 Rachel Ali Rodriguez
Rachel Ali Rodriguez Christina Joya
Christina Joya Rochelle M. Hines
Rochelle M. Hines
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 24 April 2018
Sec. Neuroplasticity and Development
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00132
This article is part of the Research Topic Synaptic Diseases: From Biology to Potential Therapy View all 32 articles
The term neurodevelopmental disorder (NDD) is an umbrella term used to group together a heterogeneous class of disorders characterized by disruption in cognition, emotion, and behavior, early in the developmental timescale. These disorders are heterogeneous, yet they share common behavioral symptomatology as well as overlapping genetic contributors, including proteins involved in the formation, specialization, and function of synaptic connections. Advances may arise from bridging the current knowledge on synapse related factors indicated from both human studies in NDD populations, and in animal models. Mounting evidence has shown a link to inhibitory synapse formation, specialization, and function among Autism, Angelman, Rett and Dravet syndromes. Inhibitory signaling is diverse, with numerous subtypes of inhibitory interneurons, phasic and tonic modes of inhibition, and the molecular and subcellular diversity of GABAA receptors. We discuss common ribs of inhibitory synapse dysfunction in the umbrella of NDD, highlighting alterations in the developmental switch to inhibitory GABA, dysregulation of neuronal activity patterns by parvalbumin-positive interneurons, and impaired tonic inhibition. Increasing our basic understanding of inhibitory synapses, and their role in NDDs is likely to produce significant therapeutic advances in behavioral symptom alleviation for interrelated NDDs.
Highlights
• Human studies and animal models need to be bridged in neurodevelopmental disorders
• Inhibitory signaling emerges as a common contributor to neurodevelopmental disorders
• Inhibitory signaling is diverse in mode, source, and target
• Systematic evaluation of inhibitory diversity is lacking in neurodevelopment
• Understanding of inhibitory signaling diversity will advance therapeutic strategies
Neurodevelopmental disorders (NDDs) are a broad class of disorders involving disruption in one or more domains including cognitive, emotional, motor, and/or behavioral function. NDDs affect up to 36.8% of children in low- and middle-income earning countries as evaluated in 2015 (Boivin et al., 2015; McCoy et al., 2016). Although NDDs have been the subject of extensive research, the common underlying mechanisms for behavioral and neurobiological symptomatology have yet to be systematically evaluated and compared among broad NDD subtypes. A basis for commonality among this complex and seemingly heterogeneous class of disorders lies in shared symptomatology (Table 1) and common genetic factors (Table 2). Further, genetic studies are beginning to indicate converging biological and phenotypic traits in complex disorders such as NDDs (Parikshak et al., 2016).

TABLE 1. Brief overview of symptom commonalities in NDDs.
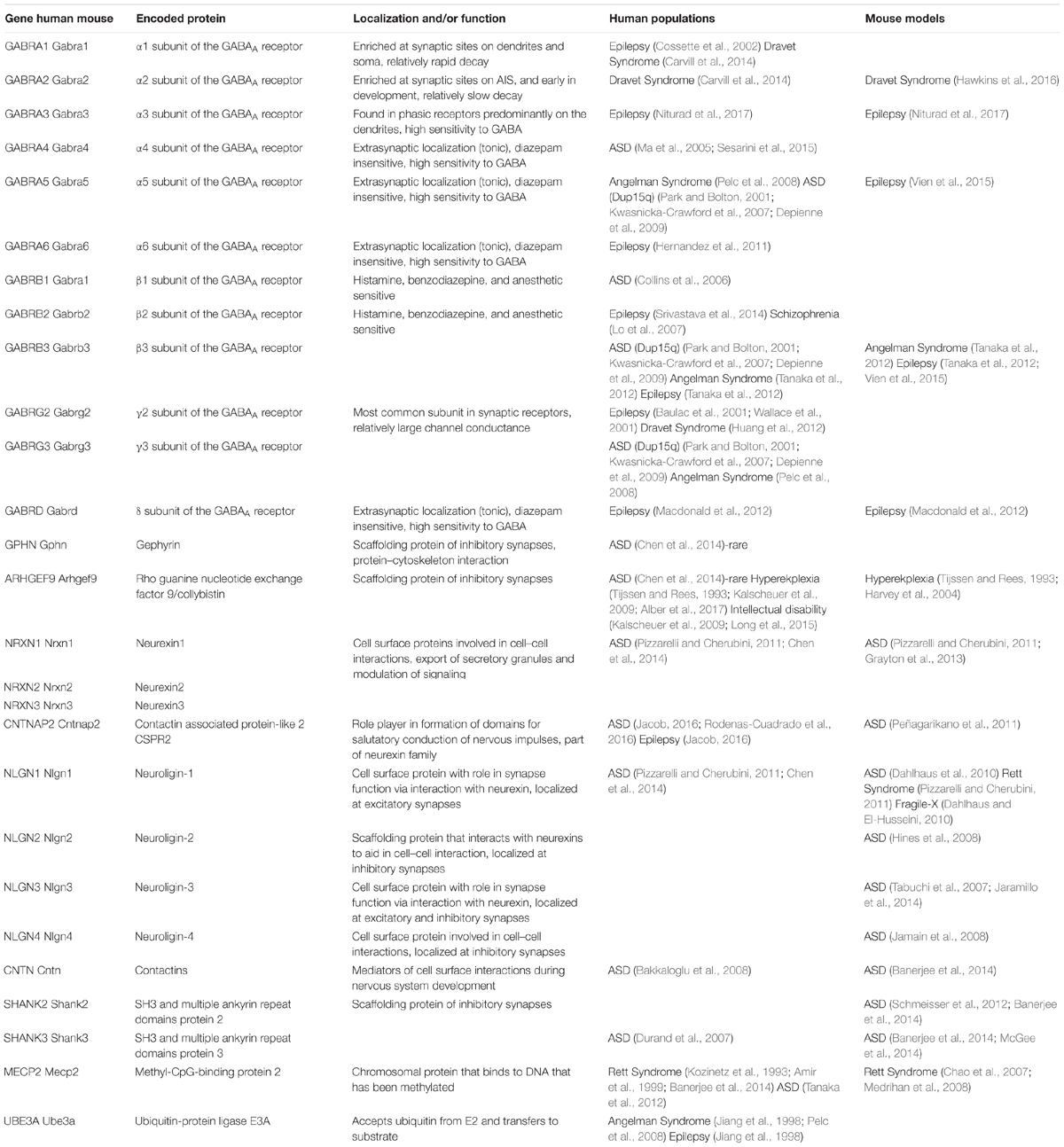
TABLE 2. Summary of genes and proteins linked to synaptic function that are implicated, either directly through mutation or indirectly as secondary effects, in neurodevelopmental and associated disorders.
Autism spectrum disorder (ASD) is a prototypical NDD, and acts as a reference point for this class of disorders, with others often being described as possessing “autistic-like features.” Features of ASD include any range of impairments in social interaction, language development, and cognition, along with stereotyped behavior, and restricted interests (Rapin and Katzman, 1998). In addition to these core features, ASD is also frequently associated with epilepsy, impaired sensory processing, hyperactivity, and disrupted brain activity patterns as characterized by electroencephalogram (EEG) recordings (Rubenstein and Merzenich, 2003). These associated symptoms, in particular, hint that impaired inhibitory signaling is a contributor to ASD.
Other NDDs with similar symptoms to ASD, including Rett Syndrome (RS) and Angelman Syndrome (AS), have a clear genetic cause (Table 2). Specifically, RS is linked to the MECP2 gene (Banerjee et al., 2012), and AS linked to UBE3A and/or GABAAR subunit dysfunction through a loss of activity within the 15q11-13 locus (Malzac et al., 1998). The monogenetic nature of these disorders raises them as attractive targets to begin investigating NDDs. This is in contrast to ASD that is linked to an extremely heterogeneous and inconsistent genetic profile (Hu and Steinberg, 2009; Hu et al., 2009). Strikingly, RS and AS often share features of ASD, including the associated symptoms indicative of impaired inhibitory signaling. RS and AS also have a high incidence of epilepsy, with the vast majority of patients being affected by seizures (Fiumara et al., 2010; Vignoli et al., 2017). Comparing and contrasting the current knowledge on these disorders stands to provide information about the common neuropathological underpinnings of NDDs. In particular, examination of their association with epilepsy and other indications of impaired inhibitory control of principal cell firing, alongside an analysis of inhibitory synapse dysfunction could advance our understanding.
In typical development of the central nervous system, excitatory and inhibitory signals work in concert achieving regulated and orchestrated signaling patterns or oscillations. This tenet is the basis for a theory originally proposed by Rubenstein and Merzenich (2003), which suggests that NDDs are the result of an atypical “balance” between excitation and inhibition. An “imbalance” is proposed to increase the signal-to-noise ratio and decrease the efficiency of processing in afflicted individuals, leaving the cortex susceptible to seizures and other dysfunctional symptoms linked to hyperexcitability (Rubenstein and Merzenich, 2003). Mounting evidence is demonstrating that a number of genetic mutations can disrupt this balance, and in the case of NDDs, can tip the balance toward excitation at the expense of inhibition, contributing to multiple symptoms including a lower seizure threshold and a high incidence of epilepsy. Although the concept of an excitatory-inhibitory balance has been useful in understanding pathological and non-pathological brain states, it is likely an oversimplification and may not be sufficient to explain the immense heterogeneity and complex etiology of NDDs.
To advance from the excitatory-inhibitory balance hypothesis we need to understand synapses more fundamentally in the brain, particularly during development. We need both more detailed understanding of the developmental regulation of specific synapse types and implications for network activity, as well as more global understanding of the functional intersection between molecular and ionic states, synapse types, and network signaling during development. The current state of knowledge on inhibitory synapse development and regulation is limited compared to that of excitatory glutamatergic synapses. Furthering research in the field of inhibitory synapse development is essential to gaining insight into how this system impacts typical and atypical neurodevelopment. This review will examine the commonalities between seemingly heterogeneous NDDs, highlighting common genetic links and evidence of inhibitory synaptic dysfunction, in pursuit of a common rib in the umbrella of NDD.
Neuronal network signaling relies not only on excitation, but also heavily on inhibition to spatially and temporally pattern principal cell activity. Inhibition in the brain is governed by interneurons that synthesize and release the neurotransmitter γ-aminobutyric acid (GABA). A large body of work has documented and characterized 16 subtypes of interneurons in the hippocampus and/or cortex that possess unique morphologies, differential protein expression patterns, and specialized connectivity, physiological function, and functional impact within neuronal networks (Figure 1; Klausberger and Somogyi, 2008; Petilla Interneuron Nomenclature Group et al., 2008). A number of these interneuron types are relevant to the investigation of inhibitory signaling in NDD, due to their enrichment in cortical circuitry, and implication in patterning network activity. The majority of hippocampal and cortical interneurons express either the calcium binding protein Parvalbumin (PV), or the neuropeptide cholecystokinin (CCK) (Klausberger and Somogyi, 2008; Petilla Interneuron Nomenclature Group et al., 2008). PV-positive axo-axonic chandelier cells innervate the axon initial segment (AIS) of principal cells at or near the site of action potential generation, and can coordinate the activity of the up to 1,200 cells that they innervate (Li et al., 1992; Klausberger and Somogyi, 2008; Petilla Interneuron Nomenclature Group et al., 2008). PV-positive and CCK-positive basket cells innervate the soma and proximal dendrites of principal cells, and again play a role in coordinating activity patterns due to proximity to the AIS (Freund and Katona, 2007; Klausberger and Somogyi, 2008; Petilla Interneuron Nomenclature Group et al., 2008). In the cortex, Martinotti cells express the neuropeptide somatostatin (SOM) and innervate the dendrites of principal cells, providing local effects on incoming excitatory input (Rudy et al., 2011). In addition, hippocampal and cortical networks also include interneurons that innervate interneurons, thereby acting in a disinhibitory manner (Figure 1). One example in the cortex is a type of bipolar cell that is positive for the neuropeptide vasoactive intestinal polypeptide (VIP), which have been shown to target the majority of PV-positive interneurons (Dávid et al., 2007).
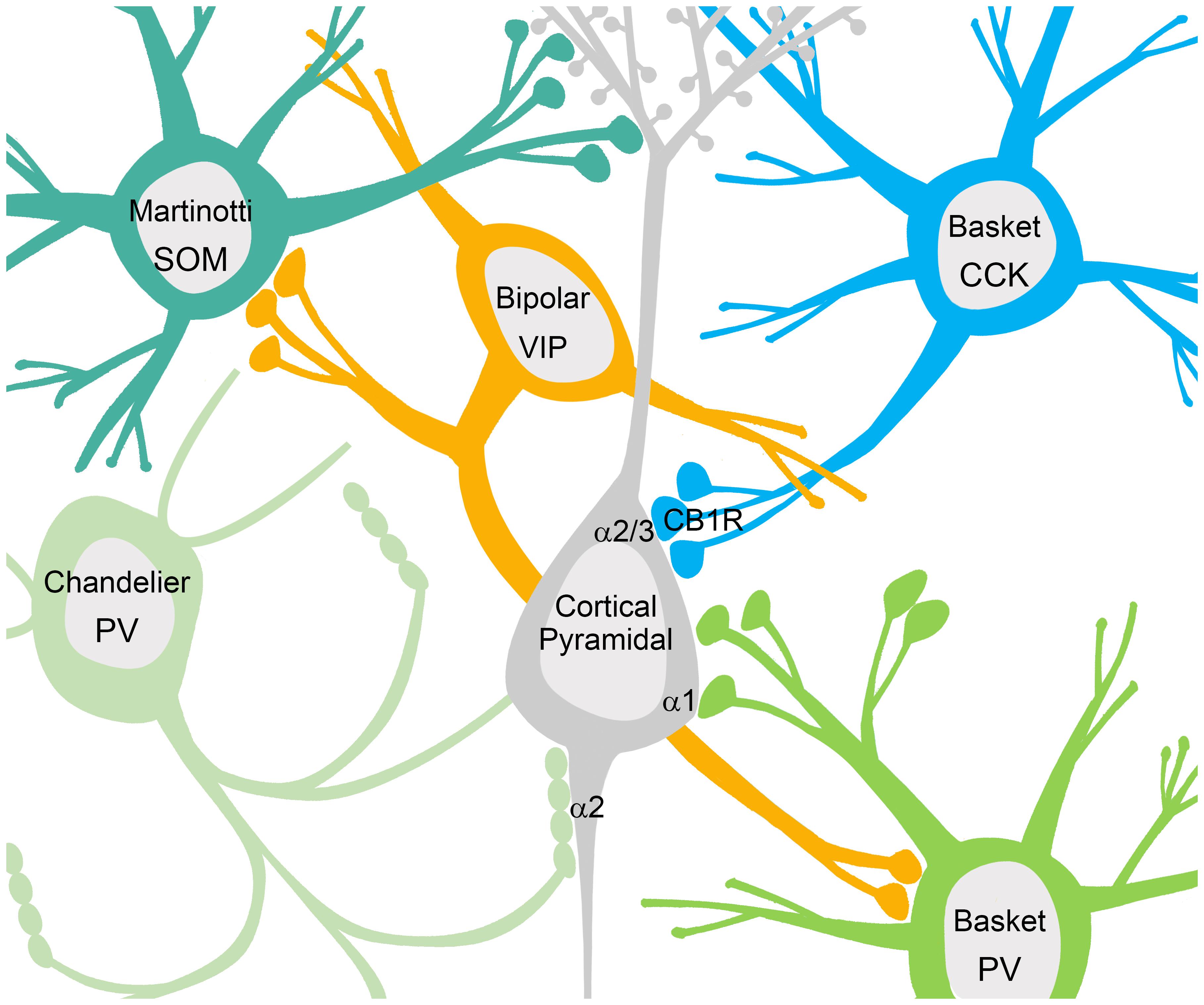
FIGURE 1. Interneurons contact multiple domains of their postsynaptic targets producing different forms of phasic inhibition. Somatostatin (SOM) positive Martinotti cells contact the dendritic domain to provide local inhibitory influences on excitatory inputs. Basket cells positive for CCK contact the soma where GABAARs containing the α2/3 subunits are enriched, while basket cells positive for parvalbumin (PV) contact the cell body at sites enriched with α1 containing GABAARs. PV positive chandelier cells project to the axon initial segment where GABAARs containing the α2 subunit are enriched. Somatic and axonal inhibition plays a role in coordinating neural activity patterns due to the proximity of the incoming graded potentials to the site of action potential generation. Disinhibitory interneurons innervate other interneurons providing net excitation, as seen with vasoactive intestinal polypeptide (VIP) positive bipolar cells.
Once released by interneurons, GABA acts on ionotropic GABA type A receptors (GABAARs; as well as metabotropic GABA type B receptors). GABAARs are heteropentamers gating a chloride channel, composed of combinations of subunits from seven families: α(1-6), β(1-3), γ(1-3), δ, ε, θ, π, and ρ(1-3) (Rudolph and Möhler, 2006). The genes and protein products for the most abundant CNS subunits are briefly described in Table 2 and depicted in Figure 2. Subtypes of GABAARs composed of different subunit combinations have specific localization patterns and diverse functional impacts. In addition, GABAARs can be found at both synaptic and extrasynaptic sites mediating functionally distinct modes of inhibition (Figure 2). The most abundant subtypes of GABAARs are synaptic, sensitive to benzodiazepines, and composed of α1-3, β1-3, and γ2 subunits (Lüscher and Keller, 2004; Rudolph and Möhler, 2006; Jacob et al., 2008). In contrast, extrasynaptic, benzodiazepine insensitive receptors typically contain α4-6, β2/3, and δ subunits (Herd et al., 2013).
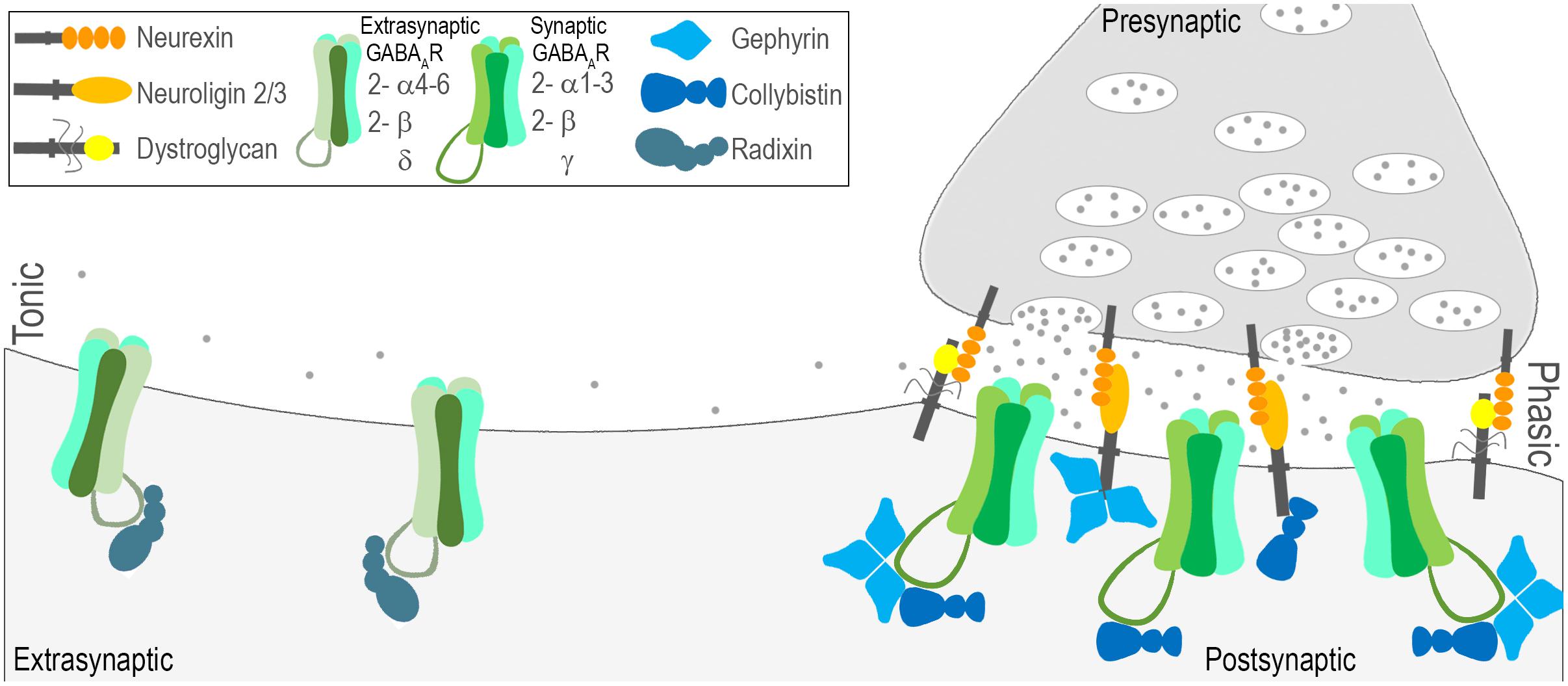
FIGURE 2. Molecular characterization of phasic and tonic modes of inhibition. Fast inhibitory synaptic transmission, also known as phasic inhibition, is governed by GABAAR’s with relatively low sensitivity to GABA densely clustered at sites opposing interneuron terminal boutons. Several general factors have been identified that play a role in establishing the contact between pre and post (neurexin-neuroligin2/3; neurexin-dystroglycan), and/or retaining receptors at these sites (gephyrin, collybistin). Phasic GABAARs are most commonly composed of 2-α1-3, 2-β, and a γ subunit and are sensitive to benzodiazepines. Tonic inhibition is governed by GABAAR’s that reside outside of synaptic sites, are highly sensitive to GABA, and are most commonly composed of 2-α4-6, 2-β, and a δ subunit. Much less is known about the mechanisms that localize extrasynaptic GABAARs, but radixin is implicated.
In addition to modulation by benzodiazepines, synaptic GABAARs are also the site of action for non-benzodiazepine Z-drugs (Dixon et al., 2015), and barbiturates, while extrasynaptic GABAARs are thought to be the site of action of intravenous and inhaled anesthetics (Chiara et al., 2016), anesthetic steroids, neurosteroids and other exogenous modulators such as ethanol (Brickley and Mody, 2012; Sigel and Steinmann, 2012; Olsen, 2015). In addition to clinically relevant ligands, there are also a number of preclinical or experimental ligands such as muscimol (synaptic), and gaboxadol (extrasynaptic), and the channel blocker picrotoxin (Sigel and Steinmann, 2012; Olsen, 2015). The diversity of localization and function, paired with the clear clinical and behavioral impact of pharmacological manipulation, make GABAARs an important target of study.
GABAARs located at synaptic contacts are activated in a transient or ‘phasic’ manner by a high concentration of GABA released from presynaptic vesicles (Figure 2, right). GABA released from the presynaptic neuron bind to GABAARs gating a channel permeable to chloride and bicarbonate ions at the postsynaptic membrane. Generally, this leads to a net inward flow of anions, and a hyperpolarizing postsynaptic response known as the inhibitory postsynaptic potential (IPSP). Defining features of this phasic mode of receptor activation are the rapid synchronous opening of a relatively small number of channels that are clustered at the synaptic junction and the short duration of the GABA transient to which the postsynaptic receptors are exposed. Phasic receptors enriched at synapses are most commonly characterized by two α(1-3), two β, and one γ subunit.
The clustering and accumulation of GABAARs at synaptic sites relies upon the interaction of multiple receptor-associated, adhesion, and scaffolding proteins diagramed in Figure 2, including gephyrin (Tretter et al., 2008, 2011; Mukherjee et al., 2011), collybistin (Saiepour et al., 2010), neuroligin 2 (Chih et al., 2005; Poulopoulos et al., 2009), neurexin (Zhang et al., 2010), and the dystroglycan, dystrophin–glycoprotein complex (Panzanelli et al., 2011; Früh et al., 2016). Despite a common pool of interacting proteins, heterogeneity exists in the complex mechanisms governing clustering of receptors containing different subunits. Composition is likely dependent upon the combination of interacting proteins involved, and their relative interaction affinities.
Inhibitory synapses that generate phasic inhibitory signals can be found on multiple compartments of the cell including the dendrites, axon, and cell body (Figure 1). For example, receptors containing α1 subunits are enriched at inhibitory synapses on the dendrites, those containing α2 subunits are preferentially enriched at inhibitory synapses on the AIS, and both α1 and α2 subunits can be found clustered at somatic inhibitory synapses (Figure 1; Nusser et al., 1996; Freund and Katona, 2007; Gao and Heldt, 2016). AIS synapses are unique to principal neurons in the hippocampus and cortex, and are thought to control principal cell activity patterns by influencing the site of action potential generation (Kuba et al., 2010; Wefelmeyer et al., 2015). As mentioned above, AIS synapses originate from chandelier cells, which are highly specialized, fast-spiking, PV-positive interneurons (Li et al., 1992; Klausberger and Somogyi, 2008; Petilla Interneuron Nomenclature Group et al., 2008). The cortex is also enriched with PV-positive, fast-spiking basket cells, and CCK positive basket cells. CCK positive basket cell terminals contact the soma of pyramidal cells where clusters of GABAARs containing the α2/3 subunits are enriched (Nyíri et al., 2001). The formation and maintenance of CCK terminals on the soma has recently been linked to dystroglycan, a protein responsible for many inherited forms of muscular dystrophy (Figure 1; Früh et al., 2016). Outside of the role for dystroglycan in CCK-basket cell contacts on the soma, detailed information about the mechanisms of other specialized subtypes of inhibitory synapses, such as those localized to the AIS is lacking.
In addition to localization and related impacts on function, subunit contents have also been shown to influence the physiological properties of GABAARs including the decay time of GABA-activated currents within single cells. Presence of a β2 subunit results in a decreased period of time in which the receptor chloride channel is open—leading to faster desensitization. In contrast, the γ2 subunit results in channels with larger conductance (Verdoorn et al., 1990). In addition, α1 subunit containing receptors show faster decay times compared to α2 containing receptors (Lavoie et al., 1997; Eyre et al., 2012).
GABAAR activation can also take place in a less spatially and temporally restricted manner to generate a ‘tonic’ conductance (Nusser et al., 1998; Farrant and Nusser, 2005; Hines et al., 2012; Lee and Maguire, 2014). Low GABA concentrations in the extracellular space escaping from the synaptic cleft can activate GABAARs distant from sites of neurotransmitter release (Figure 2, left; Farrant and Nusser, 2005; Hines et al., 2012). The tonic mode of receptor activation has one straightforward outcome: a persistent increase in the cell’s input conductance. This increase in conductance affects the magnitude and duration of the voltage response to an injected current, and increases the decrement of voltage with distance (Farrant and Nusser, 2005; Bright and Smart, 2013). Tonic GABAARs exhibit a high sensitivity to GABA and show little desensitization, allowing them to detect and respond to ambient levels of GABA at extrasynaptic sites (Nusser and Mody, 2002; Farrant and Nusser, 2005; Hines et al., 2012). Tonic receptors are most commonly characterized by two α(4-6), two β and one δ subunit (Figure 2, left). GABAARs containing δ subunits tend to be purely extrasynaptic (Farrant and Nusser, 2005; Lee and Maguire, 2014). The α4 and α6 subunits are diazepam-insensitive receptors, as their inclusion results in decreased sensitivity to complete desensitization to benzodiazepine binding on the receptor (Whiting et al., 1995). While the complex mechanisms governing localization of GABAARs at synaptic sites have been somewhat clarified, very little is known about the mechanisms that are involved in regulation of extrasynaptic GABAARs. One of the few proteins implicated in regulation of extrasynaptic GABAARs is radixin, which has been shown to influence clustering of α5 containing receptors (Figure 2, left; Loebrich et al., 2006). Further research is needed to understand how these receptors are localized and regulated, and also to understand further how their action may be harnessed to control excitation.
Another factor that can modulate the efficacy of GABAergic inhibition is the reversal potential for GABAA (EGABA) receptor-mediated current and voltage responses. EGABA is controlled by the equilibrium potential for chloride (ECl) which in turn is regulated by the expression and function of cation-chloride cotransporters (CCCs; Rivera et al., 2005; Ben-Ari et al., 2007; Ben-Ari, 2014; Kaila et al., 2014). Two key members of the CCC family for controlling neuronal ECl are the potassium chloride cotransporter (KCC2) and the sodium potassium chloride cotransporter (NKCC1) which provide outwardly and inwardly directed chloride fluxes in neuronal cells, respectively (Ben-Ari et al., 2007; Hartmann and Nothwang, 2014; Doyon et al., 2016). In mature neurons, chloride is extruded by high levels of expression and function of KCC2, using the potassium gradient in order to maintain a low intracellular chloride concentration (Ben-Ari et al., 2007; Doyon et al., 2016). A low intracellular concentration of chloride is important to maintaining the driving force of chloride, such that when GABA binds, chloride will enter the cell through the pore of the GABAAR and hyperpolarize the cell (Ben-Ari, 2014; Kaila et al., 2014). Early in development, KCC2 is low in expression and activity in neurons leading to a high intracellular chloride concentration (Ben-Ari et al., 2007; Ben-Ari, 2014). High levels of chloride inside the neuron cause depolarization in response to GABAAR activation, and under specific conditions, excitatory GABAergic transmission (Rivera et al., 2005; Ben-Ari et al., 2007; Kaila et al., 2014). Excitatory GABA is proposed to play a trophic role in early development, with important effects on the cell cycle, cell migration, and primitive oscillatory patterns thought to influence network wiring (Represa and Ben-Ari, 2005). During development there is a caudal to rostral gradient in the increase of KCC2 expression, with the cortex showing continued increases in KCC2 expression well after birth (Kaila et al., 2014). Beyond the developmental state of high intracellular chloride and depolarizing GABA, it is now established that pathological states including epilepsy and trauma are also characterized by reduced KCC2 expression and high intracellular chloride (Kaila et al., 2014). Further understanding is required about the conditions for inducing change in the expression of KCC2, and also for the implications, that this has on circuit level function.
Understanding the distinctive functional contributions of GABAergic synapses on specific subcellular compartments, phasic and tonic forms of GABAergic signaling, as well as the impact of alterations in ECl will lead to greater ability to fine tune inhibitory signaling. An understanding that is currently lacking is of the neurodevelopmental processes that establish and maintain the structurally and functionally distinctive types of GABAergic synapses, as well as the roles for tonic inhibition throughout development. Further, we are continuing to understand the roles of excitatory GABA early in development. We need to evaluate perturbations in each of these specializations of GABAergic signaling in NDDs with a subunit specific, cell-domain specific level of detail, deciphering precisely which subtypes of GABAARs are affected, and what role the transition from excitatory to inhibitory GABA may play. In the following sections, we will describe the features of specific NDDs and highlight existing studies that have assessed GABAergic signaling in either NDD populations or disease models.
Autism spectrum disorder affects boys more commonly than girls, with a ratio between 3:1 and 4:1, with a prevalence of 1 in 54 boys (Coury et al., 2014; Loomes et al., 2017). The disorder is characterized by core symptomatology that includes compulsive or repetitive behaviors and social communication deficits (American Psychiatric Association, Diagnostic and Statistical Manual 5th edn). These core symptoms are often accompanied by intellectual disability, sensory sensitivity, and seizures. Seizures (and/or epilepsy) occur in ∼30–60% of individuals with ASD (Rubenstein and Merzenich, 2003; Jacob, 2016). Historically, the relationship between ASD and abnormal EEG recordings was the first reason it was considered a brain disorder (Banerjee et al., 2014). This suggests that altered oscillatory activity is a prominent feature of ASD. The heritability of ASD has been studied in family and twin studies and shows a heritability rate of 70% (Tick et al., 2016). Accumulating genomic/genetic investigations indicate hundreds of genetic variants in the occurrence of ASD, including a range from common to rare variants (Krumm et al., 2014). Several of these identified genetic mutations producing autistic-like behaviors have been investigated in mouse models. Despite extensive research in human populations and mouse models, no unifying cause has been determined.
Multiple genetic contributors to ASD are synaptic proteins, and in particular can be classified as trans-synaptic adhesion molecules, or synaptic scaffolding proteins, or synaptic signaling proteins (Table 2). Specific genes implicated within these categories are discussed in detail below: adhesion molecules from the neurexin, neuroligin, and contactin families; synaptic scaffolding proteins from the shank family; and GABAAR subunit genes within the 15q11–13 locus.
The neurexin (NRXN/Nrxn), neuroligin (NLGN/Nlgn), and contactin (CNTN) families are cell adhesion molecules (CAMs; Table 2). In broad terms, CAMs are membrane proteins that extend into the extracellular space and facilitate physical interaction with other cells or with the extracellular matrix. NRXNs are enriched in the presynaptic compartment whereas NLGNs are enriched postsynaptically (Ichtchenko et al., 1995). The two families of proteins interact to form a transsynaptic bridge, linking pre to post, and are thought to be early contributors to synapse formation (Scheiffele et al., 2000; Levinson and El-Husseini, 2005). Within their respective compartments, these proteins also interact with components of the active zone and postsynaptic density, including components linked to neurotransmitter vesicle release, and neurotransmitter receptors and scaffolding proteins (Irie et al., 1997; Prange et al., 2004). The NRXN family includes three genes (NRXN1-3) that can be expressed via one of two promoters, termed α and β. Transcription via the α promoter yields a longer protein with six laminin/neurexin/sex hormone (LNS) binding domains, whereas transcription via the β promoter yields a shorter protein with a single LNS binding domain (Tabuchi and Südhof, 2002). NRXN proteins also contain extracellular epidermal growth factor (EGF) repeats and a PSD-95-Discs Large-zona occludens-1 (PDZ) binding motif. The NRXN family also includes the contactin associated protein (CNTNAP) genes that encode CASPR proteins that are structurally similar with LNS binding domains, EGF repeats, and a PDZ binding motif (Gennarini et al., 2017). The NLGN family includes five genes in humans (NLGN1-3, 4X, 4Y; Bolliger et al., 2001) and four genes in mice (Nlgn1-4; Bolliger et al., 2008). NLGN proteins contain an intracellular PDZ binding motif and an extracellular acetylcholinesterase domain (Levinson and El-Husseini, 2005).
Members of the NLGN family show some synapse specificity, with NLGN1 enriched at excitatory synaptic contacts, and NLGN2 at inhibitory contacts (Song et al., 1999; Varoqueaux et al., 2004; Chubykin et al., 2007). NLGN3 can be localized at either type of contact (Budreck and Scheiffele, 2007), while NLGN4 is enriched at central and peripheral inhibitory contacts (Hoon et al., 2011). This preferential enrichment pattern points to a role in synapse specificity. Indeed, Nrxn and Nlgn protein families have been shown to influence the “balance” of excitation and inhibition in vitro (Prange et al., 2004; Levinson and El-Husseini, 2005), and in animal models (Hines et al., 2008; Dahlhaus et al., 2010; Pizzarelli and Cherubini, 2011). Specific genes within NRXN and NLGN families of CAMs have been implicated in the etiology of ASD, including: NLGN1, NLGN2, NLGN3, NLGN4, NRXN1, NRXN2, CNTNAP2, and CNTNAP4 (Jamain et al., 2003; Banerjee et al., 2014; Baig et al., 2017). Alterations to NRXN and NLGN family members in ASD range from de novo mutations to common genetic variants, and include polymorphisms, variants, frameshift mutations, truncations, and deletions. Although validated in multiple independent studies these mutations are relatively rare (Jamain et al., 2003; Hedges et al., 2012; Banerjee et al., 2014). CNTNAP2 (Arking et al., 2008; Anney et al., 2012) and NRXN1 (Sanders et al., 2011) in particular have been validated through genome-wide association studies.
Several animal models have been developed to examine the contributions of Nrxn to excitatory signaling and the symptoms of ASD (Cattabeni et al., 1999; Kattenstroth et al., 2004; Grayton et al., 2013; Dachtler et al., 2014) but information about inhibitory signaling in these models is limited. Ablation of individual Nlgn genes in mouse models have somewhat subtle effects on development and behavior likely due to compensation, yet combined deletion of Nlgn1-3 results brainstem inhibitory dysfunction leading to respiratory failure (Varoqueaux et al., 2006). Detailed examination of Nlgn1 KO mice revealed an impairment in N-methyl-D-aspartate (NMDA) signaling consistent with enrichment at excitatory synapses, along with repetitive behavior and impaired spatial memory (Blundell et al., 2010). Nlgn3 KO mice show a lack of social novelty preference and reduced vocalization, accompanied by an olfactory deficit (Radyushkin et al., 2009).
Expression of one of the Nlgn3 mutations identified in autism (R451C substitution) was shown to result in impaired social interactions paralleled by an increase in inhibitory synaptic transmission, leaving excitatory synapses unaffected (Tabuchi et al., 2007). However, a follow up study was not able to reproduce the deficits in social interaction in Nlgn3R451C mice (Chadman et al., 2008). In these studies Nlgn3R451C mice were shown to have impairments in ultrasonic vocalizations, and other subtle indications of impaired development (Chadman et al., 2008). In another follow up study the Nlgn3R451C mutation was backcrossed onto a different background strain (129S2/SvPasCrl), which reproduced deficits in social interaction (Jaramillo et al., 2014). The discrepancies among these studies may be attributed to subtle differences in methodology, particularly given that the social interaction tasks in mice are very sensitive to variations in protocol. Nlgn4 loss of function mutation results in deficits in reciprocal social interaction and vocalizations (Jamain et al., 2008).
Increased expression of NLGNs has also been shown to result in several abnormalities relevant to ASD, including behavioral deficits. Increased expression of Nlgns1-4 was shown to result from KO of the eukaryotic translation initiation factor 4E-binding protein 2 (4E-BP2; Gkogkas et al., 2013). 4E-BP2 is a repressor of eukaryotic translation initiation factor 4E (eIF4E), which is downstream in the signaling cascade of mammalian target of rapamycin (mTOR). 4E-BP2 KO mice exhibit an altered ratio of excitatory to inhibitory synapses, along with deficits in social interaction, ultrasonic vocalizations, and the occurrence of repetitive behaviors (Gkogkas et al., 2013). In other studies, transgenic mice with an overexpression of Nlgn1 and Nlgn2 were generated to determine the effect of specific single Nlgns on the excitatory to inhibitory ratio (Hines et al., 2008; Dahlhaus et al., 2010). Mice with enhanced expression of Nlgn1 show deficits in memory acquisition, along with enlarged excitatory synapse morphology and enhanced excitatory signaling (Dahlhaus et al., 2010). Mice with the enhanced expression of Nlgn2 were found to have behavioral symptoms of neurological disorders similar to those seen in RS (limb clasping), decreased social interaction, repetitive behaviors, as well as decreased viability. Immunohistochemical, electron microscopic and electrophysiological analysis of the Nlgn2 overexpressing mice revealed a shift in the excitatory to inhibitory ratio toward inhibition, with an increase in inhibitory synaptic contacts and overall enhancement of inhibitory responses (Hines et al., 2008). Nlgn2 has been shown to directly interact with gephyrin, which is known to aid in localizing and stabilizing GABAARs (Pizzarelli and Cherubini, 2011). Of particular interest, a recent study has shown that the NLGN2 R705C mutation identified in autism disrupts the interaction with gephyrin and thus has consequences for GABAAR function (Nguyen et al., 2016).
Although associated with contactins (discussed below), CNTNAP proteins are members of the NRXN family based on structural similarity, and are also associated with ASD (Katidou et al., 2008; Burbach and van der Zwaag, 2009). CNTNAP genes code for CASPR proteins which act as CAMs. Similar to NRXNs, CASPR proteins contain LNS domains, and EGF-like repeats, but also contain discoidin homology domains, and an extracellular fibrinogen-like domain (Katidou et al., 2008; Burbach and van der Zwaag, 2009). Within the CASPR cytoplasmic tail is a protein 4.1 biding site and class II PDZ binding motif (Katidou et al., 2008; Burbach and van der Zwaag, 2009).
Genetic analysis of CNTNAP2 in ASD has revealed deletions producing a frameshift mutations leading to early introduction of a stop codon (Strauss et al., 2006; Watson et al., 2014). This causes a complete loss of function in the CASPR2 protein in homozygous individuals. Following this implication of CNTNAP2, several studies have shown a link between CNTNAP2 and ASD risk (Alarcón et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Vernes et al., 2008). In human populations, CNTNAP2 heterozygous mutations do not always produce symptoms of ASD, while homozygous individuals for the mutation display ASD symptoms, along with epilepsy, facial dysmorphisms, severe intellectual disability and impaired language (Rodenas-Cuadrado et al., 2016). CNTNAP2 has been shown to cluster voltage gated potassium channels at nodes of Ranvier (Poliak et al., 1999), and influence dendritic arborization, synapse strength, and plasticity (Anderson et al., 2012). In mice, homozygous KO of CNTNAP2 causes increased grooming and digging (repetitive behaviors), impaired ultrasonic vocalization, less time interacting, and epilepsy (Peñagarikano et al., 2011). The behavioral changes in CNTNAP2 KO mice are paralleled by a decrease in PV-positive interneurons, suggesting that CNTNAP2 plays role in GABAergic interneuron development (Peñagarikano et al., 2011).
Both de novo and maternally inherited deletions have been detected in ASD populations in a related family member, CNTNAP4 (O’Roak et al., 2012). CASPR4 protein is enriched in the presynaptic terminals of developing interneurons, and CNTNAP4 KO mice show a reduction in GABAergic signaling from PV-positive interneurons in the cortex (Karayannis et al., 2014). CNTNAP4 KO mice were also found to have enhanced midbrain dopamine release into the nucleus accumbens, paralleled by increased responsiveness to startle and enhanced sensory motor gating (linked to schizophrenia), along with excessive grooming behavior (Karayannis et al., 2014). Treatment of CNTNAP4 KO mice with an α1-selective positive allosteric modulator at GABAARs, indiplon, ameliorated the sensory-motor gating phenotype (Karayannis et al., 2014).
CASPR proteins interact with the contactin (CNTN) family of proteins. CNTN proteins perform diverse functions in the CNS including myelination, synapse formation, and plasticity. This group of proteins, like the NRXNs and NLGNs, is also adhered to the cell membrane (Zuko et al., 2013). Duplication or deletion mutations in CNTN4 near CNTN3 have been implicated in the development of ASD, with more rare mutations in CNTN5 and 6 also being implicated (Banerjee et al., 2014). CNTN4 plays a role in the growth and development of axons and maintenance of mature networks. In contrast to CNTNAP mutations, loss of a single functional copy of CNTN4 can cause developmental delays (Roohi et al., 2009).
Mutations in genes encoding scaffolding proteins, such as the SHANK family of proteins have also shown connection to the development of ASD (Table 2; Leblond et al., 2012; Sato et al., 2012; Monteiro and Feng, 2017). Scaffolding proteins function in clustering and anchoring synaptic proteins including neurotransmitter receptors, and serve to link post synaptic receptors with downstream signaling components. The SHANK family includes three genes (SHANK1-3), that can be alternatively spliced to generate multiple Shank protein variants (Monteiro and Feng, 2017). Full-length Shank proteins are composed of 5–6 N-terminal ankyrin repeat domains, a Src Homology 3 (SH3) domain, a PDZ domain, a proline-rich region and a C-terminal sterile alpha motif (SAM) domain (Monteiro and Feng, 2017). Based on the interactions of these domains, Shank can bind an estimated 30 synaptic partners, including other scaffold molecules, glutamatergic receptors, and cytoskeletal proteins (Ehlers, 1999; Sheng and Kim, 2000; Baron et al., 2006; Gundelfinger et al., 2006). Shank proteins are expressed in the postsynaptic compartment of glutamatergic synapses. As central regulators of receptors, signaling machinery, and the cytoskeleton at excitatory synapses, Shank proteins have been shown to be involved in spine morphogenesis, synapse formation, and glutamate receptor trafficking (Naisbitt et al., 1999; Sala et al., 2001, 2005; Gerrow et al., 2006; Verpelli et al., 2011).
Mutations in all three SHANK family members have been connected with ASD and/or associated disorders (Ting et al., 2012; Leblond et al., 2014). SHANK1 deletions have been detected in ASD males (Sato et al., 2012), while multiple studies have detected de novo mutations and deletions in SHANK2 (Berkel et al., 2010; Pinto et al., 2010; Leblond et al., 2012). SHANK3 is even more strongly implicated in ASD and related disorders. SHANK3 haploinsufficiency is responsible for Phelan McDermid syndrome, which is characterized by intellectual disability, hypotonia, epilepsy, and ASD characteristics (Phelan and McDermid, 2012). In varied ASD populations, de novo mutations, truncations and terminal deletions have been detected in SHANK3 (Durand et al., 2007; Boccuto et al., 2013; Leblond et al., 2014). SHANK3 has also been identified in genome wide association studies of ASD (Connolly et al., 2017).
Modeling of these mutations in mice has revealed that many of the mutant proteins do not achieve their synaptic localization, and are retained in the cell body or nucleus (Grabrucker et al., 2014), and that a variety of SHANK manipulations in vivo can produce ASD relevant phenotypes (Sala et al., 2015). SHANK2 mutations in mice result in upregulation of glutamate receptors and a decrease in synaptic transmission (Schmeisser et al., 2012). Behaviorally, SHANK2 mutant mice are hyperactive, exhibit repetitive grooming, and have impairments in social interactions and vocalizations (Schmeisser et al., 2012). SHANK3 KO leads to reduction in spine volume, a decrease in the thickness of the postsynaptic density, and a loss of dendritic spines (McGee et al., 2014). SHANK3 KO mice show abnormal social behaviors, and impairments in learning and memory (Bozdagi et al., 2010). SHANK3 mutations identified in patients with ASD show a modification in dendritic spine induction and morphology as well as actin accumulation in spines affecting growth cone motility which is nicely paralleled by the mouse model (Durand et al., 2007). SHANK3 duplication in mice leads to hyperactivity and spontaneous seizures much like human subjects who have small duplications in the SHANK3 locus (Han et al., 2013). KO mice for SHANK3 showed abnormal communication patterns, repetitive behaviors, and impaired learning (Banerjee et al., 2014; McGee et al., 2014). Again, these behaviors are consistent with the phenotype of ASD in humans.
SHANK3 interacts with NLGN genes as a binding partner to play role in synaptic plasticity (Meyer et al., 2004), and SHANK3 is also involved in synapse formation and this results in an increase of excitatory signals (Arons et al., 2012). Although the link of Shank proteins to excitatory synapse function is clear, studies have not thoroughly investigated the impact that SHANK mutations may have on interneuron or inhibitory synapse function, downstream of excitation. Further studies on the existing SHANK mouse models generated may be helpful in examining the relationship between excitatory synapse dysfunction and downstream impacts on inhibitory signaling.
A known unstable genetic locus, termed 15q11-q13, is located on chromosome 15 and contains UBE3A (Ubiquitin-protein ligase E3A), and three GABAAR subunit genes (GABRB3, GABRA5, and GABRG3; Table 2). The 15q11-q13 region is subject to imprinting and recombination, and is regarded as one of the most complex regions in the genome (Martin et al., 2000). The partial trisomy of chromosome 15 was first reported as a syndrome in 1975 (Hecht, 1975). The syndrome produced by duplication of 15q11-q13, termed Dup15q, includes global developmental delay, social communication deficits, intellectual disability, with a high incidence of epilepsy and/or infantile spasms (Urraca et al., 2013). Children with Dup15q generally meet the diagnostic criteria for ASD diagnosis (Urraca et al., 2013), and several studies have now shown that duplications in 15q11-q13 are one of the most common copy number variants linked with ASD, which have been replicated in genome wide association studies (Cook et al., 1997; Park and Bolton, 2001; Kwasnicka-Crawford et al., 2007; Depienne et al., 2009; Sanders et al., 2011). Duplications in this region can take one of two forms, two extra copies of the 15q11.2-q13.1 region of maternal origin on a supernumerary chromosome (isodicentric), or one or more extra copies on the q arm of chromosome 15 (DiStefano et al., 2016). 15q11-q13 loss of activity is discussed specifically below in the “Angelman Syndrome” section.
Duplication specifically of the region containing GABAARs leads to a prediction of excessive inhibitory neurotransmission due to gene dosage; however, an in vitro model of 15q duplication displayed reduced transcripts for GABRB3 (and other nearby genes) due to impaired homologous pairing (Meguro-Horike et al., 2011). The symptoms of individuals with Dup15q also are suggestive of altered GABAergic control of oscillatory activity. Multiple reports have demonstrated enhanced activity in β frequency bands (12–30 Hz) of the EEG, with spontaneous β oscillations visually evident (Urraca et al., 2013). A further study demonstrated that enhanced power in the upper β frequencies (20–30 Hz) was significantly related to epilepsy diagnosis in Dup15q (Frohlich et al., 2016). The enhanced power and spontaneous oscillations noted in Dup15q are similar to the effects seen following treatment with benzodiazepines and other allosteric modulators of GABAARs, although none of the patients were on such medication (Urraca et al., 2013; Al Ageeli et al., 2014; Frohlich et al., 2016). Further, since the EEG β phenotype is observed with both maternal and paternal duplication cases, it is likely that this phenotype results from altered GABAAR function and not the maternally expressed (non-imprinted) Ube3a gene (Frohlich et al., 2016).
Examination of the seizure phenotype in patients with Dup15q have indicated about 63% of those with isodicentric mutations have seizures, with the majority of these cases having multiple seizure types (81%), and with a relatively high rate of infantile spasms (42%; Conant et al., 2014). In contrast, only about 25% of interstitial mutation type Dup15q patients had seizures (Conant et al., 2014). This study also revealed that broad spectrum anti-epileptic drugs, and those that block voltage gated sodium channels (carbamazepine and oxcarbazepine) were typically effective, whereas typical benzodiazepines were relatively ineffective (Conant et al., 2014). Also indicative of abnormal GABAergic signaling, paradoxical increases in seizure severity have been reported in isodicentric Dup15q following treatment with pregabalin (Di Rocco et al., 2013), a drug that acts to increase extracellular GABA by increasing the density of GABA transporter proteins, increasing the rate of functional GABA transport, and increasing the enzyme responsible for making GABA (L-glutamic acid decarboxylase) as a result of high-affinity binding to the α2-δ subunit of voltage-gated calcium channels (Sze, 1979; Marks et al., 2009). Mouse models mimicking human 15q11-q13 duplication exhibit features of autism, such as poor social interaction, behavioral inflexibility, and abnormal ultrasonic vocalizations (Nakatani et al., 2009). Further, a mouse model with a point mutation altering the surface stability of the GABAAR β3 subunit (GABRB3 gene product) displays ASD-like features and enhanced susceptibility to seizure (Vien et al., 2015).
Indirectly reacted to the findings of loss of function of GABAAR in Dup15q, postmortem studies from subjects with ASD with non-specified genetic background have shown reduced levels of [3H]-muscimol binding compared to control (Blatt et al., 2001). Muscimol has a higher affinity for α4/δ-containing receptors than other GABAAR (Huh et al., 1996). Recently, expression and protein levels of subunits where found to be significantly reduced in different regions of brains from autistic subjects compared to age-matched controls. Protein expression of α1, α2, α3, α5, and β3 subunits were reduced in the parietal cortex whereas in the frontal cortex α1, α4, α5, and β1 subunits were reduced (Fatemi et al., 2009, 2010, 2013). These data would suggest a GABAergic hypofunction and in particular, a reduced tonic inhibition of patients with autism.
Other rare mutations implicated in ASD are directly or indirectly linked to GABAergic signaling, including, mutations in SLC12A5 impacting the C-terminal regulatory domain of KCC2 (Merner et al., 2015), RIM1A (Iossifov et al., 2014) which has been shown to modulate GABA release from interneurons (Lachamp et al., 2009), DLX homeobox transcription factors (International Molecular Genetic Study of Autism Consortium [IMGSAC], 2001; Liu et al., 2009) which play a role in interneuron differentiation and induction of GABA synthesis (Pleasure et al., 2000; Cobos et al., 2005; Paina et al., 2011), and ANK2 (Ankyrin B; De Rubeis et al., 2014; Iossifov et al., 2014) which creates the AIS boundary, and is involved in modifying AIS length (Galiano et al., 2012). Collectively, these genetic and animal model studies suggest that dysfunction in the structure and or function of transmembrane adhesion, scaffolding, and receptor proteins critical for creating and stabilizing synapses are major contributors to ASD. In particular, evidence of altered inhibitory synapse adhesion molecules, scaffolding proteins, and GABAAR signaling is clearly implicated in ASD. Alterations in PV-positive neuron number and/or function, and alterations in tonic inhibition are common among multiple genetic causes of ASD, warranting further study. Below we will explore monogenetic NDDs with symptoms related to ASD, to examine the indications of inhibitory synapse dysfunction more broadly.
Angelman syndrome (AS) is a NDD characterized by severe mental retardation, ataxic movements, speech impairment, and high incidence of seizures (Buiting et al., 2016). In contrast to other NDDs, people with AS display a sociable disposition and are prone to unprovoked bouts of laughter. AS is relatively rare, with an incidence of somewhere between 1:12,000 and 1:20,000, accounting for about 6% of all children with mental retardation and epilepsy (Pelc et al., 2008; Buiting et al., 2016). AS is caused by a loss of activity within the 15q11-13 locus, and about 70% of cases are due to a “de novo” interstitial deletion in the long arm region, arising on the maternally inherited chromosome (Buiting et al., 2016). The candidate gene in this region thought to be responsible for AS is UBE3A (Table 2; Kishino et al., 1997; Matsuura et al., 1997; Sutcliffe et al., 1997), although three GABAAR subunit genes are also located in this region as mentioned above. AS is a classic example of imprinting and is most commonly caused by deletion or inactivation of genes on the maternally inherited chromosome 15 while the paternal copy is imprinted or silenced in the brain producing the most severe cases, with seizures, mental and motor retardation, dysmorphic features and microcephaly phenotype (65–70% of probands; Kyllerman, 2013). The other two possible causes, uniparental disomy and imprinting center mutations (10%), and UBE3A point mutations (11%) result in more mild phenotypes (Kyllerman, 2013). The AS diagnosis is confirmed by methylation test or by mutation analysis of the UBE3A gene. Regardless of mutation type, seizures occur between ages 1 and 3 years in approximately 85% of patients and are most commonly characterized by atypical absence, erratic myoclonic, and rare tonic–clonic seizure types (Fiumara et al., 2010). Detailed EEG studies of AS patients have revealed high-amplitude δ frequency (2–3 Hz) activity, with spike and slow-wave discharges, and a circadian pattern to epileptiform discharges, showing sleep-activation (Kyllerman, 2013).
As discussed above, the GABAAR genes, GABRB3, GABRA5, and GABRG3 genes (Table 2), encoding β3, α5, and γ3 subunits, respectively, are also within the 15q11-13 locus. The presence of this GABAAR subunit gene cluster in 15q11-13 has led to the hypothesis that GABA neurotransmission may also be involved in AS (Pelc et al., 2008), as well as ASD. In particular, the GABRB3 gene is known to be associated with epilepsy in humans, and when a deficiency of UBE3A is also present, more severe symptoms result (Pelc et al., 2008).
The phenotype of maternal Ube3a-deficient mice resembles human AS with motor dysfunction, inducible seizures and a context-dependent learning deficit (Jiang et al., 1998). Using mouse models it has been demonstrated that Ube3a-deficiency results in reduced GABA-mediated tonic inhibition in the cerebellum, mechanistically caused by a GAT-1 dependent decrease of GABA concentration in the extracellular space (Egawa et al., 2012). A recent paper elegantly examined the contributions of Ube3a loss in specific populations of cells to the pathophysiology of AS (Judson et al., 2016). Using conditional deletion in mouse models it was shown that loss of Ube3a in GABAergic interneurons, but not glutamatergic principal cells, results in EEG abnormalities of enhanced δ, and seizures, similar to individuals with AS. Other studies have examined the combined Ube3a and GABRB3 effects using chromosomal deletion spanning Ube3a to GABRB3 (includes a deletion of the Atp10a gene; Jiang et al., 2010). Maternal (Ube3am-/p++), but not paternal (Ube3am+/p-), deletion mice had increased spontaneous seizure activity and abnormal EEG, along with impairments in motor function, learning and memory, ultrasonic vocalizations, and an anxiety-like phenotype demonstrating the impact of GABAAR function on the severity of phenotype (Jiang et al., 2010). The Ube3am-/p+ model has also been shown to have an inhibitory deficit resulting from impaired vesicle cycling in interneurons (Wallace et al., 2012). In addition to Ube3a, examination of GABAAR β3 subunit-deficient mouse models have also revealed a strong link between GABAARs and AS, β3 deficient mice show 90–95% neonatal mortality, with survivors displaying a phenotype resembling severe forms of human AS (Homanics et al., 1997; DeLorey et al., 1998).
The maternal deletion model of AS (Ube3am-/p+) has also been shown to result in alterations to the intrinsic properties of neurons (Kaphzan et al., 2011). In these studies researchers demonstrated alterations in resting membrane potential, threshold potential, and action potential amplitude of principal cells. The changes in intrinsic properties were paralleled by significant increases in the expression of the voltage gated sodium channel NaV1.6, and the AIS anchoring protein ankyrin-G, as well as an increase in length of the AIS (Kaphzan et al., 2011). Previous studies have shown that the AIS can adapt to the inputs that a cell is receiving, with reduced synaptic input resulting in elongation of the AIS (Kuba et al., 2010), and increased excitability moving the AIS away from the cell body (Grubb and Burrone, 2010). The AIS is also of interest in considering GABAergic signaling, as it is the postsynaptic site of PV-positive chandelier cell contact, and changes in the properties of the AIS also likely effect GABAergic control at this important site.
As with ASD, AS is also clearly linked to dysfunction of inhibitory signaling, and in particular to dysfunction of GABAAR subunit expression and function. Again, dysfunction of PV-positive neurons emerges as a feature of AS. It is interesting that despite a common biological phenotype of reduced transcripts for GABAAR subunit genes in Dup15q ASD and AS, the EEG phenotype differs between these two syndromes, suggesting the need for regional, and more specific analysis of GABAAR subunit expression and function, as well as GABAergic synapse subtypes in these disorders.
Rett syndrome (RS), is a genetic disorder that leads to severe impairments, affecting nearly every aspect of a child’s life. ASD-like features, especially stereotyped behaviors, loss of language skills, learning and memory deficits, loss of social skills, and seizures are common characteristics of RS (Chahrour and Zoghbi, 2007). RS may also include apraxia, ataxia, impaired coordination, tremor, respiratory dysrhythmias, and sometimes premature lethality (Chahrour and Zoghbi, 2007; Chao et al., 2007). Not all of the symptoms of RS are present during early postnatal development, but rather appear over stages, following a period of developmental stagnation that typically begins between 6 and 18 months of age (Chahrour and Zoghbi, 2007). The primary cause of RS is mutation of the gene encoding the transcriptional repressor methyl-CpG-binding protein 2 (MeCP2; Table 2; Kozinetz et al., 1993). The MECP2 gene is located on the X-chromosome, and since males have only one X chromosome (instead of the two in genetic females), MECP2 mutation results in more severe symptoms, with the majority of RS males dying shortly after birth (Meloni et al., 2000). In females, RS represents one of the leading causes for mental retardation (Neul and Zoghbi, 2004). Within females, the severity of RS symptoms can vary widely depending upon the pattern of x-chromosome inactivation (Dragich et al., 2000). It is estimated that approximately 60–80% of females with RS have epilepsy (Vignoli et al., 2017), with severity of the syndrome positively correlated with the prevalence of epilepsy (Tarquinio et al., 2017). Longitudinal studies have indicated an even higher cumulative lifetime prevalence of epilepsy in RS, with approximately 90% incidence evaluated using a Kaplan–Meier method (Tarquinio et al., 2017). A fascinating link between ASD, AS, and RS was identified when researchers found that MECP2 (along with other factors) is required for the homologous pairing of 15q11-q13 during neuronal differentiation during the maturation process (Meguro-Horike et al., 2011).
Researchers have developed multiple animal models to study the underlying neuropathology associated with RS. Mecp2 deficient mice, created by deleting either exon 3, or exons 3 and 4, replicate all the features of the human disease (Chen et al., 2001). Truncation mutations (Mecp2308) produce a more moderate phenotype that shows the progressive pattern of symptom development (Shahbazian et al., 2002). It is also interesting to note that in mouse models (Collins et al., 2004), and in human NDDs (del Gaudio et al., 2006), increased expression of Mecp2 also produces a progressive neurological phenotype, indicating that precisely controlled levels of Mecp2 are essential for normal nervous system function.
Conditional, neuron specific deletion of Mecp2 in mice is sufficient to produce symptoms of RS (Guy et al., 2001), and the neurologic abnormalities can be reversed by activating the Mecp2 gene later in life (Amir et al., 1999; Luikenhuis et al., 2004). Surprisingly, despite the diversity of the features seen in RS, recent findings also indicate that deleting Mecp2 only from GABAergic inhibitory neurons in mice is sufficient to replicate most of the phenotypes of the human disease (Medrihan et al., 2008). In this elegantly performed study, researchers created a Mecp2 KO model by crossing Viaat-Cre to Mecp2flox/+ mice. The result from this study was a loss of Mecp2 in male Viaat-Mecp2-/y mice from more than 90% GABAergic neurons (Chao et al., 2010). Mice with Mecp2 deficiency from GABA-releasing neurons recapitulate numerous RS and ASD features, including repetitive behaviors, ataxia, premature death, breathing abnormalities, and seizures. These mice initially exhibit normal behavior, then develop forepaw stereotyped movements, compulsive grooming, increased sociability, impaired motor coordination, learning/memory deficits, abnormal EEG hyperexcitability, severe respiratory dysrhythmias, and premature lethality (Chao et al., 2010). Specific interneuron subtypes have been shown to play distinctive roles in RS, determined by selectively deleting Mecp2 from either PV or SOM positive interneurons (Ito-Ishida et al., 2015). Deletion of MeCP2 in PV+ interneurons resulted in motor, sensory, cognitive, and social interaction deficits, whereas deletion of MeCP2 in SOM+ interneurons resulted in seizures and stereotypies (Ito-Ishida et al., 2015). Another recent study was able to genetically re-express Mecp2 solely in GABAergic neurons in a Mecp2 KO mouse, restoring inhibitory function. GABA concentrations in the striata of 8-week-old rescue mice were restored to wildtype levels. No behavioral seizures were seen in the rescue mice, while EEG recordings showed that five out of six rescue mice had normalized activity patterns (Lu et al., 2016). Another recent study demonstrated that KCC2 is a downstream target of Mecp2, and that human neurons derived from stem cells of RS patients have reduced KCC2 expression and a delayed switch in EGABA (Tang et al., 2016).
Studies examining the neuropathology of RS have identified strong depression of GABAergic neurotransmission in the ventrolateral medulla (Medrihan et al., 2008), and nucleus tractus solitarius (Chen et al., 2017) of Mecp2 null mice. Studies in the ventrolateral medulla indicated reduced GABA release paralleled by reduced levels of the vesicular inhibitory transmitter transporter and reduced levels of α2 (typically synaptic) and α4 (typically extrasynaptic) subunits of GABAARs (Medrihan et al., 2008). In the nucleus tractus solitarius, it has been shown that GABAAR δ subunit (typically extrasynaptic) expression is enhanced, paralleled by enhanced responsiveness to THIP, which selectively activates extrasynaptic GABAAR subtypes (Chen et al., 2017). Other studies have indicated reductions in the frequency of spontaneous inhibitory postsynaptic currents in hippocampal recordings from symptomatic Mecp2 null mice (Zhang et al., 2008). In addition, these studies found that Mecp2 null hippocampal circuitry was prone to hyperexcitability, as evidenced by induction of repetitive sharp wave-like discharges following a brief train of high-frequency stimulation (Zhang et al., 2008).
Similar to ASD and AS, several lines of evidence also link RS to dysfunction of inhibitory signaling. In particular, support for the role of PV-positive interneuron dysfunction in broad NDDs is apparent. In addition, indications of KCC2 dysfunction are present in both ASD and RS. Evidence is accumulating for dysfunction of specific GABAAR subunit expression and function, with regional specificity as well, this level of analysis is currently lacking in ASD and AS models. Studies in RS models have highlighted an interesting parameter for consideration in other NDD models, namely the ratio of synaptic to extrasynaptic GABAergic signaling (Medrihan et al., 2008; Chen et al., 2017). Changes in subunit expression and mode of inhibition (phasic versus tonic) as a direct result of mutation, or as a plastic or homeostatic adaptation could result in alterations in network function leading to some of the electrophysiological, EEG, and phenotypic manifestations of NDDs.
Developmental epilepsies are also considered in the umbrella of NDD, due to the high incidence of epilepsy in prototypical NDD, as well as the developmental factors known to contribute to epileptogenesis (Bozzi et al., 2012). Further, just as epilepsy has a high occurrence in NDD populations, NDD features are also commonly observed in epilepsy syndromes. In addition to the prototypical NDDs, consideration of the phenotypes of developmental epilepsy syndromes will provide insight into the cellular and circuit dysfunction in NDD. Dravet syndrome (DS), formerly known as Severe Myoclonic Epilepsy of Infancy (SMEI), is a rare form of epilepsy that begins in the first year of life with frequent and/or prolonged seizures (Han et al., 2012). At early onset, DS is characterized by both febrile and non-febrile seizures, and progresses to myoclonic and/or partial seizures, psychomotor delay, and ataxia (Selmer et al., 2009). Patients with DS also have a high risk of sudden unexpected death in epilepsy (SUDEP; Shmuely et al., 2016). DS is also characterized by cognitive impairment, motor deficits, hyperactivity and/or impulsiveness, and in rare cases can include autistic-like behaviors (Han et al., 2012). The prevalence of DS is 1:15,700 individuals, with around 75% of patients bearing a mutation in their SCN1A gene, which encodes the α1 subunit of the voltage gated sodium channel (NaV1.1; Wu et al., 2015). The remaining 25% of DS cases have been linked to mutations in GABRG2, GABRA1 (Table 2), protocadherin 19 (PCDH 19), and Syntaxin Binding Protein-1 (STXBP1; Carvill et al., 2014). Mutations in SCN1A are also responsible for Genetic Epilepsy with Febrile Seizures Plus (GEFS+), which is an autosomal dominant form of epilepsy, typically less severe than DS (Hawkins et al., 2016; Kang and Macdonald, 2016). These findings indicate additional factors beyond SCN1A mutation that contribute to the occurrence and severity of the phenotype (Hawkins et al., 2016; Kang and Macdonald, 2016). A recent human study using single and paired pulse transcranial magnetic stimulation has found an absence of short interval intracortical inhibition in DS patients, suggestive of reduced cortical GABAergic neurotransmission (Stern et al., 2017).
Mouse models of DS (Scn1a+/-) are characterized to have spontaneous seizures, including thermally evoked seizures (Yu et al., 2006; Oakley et al., 2009), and as well as behavioral phenotypes relevant to ASD (Han et al., 2012). Electrophysiological assessment of DS models has revealed a selective loss of sodium currents in GABAergic inhibitory neurons, resulting in reduced excitability of interneurons and thus reduced GABAergic neurotransmission (Yu et al., 2006; Kalume et al., 2007; Cheah et al., 2012; Han et al., 2012). Field recordings in Scn1A+/- mice have demonstrated hyperexcitability of hippocampal circuits in both the pre-epileptic and epileptic periods (Liautard et al., 2013). Similar to the human variation in epilepsy severity, mouse models of SCN1A dysfunction, have been shown to have a strain dependent epilepsy phenotype, raising the possibility of additional factors contributing to pathogenesis (Hawkins et al., 2016). Scn1a+/- mice on 129 background have no readily detectable phenotype and a normal lifespan, while Scn1a+/- F1 129/C57Bl6 hybrid mice experience spontaneous and hyperthermia-induced seizures and high rates of premature death. Genetic mapping comparing Scn1A+/- mutations on 129 versus F1 hybrid backgrounds identified multiple modifier loci, followed by RNA-Seq of a loci of interest on chromosome 5 (Hawkins et al., 2016). RNA-Seq revealed three genes with significant differences in total gene expression between Scn1A deletion on the two backgrounds, including the α2 subunit of GABAARs, associating low Gabra2 expression with the B6 allele and reduced survival of Scn1A mutants on this background (Hawkins et al., 2016). These studies also showed that clobazam, which has preferential affinity for the GABAAR α2 subunit, normalized the temperature threshold for hyperthermia-induced seizures in Scn1a+/- mice (Hawkins et al., 2016). Compared to wildtype mice, Scn1a+/- mice displayed behavioral/social deficits as shown by circling behavior and less time spent in open arms in an elevated plus maze. These stereotypical behaviors are marked as autistic traits, indicating that DS does have shared symptomatology (Han et al., 2012). PV-positive and SOM-positive fast-spiking interneurons have also been found to have reduced excitability in Scn1a+/- mice, leading to reduced GABAergic inhibition (Tai et al., 2014). Greater understanding of Scn1a localization and function may also reveal insights about pathogenesis in DS. The protein encoded by Scn1a, the α1 subunit of NaV1.1, has been localized to the AIS, particularly in GABAergic interneurons (Duflocq et al., 2008; Lorincz and Nusser, 2008). AIS dysfunction is a proposed mechanism of neuropathology in epilepsy (Wimmer et al., 2010; Kole and Stuart, 2012), and chandelier cell cartridges opposed to α2 containing GABAARs are known to mediate inhibitory control at this site (Nusser et al., 1996; Freund and Katona, 2007; Gao and Heldt, 2016), suggesting that GABAergic dysfunction at the AIS may be a key factor in epilepsy. Of particular interest, studies have also shown that the benzodiazepine clonazepam is effective at reducing the ASD-like features of the Scn1a+/- mice (Han et al., 2012).
Examination of developmental epilepsies, alongside NDDs with epilepsy as an associated symptom is useful in elucidating common mechanisms. De novo mutation of SCN2A was also recently identified using exome sequencing of ASD samples (De Rubeis et al., 2014; Iossifov et al., 2014), further suggesting that the AIS may be a key site to evaluate in NDDs. As we have seen with NDD, developmental epilepsy syndromes such as DS also highlight GABAergic dysfunction as a key pathogenic mechanism. Again a common indication of PV-positive interneuron dysfunction is revealed, drawing commonality with ASD, AS, and RS. Greater understanding of the specific roles of interneuron subtypes, subcellular domains of inhibition, and GABAAR subunits in these disorders may help to further advance our understanding, and improve therapeutic strategies in NDDs.
Insight into NDDs may also be gleaned by examining rare genetic causes and disorders. Of particular interest, mutations in the ARHGEF9 gene (Table 2) have been reported to result in NDDs with varied behavioral symptoms (Alber et al., 2017). The ARHGEF9 gene codes for the GDP/GTP exchange factor collybistin (also known as HPEM2 in humans), which is a neuron specific RhoGEF. All collybistin isoforms possess three major functional domains: an N-terminal SH3 domain, a GEF domain, and a PH domain. Through these domains, collybistin has been shown to interact with gephyrin, neuroligin-2, and the GABAAR α2 subunit, playing an important role in clustering of GABAARs at inhibitory synapses (Saiepour et al., 2010). To this point, 18 patients have been described to have either point mutations, chromosomal rearrangements and deletions involving ARHGEF9, with a range of manifestations including ASD (Bhat et al., 2016; Machado et al., 2016), behavior disorders (ADHD, anxiety, aggression; Kalscheuer et al., 2009; Lesca et al., 2011), intellectual disability (Kalscheuer et al., 2009; Lesca et al., 2011; Marco et al., 2011; Shimojima et al., 2011; de Ligt et al., 2012; Lemke et al., 2012), hyperekplexia (Harvey et al., 2004; Marco et al., 2008), and infantile epilepsy (Harvey et al., 2004; Kalscheuer et al., 2009; Lesca et al., 2011; Shimojima et al., 2011). The majority of patients identified are male, and those that are female have been shown to have a near complete X-inactivation in favor of the affected gene (Marco et al., 2008; Kalscheuer et al., 2009). Collybistin KO mice recapitulate some aspects of the behavioral syndrome including anxiety-like behavior, learning deficits, and seizures (Papadopoulos et al., 2007; Papadopoulos and Soykan, 2011). Related to the collybistin mutations, rare mutations have also been identified in gephyrin associated with diverse NDD diagnoses (Table 2), including ASD, seizures, and schizophrenia (Lionel et al., 2013). Gephyrin KO mice die shortly after birth, and exhibit a phenotype of tactile-hyperresponsivity and difficulty breathing consistent with glycinergic signaling deficits (Feng et al., 1998). In general, further information is required to understand the detailed contribution of collybistin, gephyrin and other inhibitory synapse organizers in specialized subtypes of GABAergic synapses, and NDD. For both collybistin and gephyrin, studying the effects of the human mutations in mouse models may yield more clarity into the role of these proteins in NDD. Other rare mutations implicated in NDDs are indirectly linked to GABAergic scaffolding and signaling, including NONO (Mircsof et al., 2015; Reinstein et al., 2016) which impacts the expression of both gephyrin and the GABAAR α2 subunit (Mircsof et al., 2015). The data from rare NDD syndromes solidify direct and indirect links between NDDs and the proteins responsible for inhibitory GABAAR signaling.
Throughout this review we have identified commonalities in GABAergic signaling dysfunction in diverse NDDs. Specific perturbation in GABAergic signaling are emerging as ribs of commonality in these diverse disorders. One emerging rib centers on perturbations in the developmental excitatory to inhibitory shift in EGABA, caused by the onset of KCC2 expression. Studies have now directly indicated SLC12A5 gene (KCC2) mutation in ASD, with the C-terminus being impacted (Merner et al., 2015). Recent examination of the molecular structure of KCC2 reveals the C-terminus to be important for dimerization and function (Agez et al., 2017). Modeling of ASD and RS associated mutations has also revealed a delay in the chloride reversal associated with alterations in KCC2 expression and function, producing corresponding alterations in neuronal signaling (Tang et al., 2016; Amin et al., 2017). Environmental models of ASD, including maternal immune activation and valproic acid exposure have also indicated reductions in KCC2 expression and delays in the shift to inhibitory GABA (Corradini et al., 2017; Li et al., 2017). Clinical Trials of bumetanide, a high affinity inhibitor of NKCC1, have been successful in reducing the core symptoms of ASD (Lemonnier et al., 2012, 2017). A second emerging rib is the control of excitation by PV-positive cells. PV positive cells contact sites at or near action potential generation in neurons, exerting powerful control over excitatory firing patterns (Figure 1). A number of studies have indicated perturbations in PV-positive cells in NDD populations and mouse models (Peñagarikano et al., 2011; Karayannis et al., 2014; Hashemi et al., 2017; Soghomonian et al., 2017; Vogt et al., 2017; Ariza et al., 2018). While several studies have indicated a loss of PV-positive cells, some evidence suggests that it may be a reduction in PV expression, bringing cells below the detection threshold (Filice et al., 2016). These studies are supported by the demonstration that knockout of PV in the mouse leads to behavioral phenotypes relevant to human ASD (Wöhr et al., 2015). A third emerging rib is the role of tonic inhibition (Figure 2, left), early in development and in NDDs. Studies have identified alterations in the expression of tonic GABAAR subunits in human populations (Fatemi et al., 2014), as well as deficits in tonic inhibition in mouse models of NDD (Olmos-Serrano et al., 2011; Egawa et al., 2012; Martin et al., 2014; Vien et al., 2015; Zhong et al., 2015; Bridi et al., 2017). Gaboxadol, a GABAAR agonist with preferential activity at δ-containing receptors, is being examined in clinical trials for patients with Fragile-x and AS (Ligsay et al., 2017).
Despite these emerging ribs, our understanding of the details of inhibitory signaling lags behind that which is known about excitatory signaling. In particular, further investigation of regional or subtype specific deficits in GABAergic signaling is warranted in in NDD populations and animal models. More specific information about GABAAR subunit alterations would provide insights about which modes of inhibition, and which subtypes of inhibitory synapses may be contributing to NDDs. Investigations comparing both phasic and tonic forms of inhibition across animal models of NDD would be illuminating, as tonic inhibition plays an important and powerful role in controlling brain activity but is understudied. In addition, subtypes of synaptic GABAARs, including somatic and AIS synapses, are known to have a powerful role in coordinating neuronal activity patterns, but we are just beginning to understand the roles of these synapses, how they are specified, how they develop, and their possible contributions to NDDs. In addition, there are many key adhesion, and scaffolding proteins required for the aforementioned processes at specific inhibitory synapse subtypes, including neuroligins, dystroglycan, dystrophin, gephyrin, and collybistin, among others. Increasing our understanding of these key proteins involved in the formation, maintenance, and function of specific inhibitory synapses will also advance our understanding of how subtypes impact neurodevelopment more globally.
Further, assessments of specific interneuron function also seem promising, with evidence pointing toward PV-positive interneuron dysfunction as a key factor mentioned above. In particular, it may be important to examine the mechanisms causing changes in PV expression, as well as understand the functional implications of such changes to neuronal circuit activity. In addition, analysis of markers for other subtypes of interneurons in NDD and mouse models have not been described. In particular, analysis of interneuron targeting interneurons that have disinhibiting effects on network signaling is lacking in NDD populations and models. This is of particular relevance as a recent study has shown that Nav1.2 (SCN2A), which bears an ASD related mutation (De Rubeis et al., 2014), is enriched in inhibitory cells that innervate other interneurons (Yamagata et al., 2017). Analysis of circuit level dysfunction in NDD populations and models is also currently focused on seizures and epilepsy, and more global assessment of patterned oscillatory activity may provide the next needed advance following specific inhibitory mode, interneuron, and synapse subtype assessments. In particular, systematic assessment of oscillatory activity across multiple mouse models of NDDs may provide illuminating insights into commonalities and distinctions. Also of importance is to evaluate and/or manipulate circuit activity during execution of specific behaviors relevant to NDD, allowing for further insight into the mechanisms producing symptoms, and promoting novel therapeutic approaches by modulating specific circuits to correct perturbations in oscillatory activity. Advances in our understanding of GABAergic signaling in normal development and in NDD models and populations will ultimately advance our therapeutic strategies, improving patient outcomes.
RAR contributed to the writing. CJ contributed to the writing and tables. RH conceived of the topic, wrote the manuscript, and prepared the figures.
University of Nevada, Las Vegas start-up funds supported this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agez, M., Schultz, P., Medina, I., Baker, D. J., Burnham, M. P., Cardarelli, R. A., et al. (2017). Molecular architecture of potassium chloride co-transporter KCC2. Sci. Rep. 7:16452. doi: 10.1038/s41598-017-15739-1
Al Ageeli, E., Drunat, S., Delanoë, C., Perrin, L., Baumann, C., Capri, Y., et al. (2014). Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases. Eur. J. Med. Genet. 57, 5–14. doi: 10.1016/j.ejmg.2013.10.008
Alarcón, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Alber, M., Kalscheuer, V. M., Marco, E., Sherr, E., Lesca, G., Till, M., et al. (2017). ARHGEF9 disease: phenotype clarification and genotype-phenotype correlation. Neurol. Genet. 3:e148. doi: 10.1212/NXG.0000000000000148
Amin, H., Marinaro, F., De Pietri Tonelli, D., and Berdondini, L. (2017). Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci. Rep. 7:15752. doi: 10.1038/s41598-017-15793-9
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Anderson, G. R., Galfin, T., Xu, W., Aoto, J., Malenka, R. C., and Südhof, T. C. (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. U.S.A. 109, 18120–18125. doi: 10.1073/pnas.1216398109
Anney, R., Klei, L., Pinto, D., Almeida, J., Bacchelli, E., Baird, G., et al. (2012). Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet. 21, 4781–4792. doi: 10.1093/hmg/dds301
Ariza, J., Rogers, H., Hashemi, E., Noctor, S. C., and Martínez-Cerdeño, V. (2018). The number of chandelier and basket cells are differentially decreased in prefrontal cortex in autism. Cereb. Cortex 28, 411–420. doi: 10.1093/cercor/bhw349
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164. doi: 10.1016/j.ajhg.2007.09.015
Arons, M. H., Thynne, C. J., Grabrucker, A. M., Li, D., Schoen, M., Cheyne, J. E., et al. (2012). Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 32, 14966–14978. doi: 10.1523/JNEUROSCI.2215-12.2012
Baig, D. N., Yanagawa, T., and Tabuchi, K. (2017). Distortion of the normal function of synaptic cell adhesion molecules by genetic variants as a risk for autism spectrum disorders. Brain Res. Bull. 129, 82–90. doi: 10.1016/j.brainresbull.2016.10.006
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Banerjee, A., Castro, J., and Sur, M. (2012). Rett syndrome: genes, synapses, circuits, and therapeutics. Front. Psychiatry 3:34. doi: 10.3389/fpsyt.2012.00034
Banerjee, S., Riordan, M., and Bhat, M. A. (2014). Genetic aspects of autism spectrum disorders: insights from animal models. Front. Cell. Neurosci. 8:58. doi: 10.3389/fncel.2014.00058
Baron, M. K., Boeckers, T. M., Vaida, B., Faham, S., Gingery, M., Sawaya, M. R., et al. (2006). An architectural framework that may lie at the core of the postsynaptic density. Science 311, 531–535. doi: 10.1126/science.1118995
Baulac, S., Huberfeld, G., Gourfinkel-An, I., Mitropoulou, G., Beranger, A., Prud’homme, J. F., et al. (2001). First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet. 28, 46–48. doi: 10.1038/88254
Ben-Ari, Y. (2014). The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience 279, 187–219. doi: 10.1016/j.neuroscience.2014.08.001
Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Bhat, G., LaGrave, D., Millson, A., Herriges, J., Lamb, A. N., and Matalon, R. (2016). Xq11.1-11.2 deletion involving ARHGEF9 in a girl with autism spectrum disorder. Eur. J. Med. Genet. 59, 470–473. doi: 10.1016/j.ejmg.2016.05.014
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., and Bauman, M. L. (2001). Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J. Autism Dev. Disord. 31, 537–543. doi: 10.1023/A:1013238809666
Blundell, J., Blaiss, C. A., Etherton, M. R., Espinosa, F., Tabuchi, K., Walz, C., et al. (2010). Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 30, 2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Boivin, M. J., Kakooza, A. M., Warf, B. C., Davidson, L. L., and Grigorenko, E. L. (2015). Reducing neurodevelopmental disorders and disability through research and interventions. Nature 527, S155–S160. doi: 10.1038/nature16029
Bolliger, M. F., Frei, K., Winterhalter, K. H., and Gloor, S. M. (2001). Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem. J. 356, 581–588. doi: 10.1042/bj3560581
Bolliger, M. F., Pei, J., Maxeiner, S., Boucard, A. A., Grishin, N. V., and Südhof, T. C. (2008). Unusually rapid evolution of Neuroligin-4 in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 6421–6426. doi: 10.1073/pnas.0801383105
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1:15. doi: 10.1186/2040-2392-1-15
Bozzi, Y., Casarosa, S., and Caleo, M. (2012). Epilepsy as a neurodevelopmental disorder. Front. Psychiatry 3:19. doi: 10.3389/fpsyt.2012.00019
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Bridi, M. S., Park, S. M., and Huang, S. (2017). Developmental Disruption of GABAAR-Meditated Inhibition in Cntnap2 KO Mice. eNeuro 4:ENEURO.0162-17.2017. doi: 10.1523/ENEURO.0162-17.2017
Bright, D. P., and Smart, T. G. (2013). Methods for recording and measuring tonic GABAA receptor-mediated inhibition. Front. Neural Circuits 7:193. doi: 10.3389/fncir.2013.00193
Budreck, E. C., and Scheiffele, P. (2007). Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur. J. Neurosci. 26, 1738–1748. doi: 10.1111/j.1460-9568.2007.05842.x
Buiting, K., Williams, C., and Horsthemke, B. (2016). Angelman syndrome - insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593. doi: 10.1038/nrneurol.2016.133
Burbach, J. P. H., and van der Zwaag, B. (2009). Contact in the genetics of autism and schizophrenia. Trends Neurosci. 32, 69–72. doi: 10.1016/j.tins.2008.11.002
Carvill, G. L., Weckhuysen, S., McMahon, J. M., Hartmann, C., Møller, R. S., Hjalgrim, H., et al. (2014). GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 82, 1245–1253. doi: 10.1212/WNL.0000000000000291
Cattabeni, F., Gardoni, F., and Di Luca, M. (1999). Pathophysiological implications of the structural organization of the excitatory synapse. Eur. J. Pharmacol. 375, 339–347. doi: 10.1016/S0014-2999(99)00299-X
Chadman, K. K., Gong, S., Scattoni, M. L., Boltuck, S. E., Gandhy, S. U., Heintz, N., et al. (2008). Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 1, 147–158.
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chao, H.-T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chao, H.-T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Cheah, C. S., Yu, F. H., Westenbroek, R. E., Kalume, F. K., Oakley, J. C., Potter, G. B., et al. (2012). Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U.S.A. 109, 14646–14651. doi: 10.1073/pnas.1211591109
Chen, C.-Y., Di Lucente, J., Lin, Y.-C., Lien, C.-C., Rogawski, M. A., Maezawa, I., et al. (2017). Defective GABAergic neurotransmission in the nucleus tractus solitarius in Mecp2-null mice, a model of Rett syndrome. Neurobiol. Dis. 109, 25–32. doi: 10.1016/j.nbd.2017.09.006
Chen, J., Yu, S., Fu, Y., and Li, X. (2014). Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 8:276. doi: 10.3389/fncel.2014.00276
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chiara, D. C., Jounaidi, Y., Zhou, X., Savechenkov, P. Y., Bruzik, K. S., Miller, K. W., et al. (2016). General anesthetic binding sites in human α4β3δ γ-aminobutyric acid type a receptors (GABAARs). J. Biol. Chem. 291, 26529–26539. doi: 10.1074/jbc.M116.753335
Chih, B., Engelman, H., and Scheiffele, P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328. doi: 10.1126/science.1107470
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Cobos, I., Calcagnotto, M. E., Vilaythong, A. J., Thwin, M. T., Noebels, J. L., Baraban, S. C., et al. (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068. doi: 10.1038/nn1499
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., et al. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689. doi: 10.1093/hmg/ddh282
Collins, A. L., Ma, D., Whitehead, P. L., Martin, E. R., Wright, H. H., Abramson, R. K., et al. (2006). Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics 7, 167–174. doi: 10.1007/s10048-006-0045-1
Conant, K. D., Finucane, B., Cleary, N., Martin, A., Muss, C., Delany, M., et al. (2014). A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia 55, 396–402. doi: 10.1111/epi.12530
Connolly, S., Anney, R., Gallagher, L., and Heron, E. A. (2017). A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur. J. Hum. Genet. 25, 234–239. doi: 10.1038/ejhg.2016.153
Cook, E. H., Courchesne, R., Lord, C., Cox, N. J., Yan, S., Lincoln, A., et al. (1997). Evidence of linkage between the serotonin transporter and autistic disorder. Mol. Psychiatry 2, 247–250. doi: 10.1038/sj.mp.4000266
Corradini, I., Focchi, E., Rasile, M., Morini, R., Desiato, G., Tomasoni, R., et al. (2017). Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691. doi: 10.1016/j.biopsych.2017.09.030
Cossette, P., Liu, L., Brisebois, K., Dong, H., Lortie, A., Vanasse, M., et al. (2002). Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 31, 184–189. doi: 10.1038/ng885
Coury, D. L., Swedo, S. E., Thurm, A. E., Miller, D. T., Veenstra-VanderWeele, J. M., Carbone, P. S., et al. (2014). Treating the whole person with autism: the proceedings of the Autism Speaks National Autism Conference. Curr. Probl. Pediatr. Adolesc. Health Care 44, 26–47. doi: 10.1016/j.cppeds.2013.12.002
Dachtler, J., Glasper, J., Cohen, R. N., Ivorra, J. L., Swiffen, D. J., Jackson, A. J., et al. (2014). Deletion of α-neurexin II results in autism-related behaviors in mice. Transl. Psychiatry 4:e484. doi: 10.1038/tp.2014.123
Dahlhaus, R., and El-Husseini, A. (2010). Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 208, 96–105. doi: 10.1016/j.bbr.2009.11.019
Dahlhaus, R., Hines, R. M., Eadie, B. D., Kannangara, T. S., Hines, D. J., Brown, C. E., et al. (2010). Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 20, 305–322. doi: 10.1002/hipo.20630
Dávid, C., Schleicher, A., Zuschratter, W., and Staiger, J. F. (2007). The innervation of parvalbumin-containing interneurons by VIP-immunopositive interneurons in the primary somatosensory cortex of the adult rat. Eur. J. Neurosci. 25, 2329–2340. doi: 10.1111/j.1460-9568.2007.05496.x
de Ligt, J., Willemsen, M. H., van Bon, B. W. M., Kleefstra, T., Yntema, H. G., Kroes, T., et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. doi: 10.1056/NEJMoa1206524
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
del Gaudio, D., Fang, P., Scaglia, F., Ward, P. A., Craigen, W. J., Glaze, D. G., et al. (2006). Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 8, 784–792. doi: 10.1097/01.gim.0000250502.28516.3c
DeLorey, T. M., Handforth, A., Anagnostaras, S. G., Homanics, G. E., Minassian, B. A., Asatourian, A., et al. (1998). Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J. Neurosci. 18, 8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998
Depienne, C., Moreno-De-Luca, D., Heron, D., Bouteiller, D., Gennetier, A., Delorme, R., et al. (2009). Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol. Psychiatry 66, 349–359. doi: 10.1016/j.biopsych.2009.01.025
Di Rocco, A., Loggini, A., Di Rocco, M., Di Rocco, P., Rossi, R. P., Gimelli, G., et al. (2013). Paradoxical worsening of seizure activity with pregabalin in an adult with isodicentric 15 (IDIC-15) syndrome involving duplications of the GABRB3, GABRA5 and GABRG3 genes. BMC Neurol. 13:43. doi: 10.1186/1471-2377-13-43
DiStefano, C., Gulsrud, A., Huberty, S., Kasari, C., Cook, E., Reiter, L. T., et al. (2016). Identification of a distinct developmental and behavioral profile in children with Dup15q syndrome. J. Neurodev. Disord. 8:19. doi: 10.1186/s11689-016-9152-y
Dixon, C. L., Harrison, N. L., Lynch, J. W., and Keramidas, A. (2015). Zolpidem and eszopiclone prime α1β2γ2 GABAA receptors for longer duration of activity. Br. J. Pharmacol. 172, 3522–3536. doi: 10.1111/bph.13142
Doyon, N., Vinay, L., Prescott, S. A., and De Koninck, Y. (2016). Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron 89, 1157–1172. doi: 10.1016/j.neuron.2016.02.030
Dragich, J., Houwink-Manville, I., and Schanen, C. (2000). Rett syndrome: a surprising result of mutation in MECP2. Hum. Mol. Genet. 9, 2365–2375. doi: 10.1093/hmg/9.16.2365
Duflocq, A., Le Bras, B., Bullier, E., Couraud, F., and Davenne, M. (2008). Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol. Cell. Neurosci. 39, 180–192. doi: 10.1016/j.mcn.2008.06.008
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Egawa, K., Kitagawa, K., Inoue, K., Takayama, M., Takayama, C., Saitoh, S., et al. (2012). Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci. Transl. Med. 4:163ra157. doi: 10.1126/scitranslmed.3004655
Ehlers, M. D. (1999). Synapse structure: glutamate receptors connected by the shanks. Curr. Biol. 9, R848–R850. doi: 10.1016/S0960-9822(00)80043-3
Eyre, M. D., Renzi, M., Farrant, M., and Nusser, Z. (2012). Setting the time course of inhibitory synaptic currents by mixing multiple GABA(A) receptor α subunit isoforms. J. Neurosci. 32, 5853–5867. doi: 10.1523/JNEUROSCI.6495-11.2012
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Fatemi, S. H., Folsom, T. D., Rooney, R. J., and Thuras, P. D. (2013). Expression of GABAA α2-, β1- and ε-receptors are altered significantly in the lateral cerebellum of subjects with schizophrenia, major depression and bipolar disorder. Transl. Psychiatry 3:e303. doi: 10.1038/tp.2013.64
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and protein levels for GABAAalpha4, alpha5, beta1 and GABABR1 receptors are altered in brains from subjects with autism. J. Autism Dev. Disord. 40, 743–750. doi: 10.1007/s10803-009-0924-z
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rustan, O. G., Rooney, R. J., and Thuras, P. D. (2014). Downregulation of GABAA receptor protein subunits α6, β2, δ, ε, γ2, θ, and ρ2 in superior frontal cortex of subjects with autism. J. Autism Dev. Disord. 44, 1833–1845. doi: 10.1007/s10803-014-2078-x
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., and Thuras, P. D. (2009). GABA(A) receptor downregulation in brains of subjects with autism. J. Autism Dev. Disord. 39, 223–230. doi: 10.1007/s10803-008-0646-7
Feng, G., Tintrup, H., Kirsch, J., Nichol, M. C., Kuhse, J., Betz, H., et al. (1998). Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282, 1321–1324. doi: 10.1126/science.282.5392.1321
Filice, F., Vörckel, K. J., Sungur, A. Ö., Wöhr, M., and Schwaller, B. (2016). Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 9:10. doi: 10.1186/s13041-016-0192-8
Fiumara, A., Pittalà, A., Cocuzza, M., and Sorge, G. (2010). Epilepsy in patients with Angelman syndrome. Ital. J. Pediatr. 36:31. doi: 10.1186/1824-7288-36-31
Freund, T. F., and Katona, I. (2007). Perisomatic inhibition. Neuron 56, 33–42. doi: 10.1016/j.neuron.2007.09.012
Frohlich, J., Senturk, D., Saravanapandian, V., Golshani, P., Reiter, L. T., Sankar, R., et al. (2016). A quantitative electrophysiological biomarker of duplication 15q11.2-q13.1 Syndrome. PLoS One 11:e0167179. doi: 10.1371/journal.pone.0167179
Früh, S., Romanos, J., Panzanelli, P., Bürgisser, D., Tyagarajan, S. K., Campbell, K. P., et al. (2016). Neuronal dystroglycan is necessary for formation and maintenance of functional CCK-positive basket cell terminals on pyramidal cells. J. Neurosci. 36, 10296–10313. doi: 10.1523/JNEUROSCI.1823-16.2016
Galiano, M. R., Jha, S., Ho, T. S.-Y., Zhang, C., Ogawa, Y., Chang, K.-J., et al. (2012). A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell 149, 1125–1139. doi: 10.1016/j.cell.2012.03.039
Gao, Y., and Heldt, S. A. (2016). Enrichment of GABAA receptor α-subunits on the axonal initial segment shows regional differences. Front. Cell. Neurosci. 10:39. doi: 10.3389/fncel.2016.00039
Gennarini, G., Bizzoca, A., Picocci, S., Puzzo, D., Corsi, P., and Furley, A. J. W. (2017). The role of Gpi-anchored axonal glycoproteins in neural development and neurological disorders. Mol. Cell. Neurosci. 81, 49–63. doi: 10.1016/j.mcn.2016.11.006
Gerrow, K., Romorini, S., Nabi, S. M., Colicos, M. A., Sala, C., and El-Husseini, A. (2006). A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron 49, 547–562. doi: 10.1016/j.neuron.2006.01.015
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Grabrucker, S., Proepper, C., Mangus, K., Eckert, M., Chhabra, R., Schmeisser, M. J., et al. (2014). The PSD protein ProSAP2/Shank3 displays synapto-nuclear shuttling which is deregulated in a schizophrenia-associated mutation. Exp. Neurol. 253, 126–137. doi: 10.1016/j.expneurol.2013.12.015
Grayton, H. M., Missler, M., Collier, D. A., and Fernandes, C. (2013). Altered social behaviours in neurexin 1α knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS One 8:e67114. doi: 10.1371/journal.pone.0067114
Grubb, M. S., and Burrone, J. (2010). Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465, 1070–1074. doi: 10.1038/nature09160
Gundelfinger, E. D., Boeckers, T. M., Baron, M. K., and Bowie, J. U. (2006). A role for zinc in postsynaptic density asSAMbly and plasticity? Trends Biochem. Sci. 31, 366–373. doi: 10.1016/j.tibs.2006.05.007
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Han, K., Holder, J. L., Schaaf, C. P., Lu, H., Chen, H., Kang, H., et al. (2013). SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503, 72–77. doi: 10.1038/nature12630
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Hartmann, A.-M., and Nothwang, H. G. (2014). Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Front. Cell. Neurosci. 8:470. doi: 10.3389/fncel.2014.00470
Harvey, K., Duguid, I. C., Alldred, M. J., Beatty, S. E., Ward, H., Keep, N. H., et al. (2004). The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826. doi: 10.1523/JNEUROSCI.1184-04.2004
Hashemi, E., Ariza, J., Rogers, H., Noctor, S. C., and Martínez-Cerdeño, V. (2017). The number of parvalbumin-expressing interneurons is decreased in the medial prefrontal cortex in autism. Cereb. Cortex 27, 1931–1943. doi: 10.1093/cercor/bhw021
Hawkins, N. A., Zachwieja, N. J., Miller, A. R., Anderson, L. L., and Kearney, J. A. (2016). Fine mapping of a dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-Seq. PLoS Genet. 12:e1006398. doi: 10.1371/journal.pgen.1006398
Hecht, F. (1975). Letter: Partial trisomy 15 (dup15q) syndrome and the need for precision in clinical cytogenetics. J. Pediatr. 87, 842–843. doi: 10.1016/S0022-3476(75)80340-4
Hedges, D. J., Hamilton-Nelson, K. L., Sacharow, S. J., Nations, L., Beecham, G. W., Kozhekbaeva, Z. M., et al. (2012). Evidence of novel fine-scale structural variation at autism spectrum disorder candidate loci. Mol. Autism 3:2. doi: 10.1186/2040-2392-3-2
Herd, M. B., Brown, A. R., Lambert, J. J., and Belelli, D. (2013). Extrasynaptic GABAA receptors couple presynaptic activity to postsynaptic inhibition in the somatosensory thalamus. J. Neurosci. 33, 14850–14868. doi: 10.1523/JNEUROSCI.1174-13.2013
Hernandez, C. C., Gurba, K. N., Hu, N., and Macdonald, R. L. (2011). The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6β22 and 6β2 GABA(A) receptor channel gating and expression. J. Physiol. 589, 5857–5878. doi: 10.1113/jphysiol.2011.218883
Hines, R. M., Davies, P. A., Moss, S. J., and Maguire, J. (2012). Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 22, 552–558. doi: 10.1016/j.conb.2011.10.007
Hines, R. M., Wu, L., Hines, D. J., Steenland, H., Mansour, S., Dahlhaus, R., et al. (2008). Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J. Neurosci. 28, 6055–6067. doi: 10.1523/JNEUROSCI.0032-08.2008
Homanics, G. E., DeLorey, T. M., Firestone, L. L., Quinlan, J. J., Handforth, A., Harrison, N. L., et al. (1997). Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc. Natl. Acad. Sci. U.S.A. 94, 4143–4148. doi: 10.1073/pnas.94.8.4143
Hoon, M., Soykan, T., Falkenburger, B., Hammer, M., Patrizi, A., Schmidt, K.-F., et al. (2011). Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc. Natl. Acad. Sci. U.S.A. 108, 3053–3058. doi: 10.1073/pnas.1006946108
Hu, V. W., Sarachana, T., Kim, K. S., Nguyen, A., Kulkarni, S., Steinberg, M. E., et al. (2009). Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2, 78–97. doi: 10.1002/aur.73
Hu, V. W., and Steinberg, M. E. (2009). Novel clustering of items from the autism diagnostic interview-revised to define phenotypes within autism spectrum disorders. Autism Res. 2, 67–77. doi: 10.1002/aur.72
Huang, X., Tian, M., Hernandez, C. C., Hu, N., and Macdonald, R. L. (2012). The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol. Dis. 48, 115–123. doi: 10.1016/j.nbd.2012.06.013
Huh, K. H., Endo, S., and Olsen, R. W. (1996). Diazepam-insensitive GABAA receptors in rat cerebellum and thalamus. Eur. J. Pharmacol. 310, 225–233. doi: 10.1016/0014-2999(96)00349-4
Ichtchenko, K., Hata, Y., Nguyen, T., Ullrich, B., Missler, M., Moomaw, C., et al. (1995). Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81, 435–443. doi: 10.1016/0092-8674(95)90396-8
International Molecular Genetic Study of Autism Consortium [IMGSAC] (2001). A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am. J. Hum. Genet. 69, 570–581. doi: 10.1086/323264
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Irie, M., Hata, Y., Takeuchi, M., Ichtchenko, K., Toyoda, A., Hirao, K., et al. (1997). Binding of Neuroligins to PSD-95. Science 277, 1511–1515. doi: 10.1126/science.277.5331.1511
Ito-Ishida, A., Ure, K., Chen, H., Swann, J. W., and Zoghbi, H. Y. (2015). Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct rett syndrome-like phenotypes. Neuron 88, 651–658. doi: 10.1016/j.neuron.2015.10.029
Jacob, J. (2016). Cortical interneuron dysfunction in epilepsy associated with autism spectrum disorders. Epilepsia 57, 182–193. doi: 10.1111/epi.13272
Jacob, T. C., Moss, S. J., and Jurd, R. (2008). GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343. doi: 10.1038/nrn2370
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jamain, S., Radyushkin, K., Hammerschmidt, K., Granon, S., Boretius, S., Varoqueaux, F., et al. (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. U.S.A. 105, 1710–1715. doi: 10.1073/pnas.0711555105
Jaramillo, T. C., Liu, S., Pettersen, A., Birnbaum, S. G., and Powell, C. M. (2014). Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res. 7, 264–272. doi: 10.1002/aur.1362
Jiang, Y. H., Armstrong, D., Albrecht, U., Atkins, C. M., Noebels, J. L., Eichele, G., et al. (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811. doi: 10.1016/S0896-6273(00)80596-6
Jiang, Y.-H., Pan, Y., Zhu, L., Landa, L., Yoo, J., Spencer, C., et al. (2010). Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS One 5:e12278. doi: 10.1371/journal.pone.0012278
Judson, M. C., Wallace, M. L., Sidorov, M. S., Burette, A. C., Gu, B., van Woerden, G. M., et al. (2016). GABAergic neuron-specific loss of Ube3a causes angelman syndrome-Like EEG abnormalities and enhances seizure susceptibility. Neuron 90, 56–69. doi: 10.1016/j.neuron.2016.02.040
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., et al. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68. doi: 10.1002/humu.20814
Kalume, F., Yu, F. H., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2007). Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J. Neurosci. 27, 11065–11074. doi: 10.1523/JNEUROSCI.2162-07.2007
Kang, J.-Q., and Macdonald, R. L. (2016). Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to dravet syndrome. JAMA Neurol. 73, 1009–1016. doi: 10.1001/jamaneurol.2016.0449
Kaphzan, H., Buffington, S. A., Jung, J. I., Rasband, M. N., and Klann, E. (2011). Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of angelman syndrome. J. Neurosci. 31, 17637–17648. doi: 10.1523/JNEUROSCI.4162-11.2011
Karayannis, T., Au, E., Patel, J. C., Kruglikov, I., Markx, S., Delorme, R., et al. (2014). Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature 511, 236–240. doi: 10.1038/nature13248
Katidou, M., Vidaki, M., Strigini, M., and Karagogeos, D. (2008). The immunoglobulin superfamily of neuronal cell adhesion molecules: lessons from animal models and correlation with human disease. Biotechnol. J. 3, 1564–1580. doi: 10.1002/biot.200800281
Kattenstroth, G., Tantalaki, E., Südhof, T. C., Gottmann, K., and Missler, M. (2004). Postsynaptic N-methyl-D-aspartate receptor function requires alpha-neurexins. Proc. Natl. Acad. Sci. U.S.A. 101, 2607–2612. doi: 10.1073/pnas.0308626100
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. doi: 10.1126/science.1149381
Kole, M. H. P., and Stuart, G. J. (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. doi: 10.1016/j.neuron.2012.01.007
Kozinetz, C. A., Skender, M. L., MacNaughton, N., Almes, M. J., Schultz, R. J., Percy, A. K., et al. (1993). Epidemiology of rett syndrome: a population-based registry. Pediatrics 91, 445–450.
Krumm, N., O’Roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi: 10.1016/j.tins.2013.11.005
Kuba, H., Oichi, Y., and Ohmori, H. (2010). Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature 465, 1075–1078. doi: 10.1038/nature09087
Kwasnicka-Crawford, D. A., Roberts, W., and Scherer, S. W. (2007). Characterization of an autism-associated segmental maternal heterodisomy of the chromosome 15q11-13 region. J. Autism Dev. Disord. 37, 694–702. doi: 10.1007/s10803-006-0225-8
Kyllerman, M. (2013). Angelman syndrome. Handb. Clin. Neurol. 111, 287–290. doi: 10.1016/B978-0-444-52891-9.00032-4
Lachamp, P. M., Liu, Y., and Liu, S. J. (2009). Glutamatergic modulation of cerebellar interneuron activity is mediated by an enhancement of GABA release and requires protein kinase A/RIM1alpha signaling. J. Neurosci. 29, 381–392. doi: 10.1523/JNEUROSCI.2354-08.2009
Lavoie, A. M., Tingey, J. J., Harrison, N. L., Pritchett, D. B., and Twyman, R. E. (1997). Activation and deactivation rates of recombinant GABA(A) receptor channels are dependent on alpha-subunit isoform. Biophys. J. 73, 2518–2526. doi: 10.1016/S0006-3495(97)78280-8
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. doi: 10.1371/journal.pgen.1004580
Lee, V., and Maguire, J. (2014). The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front. Neural Circuits 8:3. doi: 10.3389/fncir.2014.00003
Lemke, J. R., Riesch, E., Scheurenbrand, T., Schubach, M., Wilhelm, C., Steiner, I., et al. (2012). Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 53, 1387–1398. doi: 10.1111/j.1528-1167.2012.03516.x
Lemonnier, E., Degrez, C., Phelep, M., Tyzio, R., Josse, F., Grandgeorge, M., et al. (2012). A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2:e202. doi: 10.1038/tp.2012.124
Lemonnier, E., Villeneuve, N., Sonie, S., Serret, S., Rosier, A., Roue, M., et al. (2017). Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7:e1056. doi: 10.1038/tp.2017.10
Lesca, G., Till, M., Labalme, A., Vallee, D., Hugonenq, C., Philip, N., et al. (2011). De novo Xq11.11 microdeletion including ARHGEF9 in a boy with mental retardation, epilepsy, macrosomia, and dysmorphic features. Am. J. Med. Genet. A. 155A, 1706–1711. doi: 10.1002/ajmg.a.34004
Levinson, J. N., and El-Husseini, A. (2005). Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron 48, 171–174. doi: 10.1016/j.neuron.2005.09.017
Li, X. G., Somogyi, P., Tepper, J. M., and Buzsáki, G. (1992). Axonal and dendritic arborization of an intracellularly labeled chandelier cell in the CA1 region of rat hippocampus. Exp. Brain Res. 90, 519–525.
Li, Y., Zhou, Y., Peng, L., and Zhao, Y. (2017). Reduced protein expressions of cytomembrane GABAARβ3 at different postnatal developmental stages of rats exposed prenatally to valproic acid. Brain Res. 1671, 33–42. doi: 10.1016/j.brainres.2017.06.018
Liautard, C., Scalmani, P., Carriero, G., de Curtis, M., Franceschetti, S., and Mantegazza, M. (2013). Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 54, 1251–1261. doi: 10.1111/epi.12213
Ligsay, A., Van Dijck, A., Nguyen, D. V., Lozano, R., Chen, Y., Bickel, E. S., et al. (2017). A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J. Neurodev. Disord. 9:26. doi: 10.1186/s11689-017-9207-8
Lionel, A. C., Vaags, A. K., Sato, D., Gazzellone, M. J., Mitchell, E. B., Chen, H. Y., et al. (2013). Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 22, 2055–2066. doi: 10.1093/hmg/ddt056
Liu, X., Novosedlik, N., Wang, A., Hudson, M. L., Cohen, I. L., Chudley, A. E., et al. (2009). The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur. J. Hum. Genet. 17, 228–235. doi: 10.1038/ejhg.2008.148
Lo, W.-S., Harano, M., Gawlik, M., Yu, Z., Chen, J., Pun, F. W., et al. (2007). GABRB2 association with schizophrenia: commonalities and differences between ethnic groups and clinical subtypes. Biol. Psychiatry 61, 653–660. doi: 10.1016/j.biopsych.2006.05.003
Loebrich, S., Bähring, R., Katsuno, T., Tsukita, S., and Kneussel, M. (2006). Activated radixin is essential for GABAA receptor alpha5 subunit anchoring at the actin cytoskeleton. EMBO J. 25, 987–999. doi: 10.1038/sj.emboj.7600995
Long, P., May, M. M., James, V. M., Grannò, S., Johnson, J. P., Tarpey, P., et al. (2015). Missense mutation R338W in ARHGEF9 in a family with X-linked intellectual disability with variable macrocephaly and macro-orchidism. Front. Mol. Neurosci. 8:83. doi: 10.3389/fnmol.2015.00083
Loomes, R., Hull, L., and Mandy, W. P. L. (2017). What is the male-to-female ratio in autism spectrum disorder? a systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 56, 466–474. doi: 10.1016/j.jaac.2017.03.013
Lorincz, A., and Nusser, Z. (2008). Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci. 28, 14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008
Lu, H., Ash, R. T., He, L., Kee, S. E., Wang, W., Yu, D., et al. (2016). Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a rett syndrome mouse model. Neuron 91, 739–747. doi: 10.1016/j.neuron.2016.07.018
Luikenhuis, S., Giacometti, E., Beard, C. F., and Jaenisch, R. (2004). Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 6033–6038. doi: 10.1073/pnas.0401626101
Lüscher, B., and Keller, C. A. (2004). Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol. Ther. 102, 195–221. doi: 10.1016/j.pharmthera.2004.04.003
Ma, D. Q., Whitehead, P. L., Menold, M. M., Martin, E. R., Ashley-Koch, A. E., Mei, H., et al. (2005). Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am. J. Hum. Genet. 77, 377–388. doi: 10.1086/433195
Macdonald, R. L., Kang, J.-Q., and Gallagher, M. J. (2012). “GABAA receptor subunit mutations and genetic epilepsies,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information). Available at: http://www.ncbi.nlm.nih.gov/books/NBK98205/
Machado, F. P., Palladino, R. R. R., Damasceno, L. L., and Cunha, M. C. (2016). Appropriateness of using autism spectrum disorders screening tools in a hearing evaluation service. Folia Phoniatr. Logop. 68, 60–66. doi: 10.1159/000446984
Malzac, P., Webber, H., Moncla, A., Graham, J. M., Kukolich, M., Williams, C., et al. (1998). Mutation analysis of UBE3A in angelman syndrome patients. Am. J. Hum. Genet. 62, 1353–1360. doi: 10.1086/301877
Marco, E. J., Abidi, F. E., Bristow, J., Dean, W. B., Cotter, P., Jeremy, R. J., et al. (2008). ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J. Med. Genet. 45, 100–105. doi: 10.1136/jmg.2007.052324
Marco, E. J., Hinkley, L. B. N., Hill, S. S., and Nagarajan, S. S. (2011). Sensory processing in autism: a review of neurophysiologic findings. Pediatr. Res. 69, 48R–54R. doi: 10.1203/PDR.0b013e3182130c54
Marks, D. M., Patkar, A. A., Masand, P. S., and Pae, C.-U. (2009). Does pregabalin have neuropsychotropic effects: a short perspective. Psychiatry Investig. 6, 55–58. doi: 10.4306/pi.2009.6.2.55
Martin, B. S., Corbin, J. G., and Huntsman, M. M. (2014). Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J. Neurophysiol. 112, 890–902. doi: 10.1152/jn.00597.2013
Martin, E. R., Menold, M. M., Wolpert, C. M., Bass, M. P., Donnelly, S. L., Ravan, S. A., et al. (2000). Analysis of linkage disequilibrium in gamma-aminobutyric acid receptor subunit genes in autistic disorder. Am. J. Med. Genet. 96, 43–48. doi: 10.1002/(SICI)1096-8628(20000207)96:1<43::AID-AJMG9>3.0.CO;2-3
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S., et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77. doi: 10.1038/ng0197-74
McCoy, D. C., Peet, E. D., Ezzati, M., Danaei, G., Black, M. M., Sudfeld, C. R., et al. (2016). Early childhood developmental status in low- and middle-income countries: national, regional, and global prevalence estimates using predictive modeling. PLoS Med. 13:e1002034. doi: 10.1371/journal.pmed.1002034
McGee, A., Li, G., Lu, Z., and Qiu, S. (2014). Convergent synaptic and circuit substrates underlying autism genetic risks. Front. Biol. 9:137–150. doi: 10.1007/s11515-014-1298-y
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., et al. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121. doi: 10.1152/jn.00826.2007
Meguro-Horike, M., Yasui, D. H., Powell, W., Schroeder, D. I., Oshimura, M., Lasalle, J. M., et al. (2011). Neuron-specific impairment of inter-chromosomal pairing and transcription in a novel model of human 15q-duplication syndrome. Hum. Mol. Genet. 20, 3798–3810. doi: 10.1093/hmg/ddr298
Meloni, I., Bruttini, M., Longo, I., Mari, F., Rizzolio, F., D’Adamo, P., et al. (2000). A mutation in the rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in males. Am. J. Hum. Genet. 67, 982–985. doi: 10.1086/303078
Merner, N. D., Chandler, M. R., Bourassa, C., Liang, B., Khanna, A. R., Dion, P., et al. (2015). Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci. 9:386. doi: 10.3389/fncel.2015.00386
Meyer, G., Varoqueaux, F., Neeb, A., Oschlies, M., and Brose, N. (2004). The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology 47, 724–733. doi: 10.1016/j.neuropharm.2004.06.023
Mircsof, D., Langouët, M., Rio, M., Moutton, S., Siquier-Pernet, K., Bole-Feysot, C., et al. (2015). Mutations in NONO lead to syndromic intellectual disability and inhibitory synaptic defects. Nat. Neurosci. 18, 1731–1736. doi: 10.1038/nn.4169
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157. doi: 10.1038/nrn.2016.183
Mukherjee, J., Kretschmannova, K., Gouzer, G., Maric, H.-M., Ramsden, S., Tretter, V., et al. (2011). The residence time of GABA(A)Rs at inhibitory synapses is determined by direct binding of the receptor α1 subunit to gephyrin. J. Neurosci. 31, 14677–14687. doi: 10.1523/JNEUROSCI.2001-11.2011
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/S0896-6273(00)80809-0
Nakatani, J., Tamada, K., Hatanaka, F., Ise, S., Ohta, H., Inoue, K., et al. (2009). Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246. doi: 10.1016/j.cell.2009.04.024
Neul, J. L., and Zoghbi, H. Y. (2004). Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist 10, 118–128. doi: 10.1177/1073858403260995
Niturad, C. E., Lev, D., Kalscheuer, V. M., Charzewska, A., Schubert, J., Lerman-Sagie, T., et al. (2017). Rare GABRA3 variants are associated with epileptic seizures, encephalopathy and dysmorphic features. Brain J. Neurol. 140, 2879–2894. doi: 10.1093/brain/awx236
Nguyen, Q.-A., Horn, M. E., and Nicoll, R. A. (2016). Distinct roles for extracellular and intracellular domains in neuroligin function at inhibitory synapses. eLife 5:e19236. doi: 10.7554/eLife.19236
Nusser, Z., and Mody, I. (2002). Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 87, 2624–2628. doi: 10.1152/jn.2002.87.5.2624
Nusser, Z., Sieghart, W., Benke, D., Fritschy, J. M., and Somogyi, P. (1996). Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc. Natl. Acad. Sci. U.S.A. 93, 11939–11944. doi: 10.1073/pnas.93.21.11939
Nusser, Z., Sieghart, W., and Somogyi, P. (1998). Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J. Neurosci. 18, 1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998
Nyíri, G., Freund, T. F., and Somogyi, P. (2001). Input-dependent synaptic targeting of alpha(2)-subunit-containing GABA(A) receptors in synapses of hippocampal pyramidal cells of the rat. Eur. J. Neurosci. 13, 428–442. doi: 10.1046/j.1460-9568.2001.01407.x
Oakley, J. C., Kalume, F., Yu, F. H., Scheuer, T., and Catterall, W. A. (2009). Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. U.S.A. 106, 3994–3999. doi: 10.1073/pnas.0813330106
Olmos-Serrano, J. L., Corbin, J. G., and Burns, M. P. (2011). The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 33, 395–403. doi: 10.1159/000332884
Olsen, R. W. (2015). Allosteric ligands and their binding sites define γ-aminobutyric acid (GABA) type A receptor subtypes. Adv. Pharmacol. 73, 167–202. doi: 10.1016/bs.apha.2014.11.005
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Paina, S., Garzotto, D., DeMarchis, S., Marino, M., Moiana, A., Conti, L., et al. (2011). Wnt5a is a transcriptional target of Dlx homeogenes and promotes differentiation of interneuron progenitors in vitro and in vivo. J. Neurosci. 31, 2675–2687. doi: 10.1523/JNEUROSCI.3110-10.2011
Panzanelli, P., Gunn, B. G., Schlatter, M. C., Benke, D., Tyagarajan, S. K., Scheiffele, P., et al. (2011). Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J. Physiol. 589, 4959–4980. doi: 10.1113/jphysiol.2011.216028
Papadopoulos, T., Korte, M., Eulenburg, V., Kubota, H., Retiounskaia, M., Harvey, R. J., et al. (2007). Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 26, 3888–3899. doi: 10.1038/sj.emboj.7601819
Papadopoulos, T., and Soykan, T. (2011). The role of collybistin in gephyrin clustering at inhibitory synapses: facts and open questions. Front. Cell. Neurosci. 5:11. doi: 10.3389/fncel.2011.00011
Parikshak, N. N., Swarup, V., Belgard, T. G., Irimia, M., Ramaswami, G., Gandal, M. J., et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. doi: 10.1038/nature20612
Park, R. J., and Bolton, P. F. (2001). Pervasive developmental disorder and obstetric complications in children and adolescents with tuberous sclerosis. Autism Int. J. Res. Pract. 5, 237–248. doi: 10.1177/1362361301005003002
Pelc, K., Boyd, S. G., Cheron, G., and Dan, B. (2008). Epilepsy in Angelman syndrome. Seizure 17, 211–217. doi: 10.1016/j.seizure.2007.08.004
Peñagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Petilla Interneuron Nomenclature Group, Ascoli, G. A., Alonso-Nanclares, L., Anderson, S. A., Barrionuevo, G., Benavides-Piccione, R., et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568. doi: 10.1038/nrn2402
Phelan, K., and McDermid, H. E. (2012). The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2, 186–201.
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Pizzarelli, R., and Cherubini, E. (2011). Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011:297153. doi: 10.1155/2011/297153
Pleasure, S. J., Anderson, S., Hevner, R., Bagri, A., Marin, O., Lowenstein, D. H., et al. (2000). Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron 28, 727–740. doi: 10.1016/S0896-6273(00)00149-5
Poliak, S., Gollan, L., Martinez, R., Custer, A., Einheber, S., Salzer, J. L., et al. (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. doi: 10.1016/S0896-6273(00)81049-1
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Papadopoulos, T., et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. doi: 10.1016/j.neuron.2009.08.023
Prange, O., Wong, T. P., Gerrow, K., Wang, Y. T., and El-Husseini, A. (2004). A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. U.S.A. 101, 13915–13920. doi: 10.1073/pnas.0405939101
Radyushkin, K., Hammerschmidt, K., Boretius, S., Varoqueaux, F., El-Kordi, A., Ronnenberg, A., et al. (2009). Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 8, 416–425. doi: 10.1111/j.1601-183X.2009.00487.x
Rapin, I., and Katzman, R. (1998). Neurobiology of autism. Ann. Neurol. 43, 7–14. doi: 10.1002/ana.410430106
Reinstein, E., Tzur, S., Cohen, R., Bormans, C., and Behar, D. M. (2016). Intellectual disability and non-compaction cardiomyopathy with a de novo NONO mutation identified by exome sequencing. Eur. J. Hum. Genet. 24, 1635–1638. doi: 10.1038/ejhg.2016.72
Represa, A., and Ben-Ari, Y. (2005). Trophic actions of GABA on neuronal development. Trends Neurosci. 28, 278–283. doi: 10.1016/j.tins.2005.03.010
Rivera, C., Voipio, J., and Kaila, K. (2005). Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 562, 27–36. doi: 10.1113/jphysiol.2004.077495
Rodenas-Cuadrado, P., Pietrafusa, N., Francavilla, T., La Neve, A., Striano, P., and Vernes, S. C. (2016). Characterisation of CASPR2 deficiency disorder–a syndrome involving autism, epilepsy and language impairment. BMC Med. Genet. 17:8. doi: 10.1186/s12881-016-0272-8
Roohi, J., Montagna, C., Tegay, D. H., Palmer, L. E., DeVincent, C., Pomeroy, J. C., et al. (2009). Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 46, 176–182. doi: 10.1136/jmg.2008.057505
Rubenstein, J. L. R., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183X.2003.00037.x
Rudolph, U., and Möhler, H. (2006). GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr. Opin. Pharmacol. 6, 18–23. doi: 10.1016/j.coph.2005.10.003
Rudy, B., Fishell, G., Lee, S., and Hjerling-Leffler, J. (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61. doi: 10.1002/dneu.20853
Saiepour, L., Fuchs, C., Patrizi, A., Sassoè-Pognetto, M., Harvey, R. J., and Harvey, K. (2010). Complex role of collybistin and gephyrin in GABAA receptor clustering. J. Biol. Chem. 285, 29623–29631. doi: 10.1074/jbc.M110.121368
Sala, C., Piëch, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/S0896-6273(01)00339-7
Sala, C., Roussignol, G., Meldolesi, J., and Fagni, L. (2005). Key role of the postsynaptic density scaffold proteins Shank and Homer in the functional architecture of Ca2+ homeostasis at dendritic spines in hippocampal neurons. J. Neurosci. 25, 4587–4592. doi: 10.1523/JNEUROSCI.4822-04.2005
Sala, C., Vicidomini, C., Bigi, I., Mossa, A., and Verpelli, C. (2015). Shank synaptic scaffold proteins: keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 135, 849–858. doi: 10.1111/jnc.13232
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Scheiffele, P., Fan, J., Choih, J., Fetter, R., and Serafini, T. (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. doi: 10.1016/S0092-8674(00)80877-6
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Selmer, K. K., Lund, C., Brandal, K., Undlien, D. E., and Brodtkorb, E. (2009). SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy Behav. 16, 555–557. doi: 10.1016/j.yebeh.2009.08.021
Sesarini, C. V., Costa, L., Grañana, N., Coto, M. G., Pallia, R. C., and Argibay, P. F. (2015). Association between GABA(A) receptor subunit polymorphisms and autism spectrum disorder (ASD). Psychiatry Res. 229, 580–582. doi: 10.1016/j.psychres.2015.07.077
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/S0896-6273(02)00768-7
Sheng, M., and Kim, E. (2000). The Shank family of scaffold proteins. J. Cell Sci. 113(Pt 11), 1851–1856.
Shimojima, K., Sugawara, M., Shichiji, M., Mukaida, S., Takayama, R., Imai, K., et al. (2011). Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J. Hum. Genet. 56, 561–565. doi: 10.1038/jhg.2011.58
Shmuely, S., Sisodiya, S. M., Gunning, W. B., Sander, J. W., and Thijs, R. D. (2016). Mortality in Dravet syndrome: a review. Epilepsy Behav. 64, 69–74. doi: 10.1016/j.yebeh.2016.09.007
Sigel, E., and Steinmann, M. E. (2012). Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 287, 40224–40231. doi: 10.1074/jbc.R112.386664
Soghomonian, J.-J., Zhang, K., Reprakash, S., and Blatt, G. J. (2017). Decreased parvalbumin mRNA levels in cerebellar Purkinje cells in autism. Autism Res. 10, 1787–1796. doi: 10.1002/aur.1835
Song, J. Y., Ichtchenko, K., Südhof, T. C., and Brose, N. (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. U.S.A. 96, 1100–1105. doi: 10.1073/pnas.96.3.1100
Srivastava, S., Cohen, J., Pevsner, J., Aradhya, S., McKnight, D., Butler, E., et al. (2014). A novel variant in GABRB2 associated with intellectual disability and epilepsy. Am. J. Med. Genet. A 164, 2914–2921. doi: 10.1002/ajmg.a.36714
Stern, W. M., Sander, J. W., Rothwell, J. C., and Sisodiya, S. M. (2017). Impaired intracortical inhibition demonstrated in vivo in people with Dravet syndrome. Neurology 88, 1659–1665. doi: 10.1212/WNL.0000000000003868
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M., et al. (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 354, 1370–1377. doi: 10.1056/NEJMoa052773
Sutcliffe, J. S., Jiang, Y. H., Galijaard, R. J., Matsuura, T., Fang, P., Kubota, T., et al. (1997). The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 7, 368–377. doi: 10.1101/gr.7.4.368
Sze, P. Y. (1979). L-Glutamate decarboxylase. Adv. Exp. Med. Biol. 123, 59–78. doi: 10.1007/978-1-4899-5199-1_4
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Tabuchi, K., and Südhof, T. C. (2002). Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics 79, 849–859. doi: 10.1006/geno.2002.6780
Tai, C., Abe, Y., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, E3139–E3148. doi: 10.1073/pnas.1411131111
Tanaka, M., DeLorey, T. M., Delgado-Escueta, A., and Olsen, R. W. (2012). “GABRB3, epilepsy, and neurodevelopment,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information). Available at: http://www.ncbi.nlm.nih.gov/books/NBK98178/
Tang, X., Kim, J., Zhou, L., Wengert, E., Zhang, L., Wu, Z., et al. (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 751–756. doi: 10.1073/pnas.1524013113
Tarquinio, D. C., Hou, W., Berg, A., Kaufmann, W. E., Lane, J. B., Skinner, S. A., et al. (2017). Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain 140, 306–318. doi: 10.1093/brain/aww302
Tick, B., Bolton, P., Happé, F., Rutter, M., and Rijsdijk, F. (2016). Heritability of autism spectrum disorders: a meta-analysis of twin studies. J. Child Psychol. Psychiatry 57, 585–595. doi: 10.1111/jcpp.12499
Tijssen, M. A., and Rees, M. I. (1993). “Hyperekplexia,” in GeneReviews(®), eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. Bean, et al. (Seattle, WA: University of Washington). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1260/
Ting, J. T., Peça, J., and Feng, G. (2012). Functional consequences of mutations in postsynaptic scaffolding proteins and relevance to psychiatric disorders. Annu. Rev. Neurosci. 35, 49–71. doi: 10.1146/annurev-neuro-062111-150442
Tretter, V., Jacob, T. C., Mukherjee, J., Fritschy, J.-M., Pangalos, M. N., and Moss, S. J. (2008). The clustering of GABA(A) receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J. Neurosci. 28, 1356–1365. doi: 10.1523/JNEUROSCI.5050-07.2008
Tretter, V., Kerschner, B., Milenkovic, I., Ramsden, S. L., Ramerstorfer, J., Saiepour, L., et al. (2011). Molecular basis of the γ-aminobutyric acid A receptor α3 subunit interaction with the clustering protein gephyrin. J. Biol. Chem. 286, 37702–37711. doi: 10.1074/jbc.M111.291336
Urraca, N., Cleary, J., Brewer, V., Pivnick, E. K., McVicar, K., Thibert, R. L., et al. (2013). The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 6, 268–279. doi: 10.1002/aur.1284
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., et al. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. doi: 10.1016/j.neuron.2006.09.003
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 83, 449–456. doi: 10.1078/0171-9335-00410
Verdoorn, T. A., Draguhn, A., Ymer, S., Seeburg, P. H., and Sakmann, B. (1990). Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron 4, 919–928. doi: 10.1016/0896-6273(90)90145-6
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359, 2337–2345. doi: 10.1056/NEJMoa0802828
Verpelli, C., Dvoretskova, E., Vicidomini, C., Rossi, F., Chiappalone, M., Schoen, M., et al. (2011). Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 286, 34839–34850. doi: 10.1074/jbc.M111.258384
Vien, T. N., Modgil, A., Abramian, A. M., Jurd, R., Walker, J., Brandon, N. J., et al. (2015). Compromising the phosphodependent regulation of the GABAAR β3 subunit reproduces the core phenotypes of autism spectrum disorders. Proc. Natl. Acad. Sci. U.S.A. 112, 14805–14810. doi: 10.1073/pnas.1514657112
Vignoli, A., Savini, M. N., Nowbut, M. S., Peron, A., Turner, K., La Briola, F., et al. (2017). Effectiveness and tolerability of antiepileptic drugs in 104 girls with Rett syndrome. Epilepsy Behav. 66, 27–33. doi: 10.1016/j.yebeh.2016.10.006
Vogt, D., Cho, K. K. A., Shelton, S. M., Paul, A., Huang, Z. J., Sohal, V. S., et al. (2017). Mouse Cntnap2 and human CNTNAP2 ASD alleles cell autonomously regulate PV+ cortical interneurons. Cereb. Cortex doi: 10.1093/cercor/bhx248 [Epub ahead of print].
Wallace, M. L., Burette, A. C., Weinberg, R. J., and Philpot, B. D. (2012). Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74, 793–800. doi: 10.1016/j.neuron.2012.03.036
Wallace, R. H., Marini, C., Petrou, S., Harkin, L. A., Bowser, D. N., Panchal, R. G., et al. (2001). Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 28, 49–52. doi: 10.1038/ng0501-49
Watson, C. M., Crinnion, L. A., Tzika, A., Mills, A., Coates, A., Pendlebury, M., et al. (2014). Diagnostic whole genome sequencing and split-read mapping for nucleotide resolution breakpoint identification in CNTNAP2 deficiency syndrome. Am. J. Med. Genet. A 164A, 2649–2655. doi: 10.1002/ajmg.a.36679
Wefelmeyer, W., Cattaert, D., and Burrone, J. (2015). Activity-dependent mismatch between axo-axonic synapses and the axon initial segment controls neuronal output. Proc. Natl. Acad. Sci. U.S.A. 112, 9757–9762. doi: 10.1073/pnas.1502902112
Whiting, P. J., McKernan, R. M., and Wafford, K. A. (1995). Structure and pharmacology of vertebrate GABAA receptor subtypes. Int. Rev. Neurobiol. 38, 95–138. doi: 10.1016/S0074-7742(08)60525-5
Wimmer, V. C., Reid, C. A., So, E. Y.-W., Berkovic, S. F., and Petrou, S. (2010). Axon initial segment dysfunction in epilepsy. J. Physiol. 588, 1829–1840. doi: 10.1113/jphysiol.2010.188417
Wöhr, M., Orduz, D., Gregory, P., Moreno, H., Khan, U., Vörckel, K. J., et al. (2015). Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 5:e525. doi: 10.1038/tp.2015.19
Wu, Y. W., Sullivan, J., McDaniel, S. S., Meisler, M. H., Walsh, E. M., Li, S. X., et al. (2015). Incidence of dravet syndrome in a US population. Pediatrics 136, e1310–e1315. doi: 10.1542/peds.2015-1807
Yamagata, T., Ogiwara, I., Mazaki, E., Yanagawa, Y., and Yamakawa, K. (2017). Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: Mutually exclusive distributions of Nav1.1 and Nav1.2. Biochem. Biophys. Res. Commun. 491, 1070–1076. doi: 10.1016/j.bbrc.2017.08.013
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Zhang, C., Atasoy, D., Araç, D., Yang, X., Fucillo, M. V., Robison, A. J., et al. (2010). Neurexins physically and functionally interact with GABA(A) receptors. Neuron 66, 403–416. doi: 10.1016/j.neuron.2010.04.008
Zhang, L., He, J., Jugloff, D. G. M., and Eubanks, J. H. (2008). The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 18, 294–309. doi: 10.1002/hipo.20389
Zhong, W., Cui, N., Jin, X., Oginsky, M. F., Wu, Y., Zhang, S., et al. (2015). Methyl CpG binding protein 2 gene disruption augments tonic currents of γ-aminobutyric acid receptors in locus coeruleus neurons: impact on neuronal excitability and breathing. J. Biol. Chem. 290, 18400–18411. doi: 10.1074/jbc.M115.650465
Keywords: neurodevelopment, GABA A receptor, autism spectrum disorders, Rett Syndrome (RTT), Angelman Syndrome, Dravet Syndrome, seizures, phasic and tonic inhibition
Citation: Ali Rodriguez R, Joya C and Hines RM (2018) Common Ribs of Inhibitory Synaptic Dysfunction in the Umbrella of Neurodevelopmental Disorders. Front. Mol. Neurosci. 11:132. doi: 10.3389/fnmol.2018.00132
Received: 11 January 2018; Accepted: 03 April 2018;
Published: 24 April 2018.
Edited by:
Hansen Wang, University of Toronto, CanadaReviewed by:
Audrey Christine Brumback, University of Texas at Austin, United StatesCopyright © 2018 Ali Rodriguez, Joya and Hines. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rochelle M. Hines, cm9jaGVsbGUuaGluZXNAdW5sdi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.