 Steffen Vogelgesang1,2
Steffen Vogelgesang1,2 Marcus Niebert
Marcus Niebert Swen Hülsmann
Swen Hülsmann
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 20 February 2018
Sec. Neuroplasticity and Development
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00028
This article is part of the Research Topic Synaptic Diseases: From Biology to Potential Therapy View all 32 articles
Mutations in the transcription factor methyl-CpG-binding protein 2 (MeCP2) cause the neurodevelopmental disorder Rett syndrome (RTT). Besides many other neurological problems, RTT patients show irregular breathing with recurrent apneas or breath-holdings. MeCP2-deficient mice, which recapitulate this breathing phenotype, show a dysregulated, persistent expression of G-protein-coupled serotonin receptor 5-ht5b (Htr5b) in the brainstem. To investigate whether the persistence of 5-ht5b expression is contributing to the respiratory phenotype, we crossbred MeCP2-deficient mice with 5-ht5b-deficient mice to generate double knockout mice (Mecp2−/y;Htr5b−/−). To compare respiration between wild type (WT), Mecp2−/y and Mecp2−/y;Htr5b−/− mice, we used unrestrained whole-body plethysmography. While the breathing of MeCP2-deficient male mice (Mecp2−/y) at postnatal day 40 is characterized by a slow breathing rate and the occurrence of prolonged respiratory pauses, we found that in MeCP2-deficient mice, which also lacked the 5-ht5b receptor, the breathing rate and the number of pauses were indistinguishable from WT mice. To test for a potential mechanism, we also analyzed if the known coupling of 5-ht5b receptors to Gi proteins is altering second messenger signaling. Tissue cAMP levels in the medulla of Mecp2−/y mice were decreased as compared to WT mice. In contrast, cAMP levels in Mecp2−/y;Htr5b−/− mice were indistinguishable from WT mice. Taken together, our data points towards a role of 5-ht5b receptors within the complex breathing phenotype of MeCP2-deficient mice.
The neurodevelopmental Rett syndrome (RTT) occurs primarily in females with an incidence of 1:10,000 live births and presents as a delayed regression after 6–18 months of apparently normal development (Rett, 1966; Julu et al., 2001). The disturbances typically start with early autonomic dysfunctions, including breathing abnormalities that are considered a potential cause of sudden death (Kerr et al., 1997). The progressed phenotype manifests with neurological symptoms such as stereotypic hand movements, seizures and mental retardation with loss of language skills. RTT is caused by nonsense, missense or frameshift mutations, as well as large deletions of the human X-chromosomal gene methyl-CpG-binding protein 2 (MECP2; Amir et al., 1999; Philippe et al., 2006), which encodes the transcription factor MeCP2. MeCP2 acts in a DNA-methylation-dependent manner by repressing or activating gene transcription (Lewis et al., 1992; Nan et al., 1997; Chahrour et al., 2008). MeCP2 is considered to be important during brain development, it accumulates most abundantly in post-mitotic adult neurons, where it is thought to be indispensable for maturation and synaptogenesis (Kishi and Macklis, 2005; Guy et al., 2011).
Different types of breathing abnormalities are associated with defects of MeCP2. Female RTT patients show periods of hyperventilation alternating with prolonged periods of breath-holdings (Kerr and Julu, 1999; Julu et al., 2001). In contrast, male patients rather show hypoventilation, apneas and respiratory insufficiency soon after birth (Geerdink et al., 2002; Kankirawatana et al., 2006; Schüle et al., 2008). The male Mecp2−/y null mice, which are the original model to analyze the loss of MeCP2 in neurons, have a characteristic impairment of breathing (Guy et al., 2001), which manifests as hypoventilation, with a reduced respiratory rate and minute ventilation together with a high number of apneas (Viemari et al., 2005; Chao et al., 2010; Wegener et al., 2014). Currently, the ultimate cause of altered respiratory behavior is not yet known and might very well be different in male and female subjects. However, it is clear that breathing of MeCP2 deficient mice is influenced by many factors that include neurotransmitter systems like norepinephrine (Viemari et al., 2005) or neurotrophic factors like BDNF (Li and Pozzo-Miller, 2014) as well as cellular systems like glia cells (Lioy et al., 2011; Delépine et al., 2015) and inhibitory neurons (Hülsmann et al., 2016).
When we previously investigated the serotonergic system, we found that the serotonin receptor 5-ht5b is heavily dysregulated in the brainstem of Mecp2−/y mice (Vogelgesang et al., 2017). Rodents have been shown to possess two functional 5-ht5 receptor subtypes, 5-ht5a (Plassat et al., 1992) and 5-ht5b (Matthes et al., 1993). While their physiological role is unknown, both are expressed at low levels in several brain regions and appear to be restricted to neural tissue (Rees et al., 1994). So far, we know that 5-ht5b is expressed as both a full length as well as a truncated protein that is retained in the endosomal compartment. Yet, it is still able to interact with proteins (Vogelgesang et al., 2017), and therefore can potentially alter second messenger signaling and cAMP levels.
In wild type (WT) mice, the expression of 5-ht5b receptor gradually increases during early development, peaks around postnatal day 21 (P21) and is then down-regulated. In Mecp2−/y mice, however, 5-ht5b expression remains elevated past P21 (Vogelgesang et al., 2017). This developmental difference coincides with the appearance of the respiratory phenotype, which develops between P20 and P40 (Viemari et al., 2005; Mesuret et al., 2018).
To investigate the functional role of the persistent expression of 5-ht5b receptors in vivo we generated MeCP2-deficient mice that also lack the 5-ht5b receptor (Mecp2−/y;Htr5b−/− mice). Since our goal was to investigate the role of 5-ht5b, we chose to limit our analysis to male Mecp2−/y;Htr5b−/− mice, because the breathing phenotype of female mice is less predictable due to the variable x-chromosomal inactivation (Johnson et al., 2015). Moreover, female mice remain asymptomatic often for more than a year (Guy et al., 2001; Wegener et al., 2014). Using only male mice allowed us not only to analyze the effect of the 5-ht5b receptors on breathing but also to measure their impact on the cellular cAMP level in the medulla.
The experimental procedures were performed in accordance with European Community (EU Directive 2010/63/EU for animal experiments) and National Institutes of Health guidelines for the care and use of laboratory animals. In accordance with the German Protection of Animals Act (TierSchG §4 Abs. 3) procedures were approved by the Animal Welfare Office of University Medical Center Gottingen (file number ID T12/18).
The knockout mouse model for RTT, strain B6.129P2(C)-Mecp2tm1-1Bird (Guy et al., 2001; maintained on a C57BL/6J background) was purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Hemizygous mutant Mecp2−/y males were generated by crossing heterozygous Mecp2+/− females with C57BL/6J wild-type males. The genotyping was performed in accordance with The Jackson Laboratory genotyping protocols1.
The knockout mouse model for the 5-ht5b receptor, strain 129SvEvBrd (maintained on a 29/SvEv-C57BL/6 background) was obtained from Taconic Europe A/S (Tornbjergvej 40, Ejby, 4623 Lille Skensved, Denmark). Htr5b knockout mice were backcrossed into the C57BL/6 background for at least eight generations. Mecp2−/y;Htr5b−/−-double-knockout mice were then generated by crossbreeding female Mecp2+/−;Htr5b+/− mice with male Mecp2+/y;Htr5b−/− mice.
Tissue samples were incubated in 25 mM NaOH/0.2 mM EDTA for 3 h at 65°C. After neutralization with an equal volume of 40 mM Tris/HCl pH 5.5, 1 μL was taken as a template for subsequent PCR. The primers used for the identification of Htr5b-genotype were: WT-for (5′-ctctgcagtcggtttgatg-3′), WT-rev (5′-gtagagtcaccacaagcac-3′), KO-for (5′-gcagcgcatcgccttctatc-3′), KO-rev (5′-gtgctgggattagaagtcc-3′). The primers used for the identification of the Mecp2 genotype were: WT-for (5′-gaccccttgggactgaagtt-3′), KO-for (5′-ccatgcgataagcttgatga-3′) and WT-KO-rev (5′-ccaccctccagtttggttta-3′).
Ventilation was measured by unrestrained whole-body-plethysmography (Drorbaugh and Fenn, 1955; Bartlett and Tenney, 1970) in 40-day old mice (P40). Mice were placed in a custom-made acrylic glass chamber (300 ml) that was connected to a differential low-pressure transducer (model DP1 03, Validyne Engineering, Northridge, CA, USA). The second channel of the pressure transducer was connected to a reference chamber (300 ml). The signal from the pressure transducer was fed into a sine wave carrier demodulator (CD-15, Validyne Engineering). Animals could explore the chamber freely. For the analysis, pressure changes were band-pass filtered (1.5–500 Hz) and amplified (four times) before storing on an Apple-PC computer. For digitization (1 kHz sampling rate) an ITC-16 interface (InstruTECH/HEKA, Lambrecht) was used that was controlled by Axograph 4.8 software (Axon Instruments, Foster City, CA, USA). A bias flow of 150 ml/min was introduced using a Normocap® CO2-sensor (Datex, Instrumentarium Oy, Helsinki, Finland). Pressure changes were exported and converted to axon binary files and used for analysis. Breaths (Inspiratory flow peaks) from a period of 3 min after 12 min adaptation to the chamber were analyzed by the threshold search peak detection method of Axon Clampfit (Molecular Devices, Sunnyvale, CA, USA). We did not discriminate between respiratory cycles associated to different types of behavior e.g., sniffing. Breathing frequencies were calculated as the reciprocal of the averaged inspiratory peak interval. The number of intervals between inspiratory peaks that were longer than 1 s was determined during the 3 min as a parameter for central apneas (Stettner et al., 2008). To define the regularity of breathing, the coefficient of variation was calculated for the interval (CV = STD/mean) as well as an irregularity score (IS) was calculated: IS = 100*ABS[(Intn − Intn-1)/Intn-1] for each respiratory cycle (Barthe and Clarac, 1997; Telgkamp et al., 2002).
The cAMP concentration of murine tissue was determined using commercially available ELISA-based DetectX-kit (Arbor Assays) and performed according to manufacturer’s instructions. Absorbance was measured at 450 nm using Infinite 200 Pro reader (TECAN). Whole brains were explanted from 40 days old mice, separated and individual regions snap-frozen in liquid nitrogen.
The cAMP measurements and plethysmography were analyzed with non-parametric Kruskal-Wallis and subsequent Dunn’s post hoc test for multiple comparisons. The survival plots were analyzed using the log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests. Analyses were performed using GraphPad Prism version 5.0d for Mac OSX (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant at P < 0.05. Data are presented as the mean ± standard error of the mean (SEM; n = number of experiments).
Mecp2−/y;Htr5b−/− mice were generated by crossbreeding female Mecp2+/−; Htr5b+/− mice with male Mecp2+/y; Htr5b−/− mice. To our knowledge, Htr5b−/− mice have no observable phenotype (see www.taconic.com and Mouse Genome Database, http://www.informatics.jax.org/marker/MGI:96284). In our hands, the life span of Htr5b−/− mice was normal and body weight at P40 (19.06 ± 1.12 g) was indistinguishable from WT mice (20.60 ± 0.56 g) of the same age. Unrestrained whole-body plethysmography at postnatal day 40 (P40) revealed that Htr5b−/− mice were breathing normal, which is in line with the notion that 5-ht5b-receptors are downregulated and thus dispensable at this age (Figure 1A). Like WT mice (n = 9), Htr5b−/− mice (n = 7) did not show long (longer than 1 s) breathing arrests (Figure 1B), which are typical for Mecp2−/y, and both, breathing rate (WT 6.68 s−1 ± 0.41 vs. Htr5b−/− 6.69 s−1 ± 0.36; n.s.; Figure 1C) and IS (WT 0.23 ± 0.03 vs. Htr5b−/− 0.28 ± 0.02; n.s.; Figure 1D) were indistinguishable from WT.
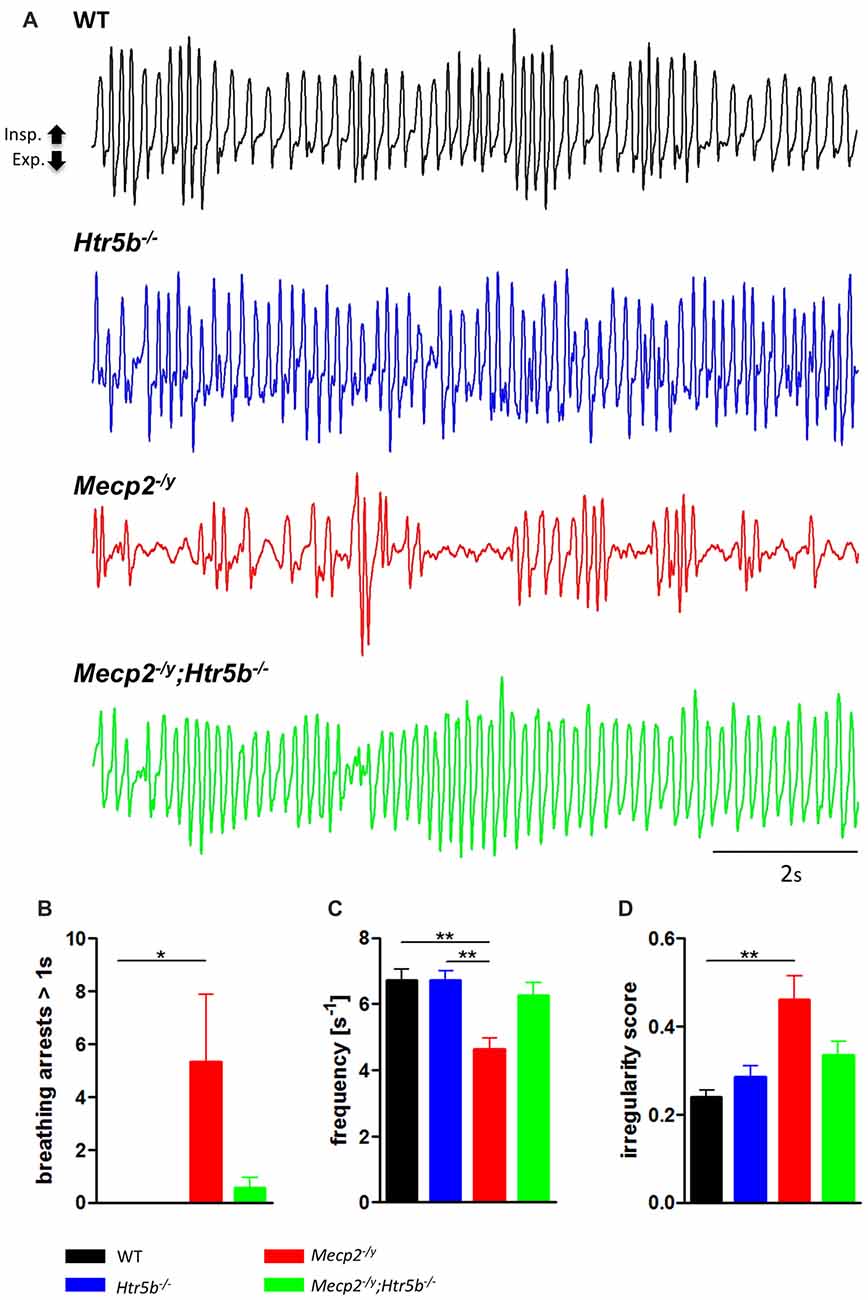
Figure 1. Plethysmographic in vivo recordings. (A) Plethysmographic in vivo recordings of wild type (WT; n = 9), Htr5b−/− (n = 7), Mecp2−/y (n = 10) and Mecp2−/y;Htr5b−/− (n = 11) mice at postnatal day 40 (P40). Recordings revealed improved breathing in Mecp2−/y;Htr5b−/− mice, which was expressed by the (B) reduction of long (>1 s) apneas, (C) respiratory frequency and (D) irregularity score (IS). Corresponding bar diagrams represent the mean value and standard error of the mean (SEM). Asterisks indicate significance (*P < 0.05, **P < 0.01; Kruskal-Wallis test with Dunn’s multiple comparisons).
When comparing breathing of Mecp2−/y;Htr5b−/− mice (n = 11) with Mecp2−/y mice (n = 10), we found significantly improved breathing parameters, yet no full rescue. Unrestrained whole-body plethysmography at P40 revealed a slow irregular breathing rate in Mecp2−/y mice with a high number of breathing arrests (5.3 ± 2.6 per 180 s, Figure 1B), low respiratory rate (4.6 ± 0.36 s−1; Figure 1C) and high IS (0.46 ± 0.06; Figure 1D). However, the breathing rhythm of Mecp2−/y;Htr5b−/− mice was more stable with intermediate values between Htr5b−/− (or WT) and Mecp2−/y mice. Mecp2−/y;Htr5b−/− mice showed a significantly lower number of breathing arrests longer than 1 s (0.5 ± 0.4 per 180 s; Figure 1B), and a higher respiratory rate (6.23 ± 0.65 s−1; Figure 1C), and the IS (0.33 ± 0.03; Figure 1D) was indistinguishable from WT and Htr5b−/− mice. Moreover, body weight of Mecp2−/y;Htr5b−/− mice (17.52 ± 0.75 g) was higher than age-matched Mecp2−/y mice (12.31 g ± 0.8; p < 0.001) but did not reach the level of WT and Htr5b−/− mice. The life span of Mecp2−/y;Htr5b−/− mice was improved compared to Mecp2−/y mice (median 80 vs. 40 days), however no Mecp2−/y;Htr5b−/− survived longer than 130 days (Figure 2).
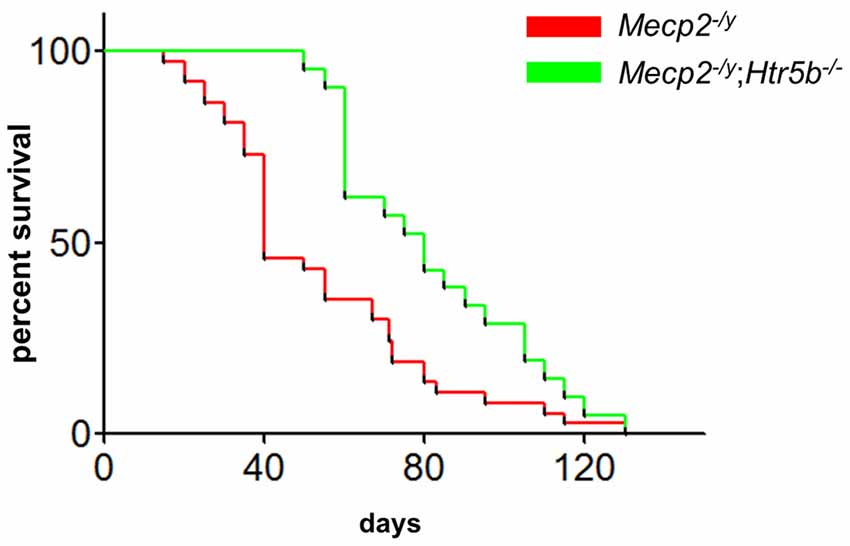
Figure 2. Mecp2−/y;Htr5b−/− mice show improved survival. The survival plot of Mecp2−/y (red; n = 37) vs. Mecp2−/y;Htr5b−/− (green; n = 21) mice revealed a significantly increased median lifespan of 40–80 days, respectively (p = 0.0058 (Mantel-Cox) or p < 0.001 (Gehan-Breslow-Wilcoxon). WT and Htr5b−/− mice showed no lethality in the timeframe indicated and were omitted for clarity.
Since 5-ht5b receptors are highly expressed in the brainstem of MeCP2-deficient mice at P40 (Vogelgesang et al., 2017) and 5-ht5b receptors are able to reduce cellular cAMP levels in vitro (Niebert et al., 2017), we measured the cAMP concentration in medullary brainstem lysates. The cAMP concentration of Mecp2−/y mice at P40 was significantly lower (73.49 ± 7.27%) when compared to WT mice (Figure 3), whose cAMP levels were set as 100%. This reduction is in line with previous reports of low cAMP in the brainstem of Mecp2−/y mice (Mironov et al., 2011). In agreement with the hypothesis of constitutive 5-ht5b-receptor signaling we found normal cellular cAMP in Mecp2−/y; Htr5b−/− mice (98.46 ± 6.34%; Figure 3). These data are in line with the concept that the constitutive activity of 5-ht5b is impairing cAMP signaling in Mecp2−/y mice (Vogelgesang et al., 2017).
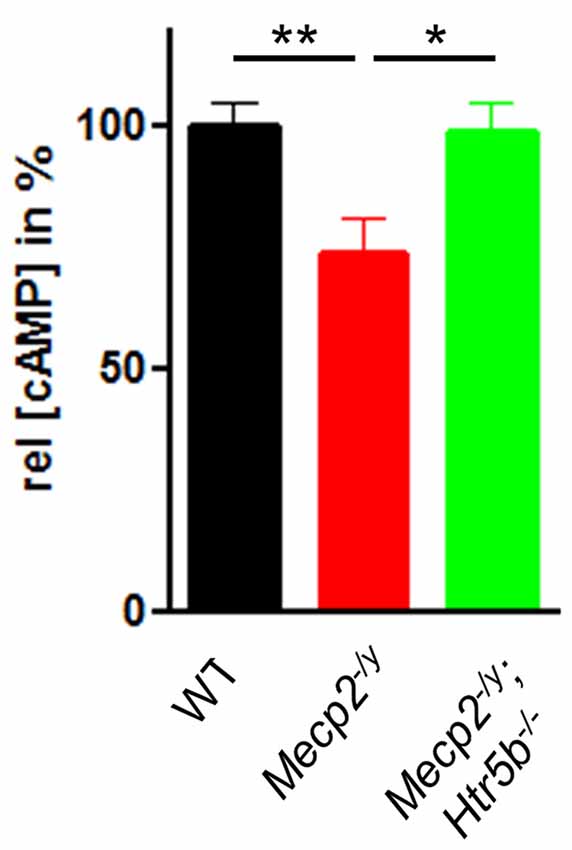
Figure 3. Effect of Htr5b expression on cAMP levels. cAMP concentration in brainstem tissue of WT (n = 5), Mecp2−/y (n = 5) and Mecp2−/y;Htr5b−/− (n = 5) mice. The bar diagram illustrates the relative cAMP concentration ([cAMP]) in brainstem tissue at P40 of Mecp2−/y knockout mice and Mecp2−/y;Htr5b−/− double knockout mice in comparison to WT mice at P40. Asterisks indicate significance (*P < 0.05, **P < 0.01; Kruskal-Wallis test with Dunn’s multiple comparisons).
The Mecp2−/y mouse model of RTT shows a prominent respiratory phenotype that develops relatively late during postnatal development. At P40, breathing is more irregular and MeCP2-deficient mice show reduced respiratory rate and an increased number of longer breathing arrests (Janc et al., 2016). At this age, 5-ht5b receptor expression in the medulla of Mecp2−/y mice is still elevated while it drops to neonatal level in the WT littermates between P21 and P40 (Vogelgesang et al., 2017). In the present study, we tried to test for a pathophysiological link between the persistence of the 5-ht5b receptor and the breathing disturbances using mouse genetics.
5-ht5b receptors are not targeted to the plasma membrane but to the endosomal compartment, where they can participate in signaling by binding to Gi proteins (Vogelgesang et al., 2017) and lowering cAMP levels (Niebert et al., 2017). Lower cAMP-levels in Mecp2−/y mice provide a simplistic rationale for the clinical use of cAMP-elevators like theophylline, as the “first choice for respiratory stimulation” in Rett patients (Julu et al., 2008). In addition, the resulting imbalance of cAMP-dependent second messenger cascades is likely to affect not only the serotonergic system (Manzke et al., 2003) but other neurotransmitters that have been implicated in the modulation of the respiratory network (Fujii et al., 2004; Lalley, 2008; Viemari, 2008; Mellios et al., 2014). In this respect, other effects on cAMP may seem contradictory: application of 5-HT1A receptor agonists, which reduces cAMP, has been reported to have positive effects on respiration in a Rett mouse model (Abdala et al., 2010, 2014) and patients (Andaku et al., 2005; Ohno et al., 2016). However, interactions between different neuromodulator pathways are manifold and even two Gi-mediated pathways can be antagonistic if restricted to different cellular compartments or located in distinct types of neurons (Manzke et al., 2010). Additionally, 5-HT1A receptors can modulate neuronal activity independently from cAMP, e.g., by modulating potassium currents (Penington et al., 1993). Although we found a net reduction of cAMP in the medulla, we cannot rule out an additional effect of the 5-ht5b dependent reduction of 5-HT1A receptor surface expression (Niebert et al., 2017). However, this mechanism appears rather unlikely to be of major importance, since 5-HT1A knockout mice have only a mild respiratory phenotype (Barrett et al., 2012). Moreover, 5-HT1A receptor agonists have diverse effects including an anxiolytic action that, in the light of a potential role of the anxiety level of Mecp2−/y mice (Ren et al., 2012), might influence respiratory rhythm without directly influencing respiratory neurons.
The fact that the improvement of breathing does not completely rescue the life expectancy of the mice is, although disappointing, in line with earlier observations, as for the restoration of MeCP2 in inhibitory neurons (Hülsmann et al., 2016). In Mecp2−/y;Htr5b−/− double-knockout mice, body weight was still reduced compared to WT and Htr5b−/− mice and, although increased, the lifespan of Mecp2−/y;Htr5b−/− double-knockout mice was still significantly shorter than the lifespan of WT mice. Since we did not find a dysregulation of the 5-ht5b-receptor expression in the hippocampus (Vogelgesang et al., 2017), any hippocampal pathology, which is e.g., involved in the increased propensity to seizures (Boison, 2012), is not cured in Mecp2−/y;Htr5b−/− mice and, thus, can manifest later during the development of Mecp2−/y mice leading to still premature death.
Although the persistent expression of 5-ht5b contributes to the respiratory phenotype in MeCP2-deficient mice, these findings cannot be immediately translated to human patients. Unlike in mice (Grailhe et al., 2001; Maekawa et al., 2010; Vogelgesang et al., 2017), it is accepted that stop codons in the human HTR5B gene prevent its expression (Grailhe et al., 2001). However, we found that 5-ht5b in mice is also expressed as a truncated protein (Vogelgesang et al., 2017). As nothing is known about the expression and potential splice variants of 5-ht5b in humans, further analysis is required to identify potential alterations of 5-ht5b receptor expression in the brainstem of patients with MECP2 mutations.
MeCP2 deficiency affects several 100 targets (Ben-Shachar et al., 2009) so any effect of 5-ht5b-receptor and cAMP signaling must be seen in the context of multiple other dysregulated genes. Our data supports the notion that 5-ht5b-receptor dysregulation is an important but probably not the only factor that contributes to respiratory problems in Mecp2−/y mice. However, currently no data is available indicating a relevance of the 5-ht5b receptor expression in the pathology of human RTT patients.
SV, AMB, TM and SH performed the experiments. TM, AMB, MN and SH analyzed the data. MN and SH wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Cluster of Excellence and Deutsche Forschungsgemeinschaft (DFG) Research Center Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB). We acknowledge support by the Open Access Publication Funds of the Göttingen University. We are grateful to Anja-Annett Grützner for technical assistance. We are especially grateful to our mentor D.W. Richter for long and fruitful discussions that helped to improve the manuscript.
Abdala, A. P. L., Dutschmann, M., Bissonnette, J. M., and Paton, J. F. R. (2010). Correction of respiratory disorders in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U S A 107, 18208–18213. doi: 10.1073/pnas.1012104107
Abdala, A. P., Lioy, D. T., Garg, S. K., Knopp, S. J., Paton, J. F. R., and Bissonnette, J. M. (2014). Effect of Sarizotan, a 5-HT1a and D2-like receptor agonist, on respiration in three mouse models of Rett syndrome. Am. J. Respir. Cell Mol. Biol. 50, 1031–1039. doi: 10.1165/rcmb.2013-0372OC
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Andaku, D. K., Mercadante, M. T., and Schwartzman, J. S. (2005). Buspirone in Rett syndrome respiratory dysfunction. Brain Dev. 27, 437–438. doi: 10.1016/j.braindev.2004.09.011
Barrett, K. T., Kinney, H. C., Li, A., Daubenspeck, J. A., Leiter, J. C., and Nattie, E. E. (2012). Subtle alterations in breathing and heart rate control in the 5-HT1A receptor knockout mouse in early postnatal development. J. Appl. Physiol. 113, 1585–1593. doi: 10.1152/japplphysiol.00939.2012
Barthe, J. Y., and Clarac, F. (1997). Modulation of the spinal network for locomotion by substance P in the neonatal rat. Exp. Brain Res. 115, 485–492. doi: 10.1007/pl00005718
Bartlett, D. J., and Tenney, S. M. (1970). Control of breathing in experimental anemia. Respir. Physiol. 10, 384–395. doi: 10.1016/0034-5687(70)90056-3
Ben-Shachar, S., Chahrour, M., Thaller, C., Shaw, C. A., and Zoghbi, H. Y. (2009). Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442. doi: 10.1093/hmg/ddp181
Boison, D. (2012). Is intrinsic hyperexcitability in CA3 the culprit for seizures in Rett syndrome? Epilepsy Curr. 12, 13–14. doi: 10.5698/1535-7511-12.1.13
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T. C., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chao, H.-T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Delépine, C., Nectoux, J., Letourneur, F., Baud, V., Chelly, J., Billuart, P., et al. (2015). Astrocyte transcriptome from the Mecp2308-truncated mouse model of Rett syndrome. Neuromolecular Med. 17, 353–363. doi: 10.1007/s12017-015-8363-9
Drorbaugh, J. E., and Fenn, W. O. (1955). A barometric method for measuring ventilation in newborn infants. Pediatrics 16, 81–87.
Fujii, M., Umezawa, K., and Arata, A. (2004). Dopaminergic modulation on respiratory rhythm in rat brainstem-spinal cord preparation. Neurosci. Res. 50, 355–359. doi: 10.1016/j.neures.2004.07.009
Geerdink, N., Rotteveel, J. J., Lammens, M., Sistermans, E. A., Heikens, G. T., Gabreëls, F. J. M., et al. (2002). MECP2 mutation in a boy with severe neonatal encephalopathy: clinical, neuropathological and molecular findings. Neuropediatrics 33, 33–36. doi: 10.1055/s-2002-23598
Grailhe, R., Grabtree, G. W., and Hen, R. (2001). Human 5-HT5 receptors: the 5-HT5A receptor is functional but the 5-HT5B receptor was lost during mammalian evolution. Eur. J. Pharmacol. 418, 157–167. doi: 10.1016/s0014-2999(01)00933-5
Guy, J., Cheval, H., Selfridge, J., and Bird, A. (2011). The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652. doi: 10.1146/annurev-cellbio-092910-154121
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hülsmann, S., Mesuret, G., Dannenberg, J., Arnoldt, M., and Niebert, M. (2016). GlyT2-Dependent preservation of MECP2-expression in inhibitory neurons improves early respiratory symptoms but does not rescue survival in a mouse model of Rett syndrome. Front. Physiol. 7:385. doi: 10.3389/fphys.2016.00385
Janc, O. A., Hüser, M. A., Dietrich, K., Kempkes, B., Menzfeld, C., Hülsmann, S., et al. (2016). Systemic radical scavenger treatment of a mouse model of rett syndrome: merits and limitations of the vitamin E derivative trolox. Front. Cell. Neurosci. 10:266. doi: 10.3389/fncel.2016.00266
Johnson, C. M., Cui, N., Zhong, W., Oginsky, M. F., and Jiang, C. (2015). Breathing abnormalities in a female mouse model of Rett syndrome. J. Physiol. Sci. 65, 451–459. doi: 10.1007/s12576-015-0384-5
Julu, P. O., Engerström, I. W., Hansen, S., Apartopoulos, F., Engerstrom, B., Pini, G., et al. (2008). Cardiorespiratory challenges in Rett’s syndrome. Lancet 371, 1981–1983. doi: 10.1016/S0140-6736(08)60849-1
Julu, P. O., Kerr, A. M., Apartopoulos, F., Al-Rawas, S., Engerström, I. W., Engerström, L., et al. (2001). Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch. Dis. Child. 85, 29–37. doi: 10.1136/adc.85.1.29
Kankirawatana, P., Leonard, H., Ellaway, C., Scurlock, J., Mansour, A., Makris, C. M., et al. (2006). Early progressive encephalopathy in boys and MECP2 mutations. Neurology 67, 164–166. doi: 10.1212/01.wnl.0000223318.28938.45
Kerr, A. M., Armstrong, D. D., Prescott, R. J., Doyle, D., and Kearney, D. L. (1997). Rett syndrome: analysis of deaths in the British survey. Eur. Child Adolesc. Psychiatry 6, 71–74.
Kerr, A. M., and Julu, P. O. (1999). Recent insights into hyperventilation from the study of Rett syndrome. Arch. Dis. Child. 80, 384–387. doi: 10.1136/adc.80.4.384
Kishi, N., and Macklis, J. D. (2005). Dissecting MECP2 function in the central nervous system. J. Child Neurol. 20, 753–759. doi: 10.1177/08830738050200091001
Lalley, P. M. (2008). Opioidergic and dopaminergic modulation of respiration. Respir. Physiol. Neurobiol. 164, 160–167. doi: 10.1016/j.resp.2008.02.004
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., et al. (1992). Purification, sequence and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914. doi: 10.1016/0092-8674(92)90610-o
Li, W., and Pozzo-Miller, L. (2014). BDNF deregulation in Rett syndrome. Neuropharmacology 76, 737–746. doi: 10.1016/j.neuropharm.2013.03.024
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Maekawa, T., Kim, S., Nakai, D., Makino, C., Takagi, T., Ogura, H., et al. (2010). Social isolation stress induces ATF-7 phosphorylation and impairs silencing of the 5-HT 5B receptor gene. EMBO J. 29, 196–208. doi: 10.1038/emboj.2009.318
Manzke, T., Guenther, U., Ponimaskin, E. G., Haller, M., Dutschmann, M., Schwarzacher, S., et al. (2003). 5-HT4a receptors avert opioid-induced breathing depression without loss of analgesia. Science 301, 226–229. doi: 10.1126/science.1084674
Manzke, T., Niebert, M., Koch, U. R., Caley, A., Vogelgesang, S., Hülsmann, S., et al. (2010). Serotonin receptor 1A-modulated phosphorylation of glycine receptor alpha3 controls breathing in mice. J. Clin. Invest. 120, 4118–4128. doi: 10.1172/JCI43029
Matthes, H., Boschert, U., Amlaiky, N., Grailhe, R., Plassat, J. L., Muscatelli, F., et al. (1993). Mouse 5-hydroxytryptamine5A and 5-hydroxytryptamine5B receptors define a new family of serotonin receptors: cloning, functional expression and chromosomal localization. Mol. Pharmacol. 43, 313–319.
Mellios, N., Woodson, J., Garcia, R. I., Crawford, B., Sharma, J., Sheridan, S. D., et al. (2014). β2-Adrenergic receptor agonist ameliorates phenotypes and corrects microRNA-mediated IGF1 deficits in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U S A 111, 9947–9952. doi: 10.1073/pnas.1309426111
Mesuret, G., Dannenberg, J., Arnoldt, M., Grützner, A.-A., Niebert, M., and Hülsmann, S. (2018). Breathing disturbances in a model of Rett syndrome: a potential involvement of the glycine receptor alpha3 subunit? Respir. Physiol. Neurobiol. 248, 43–47. doi: 10.1016/j.resp.2017.11.011
Mironov, S. L., Skorova, E. Y., and Kügler, S. (2011). Epac-mediated cAMP-signalling in the mouse model of Rett syndrome. Neuropharmacology 60, 869–877. doi: 10.1016/j.neuropharm.2011.01.002
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/s0092-8674(00)81887-5
Niebert, S., van Belle, G. J., Vogelgesang, S., Manzke, T., and Niebert, M. (2017). The serotonin receptor subtype 5b specifically interacts with serotonin receptor subtype 1A. Front. Mol. Neurosci. 10:299. doi: 10.3389/fnmol.2017.00299
Ohno, K., Saito, Y., Ueda, R., Togawa, M., Ohmae, T., Matsuda, E., et al. (2016). Effect of serotonin 1A agonists and selective serotonin reuptake inhibitors on behavioral and nighttime respiratory symptoms in Rett syndrome. Pediatr. Neurol. 60, 54.e1–59.e1. doi: 10.1016/j.pediatrneurol.2016.03.016
Penington, N. J., Kelly, J. S., and Fox, A. P. (1993). Whole-cell recordings of inwardly rectifying K+ currents activated by 5-HT1A receptors on dorsal raphe neurones of the adult rat. J. Physiol. 469, 387–405. doi: 10.1113/jphysiol.1993.sp019819
Philippe, C., Villard, L., De Roux, N., Raynaud, M., Bonnefond, J. P., Pasquier, L., et al. (2006). Spectrum and distribution of MECP2 mutations in 424 Rett syndrome patients: a molecular update. Eur. J. Med. Genet. 49, 9–18. doi: 10.1016/j.ejmg.2005.04.003
Plassat, J. L., Boschert, U., Amlaiky, N., and Hen, R. (1992). The mouse 5HT5 receptor reveals a remarkable heterogeneity within the 5HT1D receptor family. EMBO J. 11, 4779–4786.
Rees, S., den Daas, I., Foord, S., Goodson, S., Bull, D., Kilpatrick, G., et al. (1994). Cloning and characterisation of the human 5-HT5A serotonin receptor. FEBS Lett. 355, 242–246. doi: 10.1016/0014-5793(94)01209-1
Ren, J., Ding, X., Funk, G. D., and Greer, J. J. (2012). Anxiety-related mechanisms of respiratory dysfunction in a mouse model of Rett syndrome. J. Neurosci. 32, 17230–17240. doi: 10.1523/JNEUROSCI.2951-12.2012
Rett, A. (1966). On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 116, 723–726.
Schüle, B., Armstrong, D. D., Vogel, H., Oviedo, A., and Francke, U. (2008). Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clin. Genet. 74, 116–126. doi: 10.1111/j.1399-0004.2008.01005.x
Stettner, G. M., Huppke, P., Gärtner, J., Richter, D. W., and Dutschmann, M. (2008). Disturbances of breathing in Rett syndrome: results from patients and animal models. Adv. Exp. Med. Biol. 605, 503–507. doi: 10.1007/978-0-387-73693-8_88
Telgkamp, P., Cao, Y. Q., Basbaum, A. I., and Ramirez, J.-M. (2002). Long-term deprivation of substance P in PPT-A mutant mice alters the anoxic response of the isolated respiratory network. J. Neurophysiol. 88, 206–213. doi: 10.1152/jn.2002.88.1.206
Viemari, J.-C. (2008). Noradrenergic modulation of the respiratory neural network. Respir. Physiol. Neurobiol. 164, 123–130. doi: 10.1016/j.resp.2008.06.016
Viemari, J.-C., Roux, J.-C., Tryba, A. K., Saywell, V., Burnet, H., Peña, F., et al. (2005). Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J. Neurosci. 25, 11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005
Vogelgesang, S., Niebert, S., Renner, U., Mobius, W., Hulsmann, S., Manzke, T., et al. (2017). Analysis of the serotonergic system in a mouse model of rett syndrome reveals unusual upregulation of serotonin receptor 5b. Front. Mol. Neurosci. 10:61. doi: 10.3389/fnmol.2017.00061
Keywords: Rett syndrome, MeCP2, GPCR, 5-ht5b receptor, cAMP regulation, breathing disturbances
Citation: Vogelgesang S, Niebert M, Bischoff AM, Hülsmann S and Manzke T (2018) Persistent Expression of Serotonin Receptor 5b Alters Breathing Behavior in Male MeCP2 Knockout Mice. Front. Mol. Neurosci. 11:28. doi: 10.3389/fnmol.2018.00028
Received: 18 May 2017; Accepted: 22 January 2018;
Published: 20 February 2018.
Edited by:
Hansen Wang, University of Toronto, CanadaReviewed by:
Erhard Wischmeyer, University of Würzburg, GermanyCopyright © 2018 Vogelgesang, Niebert, Bischoff, Hülsmann and Manzke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcus Niebert, bW5pZWJlckBnd2RnLmRl
†These authors share senior authorship.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.