
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 14 November 2018
Sec. Molecular Signalling and Pathways
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00421
This article is part of the Research Topic Alzheimer’s Disease Proteomics and Beyond View all 7 articles
 Alessia Lasorsa1†
Alessia Lasorsa1† Idir Malki1†François-Xavier Cantrelle1
Idir Malki1†François-Xavier Cantrelle1 Hamida Merzougui1Emmanuelle Boll1
Hamida Merzougui1Emmanuelle Boll1 Jean-Charles Lambert2
Jean-Charles Lambert2 Isabelle Landrieu1*
Isabelle Landrieu1*Bridging integrator-1 (BIN1) gene is associated with an increased risk to develop Alzheimer’s disease, a tauopathy characterized by intra-neuronal accumulation of phosphorylated Tau protein as paired helical filaments. Direct interaction of BIN1 and Tau proteins was demonstrated to be mediated through BIN1 SH3 C-terminal domain and Tau (210–240) peptide within Tau proline-rich domain. We previously showed that BIN1 SH3 interaction with Tau is decreased by phosphorylation within Tau proline-rich domain, of at least T231. In addition, the BIN1/Tau interaction is characterized by a dynamic equilibrium between a closed and open conformations of BIN1 isoform 1, involving an intramolecular interaction with its C-terminal BIN1 SH3 domain. However, the role of the BIN1/Tau interaction, and its potential dysregulation in Alzheimer’s disease, is not yet fully understood. Here we showed that within Tau (210–240) peptide, among the two proline-rich motifs potentially recognized by SH3 domains, only motif P216TPPTR221 is bound by BIN1 SH3. A structural model of the complex between BIN1 SH3 and Tau peptide (213–229), based on nuclear magnetic resonance spectroscopy data, revealed the molecular detail of the interaction. P216 and P219 within the proline-rich motif were in direct contact with the aromatic F588 and W562 of the BIN1 SH3 domain. The contact surface is extended through electrostatic interactions between the positively charged R221 and K224 residues of Tau peptide and those negatively charged of BIN1 SH3, corresponding to E556 and E557. We next investigated the impact of multiple Tau phosphorylations within Tau (210–240) on its interaction with BIN1 isoform 1. Tau (210–240) phosphorylated at four different sites (T212, T217, T231, and S235), contrary to unphosphorylated Tau, was unable to compete with the intramolecular interaction of BIN1 SH3 domain with its CLAP domain. In accordance, the affinity of BIN1 SH3 for phosphorylated Tau (210–240) peptide was reduced, with a five-fold increase in the dissociation constant, from a Kd of 44 to 256 μM. This study highlights the complexity of the regulation of BIN1 isoform 1 with Tau. As abnormal phosphorylation of Tau is linked to the pathology development, this regulation by phosphorylation might have important functional consequences.
Alzheimer’s disease (AD) is the most common form of dementia and the most prevalent tauopathy. In genome-wide association studies, variants of the BIN1 gene coding for bridging integrator-1 (also known as amphiphysin 2) have been identified as the second highest genetic risk factor for AD after the ApoE gene, (Seshadri et al., 2010; Genin et al., 2011; Lambert et al., 2013). Several hypothesis have emerged on how BIN1 protein contributes to the increased risk of developing AD, which could involve both Aβ and Tau neurodegeneration pathways (Chapuis et al., 2013; Calafate et al., 2016; Ubelmann et al., 2017). The BIN1 gene is subject to extensive tissue-specific alternative splicing, leading to several isoforms with different cellular localizations and functions. BIN1 protein isoforms 1–7 are expressed in the brain, isoform 8 is expressed in the muscle, and isoforms 9 and 10 are expressed ubiquitously (Prokic et al., 2014). All BIN1 isoforms contain a N-terminal Bin/amphiphysin/Rvs (BAR) domain and a C-terminal SH3 domain (BIN1 SH3) (Prokic et al., 2014). The BAR domain has a role in membrane remodeling by inducing curvature (Frost et al., 2009), and the SH3 domain mediates BIN1’s interactions with several partners (such as dynamin). However, only the brain-specific BIN1 isoforms contain a clathrin and AP-2-binding domain (CLAP) that mediates the interaction with clathrin and AP2 – suggesting a specific role for BIN1 in clathrin-mediated endocytosis in the brain (David et al., 1996; Takei et al., 1999). BIN1 was recently shown to negatively regulate this process and it has been proposed that lower Bin1 expression may favor Tau propagation through increased endocytosis (Calafate et al., 2016). Regulation of endosomal trafficking by BIN1 is also proposed to affect Aβ42 endocytic production by negatively regulating APP and BACE1 convergence in early endosomes (Ubelmann et al., 2017). BIN1 loss of function thus prevents APP and BACE1 segregation at early endosomes and may boost Aβ accumulation.
The BIN1 gene is the first of the sporadic AD genetic determinants to have been linked to a Tau pathology (Chapuis et al., 2013). BIN1 is upregulated in the AD brain, and its expression correlates with the neurofibrillary tangle pathology caused by intraneuronal aggregates of abnormally phosphorylated Tau proteins (Chapuis et al., 2013; Holler et al., 2014). Furthermore, the BIN1 homolog AMPH mediates Tau toxicity in a Drosophila melanogaster model overexpressing Tau (Chapuis et al., 2013; Dourlen et al., 2017). BIN1 directly interacts with Tau and the BIN1/Tau complex is observed at specific locations in primary neurons (partly co-localized with the actin cytoskeleton network) (Sottejeau et al., 2015). BIN1 was indeed shown to modulate actin dynamics by both having actin bundling activity and stabilizing Tau-induced actin bundles (Dräger et al., 2017). Deregulation of BIN1/Tau complex formation may thus impact neuronal functions.
During the disease progression, Tau presents both an increased phosphorylation level and a modification of its pattern of phosphorylation. Tau is phosphorylated by various kinases, including proline-directed kinases [like CDK5 (Baumann et al., 1993; Kimura et al., 2014), GSK3 (Ishiguro et al., 1993; Cho and Johnson, 2003), and ERK2 (Drewes et al., 1992)], that mainly target the proline rich domain (PRD) of Tau (Landrieu et al., 2010; Leroy et al., 2010; Qi et al., 2016). For example, the AT8 diagnostic monoclonal antibody [pS202 + pT205 + (pS208) epitope] can be used to track the spatial progression of the pathology in the AD brain over time (Braak et al., 2006). Given that Tau has so many phosphorylation sites, characterizing the impact of these modifications on Tau functions is an analytical challenge (Lippens et al., 2012a,b). Yet, it remains of interest to decipher the functional consequences of specific Tau phosphorylation, and in the case of the present study, how it could modulate the BIN1/Tau interaction.
We have previously reported that the BIN1 SH3 binds directly to Tau PRD (Sottejeau et al., 2015). The interaction motif within the Tau PRD was narrowed down to a sequence located between amino acid residues 210–240, which contains two PXXP SH3 binding motifs (Sottejeau et al., 2015). This interaction is weakened by phosphorylation of the Tau PRD (Sottejeau et al., 2015), and in particular, phosphorylation of T231 (Sottejeau et al., 2015). Furthermore, we recently showed that the SH3 domain of the neuronal-specific BIN1 isoform 1 protein is engaged in a dynamic equilibrium between open and closed conformations (Malki et al., 2017). The closed conformation results from BIN1-SH3’s interaction with the BIN1-CLAP domain. Tau (210–240) is able to shift the equilibrium toward the intermolecular interaction – indicating a subtle regulation of the Tau/BIN1 interaction.
As studying the regulation of the BIN1/Tau interaction is an important step in understanding its specific contribution to the AD process, we further pursued this investigation. Here, we first determined which PXXP motif(s) is(are) critical for the BIN1/Tau interaction. Based on the NMR data, we proposed next a structural model of the complex between BIN1 SH3 and a Tau peptide, which provides the atomic detail of the interaction. Lastly, we looked at whether Tau (210–240) phosphorylation affects the dynamic equilibrium between open and closed conformations of BIN1 isoform 1.
The BIN1 isoform 1 and BIN1 SH3 domain were expressed and purified as previously described (Malki et al., 2017). cDNA encoding Tau (210–240) peptide SRTPSLPTPPTREPKKVAVVRTPPKSPSSAK was cloned into the pETNKI-HisSUMO3-LIC-Kan vector following a ligation-independent cloning procedure (Luna-Vargas et al., 2011). Uniformly labeled 15N and 13C or 15N protein/peptide samples were produced in M9 medium supplemented with 0.1 mM CaCl2 and 2 mM MgSO4 and containing 0.1% 15NH4Cl and 0.2% 13C-glucose (Sigma-Aldrich) or 0.4% glucose, with 0.5 g/l of 15N- and 13C-enriched or 15N-enriched ISOGROW (Sigma-Aldrich), depending of the labeling scheme. The recombinant His6-SUMO Tau (210–240) was purified using a nickel affinity chromatography column and then incubated 16 h with SENP2 protease in the presence of 2 mM DTT, at 4°C. The protein sample was buffer-exchanged using a desalting column (G25 resin, cutoff of 7 kDa; PD-10 GE Healthcare) against 50 mM sodium phosphate pH 7.3, 30 mM NaCl, 3 mM DTT, and then loaded on a nickel affinity chromatography column. The tag-cleaved peptide was then recovered in the flow-through.
NMR experiments were recorded on Bruker 950-MHz, 900-MHz, or 600-MHz spectrometers all equipped with a triple-resonance cryogenic probe. NMR measurements were performed in 50 mM sodium phosphate buffer, pH 7.3, 30 mM NaCl, 3 mM DTT and 10% D2O (NMR buffer). BIN1 SH3 domain backbone assignment has been previously reported (Malki et al., 2017). Tau (210–240) backbone assignment (0.5 mM sample) was performed at 600 MHz by recording 3D HNCA, 3D HN(CO)CA, 3D H(CA)NH, 3D HNCO, 3D HN(CA)CO 3D CBCA(CO)NH, and 3D CBCANH. For side-chain assignment, additional 3D15N NOESY-HSQC, 3D15N-edited TOCSY, 3D (H)CCH TOCSY, and 3D H(C)CH TOCSY experiments were recorded. A 3D 13C-edited NOESY was further used for side-chain assignment validation. All assignment experiments were recorded at 5°C. Chemical shift values (Supplementary Table S1) were directly transferred at 20°C, because the differences of chemical shift values were small between 5 and 20°C. An analogous set of experiments was recorded for phosphorylated Tau (210–240) backbone assignment (Supplementary Table S2). The HαCαN spectra (Wang et al., 1995) of Tau (210–240) peptide at 0.5 mM were recorded at 20°C with the standard Bruker pulse sequence in the NMR buffer containing 100% D2O. BIN1 isoform 1 1H-15N HSQC spectra were recorded at 20°C at 900 MHz (0.08 mM sample) with 3096 points (direct) and 280 points (indirect), for spectral width of 14 ppm and 26 ppm, respectively, and 256 scans. A 3D F1 13C/15N-filtered and F3 15N-edited NOESY-HSQC, with WATERGATE (WATER suppression by GrAdient-Tailored Excitation) scheme (Piotto et al., 1992), named noesyhsqcf3gpwgx13d, part of the Bruker pulse sequence library, was recorded at 20°C at 950 MHz for the detection of intermolecular NOEs to amide groups. This data was used to solve the structure of the complex between BIN1 SH3 domain and Tau (210–240). Spectra were processed using TopSpin software (Bruker) and analyzed by CcpNmr Analysis (Vranken et al., 2005).
15N-R1, 15N-R2, and {1H}-15N steady-state heteronuclear NOE spectra were recorded at 293K on 15N-labeled samples (0.12 mM) at 600 MHz for BIN1 SH3 and BIN1 SH3/Tau (210–240) complex (1:8 molar ratio). For the R1 experiments (Supplementary Figure S1), 11 data points were recorded, using relaxation delays between 50 and 1300 ms and a recycle delay of 4 s. For R2 experiments (Supplementary Figure S1), 13 data points were recorded, using relaxation delays between 10 and 150 ms (recycle delay was 4 s). The heteronuclear NOE experiment (Supplementary Figure S1) was recorded by including, or not, a 4 s period of 120° 1H saturation pulses, separated by 5 ms. Recovery delay was 4 s. Uncertainties in peak intensities were estimated from one triplicate data point.
The HADDOCK 2.2 (Dominguez et al., 2003; van Zundert et al., 2016) program was used to calculate the structure of the BIN1 SH3/Tau peptide complex, based on experimental unambiguous and ambiguous restraints. The NMR structure of BIN1 SH3 extracted from a BIN1 SH3 complex (PDB 5i22) (Tossavainen et al., 2016) was used as a starting point for the docking. In addition, a structural model of Tau (213–229) peptide was built, using the Coot program (Emsley and Cowtan, 2004), starting from the Chikungunya virus nsP3 peptide (1728–1744) in interaction with BIN1 SH3 (PDB code 5i22). The docking was next driven under defined intermolecular unambiguous restraints from a NOESY spectrum and ambiguous restraints chosen as the residues involved in intermolecular NOEs. Intermolecular NOEs were identified between BIN1 SH3 residues 557, 558, 561, 587, and 588, and Tau (213–229) residues 228/229, 226, 223, 219, and 216, respectively (Supplementary Figure S2 and Supplementary Table S3). The docking algorithm includes three consecutive steps: (i) rigid body energy minimization (1000 generated structures) (ii) semi-flexible simulated annealing of the 200 lowest energy structures from step (i) and (iii) flexible explicit solvent refinement (200 structures, 8.0 -Å shell of TIP3P water molecules). All calculations were run through the WeNMR Web portal using the expert interface. A total of 196 final models were grouped into two clusters based on their interface-ligand RMSD, using a cutoff of 5 Å. The final overall score of each cluster (Supplementary Table S4) is based on the four highest HADDOCK scores of the models in that cluster. The best cluster, cluster 1, contained 190 structures (Supplementary Table S4), z score = −1.0). The molecular structure of the complex was viewed and represented using CHIMERA.
Phosphorylation of SUMO-Tau (210–240) was obtained by incubation with recombinant CDK2/CycA3 kinase (Welburn and Endicott, 2005) (molar ratio 1/100), for 3 h at 37°C, in the presence of 2 mM ATP, 2.5 mM MgCl2, 2 mM DTT, and protease inhibitors in 50 mM HEPES, pH 8.0. The incubation was repeated once after exchange into fresh buffer and upon new addition of kinase. The His-SUMO tag was removed after phosphorylation, as described here above for the unmodified peptide. Completion of phosphorylation was assessed by NMR 2D 1H-15N HSQC of the 15N-Tau (210–240) peptide and MALDI-TOF (Matrix Assisted Laser Desorption Ionization – Time of Flight) mass spectrometry. Mass analysis was performed in ion-positive reflector mode on an ABI Voyager DE-Pro MALDI-TOF mass spectrometer (Applied Biosystems), using as matrix a saturated solution of α-cyano-4-hydroxycinnamic acid in CH3CN:H2O:CF3COOH (50:50:0.1). Phosphorylated Tau (210–240) 1H-15N HSQC spectrum was recorded at 600 MHz, peptide concentration 0.5 mM, resolution 2048 points in the direct dimension and 256 points in the indirect dimension, spectral width 14 and 17 ppm, respectively, and 16 scans. Based on the relative intensities of the NMR resonance signals (Supplementary Figure S3), full modification was observed for residues T212, T231, and S235 while residue T217 was half-phosphorylated. Results from MS analysis were in agreement (Supplementary Figure S4). All the purification steps were performed at 4°C in the presence of a protease inhibitor cocktail (Sigma-Aldrich).
Titration of 70 μM 15N-BIN1 SH3 with increasing amount of unlabeled Tau (210–240) (from 35 to 840 μM) peptides was performed, monitoring the complex formation by the gradual 1H and15N chemical shift change of the resonances in 1H,15N HSQC, at pH 7.3 (50 mM phosphate buffer) and 20°C. The weighted average chemical shift differences were calculated as described by Garrett et al. (1997) - i.e., (ΔH2 + (ΔN×0.2)2)1/2, with ΔH and ΔN the chemical shift changes for 1H and 15N, respectively. Dissociation constants were obtained by fitting the chemical shift perturbation data to the following equation Δδobs = Δδmax(a + b + Kd−((a + b + Kd)2−4ab)1/2)/2a where Δδobs is the weighted average of the chemical shifts in the free and bound states and Δδmax is the maximal signal change upon saturation. Kd is the dissociation constant, a and b are the total peptide and BIN1 SH3 concentrations, respectively. Kd were averaged based on chemical shift perturbations of 7 distinct resonances.
We have previously shown direct interaction of the BIN1 SH3 (BIN1 SH3) with the PRD of Tau [PRD, Tau (165–245)] (Sottejeau et al., 2015). The interaction motif within Tau PRD was narrowed-down to a sequence located between amino acid residues 210–240. In this previous work, 1H-15N HSQC spectra were used for chemical shift perturbation analysis. As 1H-15N HSQC experiment correlates the resonances of directly bound 1H and 15N nuclei, the majority of the amino acid residues in the peptide was represented in this analysis, except proline residues, due to the lack of the backbone amide proton. However, Tau (210–240) has eight proline residues organized in two different canonical SH3 binding sites (Feng et al., 1994; Lim et al., 1994). One of class I, PPII.1, between residues 230 to 236, and another one of class II, PPII.2, between amino acid residues 216 and 221 (Figure 1A). To investigate the role of these proline residues in Tau (210–240)/BIN1 SH3 interaction, a specific NMR strategy was used, based on Hα-15N correlation, allowing sequential assignment of molecules rich in prolines (Kanelis et al., 2000; Ahuja et al., 2016). All the proline residues in Tau (210–240) were assigned, including their minor cis conformers (Figure 1B).
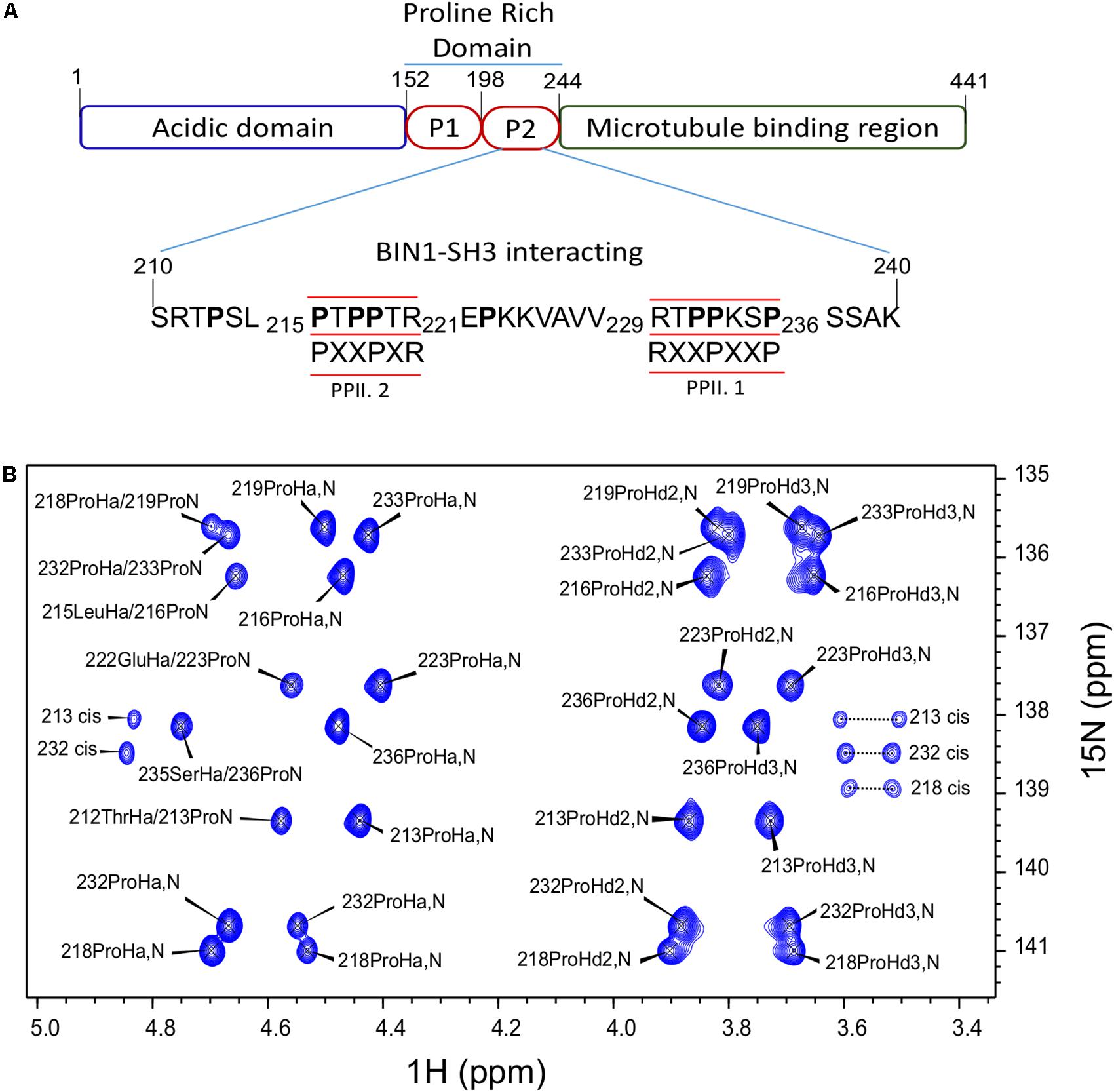
FIGURE 1. Assignment of Tau (210–240) proline residues. (A) Domain composition of the largest isoform of the neuronal Tau protein. P1 and P2 are the proline-rich regions. Tau (210–240) sequence is shown as part of the PRD. The proline residues are indicated in bold characters. The canonical SH3 class I and II binding motifs are underlined and the consensus motifs are given below. PPII.1 and PPII.2 are PPII helix motif class I and II, respectively. (B) Assigned 2D HA(CA)N spectrum of the prolines in Tau (210–240) peptide at 20°C.
After assignment of all the proline residues in Tau (210–240) fragment, we next looked at whether the two proline rich motifs of Tau (210–240) were involved in the interaction with BIN1 SH3 domain. Comparison of the 2D HA(CA)N spectra of Tau (210–240) in the absence and presence of an excess of BIN1 SH3 domain showed that all resonances corresponding to proline residues in the proline-rich motif PPII.2 disappeared in the presence of the BIN1 SH3 domain (Figure 2). Similarly, peaks corresponding to P223 at the C-terminal end of the PPII.2 were broadened beyond detection, whereas those of P213, in the N-terminal of PPII.2, were less affected and only showed minor chemical shift modifications. Resonances of proline residues in the proline-rich motif PPII.1 were barely affected, neither by broadening nor by chemical shift modification. This result clearly indicated that Tau (210–240) interacts with BIN1 SH3 domain using the PPII.2 proline-rich motif within its sequence.
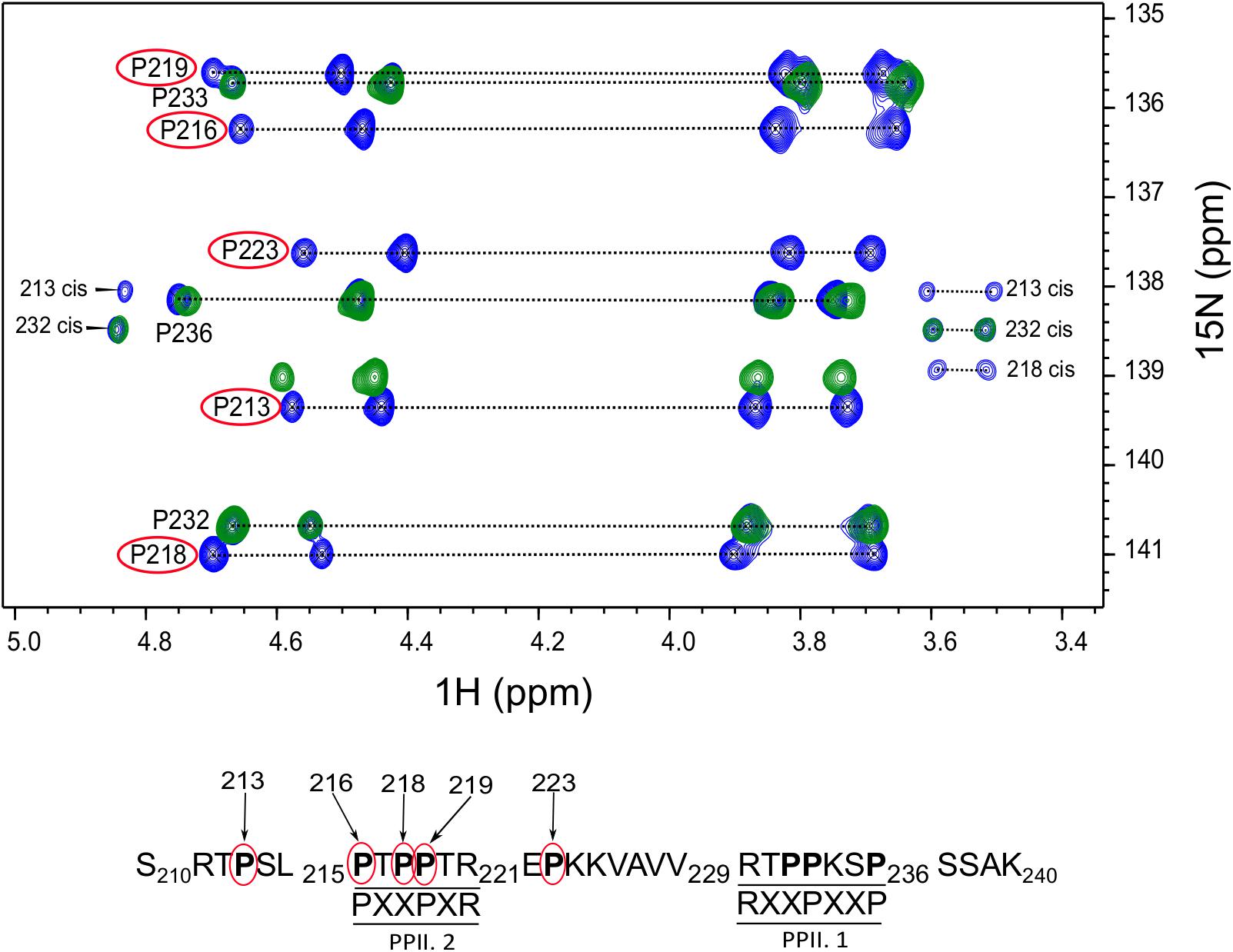
FIGURE 2. Critical proline residues involved in Tau binding to BIN1. The 2D HA(CA)N spectrum of Tau (210–240) peptide free or in the presence of BIN1 SH3 domain molar ratio 1:4 is shown in blue and green, respectively. The proline residues affected by the presence of BIN1 SH3 domain are indicated by red circles. In addition, the position of each affected proline residue within Tau (210–240) sequence is numbered in the sequence below the spectrum.
We next used these data as a starting point to obtain a structural model of BIN1 SH3 in complex with Tau (210–240) peptide. First, NMR dynamics experiments provided insights into the complex formation. 15N-relaxation measurements of 15N BIN1 SH3 showed homogenous R1 and R2 values, compatible with a compact structure (Supplementary Figure S1). A correlation time τc of 6.3 ± 0.3 ns was calculated, based on the average R1 and R2 values, as expected for a monomeric BIN1 SH3 domain in the NMR samples (Supplementary Figure S1). In the presence of the peptide, average R1 values were increased while average R2 values were decreased, corresponding to an increase in the molecular weight of the system. A correlation time τc of 9.7+/−0.4 ns was calculated for BIN1 SH3/Tau (210–240) complex, based on the average R1 and R2 values, compatible with a monomeric state of a 1:1 complex. NMR data were next used to build a model of the complex, using HADDOCK program. The docking was started from an NMR structure of BIN SH3 (Tossavainen et al., 2016) and a model of Tau (213–229) peptide. Starting from these two initial individual structures, the docking was conducted under restraints derived from the NMR experimental data (Supplementary Figure S2 and Supplementary Table S3). The four best structures resulting from the docking calculation, in the cluster of conformations showing the best statistics (cluster 1 in Supplementary Table S4), were chosen as being representative of BIN1 SH3/Tau (213–229) complex (Figures 3, 4). BIN1 SH3 accommodated the P216xxP219 consensus motif of Tau peptide (Figures 1, 2) into the canonical hydrophobic xP binding pocket of the SH3 domain (Figure 3). Specific hydrophobic interactions were observed between the side-chains of F588 on the BIN1 SH3 side and P216 on the Tau peptide, and between the side-chains of W562 and P219 (Figure 3A). In contrast, P218 side-chain pointed to the opposite direction with respect to P219 and was not interacting with BIN1 SH3 domain. Moreover, residues E556 and E557 in the n-Src loop of the specificity zone of BIN1 SH3 provided an additional anchoring point. Both E556 and E557 side-chains were oriented toward the Tau (213–229) peptide, in close proximity to K224 side-chain (Figure 3B). Mutation of K224 and K225 into alanines in Tau (210–240) peptide indeed resulted in a reduced binding affinity to the BIN1-SH3 domain, with a dissociation constant Kd of 44 ± 3 μM for Tau (210–240) peptide increasing by a factor of ten to 429 ± 94 μM for the mutated peptide (Supplementary Figure S3). Finally, at the C-terminus of the docked Tau (213–229) peptide, V228/V229 Hα protons were also in close contact with E557 backbone H-N, as observed by intermolecular NOEs (Supplementary Table S3). The position of Tau peptide on the surface of BIN1 SH3 showed a good match with the major chemical shift perturbations of BIN1 SH3 1H, 15N resonances on addition of Tau (210–240) peptide (Figures 4A,B), highlighting the β5 strand, the n-Src loop and in addition the RT loop (Figure 4B). Reporting the electrostatic potential on the BIN1 SH3 surface additionally showed positioning of the R221 and K224 side-chain on a negatively charged region (Figure 4C). The structure of the BIN1 SH3/Tau (213–229) peptide complex showed involvement in the binding of both the xP pocket and the specificity region of BIN1 SH3 (n-Src loop), mediated on the Tau side mainly by P216/P219 and R221/K224, respectively (Figure 4).
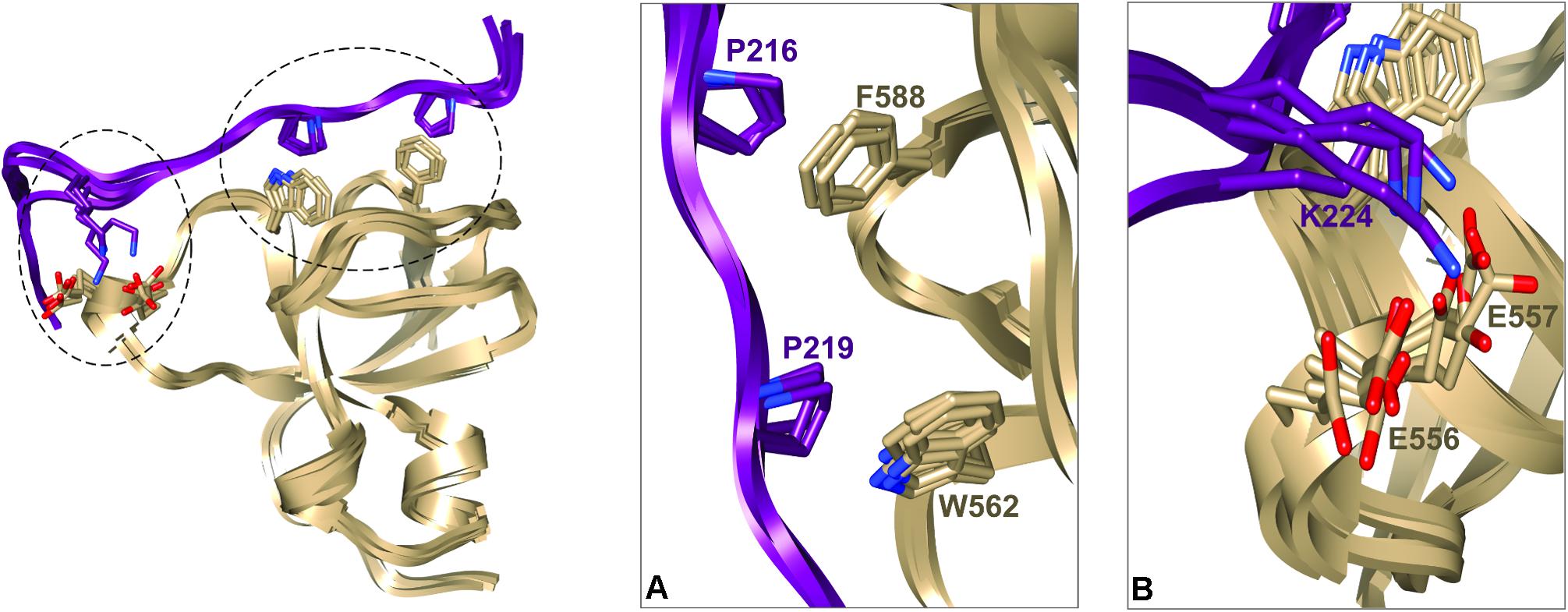
FIGURE 3. Representative structural models of BIN1 SH3/Tau (213–229) complex corresponding to the four best structures of HADDOCK calculation from cluster 1 (A). Ribbon representation of BIN1 SH3 (in gold)/Tau (213–229) (in violet). Side-chains of the main residues involved in the interaction are represented as sticks (B). Enlarged views of the complexes corresponding to the encircled regions in (A).
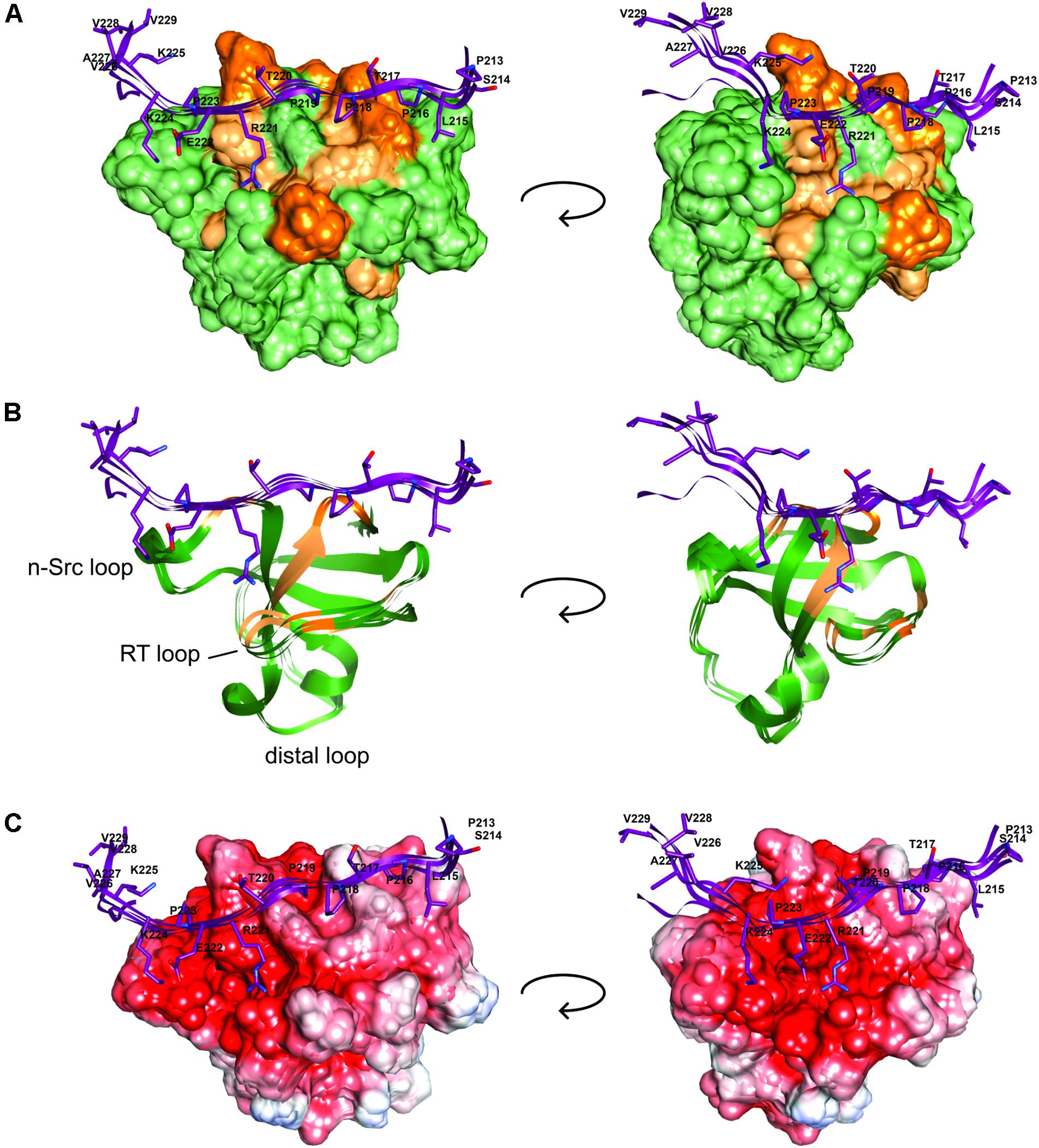
FIGURE 4. BIN1 SH3 interaction with Tau (213–229) (A). The solvent accessible molecular surface of BIN1 SH3 (in green) is highlighted with the major 1H,15N combined chemical shift perturbations observed upon addition of Tau (210–240) peptide to BIN1 SH3, dark orange > 0.3 ppm, light orange > 0.2 ppm (as in Malki et al., 2017, see Figure 6). (B) Same as A., in ribbon representation. (C) The electrostatic potential is colored on the molecular surface of BIN1 SH3 from negative values in red (-8 keV) to positive values in blue (+5 keV). Tau (213–229) is represented as ribbon on the surface (in violet), with the side-chains of Tau peptide residues shown as sticks.
We previously showed that BIN1 SH3 domain was engaged in an intramolecular interaction with BIN1 isoform 1 own CLAP domain (Malki et al., 2017). Tau (210–240) peptide was able to compete with this interaction, shifting the dynamic equilibrium from a closed to an open conformation. As NMR is sensitive to mobility, this equilibrium translated into detection of BIN1 SH3 domain signals only upon its release from the intramolecular interaction (Malki et al., 2017). We further addressed whether such equilibrium between open and closed states of BIN1 isoform 1 might be affected by phosphorylation of Tau (210–240). A 1H-15N HSQC spectrum of BIN1 isoform1 was thus acquired in the absence or presence of an excess of Tau (210–240) phosphorylated at residues T212, T217, T231, and S235 (Supplementary Figures S4), all proline-directed sites within this peptide. Addition of phosphorylated Tau (210–240) peptide to BIN1 isoform1 sample did not result in the same changes in the spectrum (Figure 5, compare A with B), although some weak signals from BIN1 SH3 were still visible. This indicated that Tau (210–240) phosphorylated at multiple sites was not able to compete with the intra-molecular interaction of BIN1 SH3 with the CLAP domain as efficiently as its unphosphorylated counterpart. Indeed, comparison of the affinity of the isolated BIN1 SH3 domain for the Tau peptide (210–240), unphosphorylated or not, showed an increase of the dissociation constant Kd, from 44 ± 3 μM to 256 ± 12 μM for the phosphorylated peptide (Figure 6 and Supplementary Figure S5). Consequently, in the presence of the phosphorylated Tau peptide (210–240), BIN1 SH3 domain remained immobilized by the intramolecular interaction with BIN1 isoform1 CLAP domain, mainly adopting the closed conformation.
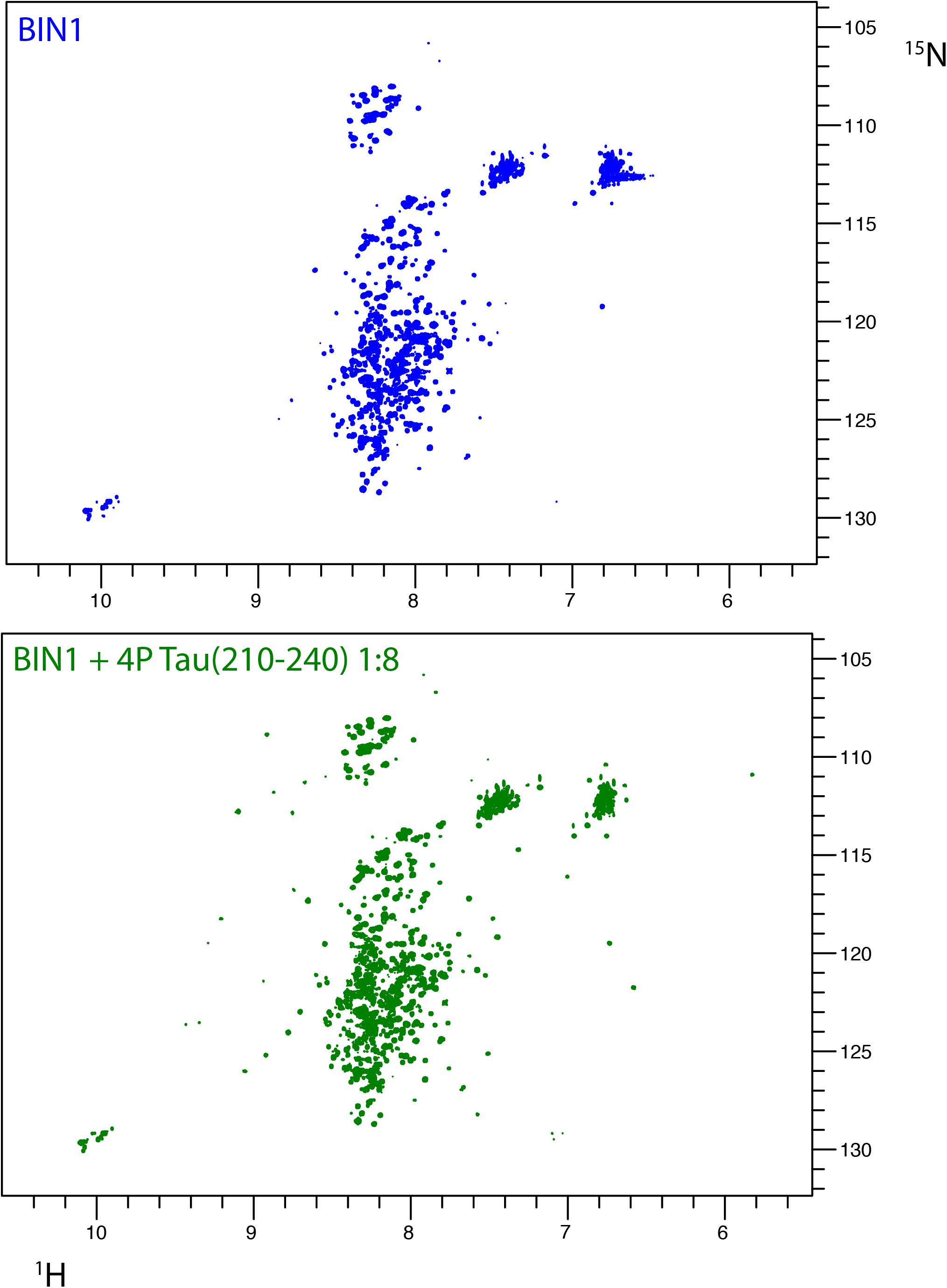
FIGURE 5. Interaction of BIN1 isoform 1 with Tau (210–240) and impact of multiple Tau phosphorylation on the interaction. (A) 1H-15N HSQC spectrum of BIN1 isoform 1 (112 KDa, homodimer). Because of the large molecular weight of BIN1, which lead to severe line broadening, its spectrum displayed, mostly in its central part, signal of low spectral dispersion, typical of disordered regions of the protein. (B) 1H-15N HSQC spectra of BIN1 isoform 1 in the presence of phospho-Tau (210–240), molar ratio 1:8. The signals corresponding to the isolated BIN1 SH3 domain were barely detected. Phospho-Tau (210–240) cannot compete with the intramolecular interaction of BIN1 SH3 with the CLAP domain.
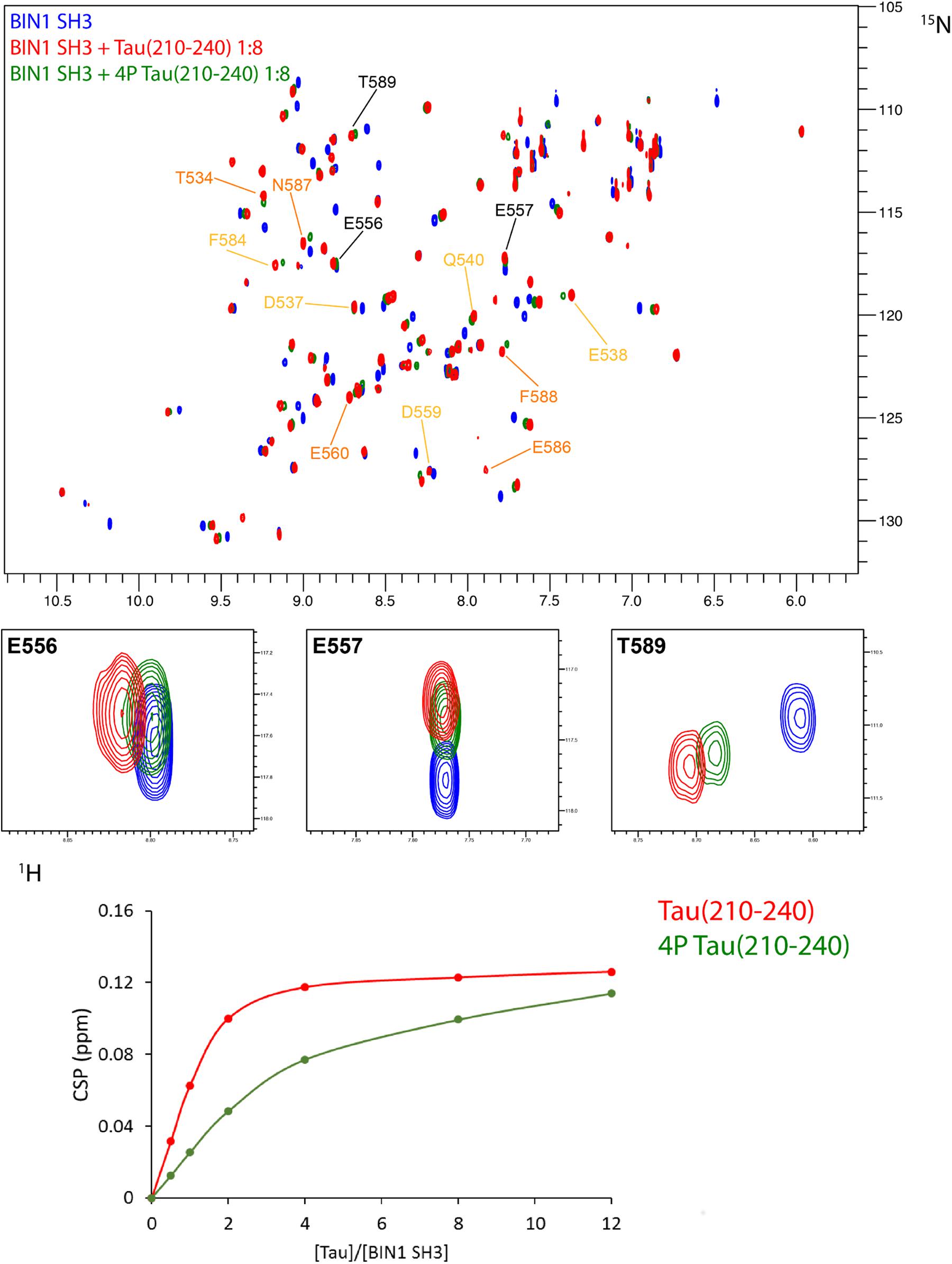
FIGURE 6. Affinity of BIN1 SH3 for Tau (210–240), phosphorylated, or not. (A) Superimposed 2D 1H-15N HSQC spectra of 70 μM 15N BIN1 SH3, free in solution (in blue) and with 8 molar amount of Tau (210–240) (overlaid in red) or phosphorylated Tau (210–240) (overlaid in green). Labeled resonances correspond to the major chemical shift perturbations, color coded as in Figure 4A. Enlarged regions of the superimposed spectra show 3 resonances involved in the interaction. (B) Gradual chemical shift perturbation of residue D537 1H, 15N resonance of BIN1 SH3 (70 μM) versus increasing amount of Tau (210–240) (red) or phosphorylated Tau (210–240) (green).
BIN1 gene was found as a genetic determinant of AD, allowing to address new patho-physiological hypothesis (Lambert et al., 2009; Seshadri et al., 2010). A first step in deciphering the role of BIN1 in AD dysfunctions was taken by the discovery of its direct interaction with Tau protein (Chapuis et al., 2013), a well-known actor of the disease. The specific function of the interaction of BIN1 with Tau, however, remained to date to be uncovered, and in this context emphasized the need to grasp its molecular detail. Here we first sought the atomic detail of the interaction of BIN1 SH3 with Tau. The BIN1 interaction motif within Tau PRD is a sequence located between amino acid residues 210–240, containing two potential SH3-binding motifs (Sottejeau et al., 2015; Malki et al., 2017). The binding site for the BIN1 SH3 within this sequence was found here to be specific for a canonical SH3 class II binding motif PXXPX+ (Feng et al., 1994; Lim et al., 1994) corresponding to P216TPPTR221. The identification was based on HA(CA)N spectra allowing for the detection of the resonances specific for the proline residues, which are absent in a classical 2D 1H,15N HSQC spectrum. Assignment of the Pro residues matched previous assignment of Pro residue resonances in peptides and fragments of Tau encompassing Tau (210–240) (Smet et al., 2004, 2005; Ahuja et al., 2016). Our analysis also confirmed that the proline residues mainly adopt the trans conformation (Smet et al., 2004, 2005; Ahuja et al., 2016). The BIN1 SH3/Tau (213-229) peptide complex structure further showed that the consensus motif is bound to the hydrophobic xP binding pocket of BIN1 SH3, in which P216 and P219 are in direct contact with the aromatic F588 and W562 residues on the domain side. The contact surface is increased through electrostatic interactions between the positively charged residues on the Tau peptide side, namely R221 and K224, and those negatively charged on the protein side within the n-Src loop, corresponding to E556 and E557. These negatively charged residues, located in the so-called specificity region of BIN SH3 (Pineda-Lucena et al., 2005), explain the importance of basic residues in the binding affinity. Further interactions involved residues V228 and V229 of Tau peptide, which appeared to be oriented toward the protein.
We have previously showed that BIN1 interaction with Tau is decreased by phosphorylation of Tau, of at least T231 (Sottejeau et al., 2015). In addition, a competition for BIN1 SH3 domain binding to Tau was shown by BIN1 Isoform 1 own CLAP domain, in an intramolecular mechanism of regulation (Malki et al., 2017). Addition of an excess of Tau to BIN1 is able to displace the intramolecular interaction, in profit of an intermolecular interaction with Tau proline-rich motif (Malki et al., 2017). These results demonstrated the complex regulation of the BIN1/Tau interaction and the need to understand all the key elements of this regulation mechanism. Here, we looked at both aspects of the regulation of this interaction: how phosphorylation of Tau (210–240) peptide on sites surrounding the proline-rich SH3-binding consensus motif (pT212, pT231, and pS235) or within this motif (pT217) would impact the competition for BIN1 SH3 domain binding. This phosphorylation corresponded to specific epitopes of Tau antibodies used to detect the pathological Tau protein, namely the AT180 epitope (pT231/pS235) (Goedert et al., 1994) and the AT100 epitope (pT212/pS214) (Zheng-Fischhofer et al., 1998).
pT231/pS235 residues affect the interaction by themselves, because full-length Tau phosphorylated on T231–S235, but not on T217, has a decreased affinity for BIN1 SH3 (Sottejeau et al., 2015). However, according to the structural model of BIN1 SH3/Tau (213–229) peptide (Figures 3, 4), T231-S235 residues did not make direct contact with BIN1 SH3 surface. Additional effects might thus be involved, and may be associated with the conformational modification of Tau induced by phosphorylation of T231 and S235 (Luna-Munoz et al., 2005; Sibille et al., 2012; Schwalbe et al., 2015). Alternatively, phosphorylation of T231 and S235 might act by a global electrostatic effect, partially neutralizing the K224-K225 basic motif that contacts the acidic BIN1 SH3 specificity region (Figure 4C). Substitution of K224 and K225 basic residues with neutral alanines indeed reduced the binding affinity by a factor of 10, confirming that K224 and/or K225 contribute significantly to the binding (Supplementary Figure S3). In addition, the structural model of BIN1 SH3/Tau (213–229) showed that T217 is accommodated by the proline-binding pocket of BIN1 SH3 (Figure 4). T217 phosphorylation in Tau (210–240) peptide may thus additionally weaken the interaction because of an unfavorable steric effect of the phosphate group.
Our results showed that conversely to Tau (210–240) peptide, phosphorylated Tau (210–240) peptide was not able to displace the intramolecular interaction of BIN1 SH3 with BIN1 CLAP domain, as its affinity for BIN1 SH3 was decreased compared to the unphosphorylated peptide. This confirmed our previous results that pT231 (together with other residues) weakens the BIN1/Tau interaction both in vitro and in primary neuronal cultures (Sottejeau et al., 2015). Here, a structure of the complex, based on NMR measurements, highlighted that T212, T231, and S235 did not make direct contact with BIN1 SH3 domain. T217, on the other hand, located in the bound proline-rich motif, but had however, its side-chain oriented in the opposite direction with respect to the domain surface (Figure 4). However, these combined phosphorylations decreased the affinity of BIN1 SH3 for the Tau peptide, either from their global electrostatic contribution, or by their impact on the peptide structure (Luna-Munoz et al., 2005; Sibille et al., 2012; Schwalbe et al., 2015).
Addition of Tau excess to BIN1 isoform 1 disfavors the intramolecular interaction whereas it favors an intermolecular interaction with a Tau proline-rich motif (Malki et al., 2017). Conversely, phosphorylation of Tau decreased the BIN1-SH3 domain’s affinity for Tau and pushed the dynamic equilibrium toward the intramolecular interaction, and might thus indirectly influence the availability of BIN1-CLAP domain. The proline-directed sites within Tau (210–240) might thus be the target of a transduction pathway; the activation of Pro-directed kinases would result in a decrease of BIN1’s affinity for Tau and (for BIN1 isoform 1) modify the availability of the CLAP domain.
AL and IM performed the biochemical experiments. AL, IM, and F-XC performed the NMR experiments. AL solved the structure of the complex. EB participated in NMR data treatment and HM in protein sample preparation. AL, IM, and IL wrote the manuscript. J-CL and IL conceived the study.
The NMR facilities were funded by the Nord Region Council, CNRS, Institut Pasteur de Lille, the European Union with the European Regional Development Fund (ERDF), the French Ministry of Research and the University of Lille and by the CTRL CPER co-funded by the European Union (ERDF), by the Hauts de France Regional Council (contrat no. 17003781), Métropole Européenne de Lille (Contract N°2016_ESR_05) and French State (Contract No. 2017-R3-CTRL-Phase 1). Financial support from the TGIR-RMN-THC Fr3050 CNRS and from FRABio (Univ. Lille, CNRS, FR-3688) for conducting the research is gratefully acknowledged. This study was supported by a grant from the LabEx (Laboratory of Excellence) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to Alzheimer’s disease), by the French government funding agency Agence Nationale de la Recherche ANR-15-CE16-0002 BinAlz and by the Alzheimer’s association (BFG-14-318355).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Nelly Morellet (ICSN, France) for assistance in using the 950 MHz spectrometer for NMR data acquisition, in Gif-sur-Yvette, France. The FP7 WeNMR (project# 261572) and H2020 West-Life (project# 675858) European e-Infrastructure projects are acknowledged for the use of their web portals.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00421/full#supplementary-material
AD, Alzheimer’s disease; BIN1, bridging integrator-1; BIN1 SH3, SH3 domain of BIN1; HSQC, heteronuclear single quantum correlation tau; NMR, nuclear magnetic resonance; NOESY, nuclear overhauser effect spectroscopy; PRD, proline-rich domain; pS, phosphoserine; pT, phosphothreonine; RMSD, root mean square deviation; SH3, Scr-homology-3.
Ahuja, P., Cantrelle, F. X., Huvent, I., Hanoulle, X., Lopez, J., Smet, C., et al. (2016). Proline conformation in a functional tau fragment. J. Mol. Biol. 428, 79–91. doi: 10.1016/j.jmb.2015.11.023
Baumann, K., Mandelkow, E. M., Biernat, J., Piwnica-Worms, H., and Mandelkow, E. (1993). Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 336, 417–424. doi: 10.1016/0014-5793(93)80849-P
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Calafate, S., Flavin, W., Verstreken, P., and Moechars, D. (2016). Loss of bin1 promotes the propagation of tau pathology. Cell Rep. 17, 931–940. doi: 10.1016/j.celrep.2016.09.063
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Cho, J. H., and Johnson, G. V. (2003). Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. differential impact on microtubule binding. J. Biol. Chem. 278, 187–193. doi: 10.1074/jbc.M206236200
David, C., McPherson, P. S., Mundigl, O., and de Camilli, P. (1996). A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc. Natl. Acad. Sci. U.S.A. 93, 331–335. doi: 10.1073/pnas.93.1.331
Dominguez, C., Boelens, R., and Bonvin, A. M. (2003). HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737. doi: 10.1021/ja026939x
Dourlen, P., Fernandez-Gomez, F. J., Dupont, C., Grenier-Boley, B., Bellenguez, C., Obriot, H., et al. (2017). Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of tau pathology. Mol. Psychiatry 22, 874–883. doi: 10.1038/mp.2016.59
Dräger, N. M., Nachman, E., Winterhoff, M., Brühmann, S., Shah, P., Katsinelos, T., et al. (2017). Bin1 directly remodels actin dynamics through its BAR domain. EMBO Rep. 18, 2051–2066. doi: 10.15252/embr.201744137
Drewes, G., Lichtenberg-Kraag, B., Doring, F., Mandelkow, E. M., Biernat, J., Goris, J., et al. (1992). Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 11, 2131–2138. doi: 10.1002/j.1460-2075.1992.tb05272.x
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr.D Biol. Crystallogr. 60, 2126–2132. doi: 10.1107/S0907444904019158
Feng, S., Chen, J. K., Yu, H., Simon, J. A., and Schreiber, S. L. (1994). Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science 266, 1241–1247. doi: 10.1126/science.7526465
Frost, A., Unger, V. M., and De Camilli, P. (2009). The BAR domain superfamily: membrane-molding macromolecules. Cell 137, 191–196. doi: 10.1016/j.cell.2009.04.010
Garrett, D. S., Seok, Y. J., Liao, D. I., Peterkofsky, A., Gronenborn, A. M., and Clore, G. M. (1997). Solution structure of the 30 kDa N-terminal domain of enzyme I of the escherichia coli phosphoenolpyruvate:sugar phosphotransferase system by multidimensional NMR. Biochemistry 36, 2517–2530. doi: 10.1021/bi962924y
Genin, E., Hannequin, D., Wallon, D., Sleegers, K., Hiltunen, M., Combarros, O., et al. (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907. doi: 10.1038/mp.2011.52
Goedert, M., Jakes, R., Crowther, R. A., Cohen, P., Vanmechelen, E., Vandermeeren, M., et al. (1994). Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: identification of phosphorylation sites in tau protein. Biochem. J. 301 (Pt 3), 871–877. doi: 10.1042/bj3010871
Holler, C. J., Davis, P. R., Beckett, T. L., Platt, T. L., Webb, R. L., Head, E., et al. (2014). Bridging Integrator 1 (BIN1) Protein expression increases in the Alzheimer’s Disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers Dis. 42, 1221–1227. doi: 10.3233/JAD-132450
Ishiguro, K., Shiratsuchi, A., Sato, S., Omori, A., Arioka, M., Kobayashi, S., et al. (1993). Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 325, 167–72. doi: 10.1016/0014-5793(93)81066-9
Kanelis, V., Donaldson, L., Muhandiram, D. R., Rotin, D., Forman-Kay, J. D., and Kay, L. E. (2000). Sequential assignment of proline-rich regions in proteins: application to modular binding domain complexes. J. Biomol. NMR 16, 253–259. doi: 10.1023/A:1008355012528
Kimura, T., Ishiguro, K., and Hisanaga, S. I. (2014). Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 7:65. doi: 10.3389/fnmol.2014.00065
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Landrieu, I., Leroy, A., Smet-Nocca, C., Huvent, I., Amniai, L., Hamdane, M., et al. (2010). NMR spectroscopy of the neuronal tau protein: normal function and implication in Alzheimer’s disease. Biochem. Soc. Trans. 38, 1006–1011. doi: 10.1042/BST0381006
Leroy, A., Landrieu, I., Huvent, I., Legrand, D., Codeville, B., Wieruszeski, J. M., et al. (2010). Spectroscopic studies of GSK3{beta} phosphorylation of the neuronal tau protein and its interaction with the N-terminal domain of apolipoprotein E. J. Biol. Chem. 285, 33435–33444.
Lim, W. A., Richards, F. M., and Fox, R. O. (1994). Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372, 375–379. doi: 10.1038/372375a0
Lippens, G., Amniai, L., Wieruszeski, J. M., Sillen, A., Leroy, A., and Landrieu, I. (2012a). Towards understanding the phosphorylation code of tau. Biochem. Soc. Trans. 40, 698–703. doi: 10.1042/BST20120050
Lippens, G., Landrieu, I., Amniai, L., Qi, H., Leroy, A., and Smet-Nocca, C. (2012b). The phosphorylation code of the neuronal protein Tau. FEBS J. 279:51.
Luna-Munoz, J., Garcia-Sierra, F., Falcon, V., Menendez, I., Chavez-Macias, L., and Mena, R. (2005). Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J. Alzheimers Dis. 8, 29–41. doi: 10.3233/JAD-2005-8104
Luna-Vargas, M. P., Christodoulou, E., Alfieri, A., van Dijk, W. J., Stadnik, M., Hibbert, R. G., et al. (2011). Enabling high-throughput ligation-independent cloning and protein expression for the family of ubiquitin specific proteases. J. Struct. Biol. 175, 113–119. doi: 10.1016/j.jsb.2011.03.017
Malki, I., Cantrelle, F.-X., Sottejeau, Y., Lippens, G., Lambert, J.-C., and Landrieu, I. (2017). Regulation of the interaction between the neuronal BIN1 isoform 1 and Tau proteins - role of the SH3 domain. FEBS J. 284, 3218–3229. doi: 10.1111/febs.14185
Pineda-Lucena, A., Ho, C. S. W., Mao, D. Y. L., Sheng, Y., Laister, R. C., Muhandiram, R., et al. (2005). A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J. Mol. Biol. 351, 182–194. doi: 10.1016/j.jmb.2005.05.046
Piotto, M., Saudek, V., and Sklenár, V. (1992). Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665. doi: 10.1007/BF02192855
Prokic, I., Cowling, B. S., and Laporte, J. (2014). Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 92, 453–463. doi: 10.1007/s00109-014-1138-1.
Qi, H., Prabakaran, S., Cantrelle, F.-X., Chambraud, B., Gunawardena, J., Lippens, G., et al. (2016). Characterization of neuronal tau protein as a target of extracellular signal-regulated kinase. J. Biol. Chem. 291, 7742–7753. doi: 10.1074/jbc.M115.700914
Schwalbe, M., Kadavath, H., Biernat, J., Ozenne, V., Blackledge, M., Mandelkow, E., et al. (2015). Structural impact of tau phosphorylation at threonine 231. Structure 23, 1448–1458. doi: 10.1016/j.str.2015.06.002
Seshadri, S., Fitzpatrick, A. L., Ikram, M. A., DeStefano, A. L., Gudnason, V., Boada, M., et al. (2010). Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840. doi: 10.1001/jama.2010.574
Sibille, N., Huvent, I., Fauquant, C., Verdegem, D., Amniai, L., Leroy, A., et al. (2012). Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered tau protein. Proteins 80, 454–462. doi: 10.1002/prot.23210
Smet, C., Sambo, A. V., Wieruszeski, J. M., Leroy, A., Landrieu, I., Buee, L., et al. (2004). The peptidyl prolyl cis/trans-isomerase Pin1 recognizes the phospho-Thr212-Pro213 site on Tau. Biochemistry 43, 2032–2040. doi: 10.1021/bi035479x
Smet, C., Wieruszeski, J. M., Buee, L., Landrieu, I., and Lippens, G. (2005). Regulation of Pin1 peptidyl-prolyl cis/trans isomerase activity by its WW binding module on a multi-phosphorylated peptide of Tau protein. FEBS Lett. 579, 4159–4164. doi: 10.1016/j.febslet.2005.06.048
Sottejeau, Y., Bretteville, A., Cantrelle, F. X., Malmanche, N., Demiaute, F., Mendes, T., et al. (2015). Tau phosphorylation regulates the interaction between BIN1’s SH3 domain and Tau’s proline-rich domain. Acta Neuropathol. Commun. 3:58. doi: 10.1186/s40478-015-0237238
Takei, K., Slepnev, V. I., Haucke, V., and De Camilli, P. (1999). Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat. Cell Biol. 1, 33–39. doi: 10.1038/9004
Tossavainen, H., Aitio, O., Hellman, M., Saksela, K., and Permi, P. (2016). Structural basis of the high affinity interaction between the alphavirus nonstructural protein-3 (nsP3) and the SH3 domain of amphiphysin-2. J. Biol. Chem. 291, 16307–16317. doi: 10.1074/jbc.M116.732412
Ubelmann, F., Burrinha, T., Salavessa, L., Gomes, R., Ferreira, C., Moreno, N., et al. (2017). Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 18, 102–122. doi: 10.15252/embr.201642738
van Zundert, G. C. P., Rodrigues, J. P. G. L. M., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., et al. (2016). The haddock2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725. doi: 10.1016/j.jmb.2015.09.014
Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., et al. (2005). The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696. doi: 10.1002/prot.20449
Wang, A. C., Grzesiek, S., Tschudin, R., Lodi, P. J., and Bax, A. (1995). Sequential backbone assignment of isotopically enriched proteins in D2O by deuterium-decoupled HA(CA)N and HA(CACO)N. J. Biomol. NMR 5, 376–382. doi: 10.1007/BF00182281
Welburn, J., and Endicott, J. (2005). Methods for preparation of proteins and protein complexes that regulate the eukaryotic cell cycle for structural studies. Methods Mol. Biol. 296, 219–235.
Zheng-Fischhofer, Q., Biernat, J., Mandelkow, E. M., Illenberger, S., Godemann, R., and Mandelkow, E. (1998). Sequential phosphorylation of tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 252, 542–552. doi: 10.1046/j.1432-1327.1998.2520542.x
Keywords: protein-protein interaction, nuclear magnetic resonance spectroscopy, Alzheimer’s disease, bridging integrator-1 BIN1, SH3 domain, tau, phosphorylation
Citation: Lasorsa A, Malki I, Cantrelle F-X, Merzougui H, Boll E, Lambert J-C and Landrieu I (2018) Structural Basis of Tau Interaction With BIN1 and Regulation by Tau Phosphorylation. Front. Mol. Neurosci. 11:421. doi: 10.3389/fnmol.2018.00421
Received: 29 June 2018; Accepted: 26 October 2018;
Published: 14 November 2018.
Edited by:
Abdul R. Asif, Georg-August-Universität Göttingen, GermanyReviewed by:
Charles Robert Harrington, University of Aberdeen, United KingdomCopyright © 2018 Lasorsa, Malki, Cantrelle, Merzougui, Boll, Lambert and Landrieu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Landrieu, aXNhYmVsbGUubGFuZHJpZXVAdW5pdi1saWxsZS5mcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.