures, defined as “subclinical” for the apparent absence of clinical correlates. These events have been named interictal epileptiform discharges (IEDs; see Figure 1; Noachtar et al., 1999; de Curtis and Avanzini, 2001; Pillai and Sperling, 2006; Fisher et al., 2014). Interictal discharges are commonly divided on the base of the electroencephalographic signature of the event into four major categories: sharp waves, spikes, sharp waves/spikes-and-slow-waves and multiple spikes-and-slow-waves (Kooi, 1966; Gotman, 1980; Kane et al., 2017). As the name indicates, IEDs are abnormal events temporally distinct from ictal events, and are not directly implicated in the genesis of ictal events (Noachtar et al., 1999; Avoli et al., 2006; Fisher et al., 2014).
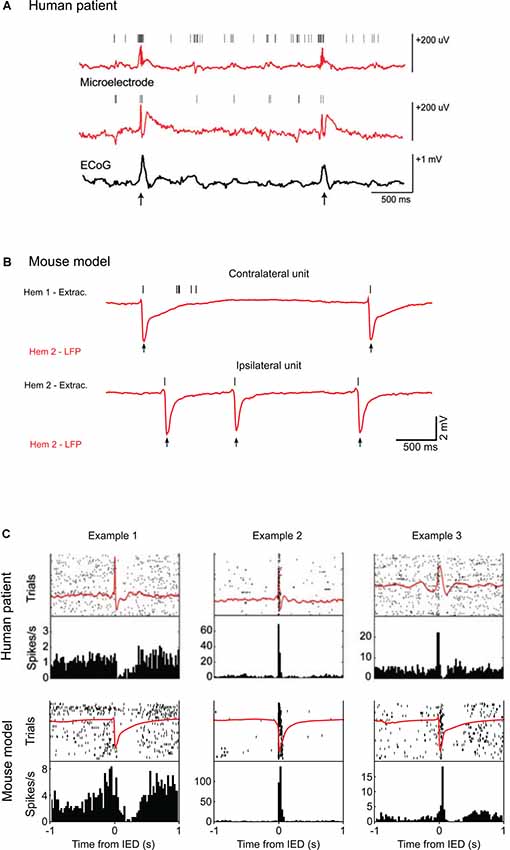
Figure 1. Single-unit activity during interictal epileptic discharges (IEDs) in humans and mice. (A) Top: example of local field potential (LFP) recordings from two microelectrodes (red) and simultaneous corticography (ECoG; black) in patients with medically intractable focal epilepsy during epilepsy monitoring. Vertical marks indicate single units recorded by the two electrodes. Action potentials from the two single units cluster around the IEDs (arrows). (B) Examples of LFP recordings (red) from layers 2/3 of the mouse visual cortex after unilateral superfusion of bicuculline methiodide. Vertical marks (black) indicate the spikes recorded in the loose patch configuration from the contralateral, untreated hemisphere (above) and from the cortical patch treated with bicuculline (below). In the IED focus, spikes are present only at the time of the IEDs (arrows) whereas in the opposite hemisphere other spikes appear around the events. (C) Top: IED-locked raster plots and peri-event time histograms for three example units from the recordings in a human patient. The average LFP is shown in red. The cells display distinct firing modulations around the hypersynchronous event. Below, the same representation is used to show average LFP and firing activity from three units recorded in the mouse during bicuculline induced IEDs. Examples 1 and 3 come from contralateral units, Example 2 from an ipsilateral unit. The firing frequency is similarly modulated by the IEDs in both in the human and mouse recordings. Human data are reproduced with permission from Keller et al. (2010). Mouse data are reproduced with permission from Petrucco et al. (2017).
IEDs are caused by the simultaneous firing of a large number of cortical neurons that produces a paroxysmal depolarization shift in neuronal cohorts (Matsumoto and Marsan, 1964; Prince, 1968; Schwartzkroin and Wyler, 1980). Since seizures are infrequent in the majority of patients, their detection by EEG is painstaking and may require prolonged recording sessions; for this reason, the detection of IEDs is often the first-line diagnostic tool. The detailed description of interictal activity provides the clinicians with insights into the nature of the associated epileptic pathology. Moreover, the different patterns and localization of interictal events are often predictive for the localization of the epileptogenic focus (Berg and Shinnar, 1991; Pillai and Sperling, 2006).
A groundbreaking study (Bridgers, 1987) determined that in a large cohort of psychiatric patients affected by depression, mania, personality disorders, suicidality without depression, nonpsychotic explosive behavior, the probability of epileptiform activity was higher than in the general population and many studies then revealed EEG anomalies in these type of patients (Cook et al., 1986; Weilburg et al., 1995; Hughes, 1996; Hayashi et al., 2010; Badrakalimuthu et al., 2011; Beletsky and Mirsattari, 2012; Gao and Penzes, 2015; Lee et al., 2017). Moreover, it was found that IEDs were present in up to 60% of school-aged children affected with hyperactivity disorders (Richer et al., 2002; Holtmann et al., 2003; Silvestri et al., 2007), while EEG alterations occurred in only 2%–3% to 6.5% of healthy children (Barkmeier and Loeb, 2009; Borusiak et al., 2010). Similarly, some studies report a high rate of EEG abnormalities, ranging from 7% to 70%, in individuals with autism spectrum disorders (ASDs), even without ictal manifestations (Spence and Schneider, 2009; Valvo et al., 2016). A natural conclusion that can be attained by these observations is that anomalies of the neural circuitry driving diverse brain dysfunctions may share some of the pathogenic mechanisms of epilepsy.
The correct regulation of inhibition is a necessary condition for normal brain processing (Van Vreeswijk and Sompolinsky, 1996; Hensch and Fagiolini, 2005; Mariño et al., 2005; Trevelyan and Watkinson, 2005; Buzsáki et al., 2007; Atallah and Scanziani, 2009; Baroncelli et al., 2011; Vogels et al., 2011; Yizhar et al., 2011; Haider et al., 2013; Nelson and Valakh, 2015; Dehghani et al., 2016; Denève et al., 2017). In the healthy brain, the recruitment of inhibitory interneurons by feedforward and feedback excitatory connections ensures that local inhibition closely follows excitation in a given cortical area (Okun and Lampl, 2008; Isaacson and Scanziani, 2011). Neuronal networks are constantly challenged by alterations of E/I and the brain employs several homeostatic mechanisms to adjust net excitability in order to maintain network activity within the physiological range and to prevent saturation (see Turrigiano, 2011). The slightest deviation from this condition can lead to dramatic outcomes: a reduction of excitation may drive the induction of a comatose state, while reduction of the inhibitory feedback can result in network hyperexcitability and epileptiform activity (Dudek and Sutula, 2007; Trevelyan et al., 2013). Even if inhibitory cells are only 25% of all neurons, their role is essential in modulating network activity (for review Schmidt-Wilcke et al., 2017). When inhibition is reduced, and the cortex becomes hyperexcitable (Dichter and Ayala, 1987), neurons may display a wide range of abnormal behaviors. These anomalies can go from a mildly diminished selectivity in the response to different stimulus features (Sillito, 1975), to massive, hypersynchronous events that entrain the population and functionally disconnect it from its afferent inputs. The interplay between E/I is essential to ensure proper gain control and normalization of neuronal activity and to refine the timing of principal cell firing (Chance et al., 2002; Isaacson and Scanziani, 2011). Obviously, in the absence of counteracting inhibition, threshold depolarization would be reached with a much weaker excitatory input, increasing the error and variability of the response. It is important to note that the maintenance of the E/I balance is not a static, hardwired mechanism but a deeply dynamic process maintained by constant plastic adjustments (Froemke et al., 2007; Dorrn et al., 2010).
Given this background, it is not surprising that a defective feedback between E/I has been recognized as one of the key factors in the insurgence of many central nervous system (CNS) pathological conditions (Fernandez and Garner, 2007; Gao and Penzes, 2015; Lee et al., 2017). Indeed, it is known that inhibitory GABAergic signaling is hampered in models of ASD (Gibson et al., 2008; Gogolla et al., 2009; Chao et al., 2010; LeBlanc and Fagiolini, 2011; Yizhar et al., 2011; Bateup et al., 2013; Selimbeyoglu et al., 2017), which justify the co-morbidity between ASD and various forms of hyperexcitability. Since the disruption of GABA signaling increases network gain, this contributes to amplify background noise thus interfering with neuronal coding. Not surprisingly, the potentiation of GABA signaling is a prime pharmacological target that can result in a rescue of the phenotype (Lewine et al., 1999; Tuchman and Rapin, 2002; Levisohn, 2007; Gatto and Broadie, 2010; Bolton et al., 2011; Pizzarelli and Cherubini, 2011; Gilby and O’Brien, 2013; Tuchman, 2013; Cellot and Cherubini, 2014; Jeste and Tuchman, 2015; Buckley and Holmes, 2016). In contrast, other studies suggest that blocking the response to GABA can ameliorate cognitive impairment such as in Down syndrome (Kleschevnikov et al., 2004; Fernandez et al., 2007; Belichenko et al., 2009), in Rett syndrome (Dani et al., 2005; Dani and Nelson, 2009), in Angelman syndrome (Mabb et al., 2011) and in phenylketonuria (De Jaco et al., 2017). These opposing scenarios could be partly explained by the fact that GABA response polarity is not univocal, but it is determined by intracellular Cl− concentration, which in turn is finely regulated by the interplay of leak channels and specific co-transporters (NKCC1 and KCC2; Kaila et al., 2014; Miles et al., 2012; Viitanen et al., 2010; Löscher et al., 2013). It is important to remember that at late embryonic/early postnatal stages intracellular Cl− is higher than 20 mM (Sulis Sato et al., 2017) and GABA causes exit of chloride from target cells leading to membrane depolarization (Ben-Ari et al., 2012). If in these pathologies the pathways underlying chloride homeostasis do not mature properly, GABA could contribute to hyperexcitablity and GABAergic agonists would exhert a paradoxycal effect. Indeed, in Down syndrome it has been demonstrated that the maturation of Cl− regulation is disrupted, thus leading to depolarizing GABA in an adult mouse model for this disorder, and the pharmacological rescue of Cl− homeostasis brought about a recovery of synaptic plasticity and memory (Deidda et al., 2015). It is tempting to speculate that this mechanisms could be shared with other forms of neurodevelopmental disorders, including ASD (Cellot and Cherubini, 2014).
Crucially, pathways involved in regulating synaptic homeostasis can also be affected by the pathology itself; for example, synaptic scaling can depress N-methyl-D-aspartate (NMDA) or AMPA receptors as a consequence of the enhancement of synaptic strength (Doyle et al., 2010; Goold and Nicoll, 2010), but the homeostatic pathways converging on these receptors can be altered by pathology. For example, at least some of the complex single gene disorders associated to ASD (Rett Syndrome, Fragile X Syndrome, Tuberous Sclerosis, Aristaless Related Homeobox associated Syndromes) results in blockade of synaptic scaling, because of the disruption of transcription and/or protein synthesis (Fernandes and Carvalho, 2016). Indeed, in Fragile X the mutation of FMRP, an RNA-binding protein that regulates dendritic protein synthesis, causes the loss of a regulator of the synthesis of AMPARs in dendrites and of their insertion at the postsynaptic site, essential for the increase in synaptic strength induced by retinoic acid or by blockade of neural activity (Soden and Chen, 2010).
Epileptiform activity is often associated to memory impairments, mental slowing, communicative and behavioral disturbances and attentional deficits both in children and adults with epilepsy (Aldenkamp et al., 1990, 2004; Dodson and Bourgeois, 1994). An important issue for the diagnosis and treatment of epilepsy is the existence of many forms of epileptiform activity that can be difficult to classify by their EEG signature. It is also challenging to correlate subtle EEG abnormalities with the behavioral readouts of specific tasks (Blume, 2001). Indeed, patients with short non-convulsive seizures are often characterized by evanescent symptoms or by frequent transitions from IEDs to seizures. In these scenarios, the understanding of the contributions of diverse phases of epileptiform activity to cognitive impairment is especially arduous. On the other hand, a spectrum of epileptic syndromes including continuous spike wave in slow-wave sleep (CSWS) or Landau-Kleffner syndrome (LKS) clearly shows how the strong activation of IEDs during sleep may hamper cognitive functions or language, respectively (Tassinari and Rubboli, 2006). It is a clinical priority to apply proper diagnostic techniques to classify the different epileptic events, and to follow the evolution of the pathology by means of longitudinal and prospective studies (Scheltens-De Boer, 2009).
In general, it is recognized that epileptiform activities can contribute to transient or permanent deficits according to many factors, e.g., their recurrence and severity, the age of the subject and the type of therapy used to prevent seizures and its efficacy. Of course, seizures and epileptiform activity have an higher impact in subjects during development rather than in a mature brain, but is hard to establish on the basis of the available evidence whether repeated interictal spikes are more detrimental than isolated or sporadic seizures in promoting aberrant connectivity (Holmes, 2014). Long-term consequences of interictal discharges accumulating over time may produce deep effects at cognitive level impacting especially on education and learning (IQ scores over time; (Siebelink et al., 1988; Brinciotti et al., 1989; Tuchman and Rapin, 1997) possibly causing life-long effects on developing children. Several mutations can lead to hyperexcitability by enrolling different mechanistic pathways (alterations of synaptic function, alterations in connectivity, impaired metabolism and homeostasis, etc…). Then, regardless of the original causal mechanisms, hyperexcitability leads to defective neuronal computation and impaired cognition (Figure 2; for review McTague et al., 2016; Staley, 2015).
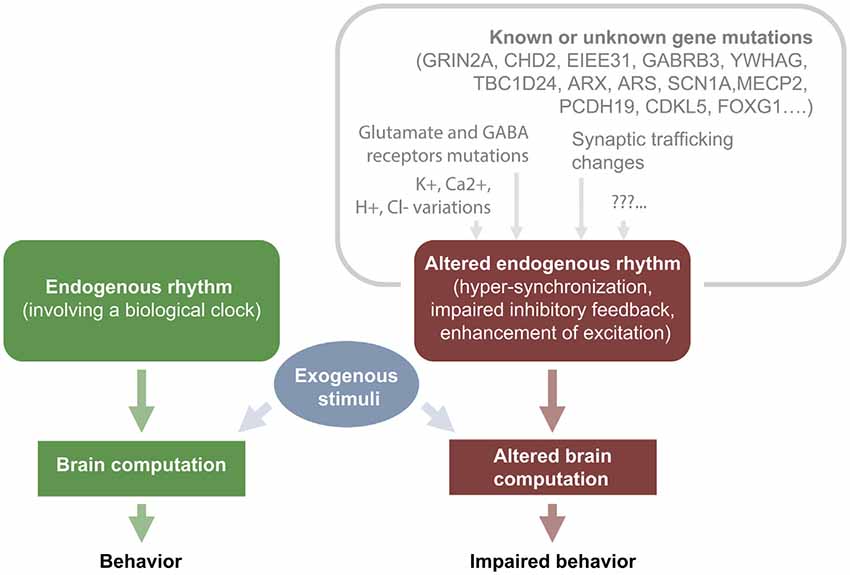
Figure 2. IEDs cause transient cognitive impairment: relationship between upstream mutations and hyperexcitability.
Epileptic encephalopathies (EE) are the most representative example of how long-lasting epileptiform activities may influence the neurodevelopmental outcome. If not properly treated, EE can cause permanent deficits due to the deleterious impact of epileptiform interictal activity and seizures on the development and organization of the immature brain (Nabbout and Dulac, 2003; Hirsch et al., 2006; Holmes and Lenck-Santini, 2006). The relationship between clinical and EEG features and the degree of cognitive deterioration remains elusive and may vary greatly between different syndromes. Thus, it is often arduous to translate into therapeutic decisions the insights arising from clinical and EEG evaluations and from our understanding of the underlying pathomechanisms.
CSWS is a paradigmatic model to understand how continuous and diffuse paroxysms, lasting months or years, may impair the neuropsychological outcome. CSWS is a disorder that appears during childhood, characterized by the presence of continuous generalized spike-wave complexes during at least 85% of slow-wave sleep. Cognitive deficits associated with impairment of neuropsychological functions, reduced IQ, reduction of language, memory and motor impairment occurring in the course of this syndrome are generally assumed to be caused by repetitive interictal discharges (Praline et al., 2003). The disruption of physiologic sleep and the interference with the slow-wave activity homeostasis, with consequent impairment of neural processes and synaptic plasticity at the site of the epileptic focus, are possible pathomechanisms underlying the neuropsychological impairment characterizing the syndrome (Tassinari and Rubboli, 2006; Tononi and Cirelli, 2006). During puberty, the electrical status epilepticus in sleep (ESES) pattern progressively vanishes and the neuropsychological assessments generally improve, although many of the acquired deficits often remain. The localization of the interictal focus seems to have a role in influencing the pattern of neuropsychological derangement. For example, linguistic impairment can be observed when abnormalities are mostly located on the temporal regions, like in the acquired auditory agnosia seen in the LKS (Galanopoulou et al., 2000), or an autistic behavior can appear in relation to frontal epileptogenic foci (Deonna and Roulet-Perez, 2010). Several studies on patients treated for CSWS (De Negri et al., 1995; Yan Liu and Wong, 2000) have shown an improvement in cognitive functions in those successfully responding to treatments and showing a significant reduction of the IEDs. This suggests that the pathological activity underlying the EEG alterations, curtailed by the pharmacological treatment, is the primary cause for the onset of the cognitive symptoms. Evidences deriving from in vitro, in vivo and computational models suggest that the change from physiological to pathological oscillation seen in ESES is related to the blocking of GABAA-mediated inhibition between reticular thalamic neurons and thalamic neurons, which is associated with a differential activation of GABAB-mediated inhibition and in consequent epileptiform activity (Smith and Fisher, 1996; Beenhakker and Huguenard, 2009). Interestingly, in rat models of atypical absence epilepsy, GABAB-receptor antagonists have been shown to rescue the associated learning impairment (Chan et al., 2006).
Another example of EE characterized by strict correlation between the severity of EEG alterations and cognitive impairment is West Syndrome. Affected children typically have clusters of epileptic spasms within the first year of life, and show a specific interictal EEG pattern (called hypsarrhythmia) of highly disorganized background activity and multifocal slow-waves and paroxysms. Both epileptic spasms and hypsarrhythmia are typically associated with developmental regression, usually beginning with impairment of visual functions and possibly linked to hemodynamic and metabolic interferences of the IEDs on cerebral homeostasis (Siniatchkin et al., 2007). An early and appropriate treatment may, in some cases, ameliorate seizures and EEG and have a profound impact on developmental progresses (McTague and Cross, 2013).
Other forms of EE, on the contrary, show poor or no correlation between clinical or EEG severity and neurobehavioral outcome. Dravet syndrome (DS) is one of the main examples of severe EE, where the cognitive outcome does not clearly reflect seizure or IED severity. DS is primarily caused (80% of cases) by heterozygous loss-of-function mutations in the SCN1A gene that encodes the brain voltage-gated sodium channel type-1 (NaV1.1; Dravet et al., 2011; Marini et al., 2011). As the exons of SCN1A only comprise 6 kb of the gene, which is over 100 kb in size, it is conceivable that the remaining patients that have normal transcript, may be affected by mutations of the non-coding portion of the gene leading to correct expression and trafficking of NaV1.1 (Catterall, 2018). Besides febrile and afebrile pharmacoresistant seizures beginning in the first year of life, and frequent episodes of status epilepticus, affected children may develop a progressive slowing of basal EEG activities and interictal asymmetrical spikes or polyspikes and waves (Dravet et al., 2011). In addition, children with DS display a progressively worsening psychomotor delay that is only partly related to severity of epilepsy or EEG features. This suggests that the cognitive defect in DS is not only a consequence of epilepsy or EEG abnormalities themselves but may in part be due to the underlying genetic pathology, i.e., a direct role of the sodium channel dysfunction (Nabbout et al., 2013). Two additional examples of epilepsy where cognitive impairment and developmental consequences are not clearly correlated with the severity and characteristics of seizures are provided by the diseases associated to the mutations of PCDH19 and STXBP1. Mutations of the PCDH19 gene in female patients causes early onset epilepsy, in part resembling DS (Dibbens et al., 2008; Specchio et al., 2011), and often lead to cognitive phenotypes ranging from borderline to severe intellectual disability (Depienne et al., 2009; Hynes et al., 2010; Marini et al., 2010). A similar scenario is found in patients affected by STXBP1 mutations that result in early-onset epilepsy, cognitive and motor disability and ASD (Saitsu et al., 2008; Stamberger et al., 2016).
Dysfunctions of GABAA receptors have been also postulated to play important roles in EE etiology. In fact, mutations or genetic variations of the genes encoding the α1, α6, β1, β2, β3, γ2, or δ subunits (GABRA1, GABRA6, GABRB1, GABRB2, GABRB3, GABRG2 and GABRD, respectively) have been associated with early-onset epilepsies with or without febrile seizures. Evidences show that the compromised hyperpolarization mediated by altered GABAA receptors is not simply caused by receptor gating abnormalities, but by complex mechanisms, including endoplasmic reticulum (ER)-associated degradation, nonsense-mediated mRNA decay, intracellular trafficking defects and ER stress (Hirose, 2014; Hernandez et al., 2016; Neske, 2016; Møller et al., 2017; Shen et al., 2017). In addition, a number of antiepileptic drugs have agonistic effects on GABAA receptors (Hirose, 2014), confirming their likely contribution in the pathomechanisms of epilepsy, and their potential role as candidate targets for new therapies.
Early studies based on EEG recordings suggested that interictal events can also lead to transitory cognitive effects in humans (Hutt et al., 1977; Aarts et al., 1984; Shewmon and Erwin, 1988a, b). These observations have been confirmed in subsequent studies employing improved video EEG recordings synchronized with behavioral assessment (Binnie et al., 1991; Krauss et al., 1997; Liu et al., 2016; Ung et al., 2017). The general idea is that IEDs transiently disrupt the functioning of the area directly involved in the generation of the epileptiform activity, as well as connected regions (Hutt et al., 1977; Aarts et al., 1984; Shewmon and Erwin, 1988a, b; Ung et al., 2017). In general, transient cognitive impairment resulting from single discharge patterns is obviously related to the location and propagation pattern of the epileptiform event. Therefore, the identification of the cognitive phenotype requires proper behavioral assessment in terms of the specific task, its duration, and the modalities of administration.
An early study that addressed the relationship between IEDs and transient cognitive deficits was performed by analyzing short-term verbal and non-verbal memory in a cohort of subjects displaying focal/asymmetrical or symmetrical generalized epileptiform activity. Half of the patients showed transient cognitive impairment associated to the presence of IEDs. IEDs cause region-specific impairments; indeed, IEDs starting in the left hemisphere caused errors in verbal tasks, while those generated in the right one produced impairments in non-verbal tests (Aarts et al., 1984; Aldenkamp et al., 2004).
Following studies demonstrated that bursts of spike-wave patterns are usually followed by a slowing of reaction times lasting several seconds, and by total amnesia for events occurred during the EEG alteration (Porter et al., 1973; Holmes et al., 1987; Krauss et al., 1997). Importantly, this transient but conspicuous deficits can be caused even by a single focal interictal event (Aarts et al., 1984; Binnie et al., 1987; Shewmon and Erwin, 1988b; Siebelink et al., 1988; Binnie, 2003; Kasteleijn-Nolst Trenité and Vermeiren, 2005; see Petrucco et al., 2017) for an animal model correlate. The dependency of cognitive impairment on the IEDs location has been shown recently in patients with seizure onset zones lateralized on the left hemisphere, since spikes that were generated outside the seizure onset area disrupted memory encoding, while those recorded inside the critical activity focus were not detrimental for cognition (Ung et al., 2017).
Many studies have observed that transient cognitive deficits critically depend on the timing between stimulus test and the appearance of the IEDs (Figure 3). Indeed, a single interictal discharge can affect the perception of a visual stimulus, e.g., the presentation of a stimulus at the time of an interictal event in visual cortex resulted in missed or delayed perception (Shewmon and Erwin, 1988b). A detailed characterization of the timing of the sensory deficit (Shewmon and Erwin, 1989) showed that the effect of the spikes started immediately before the deflection in the EEG trace and terminated at the end of the slow-wave. This finding led the authors to the conclusion that the long lasting slow-wave, and not only the paroxysmal spike, can affect the perception of the stimulus (see Figure 3A). The critical correlation of the IEDs timing and the transient cognitive deficit is also present for memory tasks. An important study was performed on patients undergoing the procedure for preoperative seizure localization that were implanted with deep electrodes and were tested for memory maintenance and retrieval during the electrophysiological recording (Kleen et al., 2013). These data showed a decrease of the likelihood of correct responses when the IEDs occurred during the memory retrieval period, thus suggesting a direct link between the pathological activity and cognitive impairment.
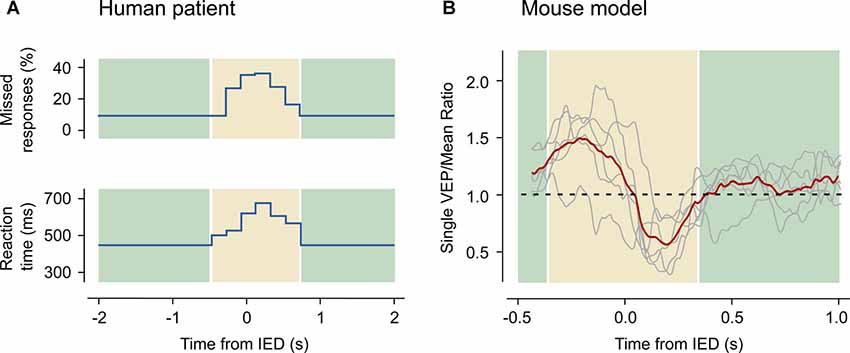
Figure 3. Transient cognitive impairment in humans and in mice. (A) IEDs interfere with a visual reaction test. Data have been acquired from a 10-year old boy suffering from partial complex seizures, tested during the occurrence of focal interictal discharges. The upper panel shows the rate of missed responses as a function of the distance between the stimulus and the nearest IED. The lower panel shows the latency of the response. Both the rate of missed stimuli and the reaction times increase in a small window around the IED event. Redrawn with data from Shewmon and Erwin (1988a). (B) Amplitude of visual evoked potentials (VEP) recorded in the anesthetized mouse as a function of the distance between the stimulus and the closest IED occurring in the contralateral hemisphere. A bi-directional alteration of the VEP amplitude (highlighted in the yellow area) appears when the stimulus occurs nearby an IED: if the spike precedes the stimulus the response is enhanced, while the contrary happens if the spike follows the stimulus. Reproduced with permission from Petrucco et al. (2017).
The dialogs between IEDs and cognitive functions are somehow bidirectional, since it has long been recognized that performing a cognitive task can affect the frequency of interictal paroxysms. For example, an old study (Schwab et al., 1941) determined that generalized spike and wave discharges could be reduced by light or sound stimuli delivered during a reaction time task. Recently, it has been shown that during a test in which images already archived in memory are recognized, there is a significant reduction in the IEDs rate in the amygdala, hippocampus and temporal cortex. Interestingly, during the visual task all these structures were interested by a loss of power in the theta, alpha and beta bands, as well as by an enhanced power in the gamma band. This suggests that the reorganization of network synchronization during the task may correlate with a negative modulation of local IEDs frequency (Matsumoto et al., 2013).
Finally, the detection of temporary cognitive impairment depends on the type and duration of the test (Mirsky and Van Buren, 1965; Aarts et al., 1984; Aldenkamp et al., 2004). Indeed, the beginning of a cognitive task is usually associated to an activating “arousal” effect which can suppress IEDs discharges, causing a global improvement of the test performance (Aarts et al., 1984; Binnie et al., 1987; Stores, 1990). Thus, simple motor tasks and other tests that do not require the processing of complex sets of information are less affected in their execution than more cognitively demanding tests.
Animal models are essential to understand the molecular and physiological mechanism linking the transient electrophysiological disturbance to the alteration of cortical processing. However, the literature on animal models is relatively poor in comparison with the wealth of data provided by the clinics, possibly because of the difficulty of generating standardized models of interictal epileptiform activity (Grone and Baraban, 2015; see Table 1).
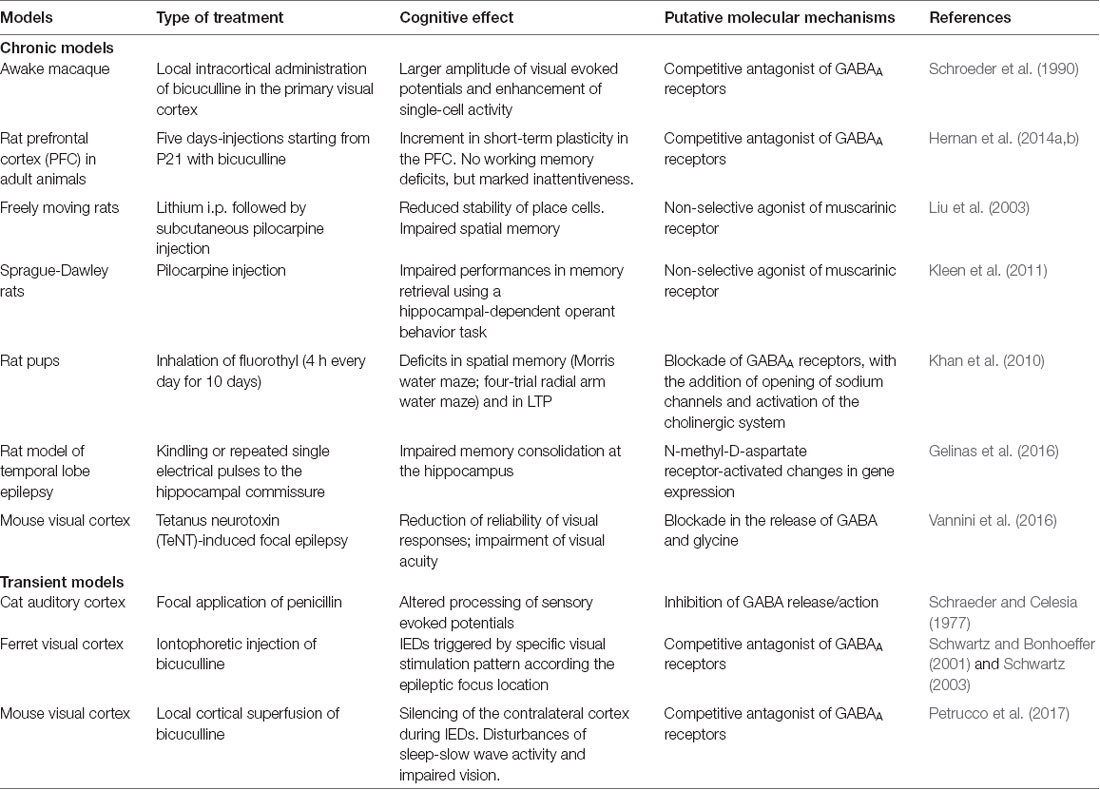
Table 1. Cognitive deficits in models of interictal epileptic discharges (IEDs).
A large body of studies on slices have employed different pharmacological strategies to induce interictal activity (Fisher, 1989; Pitkänen and McIntosh, 2006; Barkmeier and Loeb, 2009; Avoli and Jefferys, 2016): GABAA receptor blockade (Meldrum and Horton, 1971; Collins and Caston, 1979; de Curtis et al., 1998; Uva et al., 2005), low extracellular magnesium concentration (Tancredi et al., 1990), low extracellular calcium (Lian et al., 2001), kainic acid (Medvedev et al., 2000) and 4-aminopyridine (Lévesque et al., 2013). In slice studies have provided valuable information on the cellular mechanisms at the basis of IED generation and propagation, but they cannot fully recapitulate what happens in the animal brain in vivo and cannot provide a causal link between the ectopic activity and the disruption of cortical computation.
In intact animals, focal IEDs have been generated by electrical stimulation (Collins and Caston, 1979; Gelinas et al., 2016) or after systemic or focal administration of inhibitors of GABA activity (Meldrum and Horton, 1971; Schroeder et al., 1990; Schwartz and Bonhoeffer, 2001; Hirase et al., 2004; Ma et al., 2004; Hernan et al., 2014a, b), kainic acid (Berdyyeva et al., 2016) and pilocarpine (Kleen et al., 2011). Finally, a focal model of epileptic activity has been obtained by the localized microinjection of tetanus toxin (TeNT; Brener et al., 1991; Nilsen et al., 2005; Vannini et al., 2016) that causes an increase of the E/I ratio because of a strong blockade of inhibitory synapses (Ferecskó et al., 2014).
When discussing animal studies, one should make an important distinction between chronic and transient models of IED activity. The chronic models are defined by prolonged treatments that may lead to permanent EEG irregularities. In these models, the assessment of cognitive functions generally occurs days or weeks after treatment and probes the effects of the IED activity, if still present, and of the homeostatic response of the brain to the treatment. However, in chronic models, it is difficult to understand whether the observed cognitive deficits are directly due to anomalous activity or to cortical rearrangements that have occurred during the response to the induction that cause steady state alterations of cortical computation. In contrast, transient models take advantage of an acute manipulation that leads immediately to IEDs, and allow assessing their short-term effects on cognitive functions before the onset of any long-term homeostatic change.
A useful example of chronic model is provided by rat pups exposed for 10 days to a low dose of flurothyl (an inhalant chemoconvulsant) for 4 h (Khan et al., 2010). Pups developed IED activity without seizures during the treatment and activity returned to normality after inhalation ending. When the rats reached adulthood, behavioral and electrophysiological testing revealed impairments of spatial memory and long-term potentiation. Anatomical assessment of the hippocampus showed a reduction of newly born cells without an increase in apoptosis at a single time point; however, it was not clear in this study if neurogenesis or apoptosis affected excitatory or inhibitory neurons, rather than glia. In a previous study on the same model by the same group, it was reported the loss of inhibitory GABAergic interneurons, while epileptiform activity did not influence glutamatergic synapse maturation, thus supporting the hypothesis of activity independence of the development of AMPA/NMDA signaling (Isaeva et al., 2006). These studies suggest that the transient IEDs evoked by the treatment have led to long term changes in the hippocampal structure and to cognitive impairment.
Although flurothyl inhalation can be precisely regulated to provide varied degrees of ictal or interictal activity (Modica et al., 1990), its long-term effects are very complex involving a number of factors, including changes in the brain content of DNA (Wasterlain, 1976), increased expression of cyclooxygenase-2 (Kim and Jang, 2006), changes in the intrinsic excitability and postsynaptic composition (Villeneuve et al., 2000; Swann et al., 2006) and impairment of dendrite development (Nishimura et al., 2011). Thus, the interpretation of the flurothyl model is not straightforward, thus preventing a clear causal relationship between IEDs in early development and cognitive impairment in adulthood.
Other pharmacological models that affect directly the E/I balance may provide a more direct causal link between abnormal activity and cognitive deficit. The obvious target for this manipulation is the GABAA receptor, which can be modulated by a rich pharmacological toolbox. Bicuculline methiodide is a competitive agonist that, upon local delivery, causes the onset of IEDs resembling the activity appearing in lesional human epilepsy (Noachtar et al., 1999) and in idiopathic benign partial epilepsies of childhood (de Curtis et al., 1998). A recent study generated a rat model of focal IEDs caused by repeated injections of bicuculline into the prefrontal cortex (PFC) starting from P21 (Hernan et al., 2014a). Upon reaching adulthood, the treated rats showed an increment of short-term plasticity in the PFC, deficits in social behavior and marked absence of attention without significant increment of anxiety or of hyperactivity. This study concluded that the focal IED activity, caused by GABAergic blockade during early-life, disrupted the circuitry of the PFC thus leading to long-term effects on behavior after the ending of IEDs. However, the co-administration of adrenocorticotropic hormone, a drug widely used to treat seizures starting early in life (Rosati et al., 2017), produced a modest amelioration of the attention deficit in adulthood even if it did not reduce IEDs. This result suggested a partial uncoupling between the acute electrophysiological response and the long-term cognitive deficits.
The long-term effects of epileptiform activity have been recently studied in the visual cortex after focal treatment with TeNT. In this case, frequent spike bursts were followed by an upregulation of GABA markers, possibly suggesting a compensatory response. Sensory processing is a paradigmatic example of cortical computation that relies on a finely tuned negative feedback operated by inhibitory interneurons to normalize the wide range of external stimuli to the limited dynamic range of cortical coding. Interestingly, this treatment caused a dendritic rearrangement different from that one observed after flurothyl inhalation, since length complexity of dendrites increased. Visual responses were less reliable in comparison to controls, possibly because of the degradation of the signal to noise ratio of the network, as also suggested by increased firing rate in resting state (Vannini et al., 2016).
Finally, in a kindling model of temporal lobe epilepsy, the communication between hippocampus and medial PFC (mPFC) was altered by IEDs, determining the disruption of the spatial navigation memory in freely behaving rats; moreover, there was a correlation between the degree of memory impairment and the frequency of hippocampal IEDs capable to elicit spindles in the mPFC (Gelinas et al., 2016).
Transient models of activity provide the opportunity to study in a normal brain the direct effect of IEDs on cognitive functions. In these models, there are no long-term effects caused by previous treatments, and the anomalies can be ascribed only to neurons being hijacked by the hypersynchronous firing occurring at each IED. In the past, a large number of investigators has been using acute superfusion with convulsants, such as penicillin or bicuculline, to address in vivo the physiological mechanisms of IEDs (Goldenshohn and Purpura, 1963; Matsumoto and Marsan, 1964; Prince, 1968; Collins and Caston, 1979). These early studies provided us with the first detailed description of the anatomy of an IED (reviewed in Dichter and Ayala, 1987).
Acute pharmacological models are ideal to study the interaction between focal IEDs and its effects on cortical computation, since the timing and intensity of the IED activity can be carefully regulated while probing cortical computation. Indeed, similar manipulations have been used to address how the abnormal recruitment affected physiological sensory processing of both auditory (Schraeder and Celesia, 1977) or visual stimuli (Ebersole and Levine, 1975; Schwartz and Bonhoeffer, 2001; Schwartz, 2003).
A recent study from our laboratory (Petrucco et al., 2017) addressed how cortical processing can be transiently disrupted even in areas far from the interictal focus. IEDs were triggered in the visual cortex of the anesthetized mouse by strictly localized superfusion of bicuculline (Figure 1). As expected, in the treated territory computation was abolished, since all pyramidal neurons were recruited by IEDs. This effect extended to the contralateral cortex, because the IED initially facilitated firing in this area but, after about 100 ms, completely silenced the contralateral cortex for almost 300 ms. Moreover, visual evoked responses were affected depending on the relative timing between stimulus presentation and contralateral spike burst (Figure 3). Therefore, the timing of the hypersynchronous spike defines a temporal window of impaired cognition in the cortical territory recruited by the event. This was accompanied by a secondary deficit caused by the propagation of the hypersynchronous activity to connected brain areas. These rodent data correlates with findings from coupled EEG and fMRI recordings in human subjects showing changes of the BOLD signal in remote structures far from the epileptic generator (Kobayashi et al., 2005, 2006). Indeed, several of the clinical evidences discussed above indicate that, while the location of focal IEDs determines the nature of the deficit, the connected areas are also affected by the propagated spikes and contribute to the overall cognitive deficit (Ung et al., 2017).
Together, these studies prove that IEDs disrupt endogenous rhythms and affect brain information processing even in absence of the circuitry rearrangements proper of chronic epilepsy and independently from epileptic foci.
It is fair to conclude that several clinical evidence and the acute animal models suggest that IED activity interfere with brain computation in the focus and in connected areas, contributing to the overall cognitive impairment. Therefore, it is important to understand whether IEDs should receive pharmacological treatment even in absence of seizures. The answer is not obvious, since antiepileptic drugs can be associated with cognitive side effects (Ben-Menachem et al., 1989; Aldenkamp et al., 2004; Eddy et al., 2012; Perucca and Gilliam, 2012; Glauser et al., 2013) and a careful cost/benefit analysis is required for each specific case. However, curing IEDs is possible and sometimes it results in cognitive improvement, especially ameliorating educational development in children (Besag, 1995; Noachtar et al., 1999; Pressler et al., 2005; Beghi et al., 2013; Kleen et al., 2013). This is true not only for EE like CSWS, where IEDs persist over months or years thus chronically impairing the physiological patterns of EEG sleep activities, but also for epilepsies where the IEDs are less evident, especially some focal epilepsies (Aldenkamp et al., 2004; Sánchez Fernández et al., 2015). The demonstration that the effects of IEDs on neuronal processing are restricted to a brief temporal window around each event, underlines the importance of matching the behavioral readout of transient cognitive impairments to the ongoing EEG identification of IEDs. A better understanding and evaluation of the impact of IEDs on cognition will allow defining the most appropriate pharmacological strategies to treat not only seizures, but also those interictal events that may affect intellectual development and functions.
All authors contributed to the discussion and preparation of the manuscript.
This review was supported by Telethon GGP13187 to GMR, Flagship Project Nanomax to GMR, Fondazione Pisa- Project RST 148/16 and by the Italian Ministry of Health (Ricerca Corrente to FS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aarts, J. H. P., Binnie, C. D., Smit, A. M., and Wilkins, A. J. (1984). Selective cognitive impairment during focal and generalized epileptiform EEG activity. Brain 107, 293–308. doi: 10.1093/brain/107.1.293
Aldenkamp, A. P., Alpherts, W. C. J., Dekker, M. J. A., and Overweg, J. (1990). Neuropsychological aspects of learning disabilities in epilepsy. Epilepsia 31, S9–S20. doi: 10.1111/j.1528-1157.1990.tb05874.x
Aldenkamp, A. P., Arends, J., Verspeek, S., and Berting, M. (2004). The cognitive impact of epileptiform EEG-discharges; relationship with type of cognitive task. Child Neuropsychol. 10, 297–305. doi: 10.1080/09297040490909341
Atallah, B. V., and Scanziani, M. (2009). Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron 62, 566–577. doi: 10.1016/j.neuron.2009.04.027
Avoli, M., Biagini, G., and de Curtis, M. (2006). Do interictal spikes sustain seizures and epileptogenesis? Epilepsy Curr. 6, 203–207. doi: 10.1111/j.1535-7511.2006.00146.x
Avoli, M., and Jefferys, J. G. R. (2016). Models of drug-induced epileptiform synchronization in vitro. J. Neurosci. Methods 260, 26–32. doi: 10.1016/j.jneumeth.2015.10.006
Badrakalimuthu, V. R., Swamiraju, R., and de Waal, H. (2011). EEG in psychiatric practice: to do or not to do? Oncotarget 17, 114–121. doi: 10.1192/apt.bp.109.006916
Barkmeier, D. T., and Loeb, J. A. (2009). An animal model to study the clinical significance of interictal spiking. Clin. EEG Neurosci. 40, 234–238. doi: 10.1177/155005940904000405
Baroncelli, L., Braschi, C., Spolidoro, M., Begenisic, T., Maffei, L., and Sale, A. (2011). Brain plasticity and disease: a matter of inhibition. Neural Plast. 2011:286073. doi: 10.1155/2011/286073
Bateup, H. S., Johnson, C. A., Denefrio, C. L., Saulnier, J. L., Kornacker, K., and Sabatini, B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. doi: 10.1016/j.neuron.2013.03.017
Beenhakker, M. P., and Huguenard, J. R. (2009). Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron 62, 612–632. doi: 10.1016/j.neuron.2009.05.015
Beghi, E., Giussani, G., Grosso, S., Iudice, A., La Neve, A., Pisani, F., et al. (2013). Withdrawal of antiepileptic drugs: guidelines of the italian league against epilepsy. Epilepsia 54, 2–12. doi: 10.1111/epi.12305
Beletsky, V., and Mirsattari, S. M. (2012). Epilepsy, mental health disorder, or both? Epilepsy Res. Treat. 2012, 1–13. doi: 10.1155/2012/163731
Belichenko, N. P., Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Reeves, R. H., and Mobley, W. C. (2009). The “down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological and synaptic phenotypes characteristic of down syndrome. J. Neurosci. 29, 5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. doi: 10.1177/1073858412438697
Ben-Menachem, E., Persson, L. I., Schechter, P. J., Haegele, K. D., and Huebert, N. (1989). The effect of different vigabatrin treatment regimens on CSF biochemistry and seizure control in epileptic patients. Br. J. Clin. Pharmacol. 27, 79S–85S. doi: 10.1111/j.1365-2125.1989.tb03466.x
Berdyyeva, T. K., Frady, E. P., Nassi, J. J., Aluisio, L., Cherkas, Y., Otte, S., et al. (2016). Direct imaging of hippocampal epileptiform calcium motifs following kainic acid administration in freely behaving mice. Front. Neurosci. 10:53. doi: 10.3389/fnins.2016.00053
Berg, A. T., and Shinnar, S. (1991). The risk of seizure recurrence following a first unprovoked seizure: a quantitative review. Neurology 41, 965–972. doi: 10.1212/wnl.41.7.965
Besag, F. M. C. (1995). The therapeutic dilemma: treating subtle seizures or indulging in electroencephalogram cosmetics? Semin. Pediatr. Neurol. 2, 261–268. doi: 10.1016/s1071-9091(95)80005-0
Binnie, C. D. (2003). Cognitive impairment during epileptiform discharges: is it ever justifiable to treat the EEG? Lancet Neurol. 2, 725–730. doi: 10.1016/s1474-4422(03)00584-2
Binnie, C. D., Channon, S., and Marston, D. (1991). Behavioral correlates of interictal spikes. Adv. Neurol. 55, 113–126.
Binnie, C. D., Kasteleijn-Nolst Trenité, D. G. A., Smit, A. M., and Wilkins, A. J. (1987). Interactions of epileptiform EEG discharges and cognition. Epilepsy Res. 1, 239–245. doi: 10.1016/0920-1211(87)90031-3
Blume, W. T. (2001). Current trends in electroencephalography. Curr. Opin. Neurol. 14, 193–197. doi: 10.1097/00019052-200104000-00010
Bolton, P. F., Carcani-Rathwell, I., Hutton, J., Goode, S., Howlin, P., and Rutter, M. (2011). Epilepsy in autism: features and correlates. Br. J. Psychiatry 198, 289–294. doi: 10.1192/bjp.bp.109.076877
Borusiak, P., Zilbauer, M., and Jenke, A. C. W. (2010). Prevalence of epileptiform discharges in healthy children—new data from a prospective study using digital EEG. Epilepsia 51, 1185–1188. doi: 10.1111/j.1528-1167.2009.02411.x
Brener, K., Amitai, Y., Jefferys, J. G. R., and Gutnick, M. J. (1991). Chronic epileptic foci in neocortex: in vivo and in vitro effects of tetanus toxin. Eur. J. Neurosci. 3, 47–54. doi: 10.1111/j.1460-9568.1991.tb00810.x
Bridgers, S. L. (1987). Epileptiform abnormalities discovered on electroencephalographic screening of psychiatric inpatients. Arch. Neurol. 44, 312–316. doi: 10.1001/archneur.1987.00520150056022
Brinciotti, M., Matricardi, M., Paolella, A., Porro, G., and Benedetti, P. (1989). Neuropsychological correlates of subclinical paroxysmal EEG activity in children with epilepsy. 1: qualitative features (generalized and focal abnormalities). Funct. Neurol. 4, 235–239.
Buckley, A. W., and Holmes, G. L. (2016). Epilepsy and autism. Cold Spring Harb. Perspect. Med. 6:a022749. doi: 10.1101/cshperspect.a022749
Buzsáki, G., Kaila, K., and Raichle, M. (2007). Inhibition and brain work. Neuron 56, 771–783. doi: 10.1016/j.neuron.2007.11.008
Catterall, W. A. (2018). Dravet syndrome: a sodium channel interneuronopathy. Curr. Opin. Physiol. 2, 42–50. doi: 10.1016/j.cophys.2017.12.007
Cellot, G., and Cherubini, E. (2014). GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2:70. doi: 10.3389/fped.2014.00070
Chan, K. F. Y., Burnham, W. M. I., Jia, Z., Cortez, M. A., and Snead, O. C. III. (2006). GABABreceptor antagonism abolishes the learning impairments in rats with chronic atypical absence seizures. Eur. J. Pharmacol. 541, 64–72. doi: 10.1016/j.ejphar.2006.04.012
Chance, F. S., Abbott, L. F., and Reyes, A. D. (2002). Gain modulation from background synaptic input. Neuron 35, 773–782. doi: 10.1016/s0896-6273(02)00820-6
Chang, B. S., and Lowenstein, D. H. (2003). Epilepsy. N. Engl. J. Med. 349, 1257–1266. doi: 10.1056/NEJMra022308
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Collins, R. C., and Caston, T. V. (1979). Activation of cortical circuits during interictal spikes. Ann. Neurol. 6, 117–125. doi: 10.1002/ana.410060207
Cook, B. L., Shukla, S., and Hoff, A. L. (1986). EEG abnormalities in bipolar affective disorder. J. Affect. Disord. 11, 147–149. doi: 10.1016/0165-0327(86)90021-2
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. U S A 102, 12560–12565. doi: 10.1073/pnas.0506071102
Dani, V. S., and Nelson, S. B. (2009). Intact LTP but reduced connectivity between neocortical Layer 5 pyramidal neurons in a mouse model of Rett Syndrome. J. Neurosci. 29, 11263–11270. doi: 10.1523/jneurosci.1019-09.2009
de Curtis, M., and Avanzini, G. (2001). Interictal spikes in focal epileptogenesis. Prog. Neurobiol. 63, 541–567. doi: 10.1016/s0301-0082(00)00026-5
de Curtis, M., Manfridi, A., and Biella, G. (1998). Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis. J. Neurosci. 18, 7543–7551. doi: 10.1523/jneurosci.18-18-07543.1998
De Jaco, A., Mango, D., De Angelis, F., Favaloro, F. L., Andolina, D., Nisticò, R., et al. (2017). Unbalance between excitation and inhibition in phenylketonuria, a genetic metabolic disease associated with autism. Int. J. Mol. Sci. 18:E941. doi: 10.3390/ijms18050941
De Negri, M., Baglietto, M. G., Battaglia, F. M., Gaggero, R., Pessagno, A., and Recanati, L. (1995). Treatment of electrical status epilepticus by short diazepam (DZP) cycles after DZP rectal bolus test. Brain Dev. 17, 330–333. doi: 10.1016/0387-7604(95)00076-n
Dehghani, N., Peyrache, A., Telenczuk, B., Le Van Quyen, M., Halgren, E., Cash, S. S., et al. (2016). Dynamic balance of excitation and inhibition in human and monkey neocortex. Sci. Rep. 6:23176. doi: 10.1038/srep23176
Deidda, G., Parrini, M., Naskar, S., Bozarth, I. F., Contestabile, A., and Cancedda, L. (2015). Reversing excitatory GABAA R signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 21, 318–326. doi: 10.1038/nm.3827
Denève, S., Alemi, A., and Bourdoukan, R. (2017). The brain as an efficient and robust adaptive learner. Neuron 94, 969–977. doi: 10.1016/j.neuron.2017.05.016
Deonna, T., and Roulet-Perez, E. (2010). Early-onset acquired epileptic aphasia (Landau-Kleffner syndrome, LKS) and regressive autistic disorders with epileptic EEG abnormalities: the continuing debate. Brain Dev. 32, 746–752. doi: 10.1016/j.braindev.2010.06.011
Depienne, C., Bouteiller, D., Keren, B., Cheuret, E., Poirier, K., Trouillard, O., et al. (2009). Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 5:e1000381. doi: 10.1371/journal.pgen.1000381
Dibbens, L. M., Tarpey, P. S., Hynes, K., Bayly, M. A., Scheffer, I. E., Smith, R., et al. (2008). X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 40, 776–781. doi: 10.1038/ng.149
Dichter, M. A. (2009). Emerging concepts in the pathogenesis of epilepsy and epileptogenesis. Arch. Neurol. 66, 443–447. doi: 10.1001/archneurol.2009.10
Dichter, M., and Ayala, G. (1987). Cellular mechanisms of epilepsy: a status report. Science 237, 157–164. doi: 10.1126/science.3037700
Dodson, W. E., and Bourgeois, B. F. D. (1994). Pharmacology and therapeutic aspects of antiepileptic drugs in pediatrics. J. Child Neurol. 9, 2S1–2S7. doi: 10.1177/0883073894009002011
Dorrn, A. L., Yuan, K., Barker, A. J., Schreiner, C. E., and Froemke, R. C. (2010). Developmental sensory experience balances cortical excitation and inhibition. Nature 465, 932–936. doi: 10.1038/nature09119
Doyle, S., Pyndiah, S., De Gois, S., and Erickson, J. D. (2010). Excitation-transcription coupling via calcium/calmodulindependent protein kinase/ERK1/2 signaling mediates the coordinate induction of VGLUT2 and Narp triggered by a prolonged increase in glutamatergic synaptic activity. J. Biol. Chem. 285, 14366–14376. doi: 10.1074/jbc.m109.080069
Dravet, C., Bureau, M., Dalla Bernardina, B., and Guerrini, R. (2011). Severe myoclonic epilepsy in infancy (Dravet syndrome) 30 years later. Epilepsia 52, 1–2. doi: 10.1111/j.1528-1167.2011.02993.x
Dudek, F. E., and Sutula, T. P. (2007). Epileptogenesis in the dentate gyrus: a critical perspective. Prog. Brain Res. 163, 755–773. doi: 10.1016/s0079-6123(07)63041-6
Ebersole, J. S., and Levine, R. A. (1975). Abnormal neuronal responses during evolution of a penicillin epileptic focus in cat visual cortex. J. Neurophysiol. 38, 250–256. doi: 10.1152/jn.1975.38.2.250
Eddy, C. M., Rickards, H. E., and Cavanna, A. E. (2012). Behavioral adverse effects of antiepileptic drugs in epilepsy. J. Clin. Psychopharmacol. 32, 362–375. doi: 10.1097/JCP.0b013e318253a186
Ferecskó, A. S., Jiruska, P., Foss, L., Powell, A. D., Chang, W.-C., Sik, A., et al. (2014). Structural and functional substrates of tetanus toxin in an animal model of temporal lobe epilepsy. Brain Struct. Funct. 220, 1013–1029. doi: 10.1007/s00429-013-0697-1
Fernandes, D., and Carvalho, A. L. (2016). Mechanisms of homeostatic plasticity in the excitatory synapse. J. Neurochem. 139, 973–996. doi: 10.1111/jnc.13687
Fernandez, F., and Garner, C. C. (2007). Over-inhibition: a model for developmental intellectual disability. Trends Neurosci. 30, 497–503. doi: 10.1016/j.tins.2007.07.005
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10, 411–413. doi: 10.1038/nn1860
Fisher, R. S. (1989). Animal models of the epilepsies. Brain Res. Rev. 14, 245–278. doi: 10.1016/0165-0173(89)90003-9
Fisher, R. S., Scharfman, H. E., and deCurtis, M. (2014). How can we identify ictal and interictal abnormal activity? Adv. Exp. Med. Biol. 813, 3–23. doi: 10.1007/978-94-017-8914-1_1
Froemke, R. C., Merzenich, M. M., and Schreiner, C. E. (2007). A synaptic memory trace for cortical receptive field plasticity. Nature 450, 425–429. doi: 10.1038/nature06289
Galanopoulou, A. S., Bojko, A., Lado, F., and Moshé, S. L. (2000). The spectrum of neuropsychiatric abnormalities associated with electrical status epilepticus in sleep. Brain Dev. 22, 279–295. doi: 10.1016/s0387-7604(00)00127-3
Gao, R., and Penzes, P. (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167. doi: 10.2174/1566524015666150303003028
Gatto, C. L., and Broadie, K. (2010). Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Synaptic Neurosci. 2:4. doi: 10.3389/fnsyn.2010.00004
Gelinas, J. N., Khodagholy, D., Thesen, T., Devinsky, O., and Buzsáki, G. (2016). Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nat. Med. 22, 641–648. doi: 10.1038/nm.4084
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Gilby, K. L., and O’Brien, T. J. (2013). Epilepsy, autism and neurodevelopment: kindling a shared vulnerability? Epilepsy Behav. 26, 370–374. doi: 10.1016/j.yebeh.2012.11.002
Glauser, T., Ben-Menachem, E., Bourgeois, B., Cnaan, A., Guerreiro, C., Kälviäinen, R., et al. (2013). Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 54, 551–563. doi: 10.1111/epi.12074
Gogolla, N., Leblanc, J. J., Quast, K. B., Südhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 1, 172–181. doi: 10.1007/s11689-009-9023-x
Goldberg, E. M., and Coulter, D. A. (2013). Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 14, 337–349. doi: 10.1038/nrn3482
Goldenshohn, E. S., and Purpura, D. P. (1963). Intracellular potentials of cortical neurons during focal epileptogenic discharges. Science 139, 840–842. doi: 10.1126/science.139.3557.840
Goold, C. P., and Nicoll, R. A. (2010). Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68, 512–528. doi: 10.1016/j.neuron.2010.09.020
Gotman, J. (1980). Quantitative measurements of epileptic spike morphology in the human EEG. Electroencephalogr. Clin. Neurophysiol. 48, 551–557. doi: 10.1016/0013-4694(80)90290-4
Grone, B. P., and Baraban, S. C. (2015). Animal models in epilepsy research: legacies and new directions. Nat. Neurosci. 18, 339–343. doi: 10.1038/nn.3934
Haider, B., Häusser, M., and Carandini, M. (2013). Inhibition dominates sensory responses in the awake cortex. Nature 493, 97–100. doi: 10.1038/nature11665
Hayashi, K., Makino, M., Hashizume, M., Nakano, K., and Tsuboi, K. (2010). Electroencephalogram abnormalities in panic disorder patients: a study of symptom characteristics and pathology. Biopsychosoc. Med. 4:9. doi: 10.1186/1751-0759-4-9
Hensch, T. K., and Fagiolini, M. (2005). Excitatory-inhibitory balance and critical period plasticity in developing visual cortex. Prog. Brain Res. 147, 115–124. doi: 10.1016/s0079-6123(04)47009-5
Hernan, A. E., Alexander, A., Jenks, K. R., Barry, J., Lenck-Santini, P. P., Isaeva, E., et al. (2014a). Focal epileptiform activity in the prefrontal cortex is associated with long-term attention and sociability deficits. Neurobiol. Dis. 63, 25–34. doi: 10.1016/j.nbd.2013.11.012
Hernan, A. E., Alexander, A., Lenck-Santini, P. P., Scott, R. C., and Holmes, G. L. (2014b). Attention deficit associated with early life interictal spikes in a rat model is improved with ACTH. PLoS One 9:e89812. doi: 10.1371/journal.pone.0089812
Hernandez, C. C., Klassen, T. L., Jackson, L. G., Gurba, K., Hu, N., Noebels, J. L., et al. (2016). Correction: deleterious rare variants reveal risk for loss of GABAA receptor function in patients with genetic epilepsy and in the general population. PLoS One 11:e0167264. doi: 10.1371/journal.pone.0167264
Hirose, S. (2014). Mutant GABAA receptor subunits in genetic (idiopathic) epilepsy. Prog. Brain Res. 213, 55–85. doi: 10.1016/B978-0-444-63326-2.00003-X
Hirase, H., Creso, J., and Buzsáki, G. (2004). Capillary level imaging of local cerebral blood flow in bicuculline-induced epileptic foci. Neuroscience 128, 209–216. doi: 10.1016/j.neuroscience.2004.07.002
Hirsch, E., Valenti, M. P., Rudolf, G., Seegmuller, C., de Saint Martin, A., Maquet, P., et al. (2006). Landau-Kleffner syndrome is not an eponymic badge of ignorance. Epilepsy Res. 70, S239–S247. doi: 10.1016/j.eplepsyres.2006.02.010
Holmes, G. L. (2014). What is more harmful, seizures or epileptic EEG abnormalities? Is there any clinical data? Epileptic Disord. 16, S12–S22.
Holmes, G. L., and Lenck-Santini, P. P. (2006). Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 8, 504–515. doi: 10.1016/j.yebeh.2005.11.014
Holmes, G. L., McKeever, M., and Adamson, M. (1987). Absence seizures in children: clinical and electroencephalographic features. Ann. Neurol. 21, 268–273. doi: 10.1002/ana.410210308
Holtmann, M., Becker, K., Kentner-Figura, B., and Schmidt, M. H. (2003). Increased frequency of rolandic spikes in ADHD children. Epilepsia 44, 1241–1244. doi: 10.1046/j.1528-1157.2003.13403.x
Hughes, J. R. (1996). A review of the usefulness of the standard EEG in psychiatry. Clin. Electroencephalogr. 27, 35–39. doi: 10.1177/155005949602700106
Hutt, S. J., Newton, J., and Fairweather, H. (1977). Choice reaction time and EEG activity in children with epilepsy. Neuropsychologia 15, 257–267. doi: 10.1016/0028-3932(77)90034-3
Hynes, K., Tarpey, P., Dibbens, L. M., Bayly, M. A., Berkovic, S. F., Smith, R., et al. (2010). Epilepsy and mental retardation limited to females with PCDH19 mutations can present de novo or in single generation families. J. Med. Genet. 47, 211–216. doi: 10.1136/jmg.2009.068817
Isaacson, J. S., and Scanziani, M. (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. doi: 10.1016/j.neuron.2011.09.027
Isaeva, E., Isaev, D., Khazipov, R., and Holmes, G. L. (2006). Selective impairment of GABAergic synaptic transmission in the flurothyl model of neonatal seizures. Eur. J. Neurosci. 23, 1559–1566. doi: 10.1111/j.1460-9568.2006.04693.x
Jeste, S. S., and Tuchman, R. (2015). Autism spectrum disorder and epilepsy: two sides of the same coin? J. Child Neurol. 30, 1963–1971. doi: 10.1177/0883073815601501
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Kane, N., Acharya, J., Benickzy, S., Caboclo, L., Finnigan, S., Kaplan, P. W., et al. (2017). A revised glossary of terms most commonly used by clinical electroencephalographers and updated proposal for the report format of the EEG findings. Clin. Neurophysiol. Pract. 2, 170–185. doi: 10.1016/j.cnp.2017.07.002
Kasteleijn-Nolst Trenité, D. G., and Vermeiren, R. (2005). The impact of subclinical epileptiform discharges on complex tasks and cognition: relevance for aircrew and air traffic controllers. Epilepsy Behav. 6, 31–34. doi: 10.1016/j.yebeh.2004.10.005
Keller, C. J., Truccolo, W., Gale, J. T., Eskandar, E., Thesen, T., Carlson, C., et al. (2010). Heterogeneous neuronal firing patterns during interictal epileptiform discharges in the human cortex. Brain 133, 1668–1681. doi: 10.1093/brain/awq112
Khan, O. I., Zhao, Q., Miller, F., and Holmes, G. L. (2010). Interictal spikes in developing rats cause long-standing cognitive deficits. Neurobiol. Dis. 39, 362–371. doi: 10.1016/j.nbd.2010.05.002
Kim, D. K., and Jang, T. J. (2006). Cyclooxygenase-2 expression and effect of celecoxib in flurothyl-induced neonatal seizure. Int. J. Exp. Pathol. 87, 73–78. doi: 10.1111/j.0959-9673.2006.00457.x
Kleen, J. K., Scott, R. C., Holmes, G. L., Roberts, D. W., Rundle, M. M., Testorf, M., et al. (2013). Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology 81, 18–24. doi: 10.1212/wnl.0b013e318297ee50
Kleen, J. K., Scott, R. C., Holmes, G. L., and Lenck-Santini, P. P. (2011). Hippocampal interictal spikes disrupt cognition in rats. Ann. Neurol. 67, 250–257. doi: 10.1002/ana.21896
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004
Kobayashi, E., Bagshaw, A. P., Grova, C., Gotman, J., and Dubeau, F. (2006). Grey matter heterotopia: what EEG-fMRI can tell us about epileptogenicity of neuronal migration disorders. Brain 129, 366–374. doi: 10.1093/brain/awh710
Kobayashi, E., Bagshaw, A. P., Jansen, A., Andermann, F., Andermann, E., Gotman, J., et al. (2005). Intrinsic epileptogenicity in polymicrogyric cortex suggested by EEG-fMRI BOLD responses. Neurology 64, 1263–1266. doi: 10.1212/01.wnl.0000154640.23656.a3
Kooi, K. A. (1966). Voltage-time characteristics of spikes and other rapid electroencephalographic transients: semantic and morphological considerations. Neurology 16, 59–66. doi: 10.1212/wnl.16.1.59
Krauss, G. L., Summerfield, M., Brandt, J., Breiter, S., and Ruchkin, D. (1997). Mesial temporal spikes interfere with working memory. Neurology 49, 975–980. doi: 10.1212/wnl.49.4.975
LeBlanc, J. J., and Fagiolini, M. (2011). Autism: a “critical period” disorder? Neural Plast. 2011:921680. doi: 10.1155/2011/921680
Lee, E., Lee, J., and Kim, E. (2017). Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81, 838–847. doi: 10.1016/j.biopsych.2016.05.011
Lévesque, M., Salami, P., Behr, C., and Avoli, M. (2013). Temporal lobe epileptiform activity following systemic administration of 4-aminopyridine in rats Maxime. Epilepsia 54, 596–604. doi: 10.1111/epi.12041
Levisohn, P. M. (2007). The autism-epilepsy connection. Epilepsia 48, 33–35. doi: 10.1111/j.1528-1167.2007.01399.x
Lewine, J. D., Andrews, R., Chez, M., Patil, A.-A., Devinsky, O., Smith, M., et al. (1999). Magnetoencephalographic patterns of epileptiform activity in children with regressive autism spectrum disorders. Pediatrics 104, 405–418. doi: 10.1542/peds.104.3.558
Lian, J., Shuai, J., Hahn, P., and Durand, D. M. (2001). Nonlinear dynamic properties of low calcium-induced epileptiform activity. Brain Res. 890, 246–254. doi: 10.1016/s0006-8993(00)03166-8
Liu, X., Muller, R. U., Huang, L.-T., Kubie, J. L., Rotenberg, A., Rivard, B., et al. (2003). Seizure-induced changes in place cell physiology: relationship to spatial memory. J. Neurosci. 23, 11505–11515. doi: 10.1523/JNEUROSCI.23-37-11505.2003
Liu, X.-Y., Shi, T., Yin, W.-N., Ren, Z.-Y., Deng, Y.-L., and Chen, S.-D. (2016). Interictal epileptiform discharges were associated with poorer cognitive performance in adult epileptic patients. Epilepsy Res. 128, 1–5. doi: 10.1016/j.eplepsyres.2016.09.022
Löscher, W., Puskarjov, M., and Kaila, K. (2013). Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74. doi: 10.1016/j.neuropharm.2012.05.045
Ma, H., Wu, C., and Wu, J. (2004). The initiation of spontaneous epileptiform events in the rat neocortex, in vivo. J. Neurophysiol. 91, 934–945. doi: 10.1152/jn.00274.2003
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303. doi: 10.1016/j.tins.2011.04.001
Marini, C., Mei, D., Parmeggiani, L., Norci, V., Calado, E., Ferrari, A., et al. (2010). Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 75, 646–653. doi: 10.1212/WNL.0b013e3181ed9e67
Marini, C., Scheffer, I. E., Nabbout, R., Suls, A., De Jonghe, P., Zara, F., et al. (2011). The genetics of Dravet syndrome. Epilepsia 52, 24–29. doi: 10.1111/j.1528-1167.2011.02997.x
Mariño, J., Schummers, J., Lyon, D. C., Schwabe, L., Beck, O., Wiesing, P., et al. (2005). Invariant computations in local cortical networks with balanced excitation and inhibition. Nat. Neurosci. 8, 194–201. doi: 10.1038/nn1391
Matsumoto, H., and Marsan, C. A. (1964). Cortical cellular phenomena in experimental epilepsy: interictal manifestations. Exp. Neurol. 9, 286–304. doi: 10.1016/0014-4886(64)90025-1
Matsumoto, J. Y., Stead, M., Kucewicz, M. T., Matsumoto, A. J., Peters, P. A., Brinkmann, B. H., et al. (2013). Network oscillations modulate interictal epileptiform spike rate during human memory. Brain 136, 2444–2456. doi: 10.1093/brain/awt159
McTague, A., and Cross, J. H. (2013). Treatment of epileptic encephalopathies. CNS Drugs 27, 175–184. doi: 10.1007/s40263-013-0041-6
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., and Scheffer, I. E. (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15, 304–316. doi: 10.1016/S1474-4422(15)00250-1
Medvedev, A., Mackenzie, L., Hiscock, J. J., and Willoughby, J. O. (2000). Kainic acid induces distinct types of epileptiform discharge with differential involvement of hippocampus and neocortex. Brain Res. Bull. 52, 89–98. doi: 10.1016/s0361-9230(00)00239-2
Meldrum, B. S., and Horton, R. W. (1971). Convulsive effects of 4-deoxypyridoxine and of bicuculline in photosensitive baboons (Papio papio) and in rhesus monkeys (Macaca mulatta). Brain Res. 35, 419–436. doi: 10.1016/0006-8993(71)90485-9
Miles, R., Blaesse, P., Huberfeld, G., Wittner, L., and Kaila, K. (2012). “Chloride homeostasis and GABA signaling in temporal lobe epilepsy,” in Jasper’s Basic Mechanisms of the Epilepsies, 4th Edn. eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen and A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information (US)).
Mirsky, A. F., and Van Buren, J. M. (1965). On the nature of the “absence” in centrencephalic epilepsy: a study of some behavioral, electroencephalographic and autonomic factors. Electroencephalogr. Clin. Neurophysiol. 18, 334–348. doi: 10.1016/0013-4694(65)90053-2
Modica, P. A., Tempelhoff, R., and White, P. F. (1990). Pro- and anticonvulsant effects of anesthetics (Part I). Anesth. Analg. 70, 303–315. doi: 10.1213/00000539-199003000-00013
Møller, R. S., Wuttke, T. V., Helbig, I., Marini, C., Johannesen, K. M., Brilstra, E. H., et al. (2017). Mutations in GABRB3 from febrile seizures to epileptic encephalopathies. Neurology 88, 483–492. doi: 10.1212/WNL.0000000000003565
Nabbout, R., Chemaly, N., Chipaux, M., Barcia, G., Bouis, C., Dubouch, C., et al. (2013). Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J. Rare Dis. 8:176. doi: 10.1186/1750-1172-8-176
Nabbout, R., and Dulac, O. (2003). Epileptic encephalopathies: a brief overview. J. Clin. Neurophysiol. 20, 393–397. doi: 10.1097/00004691-200311000-00002
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Neske, G. T. (2016). The slow oscillation in cortical and thalamic networks: mechanisms and functions. Front. Neural Circuits 9:88. doi: 10.3389/fncir.2015.00088
Ngugi, A. K., Bottomley, C., Kleinschmidt, I., Sander, J. W., and Newton, C. R. (2010). Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia 51, 883–890. doi: 10.1111/j.1528-1167.2009.02481.x
Nilsen, K. E., Walker, M. C., and Cock, H. R. (2005). Characterization of the tetanus toxin model of refractory focal neocortical epilepsy in the rat. Epilepsia 46, 179–187. doi: 10.1111/j.0013-9580.2005.26004.x
Nishimura, M., Gu, X., and Swann, J. W. (2011). Seizures in early life suppress hippocampal dendrite growth while impairing spatial learning. Neurobiol. Dis. 44, 205–214. doi: 10.1016/j.nbd.2011.07.002
Noachtar, S., Binnie, C., Ebersole, J., Mauguière, F., Sakamoto, A., and Westmoreland, B. (1999). A glossary of terms most commonly used by clinical electroencephalographers and proposal for the report form for the EEG findings. The International Federation of Clinical Neurophysiology. Electroencephalogr. Clin. Neurophysiol. 52, 21–41.
Okun, M., and Lampl, I. (2008). Instantaneous correlation of excitation and inhibition during ongoing and sensory-evoked activities. Nat. Neurosci. 11, 535–537. doi: 10.1038/nn.2105
Perucca, P., and Gilliam, F. G. (2012). Adverse effects of antiepileptic drugs. Lancet Neurol. 11, 792–802. doi: 10.1016/S1474-4422(12)70153-9
Petrucco, L., Pracucci, E., Brondi, M., Ratto, G. M., and Landi, S. (2017). Epileptiform activity in the mouse visual cortex interferes with cortical processing in connected areas. Sci. Rep. 7:40054. doi: 10.1038/srep40054
Pillai, J., and Sperling, M. R. (2006). Interictal EEG and the diagnosis of epilepsy. Epilepsia 47, 14–22. doi: 10.1111/j.1528-1167.2006.00654.x
Pitkänen, A., and Lukasiuk, K. (2009). Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 14, 16–25. doi: 10.1016/j.yebeh.2008.09.023
Pitkänen, A., and McIntosh, T. K. (2006). Animal models of post-traumatic epilepsy. J. Neurotrauma 23, 241–261. doi: 10.1089/neu.2006.23.241
Pizzarelli, R., and Cherubini, E. (2011). Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011:297153. doi: 10.1155/2011/297153
Porter, R. J., Kiffin Penry, J., and Dreifuss, F. E. (1973). Responsiveness at the onset of spike-wave bursts. Electroencephalogr. Clin. Neurophysiol. 34, 239–245. doi: 10.1016/0013-4694(73)90251-4
Praline, J., Hommet, C., Barthez, M. A., Brault, F., Perrier, D., Passage, G. D., et al. (2003). Outcome at adulthood of the continuous spike-waves during slow sleep and landau-kleffner syndromes. Epilepsia 44, 1434–1440. doi: 10.1046/j.1528-1157.2003.08403.x
Pressler, R. M., Robinson, R. O., Wilson, G. A., and Binnie, C. D. (2005). Treatment of interictal epileptiform discharges can improve behavior in children with behavioral problems and epilepsy. J. Pediatr. 146, 112–117. doi: 10.1016/j.jpeds.2004.08.084
Prince, D. A. (1968). Inhibition in “epileptic” neurons. Exp. Neurol. 21, 307–321. doi: 10.1016/0014-4886(68)90043-5
Richer, L. P., Shevell, M. I., and Rosenblatt, B. R. (2002). Epileptiform abnormalities in children with attention-deficit-hyperactivity disorder. Pediatr. Neurol. 26, 125–129. doi: 10.1016/s0887-8994(01)00370-8
Rosati, A., Ilvento, L., Lucenteforte, E., Pugi, A., Crescioli, G., McGreevy, K. S., et al. (2017). Comparative efficacy of antiepileptic drugs in children and adolescents: a network meta-analysis. Epilepsia 59, 297–314. doi: 10.1111/epi.13981
Saitsu, H., Kato, M., Mizuguchi, T., Hamada, K., Osaka, H., Tohyama, J., et al. (2008). De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet. 40, 782–788. doi: 10.1038/ng.150
Sánchez Fernández, I., Loddenkemper, T., Galanopoulou, A. S., and Moshé, S. L. (2015). Should epileptiform discharges be treated? Epilepsia 56, 1492–1504. doi: 10.1111/epi.13108
Scheltens-De Boer, M. (2009). Guidelines for EEG in encephalopathy related to ESES/CSWS in children. Epilepsia 50, 13–17. doi: 10.1111/j.1528-1167.2009.02211.x
Schmidt-Wilcke, T., Fuchs, E., Funke, K., Vlachos, A., Müller-Dahlhaus, F., Puts, N. A. J., et al. (2017). GABA—from inhibition to cognition: emerging concepts. Neuroscientist 24, 501–515. doi: 10.1177/1073858417734530
Schraeder, P. L., and Celesia, G. G. (1977). The effects of epileptogenic activity on auditory evoked potentials in cats. Arch. Neurol. 34, 677–682. doi: 10.1001/archneur.1977.00500230047007
Schroeder, C. E., Tenke, C. E., Givre, S. J., Arezzo, J. C., and Vaughan, H. G. (1990). Laminar analysis of bicuculline-induced epileptiform activity in area 17 of the awake macaque. Brain Res. 515, 326–330. doi: 10.1016/0006-8993(90)90617-k
Schwab, R., Grunwald, A., and Sargant, W. (1941). Regulation of the treatment of epilepsy by synchronized recording of respiration and brain waves. Arch. Neurol. Psychiatry 46, 1017–1034. doi: 10.1001/archneurpsyc.1941.02280240072004
Schwartz, T. H. (2003). Optical imaging of epileptiform events in visual cortex in response to patterned photic stimulation. Cereb. Cortex 13, 1287–1298. doi: 10.1093/cercor/bhg076
Schwartz, T. H., and Bonhoeffer, T. (2001). In vivo optical mapping of epileptic foci and surround inhibition in ferret cerebral cortex. Nat. Med. 7, 1063–1067. doi: 10.1038/nm0901-1063
Schwartzkroin, P. A., and Wyler, A. R. (1980). Mechanisms underlying epileptiform burst discharge. Ann. Neurol. 7, 95–107. doi: 10.1002/ana.410070202
Selimbeyoglu, A., Kim, C. K., Inoue, M., Lee, S. Y., Hong, A. S. O., Kauvar, I., et al. (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2 -deficient mice. Sci. Transl. Med. 9:eaah6733. doi: 10.1126/scitranslmed.aah6733
Shen, D., Hernandez, C. C., Shen, W., Hu, N., Poduri, A., Shiedley, B., et al. (2017). De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 140, 49–67. doi: 10.1093/brain/aww272
Shewmon, D. A., and Erwin, R. J. (1988a). Focal spike-induced cerebral dysfunction is related to the after-coming slow wave. Ann. Neurol. 23, 131–137. doi: 10.1002/ana.410230205
Shewmon, D. A., and Erwin, R. J. (1988b). The effect of focal interictal spikes on perception and reaction time. II. Neuroanatomic specificity. Electroencephalogr. Clin. Neurophysiol. 69, 338–352. doi: 10.1016/0013-4694(88)90005-3
Shewmon, D. A., and Erwin, R. J. (1989). Transient impairment of visual perception induced by single interictal occipital spikes. J. Clin. Exp. Neuropsychol. 11, 675–691. doi: 10.1080/01688638908400924
Shorvon, S. (2011). The etiologic classification of epilepsy. Epilepsia 52, 1052–1057. doi: 10.1111/j.1528-1167.2011.03041.x
Siebelink, B. M., Bakker, D. J., Binnie, C. D., and Kasteleijn-Nolst Trenité, D. G. A. (1988). Psychological effects of subclinical epileptiform EEG discharges in children. II. General intelligence tests. Epilepsy Res. 2, 117–121. doi: 10.1016/0920-1211(88)90028-9
Sillito, A. M. (1975). The contribution of inhibitory mechanisms to the receptive field properties of neurones in the striate cortex of the cat. J. Physiol. 250, 305–329. doi: 10.1113/jphysiol.1975.sp011056
Silvestri, R., Gagliano, A., Calarese, T., Aricò, I., Cedro, C., Condurso, R., et al. (2007). Ictal and interictal EEG abnormalities in ADHD children recorded over night by video-polysomnography. Epilepsy Res. 75, 130–137. doi: 10.1016/j.eplepsyres.2007.05.007
Siniatchkin, M., Van Baalen, A., Jacobs, J., Moeller, F., Moehring, J., Boor, R., et al. (2007). Different neuronal networks are associated with spikes and slow activity in hypsarrhythmia. Epilepsia 48, 2312–2321. doi: 10.1111/j.1528-1167.2007.01195.x
Smith, K. A., and Fisher, R. S. (1996). The selective GABAB antagonist CGP-35348 blocks spike-wave bursts in the cholesterol synthesis rat absence epilepsy model. Brain Res. 729, 147–150. doi: 10.1016/0006-8993(96)00174-6
Soden, M., and Chen, L. (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J. Neurosci. 30, 16910–16921. doi: 10.1523/jneurosci.3660-10.2010
Specchio, N., Marini, C., Terracciano, A., Mei, D., Trivisano, M., Sicca, F., et al. (2011). Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 52, 1251–1257. doi: 10.1111/j.1528-1167.2011.03063.x
Spence, S. J., and Schneider, M. T. (2009). The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr. Res. 65, 599–606. doi: 10.1203/pdr.0b013e31819e7168
Staley, K. (2015). Molecular mechanisms of epilepsy. Nat. Neurosci. 18, 367–372. doi: 10.1038/nn.3947
Stamberger, H., Nikanorova, M., Willemsen, M. H., Accorsi, P., Angriman, M., Baier, H., et al. (2016). STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology 86, 954–962. doi: 10.1212/WNL.0000000000002457
Stores, G. (1990). Electroencephalographic parameters in assessing the cognitive function of children with epilepsy. Epilepsia 31, S45–S49. doi: 10.1111/j.1528-1157.1990.tb05870.x
Sulis Sato, S., Artoni, P., Landi, S., Cozzolino, O., Parra, R., Pracucci, E., et al. (2017). Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc. Natl. Acad. Sci. U S A 114, E8770–E8779. doi: 10.1073/pnas.1702861114
Swann, J. W., Le, J. T., and Lee, C. L. (2006). Recurrent seizures and the molecular maturation of hippocampal and neocortical glutamatergic synapses. Dev. Neurosci. 29, 168–178. doi: 10.1159/000096221
Tancredi, V., Hwa, G. G. C., Zona, C., Brancati, A., and Avoli, M. (1990). Low magnesium epileptogenesis in the rat hippocampal slice: electrophysiological and pharmacological features. Brain Res. 511, 280–290. doi: 10.1016/0006-8993(90)90173-9
Tassinari, C. A., and Rubboli, G. (2006). Cognition and paroxysmal EEG activities: from a single spike to electrical status epilepticus during sleep. Epilepsia 47, 40–43. doi: 10.1111/j.1528-1167.2006.00686.x
Tononi, G., and Cirelli, C. (2006). Sleep function and synaptic homeostasis. Sleep Med. Rev. 10, 49–62. doi: 10.1016/j.smrv.2005.05.002
Trevelyan, A. J. (2016). Do cortical circuits need protecting from themselves? Trends Neurosci. 39, 502–511. doi: 10.1016/j.tins.2016.06.002
Trevelyan, A. J., Bruns, W., Mann, E. O., Crepel, V., and Scanziani, M. (2013). The information content of physiological and epileptic brain activity. J. Physiol. 591, 799–805. doi: 10.1113/jphysiol.2012.240358
Trevelyan, A. J., and Watkinson, O. (2005). Does inhibition balance excitation in neocortex? Prog. Biophys. Mol. Biol. 87, 109–143. doi: 10.1016/j.pbiomolbio.2004.06.008
Tuchman, R. (2013). Autism and social cognition in epilepsy: implications for comprehensive epilepsy care. Curr. Opin. Neurol. 26, 214–218. doi: 10.1097/wco.0b013e32835ee64f
Tuchman, R. F., and Rapin, I. (1997). Regression in pervasive developmental disorders: seizures and epileptiform electroencephalogram correlates. Pediatrics 99, 560–566. doi: 10.1542/peds.99.4.560
Tuchman, R., and Rapin, I. (2002). Epilepsy in autism. Lancet Neurol. 1, 352–358. doi: 10.1016/S1474-4422(02)00160-6
Turrigiano, G. (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89–103. doi: 10.1146/annurev-neuro-060909-153238
Ung, H., Cazares, C., Nanivadekar, A., Kini, L., Wagenaar, J., Becker, D., et al. (2017). Interictal epileptiform activity outside the seizure onset zone impacts cognition. Brain 140, 2157–2168. doi: 10.1093/brain/awx143
Uva, L., Librizzi, L., Wendling, F., and De Curtis, M. (2005). Propagation dynamics of epileptiform activity acutely induced by bicuculline in the hippocampal-parahippocampal region of the isolated guinea pig brain. Epilepsia 46, 1914–1925. doi: 10.1111/j.1528-1167.2005.00342.x
Valvo, G., Baldini, S., Retico, A., Rossi, G., Tancredi, R., Ferrari, A. R., et al. (2016). Temporal lobe connects regression and macrocephaly to autism spectrum disorders. Eur. Child Adolesc. Psychiatry 25, 421–429. doi: 10.1007/s00787-015-0746-9
Van Vreeswijk, C., and Sompolinsky, H. (1996). Chaos in neuronal networks with balanced excitatory and inhibitory activity. Science 274, 1724–1726. doi: 10.1126/science.274.5293.1724
Vannini, E., Restani, L., Pietrasanta, M., Panarese, A., Mazzoni, A., Rossetto, O., et al. (2016). Altered sensory processing and dendritic remodeling in hyperexcitable visual cortical networks. Brain Struct. Funct. 221, 2919–2936. doi: 10.1007/s00429-015-1080-1
Vezzani, A. (2014). Epilepsy and inflammation in the brain: overview and pathophysiology. Epilepsy Curr. 14, 3–7. doi: 10.5698/1535-7511-14.s2.3
Viitanen, T., Ruusuvuori, E., Kaila, K., and Voipio, J. (2010). The K+-Cl− cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J. Physiol. 588, 1527–1540. doi: 10.1113/jphysiol.2009.181826
Villeneuve, N., Ben-Ari, Y., Holmes, G. L., and Gaiarsa, J. L. (2000). Neonatal seizures induced persistent changes in intrinsic properties of CA1 rat hippocampal cells. Ann. Neurol. 47, 729–738. doi: 10.1002/1531-8249(200006)47:6<729::aid-ana5>3.3.co;2-3
Vogels, T. P., Sprekeler, H., Zenke, F., Clopath, C., and Gerstner, W. (2011). Inhibitory plasticity balances excitation and inhibition in sensory pathways and memory networks. Science 334, 1569–1573. doi: 10.1126/science.1211095
Wang, B., Huang, L., Zhu, Y., Kundaje, A., Batzoglou, S., and Goldenberg, A. (2017). Vicus: exploiting local structures to improve network-based analysis of biological data. PLoS Comput. Biol. 13:e1005621. doi: 10.1371/journal.pcbi.1005621
Wasterlain, C. G. (1976). Effect of neonatal status epilepticus on rat brain development. Neurology 26, 975–986. doi: 10.1212/WNL.26.10.975
Weilburg, J. B., Schachter, S., Worth, J., Pollack, M. H., Sachs, G. S., Ives, J. R., et al. (1995). EEG abnormalities in patients with atypical panic attacks. J. Clin. Psychiatry 56, 358–362.
Yan Liu, X., and Wong, V. (2000). Spectrum of epileptic syndromes with electrical status epilepticus during sleep in children. Pediatr. Neurol. 22, 371–379. doi: 10.1016/s0887-8994(00)00132-6
Keywords: epileptic encephalopathy, cognitive impairment in mental disorder, interictal epileptiform discharges, EEG, animal models of epilepsy
Citation: Landi S, Petrucco L, Sicca F and Ratto GM (2019) Transient Cognitive Impairment in Epilepsy. Front. Mol. Neurosci. 11:458. doi: 10.3389/fnmol.2018.00458
Received: 30 January 2018; Accepted: 28 November 2018;
Published: 07 January 2019.
Edited by:
Chiara Verpelli, Istituto di Neuroscienze (IN), ItalyReviewed by:
Claudia Lodovichi, Istituto Veneto di Medicina Molecolare (VIMM), ItalyCopyright © 2019 Landi, Petrucco, Sicca and Ratto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Landi, c2lsdmlhLmxhbmRpQHNucy5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.