 Mario Bortolozzi1,2,3*
Mario Bortolozzi1,2,3*- 1Department of Physics and Astronomy G. Galilei, University of Padua, Padua, Italy
- 2Venetian Institute of Molecular Medicine (VIMM), Padua, Italy
- 3Padova Neuroscience Center (PNC), Padua, Italy
Connexin 32 (Cx32) is a fundamental protein in the peripheral nervous system (PNS) as its mutations cause the X-linked form of Charcot–Marie–Tooth disease (CMT1X), the second most common form of hereditary motor and sensory neuropathy and a demyelinating disease for which there is no effective therapy. Since mutations of the GJB1 gene encoding Cx32 were first reported in 1993, over 450 different mutations associated with CMT1X including missense, frameshift, deletion and non-sense ones have been identified. Despite the availability of a sizable number of studies focusing on normal and mutated Cx32 channel properties, the crucial role played by Cx32 in the PNS has not yet been elucidated, as well as the molecular pathogenesis of CMT1X. Is Cx32 fundamental during a particular phase of Schwann cell (SC) life? Are Cx32 paired (gap junction, GJ) channels in myelinated SCs important for peripheral nerve homeostasis? The attractive hypothesis that short coupling of adjacent myelin layers by Cx32 GJs is required for efficient diffusion of K+ and signaling molecules is still debated, while a growing body of evidence is supporting other possible functions of Cx32 in the PNS, mainly related to Cx32 unpaired channels (hemichannels), which could be involved in a purinergic-dependent pathway controlling myelination. Here we review the intriguing puzzle of findings about Cx32 function and dysfunction, discussing possible directions for future investigation.
Introduction
Connexin 32 (Cx32) is a 32 kDa protein of the connexin family, abundantly found in liver (Paul, 1986), but it is also expressed in many other tissues, including the central nervous system (CNS) and the peripheral nervous system (PNS) (Scherer et al., 1995; Rash et al., 2001). Mutations in the GJB1 gene, which encodes Cx32, are the leading cause of the X-linked dominant form of Charcot–Marie–Tooth disease (CMT1X or CMTX1), the second most common form of hereditary motor and sensory neuropathy and a disease for which there is no cure (Kleopa and Scherer, 2006; Kleopa et al., 2012). Since mutations were first reported in 1993 (Bergoffen et al., 1993), over 450 different mutations associated with CMT1X including missense, frameshift, deletion and non-sense ones have been identified according to The Human Gene Mutation Database (HGMD®; Stenson et al., 2014). In both nerves and ganglia of the PNS, Cx32 localizes only in myelinating Schwann cells (SCs), mainly to the paranodes, the periodic interruptions in the compact myelin called Schmidt–Lanterman incisures, and the two outer layers of myelin (Scherer et al., 1995; Meier et al., 2004; Procacci et al., 2008). Elucidation of the molecular function of Cx32 in myelinating SCs is a requirement for understanding how different mutations lead to the sequence of events that end in demyelination and axonal loss in CMT1X patients. Despite the availability of an incredible number of studies, mostly in vitro, focusing on normal and mutated Cx32 channel properties, an interpretative framework is still lacking.
Cx32 Gap Junction Channels in Non-compact Regions of Myelinating Schwann Cells
Minute intracellular gap junction (GJ) channels are formed in non-compact regions of SC myelin when hexamers of Cx32 (connexons or hemichannels) spanning opposite myelin layers dock end-to-end (Meier et al., 2004), providing a fast diffusive radial path between the abaxonal and adaxonal region (Balice-Gordon et al., 1998). This has led to the attractive hypothesis that Cx32 GJ channels are critical for the passage of K+ and signaling molecules across the myelin sheath of SCs, whose function is not only to myelinate axons but also to maintain their long-term functional integrity (Nave and Trapp, 2008). The overlap in the distribution of Cx32 and Cx29 at incisures and paranodes suggests that they both contribute to reflexive junctions (Altevogt et al., 2002; Li et al., 2002), even if immunogold labeling for Cx29 did not reveal ultrastructurally defined GJs at incisures or directly linking successive paranodal loops (Li et al., 2002). Furthermore Cx29 does not form junctional channels when expressed in cultured cells (Altevogt et al., 2002; Ahn et al., 2008), neither Cx29 human ortholog Cx31.3 (Sargiannidou et al., 2008). Cx43 is also highly expressed in paranodal regions of myelinating SCs of adult wild-type (WT) and Cx32-null mice (Zhao et al., 1999). In adult rat, staining of Cx43 along myelin sheath and SC bodies was observed, but lower than Cx32 (Mambetisaeva et al., 1999), whereas an immunohistochemical study of human peripheral nerves revealed that anti-Cx43 antibody stained cytoplasm around the nucleus of SCs but not myelin (Yoshimura et al., 1996). Cx26 is also expressed in myelinating SCs of neonatal WT mice (Zhao et al., 1999), but not in those of neonatal rats (Mambetisaeva et al., 1999), supporting the conclusion that connexin expression in SCs is, at least in part, species-dependent, so direct investigation in the human nerve would be needed.
Cx32 Hemichannels May Participate in the Myelination Process of Schwann Cells
In the PNS, electrical stimulation of myelinated nerves triggers axonal ATP release which induces Ca2+ increases in the cytosol and the mitochondrial matrix of the surrounding SCs via P2Y receptor activation (Lev-Ram and Ellisman, 1995; Lyons et al., 1995; Mayer et al., 1997; Stevens and Fields, 2000; Ino et al., 2015). This neuron-to-SC pathway is likely to have an important role in proper myelination as in vivo chronic suppression of the purinergic-mediated signaling inhibits correct myelin formation and causes hypomyelination (Ino et al., 2015). Cx32 hemichannels in myelinating SCs may contribute to regulate the myelination process by enhancing the intracellular and intercellular propagation of this Ca2+ signaling by a regenerative ATP-induced ATP release mechanism. The presence of functional Cx32 hemichannels was recently hypothesized based on connexin-mediated ATP release observed during electrical stimulation of mice sciatic nerves (Nualart-Marti et al., 2013). Indeed, the molecular machinery ideally suited to support a Cx32-mediated purinergic signaling throughout SCs is actually present in peripheral nerves, given that Cx32, IP3R and P2Y receptors are found together in the paranodes and in the outer layer of SCs (Martínez-Gómez and Dent, 2007; Toews et al., 2007) and Cx32 hemichannels can release ATP (Cotrina et al., 2000; Belliveau et al., 2006; De Vuyst et al., 2006; Nualart-Marti et al., 2013). Indeed, comparing SCs cultured from sciatic nerves of WT and Cx32-null mice, Cx32 was found to enhance the intercellular Ca2+ waves spreading without contribution of Cx32 GJs (Zhao et al., 1999). As the Ca2+ wave propagation was mediated by extracellular release of ATP, it can reasonably be inferred the involvement of Cx32 hemichannels. The same consideration applies to another work (Freidin et al., 2009) using primary cultures of purified SCs from sciatic nerve which suggests a link between Cx32 expression and GGF2 (a growth factor which controls SC proliferation and differentiation), which does not involve Cx32-mediated GJ communication.
Other Possible Functions of Cx32 in Myelinating Schwann Cells
GJB1 gene depletion results in a mitotic phenotype from the genome-wide phenotypic profiling performed by the Mitocheck consortium (Neumann et al., 2010). Mitotic instability and CMT1X phenotype were linked to increased CaMKII activity in both human and murine fibroblasts carrying the G12S and S26L mutations of Cx32 as normal mitosis and motor function of mutant mice were partially recovered by CaMKII inhibitors (Mones et al., 2012, 2014). Cx32-S26L hemichannel dysfunction due to altered CaMKII activity was also proposed (Mones et al., 2014), supporting the notion that a CaM-dependent pathway controls the hemichannel gating by cytosolic Ca2+ of α and β connexin isoforms (De Vuyst et al., 2006, 2009; Zhang et al., 2006; Zhou et al., 2009; Hu et al., 2018). As found in oligodendrocytes (Waggener et al., 2013), CaMKII may be also critical in SCs for the well balanced equilibrium between dynamic remodeling and kinetic stability of the actin cytoskeleton required for efficient myelination.
Recently (Fowler et al., 2013), by employing a proteomic approach in murine liver, it has been reported that Cx32 is expressed in the inner mitochondrial membrane and interacts with the outer mitochondrial membrane resident fraction of syderoflexin-1 (SFXN-1), thus suggesting a putative role for Cx32-SFXN1 axis as protein complex for mitochondrial plasma membrane tethering. In Cx32-null mice, several mitochondrial proteins are upregulated, indicating that Cx32 optimizes mitochondrial bioenergetics by restricting rates of oxidative phosphorylation (Fowler et al., 2017, communication at the International GJ Conference).
The X-Linked Form of Charcot-Marie-Tooth Disease
CMT1X patients develop progressive distal muscle weakness and amyotrophy, together with sensory abnormalities that are most pronounced in the distal extremities (Vance, 1991; Harding, 1995; Suter and Snipes, 1995). CNS disturbances, mainly episodic but in some cases including static deafness and cognitive impairment, can occur in CMT1X patients, whereas the symptoms do not appear to correlate with the stage and severity of the peripheral neuropathy (Abrams and Freidin, 2015; Wang and Yin, 2016). Nerve electrophysiological and pathological analysis show intermediate slowing of conduction and length-dependent axonal loss, with more prominent axonal degeneration than de/remyelination (Kleopa et al., 2012), which supports the hypothesis that axonal abnormalities precede demyelination (Vavlitou et al., 2010). Interestingly, these studies together show that clinical and pathophysiological features of patients lacking the entire coding region of Cx32 are similar to that of most other patients with CMT1X, suggesting that most mutations cause loss-of-function (Shy et al., 2007; Kleopa, 2011). Functional analysis of Cx32 mutations (mainly CMT1X-related) using various expression systems has revealed a plethora of alterations that could schematically subdivided in classes, not mutually exclusive, which range from mutations that cause loss of channel formation to those that retain electrical coupling but show altered permeation properties or defective gating mechanisms:
1. Cx32 protein is not synthesized (Ionasescu et al., 1996; Ainsworth et al., 1998; Abrams and Freidin, 2015).
2. Mutant Cx32 protein is normally transcribed but little protein is expressed in the cell, such as in a frameshift of Cx32 at amino acid 175 (Deschenes et al., 1997). Some GJB1 gene mutations in non-coding regions controlling Cx32 expression should belong to this category (Tomaselli et al., 2017).
3. Mutant Cx32 protein is properly synthesized but not transported to the plasma membrane, causing abnormal and possibly toxic accumulation in intracellular compartment such as the endoplasmic reticulum (ER), the Golgi apparatus or the cytoplasm. This condition results for mutations W3D (Kalmatsky et al., 2012), W3Y (Martin et al., 2000), Y7D (Kalmatsky et al., 2012), G12S (Deschenes et al., 1997; Wang et al., 2004; Kalmatsky et al., 2009; Mones et al., 2014), R32E (Fleishman et al., 2006), M34K (Yum et al., 2002), M34T (Yum et al., 2002), V38M (Yum et al., 2002), A39P and A39V (Kleopa et al., 2002), A40V (Yum et al., 2002), F51L (Abrams et al., 2017), C53S (Yoshimura et al., 1998), T55I (Kleopa et al., 2002; Sargiannidou et al., 2009; Abrams et al., 2017), R75P and R75Q (Yum et al., 2002), R75W (Yum et al., 2002; Sargiannidou et al., 2009; Abrams et al., 2013), E102del (Abrams et al., 2017), V140E (Kleopa et al., 2006), R142E (Fleishman et al., 2006), R142Q (Abrams et al., 2017), R142W (Deschenes et al., 1997; Vanslyke et al., 2000; Jeng et al., 2006; Abrams et al., 2017), L143P (Kleopa et al., 2006), E146R (Fleishman et al., 2006), R164Q (Kleopa et al., 2002; Abrams et al., 2017), R164W (Kleopa et al., 2002), C168Y (Abrams et al., 2017), P172R (Yoshimura et al., 1998), V177A (Abrams et al., 2017), E186K (Deschenes et al., 1997; Vanslyke et al., 2000), N205I (Yum et al., 2002), E208K (Deschenes et al., 1997; Martin et al., 2000; Vanslyke et al., 2000; Wang et al., 2004), Y211X and C217X (Yum et al., 2002).
4. Mutant Cx32 protein is transported and inserted in the membrane to form hemichannels but no plaques or functional GJ channels are found. This condition results for mutations W3S (Martin et al., 2000), R15W (Abrams et al., 2001), R22G and R22P (Ressot et al., 1998), C60F (Omori et al., 1996), R75D-R75E-R75P-R75Q-R75V (Abrams et al., 2013), L90H (Ressot et al., 1998), H94Y (Abrams et al., 2001), V95M (Ressot et al., 1998), V139M (Omori et al., 1996; Deschenes et al., 1997; Abrams et al., 2001, 2017), R142Q and R164W (Abrams et al., 2017), P172S (Ressot et al., 1998), N175Y (Nakagawa et al., 2011), S182T (Wang et al., 2004), E208L (Ressot et al., 1998), E208K (Castro et al., 1999), Y211X (Ressot et al., 1998; Castro et al., 1999; Wang et al., 2004), I214X (Rabadan-Diehl et al., 1994), R215Q (Castro et al., 1999), R215W (Omori et al., 1996; Castro et al., 1999), R215X (Rabadan-Diehl et al., 1994; Castro et al., 1999), C217X (Rabadan-Diehl et al., 1994).
5. Mutant Cx32 protein forms electrically conductive GJ channels and hemichannels presenting with altered properties in respect to WT channels, e.g., decreased/increased channel number, distribution, gating sensitivity or unitary permeability to physiologically crucial ions and molecules. Studies of functionality refers to mutations N2A-N2D-N2E-N2Q (Oh et al., 1999), G12S (Abrams et al., 2001), V13L (Martin et al., 2000; Wang et al., 2004), R15Q (Abrams et al., 2001; Wang et al., 2004), R22Q (Wang et al., 2004), S26L (Oh et al., 1997; Bicego et al., 2006; Mones et al., 2014), I30N (Oh et al., 1997; Wang et al., 2004), M34T (Oh et al., 1997), V35M (Oh et al., 1997; Wang et al., 2004), V38M (Oh et al., 1997), S42E (Oh et al., 1999), L56F (Ressot et al., 1998), V63I (Wang et al., 2004), Y65C (Wang et al., 2004), R75A-R75H-R75K-R75L (Abrams et al., 2013), R75Q (Wang et al., 2004), Q80R (Wang et al., 2004), S85C (Abrams et al., 2001), P87A (Oh et al., 1997), H94Q (Abrams et al., 2001), V95M (Wang et al., 2004), E102G (Oh et al., 1997; Ressot et al., 1998; Abrams et al., 2003), R107W (Wang et al., 2004), Del 111–116 (Oh et al., 1997; Ressot et al., 1998; Bicego et al., 2006), W133R (Wang et al., 2004), Y151C (Abrams et al., 2017), L156R (Wang et al., 2004), P158A-R164W-P172S (Wang et al., 2004), N175D (Gong et al., 2013), V181A (Abrams et al., 2003), V181M and R183C (Abrams et al., 2017), G199R (Wang et al., 2004), N205S (Kleopa et al., 2002; Wang et al., 2004), R215X (Rabadan-Diehl et al., 1994), C217X (Rabadan-Diehl et al., 1994; Castro et al., 1999), R220X (Rabadan-Diehl et al., 1994; Omori et al., 1996; Deschenes et al., 1997; Ressot et al., 1998; Castro et al., 1999; Revilla et al., 1999; Bicego et al., 2006; Katoch et al., 2015; Carrer et al., 2018), R223X and N226X (Rabadan-Diehl et al., 1994), R238H (Castro et al., 1999), L239I (Abrams et al., 2017), C280G (Castro et al., 1999; Kleopa et al., 2002), S281X (Castro et al., 1999).
Most of these studies were limited to testing only GJ channel electrical conductance, so minimal information is available about specific permeability to important molecules up to 1 kDa (e.g., second messengers), which could explain why some mutants appear as “functional” with respect to the WT. Limited information is also available about gating/permeability dysfunction of mutant Cx32 hemichannels, analyzed in the following studies: S26L (Mones et al., 2014), S85C (Abrams et al., 2002), D178Y (Gómez-Hernández et al., 2003), E208K-Y211X-R215X-R215W-R215Q-C217X (Castro et al., 1999), R220X (Castro et al., 1999; Carrer et al., 2018), F235C (Liang et al., 2005), R238H-R265X-C280G-S281X (Castro et al., 1999).
Figure 1 outlines a graphical summary of: (i) possible functions of Cx32 in myelinating SCs; and (ii) the available information about in vitro functional studies of Cx32 mutations. Interestingly, the expression and function of some Cx32 mutants are cell-dependent, e.g., mutants R75Q, M34T, V38M, R164W, Y211X, C217X that reach the plasma membrane in non-human non-glial Xenopus oocytes and N2A cells (Oh et al., 1997; Castro et al., 1999; Wang et al., 2004), fail to reach the plasma membrane in human cells (HeLa) and cultured rat SCs (Kleopa et al., 2002; Yum et al., 2002).
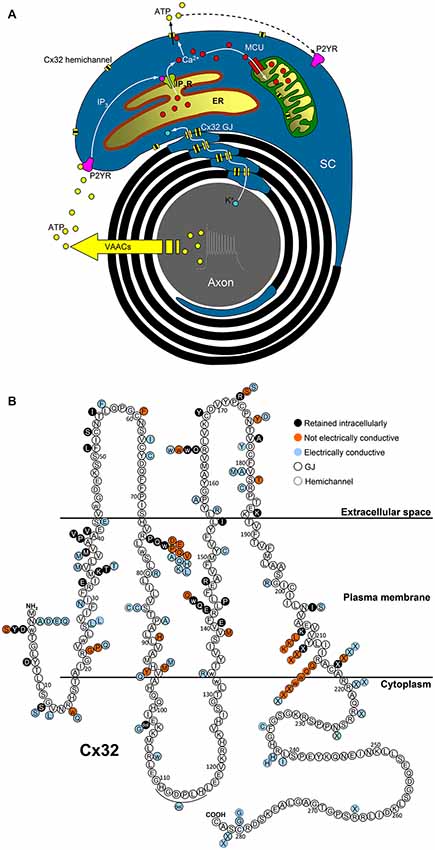
Figure 1. Possible functions of Cx32 in myelinating SCs and topology of Cx32 mutations. (A) Electrical activity of myelinated nerves triggers axonal K+ release, whose recycling could involve Cx32 GJs located in SC paranodes and Schmidt–Lanterman incisures. Axonal firing also stimulates ATP release from volume-activated anion channels (VAACs; Fields and Ni, 2010) which induces P2Y-mediated Ca2+ increases in the cytosol and the mitochondrial matrix of the surrounding SCs via IP3 receptors (IP3R) of the endoplasmic reticulum (ER) and the mitochondrial calcium uniporter (MCU), respectively. The increase in the cytosolic Ca2+ concentration ([Ca2+]i) should be sufficient to trigger Cx32 hemichannel opening and ATP release, contributing to the intracellular and intercellular propagation of the Ca2+ signal. Interaction between Cx32 hemichannels and mitochondria may play a role in cell bioenergetics as found in liver of Cx32-null mice (Fowler et al., 2013). (B) Cx32 mutations belonging to classes 3-4-5 mentioned in the text are represented as colored circles (black-red-azure, respectively) associated to the correspondent WT amino acid (white circle), where the topology of Cx32 domains is derived from the all-atom model of Cx32 connexon in Carrer et al. (2018).
Together with the Cx32-null mouse (Nelles et al., 1996; Anzini et al., 1997; Balice-Gordon et al., 1998; Scherer et al., 1998, 2005; Zhao et al., 1999; Nicholson et al., 2001; Freidin et al., 2015; Sargiannidou et al., 2015; Kagiava et al., 2016), which displays a progressive peripheral neuropathy, a limited number of CMT1X mice models has been developed, including mutations G12S (Mones et al., 2012, 2014), S26L (Mones et al., 2012, 2014), T55I (Sargiannidou et al., 2009), R75W (Sargiannidou et al., 2009), R142W (Jeng et al., 2006), 175fs (Abel et al., 1999), C280G (Huang et al., 2005), S281X (Huang et al., 2005).
Discussion
A key feature that emerges from the study of CMT1X is that Cx32 is a fundamental protein in the PNS as its dysfunction cannot be compensated by other mechanisms. In particular, loss of Cx32 in SCs of Cx32-null mice did not induce any compensatory change in the expression of other connexins (Nicholson et al., 2001). The expression of Cx32 in the PNS appears regulated by the transcription factors SOX10 and EGR2 which directly bind Cx32 promoter with synergistic action (Bondurand et al., 2001). Robust expression of glial fibrillary acidic protein (GFAP), the only non-myelinating SC marker persisting in adult Cx32-null mice, suggests that Cx32 is involved in regulating GFAP levels and coordinating the program of myelin gene expression (Nicholson et al., 2001). A recent microarray analysis of normal and injured sciatic nerves of WT and Cx32-null mice supports a crucial role for Cx32 in re-myelination of SCs (Freidin et al., 2015), both in normal axonal maintenance and regeneration following peripheral nerve injury, during which Cx32 is downregulated (Scherer et al., 1995). The same study, together with others (Kobsar et al., 2003; Groh et al., 2012; Klein et al., 2015), indicates that loss of Cx32 dysregulates several genes associated with immune response, thus contributing to the severity of the disease.
The identification of so-called “functional” CMT1X mutations, which retain the capacity to ensure normal electrical GJ coupling in vitro, suggests that permeability or gating abnormalities of Cx32 channels are per se sufficient to trigger a severe neuropathy. As Cx32 WT GJ channels are known to be permeable to Ca2+, cAMP, cGMP, IP3 and ATP (Harris, 2007), it is possible that these “functional” mutations alter second messenger or other cytoplasmic molecules signals, causing downregulation in the expression of genes that are required to maintain the myelinating state of SCs. In 1997, Oh et al. (Oh et al., 1997) hypothesized that the primary defect underlying CMT1X neuropathy in the presence of Cx32 mutants forming electrically conductive channels is the lower permeability of GJ channels to cAMP, which is involved in myelin homeostasis in SCs (LeBlanc et al., 1992). We have recently demonstrated that this theory is unlikely, at least with regard to the most studied CMT1X mutant (R220X; Carrer et al., 2018). Indeed, lack of Cx32 GJs in myelinating SCs does not appear to cause a slower radial diffusion of low molecular weight dyes along the myelin sheath of Cx32-null mouse with respect to the WT due to the presence of other connexins forming GJ channels (Balice-Gordon et al., 1998). Nonetheless, Cx32-null mice develop a late-onset peripheral neuropathy with demyelination features similar to those found in humans with Cx32 mutations (Nelles et al., 1996; Anzini et al., 1997; Scherer et al., 1998), indicating that other functions performed by Cx32 in SCs could be involved in the pathogenesis of the disease. The validity of the Cx32-null mouse as a model of Cx32 function has been strengthened by the demonstration that both transgenic and lentiviral expression of Cx32 in myelinating SCs ameliorates nerve pathology (Scherer et al., 2005; Sargiannidou et al., 2015), improving motor performance (Kagiava et al., 2016). The hypothesis that the key role of Cx32 in the myelination process does not involve GJ channels was initially suggested in Freidin et al. (2009) and supported by the recent observation that defective Cx32 GJ plaque formation in 14 CMT1X mutants correlates only with CNS abnormalities (Abrams et al., 2017).
Despite the large amount of studies on mutant GJ properties, Cx32 hemichannel dysfunction was poorly investigated. Altered hemichannel gating properties could have devastating consequences for cellular function due to loss of ionic gradients and small metabolites and increased influx of Ca2+. A growing body of evidence also indicates that ATP-mediated paracrine signaling in SCs is critical for the myelination process, which supports the hypothesis that alteration of Cx32 hemichannel opening and ATP release could underlie CMT1X (Nualart-Marti et al., 2013; Carrer et al., 2018). Little is known about the physiological mechanism that controls opening and closure of Cx32 hemichannels in response to [Ca2+]i changes (Cotrina et al., 2000; Belliveau et al., 2006; De Vuyst et al., 2006; Carrer et al., 2018), but we recently found that Cx32 hemichannels carrying the pathological C-terminus truncation R220X fail to open in response to a canonical IP3-mediated signal transduction cascade that elevates [Ca2+]i (Carrer et al., 2018). A useful combination of patch-clamp and optical fluorescence microscopy for dissecting Cx32 channel properties is described in Figure 2. Interestingly, the gating function of R220X hemichannels was completely restored by both the intracellular and extracellular application of a 12 amino acid peptide that mimics the Cx32 cytoplasmic loop, suggesting that the C-terminal domain of Cx32 is not directly involved in the gating mechanism but acts as a modulator.
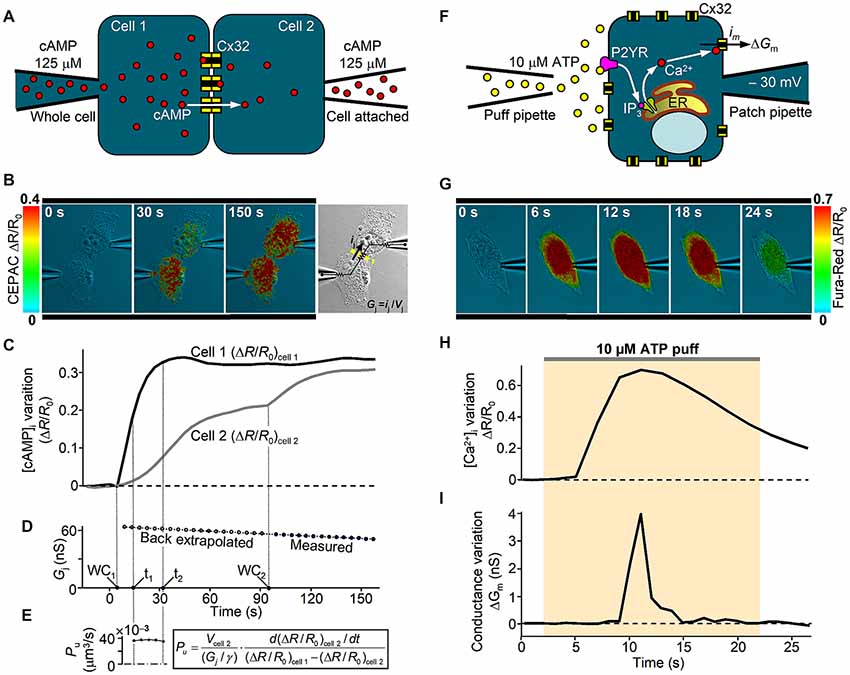
Figure 2. In vitro analysis of human Cx32 GJ and hemichannel functionality. (A) Scheme of dual patch clamp experiment to derive the unitary GJ permeability to cAMP in an isolated pair of Cx32-WT transfected HeLa cells, as in Carrer et al. (2018). At time zero, cAMP is injected in cell 1 under whole-cell recording conditions (WC1) and its intercellular transfer is monitored by FRET variation (ΔR/R0) of CEPAC sensor. (B) Three representative frames illustrate successive stages of an experiment. Around 90 s after WC1, the whole-cell configuration is achieved also in cell 2 (WC2), delivering the same concentration of cAMP and deriving the junctional conductance Gj from the current ij elicited by a 10 mV voltage difference (Vj) between the two pipettes, as illustrated in the bright field image. (C,D) Time course of the cell-averaged FRET signal and the junctional conductance Gj from the experiment in (B). (E) A confocal z-stack was performed at the end of the experiment to derive cell 2 volume (Vcell 2), which is required to compute the single channel permeability Pu to cAMP, as described in Hernandez et al. (2007), where γ is the single channel conductance. (F) Scheme of patch clamp experiment to study the hemichannel gating by [Ca2+]i in a single Cx32-WT transfected HeLa cell. An IP3-dependent [Ca2+]i transient is stimulated by an extracellular puff containing 10 μM ATP. Cx32 hemichannel opening and closure were monitored in terms of membrane conductance variation (ΔGm) computed by the periodic application (at 1 Hz) of a +10 μV voltage step lasting 100 ms. Contribution to ΔGm by other Ca2+-activated channels was kept negligible by specific blockers contained in the extracellular solution. (G) Representative frame sequence of an experiment using Fura-Red dye. (H,I) Time course from the experiment in (G) of the cell-averaged Fura-Red ΔR/R0 and the membrane conductance variation ΔGm due to opening and closure of Cx32-WT hemichannels. For further details, see Carrer et al. (2018).
Molecular determinants of permeability/gating properties investigated in GJ channels and hemichannels of any connexin isoform, including Cx32, are still debated (Peracchia, 2004; Zhou et al., 2009; Oh and Bargiello, 2015), but their elucidation could help to answer why different Cx32 mutations cause a similar phenotype equivalent to a loss-of-function in the PNS of CMT1X patients. In situ investigation of signaling pathways mediated by Cx32 in pre-myelinating and myelinated SCs is also a pre-requisite for understanding how Cx32 dysfunction deregulates axon/SC homeostasis causing the CMT1X phenotype.
Author Contributions
MB wrote the review article and prepared figures.
Funding
This work was supported by grants from the Italian Telethon Foundation (GGP12269) and the University of Padua (CPDA150248).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Prof. Camillo Peracchia and Prof. Michel Fontes for valuable discussions and constructive criticism.
References
Abel, A., Bone, L. J., Messing, A., Scherer, S. S., and Fischbeck, K. H. (1999). Studies in transgenic mice indicate a loss of connexin32 function in X-linked Charcot-Marie-Tooth disease. J. Neuropathol. Exp. Neurol. 58, 702–710. doi: 10.1097/00005072-199907000-00004
Abrams, C. K., Bennett, M. V., Verselis, V. K., and Bargiello, T. A. (2002). Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc. Natl. Acad. Sci. U S A 99, 3980–3984. doi: 10.1073/pnas.261713499
Abrams, C. K., and Freidin, M. (2015). GJB1-associated X-linked Charcot-Marie-Tooth disease, a disorder affecting the central and peripheral nervous systems. Cell Tissue Res. 360, 659–673. doi: 10.1007/s00441-014-2014-6
Abrams, C. K., Freidin, M., Bukauskas, F., Dobrenis, K., Bargiello, T. A., Verselis, V. K., et al. (2003). Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32. J. Neurosci. 23, 10548–10558. doi: 10.1523/jneurosci.23-33-10548.2003
Abrams, C. K., Freidin, M. M., Verselis, V. K., Bennett, M. V., and Bargiello, T. A. (2001). Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res. 900, 9–25. doi: 10.1016/s0006-8993(00)03327-8
Abrams, C. K., Goman, M., Wong, S., Scherer, S. S., Kleopa, K. A., Peinado, A., et al. (2017). Loss of coupling distinguishes GJB1 mutations associated with CNS manifestations of CMT1X from those without CNS manifestations. Sci. Rep. 7:40166. doi: 10.1038/srep40166
Abrams, C. K., Islam, M., Mahmoud, R., Kwon, T., Bargiello, T. A., and Freidin, M. M. (2013). Functional requirement for a highly conserved charged residue at position 75 in the gap junction protein connexin 32. J. Biol. Chem. 288, 3609–3619. doi: 10.1074/jbc.m112.392670
Ahn, M., Lee, J., Gustafsson, A., Enriquez, A., Lancaster, E., Sul, J. Y., et al. (2008). Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J. Neurosci. Res. 86, 992–1006. doi: 10.1002/jnr.21561
Ainsworth, P. J., Bolton, C. F., Murphy, B. C., Stuart, J. A., and Hahn, A. F. (1998). Genotype/phenotype correlation in affected individuals of a family with a deletion of the entire coding sequence of the connexin 32 gene. Hum. Genet. 103, 242–244. doi: 10.1007/s004390050812
Altevogt, B. M., Kleopa, K. A., Postma, F. R., Scherer, S. S., and Paul, D. L. (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22, 6458–6470. doi: 10.1523/jneurosci.22-15-06458.2002
Anzini, P., Neuberg, D. H., Schachner, M., Nelles, E., Willecke, K., Zielasek, J., et al. (1997). Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 17, 4545–4551. doi: 10.1523/jneurosci.17-12-04545.1997
Balice-Gordon, R. J., Bone, L. J., and Scherer, S. S. (1998). Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 142, 1095–1104. doi: 10.1083/jcb.142.4.1095
Belliveau, D. J., Bani-Yaghoub, M., Mcgirr, B., Naus, C. C., and Rushlow, W. J. (2006). Enhanced neurite outgrowth in PC12 cells mediated by connexin hemichannels and ATP. J. Biol. Chem. 281, 20920–20931. doi: 10.1074/jbc.m600026200
Bergoffen, J., Scherer, S. S., Wang, S., Scott, M. O., Bone, L. J., Paul, D. L., et al. (1993). Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042. doi: 10.1126/science.8266101
Bicego, M., Morassutto, S., Hernandez, V. H., Morgutti, M., Mammano, F., D’andrea, P., et al. (2006). Selective defects in channel permeability associated with Cx32 mutations causing X-linked Charcot-Marie-Tooth disease. Neurobiol. Dis. 21, 607–617. doi: 10.1016/j.nbd.2005.09.005
Bondurand, N., Girard, M., Pingault, V., Lemort, N., Dubourg, O., and Goossens, M. (2001). Human Connexin 32, a gap junction protein altered in the X-linked form of Charcot-Marie-Tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet. 10, 2783–2795. doi: 10.1093/hmg/10.24.2783
Carrer, A., Leparulo, A., Crispino, G., Ciubotaru, C. D., Marin, O., Zonta, F., et al. (2018). Cx32 hemichannel opening by cytosolic Ca2+ is inhibited by the R220X mutation that causes Charcot-Marie-Tooth disease. Hum. Mol. Genet. 27, 80–94. doi: 10.1093/hmg/ddx386
Castro, C., Gómez-Hernández, J. M., Silander, K., and Barrio, L. C. (1999). Altered formation of hemichannels and gap junction channels caused by C-terminal connexin-32 mutations. J. Neurosci. 19, 3752–3760. doi: 10.1523/jneurosci.19-10-03752.1999
Cotrina, M. L., Lin, J. H., Lopez-Garcia, J. C., Naus, C. C., and Nedergaard, M. (2000). ATP-mediated glia signaling. J. Neurosci. 20, 2835–2844. doi: 10.1523/JNEUROSCI.20-08-02835.2000
De Vuyst, E., Decrock, E., Cabooter, L., Dubyak, G. R., Naus, C. C., Evans, W. H., et al. (2006). Intracellular calcium changes trigger connexin 32 hemichannel opening. EMBO J. 25, 34–44. doi: 10.1038/sj.emboj.7600908
De Vuyst, E., Wang, N., Decrock, E., De Bock, M., Vinken, M., Van Moorhem, M., et al. (2009). Ca2+ regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium 46, 176–187. doi: 10.1016/j.ceca.2009.07.002
Deschenes, S. M., Walcott, J. L., Wexler, T. L., Scherer, S. S., and Fischbeck, K. H. (1997). Altered trafficking of mutant connexin32. J. Neurosci. 17, 9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997
Fields, R. D., and Ni, Y. (2010). Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci. Signal. 3:ra73. doi: 10.1126/scisignal.2001128
Fleishman, S. J., Sabag, A. D., Ophir, E., Avraham, K. B., and Ben-Tal, N. (2006). The structural context of disease-causing mutations in gap junctions. J. Biol. Chem. 281, 28958–28963. doi: 10.1074/jbc.M605764200
Fowler, S. L., Akins, M., Zhou, H., Figeys, D., and Bennett, S. A. (2013). The liver connexin32 interactome is a novel plasma membrane-mitochondrial signaling nexus. J. Proteome Res. 12, 2597–2610. doi: 10.1021/pr301166p
Fowler, S., Mailloux, R., Akins, M., Zhou, H., Figeys, D., Harper, M.-E., et al. (2017). “Altered proteomic and respiratory characteristics of Cx32Ko mitochondria,” in Proceedings of the 2017 International Gap Junction Conference, (Glasgow). Available online at: http://igjc2017.com/wp-content/uploads/2017/07/Book-of-Abstracts_26th-July.pdf
Freidin, M., Asche, S., Bargiello, T. A., Bennett, M. V., and Abrams, C. K. (2009). Connexin 32 increases the proliferative response of Schwann cells to neuregulin-1 (Nrg1). Proc. Natl. Acad. Sci. U S A 106, 3567–3572. doi: 10.1073/pnas.0813413106
Freidin, M., Asche-Godin, S., and Abrams, C. K. (2015). Gene expression profiling studies in regenerating nerves in a mouse model for CMT1X: uninjured Cx32-knockout peripheral nerves display expression profile of injured wild type nerves. Exp. Neurol. 263, 339–349. doi: 10.1016/j.expneurol.2014.10.014
Gómez-Hernández, J. M., de Miguel, M., Larrosa, B., González, D., and Barrio, L. C. (2003). Molecular basis of calcium regulation in connexin-32 hemichannels. Proc. Natl. Acad. Sci. U S A 100, 16030–16035. doi: 10.1073/pnas.2530348100
Gong, X. Q., Nakagawa, S., Tsukihara, T., and Bai, D. (2013). A mechanism of gap junction docking revealed by functional rescue of a human-disease-linked connexin mutant. J. Cell Sci. 126, 3113–3120. doi: 10.1242/jcs.123430
Groh, J., Weis, J., Zieger, H., Stanley, E. R., Heuer, H., and Martini, R. (2012). Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot-Marie-Tooth disease type 1X. Brain 135, 88–104. doi: 10.1093/brain/awr283
Harding, A. E. (1995). From the syndrome of Charcot, Marie and Tooth to disorders of peripheral myelin proteins. Brain 118, 809–818. doi: 10.1093/brain/118.3.809
Harris, A. L. (2007). Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 94, 120–143. doi: 10.1016/j.pbiomolbio.2007.03.011
Hernandez, V. H., Bortolozzi, M., Pertegato, V., Beltramello, M., Giarin, M., Zaccolo, M., et al. (2007). Unitary permeability of gap junction channels to second messengers measured by FRET microscopy. Nat. Methods 4, 353–358. doi: 10.1038/nmeth1031
Hu, Z., Riquelme, M. A., Wang, B., Bugay, V., Brenner, R., Gu, S., et al. (2018). Cataract-associated connexin 46 mutation alters its interaction with calmodulin and function of hemichannels. J. Biol. Chem. 393, 2573–2585. doi: 10.1074/jbc.RA117.001348
Huang, Y., Sirkowski, E. E., Stickney, J. T., and Scherer, S. S. (2005). Prenylation-defective human connexin32 mutants are normally localized and function equivalently to wild-type connexin32 in myelinating Schwann cells. J. Neurosci. 25, 7111–7120. doi: 10.1523/JNEUROSCI.1319-05.2005
Ino, D., Sagara, H., Suzuki, J., Kanemaru, K., Okubo, Y., and Iino, M. (2015). Neuronal regulation of schwann cell mitochondrial Ca2+ signaling during myelination. Cell Rep. 12, 1951–1959. doi: 10.1016/j.celrep.2015.08.039
Ionasescu, V. V., Searby, C., Ionasescu, R., Neuhaus, I. M., and Werner, R. (1996). Mutations of the noncoding region of the connexin32 gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Neurology 47, 541–544. doi: 10.1212/WNL.47.2.541
Jeng, L. J., Balice-Gordon, R. J., Messing, A., Fischbeck, K. H., and Scherer, S. S. (2006). The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol. Cell. Neurosci. 32, 283–298. doi: 10.1016/j.mcn.2006.05.001
Kagiava, A., Sargiannidou, I., Theophilidis, G., Karaiskos, C., Richter, J., Bashiardes, S., et al. (2016). Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. U S A 113, E2421–E2429. doi: 10.1073/pnas.1522202113
Kalmatsky, B. D., Batir, Y., Bargiello, T. A., and Dowd, T. L. (2012). Structural studies of N-terminal mutants of connexin 32 using 1H NMR spectroscopy. Arch. Biochem. Biophys. 526, 1–8. doi: 10.1016/j.abb.2012.05.027
Kalmatsky, B. D., Bhagan, S., Tang, Q., Bargiello, T. A., and Dowd, T. L. (2009). Structural studies of the N-terminus of Connexin 32 using 1H NMR spectroscopy. Arch. Biochem. Biophys. 490, 9–16. doi: 10.1016/j.abb.2009.07.015
Katoch, P., Mitra, S., Ray, A., Kelsey, L., Roberts, B. J., Wahl, J. K. III, et al. (2015). The carboxyl tail of connexin32 regulates gap junction assembly in human prostate and pancreatic cancer cells. J. Biol. Chem. 290, 4647–4662. doi: 10.1074/jbc.M114.586057
Klein, D., Patzkó, Á., Schreiber, D., van Hauwermeiren, A., Baier, M., Groh, J., et al. (2015). Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice. Brain 138, 3193–3205. doi: 10.1093/brain/awv240
Kleopa, K. A. (2011). The role of gap junctions in charcot-marie-tooth disease. J. Neurosci. 31, 17753–17760. doi: 10.1523/JNEUROSCI.4824-11.2011
Kleopa, K. A., Abrams, C. K., and Scherer, S. S. (2012). How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res. 1487, 198–205. doi: 10.1016/j.brainres.2012.03.068
Kleopa, K. A., and Scherer, S. S. (2006). Molecular genetics of X-linked Charcot-Marie-Tooth disease. Neuromolecular Med. 8, 107–122. doi: 10.1385/nmm:8:1:107
Kleopa, K. A., Yum, S. W., and Scherer, S. S. (2002). Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J. Neurosci. Res. 68, 522–534. doi: 10.1002/jnr.10255
Kleopa, K. A., Zamba-Papanicolaou, E., Alevra, X., Nicolaou, P., Georgiou, D. M., Hadjisavvas, A., et al. (2006). Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology 66, 396–402. doi: 10.1212/01.WNL.0000196479.93722.59
Kobsar, I., Berghoff, M., Samsam, M., Wessig, C., Mäurer, M., Toyka, K. V., et al. (2003). Preserved myelin integrity and reduced axonopathy in connexin32-deficient mice lacking the recombination activating gene-1. Brain 126, 804–813. doi: 10.1093/brain/awg072
LeBlanc, A. C., Windebank, A. J., and Poduslo, J. F. (1992). P0 gene expression in Schwann cells is modulated by an increase of cAMP which is dependent on the presence of axons. Mol. Brain Res. 12, 31–38. doi: 10.1016/0169-328x(92)90065-j
Lev-Ram, V., and Ellisman, M. H. (1995). Axonal activation-induced calcium transients in myelinating Schwann cells, sources, and mechanisms. J. Neurosci. 15, 2628–2637. doi: 10.1523/JNEUROSCI.15-04-02628.1995
Li, X., Lynn, B. D., Olson, C., Meier, C., Davidson, K. G., Yasumura, T., et al. (2002). Connexin29 expression, immunocytochemistry and freeze-fracture replica immunogold labelling (FRIL) in sciatic nerve. Eur. J. Neurosci. 16, 795–806. doi: 10.1046/j.1460-9568.2002.02149.x
Liang, G. S., de Miguel, M., Gómez-Hernández, J. M., Glass, J. D., Scherer, S. S., Mintz, M., et al. (2005). Severe neuropathy with leaky connexin32 hemichannels. Ann. Neurol. 57, 749–754. doi: 10.1002/ana.20459
Lyons, S. A., Morell, P., and McCarthy, K. D. (1995). Schwann cell ATP-mediated calcium increases in vitro and in situ are dependent on contact with neurons. Glia 13, 27–38. doi: 10.1002/glia.440130104
Mambetisaeva, E. T., Gire, V., and Evans, W. H. (1999). Multiple connexin expression in peripheral nerve, Schwann cells, and Schwannoma cells. J. Neurosci. Res. 57, 166–175. doi: 10.1002/(sici)1097-4547(19990715)57:2<166::aid-jnr2>3.3.co;2-p
Martin, P. E., Mambetisaeva, E. T., Archer, D. A., George, C. H., and Evans, W. H. (2000). Analysis of gap junction assembly using mutated connexins detected in Charcot-Marie-Tooth X-linked disease. J. Neurochem. 74, 711–720. doi: 10.1046/j.1471-4159.2000.740711.x
Martínez-Gómez, A., and Dent, M. A. (2007). Expression of IP3 receptor isoforms at the nodes of Ranvier in rat sciatic nerve. Neuroreport 18, 447–450. doi: 10.1097/WNR.0b013e32805868a6
Mayer, C., Wachtler, J., Kamleiter, M., and Grafe, P. (1997). Intracellular calcium transients mediated by P2 receptors in the paranodal Schwann cell region of myelinated rat spinal root axons. Neurosci. Lett. 224, 49–52. doi: 10.1016/s0304-3940(97)13457-7
Meier, C., Dermietzel, R., Davidson, K. G., Yasumura, T., and Rash, J. E. (2004). Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt-Lanterman incisures. J. Neurosci. 24, 3186–3198. doi: 10.1523/JNEUROSCI.5146-03.2004
Mones, S., Bordignon, B., and Fontes, M. (2012). Connexin 32 is involved in mitosis. Glia 60, 457–464. doi: 10.1002/glia.22279
Mones, S., Bordignon, B., Peiretti, F., Landrier, J. F., Gess, B., Bourguignon, J. J., et al. (2014). CamKII inhibitors reduce mitotic instability, connexon anomalies and progression of the in vivo behavioral phenotype in transgenic animals expressing a mutated Gjb1 gene. Front. Neurosci. 8:151. doi: 10.3389/fnins.2014.00151
Nakagawa, S., Gong, X. Q., Maeda, S., Dong, Y., Misumi, Y., Tsukihara, T., et al. (2011). Asparagine 175 of connexin32 is a critical residue for docking and forming functional heterotypic gap junction channels with connexin26. J. Biol. Chem. 286, 19672–19681. doi: 10.1074/jbc.M110.204958
Nave, K. A., and Trapp, B. D. (2008). Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 31, 535–561. doi: 10.1146/annurev.neuro.30.051606.094309
Nelles, E., Bützler, C., Jung, D., Temme, A., Gabriel, H. D., Dahl, U., et al. (1996). Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc. Natl. Acad. Sci. U S A 93, 9565–9570. doi: 10.1073/pnas.93.18.9565
Neumann, B., Walter, T., Hériché, J. K., Bulkescher, J., Erfle, H., Conrad, C., et al. (2010). Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727. doi: 10.1038/nature08869
Nicholson, S. M., Gomes, D., de Néchaud, B., and Bruzzone, R. (2001). Altered gene expression in Schwann cells of connexin32 knockout animals. J. Neurosci. Res. 66, 23–36. doi: 10.1002/jnr.1194
Nualart-Marti, A., Del Molino, E. M., Grandes, X., Bahima, L., Martin-Satué, M., Puchal, R., et al. (2013). Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 61, 1976–1989. doi: 10.1002/glia.22568
Oh, S., and Bargiello, T. A. (2015). Voltage regulation of connexin channel conductance. Yonsei Med. J. 56, 1–15. doi: 10.3349/ymj.2015.56.1.1
Oh, S., Ri, Y., Bennett, M. V., Trexler, E. B., Verselis, V. K., and Bargiello, T. A. (1997). Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 19, 927–938. doi: 10.1016/s0896-6273(00)80973-3
Oh, S., Rubin, J. B., Bennett, M. V., Verselis, V. K., and Bargiello, T. A. (1999). Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J. Gen. Physiol. 114, 339–364. doi: 10.1085/jgp.114.3.339
Omori, Y., Mesnil, M., and Yamasaki, H. (1996). Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol. Biol. Cell 7, 907–916. doi: 10.1091/mbc.7.6.907
Paul, D. L. (1986). Molecular cloning of cDNA for rat liver gap junction protein. J. Cell Biol. 103, 123–134. doi: 10.1083/jcb.103.1.123
Peracchia, C. (2004). Chemical gating of gap junction channels; roles of calcium, pH and calmodulin. Biochim. Biophys. Acta 1662, 61–80. doi: 10.1016/j.bbamem.2003.10.020
Procacci, P., Magnaghi, V., and Pannese, E. (2008). Perineuronal satellite cells in mouse spinal ganglia express the gap junction protein connexin43 throughout life with decline in old age. Brain Res. Bull. 75, 562–569. doi: 10.1016/j.brainresbull.2007.09.007
Rabadan-Diehl, C., Dahl, G., and Werner, R. (1994). A connexin-32 mutation associated with Charcot-Marie-Tooth disease does not affect channel formation in oocytes. FEBS Lett. 351, 90–94. doi: 10.1016/0014-5793(94)00819-1
Rash, J. E., Yasumura, T., Dudek, F. E., and Nagy, J. I. (2001). Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J. Neurosci. 21, 1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001
Ressot, C., Gomes, D., Dautigny, A., Pham-Dinh, D., and Bruzzone, R. (1998). Connexin32 mutations associated with X-linked Charcot-Marie-Tooth disease show two distinct behaviors: loss of function and altered gating properties. J. Neurosci. 18, 4063–4075. doi: 10.1523/JNEUROSCI.18-11-04063.1998
Revilla, A., Castro, C., and Barrio, L. C. (1999). Molecular dissection of transjunctional voltage dependence in the connexin-32 and connexin-43 junctions. Biophys. J. 77, 1374–1383. doi: 10.1016/s0006-3495(99)76986-9
Sargiannidou, I., Ahn, M., Enriquez, A. D., Peinado, A., Reynolds, R., Abrams, C., et al. (2008). Human oligodendrocytes express Cx31.3: function and interactions with Cx32 mutants. Neurobiol. Dis. 30, 221–233. doi: 10.1016/j.nbd.2008.01.009
Sargiannidou, I., Kagiava, A., Bashiardes, S., Richter, J., Christodoulou, C., Scherer, S. S., et al. (2015). Intraneural GJB1 gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 78, 303–316. doi: 10.1002/ana.24441
Sargiannidou, I., Vavlitou, N., Aristodemou, S., Hadjisavvas, A., Kyriacou, K., Scherer, S. S., et al. (2009). Connexin32 mutations cause loss of function in Schwann cells and oligodendrocytes leading to PNS and CNS myelination defects. J. Neurosci. 29, 4736–4749. doi: 10.1523/JNEUROSCI.0325-09.2009
Scherer, S. S., Deschenes, S. M., Xu, Y. T., Grinspan, J. B., Fischbeck, K. H., and Paul, D. L. (1995). Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 15, 8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995
Scherer, S. S., Xu, Y. T., Messing, A., Willecke, K., Fischbeck, K. H., and Jeng, L. J. (2005). Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J. Neurosci. 25, 1550–1559. doi: 10.1523/JNEUROSCI.3082-04.2005
Scherer, S. S., Xu, Y. T., Nelles, E., Fischbeck, K., Willecke, K., and Bone, L. J. (1998). Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24, 8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3
Shy, M. E., Siskind, C., Swan, E. R., Krajewski, K. M., Doherty, T., Fuerst, D. R., et al. (2007). CMT1X phenotypes represent loss of GJB1 gene function. Neurology 68, 849–855. doi: 10.1212/01.WNL.0000256709.08271.4d
Stenson, P. D., Mort, M., Ball, E. V., Shaw, K., Phillips, A., and Cooper, D. N. (2014). The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9. doi: 10.1007/s00439-013-1358-4
Stevens, B., and Fields, R. D. (2000). Response of Schwann cells to action potentials in development. Science 287, 2267–2271. doi: 10.1126/science.287.5461.2267
Suter, U., and Snipes, G. J. (1995). Biology and genetics of hereditary motor and sensory neuropathies. Annu. Rev. Neurosci. 18, 45–75. doi: 10.1146/annurev.ne.18.030195.000401
Toews, J. C., Schram, V., Weerth, S. H., Mignery, G. A., and Russell, J. T. (2007). Signaling proteins in the axoglial apparatus of sciatic nerve nodes of Ranvier. Glia 55, 202–213. doi: 10.1002/glia.20448
Tomaselli, P. J., Rossor, A. M., Horga, A., Jaunmuktane, Z., Carr, A., Saveri, P., et al. (2017). Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurology 88, 1445–1453. doi: 10.1212/WNL.0000000000003819
Vance, J. M. (1991). Hereditary motor and sensory neuropathies. J. Med. Genet. 28, 1–5. doi: 10.1136/jmg.28.1.1
Vanslyke, J. K., Deschenes, S. M., and Musil, L. S. (2000). Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol. Biol. Cell 11, 1933–1946. doi: 10.1091/mbc.11.6.1933
Vavlitou, N., Sargiannidou, I., Markoullis, K., Kyriacou, K., Scherer, S. S., and Kleopa, K. A. (2010). Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/type I Charcot-Marie Tooth neuropathy. J. Neuropathol. Exp. Neurol. 69, 945–958. doi: 10.1097/NEN.0b013e3181efa658
Waggener, C. T., Dupree, J. L., Elgersma, Y., and Fuss, B. (2013). CaMKIIβ regulates oligodendrocyte maturation and CNS myelination. J. Neurosci. 33, 10453–10458. doi: 10.1523/JNEUROSCI.5875-12.2013
Wang, H. L., Chang, W. T., Yeh, T. H., Wu, T., Chen, M. S., and Wu, C. Y. (2004). Functional analysis of connexin-32 mutants associated with X-linked dominant Charcot-Marie-Tooth disease. Neurobiol. Dis. 15, 361–370. doi: 10.1016/j.nbd.2003.11.005
Wang, Y., and Yin, F. (2016). A review of X-linked charcot-marie-tooth disease. J. Child Neurol. 31, 761–772. doi: 10.1177/0883073815604227
Yoshimura, T., Satake, M., and Kobayashi, T. (1996). Connexin43 is another gap junction protein in the peripheral nervous system. J. Neurochem. 67, 1252–1258. doi: 10.1046/j.1471-4159.1996.67031252.x
Yoshimura, T., Satake, M., Ohnishi, A., Tsutsumi, Y., and Fujikura, Y. (1998). Mutations of connexin32 in Charcot-Marie-Tooth disease type X interfere with cell-to-cell communication but not cell proliferation and myelin-specific gene expression. J. Neurosci. Res. 51, 154–161. doi: 10.1002/(sici)1097-4547(19980115)51:2<154::aid-jnr4>3.3.co;2-e
Yum, S. W., Kleopa, K. A., Shumas, S., and Scherer, S. S. (2002). Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol. Dis. 11, 43–52. doi: 10.1006/nbdi.2002.0545
Zhang, X., Zou, T., Liu, Y., and Qi, Y. (2006). The gating effect of calmodulin and calcium on the connexin50 hemichannel. Biol. Chem. 387, 595–601. doi: 10.1515/bc.2006.076
Zhao, S., Fort, A., and Spray, D. C. (1999). Characteristics of gap junction channels in Schwann cells from wild-type and connexin-null mice. Ann. N Y Acad. Sci. 883, 533–537. doi: 10.1111/j.1749-6632.1999.tb08630.x
Keywords: Cx32, Connexin 32, Schwann cell, CMT1X, CMTX1, Charcot-Marie-Tooth disease, gap junction, hemichannel
Citation: Bortolozzi M (2018) What’s the Function of Connexin 32 in the Peripheral Nervous System? Front. Mol. Neurosci. 11:227. doi: 10.3389/fnmol.2018.00227
Received: 14 March 2018; Accepted: 11 June 2018;
Published: 10 July 2018.
Edited by:
Juan Andrés Orellana, Pontificia Universidad Católica de Chile, ChileReviewed by:
Kleopas A. Kleopa, The Cyprus Institute of Neurology and Genetics, CyprusValerio Magnaghi, Università degli Studi di Milano, Italy
Copyright © 2018 Bortolozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Bortolozzi, bWFyaW8uYm9ydG9sb3p6aUB1bmlwZC5pdA==