 Saori Inafuku
Saori Inafuku Garrett Klokman
Garrett Klokman Kip M. Connor
Kip M. Connor
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 17 August 2018
Sec. Molecular Signalling and Pathways
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00278
Ischemia reperfusion (IR) injury induces retinal cell death and contributes to visual impairment. Previous studies suggest that the complement cascade plays a key role in IR injury in several systemic diseases. However, the role of the complement pathway in the ischemic retina has not been investigated. The aim of this study is to determine if the alternative complement cascade plays a role in retinal IR injury, and identify which components of the pathway mediate retinal degeneration in response to IR injury. To accomplish this, we utilized the mouse model of retinal IR injury, wherein the intraocular pressure (IOP) is elevated for 45 min, collapsing the retinal blood vessels and inducing retinal ischemia, followed by IOP normalization and subsequent reperfusion. We found that mRNA expression of complement inhibitors complement receptor 1-related gene/protein-y (Crry), Cd55 and Cd59a was down-regulated after IR. Moreover, genetic deletion of complement component 3 (C3−/−) and complement factor b (Fb−/−) decreased IR-induced retinal apoptosis. Because vascular dysfunction is central to IR injury, we also assessed the role of complement in a model of shear stress. In human retinal endothelial cells (HRECs), shear stress up-regulated complement inhibitors Cd46, Cd55, and Cd59, and suppressed complement-mediated cell death, indicating that a lack of vascular flow, commonly observed in IR injury, allows for complement mediated attack of the retinal vasculature. These results suggested that in retinal IR injury, the alternative complement system is activated by suppression of complement inhibitors, leading to vascular dysfunction and neuronal cell death.
Ischemia reperfusion (IR) injury is a complex pathophysiological phenomenon that is initiated by the loss of blood flow to a tissue and resultant ischemia followed by the subsequent return of blood flow, which results in oxidative stress and downstream cytotoxic inflammatory effects. Retinal ischemia is a major cause of damage in ocular diseases including glaucoma, central retinal vein occlusion and diabetic retinopathy (Osborne et al., 2004; Country, 2017). Current therapeutic approaches for IR injury focus on returning retinal blood flow through the use of anticoagulants, vasodilators, and laser treatment, or by alleviating oxidative stress using free radical scavengers (Osborne et al., 2004). Currently, no clinically available therapeutic modalities are focused on reperfusion-associated inflammatory damage, a major contributor to IR injury. In order to develop these important approaches to treatment, it is essential to identify which inflammatory mediators contribute to retinal IR-induced inflammatory damage.
Ocular immune activity, including activation of the complement cascade, is dramatically increased in multiple forms of ocular IR injury (Ahmed et al., 2004; Kuehn et al., 2006). The complement system is comprised of a complex network of proteins that work together to induce an inflammatory response that signals for removal of non-functional self-cells and non-self-cells. There are three complement pathways (classical, lectin, and alternative) that activate the central component of the complement system, C3, which subsequently activates a final common pathway facilitating cellular clearance (Walport, 2001b; Danese et al., 2007; Ricklin et al., 2010). Evolutionarily, the alternative complement pathway is the oldest of the complement activation pathways, so is broadly conserved (Ricklin et al., 2010). The alternative complement pathway is a key mediator of endogenous immune surveillance and tissue homeostasis. To monitor tissue homeostasis, the alternative complement pathway remains in a constant state of low-level activation, which allows for continuous probing of cells. In contrast, the classical and lectin pathways do not maintain this low-level activation (Pangburn and Müller-Eberhard, 1980; Pangburn et al., 1981; Bexborn et al., 2008; Ricklin et al., 2010).
Activation of the alternative complement pathway occurs through the spontaneous hydrolysis of an internal thioester bond within C3 (Harboe and Mollnes, 2008). Further continuation of the pathway is allowed only by complement factor b (Fb), which binds to C3b deposited in the membrane of a target cell (Walport, 2001a,b). These components form the C3 convertase enzyme, promoting the cleavage of C3 into C3a and C3b, creating a positive feedback activation loop that is down-regulated by complement inhibitors such as Crry/CD46, Cd55, and Cd59 (Hamilton et al., 1990; Rollins and Sims, 1990; Harris et al., 1999).
Soluble and cell-bound regulators of complement help to protect healthy host tissue from self-recognition and serve to prevent activation of a complement response (Song et al., 1996; Song, 2004; Ricklin et al., 2010). However, damaged or diseased host cells may down-regulate membrane bound inhibitors of complement, allowing targeted clearance (Ricklin et al., 2010). An imbalance between complement recognition and initiation on healthy host cells can lead to unregulated complement activation and subsequent cellular damage (Ricklin et al., 2010). Thus, activation of complement not only helps defend the host against pathogens, but also has the potential to affect self-tissues in a both beneficial (protective autoimmunity) and detrimental manner (autoimmunity) (Markiewski and Lambris, 2007; Ricklin et al., 2010).
We previously identified the alternative complement pathway as an important regulator of ocular health in proliferative retinopathy and retinal detachment (Sweigard et al., 2014, 2015; Kim et al., 2016). We found that within the ocular microenvironment, the alternative complement pathway maintains low levels of constitutive activity to ensure the intermittent probing of host cells. In normal conditions, host cells normally express endogenous membrane inhibitors of complement (e.g., Cd55) to protect against targeting by the complement system, these inhibitors are down-regulated in response to host cell injury allowing for their targeted removal by the complement system.
In this study, we sought to investigate the role of the alternative complement system in IR injury-induced neuronal damage and vascular dysfunction associated with IR injury. The role of the complement cascade in IR injury has been investigated in many tissues including the kidney (Zhou et al., 2000; de Vries et al., 2003), intestine (Austen et al., 1999; Satyam et al., 2017), lung (Hu et al., 2014), and heart (De Hoog et al., 2014; Chun et al., 2017). These studies suggest complement pathway inhibition as a putative therapeutic approach to alleviate IR-induced cell death and injury. However, the role of the alternative complement pathway in retinal IR injury remains incompletely understood. A previous study identified that activation of the core complement component 3 (C3), was increased following IR injury, and that deletion of C3 alleviated retinal damage and neuron loss after IR injury (Kuehn et al., 2008). This study was important in identifying a role for the complement system in neuronal IR injury, but many elements are as yet unknown, including the role of the alternative complement pathway and how specific compliment inhibitors are regulated in response to IR injury.
Ischemia is categorized by a lack of blood flow to tissue associated with vascular dysfunction (Zhao et al., 2014). Most studies of ischemic disease have focused upon the ramifications of decreased oxygen and nutrient availability. However, blood flow also causes shear stress, which is the frictional force exerted on vessel wall endothelial cells. Physiologic levels of shear stress are important for normal endothelial cell function (dela Paz and D'Amore, 2009). Shear regulates the Kruppel Like Factor 2 (KLF-2) transcription factor, which regulates molecules important to endothelial cell homeostasis including vascular endothelial growth factor (VEGF), endothelial Nitric Oxide Synthase (eNOS), and CD59 (dela Paz et al., 2012). Thus, given the role of vascular dysfunction associated with IR injury and complement's role in targeting injured self-cells we were interested in investigating the role decreased shear stress had on complement inhibitor expression in retinal endothelial cells. The down-regulation of complement inhibitors, occurring as a result of decreased shear stress in IR injury, could contribute to vascular damage and dysfunction through over activation of the complement system.
All animal procedures adhered to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. The Animal Care and Use Committee of Massachusetts Eye and Ear Infirmary approved the protocol for the experiments outlined herein. C57BL6 mice (stock no. 000664) at 8 week of age were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Fb−/− and C3−/− mice were kind gifts of Dr. John Lambris (University of Pennsylvania, Philadelphia, PA, USA). Breeding colonies were maintained at the Massachusetts Eye and Ear Infirmary (MEEI) animal facility. All animals used were between the ages of 8 and 9 weeks for all studies performed.
Mice were anesthetized with Avertin (T4, 840-2; 2,2,2 tribromoethanol; Sigma- Aldrich, St. Louis, MO, USA, in isoamyl alcohol, A730-1; ThermoScientific, Waltham, MA, USA), and deep anesthesia was confirmed by a toe pinch test. Pupils were dilated with 1% tropicamide, and topical anesthesia (1 drop of proparacaine hydrochloride) was applied to cornea. The anterior chamber of the left eye was cannulated with a 30-gauge needle connected to a line infusing sterile saline. The IOP was raised by elevating the saline reservoir to cause ischemia. After 45 min of ischemia, the needle was withdrawn to allow reperfusion. IOP was measured 0, 15, 30, and 45 min after induction of ischemia, before and after the surgery using a TonoLab tonometer (icare, Vantaa, Finland). For sham surgery, a 30-gauge needle was inserted into the anterior chamber without elevating the IOP. Mice were kept on a heating pad until fully awake, as confirmed by upright mobility. Mice were euthanized at various time points after IR, and their retinas were prepared as described below.
Fundus color video and FA during the IR surgery were taken using the Micron III Retinal Imaging Microscope (Phoenix Research Laboratories, Pleasanton, CA, USA). For FA, 0.1 ml of 2% fluorescein sodium (Akorn, Lake Forest, IL, USA) was injected intraperitoneally. Images were taken by attaching the cornea covered with 2.5% Goniovisc (HUB Pharmaceuticals, Rancho Cucamonga, CA, USA) to the Micron camera lens using StreamPix software (Phoenix Research Laboratories, Pleasanton, CA, USA).
Immediately after enucleation, mouse eyes were placed in optimum cutting temperature (OCT) compound (Tissue Tek, 4583, Torrance, CA, USA), and quickly frozen by submerging in a beaker of isopropanol chilled by dry ice. All eyes were cut into 10 μm sections with four sections per eye.
TUNEL was performed using the ApopTag Fluorescein In Situ Apoptosis Kit (S7110; Millipore, Billerica, MA, USA) following the manufacturers instructions. The sections were coverslipped with 4′,6-diamidino-2-phenylindole (DAPI) containing medium (H-1200; Vector Laboratories, CA, USA). All images were obtained using an AxioVision microscope (Zeiss, Chester, VA, USA), and the TUNEL Cell Counter plugin in Fiji image analysis software was used to automatically calculate the area and number of TUNEL-positive cells. When using the TUNEL Cell Counter plugin, the retina area was selected manually and the threshold sensitivity was changed to high in all the images, while all other settings remained unchanged. Eight images were taken in the midperiphery of each retina using a 20X objective lens.
Mice were euthanized on day 7 following IR or sham surgery, and eyes were enucleated. All eyes were fixed in 4% paraformaldehyde and paraffin embedded. Sections (6-μm thick) were cut parallel to the maximal circumference of the eye ball through the optic nerve and stained with hematoxylin and eosin (H&E). Inner nuclear layer (INL) thickness was measured in eight areas within 200–500 μm from the optic nerve, and the mean value was calculated.
Human retinal endothelial cells (HRECs) were purchased from Cell Sytems, Inc. (ACBRI 181; Kirkland, WA, USA) and grown in EGM-2 Growth Medium with SingleQuots (Bulletkit CC-3162; Lonza, Basel, Switzerland) supplemented with 1% L-glutamine and penicillin-streptomycin. They were grown to 90% confluence in T75s coated with 0.2% gelatin under the following incubator conditions: 5% CO2, 37°C, and 95% humidity. HRECs used for experiments were from passage 7–9.
Cells were seeded into three wells of a six-well plate at a density of 50,000 cells/well. For each experiment, one plate was used for each magnitude of shear stress. Cells were synchronized by culturing in starvation medium [EBM-2 basal medium (CC-3156; Lonza), 5% calf serum, 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), L-glutamine, and penicillin-streptomycin] for 24 h. After 24 h, fresh starvation medium was added and plates were exposed to orbital shear stress for 24 h using orbital shakers (Orbi-Shaker JR BT300; Benchmark Scientific, Sayreville, NJ, USA) set to either ~5 (150 rpm) or ~10 (240 rpm) dynes/cm2 inside an incubator. Cells of the same passage were placed in the same incubator as the static control. Shear stress was approximated using the formula , where α is the radius of orbital rotation (0.95 cm), ρ is the density of the medium (1.0 g/ml), η is the viscosity of the medium poise (7.5 × 10−3 dynes·s/cm2), and f is the frequency of rotation (rotations/second) (Dardik et al., 2005; dela Paz et al., 2012).
HRECs were cultured in starvation medium supplemented with 1% mouse serum, while being exposed to shear stress (0, 5, or 10 dynes/cm2) for 24 h. The percentage of dead cells was determined using the ReadyProbes Cell Viability Imaging Kit (ThermoFisher Scientific, Waltham, MA, USA). Three representative images of the peripheral area were taken per well using Zeiss Zen imaging software. Without any post processing, Fiji image analysis software was used to automatically count the number of dead cells (green) and total cells (blue) using the particle analyzer tool. The number of dead cells and total cells of the three images taken for each well were averaged to calculate the percent cell death per well.
For retinal gene expression analysis, eyes were collected 6 h after IR surgery or sham surgery, and dissected retinas were flash frozen in liquid nitrogen. For HRECs, cells were treated with trypsin, pelleted by centrifugation, and flash frozen. RNA was collected using the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturers instructions. Total RNA was measured using the NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific). cDNA was normalized prior to reverse transcription with SuperScript III and random hexamer primers (Invitrogen, Carlsbad, CA, USA).
RT-PCR was performed using 1 μl of cDNA and a StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). Primers to Crry (Mm00787529_s1, ThermoFischer Scientific), Cd55 (Mm00438377_m1, ThermoFischer Scientific), Cd59a (Mm00483149_m1, ThermoFischer Scientific) were used for mouse retinas, and primers to Cd46 (Hs00611257_m1, ThermoFischer Scientific), Cd55 (Hs00892618_m1, ThermoFischer Scientific), and Cd59 (Hs00174141_m1; ThermoFischer Scientific) were used for HRECs with TaqMan Universal PCR Master Mix (4304437, ThermoFischer Scientific). All data was normalized to β-actin.
Data were analyzed using one-way ANOVA. Results are expressed as mean ± SEM. Significance was set at P < 0.05.
For the retinal IR model, we applied a widely-used method, which causes retinal ischemia by increasing the intraocular pressure (IOP) above the ocular perfusion pressure (Zhi et al., 2012, 2015; He et al., 2013). In this model, the anterior chamber, located directly behind the cornea, is cannulated with a needle connected to a sterile saline reservoir to increase intraocular pressure and collapse retinal blood vessels, inducing retinal ischemia. The blood supply deficiency results in oxygen and nutrient deprivation of the retina, causing retinal cell death.
To confirm the obstruction of the blood flow by IR surgery, we recorded color fundus videos and fluorescein angiography (FA) videos during the IR operation (Supplementary Video 1). Immediately after the elevation of the IOP, constriction of vessels and whitening of the whole retina were observed in the color fundus video (Figure 1A and Supplementary Video 1). Utilizing FA, remarkable slowing of blood flow and a lack of choroidal filling were observed (Figure 1B and Supplementary Video 2). When the needle was removed from the anterior chamber, blood flowed in from the central retinal artery instantaneously, allowing retinal vessels to revert to their normal width and the retina to regain its original color (Figure 1C and Supplementary Video 3). FA demonstrated that blood flow reverted to a normal velocity, and that choroidal filling was restored (Figure 1D and Supplementary Video 4). We next assessed IOP during the IR operation. IOP was measured before the IR surgery, 0, 15, 30, and 45 min after the onset of IR, and immediately after the removal of the needle. Mean IOP before the surgery (13 ± 0.3 mmHg) was within normal range. By the onset of the IR, mean IOP increased to 90 ± 1 mmHg and remained over 80 mmHg during the surgery (84 ± 1mmHg at 15 min, 82 ± 1 mmHg at 30 min, and 81 ± 1 mmHg at 45 min) (Figure 1E). IOP decreased immediately upon removal of the needle from the anterior chamber. There was no significant difference in IOP between male and female animals at all time points (Supplementary Images 1A,B). Taken together, these results indicate that the IR model impaired blood supply from both choroidal and retinal circulation, and that ischemia was maintained during the 45-min surgery.
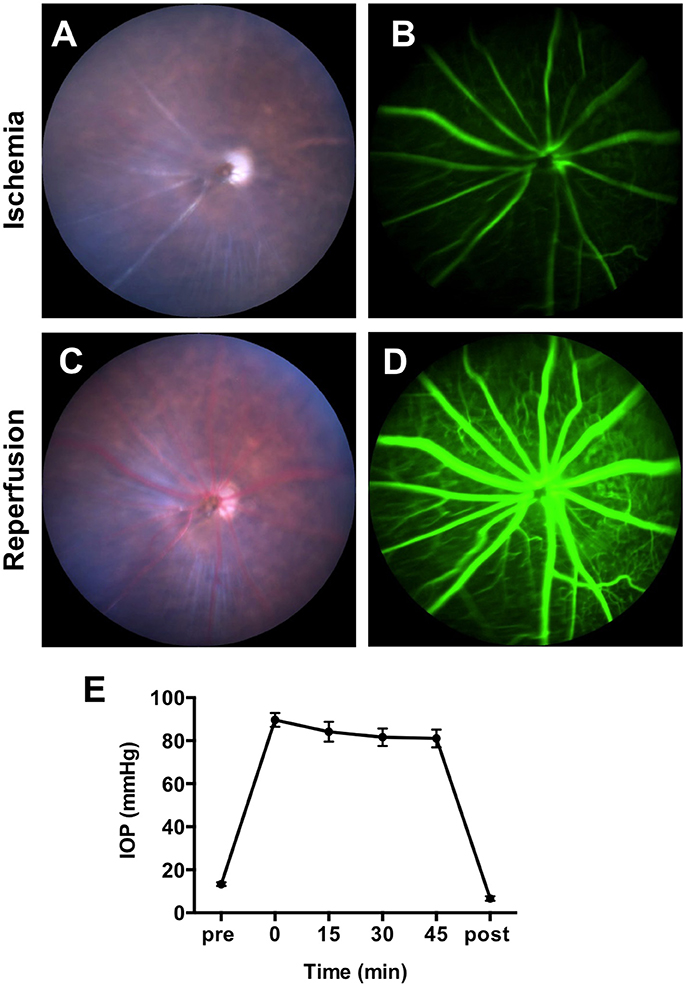
Figure 1. Obstruction of blood flow and the intraocular pressure (IOP) changes during IR surgery. Representative images of fundus color video (A) and fluorescein angiography (B) during the ischemia phase of IR surgery. Representative images of fundus color video (C) and fluorescein angiography (D) during the reperfusion phase of IR surgery. (E) IOP was measured before the IR surgery, 0, 15, 30, and 45 min after the initiation of the ischemia phase and immediately after the reperfusion. N = 12 eyes/time point. Data are presented as the mean ± SEM.
In the mouse model of IR injury the inner nuclear layer (INL) of the retina is more prone to reproducible irreversible cell loss caused by IR injury, thus we sought to assess cell death in the INL for our studies (Hughes, 1991). To assess changes in INL cell death following acute ocular hypertension, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of retinas at 3, 6, 12, 24, 48, and 72 h after reperfusion. The number of apoptotic cells peaked 24 h after reperfusion (2976 ± 252 cells/mm2), significantly higher than other time points (Figures 2A,B). There was no difference in INL TUNEL positivity between male and female mice at all the time points (Supplementary Image 2). Based on this result, we analyzed cell death 24 h after reperfusion in subsequent mechanistic studies.
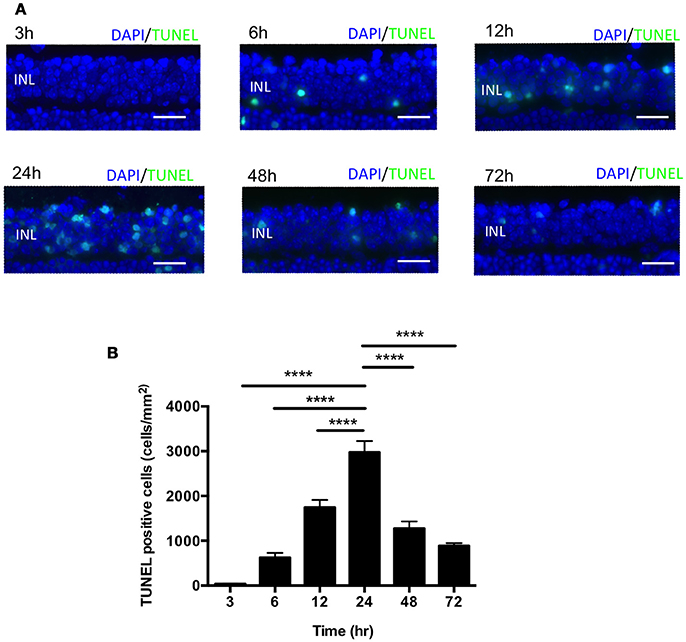
Figure 2. IR induced apoptosis peaked 24 h after reperfusion. (A) Representative images of TUNEL labeling of retinal cross-sections at 3, 6, 12, 24, 48, and 72 h after IR injury co-labeled with DAPI (blue) and TUNEL (green). Scale bars = 20 μm. INL, inner nuclear layer. (B) Quantification of TUNEL-positive cells in the INL at 3, 6, 12, 24, 48, and 72 h after IR injury. N = 8–10 eyes/time point. Data are presented as the mean ± SEM. ****P < 0.0001 vs. 24 h.
The complement system acts as an immune surveillance system to discriminate among healthy host tissue, cellular debris, apoptotic cells, varying its response in reaction to cellular injury (Ricklin et al., 2010). Generally, normal healthy host cells endogenously express membrane-bound complement inhibitors to prevent self-recognition and targeting by the complement system, thereby protecting themselves from complement-mediated cell death (Powers et al., 2006; Swirski et al., 2009; Rice et al., 2010). However, damaged host cells down-regulate complement inhibitors, allowing for targeted clearance of damaged cells (Powers et al., 2006; Pacher and Szabo, 2008; Bamboat et al., 2010). To investigate whether the complement pathway is involved in IR injury-induced retinal cell death, we evaluated the expression of complement inhibitors complement receptor 1-related gene/protein-y (Crry), Cd55 and Cd59a in C57BL6 mice, Fb−/− mice and C3−/− mice at 6 h after IR surgery or sham surgery. The complement inhibitors were significantly decreased in the retina after IR surgery in all three strains (Figures 3A–F). This result suggests that the complement cascade may be activated by suppression of complement inhibitors in IR injury.
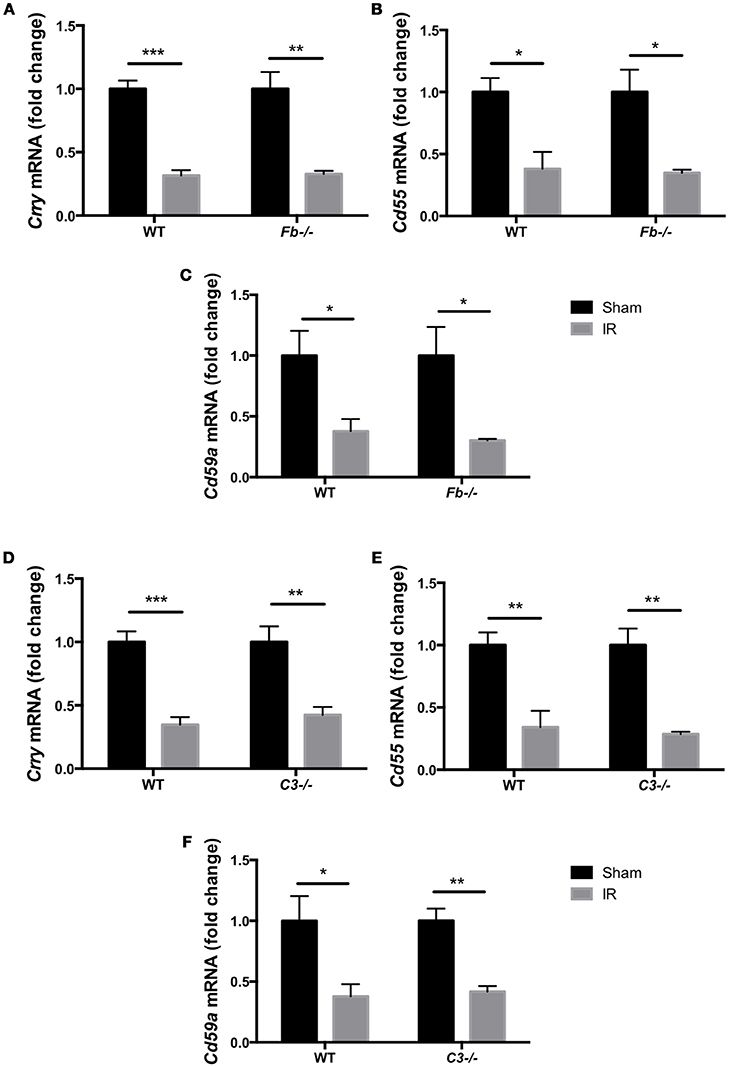
Figure 3. Complement inhibitors gene expression was down-regulated in the IR retina. The mRNA levels of complement inhibitors, Crry (A,D), Cd55 (B,E), and Cd59a (C,F) in the retina of wild type control mice (WT), complement factor b-deficient mice (Fb−/−) and complement component 3-deficient mice (C3−/−) at 6 h after IR injury were quantified by RT-PCR, and were normalized to sham surgery. N = 4 eyes/group. Data are presented as the mean ± SEM. *P < 0.05 vs. sham, **P < 0.01 vs. sham, ***P < 0.001 vs. sham.
The complement system is comprised of three pathways (classical, lectin, and alternative pathways). All three pathways activate the central complement component 3 (C3). Subsequent entry into a final terminal pathway results in membrane attack complex (MAC) formation, which creates a pore in the cell membrane and leads to cell lysis and death (Walport, 2001a; Danese et al., 2007; Ricklin et al., 2010).
To elucidate the role of the complement system in retinal cell death caused by IR, we performed retinal TUNEL staining in IR-injured C3−/− mice relative to wild-type controls. C3−/− mice lack the central C3 protein required for all three complement pathways. Cell death was quantified at 24 h after IR, the peak of cell death. The number of TUNEL-positive cells in C3−/− mice (1727 ± 418 cells/mm2) decreased by 46% compared to C57BL6 mice (3213 ± 216 cells/mm2) (Figures 4A,B).
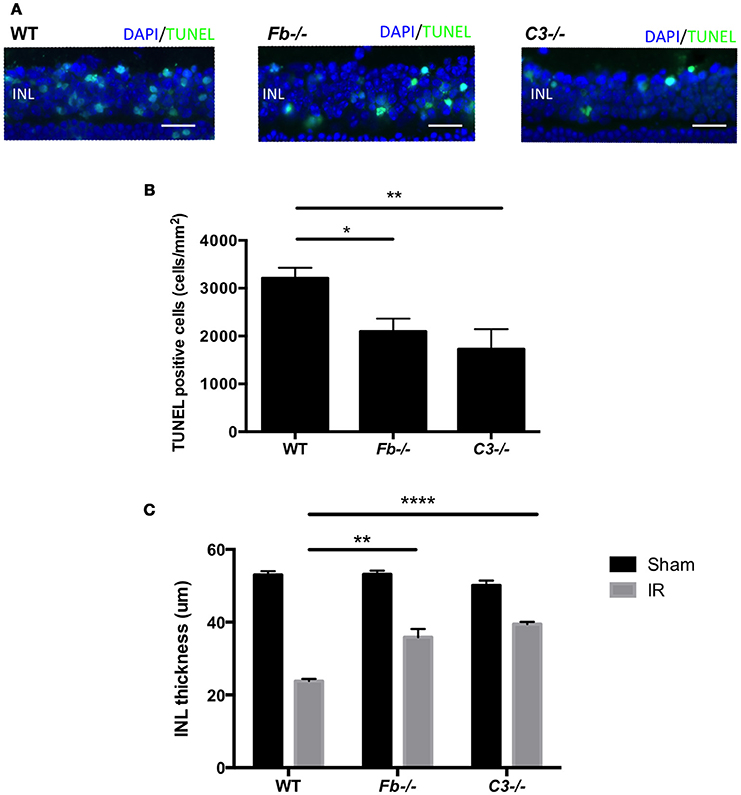
Figure 4. IR induced retinal cell death and degeneration is suppressed in complement-deficient mice. (A) Representative images of TUNEL labeling of retinal cross-sections at 24 h after IR injury in wild type control mice (WT), complement factor b-deficient mice (Fb−/−) and complement component 3-deficient mice (C3−/−), colabeled with DAPI (blue) and TUNEL (green). Scale bars = 20μm. INL, inner nuclear layer. (B) Quantification of TUNEL-positive cells in the INL of WT control, Fb−/− and C3−/− mice at 24 h after IR injury. N = 11–14 eyes/group. Data are presented as the mean ± SEM. *P < 0.05 vs. WT, **P < 0.01 vs. WT. (C) The thickness of the INL was measured 7 days after IR injury or sham surgery. N = 6 eyes/group. Data are presented as the mean ± SEM. **P < 0.01 vs. WT, ****P < 0.0001 vs. WT.
To further elucidate the role (Geijer and Bill, 1979) of the complement system in retinal IR injury, we next measured retinal cell death in complement factor b-deficient (Fb−/−) mice, which are deficient in the effector of the alternative pathway. The number of apoptotic cells in Fb−/− mice (2098 ± 268 cells/mm2) was down-regulated by 35% compared to C57BL6 mice (Figures 4A,B). There was no difference in INL TUNEL positivity between male and female mice of both strains (Supplementary Image 3).
In order to evaluate the long-term effect of IR injury, we analyzed INL thickness in C57BL6 mice, Fb−/− mice and C3−/− mice 7 days after IR surgery or sham surgery. When compared to C57BL6 mice (24 ± 1 μm), a reduction of INL thickness after IR injury was suppressed by 47% in Fb−/− (38 ± 2 μm) and by 63% in C3−/− mice (40 ± 1 μm). This demonstrated that IR injury induces morphologic changes in the retina, and revealed the long-term protective effect conferred by the lack of C3 or Fb (Figure 4C). These results indicate that the alternative complement pathway contributes to the retinal cell death induced by IR injury.
The main factor in IR injury pathology is the loss of blood flow, leading to vascular dysfunction as well as neuronal damage. Most investigations of ischemic disease have focused on the consequences of resultant oxygen and nutrient deprivation. However, loss of blood flow also results in loss of shear stress in endothelial cells lining the vessel walls, which is important for normal endothelial cell homeostasis (Ballermann et al., 1998). Therefore, we hypothesized that shear stress may also play a role in IR injury. Endothelial cells are known producers of complement, and we sought to investigate whether shear stress affected complement activation in endothelial cells.
To investigate the effect of shear stress on complement inhibitors, we evaluated the expression of Cd46, Cd55, and Cd59 in human retinal endothelial cells (HRECs). The expression of complement inhibitors was determined by real-time polymerase chain reaction (RT-PCR) after exposure to orbital shear stress (0, 5, or 10 dynes/cm2) for 24 h. All three complement inhibitors were significantly up-regulated by shear stress in proportion to the relative amount of shear stress applied (Figures 5A–C).
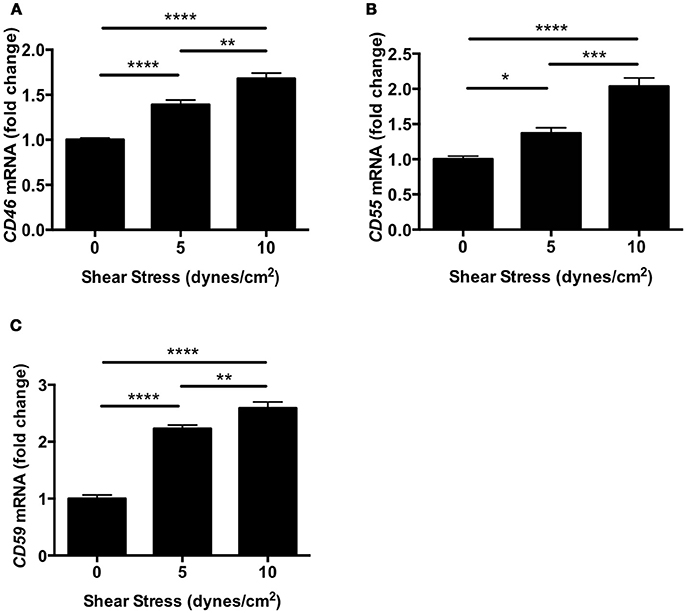
Figure 5. Complement inhibitors were up-regulated in HRECs under shear stress. (A–C) The mRNA levels of complement inhibitors, CD46 (A), Cd55 (B), and Cd59 (C) in HRECs were quantified by RT-PCR after exposure to orbital shear stress (5 or 10 dynes/cm2) for 24 h, and were normalized to control (0 dynes/cm2). The expression of complement inhibitors was increased in proportion to the magnitude of the shear stress applied. Data are the average of four independent experiments. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next assessed whether shear stress may protect against complement-mediated cell death. HRECs were exposed to shear stress (0, 5, or 10 dynes/cm2) for 24 h during incubation in media containing 1% mouse serum as a source of foreign complement. Cells were stained with a viability imaging kit, and the percentage of dead cells was calculated. The percentage of HRECs undergoing cell death was significantly suppressed by 60% in both 5 and 10 dynes/cm2 magnitudes (5 ± 1 and 5 ± 1%, respectively) compared to 0 dynes/cm2 magnitude (13 ± 1%) (Figures 6A,B). These data indicate that shear stress may contribute to protection from complement-mediated cell death by up-regulating expression of complement inhibitors.
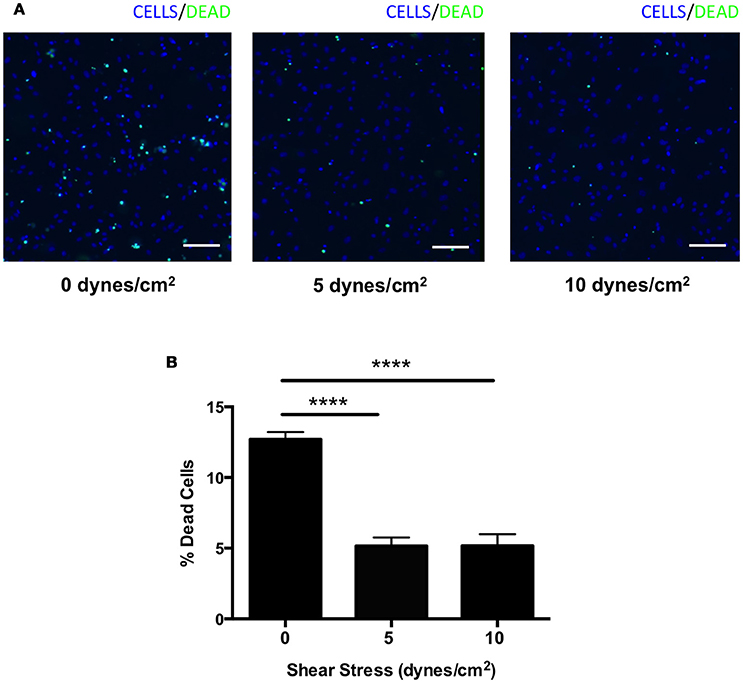
Figure 6. Complement mediated cell death was suppressed in HRECs by shear stress. (A) Representative images of HRECs stained with a cell viability imaging kit after exposure to orbital shear stress (0, 5, or 10 dynes/cm2) for 24 h during incubation in media with foreign complement. Cells (blue), dead cells (green). Scale bars = 100 μm. (B) The percentage of dead cells in HRECs after exposure to to orbital shear stress (0, 5, or 10 dynes/cm2) for 24 h with media containing foreign complement. The cell death was significantly suppressed in HRECs exposed to shear stress. Data are the average of five independent experiments. Data are presented as the mean ± SEM. ****P < 0.0001 vs. 0 dynes/cm2.
IR injury is a well-established model of retinal vascular occlusion diseases such as acute glaucoma and diabetic retinopathy (Büchi et al., 1991). Although the elevation of IOP is well-known to affect retinal perfusion (Geijer and Bill, 1979; Zhi et al., 2012, 2015; He et al., 2013), the IOP during the ischemic phase of IR surgery ranges from 60 to 120 mmHg, and the reservoir height used is not standardized, meaning that there may be significant variation between investigators (Zheng et al., 2007; Huang et al., 2013; Hu et al., 2015; Seong et al., 2017; Zanoni et al., 2017). Furthermore, ischemia and reperfusion are usually confirmed indirectly by observation of the iris, episcleral veins or the reflex from the fundus (Da and Verkman, 2004; Oharazawa et al., 2010; Yokota et al., 2011; Xiong et al., 2017). Few studies have focused on observing the ischemia and reperfusion directly, meaning that the degree of ischemia and reperfusion attained could vary between groups. In this study, we demonstrated that blood supply from both the choroidal and retinal circulation is obstructed immediately by elevating the IOP to 80 mmHg, and reperfused by the removal of the needle (Supplementary Videos 1A–D).
While several studies have reported a time course of apoptosis in the IR model, the results between investigators were variable. In our IR model, TUNEL-positive cells in INL were found as early as 3 h after the reperfusion, peaked at 24 h, and decreased gradually over subsequent time points. This result is consistent with a previous report evaluating apoptotic cells in the INL of IR-injured rat retinas (Hu et al., 2015). The earliest evaluated time point in most prior studies was 6 or 12 h post-reperfusion. We also measured retinal apoptosis 3 h post-reperfusion, and found that apoptosis began to occur earlier than previously reported (Huang et al., 2013; Hu et al., 2015).
The complement system is known to be a crucial mediator of systemic IR injury (Austen et al., 1999; de Vries et al., 2003). However, little is known about complement-mediated cell death in retinal IR injury. In the present study, we demonstrated that compliment inhibitors Crry, Cd55, and Cd59a were suppressed early on in the retina (6 h) after IR injury in C57BL6 mice, Fb−/− mice and C3−/− mice. These inhibitors are regarded as critical negative regulators of the complement system due to their ability to modulate all three complement pathways. Crry inhibits C3 convertase (Harris et al., 1999), CD55 inhibits both C3 convertase and C5 convertase, and CD59 inhibits formation of the MAC (Hamilton et al., 1990; Rollins and Sims, 1990). These results are consistent with our previous study reporting that photoreceptors are susceptible to targeted cell death due to down-regulation of complement inhibitors (Sweigard et al., 2015). Furthermore, in the mouse renal IR model, CD55-deficient and CD55/CD59-double deficient mice were found to develop more severe IR injury and tubular damage, accompanied by endothelial deposition of C3 and the MAC (Yamada et al., 2004). Our findings indicate that down-regulation of complement inhibitor expression increases activation of the complement pathway, and eventual death of retinal cells.
The complement cascade plays a well-established role in systemic IR injury (Zhou et al., 2000; Orsini et al., 2016; Chun et al., 2017; Satyam et al., 2017). Several studies have reported decreased IR injury in mice deficient in the complement system. In C3−/− mice, which lack the central C3 protein required for all three complement pathways, renal tubule damage (Zhou et al., 2000), intestinal damage (Satyam et al., 2017), optic nerve and retinal ganglion cells damage (Kuehn et al., 2008) after IR injury was decreased relative to wild-type controls. In Fb−/− mice, which are deficient in an essential rate-limiting protein for alternative pathway activation (Nielsen et al., 1992), decreased myocardial necrosis (Chun et al., 2017) and neurological deficits (Orsini et al., 2016) were observed after IR injury. In our retinal IR model, cell death in the INL was suppressed in C3−/− mice compared to wild-type (WT) mice. Furthermore, a similar reduction in the amount of retinal cell death was observed in Fb−/− mice. Consistent with these results, when the thickness of the INL was measured 7 days following IR injury in WT, C3−/− and Fb−/− mice, complement deficient strains were found to have significant protection against INL degeneration compared to WT mice in response to IR injury (Figure 4C). As factor B is exclusive to the alternative pathway, these results suggest that the complement system, especially the alternative pathway, likely contributes to IR injury-induced retinal cell death. We previously found that the classical and lectin pathways play a minor role in a oxygen-induced retinopathy model and photoreceptor cell death retinal detachment model, using classical pathway knockout mice (C1q−/−) and lectin pathway knockout mice (Mbl A/C−/−) (Sweigard et al., 2014, 2015; Kim et al., 2016). Thus, it would be interesting to assess the contribution of these pathways in this IR model in a future study.
Our current in vitro experiments showed that apoptotic cell death was suppressed by shear stress in HRECs. This result was consistent with previous studies demonstrating that shear stress protects human umbilical vein endothelial cells (HUVECs) from apoptosis (Dimmeler et al., 1996; dela Paz et al., 2012). Furthermore, we also demonstrated that complement inhibitors Cd46, Cd55, and Cd59 are up-regulated by shear stress. However, earlier work has illustrated that Cd59 expression was increased by shear stress, but no significant difference in Cd46 and Cd55 expression was observed in HUVECs (Kinderlerer et al., 2008). One possible explanation for this is the difference in model of shear stress. While the previous study group used a parallel plate flow chamber, which induces laminar shear stress, we used the orbital plate method, which induces orbital shear stress. Prior studies suggest that orbital shear stress more closely mimics disturbed or turbulent flow, and causes an increase in endothelial cell proliferation and apoptosis compared to laminar shear stress (Dardik et al., 2005). This is due to the uniformity of the shear stress in the plate. The peripheral cells are exposed to more shear stress compared to the cells in the center (Dardik et al., 2005). Endothelial cells are known producers of complement (Fischetti and Tedesco, 2006), and it is possible that the vasculature stops producing complement inhibitors in the absence of shear stress. Identifying TUNEL positivity in vessels in retinal cross-sections after IR has proven to be technically difficult, likely due to the small cross-sectional area of these structures. However, it is likely that in IR injury, like stroke, vessels become dysfunctional and injured, but are by-in-large protected from the temporary ischemic conditions. Supporting this was the finding that interruption of shear stress for 45 min did not cause significant difference in vitro. This is likely due to differences between an in vivo system and a cell culture system. The cells in culture are likely able to adapt more rapidly due to their immortalized nature and normal growing conditions of 20% oxygen.
In conclusion, we found that complement system plays a key role in IR injury-induced retinal cell death. Our data suggest that the lack of blood flow may contribute to the suppression of complement inhibitors, leading to apoptotic cell death via activation of the complement system.
All datasets for this study are included in the manuscript and the supplementary files.
SI, GK, and KC designed the study. SI and GK conducted experiments, and analyzed data. SI and KC wrote the manuscript. All authors have been involved in drafting the manuscript critically for important intellectual content.
This study was supported by a NIH/National Eye Institute Grant R01EY027303, the Massachusetts Lions Eye Research Fund; the BrightFocus Foundation (KC). Special thanks go to Department of Ophthalmology, Harvard University, and Massachusetts Eye and Ear Infirmary for supporting this research (KC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to express our gratitude to Eiichi Hasegawa, Ryo Mukai, Patricia A. D'Amore, and Jinling Yan for their technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00278/full#supplementary-material
Supplementary Image 1. No gender difference was observed in intraocular pressure (IOP) during ischemia reperfusion (IR) surgery. (A,B) Mean IOP before, after and during the 45-min IR surgery in male (A) and female (B) mice. n = 6 eyes/time point. Data are presented as the mean ± SEM.
Supplementary Image 2. No gender difference was observed in time course of ischemia reperfusion (IR) induced apoptosis. Quantification of TUNEL-positive cells in the INL at the indicated time points after IR. TUNEL, terminal deoxynucleotidyl dUTP nick end labeling; M, male; F, female. n = 4–5 eyes/time point. Data are presented as the mean ± SEM. N.S., not significant.
Supplementary Image 3. No gender difference was observed in ischemia reperfusion (IR) induced apoptosis in complement pathway-deficient mice. Quantification of TUNEL-positive cells in the INL in WT control, Fb−/− and C3−/− mice after IR. TUNEL, terminal deoxynucleotidyl dUTP nick end labeling; WT, wild type control. n = 5–8 eyes/group. Data are presented as the mean ± SEM. N.S., not significant.
Supplementary Videos 1–4. Color fundus videos (1) and (3) and fluorescein angiography (FA) videos (2) and (4) illustrating vascular collapse and reperfusion during the IR model. Color fundus (1) and FA (2) videos converting from the normal perfusion phase to the ischemic phase. Color fundus (3) and FA (4) videos converting from the ischemic phase to the reperfusion phase.
Ahmed, F., Brown, K. M., Stephan, D. A., Morrison, J. C., Johnson, E. C., and Tomarev, S. I. (2004). Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Invest. Opthalmol. Vis. Sci. 45, 1247–1254. doi: 10.1167/iovs.03-1123
Austen, W. G, Kyriakides, C., Favuzza, J., Wang, Y., Kobzik, L., Moore, F. D. Jr., et al. (1999). Intestinal ischemia-reperfusion injury is mediated by the membrane attack complex. Surgery 126, 343–348. doi: 10.1016/S0039-6060(99)70175-3
Ballermann, B. J., Dardik, A., Eng, E., and Liu, A. (1998). Shear stress and the endothelium. Kidney Int. Suppl. 54, S100–S108.
Bamboat, Z. M., Ocuin, L. M., Balachandran, V. P., Obaid, H., Plitas, G., and Dematteo, R. P. (2010). Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J. Clin. Invest. 120, 559–569. doi: 10.1172/JCI40008
Bexborn, F., Andersson, P. O., Chen, H., Nilsson, B., and Ekdahl, K. N. (2008). The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol. Immunol. 45, 2370–2379. doi: 10.1016/j.molimm.2007.11.003
Büchi, E. R., Suivaizdis, I., and Fu, J. (1991). Pressure-induced retinal ischemia in rats: an experimental model for quantitative study. Ophthalmologica 203, 138–147. doi: 10.1159/000310240
Chun, N., Haddadin, A. S., Liu, J., Hou, Y., Wong, K. A., Lee, D., et al. (2017). Activation of complement factor B contributes to murine and human myocardial ischemia/reperfusion injury. PLoS ONE 12:e0179450. doi: 10.1371/journal.pone.0179450
Country, M. W. (2017). Retinal metabolism: a comparative look at energetics in the retina. Brain Res. 1672, 50–57. doi: 10.1016/j.brainres.2017.07.025
Da, T., and Verkman, A. S. (2004). Aquaporin-4 gene disruption in mice protects against impaired retinal function and cell death after ischemia. Invest. Ophthalmol. Vis. Sci. 45, 4477–4483. doi: 10.1167/iovs.04-0940
Danese, S., Dejana, E., and Fiocchi, C. (2007). Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J. Immunol. 178, 6017–6022. doi: 10.4049/jimmunol.178.10.6017
Dardik, A., Chen, L., Frattini, J., Asada, H., Aziz, F., Kudo, F. A., et al. (2005). Differential effects of orbital and laminar shear stress on endothelial cells. J. Vasc. Surg. 41, 869–880. doi: 10.1016/j.jvs.2005.01.020
De Hoog, V. C., Timmers, L., Van Duijvenvoorde, A., De Jager, S. C., Van Middelaar, B. J., Smeets, M. B., et al. (2014). Leucocyte expression of complement C5a receptors exacerbates infarct size after myocardial reperfusion injury. Cardiovasc. Res. 103, 521–529. doi: 10.1093/cvr/cvu153
de Vries, B., Köhl, J., Leclercq, W. K., Wolfs, T. G., Van Bijnen, A. A., Heeringa, P., et al. (2003). Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J. Immunol. 170, 3883–3889. doi: 10.4049/jimmunol.170.7.3883
dela Paz, N. G., and D'Amore, P. A. (2009). Arterial versus venous endothelial cells. Cell Tissue Res. 335, 5–16. doi: 10.1007/s00441-008-0706-5
dela Paz, N. G., Walshe, T. E., Leach, L. L., Saint-Geniez, M., and D'Amore, P. A. (2012). Role of shear-stress-induced VEGF expression in endothelial cell survival. J. Cell Sci. 125, 831–843. doi: 10.1242/jcs.084301
Dimmeler, S., Haendeler, J., Rippmann, V., Nehls, M., and Zeiher, A. M. (1996). Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 399, 71–74. doi: 10.1016/S0014-5793(96)01289-6
Fischetti, F., and Tedesco, F. (2006). Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 39, 417–428. doi: 10.1080/08916930600739712
Geijer, C., and Bill, A. (1979). Effects of raised intraocular pressure on retinal, prelaminar, laminar, and retrolaminar optic nerve blood flow in monkeys. Invest. Ophthalmol. Vis. Sci. 18, 1030–1042.
Hamilton, K. K., Ji, Z., Rollins, S., Stewart, B. H., and Sims, P. J. (1990). Regulatory control of the terminal complement proteins at the surface of human endothelial cells: neutralization of a C5b-9 inhibitor by antibody to CD59. Blood 76, 2572–2577.
Harboe, M., and Mollnes, T. E. (2008). The alternative complement pathway revisited. J. Cell. Mol. Med. 12, 1074–1084. doi: 10.1111/j.1582-4934.2008.00350.x
Harris, C. L., Rushmere, N. K., and Morgan, B. P. (1999). Molecular and functional analysis of mouse decay accelerating factor (CD55). Biochem. J. 341, 821–829. doi: 10.1042/bj3410821
He, Z., Lim, J. K., Nguyen, C. T., Vingrys, A. J., and Bui, B. V. (2013). Coupling blood flow and neural function in the retina: a model for homeostatic responses to ocular perfusion pressure challenge. Physiol. Rep. 1:e00055. doi: 10.1002/phy2.55
Hu, Q., Huang, C., Wang, Y., and Wu, R. (2015). Expression of leukemia inhibitory factor in the rat retina following acute ocular hypertension. Mol. Med. Rep. 12, 6577–6583. doi: 10.3892/mmr.2015.4287
Hu, R., Chen, Z. F., Yan, J., Li, Q. F., Huang, Y., Xu, H., et al. (2014). Complement C5a exacerbates acute lung injury induced through autophagy-mediated alveolar macrophage apoptosis. Cell Death Dis. 5:e1330. doi: 10.1038/cddis.2014.274
Huang, P., Huo, Y., Lou, L. X., Li, H., Barnstable, C. J., Zhang, C., et al. (2013). CD4 positive T helper cells contribute to retinal ganglion cell death in mouse model of ischemia reperfusion injury. Exp. Eye Res. 115, 131–139. doi: 10.1016/j.exer.2013.06.015
Hughes, W. (1991). Quantation of ischemic damage in the rat retina. Exp. Eye Res. 53, 573–582. doi: 10.1016/0014-4835(91)90215-Z
Kim, C., Smith, K. E., Castillejos, A., Diaz-Aguilar, D., Saint-Geniez, M., and Connor, K. M. (2016). The alternative complement pathway aids in vascular regression during the early stages of a murine model of proliferative retinopathy. FASEB J. 30, 1300–1305. doi: 10.1096/fj.15-280834
Kinderlerer, A. R., Ali, F., Johns, M., Lidington, E. A., Leung, V., Boyle, J. J., et al. (2008). KLF2-dependent, shear stress-induced expression of CD59: a novel cytoprotective mechanism against complement-mediated injury in the vasculature. J. Biol. Chem. 283, 14636–14644. doi: 10.1074/jbc.M800362200
Kuehn, M. H., Kim, C. Y., Jiang, B., Dumitrescu, A. V., and Kwon, Y. H. (2008). Disruption of the complement cascade delays retinal ganglion cell death following retinal ischemia-reperfusion. Exp. Eye Res. 87, 89–95. doi: 10.1016/j.exer.2008.04.012
Kuehn, M. H., Kim, C. Y., Ostojic, J., Bellin, M., Alward, W. L., Stone, E. M., et al. (2006). Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp. Eye Res. 83, 620–628. doi: 10.1016/j.exer.2006.03.002
Markiewski, M. M., and Lambris, J. D. (2007). The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 171, 715–727. doi: 10.2353/ajpath.2007.070166
Nielsen, H. E., Larsen, S. O., and Vikingsdottir, T. (1992). Rate-limiting components and reaction steps in complement-mediated haemolysis. APMIS 100, 1053–1060. doi: 10.1111/j.1699-0463.1992.tb04040.x
Oharazawa, H., Igarashi, T., Yokota, T., Fujii, H., Suzuki, H., Machide, M., et al. (2010). Protection of the retina by rapid diffusion of hydrogen: administration of hydrogen-loaded eye drops in retinal ischemia-reperfusion injury. Invest. Ophthalmol. Vis. Sci. 51, 487–492. doi: 10.1167/iovs.09-4089
Orsini, F., Chrysanthou, E., Dudler, T., Cummings, W. J., Takahashi, M., Fujita, T., et al. (2016). Mannan binding lectin-associated serine protease-2 (MASP-2) critically contributes to post-ischemic brain injury independent of MASP-1. J. Neuroinflammation 13:213. doi: 10.1186/s12974-016-0684-6
Osborne, N. N., Casson, R. J., Wood, J. P., Chidlow, G., Graham, M., and Melena, J. (2004). Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog. Retina Eye Res. 23, 91–147. doi: 10.1016/j.preteyeres.2003.12.001
Pacher, P., and Szabo, C. (2008). Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol. 173, 2–13. doi: 10.2353/ajpath.2008.080019
Pangburn, M. K., and Müller-Eberhard, H. J. (1980). Relation of putative thioester bond in C3 to activation of the alternative pathway and the binding of C3b to biological targets of complement. J. Exp. Med. 152, 1102–1114. doi: 10.1084/jem.152.4.1102
Pangburn, M. K., Schreiber, R. D., and Müller-Eberhard, H. J. (1981). Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J. Exp. Med. 154, 856–867. doi: 10.1084/jem.154.3.856
Powers, K. A., Szászi, K., Khadaroo, R. G., Tawadros, P. S., Marshall, J. C., Kapus, A., et al. (2006). Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J. Exp. Med. 203, 1951–1961. doi: 10.1084/jem.20060943
Rice, T. W., Wheeler, A. P., Bernard, G. R., Vincent, J. L., Angus, D. C., Aikawa, N., et al. (2010). A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 38, 1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. doi: 10.1038/ni.1923
Rollins, S. A., and Sims, P. J. (1990). The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. J. Immunol. 144, 3478–3483.
Satyam, A., Kannan, L., Matsumoto, N., Geha, M., Lapchak, P. H., Bosse, R., et al. (2017). Intracellular activation of complement 3 is responsible for intestinal tissue damage during mesenteric ischemia. J. Immunol. 198, 788–797. doi: 10.4049/jimmunol.1502287
Seong, H., Ryu, J., Yoo, W. S., Kim, S. J., Han, Y. S., Park, J. M., et al. (2017). Resveratrol ameliorates retinal ischemia/reperfusion injury in C57BL/6J mice via downregulation of caspase-3. Curr. Eye. Res. 42, 1650–1658. doi: 10.1080/02713683.2017.1344713
Song, W. C. (2004). Membrane complement regulatory proteins in autoimmune and inflammatory tissue injury. Curr. Dir. Autoimmun. 7, 181–199. doi: 10.1159/000075693
Song, W. C., Deng, C., Raszmann, K., Moore, R., Newbold, R., Mclachlan, J. A., et al. (1996). Mouse decay-accelerating factor: selective and tissue-specific induction by estrogen of the gene encoding the glycosylphosphatidylinositol-anchored form. J. Immunol. 157, 4166–4172.
Sweigard, J. H., Matsumoto, H., Smith, K. E., Kim, L. A., Paschalis, E. I., Okonuki, Y., et al. (2015). Inhibition of the alternative complement pathway preserves photoreceptors after retinal injury. Sci. Transl. Med. 7, 297ra116. doi: 10.1126/scitranslmed.aab1482
Sweigard, J. H, Yanai, R., Gaissert, P., Saint-Geniez, M., Kataoka, K., Thanos, A., et al. (2014). The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J. 28, 3171–3182. doi: 10.1096/fj.14-251041
Swirski, F. K., Nahrendorf, M., Etzrodt, M., Wildgruber, M., Cortez-Retamozo, V., Panizzi, P., et al. (2009). Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616. doi: 10.1126/science.1175202
Walport, M. (2001a). Complement. first of two parts. N. Engl. J. Med. 344, 1058–1066. doi: 10.1056/NEJM200104053441406
Walport, M. (2001b). Complement. second of two parts. N. Engl. J. Med. 344, 1140–1144. doi: 10.1056/NEJM200104123441506
Xiong, S., Xu, Y., Ma, M., Wang, H., Wei, F., Gu, Q., et al. (2017). Neuroprotective effects of a novel peptide, FK18, under oxygen-glucose deprivation in SH-SY5Y cells and retinal ischemia in rats via the Akt pathway. Neurochem. Int. 108, 78–90. doi: 10.1016/j.neuint.2017.02.015
Yamada, K., Miwa, T., Liu, J., Nangaku, M., and Song, W. C. (2004). Critical protection from renal ischemia reperfusion injury by CD55 and CD59. J. Immunol. 172, 3869–3875. doi: 10.4049/jimmunol.172.6.3869
Yokota, H., Narayanan, S. P., Zhang, W., Liu, H., Rojas, M., Xu, Z., et al. (2011). Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Invest. Ophthalmol. Vis. Sci. 52, 8123–8131. doi: 10.1167/iovs.11-8318
Zanoni, D. S., Da Silva, G. A., Ezra-Elia, R., Carvalho, M., Quitzan, J. G., Ofri, R., et al. (2017). Histological, morphometric, protein and gene expression analyses of rat retinas with ischaemia-reperfusion injury model treated with sildenafil citrate. Int. J. Exp. Pathol. 98, 147–157. doi: 10.1111/iep.12233
Zhao, Y. Y., Chang, P. J., Yu, F., and Zhao, Y. E. (2014). Retinal vessel diameter changes induced by transient high perfusion pressure. Int. J. Ophthalmol. 7, 602–607. doi: 10.3980/j.issn.2222-3959.2014.04.03
Zheng, L., Gong, B., Hatala, D. A., and Kern, T. S. (2007). Retinal ischemia and reperfusion causes capillary degeneration- similarities to diabetes. Invest. Ophthalmol. Vis. Sci. 48, 361–367. doi: 10.1167/iovs.06-0510
Zhi, Z., Cepurna, W., Johnson, E., Jayaram, H., Morrison, J., and Wang, R. K. (2015). Evaluation of the effect of elevated intraocular pressure and reduced ocular perfusion pressure on retinal capillary bed filling and total retinal blood flow in rats by OMAG/OCT. Microvasc. Res. 101, 86–95. doi: 10.1016/j.mvr.2015.07.001
Zhi, Z., Cepurna, W. O., Johnson, E. C., Morrison, J. C., and Wang, R. K. (2012). Impact of intraocular pressure on changes of blood flow in the retina, choroid, and optic nerve head in rats investigated by optical microangiography. Biomed. Opt. Express 3, 2220–2233. doi: 10.1364/BOE.3.002220
Keywords: retina, complement system, ischemia reperfusion, endothelial cell, shear stress, neurodegeneration
Citation: Inafuku S, Klokman G and Connor KM (2018) The Alternative Complement System Mediates Cell Death in Retinal Ischemia Reperfusion Injury. Front. Mol. Neurosci. 11:278. doi: 10.3389/fnmol.2018.00278
Received: 29 March 2018; Accepted: 23 July 2018;
Published: 17 August 2018.
Edited by:
Clint L. Makino, Boston University, United StatesReviewed by:
Shintaro Nakao, Kyushu University, JapanCopyright © 2018 Inafuku, Klokman and Connor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kip M. Connor, a2lwX2Nvbm5vckBtZWVpLmhhcnZhcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.