 Mansour Haidar
Mansour Haidar Vincent Timmerman
Vincent Timmerman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 11 May 2017
Sec. Molecular Signalling and Pathways
Volume 10 - 2017 | https://doi.org/10.3389/fnmol.2017.00143
This article is part of the Research Topic The Role of the Protein Quality Control in Neurodegenerative Diseases View all 34 articles
The inherited peripheral neuropathies (IPNs) comprise a growing list of genetically heterogeneous diseases. With mutations in more than 80 genes being reported to cause IPNs, a wide spectrum of functional consequences is expected to follow this genotypic diversity. Hence, the search for a common pathomechanism among the different phenotypes has become the holy grail of functional research into IPNs. During the last decade, studies on several affected genes have shown a direct and/or indirect correlation with autophagy. Autophagy, a cellular homeostatic process, is required for the removal of cell aggregates, long-lived proteins and dead organelles from the cell in double-membraned vesicles destined for the lysosomes. As an evolutionarily highly conserved process, autophagy is essential for the survival and proper functioning of the cell. Recently, neuronal cells have been shown to be particularly vulnerable to disruption of the autophagic pathway. Furthermore, autophagy has been shown to be affected in various common neurodegenerative diseases of both the central and the peripheral nervous system including Alzheimer’s, Parkinson’s, and Huntington’s diseases. In this review we provide an overview of the genes involved in hereditary neuropathies which are linked to autophagy and we propose the disruption of the autophagic flux as an emerging common pathomechanism. We also shed light on the different steps of the autophagy pathway linked to these genes. Finally, we review the concept of autophagy being a therapeutic target in IPNs, and the possibilities and challenges of this pathway-specific targeting.
Disruption of intracellular homeostasis is at the base of most pathological conditions. Cellular health and function often relies on the maintenance of protein homeostasis by assuring that structurally abnormal proteins do not accumulate in cells, causing aggregate formation and organelle damage. The function of proteostasis is accomplished by two systems, the proteasome and the autophagy system. While the proteasome targets ubiquitinated and short-lived proteins, autophagy can degrade long-lived proteins and damaged organelles. Three main subtypes of autophagy have been identified, microautophagy, CMA, and macroautophagy. Microautophagy involves the degradation of cytosolic material inside the lysosome by direct lysosomal invagination (Mijaljica et al., 2011). CMA is a specific autophagic pathway where chaperones target proteins containing the KFERQ motif to the lysosome for degradation (Cuervo et al., 1995). Unlike CMA, macroautophagy (the main focus of this review), is a bulk degradation process characterized by the formation of double membrane vesicles, autophagosomes, which engulf cytoplasmic material and degrade their contents by fusing with the lysosomes (Yang and Klionsky, 2010). Disruption of the macroautophagy pathway can lead to failure to clear out misfolded proteins and dead organelles, or increased accumulation of autophagic structures and has been linked to neurodegeneration (Frake et al., 2015). Mutations in key autophagy regulating genes have been shown to cause neurodegeneration (Saitsu et al., 2013; Kyöstilä et al., 2015). Furthermore, mouse models with neuron-specific KO of autophagy genes frequently develop neurodegeneration (Komatsu et al., 2006). Interestingly, some of these mouse models display features of peripheral neuropathy as evident by decline in motor performance on the rota-rod, limb-clasping, and paw placement tests (Hara et al., 2006). The link between disruption at the gene level and the development of peripheral neuropathy is the main feature of IPNs. IPNs are a genetically heterogeneous disease population with over 80 affected genes discovered so far (Baets et al., 2014; Timmerman et al., 2014). While the clinical presentation of IPN patients is rather common, with length-dependent degeneration affecting the motor and/or sensory nerves, the variety of associated genes has produced various molecular phenotypes (Pareyson et al., 2014; Weis et al., 2016). Recently, more and more studies have indicated the involvement of autophagic impairment in IPN causing-mutations, and important roles for IPN related genes in autophagy. While the evidence linking IPN associated genes to autophagy can be direct or indirect, impairment of autophagy presents as an important contributor to the neuropathic phenotype given the vulnerability of neurons, especially those of the peripheral nervous system to disrupted cellular recycling and clearance. Identifying a common pathomechanism among the different IPN-related genes would provide a great therapeutic potential for targeting these neuropathies. This review discusses the advances made so far regarding the cellular and molecular mechanisms behind the different forms of IPN due to impairment of the autophagic pathway.
Macroautophagy, hereafter referred to as autophagy, is a homeostatic cellular process by which protein aggregates and cellular organelles are targeted, degraded, and recycled (Figure 1). Autophagy is a multi-step process consisting of: induction, nucleation of the isolation membrane (phagophore), elongation and expansion of the phagophore into a closed double-membraned autophagosome, lysosomal docking and fusion, and degradation of autophagic cargo. The different autophagy steps, explained below, are governed by a wide array of proteins and protein complexes, most notably the group of proteins encoded by the autophagy-related (ATG) genes (Parzych and Klionsky, 2014).
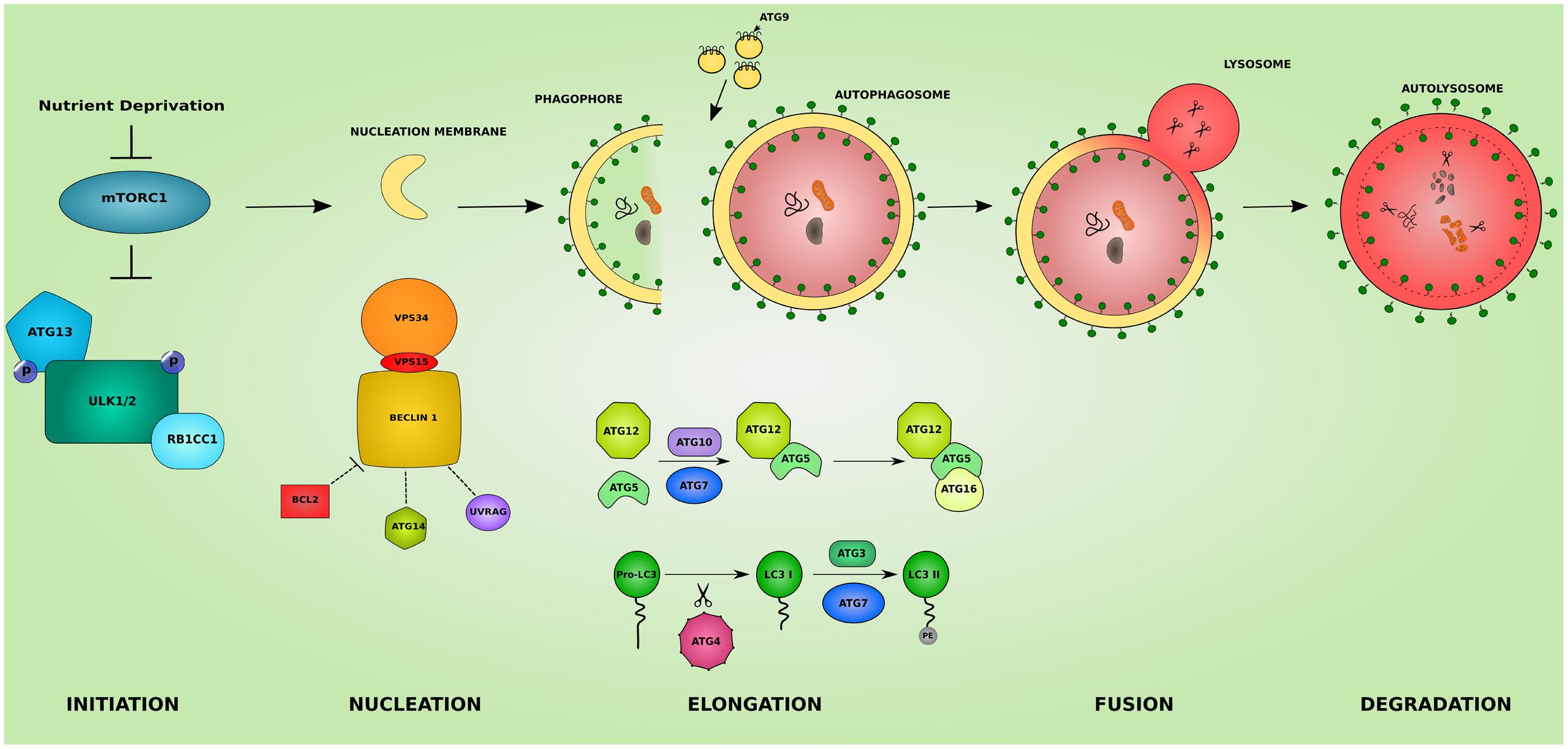
FIGURE 1. The different steps of the autophagic pathway. The autophagic pathway consists of several steps. The initiation step is governed by the initiation complex composed of ULK1 and ULK2 (unc-51 like autophagy activating kinase 1/2), RB1CC1 (RB1 inducible coiled-coil 1) and ATG13 and under the control of the nutrient sensing mechanistic target of rapamaycin (MTORC1) complex. After the initiation step, the nucleation of early autophagic membranes is controlled by the nucleation complex consisting of phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3/VPS34), beclin 1 (BECN1), and phosphoinositide-3-kinase regulatory subunit 4 (PIK3R4/VPS15). BECN1 acts as an autophagy check point by interacting with ATG14, UVRAG (UV radiation resistance associated), and apoptosis regulator BCL2 proteins. The formed phagophore then undergoes elongation to become a fully closed double-membraned autophagosome. This step involves 2 conjugation systems resulting in the formation of ATG12-ATG5-ATG16L1 complex via ATG7 and ATG10 action, and of the autophagosomal marker LC3II via the action of ATG4, ATG7, and ATG3. The delivery of membranes to forming autophagosomes is served by ATG9-containing vesicles. The completed autophagosome then fuses with a lysosome becoming an autolysosome where its cargo is subjected to lysosomal degradation. The colors and shapes of the boxes are randomly assigned and show that they are different proteins belonging to the same complexes.
The initiation of autophagy is regulated by the ULK complex composed of ULK1 and ULK2, and their stable interactors RB1CC1 and ATG13. The autophagy initiation complex is under the control of the nutrient-sensing, MTORC1, a serine–threonine kinase. During nutrient rich conditions MTORC1 binds to and phosphorylates ULK1/2 inhibiting their kinase activity. Removal of MTORC1 from the ULK1/2 complex, by nutrient starvation for example, allows for the activation of ULK1/2 and the proceeding of the autophagic process (Hosokawa et al., 2009).
The membrane nucleation step involves the conversion of PtdIns to phosphatidylinositol 3-phosphate (PtdIns3P) by the kinase complex Class III PtdIns3K. This complex consists of PIK3C3/VPS34, BECN1, and PIK3R4/VPS15 (Petiot et al., 2000). PtdIns3P is a signal for recruiting ATG proteins in the nucleation site, by recognizing and binding to PtdIns3P. BECN1 moderates the coupling of the PtdIns3K complex with various proteins, and via its binding partners serves as a check point that can inhibit or upregulate autophagy. Among the binding partners are ATG14, UVRAG, and apoptosis regulator BCL2 proteins. BCL2 binding suppresses autophagy (Pattingre et al., 2005). UVRAG binding targets the nucleation machinery to the endosomal membranes or the cell surface and can up or downregulate autophagy depending on its other binding partners (Yang and Klionsky, 2010). ATG14 binding on the other hand targets the nucleation to the omegasome (an ER resident precursor of the autophagosome where formation of autophagic vacuoles ensues) (Axe et al., 2008).
Two ubiquitin-like conjugation systems are at the heart of the expansion of the phagophore into an autophagosome. The first system concerns the formation of ATG12-ATG5-ATG16L1 complex. ATG12 is covalently conjugated to ATG5 via the E1 activating enzyme ATG7 (Tanida et al., 1999) and the E2 conjugating enzyme ATG10 (Shintani et al., 1999). The ATG12-ATG5 complex then binds ATG16L1 through ATG5. The second ubiquitin-like conjugation reaction involves the lipidation of ATG8 (LC3) to ATG8-Phospatidylenolamine (LC3II). This first requires the C-terminal cleavage of LC3 by the protease ATG4. The cleaved protein (LC3I) is then processed by the E1 activating enzyme ATG7 and the E2 conjugating enzyme ATG3 yielding LC3II. LC3II remains associated with the phagophore and the mature autophagosomes until its degradation in the lysosome (Kabeya, 2000). This association makes LC3II a good marker for the study of the autophagic activity in cell and animal models. Since lipid synthesis does not occur at the phagophore, delivery of membrane from other locations in the cell is necessary for the elongation step. The task of membrane recruitment is served by ATG9, the only transmembrane protein in the ATG family. Mainly localized to the trans-Golgi network and the late endosomes, autophagy activation drives the trafficking of membrane delivering-ATG9 to the sites of autophagosome formation (Mari et al., 2010). Lipid delivery supplied by ATG9 allow for the elongation of the phagophore into a fully closed autophagosome.
The complete autophagosome eventually moves to and fuses with a lysosome becoming an autolysosome and/or with an endosome forming an amphisome. The transport of autophagosomes to the lysosomes depends on microtubules. The mechanism of docking at and fusion with the lysosome is not well-understood (Chen and Klionsky, 2011), but it is thought to involve UVRAG, RAB7 GTPase and syntaxin-17 (STX17) of the SNARE machinery (Jäger et al., 2004; Liang et al., 2008; Jiang et al., 2014).
Upon the fusion of the autophagosome with the lysosome, the autophagic cargo is degraded by various lysosomal hydrolases and proteases. The degradation products including metabolites, amino acids and fatty acids are recovered and reutilized by the cell.
The morphological features that distinguish neurons from other cells are their post-mitotic nature, highly polarized structure, and extended cytoplasm into axons and dendrites that can stretch far from the cell body. The latter feature is more exaggerated in neurons supplying the peripheral nerves. The spatial compartmentalization of neurons makes them prone to aggregation and accumulation of dead organelles and misfolded proteins. Autophagy therefore forms an essential homeostatic process for neurons. Knocking out key autophagy genes, such as ATG7 in mouse neurons leads to neurodegeneration (Komatsu et al., 2006). Defective autophagy has also been linked with numerous neurodegenerative diseases (Frake et al., 2015). The high susceptibility of neurons to autophagic impairment could explain why mutations in ubiquitously expressed genes can cause neuron-specific pathology in inherited neuropathies.
Inherited peripheral neuropathies are a genetically heterogeneous group of disorders affecting the peripheral nerves. Depending on the affected nerves, IPNs are classified as CMT or HMSN if motor and sensory nerves are affected. If sensory and/or autonomic nerve dysfunction predominates, the neuropathy is termed (HSAN) and dHMN if motor deficits are the most prevalent (Pareyson et al., 2014).
Charcot-Marie-Tooth, the most common form of hereditary neuropathies affects about 1:2500. Patients with CMT share a common clinical presentation of slowly progressive muscle wasting and weakness ascending from the feet to reach the thighs and hands, reduced tendon reflexes, skeletal deformities, and sometimes sensory loss (Harding and Thomas, 1980b). In many cases, the age of onset is in the first to second decade, but infantile, early-onset and late-onset forms exist. CMT is further classified on the prevalence of myelin or axonal involvement and according to the mode of inheritance. CMT1, the demyelinating subgroup is associated with reduced NCVs to less than 35 m/s. CMT2, the axonal subgroup on the other hand, only shows slightly reduced NCV but with reduced CMAPs amplitudes. An intermediate CMT subgroup exists with NCV between 35 and 45 m/s. The mode of inheritance further subdivides CMT neuropathies into CMT4 for recessive demyelinating CMT, CMT2R for recessive axonal CMT, CMTDI, and CMTRI for dominant and recessive intermediate CMT respectively, and CMT-X for the X-linked CMT (Stojkovic, 2016).
The HSAN disorders are characterized by sensory deficits and the involvement of the autonomic dysfunction. Five types of HSAN are acknowledged depending on the inheritance pattern and the sensory abnormality (Thomas, 2005). HSAN type I is autosomal dominant with variable motor involvement. HSAN types II to VI are autosomal recessive and further classified according to the sensory involvement and the clinical presentation ranging from osteomyelitis in type II to respiratory difficulties in type VI. HSAN types III, IV, and V show congenital onset. HSAN with spastic paraplegia, also a recessive form of HSAN, presents as a sensory neuropathy with mild spastic paraplegia (Rotthier et al., 2012).
The dHMN are classified into seven subtypes depending on the age of onset, inheritance pattern, and the distribution of the deficits (Harding and Thomas, 1980a). The dHMN types I, II, V, and VII are autosomal dominant, while types III, IV, and VI are autosomal recessive and the X-linked dHMN has an X-linked inheritance. Minor sensory abnormalities are frequent to many dHMN forms, and an overlap with axonal CMT (CMT2), where mutations in the same gene can cause either phenotypes is common. The phenotypic characteristics of dHMN are very diverse with the pathology involving other organs such as the vocal cords or the respiratory system (Rossor et al., 2012).
Hereditary peripheral neuropathies show overlap on the genetic level with mutations in the same gene leading to different clinical subtypes. This has recently lead to attempts in classifying IPNs based on the gene function involved in neuropathy (Vallat et al., 2016). While many genes associated with neuropathy have a known function in neuronal maintenance, development, or myelination, several other genes revealed functions that were unrecognized or indirect to the caused neuropathy (Weis et al., 2016). The categorization based on gene function further highlighted the heterogeneity of inherited neuropathies. Hence, the search for a common pathomechanism among the different causal genes is starting to become a focal point in the hope of better understanding the pathology of inherited neuropathies and ultimately designing efficient therapeutic approaches. We believe that this classification is of great help for a better understanding of the molecular consequences of the disease-causative mutations. In the following sections, we shed light on the involvement of IPN-associated genes in the different steps of the autophagy pathway presenting autophagic impairment (Figure 2) as an emerging common pathomechanism (summarized in Table 1).
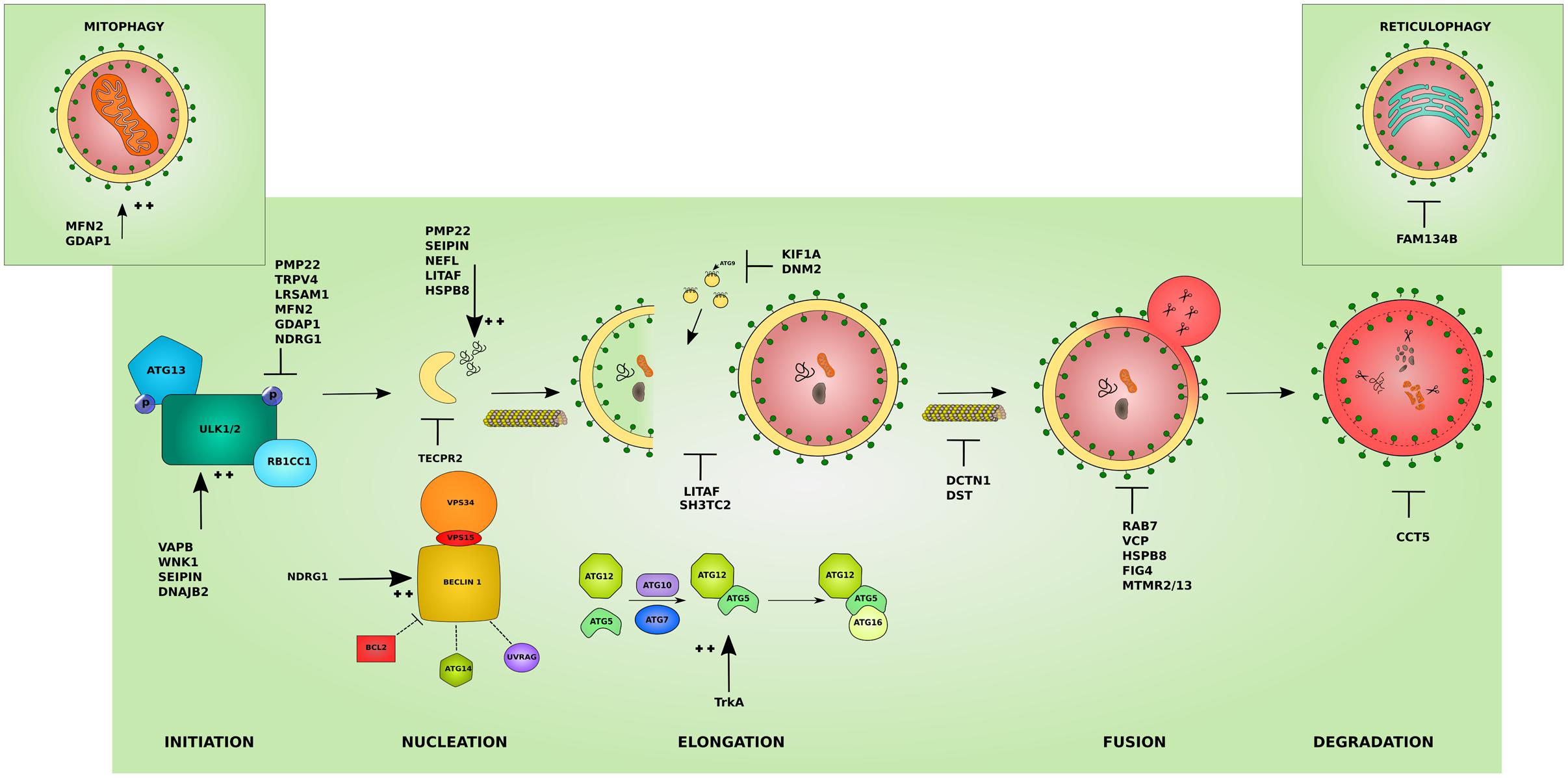
FIGURE 2. Effects of the different IPN-associated genes on the autophagy pathway. Inherited peripheral neuropathy associated genes disrupt autophagy at various levels. Several affected proteins disrupt the initiation of autophagy by inhibiting the initiation complex these include: peripheral myelin protein 22 (PMP22), involved in more than 50% of IPNs, transient receptor potential cation channel subfamily V member 4 (TRPV4), and leucine rich repeat and sterile alpha motif containing 1 (LRSAM1). Others overstimulate the initiation step such as: VAMP associated protein B (VAPB), WNK lysine deficient protein kinase 1 (WNK1), seipin lipid droplet biogenesis associated (Seipin), and DnaJ heat shock protein family (Hsp40) member B2 (DNAJB2). Mutations affecting mitochondrial proteins can inhibit autophagy by causing an abnormal increase in mitophagy and disrupting the autophagy/mitophagy balance: mitofusin 2 (MFN2) and ganglioside induced differentiation associated protein 1 (GDAP1) (left inset). Mutant ER-resident family with sequence similarity 134 member B (FAM134B) affects its role in reticulophagy (right inset). The cyto-protective N-myc downstream regulated 1 (NDRG1) inhibits the initiation complex or stimulates the nucleation complex depending on the physiological triggers. At the nucleation step, mutant tectonin beta-propeller repeat containing 2 (TECPR2) disrupts the formation of early autophagic membranes from the ER. The elongation of the phagophore into an autophagosome is disrupted by mutations that affect the supply for forming membranes from late endosomes: lipopolysaccharide induced TNF factor (LITAF) and SH3 domain and tetratricopeptide repeats 2 (SH3TC2), and from ATG9-containing vesicles as in the microtubules associated proteins: kinesin family member 1A (KIF1A) and Dynamin 2 (DNM2). This step can also be overstimulated by mutations in the tyrosine kinase A (TrkA) which lead to toxic increase in ATG12-ATG5 conjugates. The transport of autophagosomes to lysosomes is disrupted by mutations affecting cytoskeleton associated proteins such as Dynactin 1 (DCTN1) and dystonin (DST). Mutations involving Ras-related GTPase (RAB7), valosin containing protein (VCP), heat shock protein B8 (HSPB8), and the phosphatases: phosphoinositide 5-phosphatase (FIG4), and myotubularin-related proteins (MTMR2 and MTMR13) block the lysosomal fusion step in autophagy. Mutant chaperonin containing TCP1 subunit 5 (CCT5) on the other hand inhibits the degradation step. In addition, several mutant proteins lead to the formation of aggregates which basal autophagy on its own might not cope with (PMP22, Seipin, LITAF, HSPB8) and neurofilament light (NEFL). The colors and shapes of the boxes are randomly assigned and show that they are different proteins belonging to the same complexes.
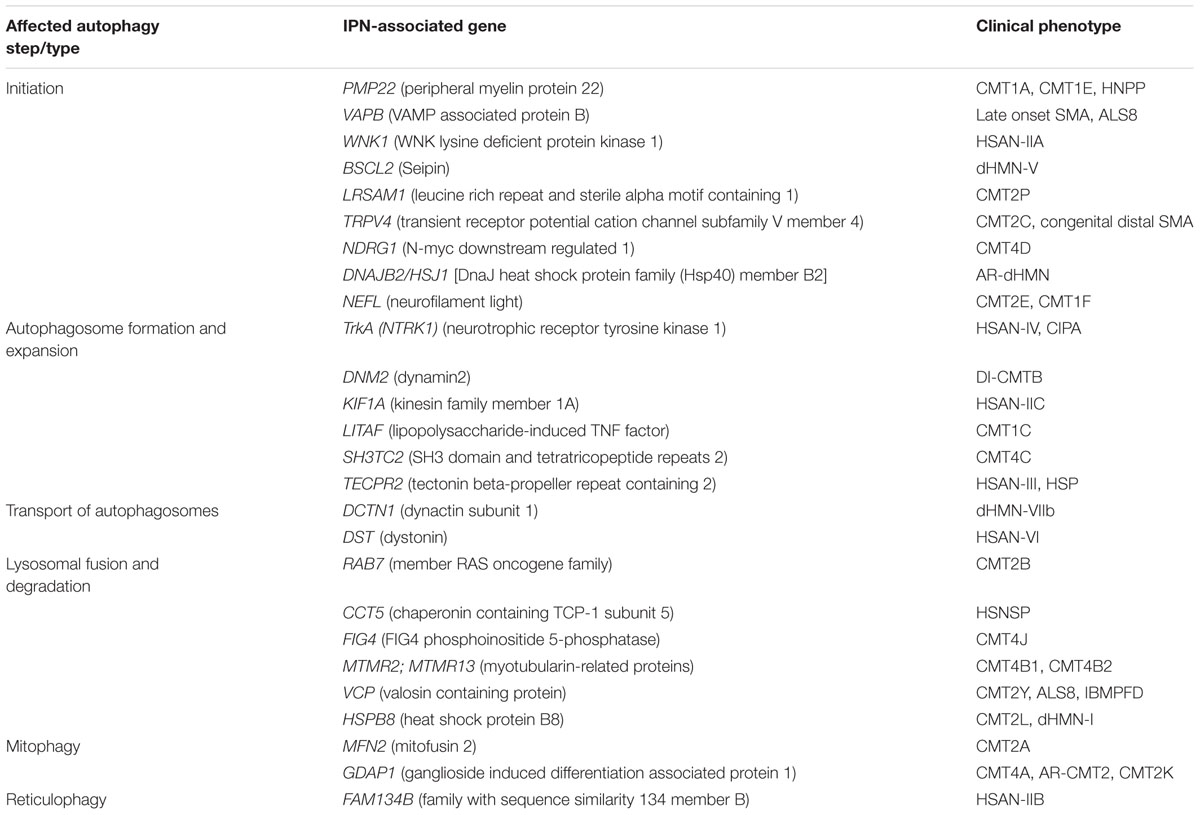
TABLE 1. Summary of IPN-associated genes, their clinical phenotype and the stage they impact the autophagy pathway.
The PMP22 is a myelin glycoprotein which plays an important role in the formation and maintenance of compact myelin hence participating in the formation of the myelin sheath (Li et al., 2012). Autophagy is also involved in myelination of sciatic nerves by regulating the structural plasticity of Schwann cells (Jang et al., 2015). Mutations in PMP22 are responsible for more than 50% of all inherited neuropathies. Duplication of PMP22 causes the most common demyelinating neuropathy, CMT1A (Lupski et al., 1992; Timmerman et al., 1992). Point mutations in PMP22 cause CMT1E neuropathy, while a heterozygous deletion cause hereditary neuropathy with liability to pressure palsies (HNPP) (Nicholson et al., 1994; Russo et al., 2011). CMT1A and CMT1E patients show variable clinical severity but the main features of the diseases include slowly progressive weakness and atrophy of the distal limb muscles, pes cavus, and reduced or absent deep tendon reflexes. In the Trembler CMT1E mouse models carrying a missense mutation in Pmp22, the mutant protein colocalizes with lysosomal markers (Notterpek et al., 1997). In the C22 mouse model of overexpression of PMP22, resembling the CMT1A phenotype, aggregate formation is seen in affected nerves. This aggregation occurs together with a reduced proteasomal activity and association of the aggregates with autophagosomes (Fortun et al., 2006). Since autophagy and the proteasome can play compensatory roles in maintaining cellular homeostasis, autophagy seems to play a part in the pathomechanism of CMT1 neuropathic mouse models. With reduced proteasomal activity, basal autophagy on its own might not be enough to clear out all the aggregating mutant proteins, but induction of autophagy by external methods can aid in the degradation of toxic products. Indeed, autophagy induction in neuronal cultures from CMT1A mouse models via nutrient deprivation or rapamycin treatment decreases the aggregate formation. Autophagy induction also improves PMP22 trafficking leading to an enhancement of remyelination and indirectly increasing the expression of myelin proteins and the abundance and length of myelin internodes (Madorsky et al., 2009; Rangaraju et al., 2010). Suppression of a key autophagy protein, ATG12 by siRNA, abolishes these effects, indicating that the phenotypic improvements are mediated by autophagy and that an intact autophagy pathway is required for proper remyelination (Rangaraju et al., 2010).
VAMP associated proteins B and C belongs to a family of ER anchored proteins and plays a role in Golgi-mediated transport, membrane trafficking and neurotransmitter release (Lev et al., 2008). Recently the tethering complex formed by VAPB (ER) and PTPIP51 (mitochondria) has been shown to regulate the induction of autophagy through a role in mitochondria-ER calcium delivery which acts on BCL2-Beclin 1 interaction (Gomez-Suaga et al., 2017). Dominant mutations in VAPB lead to complex and atypical forms of inherited motor neuron disease (Nishimura et al., 2004). In a VAPB/ALS8 knock-in mouse model, mutant VAPB translocates from the ER to the autophagosome where it is degraded (Larroquette et al., 2015). The mislocalisation of VAPB causes ER stress and can lead to loss of VAPB functions in the ER. Absence of VAPB in the ER abolishes the VAPB-PTPIP51 tethering complex. Loosening of the ER-mitochondria contacts by loss of VAPB1-PTPIP51 can lead to overstimulation of autophagy (Gomez-Suaga et al., 2017). The dysregulated autophagy may have adverse effects on neuronal homeostasis and might be a prime pathological sign in VAPB associated neuropathies.
WNK lysine deficient protein kinase 1 is a serine/threonine kinase with the canonical function of regulating ion transport across cell membranes (Moriguchi et al., 2005). Truncating mutations in WNK1 cause hereditary sensory and autonomic neuropathy type II (HSAN-II) (Rivière et al., 2004). HSAN-II is characterized by absence of pain sensations and patients suffer from ulcero-mutilating form of neuropathy. Little is known about the non-canonical functions of WNK1, but a role in autophagy was discovered recently. WNK1 exerts an inhibitory effect on basal and starvation-induced autophagy by interacting with UV Radiation Resistance-Associated Gene (UVRAG), a component of the main autophagy complex PI3KC3. WNK1 interaction reduces the activity of PI3KC3 and alters the phosphorylation status of ULK1 leading to inhibition of autophagy. Knocking-down WNK1 by siRNA in different cell lines leads to increased activation of autophagy (Gallolu Kankanamalage et al., 2016). WNK1 appears to play a regulatory role in autophagy by inhibiting the process, therefore truncating mutations in WNK1 leading to neuropathy might hinder its autophagy-related function and lead to toxic increase in autophagic flux that can have deleterious effects contributing to the neuropathology.
The BSCL2 gene encodes the protein seipin, a transmembrane protein that resides in the ER (Ito et al., 2008). The exact function of seipin remains elusive, though a role in lipid homeostasis and adipogenesis has been suggested (Cartwright and Goodman, 2012). Null mutations in BSCL2 are responsible of lipodystrophy, dominant mutations on the other hand cause distal hereditary motor neuropathy (dHMN-V) and Silver syndrome. dHMN-V patients present with uncharacteristic prominent hand muscle wasting and weakness early in the course of the disease, and mild to severe plasticity of the lower limbs (Windpassinger et al., 2004). Neuropathy-causing BSCL2 mutations have been shown to affect the glycosylation sites of BSCL2 and lead to the accumulation of the unfolded protein in the ER (Windpassinger et al., 2004). It has been confirmed that different mutations in BSCL2 can lead to the formation of perinuclear aggregates (Hsiao et al., 2016). In a neuropathic mouse model expressing mutant seipin exclusively in neurons (seipinopathy), seipin presents in intracellular aggregates. In addition, motor neurons show accumulation of autophagy marker LC3II together with a fragmented Golgi apparatus phenotype (Guo et al., 2013). The accumulation of autophagosomes was also confirmed by electron microscopy indicating that overstimulation of autophagy by mutant seipin leads to signs of degeneration (fragmented Golgi). The increased activation of autophagy was also confirmed in cell models expressing mutant seipin (Fan et al., 2015). Together these studies show that disrupted autophagy is a hallmark of seipin-related neuropathy.
Leucine rich repeat and sterile alpha motif containing 1 is a leucine-rich repeat protein and an E3 ubiquitin ligase. LRSAM1 is involved in cell adhesion and cargo sorting during receptor endocytosis (Amit et al., 2004). Dominant negative mutations in LRSAM1 cause CMT2P neuropathy (Weterman et al., 2012). CMT2P patients display features of the axonal CMT2 including mild lower-limb sensorimotor neuropathy, foot deformities, and hammer toes (Weterman et al., 2012). Early studies reported a function for LRSAM1 in endocytosis and retrovirus budding (Amit et al., 2004). Later it was shown to recognize, ubiquitinate and guide several bacteria strains to autophagy (bacteriophagy) (Huett et al., 2012). LRSAM1 is also a potential interactor of LC3 family protein GAPARAPL2 (GABA type A receptor-associated protein) (Ng et al., 2011), and is also involved in the autophagy pathway via interaction with PHD finger protein 23 (PHF23), a negative regulator of autophagy (Wang et al., 2014). A role for LRSAM1 in the general autophagic pathway is evident by the fact that LRSAM1 overexpression increases autophagic flux by increasing the formation of LC3-GFP puncta in cultured cell lines, while silencing it causes a decrease in autophagic flux (Wang et al., 2014). LRSAM1 appears to play a regulatory role in the activation of autophagy probably through the interaction with other signaling proteins. Though the exact connection between LRSAM1, interactors and general autophagy remains unclear, loss of function mutations may disrupt the role of LRSAM1 in autophagy.
Transient receptor potential cation channel subfamily V member 4 is a member of the TRP cation channels, and has an important role in sensing temperature, osmotic pressure, and mechanic stimuli (Köttgen et al., 2008). Mutations in TRPV4 cause CMT2C, a hereditary motor and sensory neuropathy with diaphragm and vocal cord paresis (Dyck et al., 1994). Alterations in TRPV4 can have toxic consequences in neuronal cells due to changes in calcium concentrations (Deng et al., 2010). TRPV4 can induce autophagy through the AKT-pathway and potentially via regulation of calcium levels and osmotic pressure (Zhan et al., 2015). Furthermore, inhibition of TRPV4 by siRNA leads to inhibition of autophagy (Zhan et al., 2015). Therefore, dysregulation of autophagy is a potential pathomechanism in TRPV4 associated neuropathies, as a direct effect of mutant TRPV4 or as one of the neurotoxic consequences due to the disruption in calcium concentrations by TRPV4 mutants.
N-myc downstream regulated 1 is mainly involved in cyto-protective stress response through regulation of p53 protein (Chen et al., 2010). In addition, it plays a role in immunity, development, differentiation and an important role in cancer (Fang et al., 2014). Mutations in NDRG1 cause CMT4D neuropathy characterized with severe reduction in NCVs, skeletal and foot deformities and sensory loss (Kalaydjieva et al., 2000). NDRG1 inhibits basal and ER Stress induced autophagy via suppression of protein kinase-like endoplasmic reticulum kinase (PERK)/eIF2α axis (Sahni et al., 2014). Upregulation of NDRG1 has also been shown to initiate BNIP3 and Beclin mediated autophagy (Han et al., 2014). Autophagy regulation seems to be a part of the cyto-protective function of NDRG1. It may therefore play a role in fine tuning autophagy levels by acting on different autophagy inducing or inhibiting pathways. Though a link between CMT4D-causing NDRG1 mutations and autophagy is yet to be established, mutations in NDRG1 might influence autophagy regulation by NDRG1.
HSJ1, a co-chaperone protein, is involved in binding ubiquitylated chaperone clients and their loading onto the Hsc70 chaperone, thus preventing aggregation and aiding proteasome sorting (Westhoff et al., 2005). HSJ1 consists of two isoforms with distinct intracellular localization. HSJ1a is cytoplasmic and nuclear, while HSJ1b localizes to the ER (Westhoff et al., 2005). In addition to its function in the proteasome system, HSJ1 has been shown to be involved in the recruitment of the autophagy marker protein LC3 to damaged mitochondria aiding in mitochondrial autophagy (mitophagy) (Rose et al., 2011) Recessive mutations in HSJ1 affecting both isoforms can cause CMT2T neuropathy or purely motor AR-dHMN (Gess et al., 2014). HSJ1a, the cytoplasmic isoform has been shown to enhance autophagy and decrease aggregation (Novoselov et al., 2013; Sanchez et al., 2016). Furthermore, deletion mutations in HSJ1b isoform, have been shown to lead to neurodegeneration and irregular increase in the autophagy marker LC3 due to increase in HSJ1a expression (Sanchez et al., 2016). These studies present HSJ1 as a moderator of autophagy and proteasome activity and show that mutations in HSJ1 could disrupt this balance leading to impaired clearance of ubiquitylated proteins and autophagy.
Neurofilament light is part of the axoskeleton (neurofilaments) of large myelinated axons of the central and peripheral nerves. The main role of neurofilaments is maintaining fiber caliber and subsequently the conduction velocities of myelinated axons (Perrot et al., 2008). Mutations in NEFL can cause CMT2E/1F neuropathy with distal weakness and wasting of the lower limbs, and occasional cerebellar dysfunction, tremor, and hearing loss (Mersiyanova et al., 2000; Pareyson et al., 2014). CMT causing NEFL mutations have been shown to cause aggregate formation in cell culture and in vitro models. Activation of PKA, alleviates the aggregation phenotype (Sasaki et al., 2006). PKA is a known regulator of autophagy mostly exerting an inhibitory effect through phosphorylation of ATG13 an essential protein in the Ulk1 autophagy-initiation complex (Stephan et al., 2009), and of LC3 (Cherra et al., 2010). Therefore, the aggregation phenotype of NEFL might be a by-product of abnormality in the autophagy pathway which is restored by the activation of PKA. Furthermore, NEFL has been shown to interact with the PI3-phosphatase MTMR2 (Previtali et al., 2003). This interaction might be essential for the potential role of MTMR2 in autophagy through its regulation of endosomal vesicle trafficking or through its interaction with MTMR13, a direct regulator of autophagosome–lysosome fusion (see IPN Genes and Lysosomal Fusion and Degradation).
Tyrosine kinase A, a nerve growth factor (NGF) receptor, is essential for neuronal survival and regulation of neuronal death (Miller and Kaplan, 2001). TrkA has been associated with regulating the interplay between autophagy and apoptosis mainly in cancer cells (Hansen et al., 2007; Dadakhujaev et al., 2009). Overexpression of TrkA leads to an increase in the amount of ATG5-ATG12 conjugates and ultimately to an increase in autophagy marker LC3II (Dadakhujaev et al., 2009). Mutations in TrkA cause HSAN-IV neuropathy with congenital insensitivity to pain (CIPA) marked by absence of reaction to noxious stimuli and self-mutilating behavior (Indo et al., 1996). Recently, mutant TrkA has been shown to cause accumulation of autophagosomes. This accumulation proved to be a result of aberrant activation of autophagy and over saturation of the lysosomes rather than a deficit in autophagosome–lysosome fusion. Neurons expressing TrkA mutant show increased cell toxicity and dystrophic neurites as a result of autophagosomes accumulation by the abnormal autophagy activation (Franco et al., 2016).
Dynamin2 is one of the three isoforms of dynamin. DNM2 is a mechano-GTPase involved in endocytosis, Golgi function and vesicle trafficking (González-Jamett et al., 2013). DNM2 has been shown to play essential roles in autophagy. One of its main roles has been the autophagic lysosomal reformation, supplying nascent protolysosomes and hence maintaining the autophagic flux (Schulze and McNiven, 2014; Klionsky et al., 2016). Recently, DNM2 has been shown to interact with another protein Bif-1 to mediate the generation of Atg9-containing vesicles delivering Atg9 to autophagosome formation sites and promoting the formation of autophagosomes (Takahashi et al., 2016). Dominant mutations in DNM2 have been linked with the intermediate form of CMT (DI-CMTB) with NCVs ranging from normal to 25 m/s (Züchner et al., 2005). A mutant DNM2 mouse model shows signs of neuropathy and a decreased autophagic flux indicated by lower levels of LC3II expression and P62-positive bodies [a selective autophagy marker also known as sequestosome1 (SQSTM1)] (Durieux et al., 2012). These studies suggest that mutations in DNM2 leading to neuropathy affect its function in the formation of autophagosomes and present autophagy impairment as a pathomechanism of DNM2-linked neuropathy.
Kinesin family member 1A is a kinesin motor protein essential for the transport of vesicles in neuronal axons in anterograde fashion (Lo et al., 2011). Mutations in KIF1A can cause hereditary sensory and autonomic neuropathy type 2 (HSAN-II) (Rivire et al., 2011). HSAN-II patients suffer from frequent occurrence of unrecognized injuries and fractures of hands and feet due to loss in all peripheral sensations including pain and temperature. Autonomic symptoms include severe feeding problems in infants and common gastroesophageal reflux. Recently, it was shown that KIF1A controls the localization of ATG-9 and regulates the spatial distribution of autophagosomes in developing neurons of Caenorhabditis elegans (Stavoe et al., 2016). Maintaining ATG9 trafficking is essential for proper autophagosome formation (Lamb et al., 2016). The effects on autophagy in mutant KIF1A patients or cell/mouse models remains to be studied, but it is not inconceivable that impairment of autophagy due to disruption in ATG9 trafficking in KIF1A mutants is one of the pathomechanisms of neuropathy due to KIF1A mutations.
Lipopolysaccharide-induced TNF factor, has been mainly linked with inflammatory functions, namely the secretion of cytokines such as TNF upon lipopolysaccharide stimulation (Tang et al., 2006). It has also been implicated in tumor suppression pathways and p53-induced apoptotic pathway (Zhou et al., 2011; Liu et al., 2012). LITAF seems to have different functions in different cell types. A role for LITAF in the positive regulation of autophagy has been shown in B cells. LITAF regulates LC3 expression and colocalizes with autophagosomes in B cells. Furthermore, overexpression of LITAF stimulates autophagy while silencing LITAF decreases the autophagic flux in these cells (Bertolo et al., 2013). Mutations in LITAF cause the dominant demyelinating CMT1C neuropathy (Street et al., 2003). In HEK293 cell models expressing CMT1C associated LITAF mutant protein, the mutant proteins cause mislocalisation of LITAF from early endosomes to the cytosol, destabilize LITAF and cause it to be aggregation prone. The aggregating mutant LITAF is then degraded by the proteasome and by autophagy (Lee et al., 2011). In neuroblastoma cell lines expressing WT and mutant forms of LITAF, CMT-causing LITAF mutants are mislocalised to the mitochondria, while the WT form traffics through the secretory pathway to the late endosome/lysosome (Lacerda et al., 2014). Late endosomes and lysosomes are essential for supplying the maturing autophagosome and for degrading the lysosomal content (Murrow et al., 2015). The trafficking function of LITAF might be related to its effect on autophagic flux, mislocalisation of mutant LITAF can therefore have a negative effect on the integrity of the endo/lysosomes and consequently on the autophagic process.
SH3TC2 encodes the Src homology 3 domain and tetratricopeptide repeats 2 protein. Little is known about the function of SH3TC2. However, the several motifs it contains suggest protein-protein interaction functions. Recessive mutations in SH3TC2 cause a form of demyelinating neuropathy, CMT4C, characterized with severe scoliosis (Senderek et al., 2003b). Wild-type SH3TC2 has been shown to localize to recycling endosomes in rat Schwann cells, while the CMT causing mutant forms mistarget SH3TC2 away from the recycling endosomes (Roberts et al., 2010). SH3TC2 interacts with the small GTPase Rab11. The SH3TC2/Rab11 interaction is disrupted in mutant SH3TC2 in cultured cell lines (Roberts et al., 2010). Interestingly, KO of SH3TC2 in a transgenic mouse model decreases the expression of Rab11 in nerves of these mice (Stendel et al., 2010). Rab11 has been described as a positive regulator of autophagy. ULK1 and ATG9 localize in part to Rab11 positive recycling endosomes. Rab11 dependent vesicular transport from the recycling endosomes contributes to the forming autophagosomes and regulates starvation induced autophagy (Longatti et al., 2012). So by disrupting Rab11 interaction and expression levels and mistargeting SH3TC2 away from the recycling endosomes, SH3TC2 CMT-associated mutations could disrupt the contribution of recycling endosomes to autophagosome formation.
Tectonin beta-propeller repeat containing 2 was initially identified as human ATG8-interacting protein (Behrends et al., 2010). TECPR2 possesses an LC3 interacting motif (LIR) through which it binds to LC3B and LC3C family proteins. TECPR2 LIR-dependent binding leads to its association with cellular trafficking components such as HOPS and SEC24D. This interaction ultimately maintains a functional ERES and efficient ER export. TECPR2 depletion causes a decrease in ERES number and a delayed ER export. Maintaining a functional ERES provides a scaffold for autophagosome formation (Stadel et al., 2015). Mutations in TECPR2 leading to the translation of a truncated and unstable version of TECPR2 cause a form of HSP (Oz-Levi et al., 2012) and hereditary sensory and autonomic neuropathy (HSAN-III) (Heimer et al., 2016). The latter presents with intellectual disability and, spasticity, and chronic respiratory disease. Skin fibroblasts from TECPR2-related HSP patients show a decreased number of LC3 and P62 proteins marking a decreased autophagic flux (Oz-Levi et al., 2012). These fibroblasts also show delayed ER export (Stadel et al., 2015), an indication that the function of TECPR2 in maintaining ER export and the formation of early autophagosome intermediates is disrupted by HSP-causing mutations, presenting autophagy disruption as a likely pathomechanism in HSP neuropathy.
Dynactin subunit 1 is the largest subunit of the dynactin complex. The dynactin complex has been associated with a large range of cellular functions including mitosis, ER-Golgi transport, lysosomes and endosomes movement as well as interacting with dynein for cargo transport (Schroer, 2004). Mutations in DCTN1 disrupt axonal transport and lead to hereditary motor neuropathy (dHMN-VIIb) with breathing difficulty due to vocal fold paralysis and progressive facial weakness (Puls et al., 2003). DCTN1 knock-down shows motor neuron degeneration associated with autophagosomes accumulation due to impaired transport of autophagosomes along the axons (Ikenaka et al., 2013). Similarly, in a mouse model of mutant DCTN1, abnormal accumulation of autophagosomes has been shown in motor neurons and linked to the motor neuropathy phenotype (Wiesner et al., 2015). This presents autophagy impairment as a pathomechanism leading to motor neuropathy for DCTN1 mutations.
The dystonin gene encodes several dystonin protein isoforms which are cytoskeletal cross-linking proteins that can interact with different organelles, microtubules, and protein complexes (Ferrier et al., 2013). Mutations in DST cause hereditary sensory and autonomic neuropathy (HSAN-VI) also known as familial dystonomia and presenting with alacrima, depressed deep tendon reflexes, and lingual fungiform papillae (Edvardson et al., 2012). In a mouse model of dystonia, mutant DST disrupts the autophagic process as evident by the accumulation of LC3 and P62 proteins. This impairment is attributed to the failure of autophagosome–lysosome fusion suggesting that dystonin is required to transport the autophagosomes to the lysosomes for maturation into autolysosomes (Ferrier et al., 2015). Interestingly, expression of the non-mutated form of dystonin restores the autophagy deficits revealing that the autophagy deficit is a direct effect of the mutated dystonin (Ferrier et al., 2015).
Ras-related protein Rab-21 is a small GTPase functioning in vesicular trafficking, more specifically in the transport from early to late endosomes and from late endosomes to lysosomes (Stenmark, 2009). RAB7 is recruited to autophagosomes and is required for the fusion of autophagosomes and lysosomes (Jäger et al., 2004). Mutations in RAB7 cause CMT2B neuropathy characterized by distal muscle wasting and weakness and frequent foot ulcers and infections (Verhoeven et al., 2003). Dominant negative mutations retard the recruitment of RAB7 to autophagosomes, and prevent the progression of autophagy by impairing autolysosome formation (Gutierrez et al., 2004).
A subunit of the TCP-1 containing chaperonin complex (CCT), an ATP-dependent chaperone responsible for folding unfolded proteins including actin and tubulin. Recessive mutations in CCT5 cause a mutilating hereditary sensory and autonomic neuropathy with spastic paraplegia (HSNSP) (Bouhouche et al., 2006). Studies have shown that CCT can prevent mutant huntingtin (htt) aggregation in vitro (Darrow et al., 2015). Recently, it was shown that the function of CCT in preventing aggregation of mutant proteins is mediated by autophagy (Pavel et al., 2016). CCT is required for lysosomal biogenesis and functioning and for autophagosome–lysosome fusion, possibly via interaction with cytoskeleton proteins. Disruption of CCT integrity by knock-down or by mutations in CCT5 disrupts autolysosome formation and cargo degradation (Pavel et al., 2016).
FIG4 phosphoinositide 5-phosphatase is a phospholipid phosphatase responsible for the generation and turnover of the PtdIns(3,5)P2 phosphoinositide (Di Paolo and De Camilli, 2006). Mutations in FIG4 cause Yunis–Varon syndrome, familial epilepsy with polymicrogyria, and the severely demyelinating Charcot-Marie-Tooth type 4J neuropathy (CMT4J) (Chow et al., 2007; Katona et al., 2011; Vaccari et al., 2015). In a FIG4 deficient neuropathic mouse model, sensory and motor neurons as well as Schwann cells seem to be affected (Katona et al., 2011; Vaccari et al., 2015). Neurons show decrease of PtdIns(3,5)P2 levels, a sign of impaired FIG4 enzymatic activity, and enlargement of late endosomes and lysosomes. Enlargement of late endosomes and lysosomes can hinder the fusion capacity of autophagosomes with lysosomes. Indeed these mice show accumulation of autophagic markers LC3II and P62 in their sciatic nerve. This accumulation is independent of an increase in the autophagic flux (Ferguson et al., 2010; Vaccari et al., 2015). Thus abolishing the enzymatic activity of FIG4 by missense mutations or haploinsufficiency seems to affect the progression of autophagic degradation by preventing the fusion between autophagosomes and the then enlarged late endosomes/lysosomes (Vaccari et al., 2015).
Myotubularin-related proteins are PI3-phosphatases consisting of catalytically active or inactive members. Mutations in MTMR2 and MTMR13 (also called SET-binding factor 2) cause the demyelinating CMT4B1 and CMT4B2 neuropathies respectively with early-onset glaucoma (Azzedine et al., 2003). MTMR2 is a catalytically active phosphatase which dephosphorylates phosphatidylinositol 3-phosphate (PI(3)P) and bisphosphate PI(3,5)P2. MTMR13 on the other hand is an inactive phosphatase but it has been shown to associate with MTMR2 (Robinson and Dixon, 2005). MTMR2 has been linked with regulation of the late endocytic pathway and vesicular transport through its putative substrate PI(3,5)P2 (Bolino et al., 2004; Lenk and Meisler, 2014). MTMR13 on the other hand acts as a RAB21 GEF required for fusion of autophagosome with the lysosome. MTMR13 GEF activity is induced upon starvation and it promotes the trafficking of VAMP8 to the lysosome where it is needed to mediate fusion with the autophagosome (Jean et al., 2015). Autophagy has not yet been studied in the context of CMT4B causing mutations in MTMRs but the roles of both MTMR2 and MTMR13 in autophagy and their functional association with each other presents autophagy impairment as a possible common pathomechanism in mutant MTMRs leading to neuropathy.
Valosin containing protein is an AAA+ ATPase associated with diverse cellular activities including the ATP-dependent remodeling of proteins to unfold them or extract them from cell structures or binding partners (Erzberger and Berger, 2006). Mutations in VCP cause a multisystem degenerative disease consisting of inclusion body myopathy, Paget’s diseases and frontotemporal dementia (IBMPFD) (Kimonis et al., 2000), familial ALS (Shaw, 2010) and can also lead to CMT2Y neuropathy with distal muscle weakness and atrophy and length-dependent sensory loss (Gonzalez et al., 2014). VCP also plays a role in lysosomal homeostasis and the lysosomal damage response. Lysosomal damage induced by lysomotropic reagents triggers a concomitant recruitment of VCP to lysosomes. VCP then cooperates with a set of cofactors to drive the degradation of ruptured lysosomes (Papadopoulos et al., 2016). Damaged lysosomes accumulate in cells expressing mutant VCP showing that its role in clearing out damaged lysosomes is compromised by neuropathy- causing mutations. VCP mutant knock-in mouse model shows accumulation of LC3II positive structures in myoblasts (Nalbandian et al., 2013). Furthermore, knock-down of VCP leads to accumulation of autophagosomes due their failure to mature into autolysosomes (Ju et al., 2009). A high percentage of the accumulated autophagic vesicles contain ubiquitin positive structures. VCP has been shown to be critical for the maturation of ubiquitin-containing autophagosomes into autolysosomes under basal conditions and under proteasome inhibition. This function of VCP in maturation of autophagosomes seems to be impaired by neuropathy-causing mutations (Lee et al., 2010).
Heat shock protein B8 belongs to the family of small heat shock proteins, ATP-independent chaperones that aid the folding of misfolded proteins by ATP-dependent chaperones (Holmgren, 2012). In addition to its function as a molecular chaperone, HSPB8 is involved in several other stress signaling functions such as removal of aggregates through CASA (Carra et al., 2008). CASA involves the recognition of substrate proteins by a complex of chaperones and co-chaperones including HSPB8 and P62 and its subsequent loading onto autophagosomes. Mutations in HSPB8 cause CMT2L and dHMN-IIa neuropathies typically presenting with paresis of the extensor muscles of the big toe and then the feet (Irobi et al., 2004; Tang et al., 2005). Expression of mutant HSPB8 in cultured cells induces aggregate formation (Irobi et al., 2004). Transcriptional induction of WT HSPB8 in mouse models of motor neuron disease aids in the clearance of misfolded proteins and aggregates (Crippa et al., 2016). Interestingly, cells from neuropathic patients carrying HSPB8 mutations also show deficits in lysosomal delivery of autophagosomes (Kwok et al., 2011). Taken together, these studies indicate that intact HSPB8 is necessary for aggregate clearance, possibly through CASA. Disruption of this role by mutations in HSPB8 contribute to the neuronal pathogenicity.
Family with sequence similarity 134 member B is a transmembrane protein localized to the cis-golgi and predominantly expressed in the sensory and autonomic ganglia. Mutations in FAM134B cause hereditary sensory and autonomic neuropathy HSAN-IIB leading to impaired nociception, autonomic dysfunction, and severe mutilations (Kurth et al., 2009). FAM134B interacts with LC3/GABARAP through an LC3 interacting motif at its C-terminal domain. This interaction is essential for selectively directing part of the ER to autophagosomes (reticulophagy). Mutations causing sensory neuropathy disrupt the LC3 interacting motif and consequently the FAM134B-LC3 binding. FAM134B KO mice develop sensory neuropathy and degeneration of sensory neuronal axons. Deficit in reticulophagy in these mice but not general autophagy disrupts ER homeostasis leading to ER expansion and inhibition of ER turnover that ultimately leads to degeneration of sensory neurons (Khaminets et al., 2015).
Mitofusin 2 was identified as a transmembrane mitochondrial GTPase required for mitochondrial fusion (Santel and Fuller, 2001). Mutations in MFN2 cause CMT2A neuropathy (Züchner et al., 2004) and are one of the most frequent CMT2 causing mutations (OMIM 609260, CMT2A ≈ 35% of CMT2). CMT2A presents as a severe predominantly motor neuropathy or motor accompanied with profound proprioception loss. In addition to its role in mitochondrial fusion, MFN2 plays multiple roles including regulation of cell survival, cell proliferation, ER stress and autophagy (reviewed in Schrepfer and Scorrano, 2016). MFN2 deletion has been shown to impair starvation-induced autophagy by disrupting the MAM (mitochondria associated membranes), a described site for autophagosome formation (Hailey et al., 2010; Hamasaki et al., 2013). MFN2 deletion in HeLa cells has also been shown to affect cell proliferation due to autophagy impairment (Ding et al., 2015). Motor neurons derived from iPSCs obtained from CMT2A patients show increase in mitophagy and mitochondrial depletion and increased expression of PINK1, PARK2 and BNIP3, known triggers for autophagic degradation of mitochondria (Rizzo et al., 2016). In a MFN2 KO mouse model, sarcopenia is seen in correlation with impaired autophagy in the muscle, accumulation of damaged mitochondria, and activation of an adaptive mitophagy pathway (Sebastián et al., 2016). Taken together, MFN2 seems to play an essential role in autophagosome formation, possibly through maintenance of mitochondrial membranes and MAMs. This role is impaired in MFN2 deficiency and in MFN2 causing CMT2A mutations, leading to autophagy impairment and disruption of balance between mitophagy and general autophagy.
Ganglioside induced differentiation associated protein 1 is an integral protein of the outer mitochondrial membrane formed of 2 glutathione-S-transferase domains (Huber et al., 2016). Dominant and recessive mutations in GDAP1 cause demyelinating (CMT4A), or axonal (AR-CMT2 or CMT2K) neuropathies (Baxter et al., 2002; Senderek et al., 2003a; Sivera et al., 2010). GDAP1 is required for the regulation of the mitochondrial network and mitochondrial integrity (Niemann et al., 2005). In a KO mouse model of GDAP1 that recapitulates the neuropathic phenotype of CMT4A, disruption of the mitochondrial network and mitochondrial calcium homeostasis is evident in neurons and associates with accumulation of autophagic vesicles (Barneo-Muñoz et al., 2015). Mitochondrial defects can lead to abnormal increase in targeting mitochondria to autophagic degradation (mitophagy). In addition, both mitochondrial interconnectivity and calcium homeostasis are essential for the proper functioning of autophagy (Gomez-Suaga et al., 2017). It remains unclear whether the increased accumulation of autophagosomes in neurons of CMT4A mouse models is a by-product of increased mitophagy or defects in the general autophagic flux. Nonetheless, autophagic dysregulation does seem to be a feature of the pathology caused by CMT-causing GDAP1 mutations and might explain the variety of clinical phenotypes presented by these mutations ranging from myelinating to axonal CMT and from dominant to recessive.
So far, finding a treatment for hereditary neuropathies remains a challenge. An important step toward a successful therapy, from both an economic and a pharmaceutical perspective, is finding a common pathomechanism that can be targeted in different neuropathic genotypes. By bridging different neuropathic phenotypes and different causal genes, autophagy impairment emerges as a good candidate for modulation by therapeutic measures. Autophagy modulation as a therapy approach has been used in several more common neurodegenerative disease models. Inducing autophagy has been shown to reduce the severity of the Huntington’s disease phenotype and improve clearance of Htt aggregates in mouse and neuronal cell models (Rose et al., 2010). In Alzheimer’s disease, accumulation of autophagosomes contributes to the pathology (Ohta et al., 2010). Inhibition of autophagy has been reported as a mode of action in drugs that decrease amyloid-β accumulation in Alzheimer’s patients (Lipinski et al., 2010). Accumulation of autophagosomes has also been attributed to defects in beclin-1 expression, hence autophagy enhancing treatments such as resveratrol and lithium can improve the pathology of Alzheimer’s disease (Vingtdeux et al., 2010; Rahvar et al., 2011). In addition, expression of beclin-1 in neuronal cells of transgenic mice improves their Parkinson’s disease phenotype by enhancing lysosomal activation (Spencer et al., 2009). Similarly, the mTOR-independent autophagy inducer trehalose improves the clearance of α-synuclein, huntingtin (Sarkar et al., 2007) and SOD1 aggregates (Castillo et al., 2013a). Furthermore, autophagy modulating compounds are already in use to treat different neurodegenerative diseases and cancers, and are currently at different phases of clinical trials (reviewed in Towers and Thorburn, 2016). Despite its promise, autophagy modulation as a treatment for hereditary neuropathies still presents many challenges. The model of treating mutants that increase autophagic activation with inhibitors of autophagy and those that inhibit autophagy with activators is oversimplified. Many autophagy modulating drugs can have off-target effects. For example, mTORC1 inhibitors can affect cell metabolism and lipid synthesis in an autophagy-independent manner (Li et al., 2014). Therefore, targeting the specific autophagy step affected by a hereditary neuropathy causing-mutation is a better and more elegant approach to enhance the autophagic status without hindering other vital cellular processes. Certain step-specific approaches already exist and include direct stimulation of autophagy by targeting beclin1 with the alkaloid isorhynchophylline (Lu et al., 2012). Targeting the lysosomal fusion and degradation is also possible using pharmacological activators of lysosomal enzymes such ambroxol (McNeill et al., 2014) or acidic nanoparticles that increase the acidity of the lysosomes (Baltazar et al., 2012). Non-pharmacological enhancement of autophagy such as caloric restriction or exercise presents another way of overcoming the pharmacological modulation of other cellular pathways. Physical exercise has already been described as a rehabilitating measure in CMT patients (Roberts-Clarke et al., 2016; Vita et al., 2016). Whether improvement of autophagy is a mediator of the positive impact of exercise remains to be investigated, but exercise has been shown to enhance autophagy in the brain (He et al., 2012) and skeletal muscles (Ferraro et al., 2014). Another caveat of autophagy directed therapy is the difficulty of tracing autophagy in vivo. In other words, there is a need for biomarkers that can be used to make sure that the intended therapy is improving the autophagic status in patients in order to alleviate their symptoms. Such markers are as of yet unavailable, but recent advances using mouse models allows following up a treatment’s effect on autophagy in mouse tissues (Castillo et al., 2013b). This strategy, combined to behavioral and electrophysiological testing in a neuropathic mouse model can permit the direct correlation between a drug’s effect on autophagy and its ultimate modulation of the neuropathic status laying out a strong case for such a drug to enter clinical trials.
The genetic heterogeneity of IPNs and the fact that they are rare disorders, highlights the need for finding common cellular and molecular pathomechanisms among the different disease-linked genes. This review complements recent work in attempting to find shared molecular mechanisms underlying IPNs (Bucci et al., 2012; Prior et al., 2017). The relevance of autophagy as a target pathway stems from the fact that it has been implicated in other disease conditions, mainly neurodegeneration, where autophagy targeting drugs have already entered clinical trials. In addition, autophagic impairment presents a unique overlap among neuropathies affecting both sensory and motor nerves on one hand, and the myelin sheath and the neuronal axon on the other. While more research is required to get a clearer view of the involvement of autophagy in neuropathic mechanisms, current evidence points out to the emergence of autophagy as a frequently affected pathway in IPN conditions.
MH wrote the manuscript and designed table and figures. VT supervised and recommended the content of the manuscript, corrected, and submitted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The research related to this review was supported in part by the University of Antwerp, the Fund for Scientific Research (FWO-Flanders), the Medical Foundation Queen Elisabeth (GSKE), the “Association Belge contre les Maladies Neuromusculaires” (ABMM), and the EC 7th Framework Programme under grant agreement number 2012-305121, ‘Integrated European–omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS).’
ALS, amyotrophic lateral sclerosis; ATG, autophagy-related gene; BCL2, B-cell lymphoma2 (BCL2) apoptosis regulator; BECN1, beclin 1; BNIP3, BCL2 interacting protein 3; BSCL2, seipin lipid droplet biogenesis associated; CASA, chaperone-assisted selective autophagy; CCT5, chaperonin containing TCP1 subunit 5; CMA, chaperone mediated autophagy; CMAP, compound muscle action potential; CMT, Charcot-Marie-Tooth; DCTN1, dynactin subunit 1; dHMN, distal hereditary motor neuropathy; DNAJB2, DnaJ heat shock protein family (Hsp40) member B2; DNM2, dynamin2; DST, dystonin; eIF2α, eukaryotic initiation factor 2-alpha; ER, endoplasmic reticulum; ERES, ER exit sites; FAM134B, family with sequence similarity 134 member B; FIG4, FIG4 phosphoinositide 5-phosphatase; GABARAP, GABA type A receptor-associated protein; GDAP1, ganglioside induced differentiation associated protein 1; GEF, guanine nucleotide exchange factor; HOPS, homotypic fusion and protein sorting; HSAN, hereditary sensory and autonomic neuropathy; HSP, hereditary spastic paraplegia; HSPB8, heat shock protein B8; IPNs, inherited peripheral neuropathies; iPSCs, induced pluripotential stem cells; KIF1A, kinesin family member 1A; KO, knock-out; LC3, microtubule associated protein 1 light chain 3 (MAP1LC3); LITAF, lipopolysaccharide induced TNF factor; LRSAM1, leucine rich repeat and sterile alpha motif containing 1; MFN2, mitofusin 2; MTMR2/13, myotubularin-related protein 2/13; MTORC1, mechanistic target of rapamycin 1; NCVs, nerve conduction velocities; NDRG1, N-myc downstream regulated 1; NEFL, neurofilament light; P62/SQSTM1, sequestosome 1; PARK2, parkin 2; PERK, protein kinase R (PKR)-like endoplasmic reticulum kinase; PHF23, PHD finger protein 23; PIK3C3/VPS34, phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4/VPS15, phosphoinositide-3-kinase regulatory subunit 4; PINK1 PTEN, induced putative kinase 1; PKA, protein kinase A; PMP22, peripheral myelin protein 22; PtdIns, phosphatidyl inositol; PTPIP51, protein tyrosine phosphatase interacting protein 51; Rab11, Ras-related protein Rab-11A; RAB21, Ras-related protein Rab-7; RAB7, Ras-related protein Rab-21; RB1CC1, RB1 inducible coiled-coil 1; SEC24D, SEC24 homolog D, COPII coat complex component; SH3TC2, SH3 domain and tetratricopeptide repeats 2; SNARE, SNAP (soluble NSF attachment protein) receptor; SOD1, superoxide dismutase 1; TECPR2, tectonin beta-propeller repeat containing 2; TrkA, tyrosine kinase A; TRPV4, transient receptor potential cation channel subfamily V member 4; ULK1/2, unc-51 like autophagy activating kinase 1/2; UVRAG, UV radiation resistance associated; VAPB, VAMP associated protein B; VCP, valosin containing protein; WNK1, WNK lysine deficient protein kinase 1; WT, wild-type.
Amit, I., Yakir, L., Katz, M., Zwang, Y., Marmor, M. D., Citri, A., et al. (2004). Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 18, 1737–1752. doi: 10.1101/gad.294904
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Azzedine, H., Bolino, A., Taïeb, T., Birouk, N., Di Duca, M., Bouhouche, A., et al. (2003). Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am. J. Hum. Genet. 72, 1141–1153. doi: 10.1086/375034
Baets, J., Jonghe, P., and De Timmerman, V. (2014). Recent advances in Charcot-Marie-Tooth disease. Curr. Opin. Neurol. 27, 532–540. doi: 10.1097/WCO.0000000000000131
Baltazar, G. C., Guha, S., Lu, W., Lim, J., Boesze-Battaglia, K., Laties, A. M., et al. (2012). Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS ONE 7:e49635. doi: 10.1371/journal.pone.0049635
Barneo-Muñoz, M., Juárez, P., Civera-Tregón, A., Yndriago, L., Pla-Martín, D., Zenker, J., et al. (2015). Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of Charcot-Marie-Tooth neuropathy. PLOS Genet. 11:e1005115. doi: 10.1371/journal.pgen.1005115
Baxter, R. V., Ben Othmane, K., Rochelle, J. M., Stajich, J. E., Hulette, C., Dew-Knight, S., et al. (2002). Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 30, 21–22. doi: 10.1038/ng796
Behrends, C., Sowa, M. E., Gygi, S. P., and Harper, J. W. (2010). Network organization of the human autophagy system. Nature 466, 68–76. doi: 10.1038/nature09204
Bertolo, C., Roa, S., Sagardoy, A., Mena-Varas, M., Robles, E. F., Martinez-Ferrandis, J. I., et al. (2013). LITAF, a BCL6 target gene, regulates autophagy in mature B-cell lymphomas. Br. J. Haematol. 162, 621–630. doi: 10.1111/bjh.12440
Bolino, A., Bolis, A., Previtali, S. C., Dina, G., Bussini, S., Dati, G., et al. (2004). Disruption of Mtmr2 CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J. Cell Biol. 167, 711–721. doi: 10.1083/jcb.200407010
Bouhouche, A., Benomar, A., Bouslam, N., Chkili, T., and Yahyaoui, M. (2006). Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 43, 441–443. doi: 10.1136/jmg.2005.039230
Bucci, C., Bakke, O., and Progida, C. (2012). Charcot-Marie-Tooth disease and intracellular traffic. Prog. Neurobiol. 99, 191–225. doi: 10.1016/j.pneurobio.2012.03.003
Carra, S., Seguin, S. J., Lambert, H., and Landry, J. (2008). HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 283, 1437–1444. doi: 10.1074/jbc.M706304200
Cartwright, B. R., and Goodman, J. M. (2012). Seipin: from human disease to molecular mechanism. J. Lipid Res. 53, 1042–1055. doi: 10.1194/jlr.R023754
Castillo, K., Nassif, M., Valenzuela, V., Rojas, F., Matus, S., Mercado, G., et al. (2013a). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. doi: 10.4161/auto.25188
Castillo, K., Valenzuela, V., Matus, S., Nassif, M., Oñate, M., Fuentealba, Y., et al. (2013b). Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis. 4, e917. doi: 10.1038/cddis.2013.421
Chen, B., Longtine, M. S., Sadovsky, Y., and Nelson, D. M. (2010). Hypoxia downregulates p53 but induces apoptosis and enhances expression of BAD in cultures of human syncytiotrophoblasts. Am. J. Physiol. Cell Physiol. 299, C968–C976. doi: 10.1152/ajpcell.00154.2010
Chen, Y., and Klionsky, D. J. (2011). The regulation of autophagy - unanswered questions. J. Cell Sci. 124, 161–170. doi: 10.1242/jcs.064576
Cherra, S. J., Kulich, S. M., Uechi, G., Balasubramani, M., Mountzouris, J., Day, B. W., et al. (2010). Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 190, 533–539. doi: 10.1083/jcb.201002108
Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K., et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72. doi: 10.1038/nature05876
Crippa, V., D’Agostino, V. G., Cristofani, R., Rusmini, P., Cicardi, M. E., Messi, E., et al. (2016). Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 6:22827. doi: 10.1038/srep22827
Cuervo, A. M., Palmer, A., Rivett, A. J., and Knecht, E. (1995). Degradation of proteasomes by lysosomes in rat liver. Eur. J. Biochem. 227, 792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x
Dadakhujaev, S., Eun, J. J., Hae, S. N., Hah, Y. S., Chang, J. K., and Deok, R. K. (2009). Interplay between autophagy and apoptosis in TrkA-induced cell death. Autophagy 5, 103–105. doi: 10.1016/j.yexcr.2008.08.013
Darrow, M. C., Sergeeva, O. A., Isas, J. M., Galaz-Montoya, J. G., King, J. A., Langen, R., et al. (2015). Structural mechanisms of mutant huntingtin aggregation suppression by the synthetic chaperonin-like CCT5 complex explained by cryoelectron tomography. J. Biol. Chem. 290, 17451–17461. doi: 10.1074/jbc.M115.655373
Deng, H.-X., Klein, C. J., Yan, J., Shi, Y., Wu, Y., Fecto, F., et al. (2010). Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 42, 165–169. doi: 10.1038/ng.509
Di Paolo, G., and De Camilli, P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. doi: 10.1038/nature05185
Ding, Y., Gao, H., Zhao, L., Wang, X., and Zheng, M. (2015). Mitofusin 2-deficiency suppresses cell proliferation through disturbance of autophagy. PLoS ONE 10:e0121328. doi: 10.1371/journal.pone.0121328
Durieux, A. C., Vassilopoulos, S., Lainé, J., Fraysse, B., Briñas, L., Prudhon, B., et al. (2012). A centronuclear myopathy - dynamin 2 mutation impairs autophagy in mice. Traffic 13, 869–879. doi: 10.1111/j.1600-0854.2012.01348.x
Dyck, P. J., Litchy, W. J., Minnerath, S., Bird, T. D., Chance, P. F., Schaid, D. J., et al. (1994). Hereditary motor and sensory neuropathy with diaphragm and vocal cord paresis. Ann. Neurol. 35, 608–615. doi: 10.1002/ana.410350515
Edvardson, S., Cinnamon, Y., Jalas, C., Shaag, A., Maayan, C., Axelrod, F. B., et al. (2012). Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann. Neurol. 71, 569–572. doi: 10.1002/ana.23524
Erzberger, J. P., and Berger, J. M. (2006). Evolutionary relationships and structural mechanisms of Aaa+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114. doi: 10.1146/annurev.biophys.35.040405.101933
Fan, H., Chen, S., Sun, Y., Xu, S., and Wu, L. (2015). Seipin mutation at glycosylation sites activates autophagy in transfected cells via abnormal large lipid droplets generation. Acta Pharmacol. Sin. 36, 497–506. doi: 10.1038/aps.2014.164
Fang, B. A., Kovačević,Ž., Park, K. C., Kalinowski, D. S., Jansson, P. J., Lane, D. J. R., et al. (2014). Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim. Biophys. Acta 1845, 1–19. doi: 10.1016/j.bbcan.2013.11.002
Ferguson, C. J., Lenk, G. M., and Meisler, M. H. (2010). PtdIns(3,5)P2 and autophagy in mouse models of neurodegeneration. Autophagy 6, 170–171. doi: 10.4161/auto.6.1.10626
Ferraro, E., Giammarioli, A. M., Chiandotto, S., Spoletini, I., and Rosano, G. (2014). Exercise-induced skeletal muscle remodeling and metabolic adaptation: redox signaling and role of autophagy. Antioxid. Redox Signal. 21, 154–176. doi: 10.1089/ars.2013.5773
Ferrier, A., Boyer, J. G., and Kothary, R. (2013). Cellular and Molecular Biology of Neuronal Dystonin. Amsterdam: Elsevier. doi: 10.1016/B978-0-12-405210-9.00003-5
Ferrier, A., De Repentigny, Y., Lynch-Godrei, A., Gibeault, S., Eid, W., Kuo, D., et al. (2015). Disruption in the autophagic process underlies the sensory neuropathy in dystonia musculorum mice. Autophagy 11, 1025–1036. doi: 10.1080/15548627.2015.1052207
Fortun, J., Go, J. C., Li, J., Amici, S. A., Dunn, W. A., and Notterpek, L. (2006). Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol. Dis. 22, 153–164. doi: 10.1016/j.nbd.2005.10.010
Frake, R. A., Ricketts, T., Menzies, F. M., and Rubinsztein, D. C. (2015). Autophagy and neurodegeneration. J. Clin. Invest. 125, 65–74. doi: 10.1172/JCI73944
Franco, M. L., Melero, C., Sarasola, E., Acebo, P., Luque, A., Calatayud-Baselga, I., et al. (2016). Mutations in TrkA causing congenital insensitivity to pain with anhidrosis (CIPA) induce misfolding, aggregation, and mutation-dependent neurodegeneration by dysfunction of the autophagic flux. J. Biol. Chem. 291, 21363–21374. doi: 10.1074/jbc.M116.722587
Gallolu Kankanamalage, S., Lee, A.-Y., Wichaidit, C., Lorente-Rodriguez, A., Shah, A. M., Stippec, S., et al. (2016). Multistep regulation of autophagy by WNK1. Proc. Natl. Acad. Sci. U.S.A. 113, 201617649. doi: 10.1073/pnas.1617649113
Gess, B., Auer-Grumbach, M., Schirmacher, A., Strom, T., Zitzelsberger, M., Rudnik-Schöneborn, S., et al. (2014). Ovid: HSJ1-related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology 83, 1726–1732. doi: 10.1212/WNL.0000000000000966
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. J. (2017). The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27, 371–385. doi: 10.1016/j.cub.2016.12.038
Gonzalez, M. A., Feely, S. M., Speziani, F., Strickland, A. V., Danzi, M., Bacon, C., et al. (2014). A novel mutation in VCP causes Charcot-Marie-Tooth Type 2 disease. Brain 137, 2897–2902. doi: 10.1093/brain/awu224
González-Jamett, A. M., Momboisse, F., Haro-Acuña, V., Bevilacqua, J. A., Caviedes, P., and Cárdenas, A. M. (2013). Dynamin-2 function and dysfunction along the secretory pathway. Front. Endocrinol. 4:126. doi: 10.3389/fendo.2013.00126
Guo, J., Qiu, W., Soh, S. L. Y., Wei, S., Radda, G. K., Ong, W.-Y., et al. (2013). Motor neuron degeneration in a mouse model of seipinopathy. Cell Death Dis. 4, e535. doi: 10.1038/cddis.2013.64
Gutierrez, M. G., Munafó, D. B., Berón, W., and Colombo, M. I. (2004). Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117, 2687–2697. doi: 10.1242/jcs.01114
Hailey, D. W., Rambold, A. S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P. K., et al. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667. doi: 10.1016/j.cell.2010.04.009
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Han, B., Li, W., Sun, Y., Zhou, L., Xu, Y., and Zhao, X. (2014). A prolyl-hydroxylase inhibitor, ethyl-3,4-Dihydroxybenzoate, induces cell autophagy and apoptosis in esophageal squamous cell carcinoma cells via up-regulation of BNIP3 and N-myc downstream-regulated gene-1. PLoS ONE 9:e107204. doi: 10.1371/journal.pone.0107204
Hansen, K., Wagner, B., Hamel, W., Schweizer, M., Haag, F., Westphal, M., et al. (2007). Autophagic cell death induced by TrkA receptor activation in human glioblastoma cells. J. Neurochem. 103, 259–275. doi: 10.1111/j.1471-4159.2007.04753.x
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Harding, A. E., and Thomas, P. K. (1980a). Hereditary distal spinal muscular atrophy: a report on 34 cases and a review of the literature. J. Neurol. Sci. 45, 337–348.
Harding, A. E., and Thomas, P. K. (1980b). Genetic aspects of hereditary motor and sensory neuropathy (types I and II). J. Med. Genet. 17, 329–336. doi: 10.1136/jmg.17.5.329
He, C., Sumpter, R., and Levine, B. (2012). Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 8, 1548–1551. doi: 10.4161/auto.21327
Heimer, G., Oz-Levi, D., Eyal, E., Edvardson, S., Nissenkorn, A., Ruzzo, E. K., et al. (2016). TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur. J. Paediatr. Neurol. 20, 69–79. doi: 10.1016/j.ejpn.2015.10.003
Holmgren, A. (2012). Molecular Biology of Small Heat Shock Proteins associated with Peripheral Neuropathies. eLS.. Chichester: John Wiley Sons Ltd, 1–10. doi: 10.1002/9780470015902.a0024294
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi: 10.1091/mbc.E08
Hsiao, C. T., Tsai, P. C., Lin, C. C., Liu, Y. T., Huang, Y. H., Liao, Y. C., et al. (2016). Clinical and molecular characterization of BSCL2 mutations in a taiwanese cohort with hereditary neuropathy. PLoS ONE 11:e0147677. doi: 10.1371/journal.pone.0147677
Huber, N., Bieniossek, C., Wagner, K. M., Elsässer, H.-P., Suter, U., Berger, I., et al. (2016). Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci. Rep. 6:36930. doi: 10.1038/srep36930
Huett, A., Heath, R. J., Begun, J., Sassi, S. O., Baxt, L. A., Vyas, J. M., et al. (2012). The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella typhimurium. Cell Host Microbe 12, 778–790. doi: 10.1016/j.chom.2012.10.019
Ikenaka, K., Kawai, K., Katsuno, M., Huang, Z., Jiang, Y. M., Iguchi, Y., et al. (2013). dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS ONE 8:e54511. doi: 10.1371/journal.pone.0054511
Indo, Y., Tsuruta, M., Hayashida, Y., Karim, M., Ohta, K., Kawano, T., et al. (1996). Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat. Genet. 13, 485–488. doi: 10.1038/ng1096-146
Irobi, J., Van Impe, K., Seeman, P., Jordanova, A., Dierick, I., Verpoorten, N., et al. (2004). Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 36, 597–601. doi: 10.1038/ng1328
Ito, D., Fujisawa, T., Iida, H., and Suzuki, N. (2008). Characterization of seipin/BSCL2, a protein associated with spastic paraplegia 17. Neurobiol. Dis. 31, 266–277. doi: 10.1016/j.nbd.2008.05.004
Jäger, S., Bucci, C., Tanida, I., Ueno, T., Kominami, E., Saftig, P., et al. (2004). Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117, 4837–4848. doi: 10.1242/jcs.01370
Jang, S. Y., Shin, Y. K., Park, S. Y., Park, J. Y., Rha, S. H., Kim, J. K., et al. (2015). Autophagy is involved in the reduction of myelinating schwann cell cytoplasm during myelin maturation of the peripheral nerve. PLoS ONE 10:e0116624. doi: 10.1371/journal.pone.0116624
Jean, S., Cox, S., Nassari, S., and Kiger, A. (2015). Starvation-induced MTMR 13 and RAB 21 activity regulates VAMP 8 to promote autophagosome – lysosome fusion. EMBO Rep. 16, 297–311. doi: 10.15252/embr.201439464
Jiang, P., Nishimura, T., Sakamaki, Y., Itakura, E., Hatta, T., Natsume, T., et al. (2014). The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 25, 1327–1337. doi: 10.1091/mbc.E13-08-0447
Ju, J. S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
Kabeya, Y. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kalaydjieva, L., Gresham, D., Gooding, R., Heather, L., Baas, F., de Jonge, R., et al. (2000). N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am. J. Hum. Genet. 67, 47–58. doi: 10.1086/302978
Katona, I., Zhang, X., Bai, Y., Shy, M. E., Guo, J., Yan, Q., et al. (2011). Distinct pathogenic processes between Fig4-deficient motor and sensory neurons. Eur. J. Neurosci. 33, 1401–1410. doi: 10.1111/j.1460-9568.2011.07651.x
Khaminets, A., Heinrich, T., Mari, M., Huebner, A. K., Akutsu, M., Grumati, P., et al. (2015). Regulation of endoplasmic reticulum turnover by FAM134B-mediated selective autophagy. Nature 522, 354–358. doi: 10.1038/nature14498
Kimonis, V. E., Kovach, M. J., Waggoner, B., Leal, S., Salam, A., Rimer, L., et al. (2000). Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet. Med. 2, 232–241. doi: 10.1097/00125817-200007000-00006
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Köttgen, M., Buchholz, B., Garcia-Gonzalez, M. A., Kotsis, F., Fu, X., Doerken, M., et al. (2008). TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 182, 437–447. doi: 10.1083/jcb.200805124
Kurth, I., Pamminger, T., Hennings, J. C., Soehendra, D., Huebner, A. K., Rotthier, A., et al. (2009). Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 41, 1179–1181. doi: 10.1038/ng.464
Kwok, A. S., Phadwal, K., Turner, B. J., Oliver, P. L., Raw, A., Simon, A. K., et al. (2011). HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J. Neurochem. 119, 1155–1161. doi: 10.1111/j.1471-4159.2011.07521.x
Kyöstilä, K., Syrjä, P., Jagannathan, V., Chandrasekar, G., Jokinen, T. S., Seppälä, E. H., et al. (2015). A missense change in the ATG4D gene links aberrant autophagy to a neurodegenerative vacuolar storage disease. PLoS Genet. 11:e1005169. doi: 10.1371/journal.pgen.1005169
Lacerda, A. F. E., Hartjes, E., and Brunetti, C. R. (2014). LITAF mutations associated with Charcot-Marie-Tooth disease 1C show mislocalization from the late endosome/lysosome to the mitochondria. PLoS ONE 9:e103454. doi: 10.1371/journal.pone.0103454
Lamb, C. A., Nühlen, S., Judith, D., Frith, D., Snijders, A. P., Behrends, C., et al. (2016). TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J. 35, 281–301. doi: 10.15252/embj.201592695
Larroquette, F., Seto, L., Gaub, P. L., Kamal, B., Wallis, D., Larivière, R., et al. (2015). Vapb/Amyotrophic lateral sclerosis 8 knock-in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response. Hum. Mol. Genet. 24, 6515–6529. doi: 10.1093/hmg/ddv360
Lee, J.-Y., Koga, H., Kawaguchi, Y., Tang, W., Wong, E., Gao, Y.-S., et al. (2010). HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980. doi: 10.1038/emboj.2009.405
Lee, S. M., Olzmann, J. A., Chin, L.-S., Li, L., Babst, M., Bendtsen, J. D., et al. (2011). Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J. Cell Sci. 124, 3319–3331. doi: 10.1242/jcs.087114
Lenk, G. M., and Meisler, M. H. (2014). Mouse Models of PI(3,5)P2 Deficiency with Impaired Lysosome Function, 1st Edn. Amsterdam: Elsevier Inc. doi: 10.1016/B978-0-12-397926-1.00014-7
Lev, S., Ben Halevy, D., Peretti, D., and Dahan, N. (2008). The VAP protein family: from cellular functions to motor neuron disease. Trends Cell Biol. 18, 282–290. doi: 10.1016/j.tcb.2008.03.006
Li, J., Kim, S. G., and Blenis, J. (2014). Rapamycin: one drug, many effects. Cell Metab. 19, 373–379. doi: 10.1016/j.cmet.2014.01.001
Li, J., Parker, B., Martyn, C., Natarajan, C., and Guo, J. (2012). The PMP22 gene and its related diseases. Mol. Neurobiol. 47, 673–698. doi: 10.1007/s12035-012-8370-x
Liang, C., Lee, J., Inn, K., Gack, M. U., Li, Q., Roberts, E. A., et al. (2008). Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10, 776–787. doi: 10.1038/ncb1740
Lipinski, M. M., Zheng, B., Lu, T., Yan, Z., Py, B. F., Ng, A., et al. (2010). Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 107, 14164–14169. doi: 10.1073/pnas.1009485107
Liu, J., Xing, H., Chen, Y., Wang, L., Wang, D., Rao, Q., et al. (2012). PIG7, transactivated by AML1, promotes apoptosis and differentiation of leukemia cells with AML1-ETO fusion gene. Leukemia 26, 117–126. doi: 10.1038/leu.2011.178
Lo, K. Y., Kuzmin, A., Unger, S. M., Petersen, J. D., and Silverman, M. A. (2011). KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 491, 168–173. doi: 10.1016/j.neulet.2011.01.018
Longatti, A., Lamb, C. A., Razi, M., Yoshimura, S. I., Barr, F. A., and Tooze, S. A. (2012). TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 197, 659–675. doi: 10.1083/jcb.201111079
Lu, J. H., Tan, J. Q., Durairajan, S. S. K., Liu, L. F., Zhang, Z. H., Ma, L., et al. (2012). Isorhynchophylline, a natural alkaloid, promotes the degradation of α-synuclein in neuronal cells via inducing autophagy. Autophagy 8, 98–108. doi: 10.4161/auto.8.1.18313
Lupski, J. R., Wise, C. A., Kuwano, A., Pentao, L., Parke, J. T., Glaze, D. G., et al. (1992). Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1, 29–33. doi: 10.1038/ng0492-29
Madorsky, I., Opalach, K., Waber, A., Verrier, J. D., Solmo, C., Foster, T., et al. (2009). Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol. Dis. 34, 146–154. doi: 10.1016/j.nbd.2009.01.002
Mari, M., Griffith, J., Rieter, E., Krishnappa, L., Klionsky, D. J., and Reggiori, F. (2010). An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 190, 1005–1022. doi: 10.1083/jcb.200912089
McNeill, A., Magalhaes, J., Shen, C., Chau, K. Y., Hughes, D., Mehta, A., et al. (2014). Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495. doi: 10.1093/brain/awu020
Mersiyanova, I. V., Perepelov, A. V., Polyakov, A. V., Sitnikov, V. F., Dadali, E. L., Oparin, R. B., et al. (2000). A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 67, 37–46. doi: 10.1086/302962
Mijaljica, D., Prescott, M., and Devenish, R. J. (2011). Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7, 673–682. doi: 10.4161/auto.7.7.14733
Miller, F. D., and Kaplan, D. R. (2001). Neurotrophin signalling pathways regulating neuronal apoptosis. Cell. Mol. Life Sci. 58, 1045–1053.
Moriguchi, T., Urushiyama, S., Hisamoto, N., Iemura, S. I., Uchida, S., Natsume, T., et al. (2005). WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases. SPAK and OSR1. J. Biol. Chem. 280, 42685–42693. doi: 10.1074/jbc.M510042200
Murrow, L., Malhotra, R., and Debnath, J. (2015). ATG12–ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 17, 300–310. doi: 10.1038/ncb3112
Nalbandian, A., Llewellyn, K. J., Badadani, M., Yin, H. Z., Nguyen, C., Katheria, V., et al. (2013). A progressive translational mouse model of human valosin-containing protein disease: the VCPR155H/+ mouse. Muscle Nerve 47, 260–270. doi: 10.1002/mus.23522
Ng, A. C. Y., Eisenberg, J. M., Heath, R. J. W., Huett, A., Robinson, C. M., Nau, G. J., et al. (2011). Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4631–4638. doi: 10.1073/pnas.1000093107
Nicholson, G. A., Valentijn, L. J., Cherryson, A. K., Kennerson, M. L., Bragg, T. L., DeKroon, R. M., et al. (1994). A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat. Genet. 6, 263–266. doi: 10.1038/ng0394-263
Niemann, A., Ruegg, M., La Padula, V., Schenone, A., and Suter, U. (2005). Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J. Cell Biol. 170, 1067–1078. doi: 10.1083/jcb.200507087
Nishimura, A. L., Mitne-Neto, M., Silva, H. C. A., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Notterpek, L., Shooter, E. M., and Snipes, G. J. (1997). Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J. Neurosci. 17, 4190–4200.
Novoselov, S. S., Mustill, W. J., Gray, A. L., Dick, J. R., Kanuga, N., Kalmar, B., et al. (2013). Molecular chaperone mediated late-stage neuroprotection in the SOD1G93A mouse model of amyotrophic lateral sclerosis. PLoS ONE 8:e73944. doi: 10.1371/journal.pone.0073944
Ohta, K., Mizuno, A., Ueda, M., Li, S., Suzuki, Y., Hida, Y., et al. (2010). Autophagy impairment stimulates PS1 expression and γ-secretase activity. Autophagy 6, 345–352. doi: 10.4161/auto.6.3.11228
Oz-Levi, D., Ben-Zeev, B., Ruzzo, E. K., Hitomi, Y., Gelman, A., Pelak, K., et al. (2012). Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 91, 1065–1072. doi: 10.1016/j.ajhg.2012.09.015
Papadopoulos, C., Kirchner, P., Bug, M., Grum, D., Koerver, L., Schulze, N., et al. (2016). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150. doi: 10.15252/embj.201695148
Pareyson, D., Saveri, P., and Piscosquito, G. (2014). Charcot-Marie-Tooth disease and related hereditary neuropathies : from gene function to associated phenotypes. Curr. Mol. Med. 14, 1009–1033. doi: 10.2174/1566524014666141010154205
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 20, 460–473. doi: 10.1089/ars.2013.5371
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Xiao, H. L., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pavel, M., Imarisio, S., Menzies, F. M., Jimenez-Sanchez, M., Siddiqi, F. H., Wu, X., et al. (2016). CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun. 7:13821. doi: 10.1038/ncomms13821
Perrot, R., Berges, R., Bocquet, A., and Eyer, J. (2008). Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol. Neurobiol. 38, 27–65. doi: 10.1007/s12035-008-8033-0
Petiot, A., Ogier-denis, E., Edward, F. C., Meijer, A. J., Chem, J. B., Blommaart, E. F. C., et al. (2000). Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275, 992–998. doi: 10.1074/jbc.275.2.992
Previtali, S. C., Zerega, B., Sherman, D. L., Brophy, P. J., Dina, G., King, R. H. M., et al. (2003). Myotubularin-related 2 protein phosphatase and neurofilament light chain protein, both mutated in CMT neuropathies, interact in peripheral nerve. Hum. Mol. Genet. 12, 1713–1723. doi: 10.1093/hmg/ddg179
Prior, R., Van Helleputte, L., Benoy, V., Den Bosch, L., and Van. (2017). Defective axonal transport: a common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. doi: 10.1016/j.nbd.2017.02.009 [Epub ahead of print].
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L. F., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Rahvar, M., Nikseresht, M., Shafiee, S. M., Naghibalhossaini, F., Rasti, M., Panjehshahin, M. R., et al. (2011). Effect of oral resveratrol on the BDNF gene expression in the hippocampus of the rat brain. Neurochem. Res. 36, 761–765. doi: 10.1007/s11064-010-0396-398
Rangaraju, S., Verrier, J. D., Madorsky, I., Nicks, J., Dunn, W. A., and Notterpek, L. (2010). Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J. Neurosci. 30, 11388–11397. doi: 10.1523/JNEUROSCI.1356-10.2010
Rivière, J. B., Verlaan, D. J., Shekarabi, M., Lafrenière, R. G., Bénard, M., Der Kaloustian, V. M., et al. (2004). A mutation in the HSN2 gene causes sensory neuropathy type II in a lebanese family. Ann. Neurol. 56, 572–575. doi: 10.1002/ana.20237
Rivire, J. B., Ramalingam, S., Lavastre, V., Shekarabi, M., Holbert, S., Lafontaine, J., et al. (2011). KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 89, 219–301. doi: 10.1016/j.ajhg.2011.06.013
Rizzo, F., Ronchi, D., Salani, S., Nizzardo, M., Fortunato, F., Bordoni, A., et al. (2016). Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Hum. Mol. Genet. 25, 4266–4281. doi: 10.1093/hmg/ddw258
Roberts, R. C., Peden, A. A., Buss, F., Bright, N. A., Latouche, M., Reilly, M. M., et al. (2010). Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Hum. Mol. Genet. 19, 1009–1018. doi: 10.1093/hmg/ddp565
Roberts-Clarke, D., Fornusek, C., Saigal, N., Halaki, M., Burns, J., Nicholson, G., et al. (2016). Relationship between physical performance and quality of life in Charcot-Marie-Tooth disease: a pilot study. J. Peripher. Nerv. Syst. 21, 357–364. doi: 10.1111/jns.12191
Robinson, F. L., and Dixon, J. E. (2005). The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-Tooth disease. J. Biol. Chem. 280, 31699–31707. doi: 10.1074/jbc.M505159200
Rose, C., Menzies, F. M., Renna, M., Acevedo-Arozena, A., Corrochano, S., Sadiq, O., et al. (2010). Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 19, 2144–2153. doi: 10.1093/hmg/ddq093
Rose, J. M., Novoselov, S. S., Robinson, P. A., and Cheetham, M. E. (2011). Molecular chaperone-mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum. Mol. Genet. 20, 16–27. doi: 10.1093/hmg/ddq428
Rossor, A. M., Kalmar, B., Greensmith, L., and Reilly, M. M. (2012). The distal hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry 83, 6–14. doi: 10.1136/jnnp-2011-300952
Rotthier, A., Baets, J., Timmerman, V., and Janssens, K. (2012). Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 8, 73–85. doi: 10.1038/nrneurol.2011.227
Russo, M., Laurá, M., Polke, J. M., Davis, M. B., Blake, J., Brandner, S., et al. (2011). Variable phenotypes are associated with PMP22 missense mutations. Neuromuscul. Disord. 21, 106–114. doi: 10.1016/j.nmd.2010.11.011
Sahni, S., Bae, D. H., Lane, D. J. R., Kovacevic, Z., Kalinowski, D. S., Jansson, P. J., et al. (2014). The metastasis suppressor, N-myc Downstream-regulated gene 1 (NDRG1), inhibits stress-induced autophagy in cancer cells. J. Biol. Chem. 289, 9692–9709. doi: 10.1074/jbc.M113.529511
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K., et al. (2013). De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449. doi: 10.1038/ng.2562
Sanchez, E., Darvish, H., Mesias, R., Taghavi, S., Firouzabadi, S. G., Walker, R. H., et al. (2016). Identification of a large DNAJB2 deletion in a family with spinal muscular atrophy and parkinsonism. Hum. Mutat. 37, 1180–1189. doi: 10.1002/humu.23055
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114, 867–874.
Sarkar, S., Davies, J. E., Huang, Z., Tunnacliffe, A., and Rubinsztein, D. C. (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem. 282, 5641–5652. doi: 10.1074/jbc.M609532200
Sasaki, T., Gotow, T., Shiozaki, M., Sakaue, F., Saito, T., Julien, J. P., et al. (2006). Aggregate formation and phosphorylation of neurofilament-L Pro22 Charcot-Marie-Tooth disease mutants. Hum. Mol. Genet. 15, 943–952. doi: 10.1093/hmg/ddl011
Schrepfer, E., and Scorrano, L. (2016). Mitofusins, from mitochondria to metabolism. Mol. Cell 61, 683–694. doi: 10.1016/j.molcel.2016.02.022
Schroer, T. A. (2004). Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779. doi: 10.1146/annurev.cellbio.20.012103.094623
Schulze, R. J., and McNiven, M. A. (2014). A well-oiled machine DNM2/dynamin 2 helps keep hepatocyte lipophagy running smoothly. Autophagy 10, 388–389. doi: 10.4161/auto.27486
Sebastián, D., Sorianello, E., Segalés, J., Irazoki, A., Ruiz-Bonilla, V., Sala, D., et al. (2016). Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 35, 1677–1693. doi: 10.15252/embj.201593084
Senderek, J., Bergmann, C., Ramaekers, V. T., Nelis, E., Bernert, G., Makowski, A., et al. (2003a). Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain 126, 642–649. doi: 10.1093/brain/awg068
Senderek, J., Bergmann, C., Stendel, C., Kirfel, J., Verpoorten, N., De Jonghe, P., et al. (2003b). Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am. J. Hum. Genet. 73, 1106–1119. doi: 10.1086/379525
Shaw, C. E. (2010). Capturing VCP: another molecular piece in the ALS jigsaw puzzle. Neuron 68, 812–814. doi: 10.1016/j.neuron.2010.11.040
Shintani, T., Mizushima, N., Ogawa, Y., Matsuura, A., Noda, T., and Ohsumi, Y. (1999). Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 18, 5234–5241. doi: 10.1093/emboj/18.19.5234
Sivera, R., Espinõs, C., Vílchez, J. J., Mas, F., Martínez-Rubio, D., Chumillas, M. J., et al. (2010). Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 15, 334–344. doi: 10.1111/j.1529-8027.2010.00286.x
Spencer, B., Potkar, R., Trejo, M., Rockenstein, E., Patrick, C., Gindi, R., et al. (2009). Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29, 13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009
Stadel, D., Millarte, V., Tillmann, K. D., Huber, J., Tamin-Yecheskel, B. C., Akutsu, M., et al. (2015). TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol. Cell 60, 89–104. doi: 10.1016/j.molcel.2015.09.010
Stavoe, A. K. H., Hill, S. E., Hall, D. H., and Colón-Ramos, D. A. (2016). KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev. Cell 38, 171–185. doi: 10.1016/j.devcel.2016.06.012
Stendel, C., Roos, A., Kleine, H., Arnaud, E., Ozcelik, M., Sidiropoulos, P. N. M., et al. (2010). SH3TC2, a protein mutant in Charcot-Marie-Tooth neuropathy, links peripheral nerve myelination to endosomal recycling. Brain 133,2462–2474. doi: 10.1093/brain/awq168
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525. doi: 10.1038/nrm2728
Stephan, J. S., Yeh, Y.-Y., Ramachandran, V., Deminoff, S. J., and Herman, P. K. (2009). The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc. Natl. Acad. Sci. U.S.A. 106, 17049–17054. doi: 10.1073/pnas.0903316106
Stojkovic, T. (2016). Hereditary neuropathies: an update. Rev. Neurol. 172, 775–778. doi: 10.1016/j.neurol.2016.06.007
Street, V. A., Bennett, C. L., Goldy, J. D., Shirk, A. J., Kleopa, K. A., Tempel, B. L., et al. (2003). Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology 60, 22–26. doi: 10.1212/WNL.60.1.22
Takahashi, Y., Tsotakos, N., Liu, Y., Young, M. M., Serfass, J., Tang, Z., et al. (2016). The Bif-1-Dynamin 2 membrane fission machinery regulates Atg9-containing vesicle generation at the Rab11-positive reservoirs. Oncotarget 7, 20855–20868. doi: 10.18632/oncotarget.8028
Tang, B. S., Zhao, G. H., Luo, W., Xia, K., Cai, F., Pan, Q., et al. (2005). Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 116, 222–224.
Tang, X., Metzger, D., Leeman, S., and Amar, S. (2006). LPS-induced TNF-α factor (LITAF)-deficient mice express reduced LPS-induced cytokine: evidence for LITAF-dependent LPS signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 103, 13777–13782. doi: 10.1073/pnas.0605988103
Tanida, I., Mizushima, N., Kiyooka, M., Ohsumi, M., Ueno, T., Ohsumi, Y., et al. (1999). Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell 10, 1367–1379. doi: 10.1091/mbc.10.5.1367
Thomas, P. D. P. K. (2005). Peripheral Neuropathy. Available at: https://www.elsevier.com/books/peripheral-neuropathy/dyck/978-0-7216-9491-7
Timmerman, V., Nelis, E., Van Hul, W., Nieuwenhuijsen, B. W., Chen, K. L., Wang, S., et al. (1992). The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat. Genet. 1, 171–175. doi: 10.1038/ng0692-171
Timmerman, V., Strickland, A. V., and Züchner, S. (2014). Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes 5, 13–32. doi: 10.3390/genes5010013
Towers, C. G., and Thorburn, A. (2016). Therapeutic targeting of autophagy. EBioMedicine 14, 15–23. doi: 10.1016/j.ebiom.2016.10.034
Vaccari, I., Carbone, A., Previtali, S. C., Mironova, Y. A., Alberizzi, V., Noseda, R., et al. (2015). Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum. Mol. Genet. 24, 383–396. doi: 10.1093/hmg/ddu451
Vallat, J. M., Goizet, C., Tazir, M., Couratier, P., Magy, L., and Mathis, S. (2016). Classifications of neurogenetic diseases: an increasingly complex problem. Rev. Neurol. 172, 339–349. doi: 10.1016/j.neurol.2016.04.005
Verhoeven, K., De Jonghe, P., Coen, K., Verpoorten, N., Auer-Grumbach, M., Kwon, J. M., et al. (2003). Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 72, 722–727. doi: 10.1086/367847
Vingtdeux, V., Giliberto, L., Zhao, H., Chandakkar, P., Wu, Q., Simon, J. E., et al. (2010). AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J. Biol. Chem. 285, 9100–9113. doi: 10.1074/jbc.M109.060061
Vita, G., La Foresta, S., Russo, M., Vita, G. L., Messina, S., Lunetta, C., et al. (2016). Sport activity in Charcot-Marie-Tooth disease: a case study of a Paralympic swimmer. Neuromuscul. Disord. 26, 614–618. doi: 10.1016/j.nmd.2016.06.002
Wang, Z., Hu, J., Li, G., Qu, L., He, Q., Lou, Y., et al. (2014). PHF23 (plant homeodomain finger protein 23) negatively regulates cell autophagy by promoting ubiquitination and degradation of E3 ligase LRSAM1. Autophagy 10, 2158–2170. doi: 10.4161/auto.36439
Weis, J., Claeys, K. G., Roos, A., Azzedine, H., Katona, I., Schroder, J. M., et al. (2016). Towards a functional pathology of hereditary neuropathies. Acta Neuropathol. 133, 493–515. doi: 10.1007/s00401-016-1645-y
Westhoff, B., Chapple, J. P., Van Der Spuy, J., Hohfeld, J., and Cheetham, M. E. (2005). HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 15, 1058–1064. doi: 10.1016/j.cub.2005.04.058
Weterman, M. A. J., Sorrentino, V., Kasher, P. R., Jakobs, M. E., van Engelen, B. G. M., Fluiter, K., et al. (2012). A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum. Mol. Genet. 21, 358–370. doi: 10.1093/hmg/ddr471
Wiesner, D., Sinniger, J., Henriques, A., Dieterlé, S., Müller, H. P., Rasche, V., et al. (2015). Low dietary protein content alleviates motor symptoms in mice with mutant dynactin/dynein-mediated neurodegeneration. Hum. Mol. Genet. 24, 2228–2240. doi: 10.1093/hmg/ddu741
Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Hörl, G., et al. (2004). Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 36, 271–276. doi: 10.1038/ng1313
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Zhan, L., Yang, Y., Ma, T. T., Huang, C., Meng, X. M., Zhang, L., et al. (2015). Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol. Cell. Biochem. 402, 9–22. doi: 10.1007/s11010-014-2298-6
Zhou, J., Yang, Z., Tsuji, T., Gong, J., Xie, J., Chen, C., et al. (2011). LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene 30, 1892–1900. doi: 10.1038/onc.2010.575
Züchner, S., Mersiyanova, I. V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E. L., et al. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451. doi: 10.1038/ng1341
Keywords: autophagy, hereditary neuropathies, Charcot-Marie-Tooth, neurodegeneration, proteostasis
Citation: Haidar M and Timmerman V (2017) Autophagy as an Emerging Common Pathomechanism in Inherited Peripheral Neuropathies. Front. Mol. Neurosci. 10:143. doi: 10.3389/fnmol.2017.00143
Received: 29 March 2017; Accepted: 26 April 2017;
Published: 11 May 2017.
Edited by:
Angelo Poletti, Università degli Studi di Milano, ItalyReviewed by:
Maurizio D’Antonio, San Raffaele Scientific Institute, ItalyCopyright © 2017 Haidar and Timmerman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent Timmerman, dmluY2VudC50aW1tZXJtYW5AdWFudHdlcnBlbi5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.