 Marwa Elamin
Marwa Elamin David N. Ruskin
David N. Ruskin Susan A. Masino
Susan A. Masino Paola Sacchetti
Paola Sacchetti- 1Neuroscience Program, Department of Biology, University of Hartford, West Hartford, CT, United States
- 2Neuroscience Program and Psychology Department, Trinity College, Hartford, CT, United States
The ketogenic diet’s (KD) anticonvulsant effects have been well-documented for nearly a century, including in randomized controlled trials. Some patients become seizure-free and some remain so after diet cessation. Many recent studies have explored its expanded therapeutic potential in diverse neurological disorders, yet no mechanism(s) of action have been established. The diet’s high fat, low carbohydrate composition reduces glucose utilization and promotes the production of ketone bodies. Ketone bodies are a more efficient energy source than glucose and improve mitochondrial function and biogenesis. Cellular energy production depends on the metabolic coenzyme nicotinamide adenine dinucleotide (NAD), a marker for mitochondrial and cellular health. Furthermore, NAD activates downstream signaling pathways (such as the sirtuin enzymes) associated with major benefits such as longevity and reduced inflammation; thus, increasing NAD is a coveted therapeutic endpoint. Based on differential NAD+ utilization during glucose- vs. ketone body-based acetyl-CoA generation for entry into the tricarboxylic cycle, we propose that a KD will increase the NAD+/NADH ratio. When rats were fed ad libitum KD, significant increases in hippocampal NAD+/NADH ratio and blood ketone bodies were detected already at 2 days and remained elevated at 3 weeks, indicating an early and persistent metabolic shift. Based on diverse published literature and these initial data we suggest that increased NAD during ketolytic metabolism may be a primary mechanism behind the beneficial effects of this metabolic therapy in a variety of brain disorders and in promoting health and longevity.
Introduction: Ketogenic Diet and Disorders of The Nervous System
A diet high in fat, low in carbohydrate and sufficient in protein will automatically shift the dependency of energy production in the body from primarily glucose to primarily ketone bodies and is termed a “ketogenic diet” (KD; Branco et al., 2016; Masino, 2017). This dietary approach was developed nearly 100 years ago as metabolic therapy to mimic the metabolic changes that occur during fasting after observing that upon halting food intake, seizures would stop in epileptic people. The KD is well-established as a treatment for epileptic seizures and variations of the diet can be used in children and adults and can be more effective than medication in stopping seizures (Pulford, 1927; Neal et al., 2008). The KD can also prevent seizure progression (epileptogenesis) in animal models and patients (Muller-Schwarze et al., 1999; Neal et al., 2008; Lusardi et al., 2015). Some patients become seizure-free, and remain so even after diet cessation (Martinez et al., 2007; Patel et al., 2010; Caraballo et al., 2011). These lasting outcomes are likely to rely on epigenetic changes (Boison, 2017).
Metabolic dysfunction is increasingly appreciated as a fundamental pathology across disease states (Zhu and Chu, 2010; García-Escudero et al., 2013; Pathak et al., 2013). In models of neurodegenerative diseases, metabolic therapy with a KD or analogous ketone-enhancing metabolic strategies have beneficial effects in cultured neurons, animal models, and in patients. The ketone body β-hydroxybutyrate (β-OHB) protected cultured dopaminergic substantia nigra cells from N-methyl-4-phenylpyridinium (MPP+) toxicity and hippocampal neurons from amyloid β toxicity (Kashiwaya et al., 2000), and improved the disease rating score in Parkinsonian patients (Vanitallie et al., 2005). In vivo and in vitro administration of ketone esters reduced histological and biochemical pathologies and improved cognition, anxiety and motor performance in mouse models of Alzheimer’s disease (Liu et al., 2011; Hui et al., 2012; Brownlow et al., 2013; Kashiwaya et al., 2013; Zhang et al., 2013; Pawlosky et al., 2017). KD improved memory of patients with mild cognitive impairment (Krikorian et al., 2012), and administration of a ketone ester or medium chain triglycerides (often a component of ketogenic treatment) enhanced memory and cognition in Alzheimer’s patients (Reger et al., 2004; Newport et al., 2015; Cunnane et al., 2016). Treatment with a KD suppressed inflammation and improved motor disabilities in a multiple sclerosis model (Kim et al., 2012), altered disease progression and improved motor performance and neuronal survival in an amyotrophic lateral sclerosis model (Zhao et al., 2006), decreased the expression of apoptotic mediators in a traumatic brain injury model (Hu et al., 2009), and improved motor outcomes in a spinal cord injury model (Streijger et al., 2013).
It is becoming apparent that beneficial effects of ketogenic therapy extend beyond epilepsy, neurodegenerative disorders, and brain/spinal cord injury. The KD is broadly effective in improving core behavioral symptoms in animal models of autism spectrum disorder (Ruskin et al., 2013b, 2017a,b; Ahn et al., 2014; Verpeut et al., 2016; Castro et al., 2017; Dai et al., 2017), and in autistic patients (Evangeliou et al., 2003; Masino et al., 2011b; Spilioti et al., 2013). The KD is receiving growing interest in oncology as tumors are highly glucose-dependent (the Warburg effect; Seyfried and Mukherjee, 2005; Zuccoli et al., 2010; Schmidt et al., 2011; Klement et al., 2016; Lussier et al., 2016; Khodadadi et al., 2017). Also, due to the high efficiency of metabolizing fat when carbohydrates are minimal (Forsythe et al., 2010), the KD has been promoted for weight reduction (Jenkins et al., 2009; Partsalaki et al., 2012; Paoli, 2014; Gomez-Arbelaez et al., 2017) and for treatment or reversal of type II diabetes and metabolic syndrome (Yancy et al., 2005; Volek et al., 2008, 2009; Westman et al., 2008; Hussain et al., 2012; Tay et al., 2015; McKenzie et al., 2017).
In addition, healthy, disease-free cells and animals can also benefit from this therapy. The use of ketone bodies as an energy source appears to be associated with a healthier metabolic phenotype that renders cells more resistant to external insults. Ketogenic treatment decreased myocardial damage after ischemic injury, reduced lung injury after hemorrhagic shock, enhanced kidney resistance to oxidative stress, and protected neurons against glutamate-induced toxicity (Zou et al., 2002; Koustova et al., 2003; Noh et al., 2006; Shimazu et al., 2013). At the cognitive level, beneficial effects on learning and memory were reported (Brownlow et al., 2017; Newman et al., 2017). KD in mice started at 8 weeks of age did not affect longevity (Douris et al., 2015); however, KD started midlife extends longevity and healthspan (Newman et al., 2017; Roberts et al., 2017).
Mechanisms of Ketogenic Therapy: Evidence for Increased NAD+
Many mechanisms have been proposed to explain the anti-seizure and neuroprotective effects of the diet, such as enhanced mitochondrial biogenesis (Bough et al., 2006), decreased formation of reactive oxygen radicals (Sullivan et al., 2004), altered transmitter levels and ion channel function (Schwartzkroin, 1999; Bough and Rho, 2007), increased adenosine (Masino et al., 2011a; Masino and Rho, 2012), and decreased DNA methylation (Kobow et al., 2013; Lusardi et al., 2015). Each one of these mechanisms could account for some of the beneficial effects of the ketogenic therapy. However, to date fundamental metabolic mechanism(s) which could explain diverse beneficial effects across numerous diseases have yet to be confirmed. If uncovered, such mechanism(s) could provide a fundamental answer to “how does the KD work?”—a lingering question and a topic of intense resurgent research efforts and clinical interest. A unifying mechanism of action could also serve as a target for the development of therapeutics that enhance cellular and metabolic health and provide the metabolic resilience necessary to prevent and combat neurological diseases.
Glucose and ketone bodies are used to provide energy in the form of ATP. Many tissues in the body—such as muscle tissues—can oxidize fatty acids to produce energy. As one exception, in the central nervous system ketolysis is expected to be the primary pathway of energy production: neurons and oligodendrocytes have a limited capacity for mitochondrial fatty acid β oxidation (Edmond, 1992; Achanta and Rae, 2017). The ketone bodies acetoacetate (AcAc) and β-OHB are therefore the main energy source in the brain during ketosis, and the metabolism of glucose versus ketone bodies results in a differential reduction rate of nicotinamide adenine dinucleotide (NAD), an essential metabolic coenzyme and signaling molecule. NAD exists in oxidized and reduced forms, NAD+ and NADH, respectively, and whereas both glucose and ketone pathways each produce two molecules of acetyl-CoA, glucose reduces four molecules of NAD+ and ketone bodies reduce either one (β-OHB), or none (AcAc) during acetyl-CoA synthesis (Lodish et al., 2000; Cotter et al., 2013; Figure 1). A decreased reduction of NAD+ in the brain would be expected to result in increased NAD+/NADH ratio, with more oxidized molecules available for bioenergetic demands.
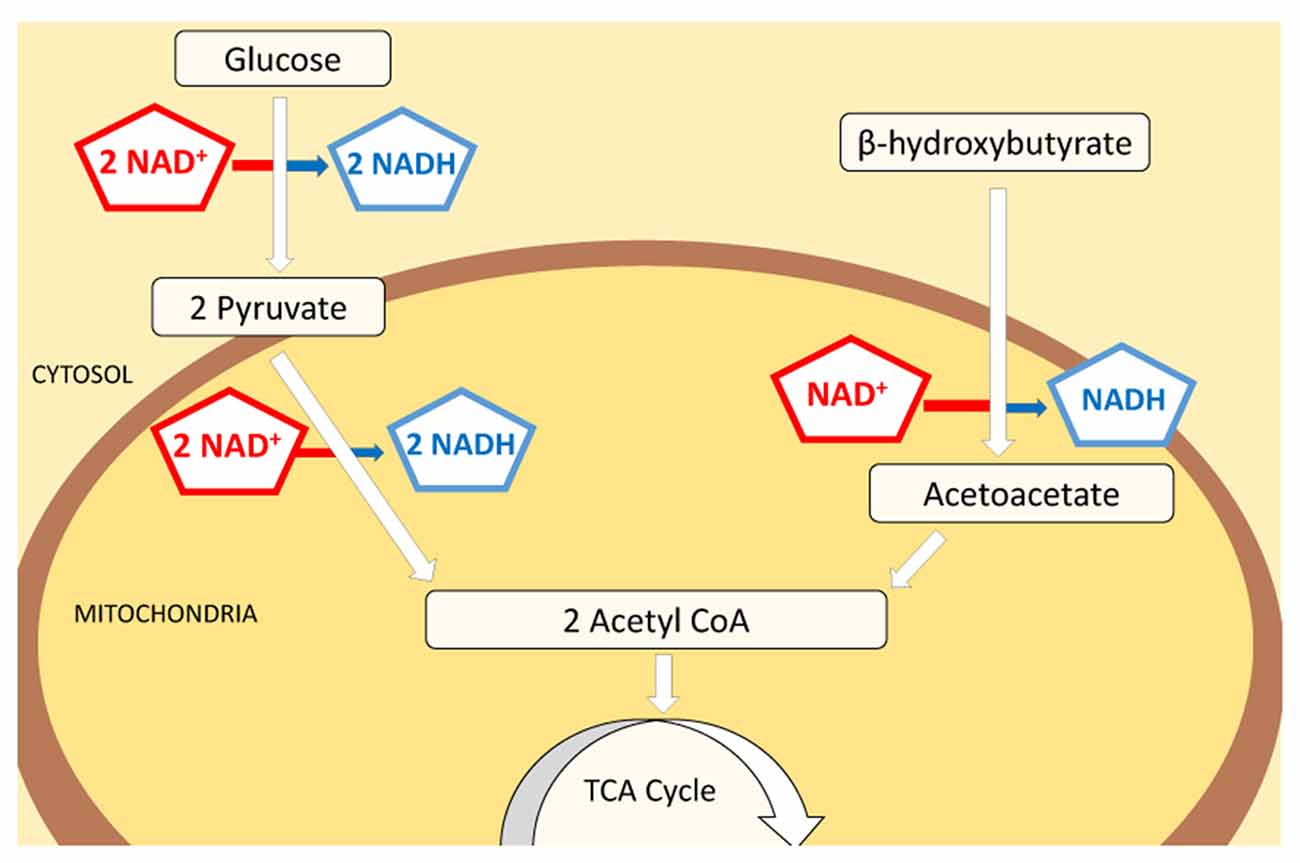
Figure 1. Schematic of NAD+ consumption during metabolism of glucose vs. ketone bodies. Both glucose and ketone bodies lead to the formation of two molecules of acetyl-CoA which subsequently enter the citric acid cycle and participate in energy production. Although glucose provides a higher final yield of ATP, the consumption of NAD+ is significantly higher in this pathway (4:1). Glucose will reduce 111 molecules of NAD+ per 1000 molecules of ATP made, while ketone bodies reduce only 41 to produce a comparable amount of ATP. Decreased use of NAD+ by ketone bodies in energy production pathways could increase the amount of free NAD+ available as substrate for enzymes and cellular signaling processes.
Increasing the NAD+/NADH ratio has multiple important implications: improved bioavailability of NAD+ molecules has been linked to anti-aging (Scheibye-Knudsen et al., 2014), longevity (van der Veer et al., 2007; Zhang et al., 2016) and other potentially beneficial effects. For example, an increased NAD+/NADH ratio was found to enhance mitochondrial function and protect against oxidative stress, and diverse research has shown that NAD molecules play an important role in cellular respiration, mitochondrial biogenesis and redox reactions (Yang and Sauve, 2016). NAD+ also serves as substrate for enzymes affecting cellular functions ranging from gene expression to post-translational protein modifications, such as deacetylation and ADP-ribosylation (Belenky et al., 2007).
We propose that the decreased reduction rate of NAD+ to NADH during ketone-based metabolism increases availability of NAD+ and thus alters the NAD+/NADH ratio. This would occur during sufficient exogenous ketone administration or during fasting or adhering to a KD, i.e., when ketones are used as a main source of energy. Considering the pivotal role of NAD+ in cellular health, and that differential NAD reduction is inherent in this metabolic pathway, we suggest that this differential rate of NAD+ reduction (and thus an increase in NAD+ availability) is a primary mechanism of ketogenic therapy: increased NAD+ can potentially be the starting point for many of the diverse benefits of this metabolic treatment.
Ketogenic Diet-Induced Increase in Brain NAD+/NADH Ratio Is Rapid, Persistent and Region Specific
As an initial test of this hypothesis, we quantified and compared KD-induced changes in blood ketones and in NAD+/NADH ratio in hippocampus and cerebral cortex of normal adult rats. Sprague-Dawley male rats (Trinity College, age 9–14 weeks, n = 20) were fed ad libitum either a standard chow diet (CD; Purina 5001; PharmaServ, Framingham, MA, USA), or a 6:1 [fat: (protein + carbohydrates); #F3666; Bio-Serv, Frenchtown, NJ, USA] KD for 2 days or 3 weeks. Analysis of trunk serum collected showed that a KD induced a significant increase in ketone bodies (β-OHB; the primary circulating ketone body; Figure 2A), consistent with previous experimental and clinical work (Nabbout et al., 2010; Ruskin et al., 2013a).
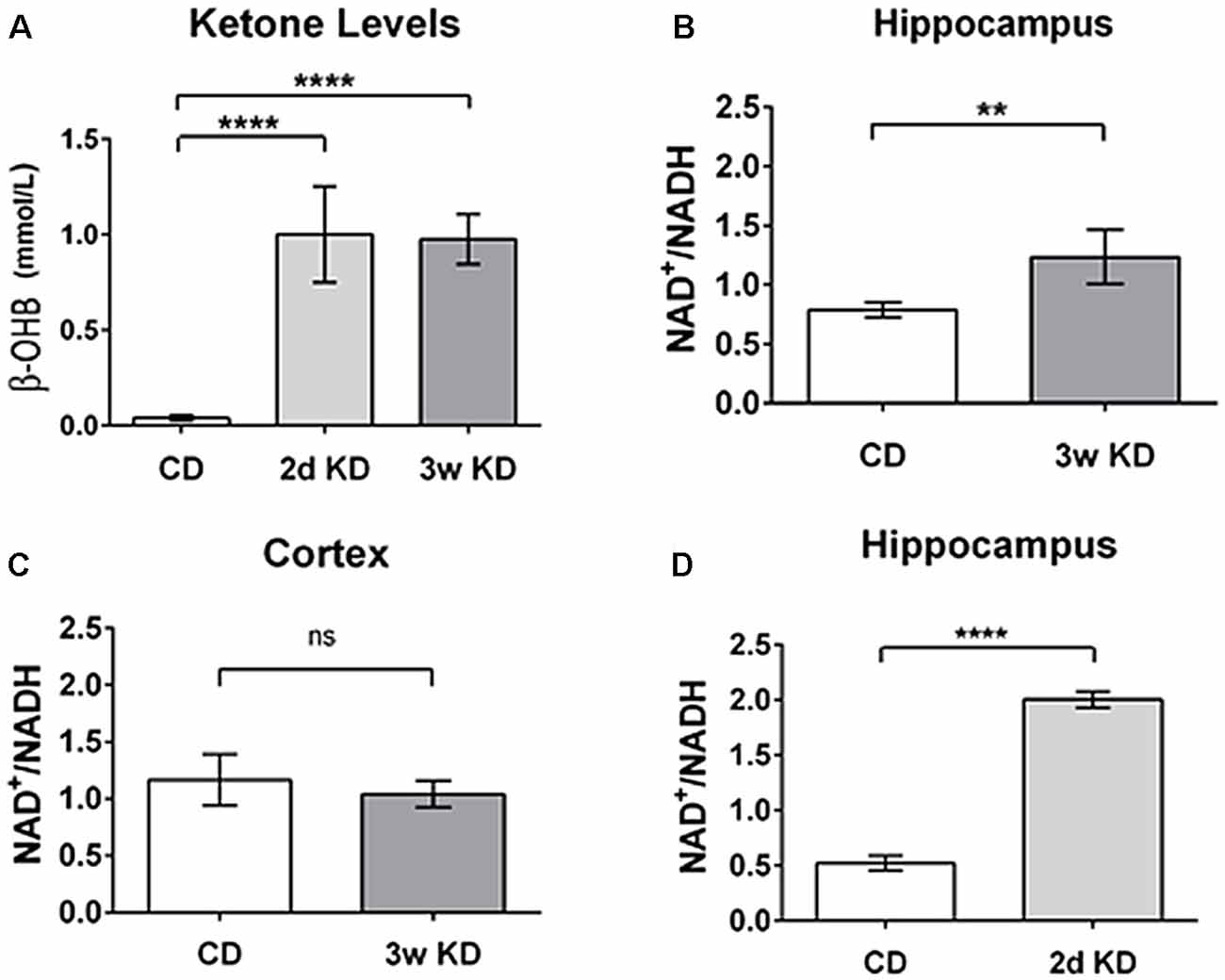
Figure 2. Changes in blood ketones and brain nicotinamide adenine dinucleotide (NAD) after ketogenic diet (KD) treatment. (A) Blood levels of β-hydroxybutyrate (β-OHB; mmol/L) after 2 days (2d KD; n = 3) and 3 weeks (3w KD; n = 4) of KD treatment vs. control chow diet (CD; n = 8; P < 0.0001). (B) Hippocampal changes in NAD+/NADH ratio after 3 weeks KD treatment. A significant increase in the NAD+/NADH ratio was quantified in the hippocampi of animals fed KD for 3 weeks (n = 4) vs. animals maintained on control diet (n = 8; P < 0.005). (C) Cortical NAD+/NADH ratio after 3 weeks KD treatment. No differences were detected in NAD+/NADH ratio in frontal cortex between the dietary groups. Control CD (n = 8); KD 3 weeks (3w KD; n = 4; P = ns). (D) NAD+/NADH ratios in the hippocampus after 2 days KD treatment. A significant increase in the NAD+/NADH ratio was quantified in hippocampi obtained from animals fed KD for 2 days (2d KD; n = 3) compared to animals maintained on control diet (CD; n = 5; P < 0.0001). All comparisons were unpaired t-tests. Data are expressed as mean ± SEM. **P < 0.005; ****P < 0.0001.
To begin to evaluate if, when and where alterations in NAD occurred in brain during ketone-based metabolism we performed NAD analysis on two regions, hippocampus and frontal cortex, after either a 2 days or a 3 weeks KD treatment. Three weeks exposure has been demonstrated to impact behavior and neuronal excitability in diverse paradigms (Hori et al., 1997; Cullingford et al., 2002; Ruskin et al., 2009; Masino et al., 2011a). As demonstrated by ketone body levels (Figure 2A), metabolic changes were significant within 2 days.
Twenty milligram samples of tissue from each region were homogenized in NAD extraction buffer, centrifuged and deproteinized using 10 kDa molecular cut-off filters. Analysis of NAD was performed using an enzymatic NAD+/NADH quantification kit (Sigma Aldrich, St. Louis, MO, USA) according to manufacturer’s instructions. Fractions of samples were incubated for 5 min at room temperature for the detection of total NAD (tNAD), while equal amounts were heated to 60°C for 30 min to decompose NAD+ and leave unaltered NADH. tNAD and NADH were quantified with a colorimetric measure. Values of NADH were subtracted from tNAD to calculate the amount of NAD+ present in the samples.
After 3 weeks of treatment, extracted hippocampi showed a significant increase in NAD+/NADH ratio in the KD-fed group compared to the CD-fed group (Figure 2B). Interestingly, no detectable changes in NAD+/NADH ratios were observed in frontal cortices of the same animals (Figure 2C).
Interestingly, a robust and similar increase in NAD+/NADH ratio was already detectable in hippocampi after 2 days of KD treatment (Figure 2D). This result aligns with previous work (Nabbout et al., 2010; Ruskin et al., 2013a) showing that metabolic changes are present within 2 days and further corroborates the evidence that ketone bodies increase NAD+ availability rapidly. Increases in NAD+/NADH ratios were due to increases in NAD+ as NADH levels were not changed significantly by diet treatments (CD: 3.25 ± 0.32 pmol/μg proteins; 2 days: 2.58 ± 0.21; 3 weeks: 2.68 ± 0.21; not significant). The changes quantified in the hippocampus are consistent with the predicted metabolic consumption, as highlighted in the metabolic pathways (Figure 1).
Perspective and Implications
Consistent with our predictions we found clear evidence that metabolic therapy with a KD increases NAD+/NADH, a mechanism that could compensate for metabolic dysregulation and serve as a common start-point for the diverse beneficial metabolic and mitochondrial effects obtained with ketogenic treatments (Bough and Rho, 2007; Masino and Geiger, 2008). As noted above, a comparison of the metabolic pathways of glucose and ketone bodies (Figure 1) suggests that the use of ketone bodies as main energy fuel requires fewer NAD+ molecules than glucose (by a factor of 4), which should lead to an increased cellular availability of this vital coenzyme. Interestingly, β-OHB or a ketone ester precursor show protective effects by counterbalancing the decrease in NAD+/NADH ratio in cases of neurotoxicity (Maalouf et al., 2007; Zhang et al., 2013), confirming the ability of β-OHB to modulate NAD+/NADH levels. Ketone ester treatment oxidizes the cytoplasmic NAD+/NADH couple in hippocampus and cortex in aged, affected Alzheimer’s disease model mice, and reverses an apparent overoxidation of the mitochondrial couple in the hippocampus (Pawlosky et al., 2017). Thus this study and our study of the whole-cell couple suggest clear positive metabolic effects of ketolytic metabolism.
In general, the hippocampus has been described as a seizure gate (Heinemann et al., 1992) and it is one of the first brain regions to be affected in Alzheimer’s type dementia; cortical changes appear later in the disease (Braak and Braak, 1998). Unexpectedly, our data show that a KD increased NAD+/NADH in the hippocampus but not in the cerebral cortex, indicating regional specificity at the time points sampled. It is possible that changes mobilized by a KD could be more rapid or pronounced in more metabolically active brain regions: the hippocampus displays a higher metabolic rate than the cerebral cortex (Feng et al., 1988). Related to this, an early decrease in metabolic rate of glucose in the hippocampus, but not in cerebral cortex, was detected in patients who received a postmortem diagnosis of Alzheimer’s disease (Mosconi et al., 2009). Because of its high metabolic rate and increased vulnerability to hypoxia, oxidative stress, and metabolic dysfunction, the hippocampus could benefit more—or more rapidly—than other regions from KD-induced metabolic changes, including increased NAD+ levels.
Rapid increases in NAD+/NADH ratio could partially explain the ability of a KD to stop seizures in many patients within a few days of KD treatment (Freeman and Vining, 1999). For example, NAD+ and NADH molecules can modulate directly the opening of ion channels important for neuronal excitability, such as ATP-sensitive and voltage-gated potassium channels (Dukes et al., 1994; Tipparaju et al., 2005). Accordingly, a rapid decrease in NAD+ availability and consequent effect on neuronal excitability should be expected upon discontinuation of treatment. Interestingly, 15% of refractory epileptic patients experienced a rapid recurrence of seizures after KD discontinuation (Martinez et al., 2007); others remain seizure-free. The differential response among patients to treatment cessation indicates the existence of multiple downstream mechanisms and epigenetic changes (Masino and Rho, 2012; Lusardi et al., 2015) implicated in seizure control. Upregulation of key ketogenic enzymes, mainly mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase, after longer periods of ketogenic treatment (Cullingford et al., 2002) might also play a role in the maintenance of the beneficial effects even after discontinuation of the diet. More work is needed on downstream and lasting effects of metabolic therapy.
Consistent with these reported effects, previous work showed that addition of ketone bodies also prevented the expected decrease in NAD+/NADH ratio induced by toxic doses of calcium and in parallel decreased the production of reactive oxygen species (ROS), a major source of cellular oxidative stress (Maalouf et al., 2007); oxidative stress is detrimental and linked to neuronal death and neurodegenerative diseases (Naoi et al., 2005). Altering the NAD+/NADH ratio can control the rate of ROS production (Kussmaul and Hirst, 2006) and impact downstream enzymatic levels and activities that regulate apoptosis and inflammation (Yeung et al., 2004; Chen et al., 2008; Zhu et al., 2011). Enhanced NAD+/NADH should thus decrease inflammation, an effect observed in KD treatment (Forsythe et al., 2008; Ruskin et al., 2009; Yang and Cheng, 2010; Dupuis et al., 2015; Nandivada et al., 2016). Overall, increasing the NAD+/NADH ratio through a number of ketone-enhancing treatments should protect against oxidative stress and enhance mitochondrial and cellular health.
Regarding gene expression, consuming a KD has been found to impact expression patterns of genes modulating neuroinflammation, proliferation, and apoptosis such as cyclooxygenase, tumor necrosis factor-α, and insulin-like growth factor 1 (Cheng et al., 2003; Jeong et al., 2011). Although increased NAD+ cannot exert these effects directly, NAD+ can impact gene expression through the action of sirtuin enzymes. Increasing NAD+ availability or the NAD+/NADH ratio can increase the activity of the NAD-dependent SIRT1 enzyme (the most abundant member of the sirtuin family; Landry et al., 2000; Chen et al., 2008). The main function of SIRT1 is deacetylation of targets that regulate apoptosis and transcription factors such as peroxisome proliferator-activated receptor-γ and the tumor suppressor protein p53 (Yeung et al., 2004; Zhu et al., 2011). Hence, some of the expected downstream consequences of increasing NAD+ with ketogenic treatment are decreased cell death, inhibition of inflammation, and modulation of gene expression and epigenetic changes through activation of sirtuin enzymes (Janke et al., 2015).
Conclusion
Here we outline the overall implications and evidence for a rapid and region-specific change in NAD+/NADH as a direct result of consuming a KD. We hypothesize this as a new and fundamental addition to potential key mechanisms underlying beneficial antiseizure, neuroprotective and disease-modifying effects of KD. Because NAD+ can modulate ion channels, enhance mitochondrial health, decrease oxidative stress, and impact gene expression, an increase in NAD+ and/or NAD+/NADH ratio is a mechanism that can account for several diverse (and seemingly-unrelated) effects of ketogenic therapy. Furthermore, benefits of increasing NAD+ such as life-span extension and enhancing cellular health have long been documented (Lin et al., 2000), and pharmaceutical companies are currently manufacturing and selling supplements that contain NAD+ precursors such as nicotinamide or nicotinamide riboside in an attempt to increase endogenous NAD+ levels and enhance metabolic resilience—an outcome that may also be achieved physiologically by ketogenic strategies.
At this time more research is needed to further identify where and when ketogenic therapy increases the NAD+/NADH ratio, and to delineate specific downstream effects. It is also important to ascertain if changes in the NAD+/NADH ratio are caused by changes in NAD+ or NADH, as levels of these two redox molecules can also vary independently (Wilhelm and Hirrlinger, 2011). Regardless, diverse lines of evidence place NAD+ at the center of metabolic health and disease, and evidence from our work and others supports the hypothesis that increased NAD+ is a fundamental molecular mechanism of the KD. Our findings also indicate the potential for a greater role for this metabolic therapy in areas with high metabolic demand and vulnerability to environmental insults and oxidative stress such as the hippocampus. Taken together, increasing brain NAD+ levels—either by consuming a KD or by other ketone-enhancing treatments—might serve as a rapid and enduring strategy to halt or even reverse disease progression.
Ethics Statement
This study was carried out in accordance with the recommendations of National Institutes of Health (NIH) Guides and Trinity College Animal Care and Use Committee. The protocol was approved by the Trinity College Animal Care and Use Committee.
Author Contributions
ME, DNR, SAM and PS: conception and design of experiments; analysis and/or interpretation of data; drafting the manuscript; revising the manuscript critically for important intellectual content and approval of the version of the manuscript to be published. ME and DNR: acquisition of data.
Funding
This research was supported by National Institutes of Health (NIH; NS065957, NS066392, SAM; AT008742, DNR), Trinity College, the Dorothy Goodwin Scholars Program—University of Hartford Women’s Advancement Initiative (ME, PS) and the College of Arts and Sciences at the University of Hartford (PS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Ece Karhan, Susanne Bahnan, Kristina McMurry and Samantha DelVecchio for invaluable technical assistance.
References
Achanta, L. B., and Rae, C. D. (2017). β-Hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem. Res. 42, 35–49. doi: 10.1007/s11064-016-2099-2
Ahn, Y., Narous, M., Tobias, R., Rho, J. M., and Mychasiuk, R. (2014). The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev. Neurosci. 36, 371–380. doi: 10.1159/000362645
Belenky, P., Bogan, K. L., and Brenner, C. (2007). NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19. doi: 10.1016/j.tibs.2006.11.006
Boison, D. (2017). New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 30, 187–192. doi: 10.1097/wco.0000000000000432
Bough, K. J., and Rho, J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58. doi: 10.1111/j.1528-1167.2007.00915.x
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., et al. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. doi: 10.1002/ana.20899
Braak, H., and Braak, E. (1998). Evolution of neuronal changes in the course of Alzheimer’s disease. J. Neural Transm. Suppl. 53, 127–140. doi: 10.1007/978-3-7091-6467-9_11
Branco, A. F., Ferreira, A., Simões, R. F., Magalhães-Novais, S., Zehowski, C., Cope, E., et al. (2016). Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur. J. Clin. Invest. 46, 285–298. doi: 10.1111/eci.12591
Brownlow, M. L., Benner, L., D’Agostino, D., Gordon, M. N., and Morgan, D. (2013). Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS One 8:e75713. doi: 10.1371/journal.pone.0075713
Brownlow, M. L., Jung, S. H., Moore, R. J., Bechmann, N., and Jankord, R. (2017). Nutritional ketosis affects metabolism and behavior in sprague-dawley rats in both control and chronic stress environments. Front. Mol. Neurosci. 10:129. doi: 10.3389/fnmol.2017.00129
Caraballo, R., Vaccarezza, M., Cersósimo, R., Rios, V., Soraru, A., Arroyo, H., et al. (2011). Long-term follow-up of the ketogenic diet for refractory epilepsy: multicenter Argentinean experience in 216 pediatric patients. Seizure 20, 640–645. doi: 10.1016/j.seizure.2011.06.009
Castro, K., Baronio, D., Perry, I. S., Riesgo, R. D. S., and Gottfried, C. (2017). The effect of ketogenic diet in an animal model of autism induced by prenatal exposure to valproic acid. Nutr. Neurosci. 20, 343–350. doi: 10.1080/1028415x.2015.1133029
Chen, D., Bruno, J., Easlon, E., Lin, S. J., Cheng, H. L., Alt, F. W., et al. (2008). Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757. doi: 10.1101/gad.1650608
Cheng, C. M., Kelley, B., Wang, J., Strauss, D., Eagles, D. A., and Bondy, C. A. (2003). A ketogenic diet increases brain insulin-like growth factor receptor and glucose transporter gene expression. Endocrinology 144, 2676–2682. doi: 10.1210/en.2002-0057
Cotter, D. G., Schugar, R. C., and Crawford, P. A. (2013). Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 304, H1060–H1076. doi: 10.1152/ajpheart.00646.2012
Cullingford, T. E., Eagles, D. A., and Sato, H. (2002). The ketogenic diet upregulates expression of the gene encoding the key ketogenic enzyme mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in rat brain. Epilepsy Res. 49, 99–107. doi: 10.1016/s0920-1211(02)00011-6
Cunnane, S. C., Courchesne-Loyer, A., St-Pierre, V., Vandenberghe, C., Pierotti, T., Fortier, M., et al. (2016). Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann. N Y Acad. Sci. 1367, 12–20. doi: 10.1111/nyas.12999
Dai, Y., Zhao, Y., Tomi, M., Shin, B.-C., Thamotharan, S., Mazarati, A., et al. (2017). Sex-Specific life course changes in the neuro-metabolic phenotype of Glut3 null heterozygous mice: ketogenic diet ameliorates electroencephalographic seizures and improves sociability. Endocrinology 158, 936–949. doi: 10.1210/en.2016-1816
Douris, N., Melman, T., Pecherer, J. M., Pissios, P., Flier, J. S., Cantley, L. C., et al. (2015). Adaptive changes in amino acid metabolism permit normal longevity in mice consuming a low-carbohydrate ketogenic diet. Biochim. Biophys. Acta 1852, 2056–2065. doi: 10.1016/j.bbadis.2015.07.009
Dukes, L. D., McIntyre, M. S., Mertz, R. J., Philipson, L. H., Roe, M. W., Spencer, B., et al. (1994). Dependence on NADH produced during glycolysis for β-cell glucose signaling. J. Biol. Chem. 269, 10979–10982.
Dupuis, N., Curatolo, N., Benoist, J.-F., and Auvin, S. (2015). Ketogenic diet exhibits anti-inflammatory properties. Epilepsia 56, e95–e98. doi: 10.1111/epi.13038
Edmond, J. (1992). Energy metabolism in developing brain cells. Can. J. Physiol. Pharmacol. 70, S118–S129. doi: 10.1007/978-1-4684-1209-3_17
Evangeliou, A., Vlachonikolis, I., Mihailidou, H., Spilioti, M., Skarpalezou, A., Makaronas, N., et al. (2003). Application of a ketogenic diet in children with autistic behavior: pilot study. J. Child Neurol. 18, 113–118. doi: 10.1177/08830738030180020501
Feng, Z.-C., Roberts, E. L., Sick, T. J., and Rosenthal, M. (1988). Depth profile of local oxygen tension and blood flow in rat cerebral cortex, white matter and hippocampus. Brain Res. 445, 280–288. doi: 10.1016/0006-8993(88)91190-0
Forsythe, C. E., Phinney, S. D., Feinman, R. D., Volk, B. M., Freidenreich, D., Quann, E., et al. (2010). Limited effect of dietary saturated fat on plasma saturated fat in the context of a low carbohydrate diet. Lipids 45, 947–962. doi: 10.1007/s11745-010-3467-3
Forsythe, C. E., Phinney, S. D., Fernandez, M. L., Quann, E. E., Wood, R. J., Bibus, D. M., et al. (2008). Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids 43, 65–77. doi: 10.1007/s11745-007-3132-7
Freeman, J. M., and Vining, E. P. G. (1999). Seizures decrease rapidly after fasting: preliminary studies of the ketogenic diet. Arch. Pediatr. Adolesc. Med. 153, 946–949. doi: 10.1001/archpedi.153.9.946
García-Escudero, V., Martín-Maestro, P., Perry, G., and Avila, J. (2013). Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013:162152. doi: 10.1155/2013/162152
Gomez-Arbelaez, D., Bellido, D., Castro, A. I., Ordoñez-Mayan, L., Carreira, J., Galban, C., et al. (2017). Body composition changes after very low-calorie-ketogenic diet in obesity evaluated by three standardized methods. J. Clin. Endocrinol. Metab. 102, 488–498. doi: 10.1210/jc.2016-2385
Heinemann, U., Beck, H., Dreier, J. P., Ficker, E., Stabel, J., and Zhang, C. L. (1992). The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res. Suppl. 7, 273–280.
Hori, A., Tandon, P., Holmes, G. L., and Stafstrom, C. E. (1997). Ketogenic diet: effects on expression of kindled seizures and behavior in adult rats. Epilepsia 38, 750–758. doi: 10.1111/j.1528-1157.1997.tb01461.x
Hui, L., Chen, X., Bhatt, D., Geiger, N. H., Rosenberger, T. A., Haughey, N. J., et al. (2012). Ketone bodies protection against HIV-1 Tat-induced neurotoxicity. J. Neurochem. 122, 382–391. doi: 10.1111/j.1471-4159.2012.07764.x
Hu, Z. G., Wang, H. D., Jin, W., and Yin, H. X. (2009). Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann. Clin. Lab. Sci. 39, 76–83. Available online at: http://www.annclinlabsci.org/content/39/1/76.full
Hussain, T. A., Mathew, T. C., Dashti, A. A., Asfar, S., Al-Zaid, N., and Dashti, H. M. (2012). Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes. Nutrition 28, 1016–1021. doi: 10.1016/j.nut.2012.01.016
Janke, R., Dodson, A. E., and Rine, J. (2015). Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol. 31, 473–496. doi: 10.1146/annurev-cellbio-100814-125544
Jenkins, D. J. A., Wong, J. M. W., Kendall, C. W. C., Esfahani, A., Ng, V. W. Y., Leong, T. C. K., et al. (2009). The effect of a plant-based low-carbohydrate (“Eco-Atkins”) diet on body weight and blood lipid concentrations in hyperlipidemic subjects. Arch. Intern. Med. 169, 1046–1054. doi: 10.1001/archinternmed.2009.115
Jeong, E. A., Jeon, B. T., Shin, H. J., Kim, N., Lee, D. H., Kim, H. J., et al. (2011). Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 232, 195–202. doi: 10.1016/j.expneurol.2011.09.001
Kashiwaya, Y., Bergman, C., Lee, J.-H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., and Veech, R. L. (2000). D-β-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 97, 5440–5444. doi: 10.1073/pnas.97.10.5440
Khodadadi, S., Sobhani, N., Mirshekar, S., Ghiasvand, R., Pourmasoumi, M., Miraghajani, M., et al. (2017). Tumor cells growth and survival time with the ketogenic diet in animal models: a systematic review. Int. J. Prev. Med. 8:35. doi: 10.4103/2008-7802.207035
Kim, D. Y., Hao, J., Liu, R., Turner, G., Shi, F.-D., and Rho, J. M. (2012). Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS One 7:e35476. doi: 10.1371/journal.pone.0035476
Klement, R. J., Champ, C. E., Otto, C., and Kämmerer, U. (2016). Anti-tumor effects of ketogenic diets in mice: a meta-analysis. PLoS One 11:e0155050. doi: 10.1371/journal.pone.0155050
Kobow, K., Kaspi, A., Harikrishnan, K. N., Kiese, K., Ziemann, M., Khurana, I., et al. (2013). Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 126, 741–756. doi: 10.1007/s00401-013-1168-8
Koustova, E., Rhee, P., Hancock, T., Chen, H., Inocencio, R., Jaskille, A., et al. (2003). Ketone and pyruvate Ringer’s solutions decrease pulmonary apoptosis in a rat model of severe hemorrhagic shock and resuscitation. Surgery 134, 267–274. doi: 10.1067/msy.2003.245
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., and Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33, 425.e19–425.e27. doi: 10.1016/j.neurobiolaging.2010.10.006
Kussmaul, L., and Hirst, J. (2006). The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U S A 103, 7607–7612. doi: 10.1073/pnas.0510977103
Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L., et al. (2000). The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U S A 97, 5807–5811. doi: 10.1073/pnas.110148297
Lin, S. H., Vincent, A., Shaw, T., Maynard, K. I., and Maiese, K. (2000). Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J. Cereb. Blood Flow Metab. 20, 1380–1391. doi: 10.1097/00004647-200009000-00013
Liu, X., Lee, Y. J., Liou, L.-C., Ren, Q., Zhang, Z., Wang, S., et al. (2011). Alpha-synuclein functions in the nucleus to protect against hydroxyurea-induced replication stress in yeast. Hum. Mol. Genet. 20, 3401–3414. doi: 10.1093/hmg/ddr246
Lodish, H., Berk, A., Matsudaira, P., Zipursky, L., Baltimore, D., and Darnell, J. (2000). “Section 16.1 Oxidation of Glucose and Fatty Acids to CO2,” in Molecular Cell Biology, (4th Edn), New York, NY: W.H. Freeman.
Lusardi, T. A., Akula, K. K., Coffman, S. Q., Ruskin, D. N., Masino, S. A., and Boison, D. (2015). Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 99, 500–509. doi: 10.1016/j.neuropharm.2015.08.007
Lussier, D. M., Woolf, E. C., Johnson, J. L., Brooks, K. S., Blattman, J. N., and Scheck, A. C. (2016). Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer 16:310. doi: 10.1186/s12885-016-2337-7
Maalouf, M., Sullivan, P. G., Davis, L., Kim, D. Y., and Rho, J. M. (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. doi: 10.1016/j.neuroscience.2006.11.065
Martinez, C. C., Pyzik, P. L., and Kossoff, E. H. (2007). Discontinuing the ketogenic diet in seizure-free children: recurrence and risk factors. Epilepsia 48, 187–190. doi: 10.1111/j.1528-1167.2006.00911.x
Masino, S. A. (2017). Ketogenic Diet and Metabolic Therapies: Expanded Roles in Health and Disease. New York, NY: Oxford University Press.
Masino, S. A., and Geiger, J. D. (2008). Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci. 31, 273–278. doi: 10.1016/j.tins.2008.02.009
Masino, S. A., Li, T., Theofilas, P., Sandau, U. S., Ruskin, D. N., Fredholm, B. B., et al. (2011a). A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Invest. 121, 2679–2683. doi: 10.1172/JCI57813
Masino, S. A., Svedova, J., Kawamura, M. Jr., DiMario, F. D. Jr., and Eigsti, I.-M. (2011b). “Adenosine and autism—recent research and a new perspective,” in Autism—A Neurodevelopmental Journey from Genes to Behaviour, ed. V. Eapen (Rijeka: InTech), 103–122.
Masino, S. A., and Rho, J. M. (2012). “Mechanism of ketogenic diet action,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Oslen, and A. V. Delgado-Escueta, (4th Edn.) (Bethesda, MD: National Center for Biotechnology Information), 1003–1018.
McKenzie, A. L., Hallberg, S. J., Creighton, B. C., Volk, B. M., Link, T. M., Abner, M. K., et al. (2017). A novel intervention including individualized nutritional recommendations reduces hemoglobin a1c level, medication use, and weight in type 2 diabetes. JMIR Diabetes 2:e5. doi: 10.2196/diabetes.6981
Mosconi, L., Mistur, R., Switalski, R., Tsui, W. H., Glodzik, L., Li, Y., et al. (2009). FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 36, 811–822. doi: 10.1007/s00259-008-1039-z
Muller-Schwarze, A. B., Tandon, P., Liu, Z., Yang, Y., Holmes, G. L., and Stafstrom, C. E. (1999). Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. Neuroreport 10, 1517–1522. doi: 10.1097/00001756-199905140-00023
Nabbout, R., Mazzuca, M., Hubert, P., Peudennier, S., Allaire, C., Flurin, V., et al. (2010). Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 51, 2033–2037. doi: 10.1111/j.1528-1167.2010.02703.x
Nandivada, P., Fell, G. L., Pan, A. H., Nose, V., Ling, P.-R., Bistrian, B. R., et al. (2016). Eucaloric ketogenic diet reduces hypoglycemia and inflammation in mice with endotoxemia. Lipids 51, 703–714. doi: 10.1007/s11745-016-4156-7
Naoi, M., Maruyama, W., Shamoto-Nagai, M., Yi, H., Akao, Y., and Tanaka, M. (2005). Oxidative stress in mitochondria: decision to survival and death of neurons in neurodegenerative disorders. Mol. Neurobiol. 31, 81–93. doi: 10.1385/mn:31:1-3:081
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. doi: 10.1016/s1474-4422(08)70092-9
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C.-P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26, 547.e8–557.e8. doi: 10.1016/j.cmet.2017.08.004
Newport, M. T., Vanitallie, T. B., Kashiwaya, Y., King, M. T., and Veech, R. L. (2015). A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 11, 99–103. doi: 10.1016/j.jalz.2014.01.006
Noh, H. S., Hah, Y.-S., Nilufar, R., Han, J., Bong, J.-H., Kang, S. S., et al. (2006). Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res. 83, 702–709. doi: 10.1002/jnr.20736
Paoli, A. (2014). Ketogenic diet for obesity: friend or foe? Int. J. Environ. Res. Public Health 11, 2092–2107. doi: 10.3390/ijerph110202092
Partsalaki, I., Karvela, A., and Spiliotis, B. E. (2012). Metabolic impact of a ketogenic diet compared to a hypocaloric diet in obese children and adolescents. J. Pediatr. Endocrinol. Metab. 25, 697–704. doi: 10.1515/jpem-2012-0131
Patel, A., Pyzik, P. L., Turner, Z., Rubenstein, J. E., and Kossoff, E. H. (2010). Long-term outcomes of children treated with the ketogenic diet in the past. Epilepsia 51, 1277–1282. doi: 10.1111/j.1528-1167.2009.02488.x
Pathak, D., Berthet, A., and Nakamura, K. (2013). Energy failure: does it contribute to neurodegeneration? Ann. Neurol. 74, 506–516. doi: 10.1002/ana.24014
Pawlosky, R. J., Kemper, M. F., Kashiwaya, Y., King, M. T., Mattson, M. P., and Veech, R. L. (2017). Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 195–207. doi: 10.1111/jnc.13958
Reger, M. A., Henderson, S. T., Hale, C., Cholerton, B., Baker, L. D., Watson, G. S., et al. (2004). Effects of β-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 25, 311–314. doi: 10.1016/s0197-4580(03)00087-3
Roberts, M. N., Wallace, M. A., Tomilov, A. A., Zhou, Z., Marcotte, G. R., Tran, D., et al. (2017). A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 26, 539.e5–546.e5. doi: 10.1016/j.cmet.2017.08.005
Ruskin, D. N., Fortin, J. A., Bisnauth, S. N., and Masino, S. A. (2017a). Ketogenic diets improve behaviors associated with autism spectrum disorder in a sex-specific manner in the EL mouse. Physiol. Behav. 168, 138–145. doi: 10.1016/j.physbeh.2016.10.023
Ruskin, D. N., Murphy, M. I., Slade, S. L., and Masino, S. A. (2017b). Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS One 12:e0171643. doi: 10.1371/journal.pone.0171643
Ruskin, D. N., Kawamura, M., and Masino, S. A. (2009). Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS One 4:e8349. doi: 10.1371/journal.pone.0008349
Ruskin, D. N., Suter, T. A. C. S., Ross, J. L., and Masino, S. A. (2013a). Ketogenic diets and thermal pain: dissociation of hypoalgesia, elevated ketones, and lowered glucose in rats. J. Pain 14, 467–474. doi: 10.1016/j.jpain.2012.12.015
Ruskin, D. N., Svedova, J., Cote, J. L., Sandau, U., Rho, J. M., Kawamura, M., et al. (2013b). Ketogenic diet improves core symptoms of autism in BTBR mice. PLoS One 8:e65021. doi: 10.1371/journal.pone.0065021
Scheibye-Knudsen, M., Mitchell, S. J., Fang, E. F., Iyama, T., Ward, T., Wang, J., et al. (2014). A high-fat diet and NAD+ activate sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855. doi: 10.1016/j.cmet.2014.10.005
Schmidt, M., Pfetzer, N., Schwab, M., Strauss, I., and Kämmerer, U. (2011). Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: a pilot trial. Nutr. Metab. (Lond) 8:54. doi: 10.1186/1743-7075-8-54
Schwartzkroin, P. A. (1999). Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res. 37, 171–180. doi: 10.1016/s0920-1211(99)00069-8
Seyfried, T. N., and Mukherjee, P. (2005). Targeting energy metabolism in brain cancer: review and hypothesis. Nutr. Metab. 2:30. doi: 10.1186/1743-7075-2-30
Shimazu, T., Hirschey, M. D., Newman, J., He, W., Shirakawa, K., Le Moan, N., et al. (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. doi: 10.1126/science.1227166
Spilioti, M., Evangeliou, A. E., Tramma, D., Theodoridou, Z., Metaxas, S., Michailidi, E., et al. (2013). Evidence for treatable inborn errors of metabolism in a cohort of 187 Greek patients with autism spectrum disorder (ASD). Front. Hum. Neurosci. 7:858. doi: 10.3389/fnhum.2013.00858
Streijger, F., Plunet, W. T., Lee, J. H. T., Liu, J., Lam, C. K., Park, S., et al. (2013). Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS One 8:e78765. doi: 10.1371/journal.pone.0078765
Sullivan, P. G., Rippy, N. A., Dorenbos, K., Concepcion, R. C., Agarwal, A. K., and Rho, J. M. (2004). The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 55, 576–580. doi: 10.1002/ana.20062
Tay, J., Luscombe-Marsh, N. D., Thompson, C. H., Noakes, M., Buckley, J. D., Wittert, G. A., et al. (2015). Comparison of low- and high-carbohydrate diets for type 2 diabetes management: a randomized trial. Am. J. Clin. Nutr. 102, 780–790. doi: 10.3945/ajcn.115.112581
Tipparaju, S. M., Saxena, N., Liu, S.-Q., Kumar, R., and Bhatnagar, A. (2005). Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am. J. Physiol. Cell Physiol. 288, C366–C376. doi: 10.1152/ajpcell.00354.2004
van der Veer, E., Ho, C., O’Neil, C., Barbosa, N., Scott, R., Cregan, S. P., et al. (2007). Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 282, 10841–10845. doi: 10.1074/jbc.c700018200
Vanitallie, T. B., Nonas, C., Di Rocco, A., Boyar, K., Hyams, K., and Heymsfield, S. B. (2005). Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology 64, 728–730. doi: 10.1212/01.WNL.0000152046.11390.45
Verpeut, J. L., DiCicco-Bloom, E., and Bello, N. T. (2016). Ketogenic diet exposure during the juvenile period increases social behaviors and forebrain neural activation in adult Engrailed 2 null mice. Physiol. Behav. 161, 90–98. doi: 10.1016/j.physbeh.2016.04.001
Volek, J. S., Fernandez, M. L., Feinman, R. D., and Phinney, S. D. (2008). Dietary carbohydrate restriction induces a unique metabolic state positively affecting atherogenic dyslipidemia, fatty acid partitioning, and metabolic syndrome. Prog. Lipid Res. 47, 307–318. doi: 10.1016/j.plipres.2008.02.003
Volek, J. S., Phinney, S. D., Forsythe, C. E., Quann, E. E., Wood, R. J., Puglisi, M. J., et al. (2009). Carbohydrate restriction has a more favorable impact on the metabolic syndrome than a low fat diet. Lipids 44, 297–309. doi: 10.1007/s11745-008-3274-2
Westman, E. C., Yancy, W. S. Jr., Mavropoulos, J. C., Marquart, M., and McDuffie, J. R. (2008). The effect of a low-carbohydrate, ketogenic diet versus a low-glycemic index diet on glycemic control in type 2 diabetes mellitus. Nutr. Metab. 5:36. doi: 10.1186/1743-7075-5-36
Wilhelm, F., and Hirrlinger, J. (2011). The NAD+/NADH redox state in astrocytes: independent control of the NAD+ and NADH content. J. Neurosci. Res. 89, 1956–1964. doi: 10.1002/jnr.22638
Yancy, W. S. Jr., Foy, M., Chalecki, A. M., Vernon, M. C., Westman, E. C., and Westman, E. C. (2005). A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutr. Metab. 2:34. doi: 10.1186/1743-7075-2-34
Yang, X., and Cheng, B. (2010). Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 42, 145–153. doi: 10.1007/s12031-010-9336-y
Yang, Y., and Sauve, A. A. (2016). NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 1864, 1787–1800. doi: 10.1016/j.bbapap.2016.06.014
Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., Frye, R. A., et al. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380. doi: 10.1038/sj.emboj.7600244
Zhang, J., Cao, Q., Li, S., Lu, X., Zhao, Y., Guan, J.-S., et al. (2013). 3-Hydroxybutyrate methyl ester as a potential drug against Alzheimer’s disease via mitochondria protection mechanism. Biomaterials 34, 7552–7562. doi: 10.1016/j.biomaterials.2013.06.043
Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., et al. (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. doi: 10.1126/science.aaf2693
Zhao, Z., Lange, D. J., Voustianiouk, A., MacGrogan, D., Ho, L., Suh, J., et al. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7:29. doi: 10.1186/1471-2202-7-29
Zhu, J., and Chu, C. T. (2010). Mitochondrial dysfunction in Parkinson’s disease. J. Alzheimers Dis. 20, S325–S334. doi: 10.3233/JAD-2010-100363
Zhu, X., Liu, Q., Wang, M., Liang, M., Yang, X., Xu, X., et al. (2011). Activation of Sirt1 by resveratrol inhibits TNF-α induced inflammation in Fibroblasts. PLoS One 6:e27081. doi: 10.1371/journal.pone.0027081
Zou, Z., Sasaguri, S., Rajesh, K. G., and Suzuki, R. (2002). dl-3-Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 283, H1968–H1974. doi: 10.1152/ajpheart.00250.2002
Keywords: ketone bodies, metabolism, hippocampus, epilepsy, neurodegeneration, Alzheimer’s disease, nicotinamide adenine dinucleotide, longevity
Citation: Elamin M, Ruskin DN, Masino SA and Sacchetti P (2017) Ketone-Based Metabolic Therapy: Is Increased NAD+ a Primary Mechanism? Front. Mol. Neurosci. 10:377. doi: 10.3389/fnmol.2017.00377
Received: 24 July 2017; Accepted: 30 October 2017;
Published: 14 November 2017.
Edited by:
Alessandro Prigione, Max Delbrück Center for Molecular Medicine (HZ), GermanyReviewed by:
Anna Maria Giudetti, University of Salento, ItalyAlba Di Pardo, Centre for Neurogenetics and Rare Diseases, Italy
Copyright © 2017 Elamin, Ruskin, Masino and Sacchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan A. Masino, c3VzYW4ubWFzaW5vQHRyaW5jb2xsLmVkdQ==
Paola Sacchetti, psacchett@hartford.edu