 Alexandre Henriques1,2,3*
Alexandre Henriques1,2,3* Vincent Croixmarie4
Vincent Croixmarie4 Alexandra Bouscary1,2
Alexandra Bouscary1,2 Althéa Mosbach1,2
Althéa Mosbach1,2 Céline Keime5Claire Boursier-Neyret6Bernard Walter6
Céline Keime5Claire Boursier-Neyret6Bernard Walter6 Michael Spedding3
Michael Spedding3 Jean-Philippe Loeffler1,2*
Jean-Philippe Loeffler1,2*- 1Université de Strasbourg, UMR_S 1118, Fédération de Médecine Translationnelle, Strasbourg, France
- 2INSERM, U1118, Mécanismes Centraux et Périphériques de la Neurodégénérescence, Strasbourg, France
- 3Spedding Research Solutions SAS, Le Vesinet, France
- 4Institut de Recherche International Servier, Suresnes, France
- 5Institut de Génétique et de Biologie Moléculaire et Cellulaire, INSERM, U964, CNRS, UMR7104, Université de Strasbourg, Illkirch, France
- 6Technologie Servier, Orleans, France
Lipid metabolism is drastically dysregulated in amyotrophic lateral sclerosis and impacts prognosis of patients. Animal models recapitulate alterations in the energy metabolism, including hypermetabolism and severe loss of adipose tissue. To gain insight into the molecular mechanisms underlying disease progression in amyotrophic lateral sclerosis, we have performed RNA-sequencing and lipidomic profiling in spinal cord of symptomatic SOD1G86R mice. Spinal transcriptome of SOD1G86R mice was characterized by differential expression of genes related to immune system, extracellular exosome, and lysosome. Hypothesis-driven identification of metabolites showed that lipids, including sphingomyelin(d18:0/26:1), ceramide(d18:1/22:0), and phosphatidylcholine(o-22:1/20:4) showed profound altered levels. A correlation between disease severity and gene expression or metabolite levels was found for sphingosine, ceramide(d18:1/26:0), Sgpp2, Sphk1, and Ugt8a. Joint-analysis revealed a significant enrichment of glycosphingolipid metabolism in SOD1G86R mice, due to the down-regulation of ceramide, glucosylceramide, and lactosylceramide and the overexpression of genes involved in their recycling in the lysosome. A drug-gene interaction database was interrogated to identify potential drugs able to modulate the dysregulated genes from the signaling pathway. Our results suggest that complex lipids are pivotally changed during the first phase of motor symptoms in an animal model of amyotrophic lateral sclerosis.
Introduction
Amyotrophic lateral sclerosis (ALS) is a non-cell autonomous disease characterized by a severe muscle denervation and degeneration of upper and lower motor neurons. ALS has prevalence close to 7 in 100,000. Death usually occurs 1–5 years after diagnosis, but survival can exceed 10 years for 10% of patients. Etiology remains unclear for the majority of patients even though genetic mutations are known to trigger or to increase the risk of ALS. The main ALS-linked genetic mutations concern SOD1, TARBP, FUS genes. Expansion repeats in C9ORF72 is also an important risk factor for ALS (Taylor et al., 2016). Altered RNA metabolism, oxidative stress, impaired axonal transport, protein misfolding, and inflammation participate to disease severity. Mutation on the SOD1 gene was the first identified genetic cause for ALS (Rosen et al., 1993). Transgenic mice harboring mutations in the Sod1 gene are used as models of ALS, such as the SOD1G93A and the SOD1G86R models. They recapitulate the main symptoms of ALS, including motor neuron degeneration, muscle denervation associated with severe paralysis and death (Ripps et al., 1995).
Multiple stages of energy and lipid metabolism dysregulations exist in ALS. Incidences of hypermetabolism and glucose insensitivity are higher in ALS and severe loss of body mass negatively affects patient's prognosis (Schmitt et al., 2014). High adiposity (Paganoni et al., 2011), dyslipidaemia (Dupuis et al., 2008; Mariosa et al., 2017), and circulating metabolic markers (Henriques et al., 2015a) are associated with survival. High caloric diets stabilize weight loss and extend life expectancy of patients under gastrostomy (Wills et al., 2014; Dorst et al., 2015). Hypermetabolism and preferential use of lipids as nutrients have been reported in SOD1 mice (Dupuis et al., 2004; Palamiuc et al., 2015). The causes of the metabolic dysfunctions in ALS remain unknown and could result from central pathologies combined with peripheral alterations. Early metabolic shift from glycolysis to beta-oxidation has been evidenced in skeletal muscles in SOD1G86R mice (Palamiuc et al., 2015). SOD1G93A present with glucose intolerance and impaired muscle glucose metabolism, which could be reversed by physical exercise (Desseille et al., 2017). Fibroblast from sporadic or familial ALS patients exhibits a metabolic shift from oxidative phosphorylation to glycolysis (Raman et al., 2015). Hypothalamus is a cerebral structure key for the integration of central and peripheral signals related to energy balance. Neuronal loss and altered melanocortin pathway have been described in the hypothalamus of ALS patients and can account for weight unbalance and defective energy metabolism (Vercruysse et al., 2016).
The nervous system is remarkably enriched in lipids compared to other tissues. They range from simple fatty acids to glycosphingolipids. Lipids directly contribute to cell signaling, neuronal energy balance during stress, and membrane stability/fluidity. Different research groups have reported dysregulations of lipids in the central nervous system of neurodegenerative diseases. Elevated diacylglycerols have been reported in the frontal cortex of patients diagnosed with Alzheimer's disease (Wood et al., 2016) and levels of sphingolipids, phospholipids, and neutral lipids were reported as significantly dysregulated in the central nervous system (CNS) of patients diagnosed with Parkinson's disease (Cheng et al., 2011). Genetic mutations causing disrupted lipid metabolism have been linked to neurological and neuromuscular phenotypes in human diseases, like hereditary spastic paraplegia (Dodge, 2017).
In ALS, accumulation of lipid metabolites (i.e., ceramides and cholesterol esters), occurred in the post-mortem spinal cord samples of ALS patients (Cutler et al., 2002). Accumulation of ceramide species in ALS post-mortem spinal samples was confirmed, by targeted metabolomics, in a second study (Dodge et al., 2015). Using this approach with spinal tissues from SOD1G93A mice, one ceramide metabolite was upregulated at disease endpoint in this animal model of ALS. Nevertheless, at earlier disease stage, ceramides, and other sphingolipids showed marked dysregulations. We have recently reported that almost all lipid classes (e.g., triglycerides, phospholipids, sphingolipids) were strongly dysregulated at both presymptomatic and late disease stages, with metabolites showing either with an upregulation or downregulation in the spinal cord of SOD1G86R mice (Henriques et al., 2015b).
Gene expression analysis by RNA-sequencing gives information on the expression levels of genes in specific cells or tissues. In ALS, several molecular pathways have been already detected as altered at transcriptional levels in the CNS of ALS patients and animal models (Henriques and Gonzalez De Aguilar, 2011). A recent study has reported that targeted metabolomics (limited to 188 metabolites) combined to targeted gene expression analysis (limited to 84 genes) was able to provide information on the regulation of branched-chain amino acid metabolism in SOD1G93A mice (Patin et al., 2016). We present for the first time an integrated analysis of RNA-sequencing and lipidomic data from the spinal cord of symptomatic SOD1G86R mice. These results allow an unbiased vision of the metabolic changes in the course of ALS, and pinpoint specific pathways which are pathologically important.
Materials and Methods
Animals
Experiments were performed by authorized investigator (A67-402 to A.H.), after approval of experimental procedure by the ethic committee of the University of Strasbourg and by the ministry of higher education and research (APAFIS#2255; AL/25/32/02/13). They followed current European Union regulations (Directive 2010/63/EU). FVB/N female mice, overexpressing the SOD1G86R protein (Ripps et al., 1995), were generated by breeding male SOD1G86R mice with non-transgenic FVB/N female mice in our animal facility. Genotypage was performed as previously described (Ripps et al., 1995). Mice were maintained in our animal facility at 23°C with a 12 h light/dark cycle. Mice had access to water and to regular A04 rodent chow ad libitum. Body mass and muscle strength were analyzed on a daily basis to access disease onset. For muscle strength measurements (mean of three tests, grip test, Bioseb, Chaville, France), a mouse was placed on grid where they spontaneously gripped it with their four paws. They were then gently pulled back by the experimenter until they released the grid (mean of three tests, grip test, Bioseb, Chaville, France). A strength-meter recorded the peak grip strength of the mouse. Age and litter matched non-transgenic female mice served as control. At 95 days of age, mice were sacrificed by intraperitoneal injection with ketamine chlorohydrate (100 mg/kg) and xylazine (5 mg/kg) and intracardially perfused with PBS at 4°C. Lumbar spinal cords were quickly dissected, fresh frozen, and kept at −80°C until further analysis.
RNA-Sequencing
RNA-sequencing was performed as previously described (Henriques et al., 2017). Briefly, total RNA was extracted from frozen samples of spinal cord from 95 days old mice (n = 5/group). Libraries of template molecules suitable for high throughput DNA sequencing were created and reads were mapped onto mm10 assembly of mouse genome using Tophat v2.0.14 (Kim et al., 2013). Quantification of gene expression was performed using HTSeq v0.6.1 (Anders and Huber, 2010) and Ensembl release 81 database. Supervised statistical analysis for differential gene expression has been performed using R (3.3.2) and the DESeq2 Bioconductor (v3.2) library. Multiple testing was adjusted by Benjamini and Hochberg FDR correction (Benjamini and Hochberg, 1995).
After normalization and rlog transformation, hierarchical clustering (single method), and PCA (principal component analysis) was performed with the indicated subset of genes, accordingly to KEGG pathways, using JMP 11.0.0. Validation of gene expression was assessed by qPCR with a CFX96 using SYBR green Supermix reagent (BioRad), with the samples used for RNA-sequencing. Relative quantification of each gene was determined using the Biorad software and normalized to reference genes (Pol2, TBP, and 18S). Primers sequences are provided in Supplementary Table 1. Comparison between groups was studied with student's t-test and p-value < 0.05 were considered significant (PRISM 6.0b, GraphPad, San Diego, CA). Data are expressed as the mean ± SEM. RNA-sequencing data are available at the Gene Expression Omnibus (GEO) database repository, under the accession number GSE106364.
Metabolomic Analysis
Lipid extraction and UPLC/TOF-MS (ultra performance liquid chromatography/time-of-flight mass spectrometry) was performed as previously described (Henriques et al., 2015b). Briefly, spinal cord samples from 95 days old mice were homogenized in precooled methanol (n = 7, for SOD1G86R mice; n = 8, for wild type). Chloroform was added and after centrifugation, organic phase was collected and evaporated. Residues were solubilized with acetonitrile/isopropanol. Chromatography was performed on an Acquity UPLC system using an Acquity BEH C18 column. The chromatographic system was coupled to a LCT Synapt G2S mass spectrometer (Waters Corporation), equipped with an electrospray source operating in positive or negative ion mode with a lockspray interface for accurate mass measurements. Refiner MS 6.0 (Genedata, Basel, Switzerland) was use to normalize metabolomics data. Data with retention times between 3 and 15 min, with a mass range between 300 and 1,000 and peak intensity distinct from zero were retained and normalized to fresh tissue mass. Data were submitted to PARETO transformation before statistical analysis. Statistical analysis for differential lipid metabolite level was performed by using supervised multivariate orthogonal partial least-squares discriminant analysis (OPLS-DA), as implemented in SIMCA-P. A difference was considered significant when the corresponding variable |correlation coefficient| was greated than or egal to 0.7, according to the p(corr) coordinate in the S-plot built after the OPLS-DA model. Molecular features with significant changes were associated with theoretically identified metabolites based on their atomic mass (m/z), by using online HMDB 2.5 databases. Together with lipid extraction from samples and liquid chromatography gradient, the brut formula associated with the exact mass-over-ionization-state ratio allowed tentative lipid identification.
Pathway Analysis
Gene enrichment analysis was performed with dysregulated genes (padj < 0.01) by using Gene Ontology/Panther (Ashburner et al., 2000; The Gene Ontology Consortium, 2015). Only the most specific subclass was considered per parent items. Given p-value were adjusted using the Bonferroni correction. For lipid metabolites, over-representation analysis was carried out with consensuspathDB (Kamburov et al., 2009, 2011b). All significantly dysregulated and identified lipid metabolites were considered for this analysis. Parameters were set to a minimum overlap of four distinct metabolites and a p-value cutoff of 0.001. Pathway over-representation analysis was conducted by using IMPaLA (Kamburov et al., 2011a) with genes and lipid metabolites whose levels were altered in SOD1G86R mice. Pathways represented by at least 3 genes and 3 lipid metabolites and with q-value for enrichment below 0.05 were considered as significantly altered.
Results
Gene Regulation in the Spinal Cord of SOD1G86R Mice
To gain insight into the nature and extent of metabolic reorientation occurring in ALS, the spinal cord transcriptomes of five 95 days old SOD1G86R and five age- and litter-matched non-transgenic wild type mice were studied by RNA-sequencing. This time point corresponds to the symptomatic disease stage of this animal model. At 95 days of age, all SOD1G86R mice present with motor symptoms and muscle denervation, even if the severity of symptoms may vary (Supplementary Figure 1). Disease onset, defined as a loss of more than 10% of muscle strength is taking place around 90 days of age (Henriques et al., 2017). Disease end-stage, corresponding to full paralysis, occurs later in this mouse line, around 105 days of age.
Group comparison identified a total of 751 genes significantly dysregulated (adjusted p-value < 0.01), including 660 up-regulations and 91 down-regulations. Table 1 presents all genes found either up- or down-regulated in SOD1G86R mice with a fold change higher than 2 (adjusted p-value < 0.01, |log2 fold change| greater than 1). Genes that were the most down-regulated were Tram1l1 (translocation associated membrane protein 1-like 1), Rwdd3 (RWD domain containing 3), Mettl14 (methyltransferase like 14), and Prss12 (protease, serine 12 neurotrypsin; or motopsin). The highest up-regulation concerned Sod1 (superoxide dismutase 1, soluble), which is expected due to the presence of the Sod1G86R transgene. Gm8566 is the second most dysregulated gene. Its altered expression could result from a real difference between SOD1G86R and WT mice, although we cannot completely rule out the possibility of an artifact as Gm8566 shares 98% homology with the Sod1 transcript. Aside from the Sod1 gene and Gm8566, highly dysregulated transcripts with known functions were Cd180 (CD180 antigen), Calca (calcitonin/calcitonin-related polypeptide, alpha), Slc15a3 (solute carrier family 15, member 3), and Runx1 (runt related transcription factor 1). Differential regulation of gene expression was assessed by quantitative PCR to confirm RNA-sequencing. Selected genes were Tram1l1, Mettl14, Cd180, Calca, Slc15a3, Runx1, Rwdd3, and Prss12. These genes were selected because they were either the most up-regulated or down-regulated (adjusted p-value < 0.01, |log2 fold change| greater than 1) and had known biological functions. Expression levels of the selected transcripts were highly similar when comparing qPCR and RNA-sequencing analysis (Figure 1).
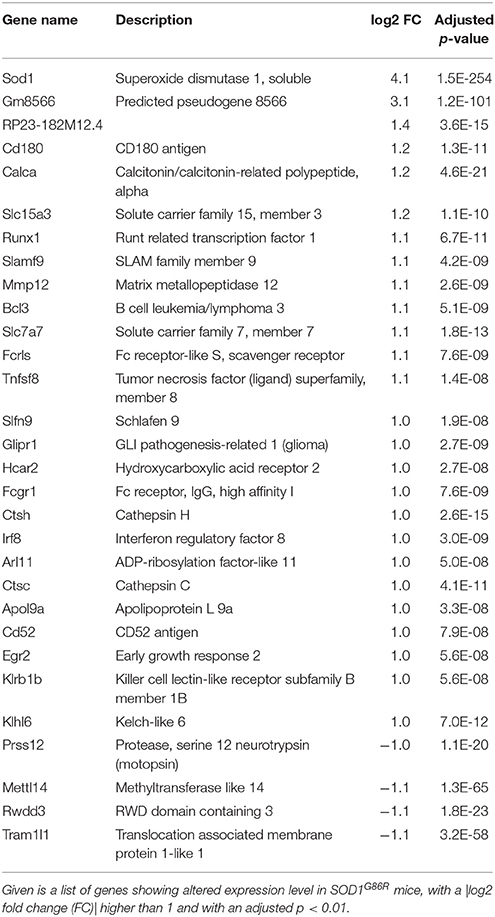
Table 1. Transcriptomic dysregulation in the spinal cord of SOD1G86R mice.
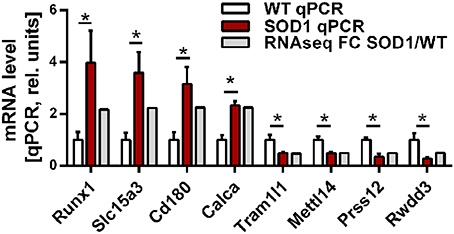
Figure 1. Expression level of selected genes as determined by RNA-sequencing and qPCR. Validation of differential gene expression by qPCR and compared to the fold change determined by RNA sequencing. qPCR values were normalized to the mean of the respective WT group. qPCR data is presented as mean ± standard variation of the mean. FC, fold change; *p < 0.05.
Next, in an attempt to identify biological processes and cellular components affected in the diseased mice, gene ontology (GO) analysis was performed with significantly dysregulated genes. Immune system process was the highest over-represented biological process with a fold enrichment (FE) of 3.9 and an adjusted p-value (padj) below 4.4.10−77. In SOD1G86R mice, 248 genes were associated to this particular biological process, which corresponds to almost of a third of all transcriptomic reorientations. Notably, “positive regulation of signal transduction” (126 genes; FE = 2.87; padj < 2.63.10−22), “regulation of locomotion” (96 genes; FE = 3.4; padj < 3.97.10−21), “regulation of apoptotic process” (122 genes; FE = 2.5; padj < 3.99.10−17), and “response to lipid” (72 genes; FE = 3.7; padj < 4.5.10−17) showed strong enrichment in SOD1G86R mice. A putative location of the products of genes showing altered expression was investigated. Over-represented cellular components were mainly associated to membranes or vesicles, such as “extracellular exosome” (170 genes; FE = 2.0; padj < 6.6.10−16), “lysosome” (47 genes; FE = 3.0; padj < 6.2.10−08), “external side of plasma membrane” (42 genes; FE = 2.89; padj < 2.72.10−06), and “membrane raft” (36 genes; FE = 3.18; padj < 3.57.10−06). Accordingly, unsupervised principal component analysis (PCA) plots performed with genes from the GO terms “lysosome,” “membrane raft,” and “external side of plasma membrane,” revealed a strong influence of genotype on the distribution of samples (Figures 2A–C). These results suggest that these pathways are strongly impacted in symptomatic SOD1G86R mice.
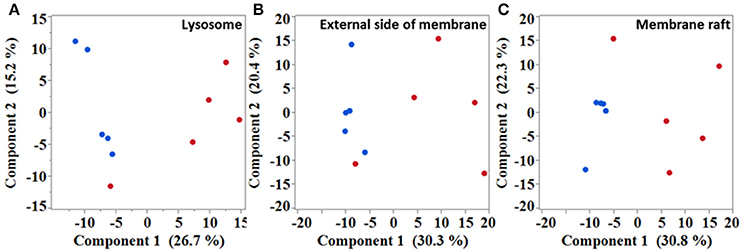
Figure 2. Multivariate analysis showing samples distribution based on different subset of genes. Unsupervised principal component analysis performed with genes isolated from “lysosome” (A), “external side of membrane” (B), and “membrane raft” (C) gene ontology terms. Blue dots, wild type; red dots, SOD1G86R.
Lipidomic Changes in the Spinal Cord of SOD1G86R Mice
In order to better characterize metabolomic changes at disease onset, lipid metabolites were studied in seven symptomatic SOD1G86R mice and eight age-matched non-transgenic wild type mice. Analysis of spinal tissue by mass spectrometry identified a total of 7,603 distinct peaks after positive ionization. A total of 293 metabolites was found altered according to the chosen threshold in the SOD1G86R mice compared to wild type controls. A hypothesis-driven identification was attributed to 196 of them accordingly to their atomic mass. All tentatively identified metabolites were down-regulated. An enrichment analysis was performed with the identified and altered metabolites using ConsensuspathDB (Kamburov et al., 2009, 2011b). According to three independent databases, “sphingolipid metabolism” (KEGG database, padj < 3.00.10−05; and Reactom database, padj < 3.9.10−05) as well as “glycosphingolipid metabolism” (EHMN database; padj < 2.5.10−04) were the metabolic pathways showing the most significant enrichment. Indeed, many metabolites showing altered levels were identified as sphingolipids. Sphingomyelin (d18:0/26:1), ceramide-P (d18:1/26:1), and ceramide (d18:1/22:0) were among the most down-regulated lipid metabolites (Table 2). Notably, several phospholipid metabolites, such as phosphatidylcholine(o-22:1/20:4), presented with significant down-regulation (Table 3). Other subclasses of dysregulated lipids included di- and triglycerides. Saturation and unsaturation of acyl chains directly influence biophysical properties of complex lipids with repercussion on membrane fluidity. According to HMDB identification, down-regulated sphingolipid metabolites mainly quasi-exclusively contained saturated and monounsaturated fatty acids. This suggests that sphingolipids having polyunsaturated acyl chains were not significantly affected in SOD1G86R mice. This difference in acyl chains could reflect enzymatic activities involved in the synthesis, transformation or degradation of fatty acids and lipids.
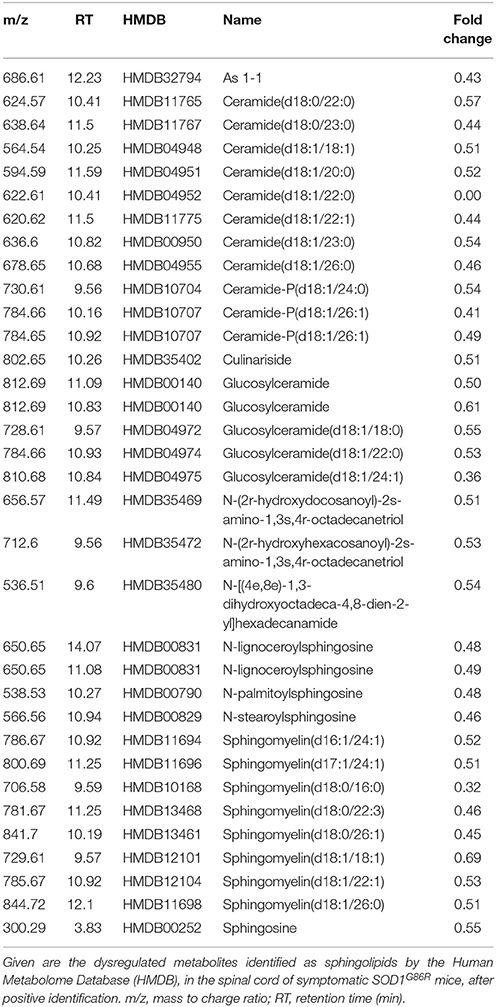
Table 2. Dysregulation of sphingolipid metabolites in the spinal cord of SOD1G86R mice, after positive ionization, and tentative identification.
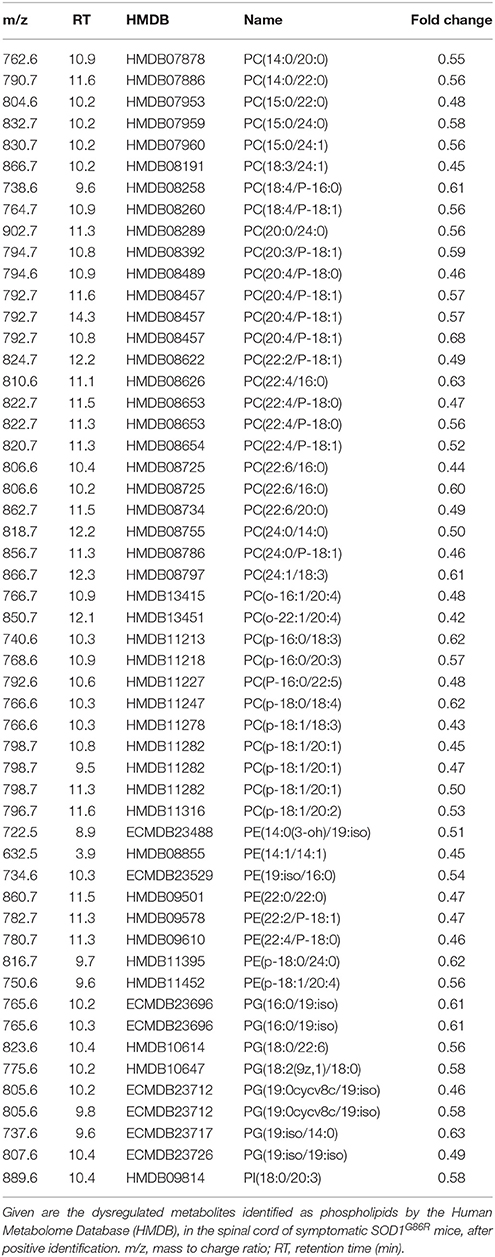
Table 3. Dysregulation of phospholipids metabolites in the spinal cord of SOD1G86R mice, after positive ionization, and tentative identification.
When considering negative ionization, similar results were obtained. HMDB identification was attributed to 165 lipids metabolites showing significantly altered levels in SOD1G86R mice. A majority of lipid metabolites were down-regulated. Main lipid subclasses were fatty acids, phospholipids (mainly phosphatidylcholine), sphingolipids (e.g., ceramide), and tri-, di-, or mono-acylglycerides (Table 4). Notably, several significantly altered lipid metabolites received an identical identification after both positive and negative ionizations. These lipids were sphingolipids [Cer(d18:1/22:1); Cer-P(d18:1/26:1); Cer(d18:1/18:0)], triglycerides [TG(18:4/15:0/18:4); TG(14:1/20:5/14:1)] or from other lipid subclasses [5-Hydroxy-7-methoxy-2-tritriacontylchromone; N-oleoylethanolamine; Randilongin; Persenone a].
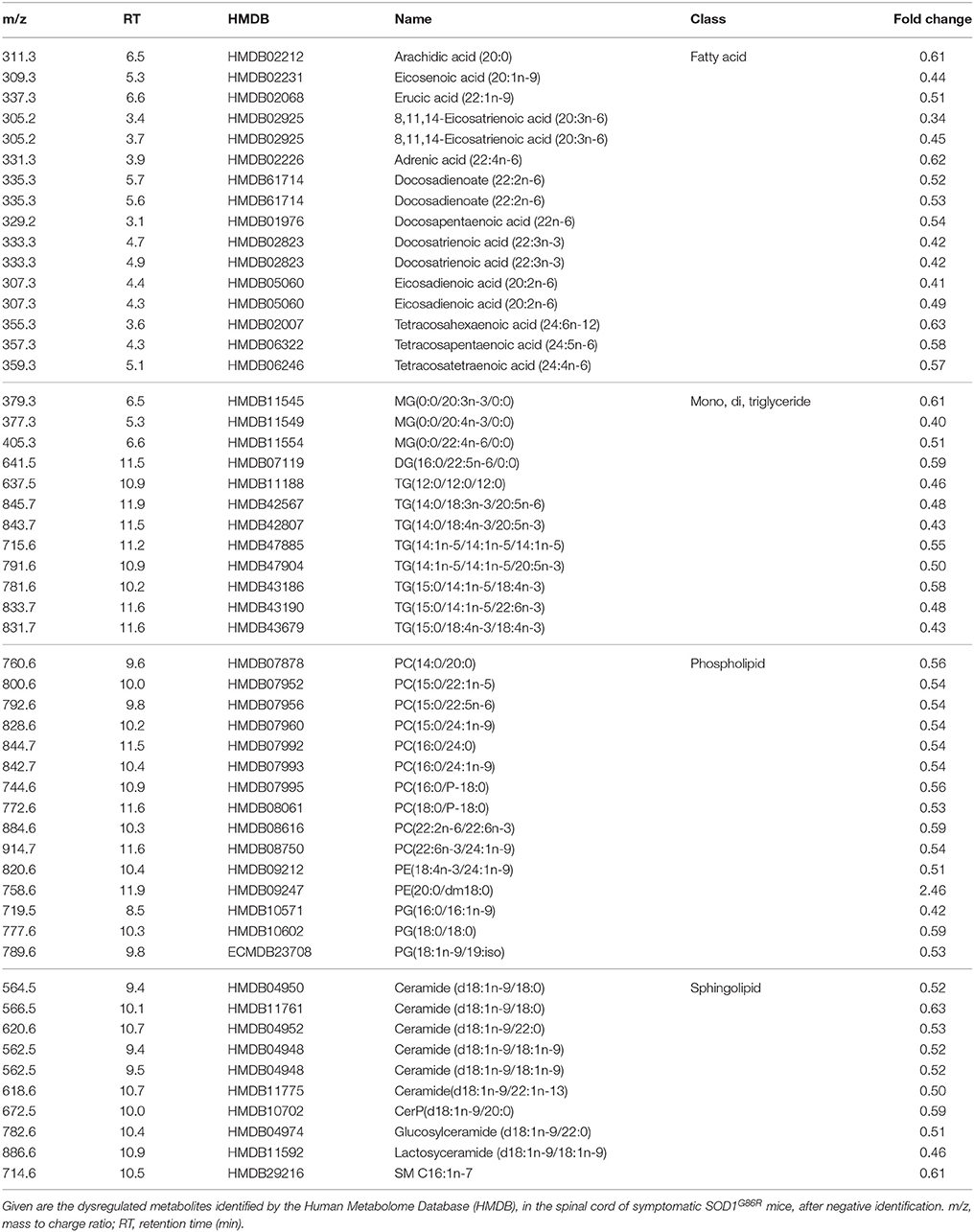
Table 4. Dysregulation of lipid metabolites in the spinal cord of SOD1G86R mice, after negative ionization, and tentative identification.
Sphingolipid Metabolism Is Altered at Transcriptomic and Lipidomic Levels in the Spinal Cord of SOD1G86R Mice
To investigate the relationship between transcriptomic and lipidomic changes in SOD1G86R mice, a joint inter-omics analysis was performed using significantly dysregulated genes and metabolites (Kamburov et al., 2011a). The objective was to identify molecular pathways associated with ALS, with a special regard toward lipid metabolism. Three distinct databases have identified “sphingolipid,” “glycosphingolipid metabolism,” and “sphingolipid signaling” pathways as significantly enriched in the spinal cord of SOD1G86R mice (Table 5, Supplementary Table 2). Overlapping genes are those coding for lysosomal proteins involved in the recycling of glycosphingolipids (e.g., Hexb, Glb1, Asah1), for ceramide kinase (Cerk), for Sgpl1, an enzyme involved in the degradation of sphingosine-1-phosphate. Overlapping lipid metabolites were ceramide, mono-hexosylceramide (glucosylceramide or galactosylceramide), lactosylceramide, and sphingomyelin. Pathways related to other lipids, such as phospholipids or triglycerides, were not detected as significantly enriched.
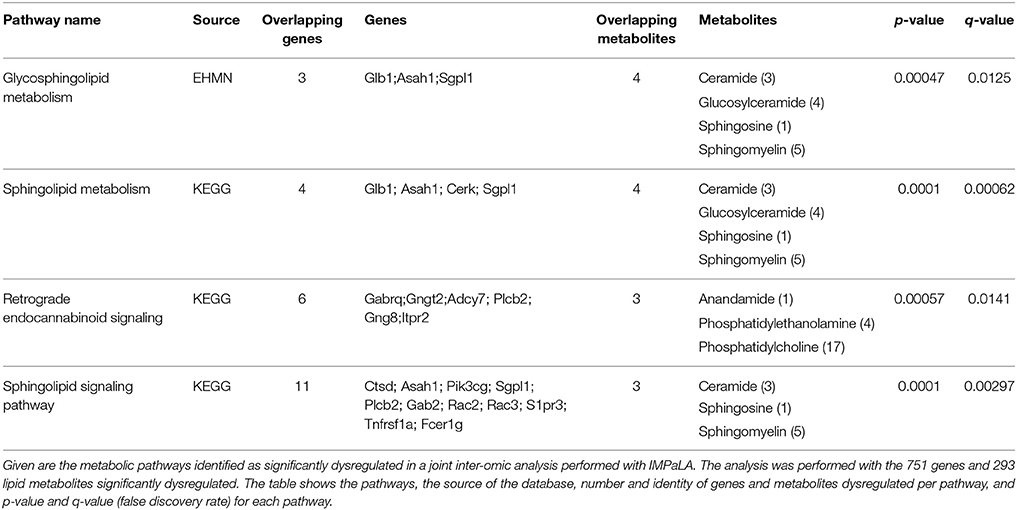
Table 5. Integrated pathway analysis of spinal transcriptomic and lipidomic data of SOD1G86R mice.
Next, hierarchical clustering was performed using all genes, dysregulated or not, listed as part of the sphingolipid metabolism by KEGG. Genotype had a strong influence on the distribution of samples on unsupervised hierarchical clustering plot (Figure 3A). A cluster of genes, which are associated with the breakdown of glycosphingolipids, showed a clear differential pattern of expression in SOD1G86R mice compared to wild type controls. Differential expression for genes (i.e., HexB, Asah1, Cerk, Glb1, and Arsb) involved in the breakdown of glycosphingolipids was confirmed by qPCR (Figure 3B). Muscle weakness is an early event in SOD1G86R mice and ALS patients. SOD1G86R spinal samples were collected at symptomatic disease stage, when motor weakness was established and ranged from mild to severe. In an attempt to identify biological processes influenced by disease severity, correlation between levels of genes or lipid metabolites from the “sphingolipid metabolism” and muscle strength was studied. Expression levels of Ugt8a (UDP galactosyltransferase 8A), Sgpp2 (sphingosine-1-phosphate phosphatase 2), and Sphk1 (sphingosine kinase 1) were significantly correlated or anti-correlated to muscle strength (Figures 3C–E). Ugt8a codes for the galactosylceramide synthase. SGPP2 and SPHK1 are two enzymes involved respectively in the synthesis and degradation of sphingosine-1-phosphate.
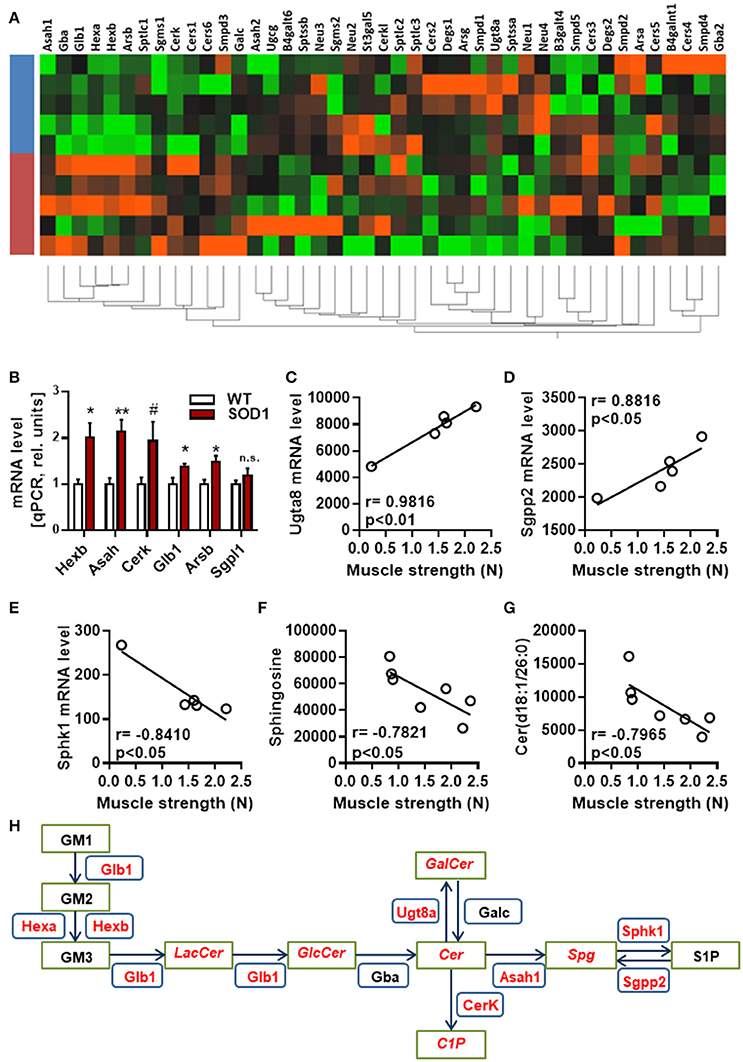
Figure 3. Regulation and interaction of genes related to glycosphingolipid pathway. (A) Unsupervised hierarchical clustering, performed with genes from the KEGG pathway “sphingolipid metabolism pathway,” showing distribution of SOD1G86R and wild type samples (blue, wild type; red, SOD1G86R). (B) Validation by qPCR of differential expression level of genes from the sphingolipid pathway previously found dysregulated by RNAseq. (C–G) Pearson correlations between muscle strength and gene expression levels of Ugt8a (C), Sgpp2 (D), Sphk1 (E), and metabolite levels of sphingosine (F) and ceramide(d18:1/26:0) (G). (H) Schematic representation of the main dysregulations related to glycosphingolipids, ceramide, and sphingosine. Green boxes represent metabolites and blue boxes refer to genes. In red are given genes and metabolites showing either altered levels or a correlation with disease severity. LacCer, lactosylceramide; GlcCer, glucosylceramide; GalCer, galatosylceramide; Cer, ceramide; Spg, sphingosine; S1P, sphingosine 1-phosphate, C1P, ceramide 1-phosphate. Gene abbreviation follows the HUGO Gene Nomenclature Committee-approved gene nomenclature. Data are presented as mean ± standard variation of the mean. n.s, not significant; #p < 0.1; *p < 0.05; **p < 0.01.
Interestingly, muscle strength showed a significant correlation between the intensity level of ceramide(d18:1/26:0) and sphingosine, product and/or substrate of Ugt8a, Sgpp2, and Sphk1 (Figures 3F,G). Collectively, our results suggest that the metabolism of sphingolipids, and particularly the glycosphingolipid and the axis “ceramide, sphingosine, sphingosine-1-phosphate” is impaired at transcriptomic and lipidomic levels, and vary depending on disease progression (Figure 3H). Several sphingolipids, such as sphingosine-1-P, ceramide and sphingomyelin, are bioactive molecules able to module cell response to stress, survival, and neuroinflammation. KEGG identified “sphingolipid signaling pathway” as dysregulated, suggesting that altered levels of sphingolipids in ALS could translate into changes in cell signaling.
DGIdb database was used to search for potential pharmaceutical modulators of the “sphingolipid signaling pathway” using genes identified as dysregulated in this pathway (Wagner et al., 2016). A total of 67 compounds were identified, including 10 negative modulators of TNF-a signaling, and 5 modulators of sphingosine-1-phosphate. Notably, 6 of them are currently investigated or were tested at preclinical and/or clinical level in ALS (Pyrimethamine, fingolimod phosphate, pentoxifylline, celastrol, lenalidomide, thalidomide; Supplementary Table 3). These findings indicate that the metabolism of sphingolipid shows dysregulation at gene and metabolites levels, in the spinal cord of symptomatic SOD1G86R mice.
Discussion
ALS is a fatal condition characterized by degeneration of motor neurons. Several lines of evidence suggest that gene expression and lipid metabolism are differentially regulated in ALS, and could contribute to disease progression. Here, we report a joint analysis of lipidomic and transcriptomic changes in the spinal cord of symptomatic SOD1G86R mice.
Transcriptomic Changes in Spinal Cord from SOD1G86R Mice
For the first time, we report RNAseq analysis of spinal tissue from SOD1G86R mice. This specific mouse line presents with the advantage to overexpress a mutated SOD1 protein without dismutase activity. Based on our transcriptomic analysis, spinal cord of SOD1G86R mice was characterized by the dysregulation of immune system, cell death and regulation of membrane and vesicles. “Immune system” was the biological pathway showing the greatest enrichment in SOD1G86R mice. Numerous studies pin point neuroinflammation in ALS. Activation of glial cells has been linked to degeneration of motor neurons (Yamanaka et al., 2008; Endo et al., 2015; Cooper-knock et al., 2017) or to defects in the integrity of peripheral motor axons (Nardo et al., 2016). Infiltration of peripheral monocytes and lymphocytes into CNS was also demonstrated in ALS (Hooten et al., 2015). Among dysregulated genes identified in our RNAseq analysis, several genes are directly connected to a pro-inflammation state. For instance, Cd180, one of the most up-regulated gene in the spinal cord of SOD1G86R mice, is a pro-inflammatory gene participating in the activation of TLR4 receptors (Bastiaansen et al., 2014). Our results also show that Gfap (log2FC: 0.54, adjusted p-value < 0.01) and Aif-1/Iba1 (FC: 0.77, adjusted p-value < 0.01), markers for astrocyte and microglial activations, were both up-regulated in our study. Interestingly, we noted an up-regulation for Htr2b (log2FC: 0.34; adjusted p-value < 0.05), gene coding for 5-HT2b receptor, in SOD1G86R mice. A recent report connected serotonin signaling in microglial cells to neuroinflammatory process and neurodegeneration in ALS patients and animal models (El Oussini et al., 2016). Csf3r (log2FC: 0.96; adjusted p-value < 0.01) and Csf2rb (log2FC: 0.96; adjusted p-value < 0.01), coding for the receptors of granulocyte (G-CSF) and granulocyte-macrophage stimulating factor (GM-CSF), were significantly up-regulated the SOD1G86R group. The hematopoietic factor G-CSF, ligand of Csf3r, is neuroprotective in SOD1G93A mice (Henriques et al., 2011) and is able to re-adjust gene expression in lumbar motor neurons (Henriques et al., 2015c).
Our analysis also identified genes whose regulation could promote neurodegeneration or impair neuronal function in SOD1G86R mice. Calca, encoding for calcitonin gene-related peptide, is up-regulated in our RNAseq analysis. This gene is tightly associated to neurodegeneration and to motor neuron function (Enjin et al., 2010). In spinal bulbar muscular atrophy, its overexpression triggered cell damage while its inhibition reduced, or even abolished, neurodegeneration in an animal model of this disease (Minamiyama et al., 2012). High expression level of Calca has been also associated to higher motor neuron loss in SOD1G93A mice (Ringer et al., 2012). Prss12, or motopsin, is a neuronal protease which positively regulates axonal plasticity (Mitsui et al., 2013). Prss12 expression is known to be down-regulated during acute stress (Numajiri et al., 2006) and in laser-captured SOD1G93A motoneurons (Henriques et al., 2015c). Down-regulation of Prss12 in SOD1G86R mice could therefore relate to axonal dysfunction. Rwdd3 is a gene coding for RSUME, a protein taking part in HIF-1 alpha signaling (Carbia-nagashima et al., 2007) and known to be activated during cell stress. RSUME modulates the sumoylation of the glucocorticoid receptor (Druker et al., 2013), connects to immune system, energy metabolism and axonal plasticity. RSUME down-regulation in spinal tissues of SOD1G86R mice could negative impact motor units through these biological processes. Runx1 is a transcription factor stimulating neuronal differentiation and axonal plasticity and reduces BMP signaling and expression of Calca (Yoshikawa et al., 2015, 2016; Halevy et al., 2016). Runx1 is a positive regulator of the expression of genes related to sphingolipid metabolism (Kilbey et al., 2010). Thus, the up-regulation of Runx1 in the spinal tissue of SOD1G86R mice could represent a compensatory neuroprotective mechanism.
Main altered cellular component were “extracellular exosome” and “lysosome.” Exosomes are vesicles releasing proteins, lipids and RNA in the extracellular environment. Involvement of exosome in ALS has been recently a subject of extensive studies. Exosomes participate in the propagation of misfolded SOD1 and TDP-43 proteins (Nonaka et al., 2013; Grad et al., 2014), in aberrant phenotype of immune cells (Pinto et al., 2017) and could impair synaptic plasticity in neurodegenerative diseases (Wang et al., 2017). In fly neuromuscular junctions, presynaptic exosomes transfer synaptotagmin 4 to post-synaptic cells, thereby enabling retrograde signaling in activity-dependant synaptic growth (Korkut et al., 2013). Physiological adaptations to exercise have been proposed to be mediated, at least partially, by muscle-derived exosomes (Safdar et al., 2016). Lysosomes are intracellular organelles with important function in recycling of macromolecules, including complex lipids. Lysosomes interact with autophagosomes to promote autophagy. Defect in lysosomal function results in pathological accumulation of biomolecules, such as sphingolipids, and cause lysosomal storage diseases, such as Gaucher's disease. In ALS, activity of lysosomal enzymes is increased in post-mortem spinal tissues of ALS patients (Dodge et al., 2015) and mutations on genes related to autophagy are causing genetic forms of ALS (Lee et al., 2015).
Lipidomic Changes in Spinal Cord from SOD1G86R Mice
At disease onset, SOD1G86R mice presented with clear rearrangement of lipid metabolites, such as phospholipids and sphingolipids. We and other previously reported that nervous tissue of SOD1 G86R and SOD1G93A mice had altered levels for lipid metabolites, at presymptomatic, or late-symptomatic disease stages (Cutler et al., 2002; Dodge et al., 2015; Henriques et al., 2015b; Patin et al., 2016). Due to technical restrictions, most of these studies focussed on a limited number of lipid metabolites. In the present study, we have detected more than 7,500 metabolites which covered main lipid subclasses (fatty acids, triglycerides, phospholipids, sphingolipids, and sterols). Shortage in energetic lipids (e.g., triglyceride) is compatible with increased energy expenditure which negatively impact prognosis in ALS patients (Jésus et al., 2018). We have previously reported that triglycerides were massively depleted in spinal cord, muscle, and plasma samples, showing the scale of the metabolic dysfunction in this animal model of ALS (Henriques et al., 2015b). Our data also shows that sphingolipids and phospholipids are under-represented. These lipid classes are key components of the cell membrane.
Changes in proportion of phospholipids could result from a defect in synthesis or higher degradation by phospholipases. Interestingly, we noted an up-regulation of lysosomal phospholipase A2 (Pla2g15; log2FC: 2.56; adjusted p-value < 0.01), phospholipase C, beta 2 (Plcb2; log2FC: 1.75; adjusted p-value < 0.001), phospholipase C, epsilon 1 (Plce1; log2FC: 1.67; adjusted p-value < 0.001), phospholipase C, gamma 2 (log2FC: 2.38; adjusted p-value < 0.01), and phospholipase D4 (log2FC: 1.04; adjusted p-value < 0.001). It is therefore plausible that the loss in phospholipids could be due, in part, to higher expression, and potentially higher activity, of phospholipases. We noted that sphingolipids with saturated and mono-unsaturated acyl chain were prone to be dysregulated. The nature of acyl chains in complex lipids may reflect dietary habits as well as the state of peripheral energy metabolism. High content in saturated fatty acids is often associated with increased basal metabolism. Lipidomic analysis in yeast revealed a regulatory role for glucose-sensing pathways in acyl chains in phospholipids (da Silveira Dos Santos et al., 2014). Altered levels of saturated fatty acids were described in the cell fraction of blood of ALS patients and higher content in monounsaturated fatty acids was associated with higher survival (Henriques et al., 2015a). Unsaturation impacts membrane fluidity and low proportion in monounsaturated acyl chains leads to higher stiffness. Disturbance in membrane fluidity is known to impair cellular function such as cell signaling.
Sphingolipids in Spinal Cord from SOD1G86R Mice
Here, we describe, for the first time, common dysregulations in the spinal cord of an animal model of ALS, at gene and lipid levels. Our analysis identified sphingolipid metabolism as the most dysregulated biological pathway. Sphingolipids are complex lipids derived from sphingosine.
These lipids act as structural lipids, signaling molecules (e.g., ceramide, ceramide-phosphate) or as ligands for cell membrane receptors (e.g., spingosine-1-P, lysophosphatidic acid). The causes behind the modifications of sphingolipid metabolism could be multiple. Sphingolipids are involved in key pathways for ALS, such as autophagy and protein clearance, cell survival, energy metabolism, and neuroinflammation. In yeast, the ceramide synthase genes are termed “lag-genes,” for “longevity-associated genes” regulating nutrient sensing, autophagy and lifespan. However, excess in ceramide in rodents promotes accumulation of lipids, triggers endoplasmic reticulum, and lipotoxic stress (Martínez-Sánchez et al., 2017). In multiple sclerosis, increased level of ceramides is suspected to contribute to mitochondrial dysfunctions and axonal damage (Vidaurre et al., 2014). Loss of ceramide in the CNS of SOD1G86R mice could therefore directly impact autophagy process, cell bioenergetics and neuronal integrity. Sphingosine-1-phosphate exerts trophic effects on neuron (Miguez et al., 2015; Geffin et al., 2017) but also on muscle during acute stress such as denervation (Danieli-Betto et al., 2005; Zanin et al., 2008).
Similarly, ceramide-phosphate promotes cell survival through the PI3-K/PKB pathway (Gómez-Muñoz et al., 2005; Gómez-Muñoz, 2006). Interestingly, the upregulation of Cerk and Sphk1, two genes involved in the synthesis of ceramide-phosphate and sphingosine-1-phosphate could be a protective, or compensatory, mechanism for counteracting neurodegeneration. Our results reinforce the critical role of complex lipids, which are dysregulated, in neuronal stress.
At disease onset, SOD1G86R mice present with marked down-regulation of monohexosylceramide (glucosylceramide or galactosylceramide), and lactosylceramide. Similarly to phospholipids, lower levels of these lipids could be due to lower synthesis or higher degradation/conversion. Ugt8a codes for the galactosylceramide synthase and was found down-regulated in SOD1G86R mice. Interestingly, its expression level correlated to disease severity in SOD1G86R mice. It suggests a reduced synthesis of galactosylceramide in SOD1G86R mice, and depends on disease severity. Galactosylceramide and glucosylceramide are precursors of sulfatides and gangliosides, which are particularly abundant in the CNS. Gangliosides in particular are enriched in lipid rafts and contribute to signal transduction. For instance, ganglioside GM1a interacts with TrkA and potentiates BDNF signaling and neuroprotection. Defect in GM1a-TrkA signaling could lead to serious neuronal phenotypes as this complex has been proposed as a binding site for toxins from clostridium perfringens (Oda et al., 2012). Dysregulated glycosphingolipid metabolism could therefore participate in the progression of ALS symptoms in SOD1G86R mice.
Clinical Perspectives for ALS
One issue with data obtained with the SOD1 mice is the translatability to clinic. Many clinical trials, based on preclinical data obtained with the SOD1 models, have failed in ALS. This has raised concerns regarding the reliability of the SOD1 mice as models for ALS. In our opinion, the SOD1G86R line remains a good experimental tool for studying ALS. Indeed, these mice present with the main motor and metabolic symptoms found in ALS patients, although only 2% of patients harbor mutations on the Sod1 gene. Regarding lipid metabolism, both ALS patients and SOD1G86R mice present higher incidence of hypermetabolism (Dupuis et al., 2004; Funalot et al., 2009) and with similar increases in glycosphingolipids in the CNS, such as GM1a (Dodge et al., 2015; Henriques et al., 2017).
Riluzole and edaravone are two drugs approved for the treatment of ALS in the USA. They both have beneficial effects on motor functions and survival in the SOD1G93A mice (Gurney et al., 1996; Waibel et al., 2004; Ito et al., 2008; Del Signore et al., 2009; Shin et al., 2012), although riluzole's effects in SOD1 mice are questioned as they were not confirmed in other studies (Scott et al., 2009; Li et al., 2013; McAllum et al., 2013). Discrepancies between studies could originate from the experimental protocols (e.g., dose, route of administration) or from low rigorousness in study design (e.g., sample size, blinding of experimenters) (Scott et al., 2009; Ludolph et al., 2010). Nutritional interventions, which have shown promising results in ALS patients (Dorst et al., 2013; Wills et al., 2014), extend significantly the survival of SOD1G86R mice (Dupuis et al., 2004). Therefore, we believe that the SOD1 models, and particularly the SOD1G86R model, remain predictive tools for studying pathological processes of ALS.
Our study highlights dysregulations of sphingolipid metabolism in SOD1G86R mice at symptomatic disease stage. As bioactive molecules, sphingolipids interplay with many cell pathways that could be targeted with drug candidates. Pharmacological modulators targeting the metabolism of sphingolipids exist and have been proposed for the treatment of human diseases (Canals et al., 2011). In silico analysis has identified fingolimod and pyrimethamine as tentative modulators for ALS. These two drugs modulate sphingosine-1-phosphate signaling and have been already tested in ALS. Fingolimod improved motor functions and survival of SOD1G93A mice and has been given to ALS patients in a clinical phase 2a safety study. Pyrimethamine has been shown to successfully lower mutant SOD1 proteins in the cerebrospinal fluid of ALS patients carrying SOD1 mutations. Other potential drug candidates have been tentatively identified and most of them have not been investigated in ALS. The facts that our data-driven analysis identified pharmacological compounds already tested in ALS reinforce the relevance of our results.
To conclude, spinal cord of SOD1G86R mice present with profound dysregulations at transcriptomic and metabolomic levels that point to sphingolipids. Our results complement existing data on dysregulation of complex lipids in ALS and could open new therapeutic strategies linked to sphingolipids.
Author Contributions
AH, VC, CK, MS, J-PL, and BW: Conceived and designed the experiments; AH, VC, AB, AM, and CK: Performed the experiments; AH, VC, AB, AM, CK, BW, CB-N, MS, and J-PL: Analysis and interpretation of the data; VC, CB-N, BW, CK, MS, and J-PL: Contributed reagents, materials, and analysis tools; AH, VC, AB, AM, CK, BW, CB-N, MS, and J-PL: Wrote, discussed and approved the final version of the manuscript.
Funding
This work was supported by the European Community's Health Seventh Framework Programme under grant agreement No. 259867 (Euro-MOTOR), Target ALS, “Association André combat la SLA” and “Association pour la Recherche et le Développement de Moyens de Lutte contre les Maladies Neurodégénératives” (AREMANE). RNA-sequencing was performed by the IGBMC GenomEast platform, a member of the “France Génomique” consortium (ANR-10-INBS-0009).
Conflict of Interest Statement
AH and MS are employees of Spedding Research Solutions SAS. VC, BW, and CB-N are employees of Les Laboratoires Servier.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2017.00433/full#supplementary-material
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology : tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bastiaansen, A. J., Karper, J. C., Wezel, A., de Boer, H. C., Welten, S. M., de Jong, R. C., et al. (2014). TLR4 accessory molecule RP105 (CD180) regulates monocyte-driven arteriogenesis in a murine hind limb ischemia model. PLoS ONE 9:e99882. doi: 10.1371/journal.pone.0099882
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300.
Canals, D., Perry, D. M., Jenkins, R. W., and Hannun, Y. A. (2011). Drug targeting of sphingolipid metabolism : sphingomyelinases and ceramidases. Br. J. Pharmacol. 163, 694–712. doi: 10.1111/j.1476-5381.2011.01279.x
Carbia-nagashima, A., Gerez, J., Perez-castro, C., Paez-pereda, M., Silberstein, S., Stalla, K., et al. (2007). RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1 a during Cell 13, 309–323. doi: 10.1016/j.cell.2007.07.044
Cheng, D., Jenner, A. M., Shui, G., Cheong, W. F., Mitchell, T. W., Jessica, R., et al. (2011). Lipid pathway alterations in Parkinson ' s disease primary visual cortex. PLoS ONE 6:e17299. doi: 10.1371/journal.pone.0017299
Cooper-knock, J., Green, C., Altschuler, G., Wei, W., Bury, J. J., Heath, P. R., et al. (2017). A data-driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 5:23. doi: 10.1186/s40478-017-0424-x
Cutler, R. G., Pedersen, W. A., Camandola, S., Rothstein, J. D., and Mattson, M. P. (2002). Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress – induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 52, 448–457. doi: 10.1002/ana.10312
Danieli-Betto, D., Germinario, E., Esposito, A., Megighian, A., Midrio, M., Ravara, B., et al. (2005). Sphingosine 1-phosphate protects mouse extensor digitorum longus skeletal muscle during fatigue. Am. J. Physiol. Cell Physiol. 288, 1367–1373. doi: 10.1152/ajpcell.00246.2004
Del Signore, S. J., Amante, D. J., Kim, J., Stack, E. C., Goodrich, S., Cormier, K., et al. (2009). Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph. Lateral Scler. 10, 85–94. doi: 10.1080/17482960802226148
Desseille, C., Deforges, S., Biondi, O., Houdebine, L., D'amico, D., Lamazière, A., et al. (2017). Specific physical exercise improves energetic metabolism in the skeletal muscle of amyotrophic-lateral- sclerosis mice. Front. Mol. Neurosci. 10:332. doi: 10.3389/fnmol.2017.00332
Dodge, J. C. (2017). lipid involvement in neurodegenerative diseases of the motor system : insights from lysosomal storage diseases. Front. Mol. Neurosci. 10:356. doi: 10.3389/fnmol.2017.00356
Dodge, J. C., Treleaven, C. M., Pacheco, J., Cooper, S., Bao, C., Abraham, M., et al. (2015). Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 1:201508767. doi: 10.1073/pnas.1508767112
Dorst, J., Cypionka, J., and Ludolph, A. C. (2013). High-caloric food supplements in the treatment of amyotrophic lateral sclerosis: a prospective interventional study. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 533–536. doi: 10.3109/21678421.2013.823999
Dorst, J., Dupuis, L., Petri, S., Kollewe, K., Abdulla, S., Wolf, J., et al. (2015). Percutaneous endoscopic gastrostomy in amyotrophic lateral sclerosis: a prospective observational study. J. Neurol. 262, 849–858. doi: 10.1007/s00415-015-7646-2
Druker, J., Liberman, A. C., Antunica-Noguerol, M., Gerez, J., Paez-pereda, M., Rein, T., et al. (2013). RSUME enhances glucocorticoid receptor SUMOylation and transcriptional activity. Mol. Cell Biol. 33, 2116–2127. doi: 10.1128/MCB.01470-12
Dupuis, L., Corcia, P., Fergani, A., Gonzalez De Aguilar, J.-L., Bonnefont-Rousselot, D., Bittar, R., et al. (2008). Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27
Dupuis, L., Oudart, H., René, F., Gonzalez de Aguilar, J.-L., and Loeffler, J.-P. (2004). Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. U.S.A. 101, 11159–11164. doi: 10.1073/pnas.0402026101
El Oussini, H., Bayer, H., Scekic-Zahirovic, J., Vercruysse, P., Sinniger, J., and Dirrig-Grosch, S. (2016). Serotonin 2B receptor slows disease progression and prevents degeneration of spinal cord mononuclear phagocytes in amyotrophic lateral sclerosis. Acta Neuropathol. 131, 465–480. doi: 10.1007/s00401-016-1534-4
Endo, F., Komine, O., Wyss-coray, T., Yamanaka, K., Endo, F., Komine, O., et al. (2015). Astrocyte-derived TGF-β1 accelerates disease progression in ALS Mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 11, 592–604. doi: 10.1016/j.celrep.2015.03.053
Enjin, A., Rabe, N., Nakanishi, S. T., Vallstedt, A., Gezelius, H., Memic, F., et al. (2010). Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as markers for fast motor neurons and partition cells. J. Comp. Neurol. 518, 2284–2304. doi: 10.1002/cne.22332
Funalot, B., Desport, J.-C., Sturtz, F., Camu, W., and Couratier, P. (2009). High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 113–117. doi: 10.1080/17482960802295192
Geffin, R., Martinez, R., de Las Pozas, A., Issac, B., and Mccarthy, M. (2017). Fingolimod induces neuronal-specific gene expression with potential neuroprotective outcomes in maturing neuronal progenitor cells exposed to HIV. J. Neurovirol. 23, 808–824. doi: 10.1007/s13365-017-0571-7
Gómez-Muñoz, A. (2006). Ceramide 1-phosphate / ceramide, a switch between life and death. Biochem. Biophys. Acta 1758, 2049–2056. doi: 10.1016/j.bbamem.2006.05.011
Gómez-Muñoz, A., Kong, J., Parhar, K., Wang, S., Gangoiti, P., Gonzalez, M., et al. (2005). Ceramide-1-phosphate promotes cell survival through activation of the phosphatidylinositol 3-kinase / protein kinase B pathway. FEBS Lett. 579, 3744–3750. doi: 10.1016/j.febslet.2005.05.067
Grad, L. I., Yerbury, J. J., Turner, B. J., Guest, W. C., Pokrishevsky, E., and O'Neill, M. A. (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 111, 3620–3625. doi: 10.1073/pnas.1312245111
Gurney, M. E., Cutting, F. B., Zhai, P., Doble, A., Taylor, C. P., Andrus, P. K., et al. (1996). Benefit of Vitamin E, Riluzole, and Gabaentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol. 39, 147–157. doi: 10.1002/ana.410390203
Halevy, T., Biancotti, J., Yanuka, O., Golan-lev, T., and Benvenisty, N. (2016). Molecular Characterization of down syndrome embryonic stem cells reveals a role for RUNX1 in neural differentiation. Stem Cell Rep. 7, 777–786. doi: 10.1016/j.stemcr.2016.08.003
Henriques, A., Blasco, H., Fleury, M.-C., Corcia, P., Echaniz-LAguna, A., Robelin, L., et al. (2015a). Blood cell palmitoleate-palmitate ratio is an independent prognostic factor for amyotrophic lateral sclerosis. PLoS ONE 10:e0131512. doi: 10.1371/journal.pone.0131512
Henriques, A., Croixmarie, V., Priestman, D. A., Rosenbohm, A., Dirrig-Grosch, S., D'Ambra, E., et al. (2015b). Amyotrophic lateral sclerosis and denervation alter sphingolipids and up-regulate glucosylceramide synthase. Hum. Mol. Genet. 24, 7390–7405. doi: 10.1093/hmg/ddv439
Henriques, A., and Gonzalez De Aguilar, J.-L. (2011). Can transcriptomics cut the gordian knot of amyotrophic lateral sclerosis? Curr. Genomics 12, 506–515. doi: 10.2174/138920211797904043
Henriques, A., Huebecker, M., Blasco, H., Keime, C., Andres, C. R., Corcia, P., et al. (2017). Inhibition of β - Glucocerebrosidase activity preserves motor unit integrity in a mouse model of amyotrophic lateral sclerosis. Sci. Rep. 7:5235. doi: 10.1038/s41598-017-05313-0
Henriques, A., Kastner, S., Chatzikonstantinou, E., Pitzer, C., Plaas, C., Kirsch, F., et al. (2015c). Gene expression changes in spinal motoneurons of the SOD1 G93A transgenic model for ALS after treatment with. Front. Cell. Neurosci. 8:464. doi: 10.3389/fncel.2014.00464
Henriques, A., Pitzer, C., Dittgen, T., Klugmann, M., Dupuis, L., and Schneider, A. (2011). CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol. Ther. 19, 284–292. doi: 10.1038/mt.2010.271
Hooten, K. G., Beers, D. R., Zhao, W., and Appel, S. H. (2015). Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics 12, 364–375. doi: 10.1007/s13311-014-0329-3
Ito, H., Wate, R., Zhang, J., Ohnishi, S., Kaneko, S., Ito, H., et al. (2008). Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 213, 448–455. doi: 10.1016/j.expneurol.2008.07.017
Jésus, P., Fayemendy, P., Nicol, M., Lautrette, G., Sourisseau, H., Preux, P.-M., et al. (2018). Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 25, 97–104. doi: 10.1111/ene.13468
Kamburov, A., Cavill, R., Ebbels, T. M. D., Herwig, R., and Keun, H. C. (2011a). Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 27, 2917–2918. doi: 10.1093/bioinformatics/btr499
Kamburov, A., Pentchev, K., Galicka, H., Wierling, C., Lehrach, H., and Herwig, R. (2011b). ConsensusPathDB : toward a more complete picture of cell biology. Nucleic Acids Res. 39, 712–717. doi: 10.1093/nar/gkq1156
Kamburov, A., Wierling, C., Lehrach, H., and Herwig, R. (2009). ConsensusPathDB — a database for integrating human functional interaction networks. Nucleic Acids Res. 37, 623–628. doi: 10.1093/nar/gkn698
Kilbey, A., Terry, A., Jenkins, A., Borland, G., Zhang, Q., Wakelam, M. J., et al. (2010). Runx regulation of sphingolipid metabolism and survival signaling. Cancer Res. 70, 5860–5870. doi: 10.1158/0008-5472.CAN-10-0726
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Korkut, C., Li, Y., Koles, K., Brewer, C., Ashley, J., Yoshihara, M., et al. (2013). Report regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron 77, 1039–1046. doi: 10.1016/j.neuron.2013.01.013
Lee, J. K., Shin, J. H., Lee, J. E., and Choi, E.-J. (2015). Role of autophagy in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1852, 2517–2524. doi: 10.1016/j.bbadis.2015.08.005
Li, J., Sung, M., and Rutkove, S. B. (2013). Electrophysiologic biomarkers for assessing disease progression and the effect of Riluzole in SOD1 G93A ALS Mice. PLoS ONE 8:e65976. doi: 10.1371/journal.pone.0065976
Ludolph, A. C., Bendotti, C., Blaugrund, E., Chio, A., Greensmith, L., Loeffler, J., et al. (2010). Guidelines for preclinical animal research in ALS / MND : a consensus meeting. Amyotroph. Lateral Scler. 11, 38–45. doi: 10.3109/17482960903545334
Mariosa, D., Hammar, N., Malmström, H., Ingre, C., Jungner, I., and Ye, W. (2017). Blood Biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis : a more than 20-year follow-up of the Swedish AMORIS Cohort. Ann. Neurol. 81, 718–728. doi: 10.1002/ana.24936
Martínez-Sánchez, N., Seoane-Collazo, P., Contreras, C., Varela, L., Villarroya, J., Rial-Pensado, E., et al. (2017). Hypothalamic AMPK-ER stress-JNK1 axis mediates the central actions of thyroid hormones on energy article axis mediates the central actions of thyroid hormones on energy balance. Cell Metab. 26, 212–229. doi: 10.1016/j.cmet.2017.06.014
McAllum, E. J., Lim, N. K.-H., Hickey, J. L., Paterson, B. M., Donnelly, P. S., Li, Q.-X., et al. (2013). Therapeutic effects of Cu (atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 586–590. doi: 10.3109/21678421.2013.824000
Miguez, A., Barriga, G. G., Brito, V., Straccia, M., Giralt, A., Ginés, S., et al. (2015). Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington' s disease by preventing p75 NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 24, 4958–4970. doi: 10.1093/hmg/ddv218
Minamiyama, M., Katsuno, M., Adachi, H., Doi, H., Kondo, N., Iida, M., et al. (2012). Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract. Nat. Med. 18, 1531–1538. doi: 10.1038/nm.2932
Mitsui, S., Hidaka, C., Furihata, M., Osako, Y., and Yuri, K. (2013). A mental retardation gene, motopsin / prss12, modulates cell morphology by interaction with seizure-related gene 6. Biochem. Biophys. Res. Commun. 436, 638–644. doi: 10.1016/j.bbrc.2013.04.112
Nardo, G., Trolese, M. C., Vito, G., De Cecchi, R., Riva, N., Dina, G., et al. (2016). Immune response in peripheral axons delays disease progression in SOD1 G93A mice. J. Neuroinflammation 13:261. doi: 10.1186/s12974-016-0732-2
Nonaka, T., Masuda-suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Article prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Numajiri, T., Mitsui, S., Hisa, Y., and Ishida, T. (2006). The expression of a motoneuron-specific serine protease, motopsin (PRSS12), after facial nerve axotomy in mice. J. Plast. Reconstr. Aesthetic Surg. 59, 393–397. doi: 10.1016/j.bjps.2005.04.018
Oda, M., Kabura, M., Takagishi, T., Suzue, A., Tominaga, K., Urano, S., et al. (2012). Clostridium perfringens alpha-toxin recognizes the GM1a-TrkA complex. J. Biol. Chem. 287, 33070–33079. doi: 10.1074/jbc.M112.393801
Paganoni, S., Deng, J., Jaffa, M., Cudkowicz, M. E., and Wills, A.-M. (2011). Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 44, 20–24. doi: 10.1002/mus.22114
Palamiuc, L., Schlagowski, A., Ngo, S. T., Vernay, A., Dirrig-Grosch, S., Henriques, A., et al. (2015). A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 7, 526–546. doi: 10.15252/emmm.201404433
Patin, F., Baranek, T., Vourc'h, P., Nadal-Desbarats, L., Goossens, J.-F., Marouillat, S., et al. (2016). Combined metabolomics and transcriptomics approaches to assess the IL-6 blockade as a therapeutic of ALS: deleterious alteration of lipid metabolism. Neurotherapeutics 13, 905–917. doi: 10.1007/s13311-016-0461-3
Pinto, S., Cunha, C., Barbosa, M., Vaz, A. R., and Brites, D. (2017). Exosomes from NSC-34 Cells transfected with hSOD1-G93A Are Enriched in miR-124 and drive alterations in microglia phenotype. Front. Neurosci. 11:273. doi: 10.3389/fnins.2017.00273
Raman, R., Allen, S. P., Goodall, E. F., Kramer, S., Ponger, L. L., Heath, P. R., et al. (2015). Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 41, 201–226. doi: 10.1111/nan.12147
Ringer, C., Weihe, E., and Schütz, B. (2012). Calcitonin gene-related peptide expression levels predict motor neuron vulnerability in the superoxide dismutase 1-G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 45, 547–554. doi: 10.1016/j.nbd.2011.09.011
Ripps, M. E., Huntleyt, G. W., Hof, P. R., Morrisontt, J. H., and Gordont, J. W. (1995). Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 92, 689–693. doi: 10.1073/pnas.92.3.689
Rosen, D., Siddique, T., Patterson, D., Figlewicz, D., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Safdar, A., Saleem, A., and Tarnopolsky, M. A. (2016). The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat. Rev. Endocrinol. 12, 504–517. doi: 10.1038/nrendo.2016.76
Schmitt, F., Hussain, G., Dupuis, L., Loeffler, J.-P., and Henriques, A. (2014). A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front. Cell. Neurosci. 8:25. doi: 10.3389/fncel.2014.00025
Scott, S., Kranz, J. E., Cole, J., Lincecum, J. M., Thompson, K., Kelly, N., et al. (2009). Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. 9, 4–15. doi: 10.1080/17482960701856300
Shin, J. H., Lee, Y. A., Lee, J. K., Lee, Y. B., Cho, W., Im, D. S., et al. (2012). Concurrent blockade of free radical and microsomal prostaglandin E synthase-1-mediated PGE2 production improves safety and efficacy in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 122, 952–961. doi: 10.1111/j.1471-4159.2012.07771.x
Taylor, J. P., Brown Jr, R. H., and Cleveland, D. W. (2016). Decoding ALS : from genes to mechanism. Nature 539:197. doi: 10.1038/nature20413
The Gene Ontology Consortium (2015). Gene Ontology Consortium : going forward. Nucleic Acids Res. 43, 1049–1056. doi: 10.1093/nar/gku1179
Vercruysse, P., Sinniger, J., El Oussini, H., Scekic-Zahirovic, J., Dieterlé, S., Dengler, R., et al. (2016). Alterations in the hypothalamic melanocortin pathway in amyotrophic lateral sclerosis. Brain 139, 1106–1122. doi: 10.1093/brain/aww004
Vidaurre, O. G., Haines, J. D., Katz Sand, I., Adula, K. P., Huynh, J. L., Mcgraw, C. A., et al. (2014). Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain 137, 2271–2286. doi: 10.1093/brain/awu139
Wagner, A. H., Coffman, A. C., Ainscough, B. J., Spies, N. C., Skidmore, Z. L., Campbell, K. M., et al. (2016). DGIdb 2. 0 : mining clinically relevant drug – gene interactions. 44, 1036–1044. doi: 10.1093/nar/gkv1165
Waibel, S., Reuter, A., Malessa, S., Blaugrund, E., and Ludolph, A. C. (2004). Rasagiline alone and in combination with riluzole prolongs survivald in an ALS mouse model. J. Neurol. 251, 1080–1084. doi: 10.1007/s00415-004-0481-5
Wang, J. K. T., Langfelder, P., Horvath, S., and Palazzolo, M. (2017). Exosomes and homeostatic synaptic plasticity are linked to each other and to Huntington's, Parkinson's and other neurodegenerative diseases by database-enabled analyses of comprehensively curated datasets. Front. Neurosci. 11:149. doi: 10.3389/fnins.2017.00149
Wills, A.-M., Hubbard, J., Macklin, E. A., Glass, J., Tandan, R., Simpson, E. P., et al. (2014). Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet 383, 2065–2072. doi: 10.1016/S0140-6736(14)60222-1
Wood, P. L., Medicherla, S., Sheikh, N., Terry, B., Phillipps, A., Kaye, J. A., et al. (2016). Targeted lipidomics of fontal cortex and plasma Diacylglycerols (DAG) in Mild Cognitive Impairment (MCI) and Alzheimer's disease: validation of DAG Accumulation early in the pathophysiology of Alzheimer's disease. J. Alzheimers Dis. 48, 537–546. doi: 10.3233/JAD-150336
da Silveira Dos Santos, A. X., Riezman, I., Aguilera-romero, M., David, F., Piccolis, M., and Loewith, R. (2014). Systematic lipidomic analysis of yeast protein kinase and phosphatase mutants reveals novel insights into regulation of lipid homeostasis. Mol. Biol. Cell 25, 3234–3246. doi: 10.1091/mbc.E14-03-0851
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 2007–2009. doi: 10.1038/nn2047
Yoshikawa, M., Hirabayashi, M., Ito, R., Ozaki, S., Aizawa, S., Masuda, T., et al. (2015). Contribution of the Runx1 transcription factor to axonal pathfinding and muscle innervation by hypoglossal motoneurons. Dev. Neurobiol. 75, 1295–1314. doi: 10.1002/dneu.22285
Yoshikawa, M., Masuda, T., Kobayashi, A., Senzaki, K., and Ozaki, S. (2016). Molecular and cellular neuroscience Runx1 contributes to the functional switching of bone morphogenetic protein 4 (BMP4) from neurite outgrowth promoting to suppressing in dorsal root ganglion. Mol. Cell. Neurosci. 72, 114–122. doi: 10.1016/j.mcn.2016.02.001
Keywords: amyotrophic lateral sclerosis, RNA-sequencing, transcriptomics, metabolomics, SOD1 mice, sphingolipids, therapy, lipids
Citation: Henriques A, Croixmarie V, Bouscary A, Mosbach A, Keime C, Boursier-Neyret C, Walter B, Spedding M and Loeffler J-P (2018) Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 10:433. doi: 10.3389/fnmol.2017.00433
Received: 31 October 2017; Accepted: 15 December 2017;
Published: 04 January 2018.
Edited by:
Isabel Varela-Nieto, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Yolanda Leon, Universidad Autonoma de Madrid, SpainAdam Keith Walker, Macquarie University, Australia
Chiara Donati, University of Florence, Italy
Copyright © 2018 Henriques, Croixmarie, Bouscary, Mosbach, Keime, Boursier-Neyret, Walter, Spedding and Loeffler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Henriques, aGVucmlxdWVzLmFAZ214LmNvbQ==
Jean-Philippe Loeffler, bG9lZmZsZXJAdW5pc3RyYS5mcg==