 Rafael Falcón-Moya
Rafael Falcón-Moya Pilar Losada-Ruiz
Pilar Losada-Ruiz Talvinder S. Sihra
Talvinder S. Sihra Antonio Rodríguez-Moreno
Antonio Rodríguez-Moreno- 1Laboratorio de Neurociencia Celular y Plasticidad, Departamento de Fisiología, Anatomía y Biología Celular, Universidad Pablo de Olavide, Sevilla, Spain
- 2Department of Physiology, Pharmacology and Neuroscience, University College London, London, United Kingdom
We elucidated the mechanisms underlying the kainate receptor (KAR)-mediated facilitatory modulation of synaptic transmission in the cerebellum. In cerebellar slices, KA (3 μM) increased the amplitude of evoked excitatory postsynaptic currents (eEPSCs) at synapses between axon terminals of parallel fibers (PF) and Purkinje neurons. KA-mediated facilitation was antagonized by NBQX under condition where AMPA receptors were previously antagonized. Inhibition of protein kinase A (PKA) suppressed the effect of KA on glutamate release, which was also obviated by the prior stimulation of adenylyl cyclase (AC). KAR-mediated facilitation of synaptic transmission was prevented by blocking Ca2+ permeant KARs using philanthotoxin. Furthermore, depletion of intracellular Ca2+ stores by thapsigargin, or inhibition of Ca2+-induced Ca2+-release by ryanodine, abrogated the synaptic facilitation by KA. Thus, the KA-mediated modulation was conditional on extracellular Ca2+ entry through Ca2+-permeable KARs, as well as and mobilization of Ca2+ from intracellular stores. Finally, KAR-mediated facilitation was sensitive to calmodulin inhibitors, W-7 and calmidazolium, indicating that the increased cytosolic [Ca2+] sustaining KAR-mediated facilitation of synaptic transmission operates through a downstream Ca2+/calmodulin coupling. We conclude that, at cerebellar parallel fiber-Purkinje cell synapses, presynaptic KARs mediate glutamate release facilitation, and thereby enhance synaptic transmission through Ca2+-calmodulin dependent activation of adenylyl cyclase/cAMP/protein kinase A signaling.
Introduction
Kainate-type glutamate receptors are well established mediators of canonical, ionotropic postsynaptic synaptic transmission and, presynaptically, the receptors support a modulatory regulation of neurotransmitter release. In the latter regard, kainate receptors (KARs) evince a non-canonical metabotropic capacity, through which they effect the control of both glutamate and GABA release (for review see Rodríguez-Moreno and Sihra, 2007a,b; Jane et al., 2009; Shira and Rodríguez-Moreno, 2013; Valbuena and Lerma, 2016). At several excitatory glutamatergic synapses, the KAR-mediated modulation is found to be biphasic, such that low agonist concentrations facilitate glutamate release, as opposed to higher agonist concentrations, which inhibit neurotransmitter release (for review see Rodríguez-Moreno and Sihra, 2007a,b, 2013; Lerma and Marques, 2013). How this diametrically opposite modulation is mechanistically manifest is subject of considerable debate and investigation, as is the question of the subcellular location of KARs responsible for presynaptic modulation (for review see Rodríguez-Moreno and Sihra, 2007a,b, 2013; Lerma and Marques, 2013).
KARs are expressed in the cerebellar cortex in the axons of cerebellar granule cells that form parallel fibers (PF), and make excitatory synapses with Purkinje cells (PuC, Smith et al., 1999). Messenger RNA transcripts encoding for different KAR subunits (GluK1, GluK2 and GluK5) have been detected in granule cells and functional expression of KAR subtypes has been reported (Bettler et al., 1990; Herb et al., 1992; Bahn et al., 1994; Petralia et al., 1994). The subunits GluK1 and GluK2 have been detected on parallel fibers, Petralia et al., 1994). Biophysical studies with single-channel recordings have shown GluK1 activity (Swanson et al., 1996), suggesting these KARs are Ca2+-permeable. A biphasic action of KARs, activated by the agonist domoate, has been shown previously at PF-PuC synapse, with low agonist concentrations facilitating synaptic transmission, and higher concentrations depressing synaptic transmission (Delaney and Jahr, 2002). However, the precise mechanism of action by which KARs mediate potentiation (and depression) of synaptic transmission at PF-PuC synapses is completely unknown. Here, we have examined the mechanism underpinning the facilitatory effect of KA in cerebellar slices at synapses between granule cell terminals and Purkinje cells.
First, establishing mechanistic features of the modulation, we found that the KAR-mediated facilitation of glutamate release and synaptic transmission has an obligatory dependency on adenylyl cyclase (AC) and cAMP-mediated protein kinase A (PKA) activity. Furthermore, the KAR-mediated modulation of transmission is reliant on both external Ca2+ entry via Ca2+-permeant KARs, and functional intracellular Ca2+-stores. Finally, obviation of facilitation by calmodulin inhibition invokes a mechanistic coupling of KARs through Ca2+-calmodulin/AC/cAMP/PKA signaling, at PF-PuC synapses in the cerebellum.
Materials and Methods
Animals
The experiments were performed on 4–6 week old C57Bl/6 male mice obtained from Harlan Laboratories (Spain). Experiments were conducted in accordance with the European Union directive for the use of laboratory animals in acute experiments and were approved by the local Ethical Committee (Junta de Andalucía and University Pablo de Olavide, Sevilla, Spain).
Slice Preparation
Cerebellar parasagittal slices were prepared. Briefly, after decapitation, the whole brain was removed under ice-cold buffered salt solution consisting of (in mM) 124 NaCl, 2.69 KCl, 1.25 KH2PO4, 2 MgSO4, 1.8 CaCl2, 26 NaHCO3, and 10 glucose (pH 7.2, 300 mOsm), and positioned on the stage of a vibratome slicer (Leica 1000S), and cut to obtain cerebellar slices (350 μm thick) containing parallel fibers-Purkinje cells synapses. Slices were maintained continuously oxygenated for at least 1 h before use. All experiments were carried out at room temperature (22–25°C). During experiments, slices were continuously perfused with buffered salt solution as detailed above.
Electrophysiological Recordings
Whole-cell patch-clamp recordings were made from Purkinje neurons. NMDA receptor-mediated evoked excitatory postsynaptic currents (eEPSCs) were recorded at +40 mV from these neurons visually identified by infrared-differential interference contrast (IR-DIC) microscopy using a 40× water immersion objective. Perfusion solution contained GYKI53655 (30 μM), to block AMPA receptors, and bicuculline (10 μM), to block GABAA receptors. In experiments involving AMPA receptor-mediated currents, performed at −70 mV, no GYKI53655 was used, but D-AP5 (50 μM) was included to block NMDA receptors. To evoke eEPSCs, electrical pulses were delivered to granule cells axons (parallel fibers) using a monopolar electrode placed in the molecular layer at a frequency of 0.2 Hz. Patch electrodes were made from borosilicate glass and had a resistance of 4–7 MΩ when filled with (mM): 120 CsCl, 8 NaCl, 1 MgCl2, 0.2 CaCl2, 10 HEPES, 2 EGTA and 20 QX-314 (pH 7.2, 290 mOsm). A 40 ms paired-pulse stimulation protocol was used for pair pulse ratio (PPR) analysis. Neurons were voltage clamped, using a Multiclamp 700B amplifier (Molecular Devices, Foster City, CA, USA). Access resistance was regularly monitored during recordings, and cells were rejected if it changed >15% during the experiment. Data were filtered at 2 kHz, digitized at 10 kHz, and stored on a computer using pClamp software (Molecular Devices). Synaptic failures were identified as the lack of synaptic responses after presynaptic stimulation with the amplitude of these responses being no different from basal noise amplitude.
Data Analysis
Data were normalized taking the control as 100% of the response and presented as means ± SEM. Signals were averaged every 12 traces. Effects of KA were measured at peak (maximum) compared to averaged 10 min baseline points. Significance was assessed at P < 0.05. Statistical comparisons were made using two-tailed Student’s t-test for comparison of two data sets and ANOVA for comparison of multiple data set using the Bonferroni as a post hoc test.
Compounds
Salts and general reagents were purchased from Sigma (St. Louis, MO, USA); GYKI 53655, D-AP5, NBQX, bicuculline, Rp-Br-cAMP, H-89, forskolin, philanthotoxin, ryanodine, thapsigargin, kainate, Pertussis toxin CMZ and W-7 were obtained from Tocris (Bristol, UK).
Results
The Activation of Kainate Receptors By 3 μM KA Produces an Increase in the Amplitude of NMDA-Evoked Postsynaptic Currents at PF-PuC Synapses
Following the observation that glutamatergic transmission at PF-PuC synapses of juvenile rats pups is modulated by KARs in a biphasic manner (Delaney and Jahr, 2002), as is also the case in the hippocampus (for review see Rodríguez-Moreno and Sihra, 2007a,b, 2013; Lerma and Marques, 2013), we established the parallel fiber-Purkinje (PF-PuC) synapse paradigm in slices from early adult mouse cerebellum.
The experimental paradigm we used was the stimulation of parallel fiber axons while measuring NMDA receptor-mediated eEPSCs in PuCs, by whole-cell patch clamp recordings, with the membrane potential held at +40 mV. Recording were made in the presence of 30 μM GYKI53655, in order to obviate AMPA receptor activation, as well as the presence of 10 μM bicuculline, to antagonize GABAA receptors. In our experiments, young adult cerebellar synapses evince detectable facilitation of NMDA receptor-mediated eEPSC amplitudes at 3 μM KA (138 ± 11%, n = 10, Figures 1A,B), with 0.3 μM and 1 μM agonist concentrations having smaller effects (115 ± 2%, n = 6, 117 ± 6%, n = 6, respectively). With 3 μM KA, synaptic facilitation was followed by a 36 ± 8% (to 64 ± 8% of baseline, n = 10) decrease in the eEPSC amplitude (Figure 1B). To analyze the mechanistic details of the KAR-mediated facilitation of glutamatergic transmission, we hereafter utilized 3 μM KA in subsequent electrophysiological experiments as 3 μM KA produced the maximum level of facilitation observable (Figures 1A,B).
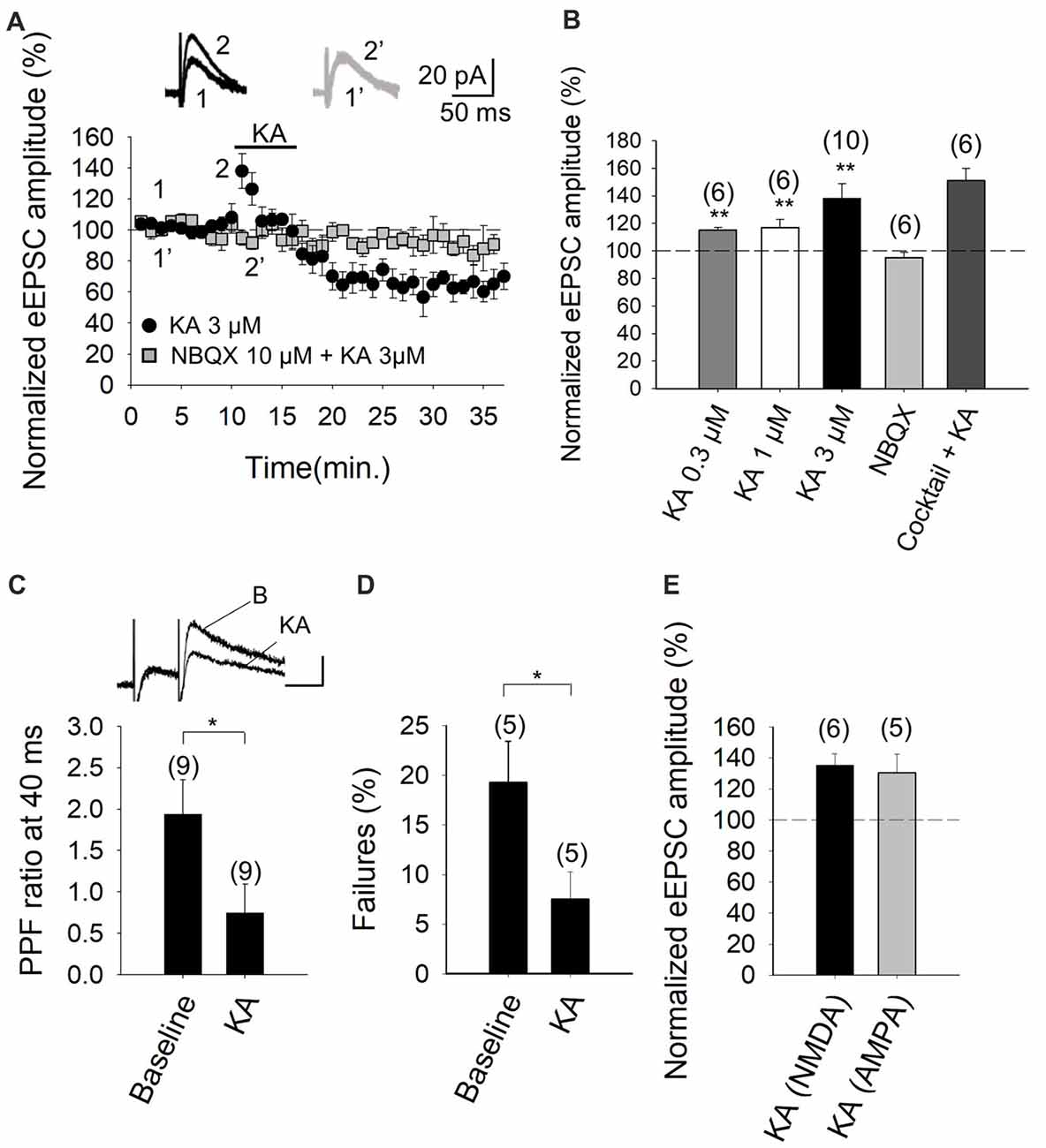
Figure 1. Kainate (KA) increases the evoked excitatory postsynaptic currents (eEPSCs) amplitude at parallel fibers-Purkinje cells (PF-PuC) synapses of the cerebellum. (A) Time course of KA (3 μM) effect on eEPSCs amplitude in the absence (circles) and presence of NBQX (squares). Inset show traces before and after 4 min KA perfusion in the absence (1, 2) and in the presence of 10 μM NBQX (1′, 2′). (B) Quantification of modulation observed in (A) and dose dependency. (C) KA (3 μM) perfusion produces a decrease of the paired pulse ratio, inset shows scaled representative traces. (D) Effect of KA on the number of failures of NMDA receptor-mediated currents. (E) Effect of KA (3 μM) on NMDA and AMPA receptor-mediated currents, respectively. Note that the effect of kainate on these currents is indistinguishable. The number of slices (from two to three mice) is indicated in parenthesis at the top of each bar. Results are expressed as means ± SEM (*P < 0.05, **P < 0.01, Student’s t-test).
To determine whether the effect of KA recorded from Purkinje neurons in slices was mediated by the activation of KARs, analogous to that observed in other brain regions, such as the hippocampus and cortex (Lauri et al., 2001a,b, 2003; Schmitz et al., 2001; Ji and Stäubli, 2002; Contractor et al., 2003; Breustedt and Schmitz, 2004; Rodríguez-Moreno and Sihra, 2004, 2013; Campbell et al., 2007; Pinheiro et al., 2007; Scott et al., 2008; Fernandes et al., 2009; Jouhanneau et al., 2011; Andrade-Talavera et al., 2012, 2013), we performed experiments in the presence of NBQX. We showed that the 3 μM KA biphasic effect on the eEPSC amplitude was abolished in the presence of 10 μM NBQX (95 ± 4%, n = 6, Figures 1A,B). In these experiments, because AMPA receptors are antagonized in the presence of the selective blocker GYKI53655 in the bath, the observation of full antagonism by NBQX invokes the modulation to be due to KARs specifically. Further, in line with the notion that the facilitation (and the depression) of synaptic transmission observed is exclusively contingent on KAR activation. KA-mediated facilitation was retained (151 ± 9% increase of eEPSCs amplitude, n = 6) when other transmitter influences were obviated by the inclusion of a cocktail of inhibitors including the receptor antagonists: MCPG and MPPG (1.5 mM), naloxone (100 μM), bicuculline (20 μM), 2-OH-saclofen (150 μM), atropine sulfate (50 μM) and DPCPX (0.1 μM), to block metabotropic glutamate, opioid, GABAA, GABAB, muscarinic and adenosine receptors, respectively. Indeed, the synaptic depression that followed the facilitation of EPSCs, was also present in the presence of the inhibitor cocktail (to 65 ± 8% of the baseline, n = 6). These data therefore exclude the possibility that KA-mediated modulation was a secondary consequence of the synaptic release of diverse neurotransmitters, but rather, support the hypothesis that a direct effect of KA on KARs at cerebellar synapses underpins the observed modulation.
Previously, the facilitatory action of KA at the PF-PuC synapses have been attributed to presynaptic regulation (Delaney and Jahr, 2002). In our studies, we confirmed a presynaptic locus of action by using several approaches. First, we performed paired-pulse recordings and measured the paired-pulse ratio (PPR; pair-pulse depression was observed at 40 ms pulse interval). PPR was 1.9 ± 0.4 (n = 9) under control/baseline conditions. After KA treatment, PPR decreased to 0.7 ± 0.3 (n = 9; Figure 1C), implying an effect on release probability (Manabe et al., 1993), thereby corroborating the presynaptic origin of the KA receptor-mediated regulation. Second, we determined the proportion of synaptic failures in presence of KA. Under control conditions, synaptic failure rate was 19 ± 4%, n = 5. Following the application of KA, the failure rate was measurably decreased (to 7 ± 3%, n = 5, Figure 1D), again indicating a presynaptic locus of KA action. Finally, we compared the KA-mediated modulation of NMDA receptor-mediated eEPSCs (with GYKI53655 present) and AMPA receptor-mediated eEPSCs recorded at −70 mV (without GYKI53655, but with D-AP5 and bicuculline, to respectively block NMDA and GABAA receptors). KA mediated a comparable increase in the NMDA receptor-mediated eEPSCs (135 ± 7%, n = 6, Figure 1E) and the AMPA receptor-mediated eEPSCs (130 ± 12%, n = 5, Figure 1E). This congruent facilitation of NMDA and AMPA receptor-mediated eEPSC amplitudes intimates that KA-modulation acts upstream of postsynaptic receptor activation, i.e., at the level of presynaptic terminal, through increased glutamate release. Together the preceding evaluation reliably evinces a presynaptic locus of action of KA at the PF-PuC synapses under investigation. However, it remains to be seen where the respondent KARs are physically located i.e., terminal, axonal or somatodendritic presynaptic compartments.
KAR-Mediated Facilitation of Glutamatergic Transmission at PF-PuC Is Contingent on cAMP-Dependent Signaling
With the selectivity of the action of KA verified, in subsequent experiments we examined the second messenger system that mediates the facilitation of eEPSCs. First, we tested whether PKA was involved in the increased eEPSCs, by inhibiting either the cAMP-mediated activation of PKA, or the catalytic activity of the kinase, by respectively treating slices with the inhibitors cAMP-Rp or H-89. With the addition of 100 μM cAMP-Rp or 2 μM H-89, the facilitation of the eEPSC amplitude by 3 μM KA was eliminated (88 ± 3%, n = 7 after cAMP-Rp and 92 ± 6%, n = 7 after H-89, vs. KA 3 μM, 131 ± 9%, n = 13 Figures 2A,B). cAMP-Rp and H-89 alone produced only small decrements in the eEPSC amplitude (7 ± 3% and 10 ± 4%, respectively, n = 5, data not shown). The data together point to PKA playing an obligatory part in the observed KA-mediated modulation of PF-PuC cerebellar glutamatergic transmission.
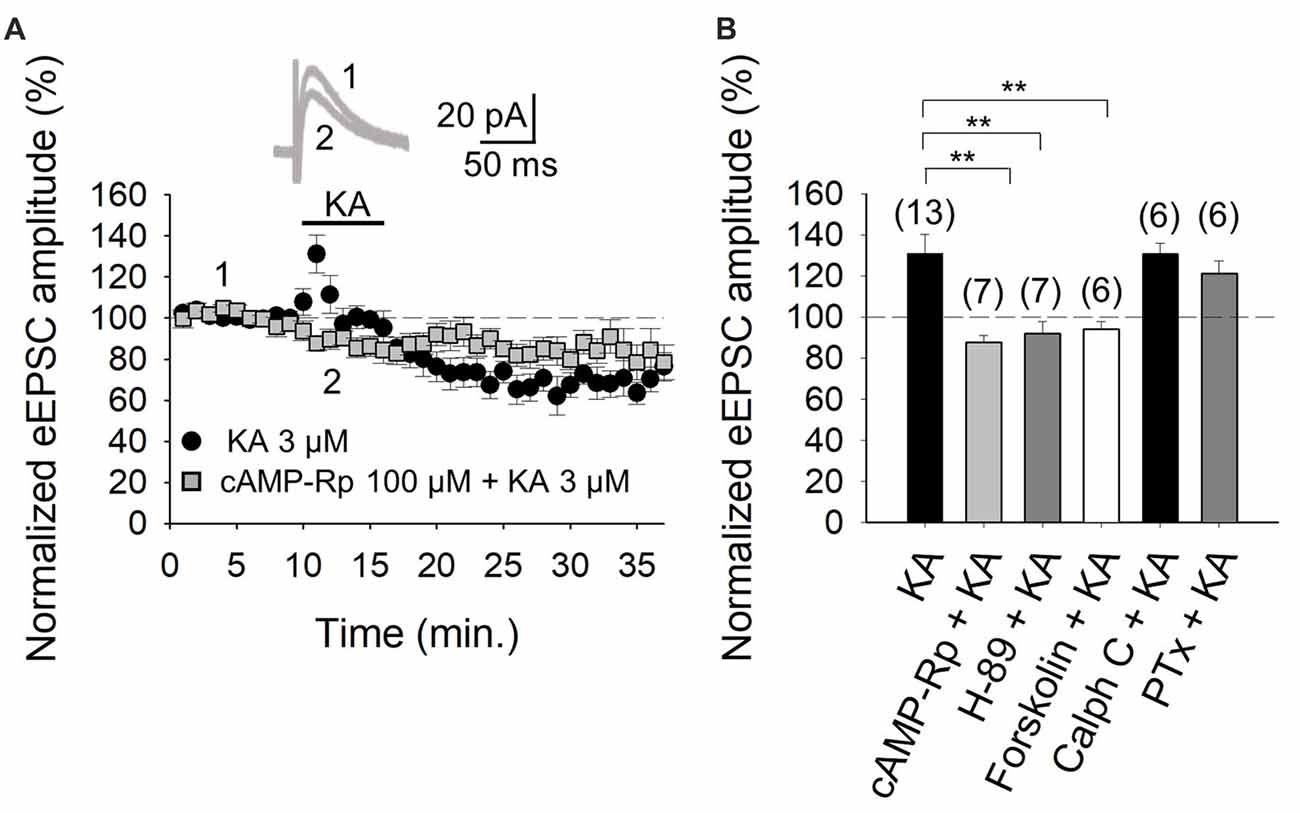
Figure 2. Activation of adenylyl cyclase (AC) and downstream protein kinase A (PKA) underlies the kainate-mediated facilitation of glutamate release in PF-PuC synapses. (A) Time-course of the effect of KA on eEPSC amplitude in cAMP-Rp treated slices. Inset shows representative traces showing that KA (3 μM) does not increase the amplitude of the eEPSCs in cAMP-Rp treated slices. (B) Inhibition of PKA by cAMP-Rp (100 μM) or H-89 (2 μM) and activation of AC by forskolin (30 μM) prevented the facilitatory action of KA. Inhibition of protein kinase c (PKC) with calphostin C (1 μM) has no effect on the KA enhancement of the eEPSC amplitude. The facilitatory effect of KA is not affected in slices treated with pertussis toxin. The number of slices (from two to three mice) is indicated in parenthesis at the top of each bar. Results are expressed as means ± SEM (**P < 0.01, ANOVA test).
For further corroboration, we next examined the KA-mediated facilitation of eEPSCs in presence of direct AC activation by forskolin. Given that forskolin produces long-lasting effects (Tong et al., 1996), slices were preincubated for 1 h with the diterpene in these experiments. KA application to forskolin (30 μM)-treated slices failed to alter eEPSC amplitudes (95 ± 4%, n = 6, Figure 2B). This observation evinces that the previous AC activation by forskolin, obviates KAR-mediated regulation at PC-PuC synapses. To ensure that the forskolin-mediate abrogation of KAR regulation was indeed due the elevation of cAMP production, rather than a non-specific effect, we used 1,9-dideoxyforskolin, the inactive diterpine analog of forskolin, in control experiments. With dideoxyforskolin (100 μM) incubation of slices, KA (3 μM) invoked a facilitation of 42 ± 5% (to 142 ± 5% of baseline, n = 5) in the amplitude of the eEPSCs, comparable to KA controls. This corroborates that occlusion of the KA receptor-mediated modulation by forskolin can indeed be ascribed to an increase in cAMP levels due to a pharmacological stimulation of AC. In sum, these data support the hypothesis that forskolin, through an AC/cAMP/PKA pathway, occludes the metabotropic, facilitatory and presynaptic action of KARs at PF-PuC synapses.
The aforementioned data in Figure 2 indicate that the KA receptor-mediated facilitation of glutamatergic transmission at these cerebellar PF-PuC synapses is manifest through an AC/cAMP/PKA signaling pathway. However, given that in other slice preparations, protein kinase C (PKC) has also been implicated in aspects of the KAR-mediated modulation (for reviews see Rodríguez-Moreno and Sihra, 2007a,b), we examined whether this kinase plays a role in the modulation of the PF-PuC cerebellar synapse by KA. In slice experiments using calphostin C (1 μM) to specifically inhibit PKC, no significant effect on the KA-mediated facilitation was observed (131 ± 5%, n = 6, Figure 2B), this result therefore obviating involvement of PKC in the modulation observed.
Next, we determined whether the facilitatory effect of presynaptic KAR activation involves G-protein function, by examining the effect of KA on slices treated with Pertussis toxin (PTx, 5 μg/ml). Intriguingly, in the presence of PTx, the KAR-mediated facilitation of synaptic transmission was retained, unaffected by G-protein block (122 ± 6%, n = 6, vs. 131 ± 9, n = 13 without PTx, Figure 2B). Interestingly, and as an important positive control for the activity of PTx, we found that the inhibitory effect of KA at the same synapse was indeed suppressed by PTx; implying selective G-protein involvement in inhibitory modulation by KAR in the preparation under study (90 ± 8%, n = 6; vs. control 65 ± 7%, n = 13, not shown).
Facilitation of Synaptic Transmission/Glutamate Release at PF-PuC Synapses Is Mediated by a Ca2+ Permeant Presynaptic KAR: Contingency on the Mobilization of Intracellular Ca2+ Stores and Ca2+/Calmodulin Dependence
The role of Ca2+ in mediating KAR-mediated synaptic facilitation has been subject of debate and controversy, for instance, at the hippocampal mossy fiber-CA3 (MF-CA3) synapses. Some studies suggest that permeation of Ca2+ through KARs and subsequent Ca2+-induced Ca2+ release from intracellular stores is obligatory for short-term and long-term plasticity at MF-CA3 synapses (Lauri et al., 2003; Scott et al., 2008). Others have registered no effect of KA on cytosolic [Ca2+] (Kamiya et al., 2002) and yet others advocate that a decrement Ca2+ concentration underpins the modulation due to KAR activation (Kamiya and Ozawa, 1998, 2000). To examine the former possibility and the presence of Ca2+ permeant KARs at these cerebellar synapses, we investigated the effect of KA on the eEPSC amplitudes in the presence of philanthotoxin, a toxin shown to block unedited, Ca2+ permeable KARs (Fletcher and Lodge, 1996; Scott et al., 2008). After treatment of slices with 3 μM philanthotoxin, the synaptic facilitation mediated by 3 μM KA was completely abrogated (to 75 ± 5% of initial amplitude, n = 7 vs. 147 ± 17%, n = 6 observed in interleaved slices, Figures 3A,B). These results evince that Ca2+ permeation through KAR is essential for the synaptic facilitation observed at PF-PuC synapses. To establish whether the aforementioned Ca2+ signal produced by KAR activation required amplification by Ca2+-induced intracellular Ca2+-store mobilization, we examined the effect of KA following depletion of intracellular Ca2+-stores, using thapsigargin to inhibit the SERCA pump responsible for Ca2+ accumulation into the stores. Treatment with thapsigargin (2 μM) eliminated the facilitatory effect of 3 μM KA, and, indeed, rather produced a depression of the response (77 ± 5%, n = 6, with thapsigargin vs. 138 ± 13%, n = 8, without thapsigargin, in interleaved slices; Figures 3C,D). These data unequivocally demonstrate the mandatory requirement for intracellular Ca2+ stores in the modulation elicited by KA. However, the question remains whether KA treatment mobilizes these intracellular Ca2+ stores by first triggered an initial rise of cytosolic Ca2+ concentrations due to extracellular Ca2+ influx. To answer this, we examined the effect of ryanodine, which selectively inhibits Ca2+-induced Ca2+ release (Berridge, 1998), to elucidate whether this underpins KAR-mediated facilitation. Ryanodine (10 μM) treatment eliminated the KAR-mediated facilitation of transmission at these PF-PuC synapses (82 ± 4%, n = 7, with ryanodine vs. 138 ± 13%, n = 7, without ryanodine, in interleaved slices; Figure 3D). Taken together, the foregoing results support the hypothesis that presynaptic KARs at PF-PuC synapses are Ca2+ permeable, and that Ca2+ entry effected by these KARs and Ca2+ channels, triggers Ca2+-induced Ca2+-release from intraterminal Ca2+-stores to produce synaptic facilitation.
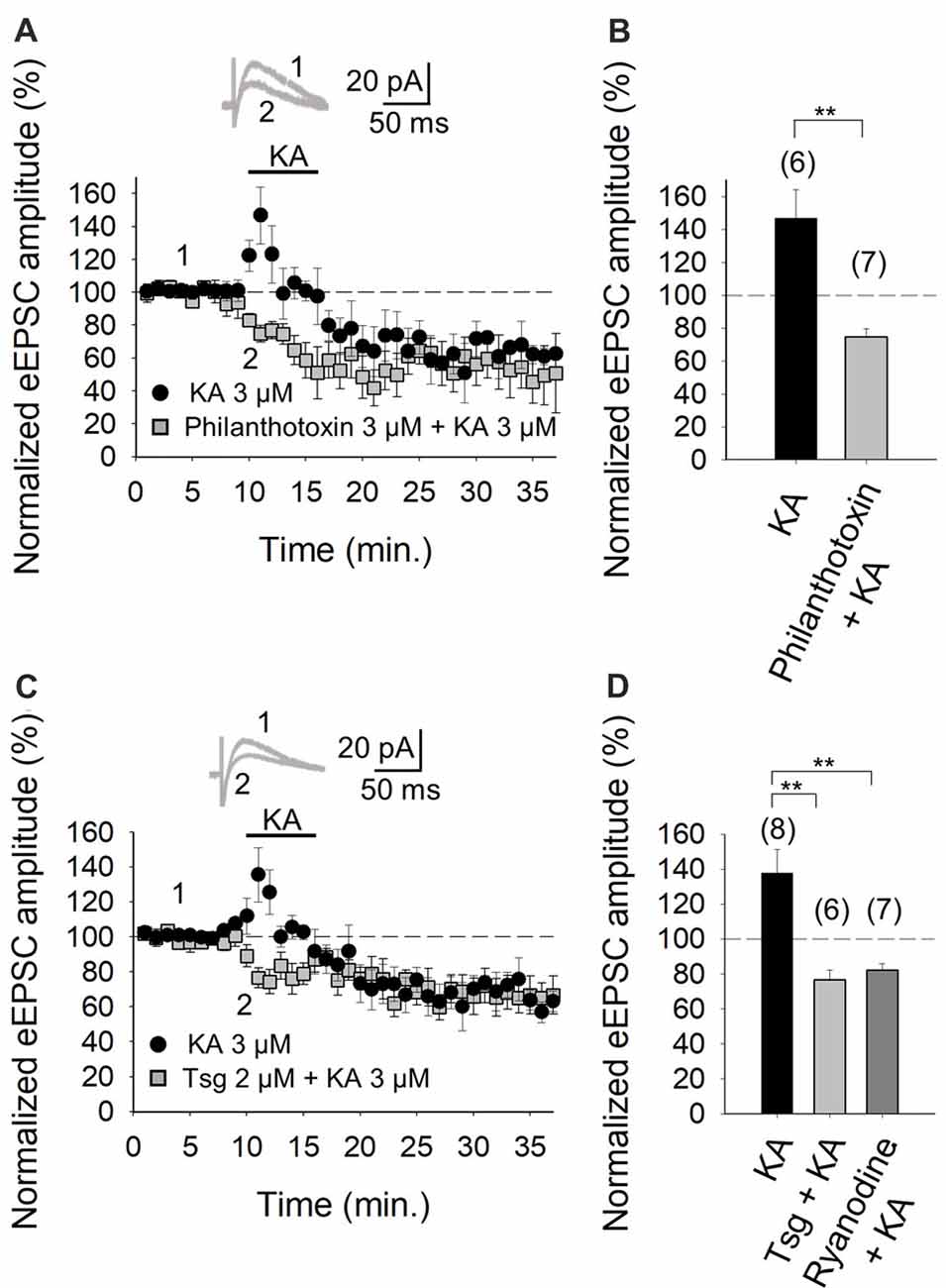
Figure 3. Facilitation of glutamate release mediated by presynaptic kainate receptor (KAR) activation requires an increase of Ca2+ in the cytosol at PF-PuC synapses. (A) Time-course of KA (3 μM) effect on eEPSCs amplitude in control condition (circles) and in slices treated with philanthotoxin (squares). (B) Quantification of modulation observed in (A). (C) Time-course of the effect of KA on eEPSCs amplitude in control slices (circles) and in thapsigargin-treated slices (squares). (D) In slices treated with thapsigargin or ryanodine, the increase of eEPSCs amplitude induced by KA is prevented. The number of slices (from two to three mice) is indicated in parenthesis at the top of each bar. Results are expressed as means ± SEM (**P < 0.01, Student’s t-test and ANOVA).
It is evident from the foregoing data that an entry of Ca2+ via KARs is obligatory for the mediation of the facilitation invoked by KA. Given that in the hippocampus and the cortex, where we have previously examined KAR-mediated modulation, the activation of a Ca2+-calmodulin complex was shown to be mandatory for AC activation (Andrade-Talavera et al., 2012, 2013), we examined this regulatory pathway at the cerebellar PF-PuC synapse by treating slices with the calmodulin antagonist, W-7, before recording eEPSCs. With W-7 (25 μM) present, KA (3 μM)-mediated facilitation was convincingly blocked (86 ± 3%, n = 6, with W-7 vs. 136 ± 13%, n = 8 without W7, in interleaved slices; Figures 4A,B). Corroboration of calmodulin dependence of the modulation, was also evident from experiments performed in the presence of calmidazolium (CMZ, 1 μM), an alternative calmodulin antagonist. As with W-7, in presence of CMZ, KA (3 μM)-mediated facilitation of synaptic transmission was abograted (78 ± 11, n = 6, Figure 4B). These data support the postulate that a presynaptic Ca2+-calmodulin complex is obligatory for the KAR-mediated synaptic regulation and operates upstream of the activation of AC/cAMP/PKA signaling.
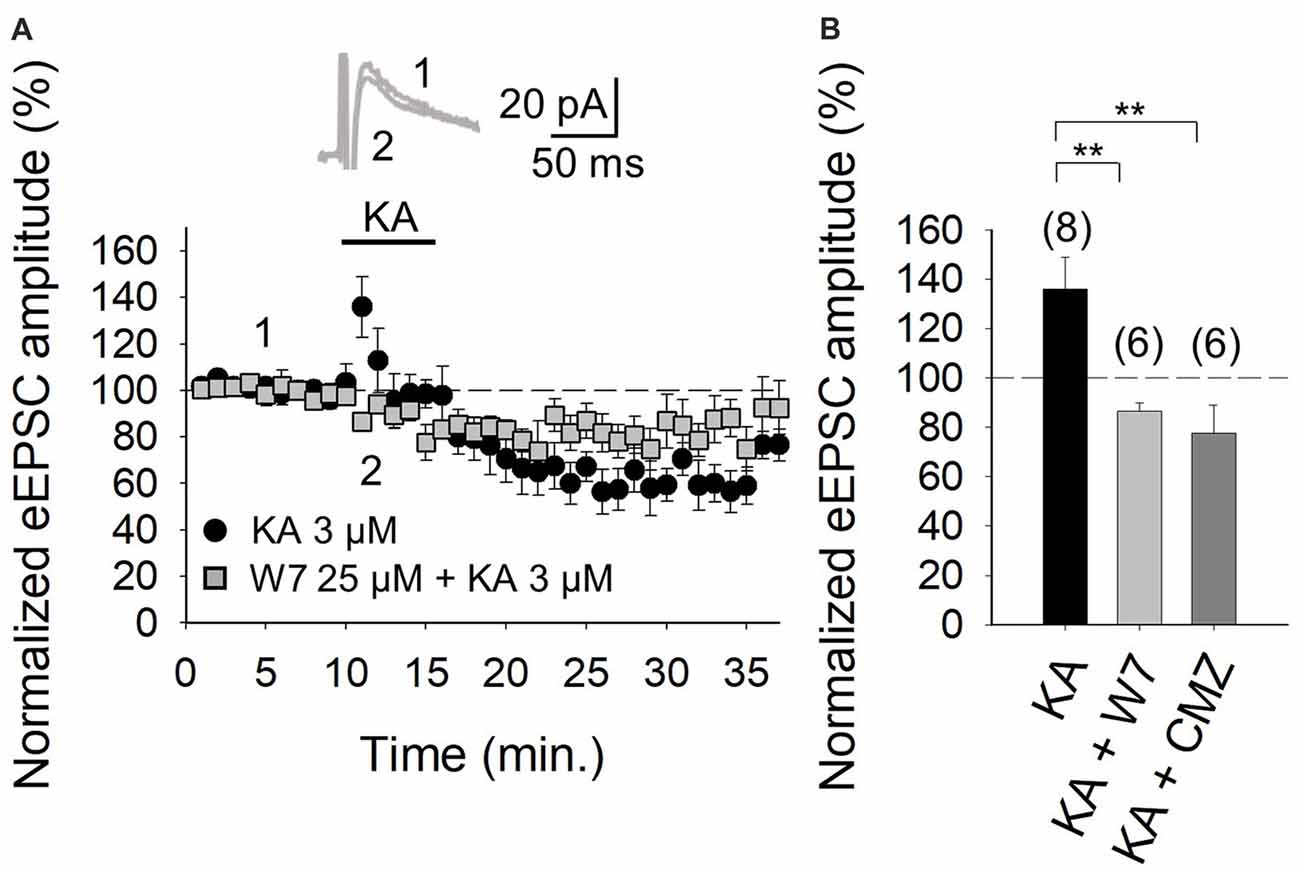
Figure 4. Facilitation of glutamate release mediated by presynaptic KAR activation requires Ca2+-calmodulin at PF-PuC synapses. (A) Time-course of KA (3 μM) effect on eEPSCs amplitude in control condition (circles) and in the slices treated with 25 μM W-7 (squares). Inset show traces before and after 4 min KA perfusion from W-7 treated slices. (B) Quantification of modulation observed in (A) and in the presence of 1 μM CMZ. The number of slices (from two to three mice) is indicated in parenthesis at the top of each bar. Results are expressed as means ± SEM (**P < 0.01, ANOVA test).
Discussion
The results in this study, utilizing electrophysiological experiments in cerebellar slices, show that the activation of presynaptic KARs in the cerebellum, at PF-PuC synapses, invokes a facilitation of synaptic transmission/glutamate release. Analysis of this modulation suggests a mechanistic coupling of KARs through Ca2+-calmodulin/AC/cAMP/PKA activity, but independently of G-protein activation.
We would hypothesize that the observed KA-mediated enhancement of the eEPSPs at PF-PuC synapses is due to increased glutamate release, which could be monitored by NMDA receptor-mediated currents (with AMPA receptors antagonized by GYKI53655), and blocked by the KAR/AMPA receptor antagonist NBQX. Under conditions where AMPA receptor activation is obviated by GYKI53655, this therefore delineates the specific role of KARs in the facilitatory regulation, particularly given that a receptor antagonist cocktail, formulated to eliminate the presynaptic effects of neurotransmitters that might be secondarily released, also had no effect on the observed synaptic modulation due to KA.
In assessing synaptic regulation, it is indeed of utmost importance to identify the subcellular location of the KAR postulated. We corroborated the presynaptic presence of KARs by electrophysiological analysis of a presynaptically manifest parameter, i.e., the PPR (pair-pulse ratio) of consecutive eEPSCs mediated by neurotransmitter release. A clear decrease in the PPR of eEPSCs observed with KA application in our experiments, suggested a change in release probability (by definition a presynaptic property in synaptic transmission). Secondly, we assessed the proportion of synaptic failures in response to KA application. With KA application, the failure proportion was evidently diminished, supportive of an increase in the probability of presynaptic transmitter release and corroborative of the observed facilitation occurring through KAR activation. Finally, importantly, the effects of KA observed on NMDA and AMPA receptor-mediated currents were similar. Given that no such equivalence would be predicted if the observed modulation was postsynaptic, the data here are supportive of a presynaptic mode of action for KARs being activated. Altogether, three independent analyses mutually corroborate and emphasize a presynaptic locus of action of KARs functioning at PF-PuC synapses.
It remains to be elucidated whether the presynaptic regulation by KA at PF-PuCs reports the activity of KARs subcellularly localized at nerve terminal/axonal or somatodendritic compartments. The technically challenging paradigms needed to address this question is beyond the scope of the present paper. However, to directly elucidate the presynaptic compartmentalization of KARs, future work necessitates: (i) high resolution immunolocalization (immunogold-based) of the receptor (contingent on the availability of high affinity antibodies with appropriate KAR subunit-specificity); and (ii) targeted blockade of KARs using caged-antagonists (contingent on the pending development of reagents) (see NMDA receptor studies, Rodríguez-Moreno et al., 2011; Reeve et al., 2012).
Corresponding with our previous studies in the hippocampus and cortex (Rodríguez-Moreno and Sihra, 2004, 2013; Andrade-Talavera et al., 2012, 2013), PKA inhibition by the cell-permeant cyclic nucleotide analog cAMP-Rp, also led to the abrogation of KA-mediated enhancement of synaptic transmission/glutamate release at PF-PuC synapses. The congruence of the mechanism between synapses was also highlighted in the current studies, by the observation that the inhibition PKA catalytic activity by H-89 also eliminated the KA-mediated facilitation. Similarly and congruently, direct activation of AC by preincubation with forskolin, produced refractoriness of the facilitatory effect of KA. Collectively, these results consistently suggest that AC/cAMP/PKA signaling underpins the facilitatory modulation of synaptic transmission/glutamate release at the synapse under study here.
Consistently, there has been a notable absence of any evidence to support G-protein mediated initiation/transduction of the AC/cAMP/PKA cascade posited to be involved in presynaptic KAR-mediated enhancement of glutamate release. We therefore addressed the conceivable role of Ca2+ as the initiator of a G-protein independent signaling cascade, in the modulation being investigated at defined cerebellar PF-PuC synapses. In the canonical context, KARs may mediate external Ca2+ entry through an ionotropic activity which would depolarize nerve terminals (Perkinton and Sihra, 1999) and thus activate voltage-gated Ca2+ channels. Non-canonically, direct Ca2+ influx via Ca2+ permeable KARs per se is also possible (Fletcher and Lodge, 1996; Scott et al., 2008). Notably in the studies reported herein, a blockade of the Ca2+ permeable KARs by the selective inhibitor, philanthotoxin, abrogated the KA-mediated synaptic facilitation in the current studies, pointing to a strict requirement for external Ca2+ entering via KARs to support facilitatory modulation. Interestingly therefore, it would appear that, although it is thought that unedited Ca2+ permeable KARs typically prevail earlier during neuronal development, these receptors evidently persevere with activity in the cerebellum of the early adult, mouse brain.
We extended the analysis of properties of KAR-mediated regulation by examining the hypothesis that the essential core, albeit perhaps limited, Ca2+ entry via KARs may be amplified by Ca2+ mobilized from intraterminal stores, as reported for similar modulation at hippocampal synapses (Lauri et al., 2003; Scott et al., 2008). Emphatically supporting a critical role for intraterminal Ca2+ stores, were our results showing that thapsigargin treatment, to deplete intracellular Ca2+ stores (Irving et al., 1992), abolished the facilitatory regulation by KARs. Again, in corroboration of the hypothesis, use of ryanodine to selectively inhibit Ca2+-induced Ca2+-release (Berridge, 1998), evinced that Ca2+ entry via KARs induces Ca2+ mobilization from intraterminal Ca2+ stores to invoke the modulation seen herein.
Having demonstrated that Ca2+ entering via KARs and subsequence Ca2+ increases due to mobilization of intraterminal Ca2+ stores is actually obligatory for the KA facilitation of synaptic transmission at PF-PuC synapse, we questioned how might such an increase in cytosolic [Ca2+] couple to the postulated AC/cAMP/PKA signaling mediating the facilitation by KARs. From previous studies, it is plausible that the rise in cytosolic [Ca2+] activates Ca2+-dependent ACs present in parallel fiber terminals. A number of ACs have been described, however, AC1 and AC8 are two members of the family, that have been shown to be activated by Ca2+-calmodulin and are abundant in the central nervous system (for reviews see Cooper, 2003; Wang and Storm, 2003). Interestingly, supporting their significance to the hypothesis, studies with double knockouts of AC1 and AC8, have shown that the Ca2+-calmodulin ACs are indeed essential for the widely reported Ca2+-dependent elevation of cAMP (Wong et al., 1999). Using the calmodulin antagonists W-7 and CMZ, our data shows that inhibiting Ca2+-calmodulin function, abolishes the presynaptic KAR-mediated modulation in cerebellar slices. This supports the hypothesis that, following KAR activation and elevation of cytosolic [Ca2+], a Ca2+-calmodulin-dependent coupling may activate AC1 and/or AC8, and thereby extend to the initiation of the AC/cAMP/PKA cascade, hence promoting synaptic facilitation through increased neurotransmitter release at PF-PuCs synapses.
This report shows the PF-PuC synapse is a reliable and robust model for the study of KAR mediated modulation. Interestingly, however, we observed a facilitation of synaptic transmission/glutamate release at 3 μM KA, contrary to previous work with the same synapse, where 500 nM agonist induced a depression in glutamate release (Delaney and Jahr, 2002). The discrepancy may reflect the notably different agonist concentrations employed, and indeed the difference in the age of animals, and perhaps the species of animals used in the previous experiments (Delaney and Jahr, 2002 utilized P13-P17 rats). The higher concentration of KA necessary for the activation of KARs at young adult synapses may indeed be indicative of the different efficacy of KA at KARs at this age, perhaps dependent on the expression of specific glutamate receptor subunit subtypes composing the resident KARs.
From our results, it is clear that KAR function is preserved in early adult animals at PF-PuC synapses and is not temporally limited to the two firsts postnatal weeks as reported previously (Delaney and Jahr, 2002). KARs have an autoreceptor role in developing animals, with the concentration of agonist determining presynaptic modulation: facilitation (at low [KA]) and depression (at high [KA]) and, thereby, putatively determining synapse consolidation and stability. Although the precise role(s) of these KARs in adult animals remains to be explicated, the modulation of presynaptic function reported herein might manifest some forms of plasticity. For instance, KARs have been shown to be involved in plasticity at PF-PuCs synapses (for review see Hirano, 2013; Sihra et al., 2014).
Our experiments here do show that KAR activation has a biphasic effect at PF-PuC synapses as previously reported (Delaney and Jahr, 2002); inducing a depression of glutamate release at relatively high concentrations of KA (>1–3 μM) rather than facilitation. The aim of the current study was to elucidate the mechanistic details of the observed facilitation, rather than the factors underpinning in the synaptic depression. However, notably, our results did reveal that the transient synaptic depression observed with high KA concentrations is abolished in the presence of cAMP-Rp (inhibition of PKA activation), but not affected by any of the experimental manipulations speaking to regulation of cytosolic [Ca2+] or function thereof, i.e., philantotoxin, thapsigargin, W-7 or CMZ. These observations point to the synaptic depression seen likely involving an AC/cAMP/PKA pathway, as described for facilitation, but without the proposed Ca2+-AC coupling. The indications are therefore, that KARs have alternative mechanistic modes for facilitatory and depressive action. Indeed, KAR coupling to an increase in cAMP concentrations (and subsequent enhancement of PKA activity), or to a decrease in cAMP concentrations (and subsequent diminution of PKA activity), has been reported in studies investigating the mossy fiber-CA3 synapse of the hippocampus (Negrete-Díaz et al., 2006, 2007; Andrade-Talavera et al., 2012), the amygdala (Negrete-Díaz et al., 2012) and in the cortex (Andrade-Talavera et al., 2013). The key difference that our current results point to is the differing direction and means of regulation of the AC/cAMP/PKA cascade in the bimodal regulation by KARs. Whereas, presynaptic facilitatory function by KARs evidently involves an increase in AC/cAMP/PKA signaling instigated by the Ca2+-calmodulin complex, KARs appear to be negatively coupled to the AC/cAMP/PKA pathway to effect depression synaptic transmission. Previous studies at MF-CA3 synapses and thalamocortical synapses (and as confirmed here) have reported that the depression of presynaptic function occurs through a negative coupling to AC/cAMP/PKA and is actually invoked by the action of a PTx sensitive G-protein (Negrete-Díaz et al., 2006; Andrade-Talavera et al., 2013). Notwithstanding the postulated differential upstream coupling to AC to achieve facilitation and depression, it is also plausible that the diametric mechanisms reflect the operation of two distinct types of KARs. Future studies will elucidate the exact instruments involved in the observed KAR-mediated modulation and address the question as to whether different populations of presynaptic KARs reside at the PF-PuC synapse.
In conclusion, our studies show that presynaptic KARs activation by KA, at PF-PuC synapses produces a facilitation of synaptic transmission consistent with an increase in neurotransmitter release. We postulate that, mechanistically, KAR-mediated presynaptic facilitation involves an increase in cytosolic [Ca2+], first by external Ca2+-entry via Ca2+-permeable KARs, and second by this then triggering the mobilization of intracellular Ca2+ from stores in granule cells terminals. The raised Ca2+ binds to calmodulin to form a Ca2+-calmodulin complex, which we postulate activates AC1 or AC8 to elevate cAMP levels and thus effect PKA stimulation. The latter invokes an enhancement of glutamate release and hence synaptic transmission at the PF-PuC synapse in the cerebellum.
Author Contributions
RF-M and PL-R performed the experiments. TS and AR-M wrote the manuscript. AR-M designed the study.
Funding
This study was supported by Grants BFU2006-1455 (from the Ministerio de Economía y Competitividad) and a grant from the “Eugenio Rodríguez-Pascual Foundation” 2011 to AR-M.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Andrade-Talavera, Y., Duque-Feria, P., Negrete-Díaz, J. V., Sihra, T. S., Flores, G., and Rodríguez-Moreno, A. (2012). Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin at mossy fiber-CA3 synapses. J. Neurochem. 122, 891–899. doi: 10.1111/j.1471-4159.2012.07844.x
Andrade-Talavera, Y., Duque-Feria, P., Sihra, T. S., and Rodríguez-Moreno, A. (2013). Presynaptic kainate-receptor-mediated facilitation of glutamate reléase involves PKA and Ca2+ -calmodulin at thalamocortical synapses. J. Neurochem. 126, 565–578. doi: 10.1016/j.febslet.2013.01.071
Bahn, S., Volk, B., and Wisden, W. (1994). Kainate receptor gene expression in the developing rat brain. J. Neurosci. 14, 5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994
Berridge, M. J. (1998). Neuronal calcium signalling. Neuron 21, 13–26. doi: 10.1016/S0896-6273(00)80510-3
Bettler, B., Boulter, J., Hermans-Borgmeyer, I., O’Shea-Greenfield, A., Deneris, E. S., Moll, C., et al. (1990). Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron 5, 583–595. doi: 10.1016/0896-6273(90)90213-y
Breustedt, J., and Schmitz, D. (2004). Assesing the role of GLUK5 and GLUK6 at hippocampal mossy fiber synapses. J. Neurosci. 24, 10093–10098. doi: 10.1523/JNEUROSCI.3078-04.2004
Campbell, S. L., Mathew, S. S., and Hablitz, J. J. (2007). Pre-and postsynaptic effects of kainate on layer II/III pyramidal cells in rat neocortex. Neuropharmacology 53, 37–47. doi: 10.1016/j.neuropharm.2007.04.008
Contractor, A., Sailer, A., Darstein, M., Maron, C., Xu, J., Swanson, G., et al. (2003). Loss of kainate receptor-mediated heterosynaptic facilitation at mossy-fiber synapses in KA2−/− mice. J. Neurosci. 23, 422–429. doi: 10.1523/JNEUROSCI.23-02-00422.2003
Cooper, D. M. (2003). Regulation and organization of adenylyl cyclases and cAMP. Biochem. J. 375, 517–529. doi: 10.1042/bj20031061
Delaney, A. J., and Jahr, C. E. (2002). Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron 36, 475–482. doi: 10.1016/s0896-6273(02)01008-5
Fernandes, H. B., Catches, J. S., Petralia, R. S., Copits, B. A., Xu, J., Russell, T. A., et al. (2009). High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 63, 818–829. doi: 10.1016/j.neuron.2009.08.010
Fletcher, E. J., and Lodge, D. (1996). New developments in the molecular pharmacology of AMPA and kainate receptors. Pharmacol. Ther. 70, 65–89. doi: 10.1016/0163-7258(96)00014-9
Herb, A., Burnashev, N., Werner, P., Sakmann, B., Wisden, W., and Seeburg, P. H. (1992). The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 8, 775–785. doi: 10.1016/0896-6273(92)90098-x
Hirano, T. (2013). Long-term depression and other synaptic plasticity in the cerebellum. Proc. Jpn. Acad. Ser. B Phys. Biol. 89, 183–195. doi: 10.2183/pjab.89.183
Irving, A. J., Collingridge, G. L., and Schofield, J. G. (1992). Interactions between Ca2+ mobilising mechanisms in cultured rat cerebellar granule cells. J. Physiol. 456, 667–680. doi: 10.1113/jphysiol.1992.sp019360
Jane, D. E., Lodge, D., and Collingridge, G. L. (2009). Kainate receptors: pharmacology, function and therapeutical potential. Neuropharmacology 56, 90–113. doi: 10.1016/j.neuropharm.2008.08.023
Ji, Z., and Stäubli, U. (2002). Presynaptic kainate receptors play different physiological roles in mossy fiber and associational-commissural synapses in CA3 of hippocampus from adult rats. Neurosci. Lett. 331, 71–74. doi: 10.1016/s0304-3940(02)00865-0
Jouhanneau, J. S., Ball, S. M., Molnár, E., and Isaac, J. T. R. (2011). Mechanisms of bi-directional modulation of thalamocortical transmission in barrel cortex by presynaptic kainite receptors. Neuropharmacology 60, 832–841. doi: 10.1016/j.neuropharm.2010.12.023
Kamiya, H., and Ozawa, S. (1998). Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J. Physiol. 509, 833–845. doi: 10.1111/j.1469-7793.1998.833bm.x
Kamiya, H., and Ozawa, S. (2000). Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre. J. Physiol. 523, 653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x
Kamiya, H., Umeda, K., Ozawa, S., and Manabe, T. (2002). Presynaptic Ca2+ entry is unchanged during hippocampal mossy fiber long-term potentiation. J. Neurosci. 22, 10524–10528. doi: 10.1523/jneurosci.22-24-10524.2002
Lauri, S. E., Bortolotto, Z. A., Bleakman, D., Ornstein, P. L., Lodge, D., Isaac, J. T. R., et al. (2001a). A critical role of a facilitatory kainate autoreceptor in mossy fiber LTP. Neuron 32, 697–709. doi: 10.1016/s0896-6273(01)00511-6
Lauri, S. E., Delany, C., Clarke, V. E. J., Bortolotto, Z. A., Ornstein, P. I., Isaac, J. T., et al. (2001b). Synaptic activation of a presynaptic kainate receptor facilitates AMPA receptor-mediated synaptic transmission at hippocampal mossy fibre synapses. Neuropharmacology 41, 907–915. doi: 10.1016/s0028-3908(01)00152-6
Lauri, S. E., Bortolotto, Z. A., Nistico, R., Bleakman, D., Ornstein, P. L., Lodge, D., et al. (2003). A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron 39, 327–341. doi: 10.1016/s0896-6273(03)00369-6
Lerma, J., and Marques, J. M. (2013). Kainate receptors in health and disease. Neuron 80, 292–311. doi: 10.1016/j.neuron.2013.09.045
Manabe, T., Willey, D. J., Perkel, D. J., and Nicoll, R. A. (1993). Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol. 70, 1451–1459. doi: 10.1152/jn.1993.70.4.1451
Negrete-Díaz, J. V., Duque-Feria, P., Andrade-Talavera, Y., Carrión, M., Flores, G., and Rodríguez-Moreno, A. (2012). Kainate receptor-mediated depression of glutamatergic transmission involving protein kinase A in the lateral amygdala. J. Neurochem. 121, 36–43. doi: 10.1111/j.1471-4159.2012.07665.x
Negrete-Díaz, J. V., Sihra, T. S., Delgado-García, J. M., and Rodríguez-Moreno, A. (2006). Kainate receptor-mediated inhibition of glutamate release involves protein kinase A in the mouse hippocampus. J. Neurophysiol. 96, 1829–1837. doi: 10.1152/jn.00280.2006
Negrete-Díaz, J. V., Sihra, T. S., Delgado-García, J. M., and Rodríguez-Moreno, A. (2007). Kainate receptor-mediated presynaptic inhibition converges with presynaptic inhibition mediated by Group II mGluRs and long-term depression at the hippocampal mossy fiber-CA3 synapse. J. Neural Transm. Vienna 114, 1425–1431. doi: 10.1007/s00702-007-0750-4
Perkinton, M. S., and Sihra, T. S. (1999). A high-affinity presynaptic kainate-type glutamate receptor facilitates glutamate exocytosis from cerebral cortex nerve terminals (synaptosomes). Neuroscience 90, 1281–1292. doi: 10.1016/s0306-4522(98)00573-9
Petralia, R. S., Wang, Y. X., and Wenthold, R. J. (1994). Histological and ultrastructural localization of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. J. Comp. Neurol. 349, 85–110. doi: 10.1002/cne.903490107
Pinheiro, P. S., Perrais, D., Coussen, F., Barhanin, J., Bettler, B., Mann, J. R., et al. (2007). GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. U S A 1004, 12181–12186. doi: 10.1073/pnas.0608891104
Reeve, J. E., Kohl, M. M., Rodríguez-Moreno, A., Paulsen, O., and Anderson, H. L. (2012). Caged intracellular NMDA receptor blockers for the study of subcellular ion channel function. Commun. Integr. Biol. 5, 240–242. doi: 10.4161/cib.19400
Rodríguez-Moreno, A., Kohl, M. M., Reeve, J. E., Eaton, T. R., Collins, H. A., Anderson, H. L., et al. (2011). Presynaptic induction and expression of timing-dependent long-term depression demonstrated by compartment-specific photorelease of a use-dependent NMDA receptor antagonist. J. Neurosci. 31, 8564–8569. doi: 10.1523/JNEUROSCI.0274-11.2011
Rodríguez-Moreno, A., and Sihra, T. S. (2004). Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus. J. Physiol. 557, 733–745. doi: 10.1113/jphysiol.2004.065029
Rodríguez-Moreno, A., and Sihra, T. S. (2007a). Metabotropic actions of kainate receptors in the CNS. J. Neurochem. 103, 2121–2135. doi: 10.1111/j.1471-4159.2007.04924.x
Rodríguez-Moreno, A., and Sihra, T. S. (2007b). Kainate receptors with a metabotropic modus operandi. Trends Neurosci. 30, 630–637. doi: 10.1016/j.tins.2007.10.001
Rodríguez-Moreno, A., and Sihra, T. S. (2013). Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin and PKA in cerebrocortical synaptosomes. FEBS Lett. 587, 788–792. doi: 10.1016/j.febslet.2013.01.071
Schmitz, D., Mellor, J., and Nicoll, R. A. (2001). Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 291, 1972–1976. doi: 10.1126/science.1057105
Scott, R., Lalic, T., Kullmann, D. M., Capogna, M., and Rusakov, D. A. (2008). Target-cell specificity of kainate autoreceptor and Ca2+ store-dependent short-term plasticity at hippocampal mossy fibers. J. Neurosci. 28, 13139–13149. doi: 10.1523/JNEUROSCI.2932-08.2008
Shira, T. S., and Rodríguez-Moreno, A. (2013). Presynaptic kainate receptor-mediated bidirectional modulatory actions: mechanisms. Neurochem. Int. 62, 982–987. doi: 10.1016/j.neuint.2013.03.012
Sihra, T. S., Flores, G., and Rodríguez-Moreno, A. (2014). Kainate receptors: multiple roles in neuronal plasticity. Neuroscientist 20, 29–43. doi: 10.1177/1073858413478196
Smith, T. C., Wang, L. Y., and Howe, J. R. (1999). Distinct kainate receptor phenotypes in immature and mature mouse cerebellar granule cells. J. Physiol. 517, 51–58. doi: 10.1111/j.1469-7793.1999.0051z.x
Swanson, G. T., Feldmeyer, D., Kaneda, M., and Cull-Candy, S. G. (1996). Effect of RNA editing and subunit co-assembly single-channel properties of recombinant kainate receptors. J. Physiol. 492, 129–142. doi: 10.1113/jphysiol.1996.sp021295
Tong, G., Malenka, R. C., and Nicoll, R. A. (1996). Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron 16, 1147–1157. doi: 10.1016/s0896-6273(00)80141-5
Valbuena, S., and Lerma, J. (2016). Non-canonical signalling, the hidden life of ligand-gated ion channels. Neuron 92, 316–329. doi: 10.1016/j.neuron.2016.10.016
Wang, H., and Storm, D. R. (2003). Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol. Pharmacol. 63, 463–468. doi: 10.1124/mol.63.3.463
Keywords: kainate receptors, glutamate, presynaptic, Ca2+-calmodulin, PKA, slices
Citation: Falcón-Moya R, Losada-Ruiz P, Sihra TS and Rodríguez-Moreno A (2018) Cerebellar Kainate Receptor-Mediated Facilitation of Glutamate Release Requires Ca2+-Calmodulin and PKA. Front. Mol. Neurosci. 11:195. doi: 10.3389/fnmol.2018.00195
Received: 17 March 2018; Accepted: 16 May 2018;
Published: 06 June 2018.
Edited by:
Inmaculada M. Gonzalez-Gonzalez, University of Central Lancashire, United KingdomReviewed by:
James P. Clement, Jawaharlal Nehru Centre for Advanced Scientific Research, IndiaElek Molnar, University of Bristol, United Kingdom
Copyright © 2018 Falcón-Moya, Losada-Ruiz, Sihra and Rodríguez-Moreno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Rodríguez-Moreno, YXJvZG1vckB1cG8uZXM=