 Rafael Falcón-Moya
Rafael Falcón-Moya Talvinder S. Sihra
Talvinder S. Sihra Antonio Rodríguez-Moreno
Antonio Rodríguez-Moreno- 1Laboratory of Cellular Neuroscience and Plasticity, Department of Physiology, Anatomy and Cell Biology, University Pablo de Olavide, Sevilla, Spain
- 2Department of Physiology, Pharmacology and Neuroscience, University College London, London, United Kingdom
Kainate (KA) is a potent neurotoxin that has been widely used experimentally to induce acute brain seizures and, after repetitive treatments, as a chronic model of temporal lobe epilepsy (TLE), with similar features to those observed in human patients with TLE. However, whether KA activates KA receptors (KARs) as an agonist to mediate the induction of acute seizures and/or the chronic phase of epilepsy, or whether epileptogenic effects of the neurotoxin are indirect and/or mediated by other types of receptors, has yet to be satisfactorily elucidated. Positing a direct involvement of KARs in acute seizures induction, as well as a direct pathophysiological role of KARs in the chronic phase of TLE, recent studies have examined the specific subunit compositions of KARs that might underly epileptogenesis. In the present mini-review, we discuss the use of KA as a convulsant in the experimental models of acute seizures of TLE, and consider the involvement of KARs, their subunit composition and the mode of action in KAR-mediated epilepsy. In acute models, evidence points to epileptogenesis being precipitated by an overall depression of interneuron GABAergic transmission mediated by GluK1 containing KARs. On glutamatergic principal cell in the hippocampus, GluK2-containing KARs regulate post-synaptic excitability and susceptibility to KA-mediated epileptogenesis. In chronic models, a role GluK2-containing KARs in the hippocampal CA3 region provokes limbic seizures. Also observed in the hippocampus, is a ‘reactive plasticity’, where MF sprouting is seen with target granule cells at aberrant synapses recruiting de novo GluR2/GluR5 heteromeric KARs. Finally, in human epilepsy and animal models, astrocytic expression of GluK1, 2, 4, and 5 is reported.
Introduction
The analog of glutamate kainic acid/kainate (KA) is a potent neurotoxin derived from the alga Digenea simplex. The word “Kainic” is derived from the Japanese “Kaininso” (“Makuri”), which means “the ghost of the sea”. KA was first isolated in 1953, from D. simplex, and therapeutically utilized, given its toxicity towards intestinal worms. Thereafter, KA was used to induce behavioral and electrophysiological seizures, establishing a model of temporal lobe epilepsy (TLE), where seizures originated in the hippocampal CA3 region (Nadler, 1981). Indeed, the toxin produces tissue damage and lesions that are reminiscent of those observed in human patients with TLE. However, while the acute effect of KA in producing seizures is well-established, its cogency in the clinically relevant chronic phase of TLE remains to be determined.
Kainate actions are largely mediated by the activation of kainate receptors (KARs) at which KA is a high affinity agonist. KARs, together with AMPA and NMDA receptors, comprise the ionotropic receptors family of glutamate receptors. Additionally, there is now considerable evidence that KARs have metabotropic/non-canonical actions (Rodríguez-Moreno and Sihra, 2007a,b). Given that metabotropic actions have also been described for AMPA, and recently for NMDA receptors (Valbuena and Lerma, 2016; Bouvier et al., 2018), the distinction between the signaling mediated by ionotropic and metabotropic glutamate receptors is increasingly blurred. KARs are specifically comprised of GluK1-GluK5 subunits. In expression systems, GluK1, GluK2, and GluK3 may form homomeric receptors, while GluK4 and GluK5 form heteromeric receptors in conjunction with GluK1–3 subunits. Native KARs are widely distributed in the brain (Bahn et al., 1994), with high densities of receptors found in the hippocampus, a key structure featuring in TLE. Hippocampal KARs containing GluK1 subunits are mostly expressed in interneurons, where the reduction of GABA release results in the increased excitability of glutamatergic principal neurons (Clarke et al., 1997; Rodríguez-Moreno et al., 1997, 2000; Rodríguez-Moreno and Lerma, 1998), or enhance inhibition by increasing GABA release (Cossart et al., 1998; Frerking et al., 1998; Jiang et al., 2001). GluK2 subunit containing KARs are mostly located in excitatory neurons, where they modulate glutamate release presynaptically (Chittajallu et al., 1996; Vignes et al., 1998; Contractor et al., 2000, 2001, 2003, Kamiya and Ozawa, 2000; Schmitz et al., 2000, 2001; Lauri et al., 2001; Rodríguez-Moreno and Sihra, 2004, 2013; Negrete-Díaz et al., 2006, 2007, 2012, Pinheiro et al., 2007; Scott et al., 2008; Fernandes et al., 2009; Lyon et al., 2011; Andrade-Talavera et al., 2012, 2013; reviewed in Sihra and Rodríguez-Moreno, 2013), and post-synaptically, underpin part of the synaptic component of excitatory transmission (Castillo et al., 1997; Vignes et al., 1997). This differential subcellular localization allows KARs to regulate the actions of neural circuits in the hippocampus.
Epilepsy is a chronic neurological disorder characterized by the occurrence of spontaneous recurrent seizures (prolonged and synchronized neuronal discharges). TLE is the most common form of human epilepsy. Recurrent seizures originate from diverse structures, usually the hippocampus, and sometimes may propagate to the amygdala and the entorhinal cortex (Lothman et al., 1991). Current antiepileptic drugs that are effective in controlling seizures in patients with TLE, either act on voltage- gated Na+ channels and Ca2+ channels, to modify neuronal excitability, or enhance GABAergic inhibition. However, although this anticonvulsant drug therapy reduces overall neuronal excitability in the brain, there are unwanted side effects (Perucca and Meador, 2005; Goldenberg, 2010). And, indeed, in many cases where TLE is severe, it is intractable to current drug therapy. Looking to alternative antiepileptic drug targets, while the GluK1 subunit-containing KAR represents a potential candidate (Rogawski et al., 2003), there is no anticonvulsant therapy based on KARs signaling that is currently in use in humans.
Models for seizures involving KAR activation can be acute or chronic. KA or pilocarpine injections represent acute models of epileptiform seizures. In chronic models, systemic administration of the aforementioned agents is continued for 1–3 days. In a high proportion of injected animals treated in this way, after a lag of some days or weeks without any seizures, a chronic phase of spontaneous recurrent limbic seizures is precipitated. These seizures increase in frequency and do not resolve (Ben-Ari, 1985; Leite et al., 1990). KA-induced seizures in animals produce patterns of activity-induced neuronal and cell loss, astrogliosis and hippocampal sclerosis of CA1 and CA3 pyramidal cells (PyC), that resemble characteristics of human TLE (Nadler et al., 1978; Ben-Ari, 1985).
Kar Role in Acute Seizures
The majority of studies of KARs related to epilepsy have investigated the acute effects of KA-induced seizures (Rodríguez-Moreno et al., 1997; Mulle et al., 1998; Smolders et al., 2002; Fritsch et al., 2014). KA injection produces an acute epileptogenesis mediated by KAR-mediated suppression of presynaptic GABA release, together with post-synaptic KAR activation of glutamatergic neurons (Rodríguez-Moreno et al., 1997; reviewed in Lerma and Marques, 2013). However, to date, there is no explanation for the chronic effect of KA that continues some months after the KA treatment. Hippocampal interneurons in the CA1 region of the hippocampus possess GluK1 subunit containing KARs in the axonal compartment, as well as in the somatodendritic compartment (Paternain et al., 2000; Rodríguez-Moreno et al., 2000). KA at interneuron-interneuron synapses facilitates GABA release, and thus inhibitory drive (Cossart et al., 1998). At interneurons-principal cells synapses, hippocampal interneurons manifest a biphasic effect of KA. Activation of KARs by “high” doses of KA suppresses GABA release (Clarke et al., 1997; Rodríguez-Moreno et al., 1997), while stimulation of KARs by “low” KA concentrations, or ATPA (an agonist of GluK5 subunit containing KARs), facilitates GABA release (Jiang et al., 2001; Khalilov et al., 2002). However, contrary to what would be predicted by the latter observation, in vivo, the systemic administration of ATPA actually induced seizures in the hippocampus and the amygdala. This effect was contingent on the presence of GluK1 given that it was abolished in GluK1-/- mice (Fritsch et al., 2014). Consistent with this, in vivo antagonism of KARs containing the GluK1 subunit, blocked seizures induced by the muscarinic receptor agonist pilocarpine (Smolders et al., 2002). These observations indicate that the regulation of inhibition is key for the potential action of KA on KARs and epilepsy. Thus, in vivo, the net effect of the activation of KARs seems to be an overall depression of GABA release leading to increased excitation and thereby epileptiform activity. This is, to date, one of the best pathophysiological demonstrations of the net effects of KA in epileptgenesis (Rodríguez-Moreno et al., 1997; Figure 1A). Post-synaptic KARs have also been found in the principal cells of the hippocampus. Here the KARs contain at least one GluK2 subunit, and their activation mediates the regulation of afterhyperpolarization. Alteration of the latter affects neuronal excitability and may therefore contribute to susceptibility to the pro-convulsant effects of KA (Melyan et al., 2002, 2004).
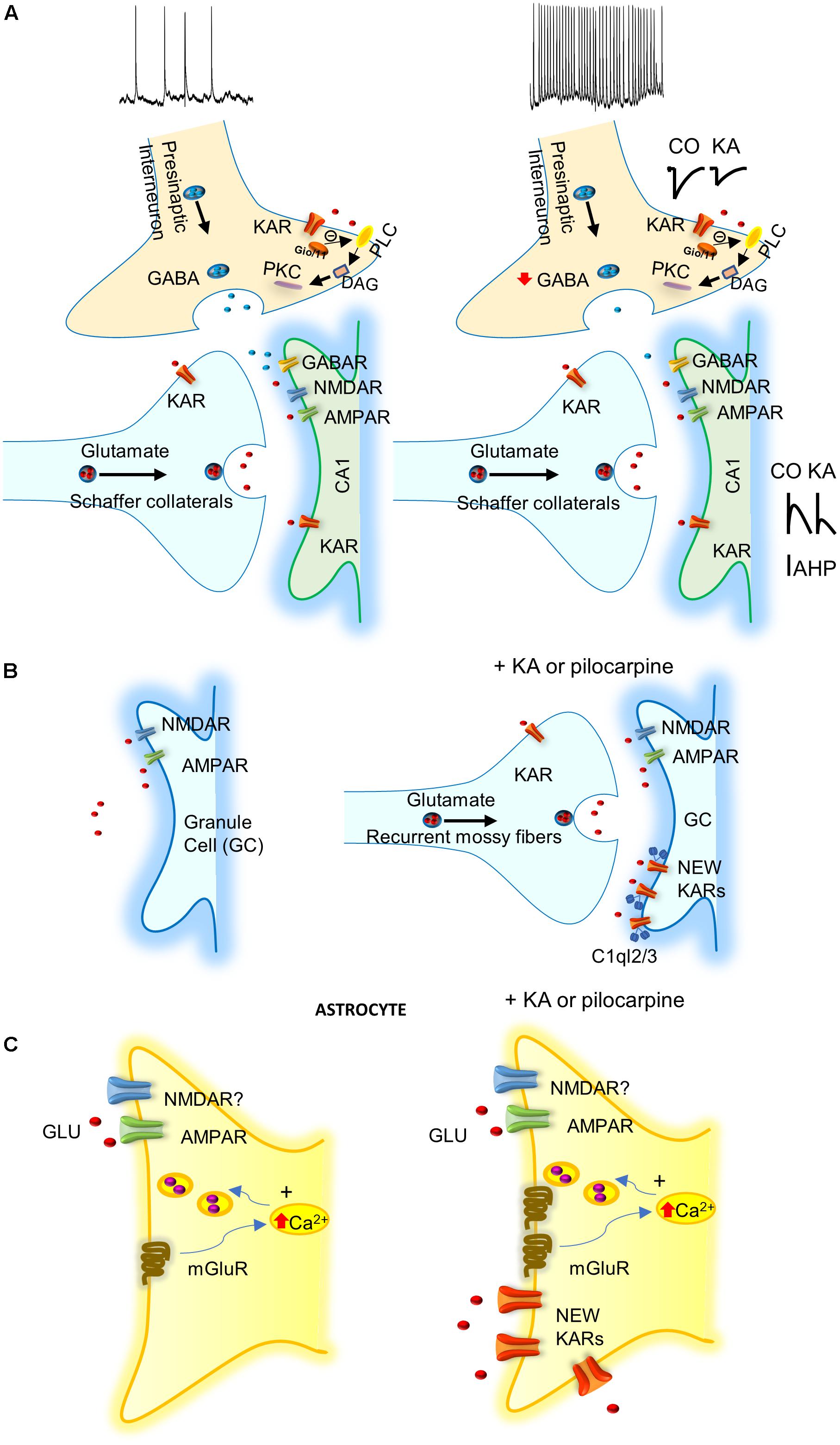
FIGURE 1. Possible mechanisms involving KARs in epilepsy. (A) KARs activation depress GABA release what together with the activation of post-synaptic KARs increase excitability of principal cells. IAHP, afterhyperpolarization potassium current is decreased by KARs activation increasing neuronal excitability. (B) KARs induce “reactive plasticity” and mediate the insertion of new post-synaptic KARs at granule cells (GCs) of dentate gyrus altering spiking. C1ql2/3 (C1q-like proteins, related to the C1q complement component) recruit KARs at GCs after pilocarpine treatment. (C) KA or pilocarpine treatments mediate the insertion of KARs in astrocytes that probably releases gliotransmitter’s to participate in protection of induction of epilepsy.
In agreement with a role of KARs containing GluK1 subunit in seizure induction, in a study of members of a family affected by idiopathic juvenile absence epilepsy, there were increased levels of Grik1 polymorphisms (Sander et al., 1997). Additionally data from TLE patients also showed that the GluK1 subunit is upregulated (Li et al., 2010), indicating that there is KAR reorganization occurring in human epilepsy. Mitigating against this, the AMPA/KAR antagonist NS1209, has been shown, in clinical Phase II studies, to alleviate refractory status epilepticus (Swanson, 2009).
In summary, in ex vivo and in in vivo models, in the hippocampus and the amygdala, KA has been found to reduce GABA release. This attenuates the inhibition of hippocampal pyramidal cells and is thereby posited to provoke epileptic activity. It has also been shown that the use of antagonists of GluK1 subunit-containing KARs prevented epileptic activity, thus corroborating a key role for synaptic inhibition in the KAR-induced seizures. Post-synaptic KARs activation mediate an increase in excitability. As a general mechanism then, the proposal is that the activation of KARs orchestrates an imbalance between excitation and inhibition, and this represent one way that KA activating KARs induces acute seizures (Figure 1A).
Kars Role in Chronic Seizures
Since the 1970s, it has been known that the CA3 region of the hippocampus is a key area related to the origin of seizures (reviewed in Ben-Ari and Cossart, 2000). KARs containing the GluK2 subunit have been linked to limbic epilepsies because of its specific distribution in CA3 pyramidal neurons (Werner et al., 1991). Consistent with a direct role of KARs in the induction of epilepsy in this region, the ablation of GluK2 subunits in knockout studies reduced the sensitivity of the mice to develop seizures after KA injection (Mulle et al., 1998).
In animal models of TLE and human patients, neuronal tissue undergoes major reorganization, where some neurons die and others sprout and make aberrant connections (Ben-Ari et al., 2008). This “reactive plasticity” is well-described in the dentate gyrus of the hippocampus. Here, mossy fibers (MFs), the axons of granule cells (GCs) from the dentate gyrus and where KARs are highly expressed, undergo sprouting after KA injections and form a functional recurrent MF network (Tauck and Nadler, 1985; Represa et al., 1987; Sutula and Dudek, 2007). MF sprouting is therefore considered as one of the pathological characteristics of TLE in humans and animal models. Interestingly, at these aberrant synapses developed under pathological conditions, ‘de novo’ KARs are expressed in the GCs of the dentate gyrus, and these likely participate in the pathogenesis of TLE (Epsztein et al., 2005; Artinian et al., 2011, 2015). KA mediates glutamatergic currents at MF-GC synapses, and some half of the glutamatergic transmission has been proposed to be due to the insertion of these new KARs (Epsztein et al., 2005; Peret et al., 2014; reviewed in Vincent and Mulle, 2009; Crépel and Mulle, 2015). The consequence of the insertion of KARs in GC would be the generation of a hyperexcitable circuit in the hippocampal dentate gyrus. This may ultimately produce epileptiform activity, particularly when inhibition by GABAergic transmission is simultaneously depressed (Epsztein et al., 2005).
The aforementioned aberrantly inserted post-synaptic KARs are likely heteromeric receptors containing GluK2/GluK5 subunits, that can be blocked by the specific antagonist for post-synaptic MF KARs, UBP310 (Pinheiro et al., 2013). GluK2, 4, and 5 subunits have been found at post-synaptic sites (Mulle et al., 1998; Contractor et al., 2003; Fernandes et al., 2009), and are involved in generation of recurrent seizures in chronic epilepsy. Presynaptic MF KARs comprise GluK2 and GluK3 subunits, and mediate facilitation or depression of glutamate release, and are thereby involved in presynaptic plasticity (reviewed in Sihra and Rodríguez-Moreno, 2013; Sihra et al., 2014). The de novo KARs present in dentate gyrus GC impair temporal precision of EPSP-spike coupling and thus induce seizures by altering the basally sparse firing rate of dentate gyrus GCs (Artinian et al., 2011, 2015). Additionally it is known that the CA3 synaptic silencing attenuated KA induced seizures and network oscillations in the hippocampus (Yu et al., 2016).
The study of the role of the aforementioned new/aberrant KARs in the chronic phase of TLE in the pilocarpine model of chronic TLE showed that the inter-ictal and ictal events were reduced in mice lacking GluK2 subunit, but not those lacking GluK1, or with the use of a GluK2/GluK5 antagonist (Peret et al., 2014). These results suggest that these KARs may represent targets for antiepileptic drugs (Figure 1B). KARs also modulate intrinsic conductance by a metabotropic action at MF-CA3 synapses (Fisahn et al., 2004). It remains to be elucidated how aberrant KARs are recruited to the membrane and whether these abnormally expressed post-synaptic KARs in dentate gyrus GCs are activated by endogenous glutamate, to develop and propagate seizures in the hippocampus. However, it has recently been shown that C1q-like proteins (related to the C1q complement component) may have a role at MFs (Matsuda et al., 2016). C1ql2 and C1ql3 proteins produced by MFs have been demonstrated to serve as extracellular organizers to recruit functional post-synaptic KAR complexes to the CA3 pyramidal neurons and GCs (Figure 1B).
Possible Role of Astrocytes in Ka Induced Seizures
In the recent years, evidence has accumulated to suggest that astrocytes may have an important role in KA-induced seizures. In tissue from patients with refractory TLE, an increase of KARs subunits GluK4 and GluK5 have been reported (Das et al., 2012). Surprisingly, KARs subunits GluK1, 2, 4, and 5 are all expressed in astrocytes, 1 week after the induction of status epilepticus in the CA1 region of the hippocampus of treated animals, but not in naïve animals. Further, GluK1 and GluK5 subunits remain enhanced in the chronic phase of epilepsy when spontaneous seizures occurs (Vargas et al., 2013). The role of the newly expressed KARs in the astrocytes is currently unclear. Indeed, it is unknown whether this recruitment of KARs in astrocytes impinges on the induction and propagation of seizures (as activation of these KARs might mediate the release of glutamate from astrocytes), or have just the opposite effect, viz. a protective role. Future work will be needed to determine the exact role of astrocytes and astrocytic KARs in epilepsy (Figure 1C).
Future Directions
One important aspect that awaits resolution is whether the KARs involved in the induction of acute or chronic seizures are activated directly by endogenously released glutamate. Further, the precise mechanism by which the loss of excitatory/inhibitory balance induces seizures and their propagation requires evaluation. Importantly, the role of “reactive plasticity” in epilepsy, as a general or basic mechanism for epilepsy, needs to be expounded and clarified. Finally, the function of astrocytic KARs and astrocytes in epilepsy requires explication, as to whether their involvement is pro- or anti-epileptogenic.
In conclusion, three approximations are promising mechanisms in determining the role of KARs in epilepsy: (i) depression of inhibitory synaptic transmission and increase in excitatory transmission (i.e., disruption of excitation/inhibition balance); (ii) induction of reactive plasticity with new KARs changing network properties; and (iii) the emerging role of astrocytes in epilepsy. These three working hypotheses await further development to understand the exact role of the intriguing KARs in epilepsy etiology, and facilitate the elaboration of novel, and hopefully more efficacious, therapies for epilepsy.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Work in ARM laboratory was supported by BFU2006-1455 and BFU2012-38208 (from the Spanish Ministry of Economy and Competitiveness).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Andrade-Talavera, Y., Duque-Feria, P., Negrete-Díaz, J. V., Flores, G., Sihra, T. S., and Rodríguez-Moreno, A. (2012). Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin at mossy fiber-CA3 synapses. J. Neurochem. 122, 891–899. doi: 10.1111/j.1471-4159.2012.07844.x
Andrade-Talavera, Y., Duque-Feria, P., Sihra, T. S., and Rodríguez-Moreno, A. (2013). Pre-synaptic kainate-receptor-mediated facilitation of glutamate reléase involves PKA and Ca2+-calmodulin at thalamocortical synapses. J. Neurochem. 126, 565–578. doi: 10.1111/jnc.12310
Artinian, J., Peret, A., Marti, G., Epsztein, J., and Crépel, V. (2011). Synaptic kainate receptors in interplay with INaP shift the sparse firing of dentate granule cells to a sustained rhythmic mode in temporal lobe epilepsy. J. Neurosci. 31, 10811–10818. doi: 10.1523/JNEUROSCI.0388-11.2011
Artinian, J., Peret, A., Mircheva, Y., Marti, G., and Crépel, V. (2015). Impaired neuronal operation through aberrant intrinsic plasticity in epilepsy. Ann. Neurol. 77, 592–606. doi: 10.1002/ana.24348
Bahn, S., Volk, B., and Wisden, W. (1994). Kainate receptor gene expression in the developing rat brain. J. Neurosci. 14, 5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994
Ben-Ari, Y. (1985). Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14, 375–403. doi: 10.1016/0306-4522(85)90299-4
Ben-Ari, Y., and Cossart, R. (2000). Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 23, 580–587. doi: 10.1016/S0166-2236(00)01659-3
Ben-Ari, Y., Crepel, V., and Represa, A. (2008). Seizures beget seizures in temporal lobe epilepsies: the boomerang effects of newly formed aberrant kainatergic synapses. Epilepsy Curr. 8, 68–72. doi: 10.1111/j.1535-7511.2008.00241.x
Bouvier, G., Larsen, R. S., Rodríguez-Moreno, A., Paulsen, O., and Sjostrom, P. J. (2018). Towards resolving the presynaptic NMDA receptor debate. Curr. Opin. Neurobiol. 51, 1–7. doi: 10.1016/j.conb.2017.12.020
Castillo, P. E., Malenka, R. C., and Nicoll, R. A. (1997). Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388, 182–186.
Chittajallu, R., Vignes, M., Dev, K. K., Barnes, J. M., Collingridge, G. L., and Henley, J. M. (1996). Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature 379, 78–81. doi: 10.1038/379078a0
Clarke, V. R., Ballyk, B. A., Hoo, K. H., Mandelzys, A., Pellizzari, A., Bath, C. P., et al. (1997). A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature 389, 599–603. doi: 10.1038/39315
Contractor, A., Sailer, A. W., Darstein, M., Maron, C., Xu, J., Swanson, G. T., et al. (2003). Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2-/- mice. J. Neurosci. 23, 422–429. doi: 10.1523/JNEUROSCI.23-02-00422.2003
Contractor, A., Swanson, G., and Heinemann, S. F. (2001). Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron 29, 209–216. doi: 10.1016/S0896-6273(01)00191-X
Contractor, A., Swanson, G. T., Sailer, A., O′Gorman, S., and Heinemann, S. F. (2000). Identification of the kainate receptors subunit underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J. Neurosci. 20, 8269–8278. doi: 10.1523/JNEUROSCI.20-22-08269.2000
Cossart, R., Esclapez, M., Hirsch, J. C., Bernard, C., and Ben-Ari, Y. (1998). GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat. Neurosci. 1, 470–478.
Crépel, V., and Mulle, C. (2015). Physiopathology of kainite receptors in epilepsy. Curr. Opin. Pharmacol. 20, 83–88. doi: 10.1016/j.coph.2014.11.012
Das, A., Wallace, G. C. IV, Holmes, C., McDowell, M. L., Smith, J. A., Marshall, J. D., et al. (2012). Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220, 237–246. doi: 10.1016/j.neuroscience.2012.06.002
Epsztein, J., Represa, A., Jorquera, I., Ben-Ari, Y., and Crépel, V. (2005). Recurrent mossy fibers establish aberrant kainate receptor-operated synapses on granule cells from epileptic rats. J. Neurosci. 25, 8229–8239. doi: 10.1523/JNEUROSCI.1469-05.2005
Fernandes, H. B., Catches, J. S., Petralia, R. S., Copits, B. A., Xu, J., Russell, T. A., et al. (2009). High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 63, 818–829. doi: 10.1016/j.neuron.2009.08.010
Fisahn, A., Contractor, A., Traub, R. D., Buhl, E. H., Heinemann, S. F., and McBain, C. J. (2004). Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J. Neurosci. 24, 9658–9668. doi: 10.1523/JNEUROSCI.2973-04.2004
Frerking, M., Malenka, R. C., and Nicoll, R. A. (1998). Synaptic activation of kainate receptors on hippocampal interneurons. Nat. Neurosci. 1, 479–486.
Fritsch, B., Reis, J., Gasior, M., Kaminski, R. M., Michael, A., and Rogawski, M. A. (2014). Role of GluK1 kainate receptors in seizures, epileptic discharges, and epileptogenesis. J. Neurosci. 34, 5765–5775. doi: 10.1523/JNEUROSCI.5307-13.2014
Goldenberg, M. M. (2010). Overview of drugs used for epilepsy and seizures etiology, diagnosis, and treatment. P T 35, 392–415.
Jiang, L., Xu, J., Nedergaard, M., and Kang, J. (2001). A kainate receptor increases the efficacy of GABAergic synapses. Neuron 30, 503–513. doi: 10.1016/S0896-6273(01)00298-7
Kamiya, H., and Ozawa, S. (2000). Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre. J. Physiol. 523, 653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x
Khalilov, I., Hirsch, J., Cossart, R., and Ben-Ari, Y. (2002). Paradoxical anti-epileptic effects of a GluR5 agonist of kainate receptors. J. Neurophysiol. 88, 523–527. doi: 10.1152/jn.2002.88.1.523
Lauri, S. E., Bortolotto, Z. A., Bleakman, D., Ornstein, P. L., Lodge, D., Isaac, J. T. R., et al. (2001). A critical role of a facilitatory kainate autoreceptor in mossy fiber LTP. Neuron 32, 697–709. doi: 10.1016/S0896-6273(01)00511-6
Leite, J. P., Bortolotto, Z. A., and Cavalheiro, E. A. (1990). Spontaneous recurrent seizures in rats: an experimental model of partial epilepsy. Neurosci. Biobehav. Rev. 14, 511–517. doi: 10.1016/S0149-7634(05)80076-4
Lerma, J., and Marques, J. (2013). Kainate receptors in health and disease. Neuron 80, 292–311. doi: 10.1016/j.neuron.2013.09.045
Li, J. M., Zeng, Y. J., Peng, F., Li, L., Yang, T. H., Hong, Z., et al. (2010). Aberrant glutamate receptor 5 expression in temporal lobe epilepsy lesions. Brain Res. 1311, 166–174. doi: 10.1016/j.brainres.2009.11.024
Lothman, E. W., Bertram, E. H. III, and Stringer, J. L. (1991). Functional anatomy of hippocampal seizures. Prog. Neurobiol. 37, 1–82. doi: 10.1016/0301-0082(91)90011-O
Lyon, L., Borel, M., Carrión, M., Kew, J. N. C., Corti, C., Harrison, P. J., et al. (2011). Hippocampal mossy fiber long-term depression in Grm2/3 double knockout mice. Synapse 65, 945–954. doi: 10.1002/syn.20923
Matsuda, K., Budisantoso, T., Mitakidis, N., Sugaya, Y., Miura, E., Kakegawa, W., et al. (2016). Transsynaptic modulation of kainate receptor functions by C1q-like proteins. Neuron 90, 752–767. doi: 10.1016/j.neuron.2016.04.001
Melyan, Z., Lancaster, B., and Wheal, H. V. (2004). Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J. Neurosci. 24, 4530–4534. doi: 10.1523/JNEUROSCI.5356-03.2004
Melyan, Z., Wheal, H. V., and Lancaster, B. (2002). Metabotropic-mediated kainate receptor regulation of isAHP and excitability in pyramidal cells. Neuron 34, 107–114. doi: 10.1016/S0896-6273(02)00624-4
Mulle, C., Sailer, A., Pérez-Otaño, I., Dickinson-Anson, H., Castillo, P. E., Bureau, I., et al. (1998). Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392, 601–605.
Nadler, J. V. (1981). Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 29, 2031–2042. doi: 10.1016/0024-3205(81)90659-7
Nadler, J. V., Perry, B. W., and Cotman, C. W. (1978). Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature 271, 676–677. doi: 10.1038/271676a0
Negrete-Díaz, J. V., Duque-Feria, P., Andrade-Talavera, Y., Carrión, M., Flores, G., and Rodríguez-Moreno, A. (2012). Kainate receptor-mediated depression of glutamatergic transmission involving protein kinase A in the lateral amygdala. J. Neurochem. 121, 36–43. doi: 10.1111/j.1471-4159.2012.07665.x
Negrete-Díaz, J. V., Sihra, T. S., Delgado-García, J. M., and Rodríguez-Moreno, A. (2006). Kainate receptor-mediated inhibition of glutamate release involves protein kinase A in the mouse hippocampus. J. Neurophysiol. 96, 1829–1837. doi: 10.1152/jn.00280.2006
Negrete-Díaz, J. V., Sihra, T. S., Delgado-García, J. M., and Rodríguez-Moreno, A. (2007). Kainate receptor-mediated presynaptic inhibition converges with presynaptic inhibition mediated by Group II mGluRs and long-term depression at the hippocampal mossy fiber-CA3 synapse. J. Neural Transm. 114, 1425–1431. doi: 10.1007/s00702-007-0750-4
Paternain, A. V., Herrera, M. T., Nieto, M. A., and Lerma, J. (2000). GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J. Neurosci. 20, 196–205. doi: 10.1523/JNEUROSCI.20-01-00196.2000
Peret, A., Christie, L. A., Ouedraogo, D. W., Gorlewicz, A., Epsztein, J., Mulle, C., et al. (2014). Contribution of aberrant GluK2-containing kainate receptors to chronic seizures in temporal lobe epilepsy. Cell Rep. 8, 347–354. doi: 10.1016/j.celrep.2014.06.032
Perucca, E., and Meador, K. J. (2005). Adverse effects of antiepileptic drugs. Acta Neurol. Scand. Suppl. 181, 30–35. doi: 10.1111/j.1600-0404.2005.00506.x
Pinheiro, P. S., Lanore, F., Veran, J., Artinian, J., Blanchet, C., Crépel, V., et al. (2013). Selective block of postsynaptic kainate receptors reveals their function at hippocampal mossy fiber synapses. Cereb. Cortex 23, 323–331. doi: 10.1093/cercor/bhs022
Pinheiro, P. S., Perrais, D., Coussen, F., Barhanin, J., Bettler, B., Mann, J. R., et al. (2007). GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. U.S.A. 104, 12181–12186. doi: 10.1073/pnas.0608891104
Represa, A., Tremblay, E., and Ben-Ari, Y. (1987). Kainate binding sites in the hippocampal mossy fibers: localization and plasticity. Neuroscience 20, 739–748. doi: 10.1016/0306-4522(87)90237-5
Rodríguez-Moreno, A., Herreras, O., and Lerma, J. (1997). Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron 19, 893–901. doi: 10.1016/S0896-6273(00)80970-8
Rodríguez-Moreno, A., and Lerma, J. (1998). Kainate receptor modulation of GABA release involves a metabotropic function. Neuron 20, 1211–1218. doi: 10.1016/S0896-6273(00)80501-2
Rodríguez-Moreno, A., López-García, J. C., and Lerma, J. (2000). Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc. Natl. Acad. Sci. U.S.A. 97, 1293–1298. doi: 10.1073/pnas.97.3.1293
Rodríguez-Moreno, A., and Sihra, T. S. (2004). Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus. J. Physiol. 557, 733–745. doi: 10.1113/jphysiol.2004.065029
Rodríguez-Moreno, A., and Sihra, T. S. (2007a). Kainate receptors with a metabotropic modus operandi. Trends Neurosci. 30, 630–637.
Rodríguez-Moreno, A., and Sihra, T. S. (2007b). Metabotropic actions of kainate receptors in the CNS. J. Neurochem. 103, 2121–2135.
Rodríguez-Moreno, A., and Sihra, T. S. (2013). Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin and PKA in cerebrocortical synaptosomes. FEBS Lett. 587, 788–792. doi: 10.1016/j.febslet.2013.01.071
Rogawski, M. A., Gryder, D., Castaneda, D., Yonekawa, W., Banks, M. K., and Li, H. (2003). GluR5 kainate receptors, seizures, and the amygdala. Ann. N. Y. Acad. Sci. 985, 150–162. doi: 10.1111/j.1749-6632.2003.tb07079.x
Sander, T., Hildmann, T., Kretz, R., Fürst, R., Sailer, U., Bauer, G., et al. (1997). Allelic association of juvenile absence epilepsy with a GluR5 kainate receptor gene (GRIK1) polymorphism. Am. J. Med. Genet. 74, 416–421. doi: 10.1002/(SICI)1096-8628(19970725)74:4<416::AID-AJMG13>3.0.CO;2-L
Schmitz, D., Frerking, M., and Nicoll, R. A. (2000). Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron 27, 327–338. doi: 10.1016/S0896-6273(00)00040-4
Schmitz, D., Mellor, J., and Nicoll, R. A. (2001). Presynaptic kainite receptor mediation of frequency facilitation at mossy fiber synapses. Science 291, 1972–1976. doi: 10.1126/science.1057105
Scott, R., Lalic, T., Kullmann, D. M., Capogna, M., and Rusakov, D. A. (2008). Target-cell specificity of kainate autoreceptor and Ca2+ store-dependent short-term plasticity at hippocampal mossy fibers. J. Neurosci. 28, 13139–13149. doi: 10.1523/JNEUROSCI.2932-08.2008
Sihra, T. S., Flores, G., and Rodríguez-Moreno, A. (2014). Kainate receptors: multiple roles in neuronal plasticity. Neuroscientist 20, 29–43. doi: 10.1177/1073858413478196
Sihra, T. S., and Rodríguez-Moreno, A. (2013). Presynaptic kainate receptor-mediated bidirectional modulatory actions: mechanisms. Neurochem. Int. 62, 982–987. doi: 10.1016/j.neuint.2013.03.012
Smolders, I., Bortolotto, Z. A., Clarke, V. R., Warre, R., Khan, G. M., O’Neill, M. J., et al. (2002). Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat. Neurosci. 5, 796–804. doi: 10.1038/nn880
Sutula, T. P., and Dudek, F. E. (2007). Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emerging property of a complex system. Prog. Brain Res. 163, 541–563. doi: 10.1016/S0079-6123(07)63029-5
Swanson, G. T. (2009). Targeting AMPA and kainate receptors in neurological disease: therapies on the horizon? Neuropsychopharmacology 34, 249–250. doi: 10.1038/npp.2008.158
Tauck, D. L., and Nadler, J. V. (1985). Evidnece of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J. Neurosci. 5, 1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985
Valbuena, S., and Lerma, J. (2016). Non-canonical signaling, the hidden life of ligand-gated ion channels. Neuron 92, 316–329. doi: 10.1016/j.neuron.2016.10.016
Vargas, J. R., Takahasi, K., Thomson, K. E., and Wilcox, K. (2013). The expression of kainate receptor subunits in hippocampal astrocytes after experimentally induced status epilepticus. J. Neuropathol. Exp. Neurol. 72, 919–932. doi: 10.1097/NEN.0b013e3182a4b266
Vignes, M., Bleakman, D., Lodge, D., and Collingridge, G. L. (1997). The synaptic activation of the GluR5 subtype of kainate receptor in area CA3 of the rat hippocampus. Neuropharmacology 36, 1477–1481. doi: 10.1016/S0028-3908(97)00158-5
Vignes, M., Clarke, V. J. R., Parry, M. J., Bleakman, D., Lodge, D., Ornstein, P. L., et al. (1998). The GluR5 subtype of kainate receptor regulates excitatory synaptic transmission in areas CA1 and CA3 of the rat hippocampus. Neuropharmacology 37, 1269–1277. doi: 10.1016/S0028-3908(98)00148-8
Vincent, P., and Mulle, C. (2009). Kainate receptors in epilepsy and excitotoxicity. Neuroscience 158, 309–323. doi: 10.1016/j.neuroscience.2008.02.066
Werner, P., Voigt, M., Keinamen, K., Wisden, W., and Seeburg, P. H. (1991). Cloning of putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature 353, 742–744. doi: 10.1038/351742a0
Keywords: KAR, epilepsy, CA1, MF, dentate gyrus, recurrent mossy fibers, astrocyte
Citation: Falcón-Moya R, Sihra TS and Rodríguez-Moreno A (2018) Kainate Receptors: Role in Epilepsy. Front. Mol. Neurosci. 11:217. doi: 10.3389/fnmol.2018.00217
Received: 20 March 2018; Accepted: 04 June 2018;
Published: 22 June 2018.
Edited by:
Inmaculada Maria Gonzalez-Gonzalez, University of Central Lancashire, United KingdomReviewed by:
Hana Kubova, Institute of Physiology, Academy of Sciences of the Czech Republic, CzechiaValérie Crépel, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2018 Falcón-Moya, Sihra and Rodríguez-Moreno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Rodríguez-Moreno, YXJvZG1vckB1cG8uZXM=