
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Mol. Neurosci. , 31 May 2018
Sec. Neuroplasticity and Development
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00184
This article is part of the Research Topic Molecular Mechanisms Underlying Psychiatric Disorders View all 18 articles
 Francesco Rusconi1*
Francesco Rusconi1* Elena Battaglioli1,2*
Elena Battaglioli1,2*Psychiatric disorders entail maladaptive processes impairing individuals’ ability to appropriately interface with environment. Among them, depression is characterized by diverse debilitating symptoms including hopelessness and anhedonia, dramatically impacting the propensity to live a social and active life and seriously affecting working capability. Relevantly, besides genetic predisposition, foremost risk factors are stress-related, such as experiencing chronic psychosocial stress—including bullying, mobbing and abuse—, and undergoing economic crisis or chronic illnesses. In the last few years the field of epigenetics promised to understand core mechanisms of gene-environment crosstalk, contributing to get into pathogenic processes of many disorders highly influenced by stressful life conditions. However, still very little is known about mechanisms that tune gene expression to adapt to the external milieu. In this Perspective article, we discuss a set of protective, functionally convergent epigenetic processes induced by acute stress in the rodent hippocampus and devoted to the negative modulation of stress-induced immediate early genes (IEGs) transcription, hindering stress-driven morphostructural modifications of corticolimbic circuitry. We also suggest that chronic stress damaging protective epigenetic mechanisms, could bias the functional trajectory of stress-induced neuronal morphostructural modification from adaptive to maladaptive, contributing to the onset of depression in vulnerable individuals. A better understanding of the epigenetic response to stress will be pivotal to new avenues of therapeutic intervention to treat depression, especially in light of limited efficacy of available antidepressant drugs.
Molecular psychiatry mainly recognizes three typologies of stressful events, namely positive stress, tolerable stress and toxic stress (McEwen, 2017). “Positive” stress (also known as eustress; Selye, 1998) entails reward-associated paradigms including whatever hard paths to meet our life expectations (job promotions and achievements in general). On the other hand, we can recognize two typologies of negative stress (also known as distress; Selye, 1998), namely “tolerable” and “toxic” stress. “Tolerable” stress has to do with negative experiences, prototypically represented by loss of beloved persons, but also related to personal, economic or health crisis (McEwen, 2017). Regardless how hard this kind of stress can be, it is identified by those experiences that most people have the mental instruments to cope with, also thanks to supportive relationships (McEwen et al., 2015). However, behavioral outcome of negative contingencies can vary in an individual-specific manner accordingly to the set of previous life experiences and with respect to genetic background in the frame of genotype × environment interactions (GxE; de Kloet et al., 2005; Caspi and Moffitt, 2006; Joëls and Baram, 2009; McEwen, 2012; Sun et al., 2013; Calhoon and Tye, 2015). Indeed, also a negative stress theoretically predictable as tolerable could lead to psychiatric issues in a subset of the general population referred to as stress-vulnerable individuals. Beside the intensity and aversiveness of the stressful stimuli, chronic reiteration or duration of a negative experience can change their outcome often leaving long-term mood effects (McEwen, 2003; Cohen et al., 2007). “Toxic” stress is represented by those experiences featuring recurrent parameters of unpredictability and inescapability (terrorist attacks, earthquake, military operations; McEwen, 2017). This form of stress is considered detrimental for the majority of population and can lead to post traumatic stress disorder (PTSD) and other neuropsychiatric issues (McEwen, 2005; Nagy et al., 2017). Nonetheless, also in case of toxic stress, there are individuals displaying resiliency (McEwen et al., 2015). Consistently, a relevant open question in neurobiology of stress response is related to molecular underpinnings of vulnerability or resiliency. Tolerable stress with a positive behavioral outcome can be modeled in rodents using a single acute stressful paradigm experienced by naïve mice or rats, since such a challenge does not elicit long-term behavioral effects on a cognitive or emotional point of view. On the contrary, chronic administration of the same forms of stress can precipitate mood and cognition-related issues in a subset of susceptible individuals (Golden et al., 2011; McEwen et al., 2015).
The most studied pathway of stress response is referred to as the hypothalamic-pituitary-adrenal (HPA) axis. This system allows environmental adaptation via a complex interplay between two sets of processes acting at the molecular and cellular level and influencing behavioral responses. The first set underlies the arousal phase of stress response (primary response), which includes reactions leading to the required wakefulness to respond to threat, invigorating physical strength and cognitive acuity (Davis et al., 2003). The second set is related to stress termination and more in general to homeostatic processes in the body aimed at restraining excessive reactions (secondary response). Glucocorticoid hormone is an important final effector of stress signals mainly involved in HPA axis homeostasis and feedback (Cohen et al., 2012). Stress termination is largely operated at the hippocampal level, brain area that is involved in stress-response, and that expresses a high level of glucocorticoid receptors. Besides HPA axis, a core process selected by evolution to survive to environmental changes, other systems contribute to cope with stress through a fine-tuning of glutamate response. A well-known pathway that helps responding to stress via glutamate signaling regulation is the endocannabinoid system (ECS). In this case, the primary response is represented by stress-induced glutamate release in brain areas that are activated by stress, which in turn promotes through ECS, depolarization-induced suppression of excitation (DSE) as the secondary response. ECS is highly effective in restraining the excitotoxic consequences of stress-induced glutamate, contrasting toxic behavioral correlates of environmental stress (Lutz et al., 2015; Morena et al., 2016).
A single stress event induces temporary activation of the stress-response machinery, and since they are not associated to long-lasting behavioral alterations, related modifications on neuronal physiology can be defined as adaptive (Hunter et al., 2009, 2012; Rusconi et al., 2016; Saunderson et al., 2016; McEwen, 2017). Interestingly, it is generally accepted that psychopathology must result from a failure in a correct functionality of HPA axis and ECS (McEwen, 2003; Weaver et al., 2004; Lutz et al., 2015; McEwen et al., 2015; Morena et al., 2016). In particular, excessive or reiterated engagement of stress-coping molecular mechanisms (including but not limited to the action of stress hormones) in a chronic manner, could lead to allostatic overload via desensitization of stress response pathways (Joëls and Baram, 2009; McEwen, 2017).
In this Perspective article, we shed new light on a further mechanism of stress-response based on epigenetic modifications of gene expression that cooperates at the nuclear level with HPA axis and ECS. We further emphasize the relevance of these adaptive mechanisms in response to acute stress as their impairment might represent a proxy for the onset of stress-related psychopathology (Borrelli et al., 2008; McEwen, 2012; Nestler, 2014; Nagy et al., 2017).
Stress induces specific brain plasticity-modifying transcriptional programs in corticolimbic circuitry including the medial prefrontal cortex (mPFC), the ventral hippocampus (vHIP) and the amygdala (Felix-Ortiz et al., 2013; Janak and Tye, 2015; Laine et al., 2017). In these areas, behavioral stress has been recognized as a powerful modifier of neuronal structural plasticity (McEwen et al., 2012; Chattarji et al., 2015). Either acute or chronic stress can transiently or stably modify dendritic spine density and arborization (Golden et al., 2013; Maras et al., 2014; Chattarji et al., 2015; Janak and Tye, 2015), suggesting that adaptive structural modification of corticolimbic areas can also correspond to a neutral behavioral response. The magnitude and direction of stress-mediated neuroplastic remodeling vary according to the peculiar structure observed, generally increasing amygdalar neuroplasticity (enhancing fear reactions) and decreasing hippocampal and prefrontal cortex functionality (leading to impaired control over affective manifestation; McEwen et al., 2012; Felix-Ortiz et al., 2013; Chattarji et al., 2015). This bidirectional corticolimbic unbalance underlies aberrant top-down inhibition of the limbic system, a core symptom of neuropsychiatric disorders (Martin et al., 2009; Franklin et al., 2012; Calhoon and Tye, 2015).
Neurons are able to modify synaptic activity in response to environmental inputs—including stress—through epigenetic modifications allowing to finely and steadily perturb gene expression (Borrelli et al., 2008). Stress can engrave chromatin with a peculiar alphabet, made up of histone post-translational modifications (PTMs) and DNA methylation (Tsankova et al., 2006; Borrelli et al., 2008). The study of epigenetic modifications—ultimately shaping the intimacy of DNA-histone interactions regulating the accessibility and processivity of basal transcription machinery—operated by different sources of stress in the brain has just begun, and much more work is required to clarify the typology and relative behavioral relevance of specific epigenetic modulations.
We here describe different examples of epigenetic processes that occur in response to acute “negative stress.” Immediate response to stress has been widely reported to induce glutamate-driven MAPK kinase pathway activation and consequent transient wave of plasticity-gene transcription including the immediate early genes (IEGs; primary response, Chwang et al., 2006; Gutièrrez-Mecinas et al., 2011; Figure 1). IEGs transcription should be instrumental to the balanced memorization of stressful event aimed at engaging a protective behavioral response against similar threatening situations, a response that however, does not have to excessively invigorate fear and anxiety. This can be achieved thanks to a delayed set of stress response mechanisms (secondary responses) that has to do with: (1) switching off immediate early transcriptional induction; and (2) increasing the threshold of IEGs transcriptional activation in the same circuitry for a limited time window (Figure 1). This second point, albeit observed, has never been clearly formalized on a functional point of view. Here, we will discuss examples of stress-induced secondary responses sharing a common epigenetic nature and predicted to be instrumental to adaptive molecular stress response.
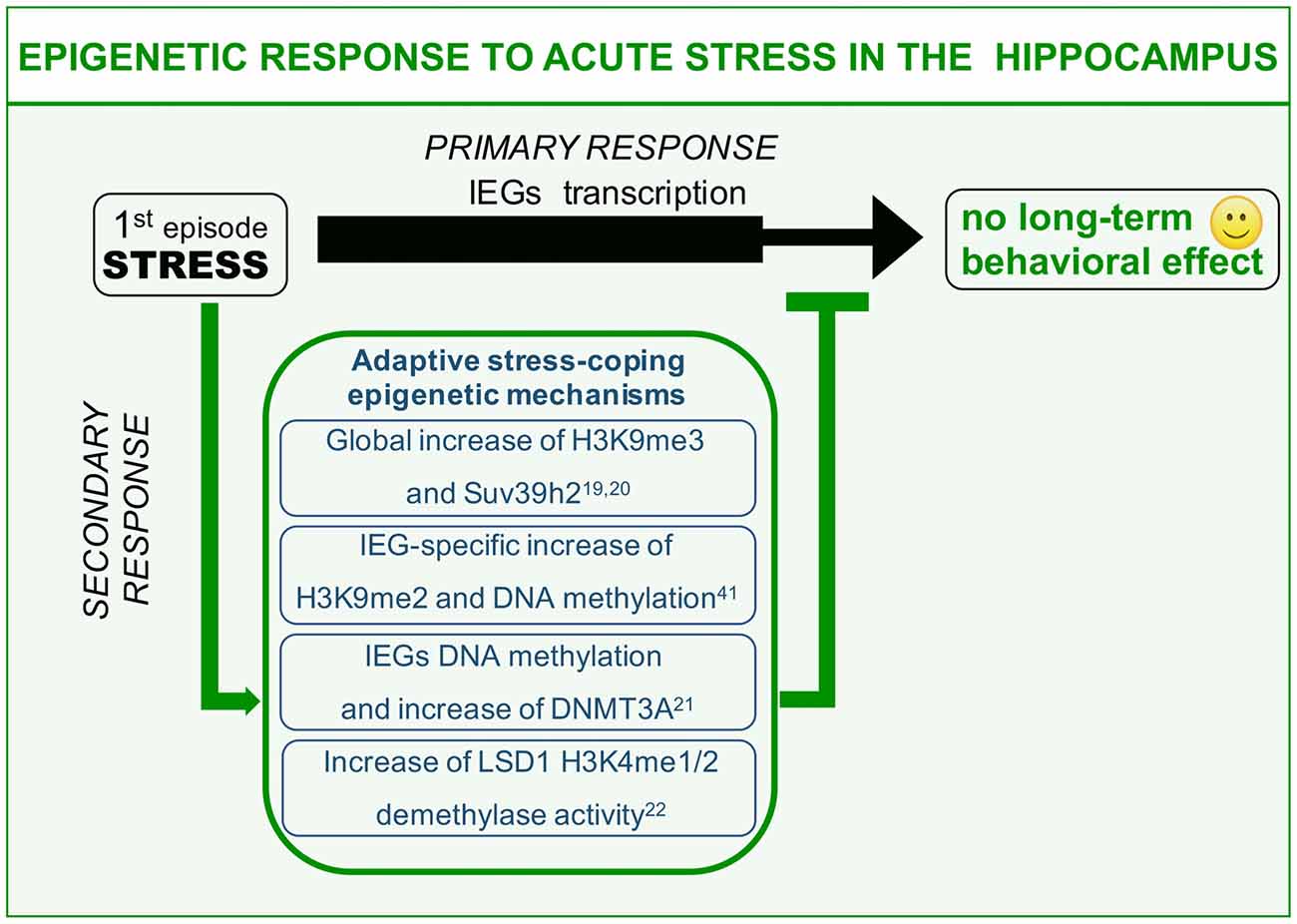
Figure 1. Epigenetic mechanisms of acute stress allostasis. Tolerable stress with a positive behavioral outcome elicits plasticity-gene transcription in the hippocampus instrumental to memorizing threat-related aspects of the negative experience with a protective valence. Meanwhile a set of epigenetic mechanisms buffer stress-induced transcription retaining its functional outcome within adaptive range. These mechanisms include global increase of H3K9me3 repressive histone mark, increased levels of DNA methyltransferase Dnmt3a as well as its association to the Immediate Early Genes (IEGs) promoters and increased Lysine-Specific Demethylase 1 (LSD1)-related repressive potential towards the same gene targets. This set of acute stress-induced epigenetic modifications contribute to counteract long-term behavioral effects on a cognitive or emotional point of view.
It has been shown that acute restraint stress elicits significant global increase of repressive histone mark H3K9me3, together with increased levels of specific methyltransferase Suv39h2 (Hunter et al., 2009, 2012). These modulations represent hippocampus-specific secondary response at the level of CA1 and DG sub area, witnessing interplay between stress and gene expression. It is worth noting that chronic restraint stress administered for 7 days does not entail any modification of H3K9me3 levels in the same areas (Hunter et al., 2009). These data suggest that H3K9me3 global increase after acute stress might represent an allostatic mechanism that can be triggered only by a limited number of stressful events of the same nature. It is conceivable to hypothesize that globally increased gene repression (H3K9me3, heterochromatin formation) is related to adaptive acute stress-coping strategies aimed at pausing gene transcription to buffer acquired neuroplasticity. Moreover, this secondary response may be diminished or degraded by chronic stressful experiences (Hunter et al., 2009). Interestingly fluoxetine—a selective serotonin reuptake inhibitor (SSRI) commonly used as antidepressant—is highly effective in restoring global H3K9me3 increase during chronic stress. Relevantly, fluoxetine has also been reportedly shown to interfere with chronic stress-induced structural and behavioral correlates (Magariños et al., 1999; Czéh et al., 2007; Bessa et al., 2009). Hence, it is plausible that H3K9me3 increase upon SSRI treatment is related to the antidepressant potential of this drug. In other words, one of the effects of fluoxetine is to reinstate an allostatic process in response to chronic stress that is normally restricted to acute stress. Thus, to restore the chronic stress corrupted epigenetic-based secondary response, could represent a strategy to counteract toxic behavioral effects of repeated sessions of environmental stress.
The foot shock paradigm, widely used to assess associative memory in rodents, also represents a highly negative experience scored as able to strongly induce molecular mechanisms of stress response in the hippocampus (Schöner et al., 2017). This paradigm can reproduce some of the core symptoms of PTSD including avoidance and anxiety behavior (Schöner et al., 2017). Interestingly, foot shock paradigm induces in the hippocampus of stressed animals IEGs transcription as primary response, which is supported by increased levels of the euchromatin-associated histone mark H3K4me3 at the level of the IEG egr1 (Gupta et al., 2010). Such an activity-dependent transcription is instrumental to promote stress-plasticity as a form of fear-related memory of the negative experience. Meanwhile, a seemingly concomitant secondary response triggers increased H3K9me2 levels, a chromatin modification related to gene silencing. Moreover, also increased methylation at multiple CpGs at the level of egr1 promoter region (Gupta et al., 2010) can be scored. Interestingly, treating mice with the HDAC inhibitor sodium butyrate before the stressful paradigm, along with significantly decreasing the global level of H3K9me2 methylation, also worsen the phenotypic read-out of foot shock, increasing the freezing behavior (Gupta et al., 2010). A possible interpretation of these evidences can be related to adaptive stress-coping strategies. In particular, concomitant induction of permissive (H3K4me3) and inhibitory (H3K9me2 and DNA methylation) chromatin modifications in the frame of a tight egr1 transcriptional control can allow the perfect expression balance of plasticity genes, leading to a correct and protective memorization of the dangerous context, preventing in the meantime exaggerated behavioral arousals to future homotypic stressful experiences.
We learned from the previous example that not only histone, but also DNA methylation at specific CpGs plays an important role in the behavioral responses to stressful situations. Recently, it has been shown that promoting DNA methylation of c-fos and egr1 promoters via S-adenosyl methionine (SAM) administration, the endogenous methyl-donor for DNA methylation, it is possible to significantly hamper the consolidation of immobility behavior after forced swim (FS) paradigm in rats (Saunderson et al., 2016). Immobility behavior represents a phenotypical read-out of stress response whose interpretation is debated, being reported as either adaptive (Trollope et al., 2012), i.e., to conserve energies to survive, or depressive (Ramaker and Dulawa, 2017), in the sense that the animal refuses to make all possible attempts to exit the water. A first FS training induces IEG transcriptional activation as a primary response to stress, together with a permissive DNA demethylation at specific CpGs in c-fos and egr1 promoters (Saunderson et al., 2016). These pro-transcriptional epigenetic modifications again are devoted to memorization of the stressful experience. Interestingly, in the same time window, the authors also observed an apparently paradoxical increase of DNA Methyl Transferase 3a (Dnmt3a) at both the transcriptional level and in terms of chromatin association to IEGs promoters. As expected, in a second FS test (re-test) animals significantly increase their immobility time, representing this phenotype a modified behavioral readout (cognitive and emotional), which depends on the first stress exposure in the frame of a behavioral sensitization. Interestingly, administration of SAM before the first stressful paradigm, blocks the behavioral outcome of the re-test, leaving immobility time unaltered. This is due to the fact that SAM, favoring the activity of physiologically increased Dnmt3a via increased substrate concentration, counteracts IEG transactivation in response to the first FS test, thus blocking consolidation of stress-induced plasticity, and reflecting on a decreased behavioral arousal in response to the second homotypic stress. In other words, stress-increased Dnmt3a repressive activity towards IEGs should represent another example of adaptive acute stress-coping process based on transcriptional constrain of plasticity genes in the hippocampus.
Another example of epigenetic modification elicited by acute stress involves the chromatin modifier Lysine-Specific Demethylase 1 (LSD1), also known as KDM1A, a transcriptional corepressor responsible for demethylation of histone H3K4me1/2 (Shi et al., 2004). Recently it has been reported that in the hippocampus, in response to a psychosocial stress paradigm, the social defeat stress (SDS), LSD1 repressive potential is increased following a single session of stress. In particular it was unveiled that LSD1 increase is the results of a transient reduction of neuroLSD1, a dominant negative LSD1 isoform unable to repress transcription, via neuro-specific alternative splicing mechanism (Wang et al., 2015; Rusconi et al., 2016). LSD1 and neuroLSD1, in association with the transcription factor SRF, control transcriptional proneness of the IEGs (Toffolo et al., 2014; Wang et al., 2015; Rusconi et al., 2016). Therefore, reported modification of the balance between the two isoforms in response to an acute stress, should negatively impact IEG transcription. Consistently, in a genetic model of neuroLSD1 haploinsufficiency (neuroLSD1HET)—mimicking the above described stress-induced neuroLSD1 decrease and increase of LSD1—stress-evoked transactivation of c-fos and egr1 genes in the hippocampus is impaired (Rusconi et al., 2016). Interestingly, neuroLSD1KO and heterozygous mice are also characterized by a very peculiar phenotype, a low anxiety-like behavior (Rusconi et al., 2016). Given that stress is highly effective in increasing the level of anxiety, the research group proposed that LSD1 mediated occlusion of stress-induced IEG transcription, must represent a secondary epigenetic response with adaptive meaning aimed at buffering excessive consolidation of stress plasticity in terms of anxiety (Rusconi et al., 2016, 2017). Relevantly, low anxiety of neuroLSD1 mutant mice can be restored to wild type levels enhancing IEG expression through administration of class I HDAC inhibitor suberoylanilide hydroxamic acid (SAHA; Rusconi et al., 2016). These results indicate a role for LSD1 in controlling IEGs expression in response to acute stress, participating together with the other epigenetic modifiers to homeostatic control of stress-response.
It is worth mentioning that although many brain areas participate to stress response (as described above) to the best of our knowledge, all the examples of epigenetic modulation following acute stress focused on the hippocampus as the brain area of interest. This does not mean that similar mechanisms cannot be triggered in other areas. However, described data further support an important role for hippocampus in stress adaptation.
The four above described examples can be clustered into a new category of protective stress response mechanisms collectively referred to as epigenetic stress response (ESR).
The cases described above shed new light on the existence of a previously unrecognized layer of stress response mechanisms the ESR. We retrieved and conceptually linked from the literature a set of epigenetic modifications occurring to control stress-induced transcription, representing novel examples of homeostatic processes elicited in rodent models by different environmental stressful events. Interestingly, in three out of four sets of data described, the IEGs were included among those genes that show negative epigenetic transcriptional modulation as a secondary response to environmental stress. Increase of DNMT3a (Saunderson et al., 2016), LSD1 (Rusconi et al., 2016) and Suv39H2 (Hunter et al., 2012), together with global increase of H3K9me2/3 (Hunter et al., 2009; Gupta et al., 2010) in the hippocampus in response to a single stress could represent concerted and functionally converging mechanisms aimed at decreasing IEGs stimulus-induced expression in response to a further homotypic stress. Given IEGs involvement in memory trace formation, reducing their transcriptional responsiveness via modification of promoters’ chromatin structure might associatively influence the emotional response to another similar experience, possibly participating to stress habituation (Figure 2). Epigenetic modifications have the intrinsic feature to last longer than a given stimulus per se, representing initial, chromatin encoded early step of memorization also in case of stressful events (Sweatt, 2016). However, this form of chromatin-based memory has to be transient, likewise all other circuitry modulations that concur to allostatic stress-response.
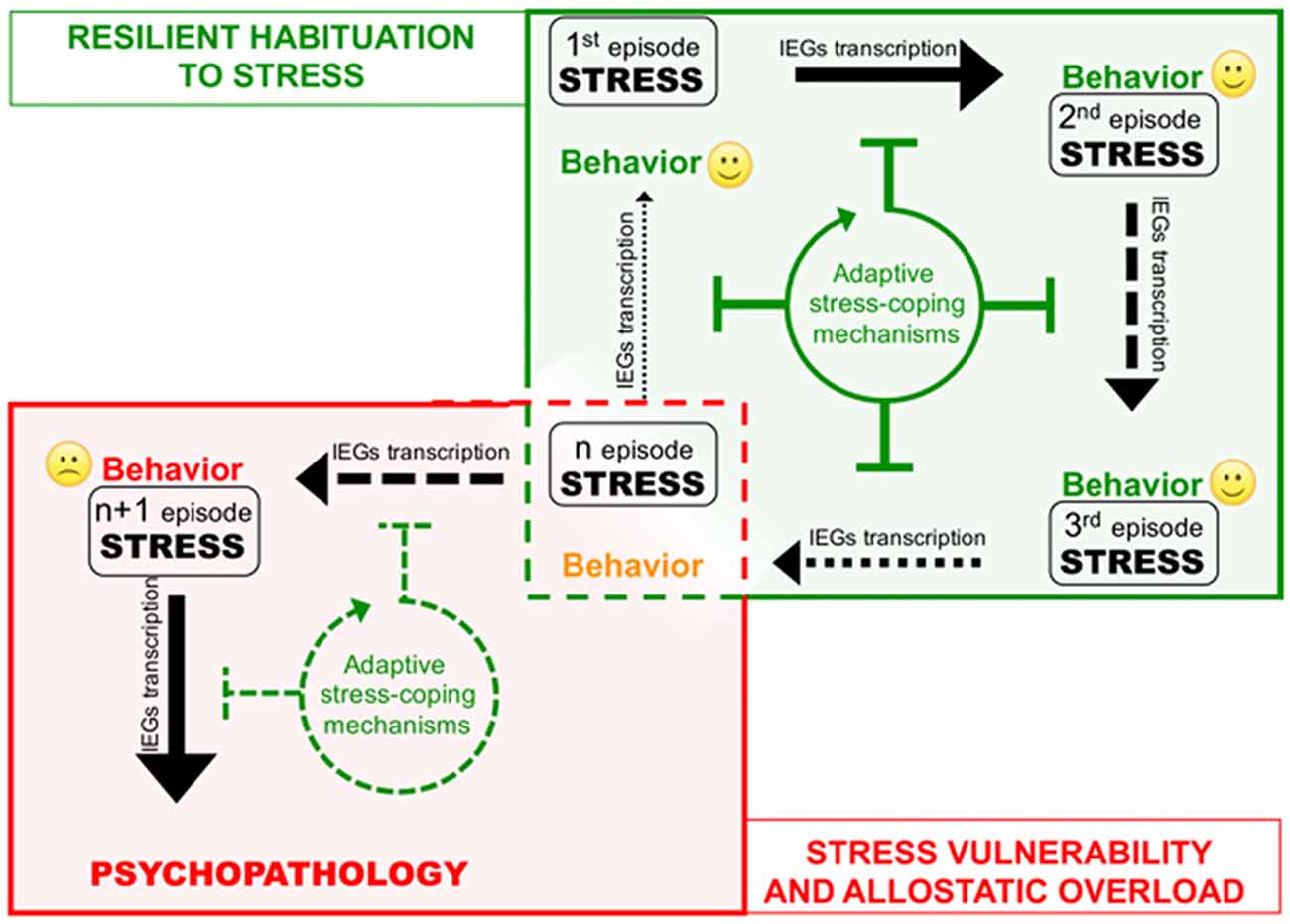
Figure 2. Corruption of epigenetic coping strategies (allostatic epigenetic overload) upon chronic stress reiteration might represent a cue to the onset of stress-related psychopathology. Consequential administration of homotypic stressful events (from stress 1 to n) induces plasticity-related transcription indicated by black arrows of different thickness depending on the transcription rate. Each stress event engages epigenetic allostatic mechanisms aimed at buffering stress-induced transcription and participating to stress habituation in resilient individuals. Stress habituation consists in a progressive reduction of the transcriptional response to stress (green box). However (red box), in vulnerable individuals, after a number of stressful events (n episode), chronic overuse of this protection system can lead to disruption of allostatic epigenetic processes. Loss of adaptive stress-coping mechanisms entails increasing stress-evoked transcription and consequent neuroplastic modifications precipitating psychopathology.
Acute stress is able to induce molecular modifications at the chromatin, transcriptional, structural and circuitry levels (Sun et al., 2013). However, acute stressful insults do not usually lead to long-term issues on a behavioral point of view (except for what concerns PTSD-inducing stimuli; Shvil et al., 2013). Therefore, it should be possible to hypothesize that, when the stress is tolerable, all different stress response-related allostatic mechanisms (HPA axis, ECS and ESR) represent important examples of adaptive molecular processes aimed at a correct interpretation and balanced memorization of stressful experiences (Lupien et al., 2009; Lutz et al., 2015; Rusconi et al., 2017).
On the other hand, excessive exposure to stress such as in case of chronic stress can cause desensitization and/or deterioration of the same allostatic pathways engaged by acute stress contributing to psychopatology (de Kloet et al., 2005). For instance, it is well known how chronic activation of the HPA axis leads to a deregulation of inflammation control in chronically stressed individuals (Cohen et al., 2012). A similar deterioration occurs at the level of the ECS, where CB1 desensitization blocking ECS-mediated buffering of stress-induced glutamate release and fosters the negative behavioral effects of stress (Lutz et al., 2015; Morena et al., 2016). In this Perspective article, we propose that chronic stress fosters the pathogenesis of stress-related depression also via disruption of epigenetic mechanisms of stress response. Consistently, in the first example reported above, chronic stress—contrary to acute—does not elicit protective epigenetic processes such as global increase of H3K9me3 levels in the hippocampus (Hunter et al., 2009, 2012). It would be interesting to understand whether also the acute stress-related molecular mechanisms underlying: (i) increase of LSD1-mediated H3K4me1/2 demethylase activity; and (ii) Dnmt3a upregulation and recruitment to IEG promoters, can be similarly less efficiently engaged upon chronic stress (Figure 2). In particular, we support the hypothesis that loss of epigenetic control over IEGs transcription in the hippocampus along with chronic stress, allowing exaggerated plasticity-related transcription, leads to vulnerability-associated corruption of corticolimbic circuitry. This hypothesis is supported by a further set of data demonstrating that IEGs increase in the vHIP, is related to chronic stress vulnerability (Bagot et al., 2015). Relevantly, in the same stress vulnerable mice, optogenetically reducing neuroplasticity by inducing LTD in the vHIP-NAc circuit, represents an effective treatment against the pro-depressive traits induced by chronic SDS (Bagot et al., 2015).
In conclusion, the novel described mechanisms of ESR that we propose to be protective against depression via tight control towards stress-induced IEGs transcription, together with the notion that in models of stress vulnerability IEGs are stably overexpressed (Bagot et al., 2015), clearly indicates an urgency to further elucidate ESR with the ultimate goal to understand the molecular basis of stress-induced depression, opening new avenues of interventions.
FR and EB conceived the idea and wrote the manuscript.
This work was supported by the Ministero dell’Istruzione, dell’Università e della Ricerca (award no. Epigenomics Flagship Project), Telethon Foundation project (award no. GGP14074) and Fondazione Cariplo (award no. 2016-0204) to EB and (award no. 2014-0972) to FR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Tiziana Rubino and Maurizio Popoli for stimulating discussions and critical revision of the manuscript. We also thank all the lab members for discussing important aspects of the article.
Bagot, R. C., Parise, E. M., Peña, C. J., Zhang, H. X., Maze, I., Chaudhury, D., et al. (2015). Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat. Commun. 6:7062. doi: 10.1038/ncomms8626
Bessa, J. M., Ferreira, D., Melo, I., Marques, F., Cerqueira, J. J., Palha, J. A., et al. (2009). The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol. Psychiatry 14, 764–773, 739. doi: 10.1038/mp.2008.119
Borrelli, E., Nestler, E. J., Allis, C. D., and Sassone-Corsi, P. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. doi: 10.1016/j.neuron.2008.10.012
Calhoon, G. G., and Tye, K. M. (2015). Resolving the neural circuits of anxiety. Nat. Neurosci. 18, 1394–1404. doi: 10.1038/nn.4101
Caspi, A., and Moffitt, T. E. (2006). Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat. Rev. Neurosci. 7, 583–590. doi: 10.1038/nrn1925
Chattarji, S., Tomar, A., Suvrathan, A., Ghosh, S., and Rahman, M. M. (2015). Neighborhood matters: divergent patterns of stress-induced plasticity across the brain. Nat. Neurosci. 18, 1364–1375. doi: 10.1038/nn.4115
Chwang, W. B., O’Riordan, K. J., Levenson, J. M., and Sweatt, J. D. (2006). ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn. Mem. 13, 322–328. doi: 10.1101/lm.152906
Cohen, S., Janicki-Deverts, D., Doyle, W. J., Miller, G. E., Frank, E., Rabin, B. S., et al. (2012). Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc. Natl. Acad. Sci. U S A 109, 5995–5999. doi: 10.1073/pnas.1118355109
Cohen, S., Janicki-Deverts, D., and Miller, G. E. (2007). Psychological stress and disease. JAMA 298, 1685–1687. doi: 10.1001/jama.298.14.1685
Czéh, B., Müller-Keuker, J. I., Rygula, R., Abumaria, N., Hiemke, C., Domenici, E., et al. (2007). Chronic social stress inhibits cell proliferation in the adult medial prefrontal cortex: hemispheric asymmetry and reversal by fluoxetine treatment. Neuropsychopharmacology 32, 1490–1503. doi: 10.1038/sj.npp.1301275
Davis, F. J., Gupta, M., Camoretti-Mercado, B., Schwartz, R. J., and Gupta, M. P. (2003). Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J. Biol. Chem. 278, 20047–20058. doi: 10.1074/jbc.M209998200
de Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
Felix-Ortiz, A. C., Beyeler, A., Seo, C., Leppla, C. A., Wildes, C. P., and Tye, K. M. (2013). BLA to vHPC inputs modulate anxiety-related behaviors. Neuron 79, 658–664. doi: 10.1016/j.neuron.2013.06.016
Franklin, T. B., Saab, B. J., and Mansuy, I. M. (2012). Neural mechanisms of stress resilience and vulnerability. Neuron 75, 747–761. doi: 10.1016/j.neuron.2012.08.016
Golden, S. A., Christoffel, D. J., Heshmati, M., Hodes, G. E., Magida, J., Davis, K., et al. (2013). Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med. 19, 337–344. doi: 10.1038/nm.3090
Golden, S. A., Covington, H. E. III., Berton, O., and Russo, S. J. (2011). A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 6, 1183–1191. doi: 10.1038/nprot.2011.361
Gupta, S., Kim, S. Y., Artis, S., Molfese, D. L., Schumacher, A., Sweatt, J. D., et al. (2010). Histone methylation regulates memory formation. J. Neurosci. 30, 3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010
Gutièrrez-Mecinas, M., Trollope, A. F., Collins, A., Morfett, H., Hesketh, S. A., Kersanté, F., et al. (2011). Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc. Natl. Acad. Sci. U S A 108, 13806–13811. doi: 10.1073/pnas.1104383108
Hunter, R. G., McCarthy, K. J., Milne, T. A., Pfaff, D. W., and McEwen, B. S. (2009). Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc. Natl. Acad. Sci. U S A 106, 20912–20917. doi: 10.1073/pnas.0911143106
Hunter, R. G., Murakami, G., Dewell, S., Seligsohn, M., Baker, M. E., Datson, N. A., et al. (2012). Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc. Natl. Acad. Sci. U S A 109, 17657–17662. doi: 10.1073/pnas.1215810109
Janak, P. H., and Tye, K. M. (2015). From circuits to behaviour in the amygdala. Nature 517, 284–292. doi: 10.1038/nature14188
Joëls, M., and Baram, T. Z. (2009). The neuro-symphony of stress. Nat. Rev. Neurosci. 10, 459–466. doi: 10.1038/nrn2632
Laine, M. A., Sokolowska, E., Dudek, M., Callan, S. A., Hyytiä, P., and Hovatta, I. (2017). Brain activation induced by chronic psychosocial stress in mice. Sci. Rep. 7:15061. doi: 10.1038/s41598-017-15422-5
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445. doi: 10.1038/nrn2639
Lutz, B., Marsicano, G., Maldonado, R., and Hillard, C. J. (2015). The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 16, 705–718. doi: 10.1038/nrn4036
Magariños, A. M., Deslandes, A., and McEwen, B. S. (1999). Effects of antidepressants and benzodiazepine treatments on the dendritic structure of CA3 pyramidal neurons after chronic stress. Eur. J. Pharmacol. 371, 113–122. doi: 10.1016/s0014-2999(99)00163-6
Maras, P. M., Molet, J., Chen, Y., Rice, C., Ji, S. G., Solodkin, A., et al. (2014). Preferential loss of dorsal-hippocampus synapses underlies memory impairments provoked by short, multi-modal stress. Mol. Psychiatry 19:745. doi: 10.1038/mp.2014.64
Martin, E. I., Ressler, K. J., Binder, E., and Nemeroff, C. B. (2009). The neurobiology of anxiety disorders: brain imaging, genetics and psychoneuroendocrinology. Psychiatr. Clin. North Am. 32, 549–575. doi: 10.1016/j.psc.2009.05.004
McEwen, B. S. (2012). Brain on stress: how the social environment gets under the skin. Proc. Natl. Acad. Sci. U S A 109, 17180–17185. doi: 10.1073/pnas.1221399110
McEwen, B. S. (2003). Mood disorders and allostatic load. Biol. Psychiatry 54, 200–207. doi: 10.1016/s0006-3223(03)00177-x
McEwen, B. S. (2005). Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metab. Clin. Exp. 54, 20–23. doi: 10.1016/j.metabol.2005.01.008
McEwen, B. S. (2017). Redefining neuroendocrinology: epigenetics of brain-body communication over the life course. Front. Neuroendocrinol. doi: 10.1016/j.yfrne.2017.11.001 [Epub ahead of print].
McEwen, B. S., Eiland, L., Hunter, R. G., and Miller, M. M. (2012). Stress and anxiety: structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology 62, 3–12. doi: 10.1016/j.neuropharm.2011.07.014
McEwen, B. S., Gray, J., and Nasca, C. (2015). Recognizing resilience: learning from the effects of stress on the brain. Neurobiol. Stress 1, 1–11. doi: 10.1016/j.ynstr.2014.09.001
Morena, M., Patel, S., Bains, J. S., and Hill, M. N. (2016). Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 41, 80–102. doi: 10.1038/npp.2015.166
Nagy, C., Vaillancourt, K., and Turecki, G. (2017). A role for activity-dependent epigenetics in the development and treatment of major depressive disorder. Genes Brain Behav. 17:e12446. doi: 10.1111/gbb.12446
Nestler, E. J. (2014). Epigenetic mechanisms of depression. JAMA Psychiatry 71, 454–456. doi: 10.1001/jamapsychiatry.2013.4291
Ramaker, M. J., and Dulawa, S. C. (2017). Identifying fast-onset antidepressants using rodent models. Mol. Psychiatry 22, 656–665. doi: 10.1038/mp.2017.36
Rusconi, F., Grillo, B., Ponzoni, L., Bassani, S., Toffolo, E., Paganini, L., et al. (2016). LSD1 modulates stress-evoked transcription of immediate early genes and emotional behavior. Proc. Natl. Acad. Sci. U S A 113, 3651–3656. doi: 10.1073/pnas.1511974113
Rusconi, F., Grillo, B., Toffolo, E., Mattevi, A., and Battaglioli, E. (2017). NeuroLSD1: splicing-generated epigenetic enhancer of neuroplasticity. Trends Neurosci. 40, 28–38. doi: 10.1016/j.tins.2016.11.002
Saunderson, E. A., Spiers, H., Mifsud, K. R., Gutierrez-Mecinas, M., Trollope, A. F., Shaikh, A., et al. (2016). Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc. Natl. Acad. Sci. U S A 113, 4830–4835. doi: 10.1073/pnas.1524857113
Schöner, J., Heinz, A., Endres, M., Gertz, K., and Kronenberg, G. (2017). Post-traumatic stress disorder and beyond: an overview of rodent stress models. J. Cell. Mol. Med. 21, 2248–2256. doi: 10.1111/jcmm.13161
Selye, H. (1998). A syndrome produced by diverse nocuous agents. 1936. J. Neuropsychiatry Clin. Neurosci. 10, 230–231. doi: 10.1176/jnp.10.2.230a
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. doi: 10.1016/j.cell.2004.12.012
Shvil, E., Rusch, H. L., Sullivan, G. M., and Neria, Y. (2013). Neural, psychophysiological, and behavioral markers of fear processing in PTSD: a review of the literature. Curr. Psychiatry Rep. 15:358. doi: 10.1007/s11920-013-0358-3
Sun, H., Kennedy, P. J., and Nestler, E. J. (2013). Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38, 124–137. doi: 10.1038/npp.2012.73
Sweatt, J. D. (2016). Neural plasticity and behavior—sixty years of conceptual advances. J. Neurochem. 139, 179–199. doi: 10.1111/jnc.13580
Toffolo, E., Rusconi, F., Paganini, L., Tortorici, M., Pilotto, S., Heise, C., et al. (2014). Phosphorylation of neuronal Lysine-Specific Demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J. Neurochem. 128, 603–616. doi: 10.1111/jnc.12457
Trollope, A. F., Gutièrrez-Mecinas, M., Mifsud, K. R., Collins, A., Saunderson, E. A., and Reul, J. M. (2012). Stress, epigenetic control of gene expression and memory formation. Exp. Neurol. 233, 3–11. doi: 10.1016/j.expneurol.2011.03.022
Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., and Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525. doi: 10.1038/nn1659
Wang, J., Telese, F., Tan, Y., Li, W., Jin, C., He, X., et al. (2015). LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 18, 1256–1264. doi: 10.1038/nn.4069
Keywords: epigenetics mechanisms of plasticity, depressive disorder, stress, psychological, LSD1, hippocampus
Citation: Rusconi F and Battaglioli E (2018) Acute Stress-Induced Epigenetic Modulations and Their Potential Protective Role Toward Depression. Front. Mol. Neurosci. 11:184. doi: 10.3389/fnmol.2018.00184
Received: 06 February 2018; Accepted: 14 May 2018;
Published: 31 May 2018.
Edited by:
Chiara Verpelli, Istituto di Neuroscienze (IN), ItalyReviewed by:
Wladyslaw-Lason, Institute of Pharmacology PAS in Krakow, PolandCopyright © 2018 Rusconi and Battaglioli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Rusconi, ZnJhbmNlc2NvLnJ1c2NvbmlAdW5pbWkuaXQ=
Elena Battaglioli, ZWxlbmEuYmF0dGFnbGlvbGlAdW5pbWkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.