 Serena Barral
Serena Barral Manju A. Kurian
Manju A. Kurian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 06 September 2016
Sec. Methods and Model Organisms
Volume 9 - 2016 | https://doi.org/10.3389/fnmol.2016.00078
This article is part of the Research Topic Molecular, cellular and model organism approaches for understanding the basis of neurological disease View all 14 articles
The study of neurological disorders often presents with significant challenges due to the inaccessibility of human neuronal cells for further investigation. Advances in cellular reprogramming techniques, have however provided a new source of human cells for laboratory-based research. Patient-derived induced pluripotent stem cells (iPSCs) can now be robustly differentiated into specific neural subtypes, including dopaminergic, inhibitory GABAergic, motorneurons and cortical neurons. These neurons can then be utilized for in vitro studies to elucidate molecular causes underpinning neurological disease. Although human iPSC-derived neuronal models are increasingly regarded as a useful tool in cell biology, there are a number of limitations, including the relatively early, fetal stage of differentiated cells and the mainly two dimensional, simple nature of the in vitro system. Furthermore, clonal variation is a well-described phenomenon in iPSC lines. In order to account for this, robust baseline data from multiple control lines is necessary to determine whether a particular gene defect leads to a specific cellular phenotype. Over the last few years patient-derived neural cells have proven very useful in addressing several mechanistic questions related to central nervous system diseases, including early-onset neurological disorders of childhood. Many studies report the clinical utility of human-derived neural cells for testing known drugs with repurposing potential, novel compounds and gene therapies, which then can be translated to clinical reality. iPSCs derived neural cells, therefore provide great promise and potential to gain insight into, and treat early-onset neurological disorders.
Over the last decade, significant advances such as whole exome and genome sequencing have facilitated genetic screening of patients, resulting in an ever-increasing number of inherited human diseases. Despite this genetic revolution, the molecular mechanisms downstream of a specific gene mutation or genetic variant remain yet to be fully elucidated for the majority of diseases. Future research priorities must therefore lie in studying such disorders in more depth, to not only understand the disease, but also to develop novel therapies for clinical translation.
To date, transgenic animal models and transformed cell lines, have allowed clarification of pathophysiological pathways affected by genetic mutations. Despite their benefits, both methods have a number of limitations, in that they often do not fully mimic human physiology, only partially recapitulate progression of disease, and do not accurately recapitulate human metabolism and homeostasis. It has long been recognized that patient derived cells are a potentially better in vitro tool for studying human disease. However, human tissue is often either unavailable or simply not accessible. This is clearly exemplified by neurological disorders, where accessing the brain and neuronal tissue for cell culture and future study is near impossible. Since the first human embryonic stem cells (ESCs) were isolated in 1998 (Thomson et al., 1998), the use of pluripotent stem cells (PSCs) has become a new reality in the study of human diseases, offering a challenging but incredibly useful model to move from clinic to bench and potentially vice versa.
The discovery of cellular reprogramming techniques has been a major step forward in the in vitro modeling of human disease, theoretically allowing the study of all genetic disease with specific patient cells as the starting point. Yamanaka and colleagues (Takahashi et al., 2007) elegantly reprogrammed adult dermal fibroblasts to a pluripotent state, by inducing ectopic expression of four factors: Oct4, Sox2, Klf4, and cMyc (Takahashi et al., 2007; Takahashi and Yamanaka, 2016). The induced PSCs generated are highly similar to human ESCs, with the ability to indefinitely proliferate and differentiate in cells derived from the three germ layers. Since publication of Yamanaka’s landmark article, reprogramming techniques have been further refined, and many new strategies have been developed to effectively reprogram somatic cells into pluripotency. Integrating retroviruses and lentiviruses have been superseded by the use of non-integrating systems, including adenovirus, Sendai virus, mRNA, episomal vectors, proteins and small molecules (Fusaki et al., 2009; Kim et al., 2009; Zhou and Freed, 2009; Warren et al., 2010; Okita et al., 2011; Bar-Nur et al., 2014).
Neural stem cells (NSCs) have been successfully derived from PSCs and several protocols for PSCs differentiation into a broad variety of mature neurons and glial cell subtypes have been published (Srikanth and Young-Pearse, 2014). Patient-derived neural cells have the specific advantage of retaining the genetic background of the donor and thus offer a unique in vitro neuronal disease model. They are an unlimited source of cells that allows the analysis of the cellular mechanisms involved in disease. Furthermore, they provide a novel platform to test new drugs and genetic therapies as well as a source of cells that could potentially be used for cell replacement therapy (Figure 1).
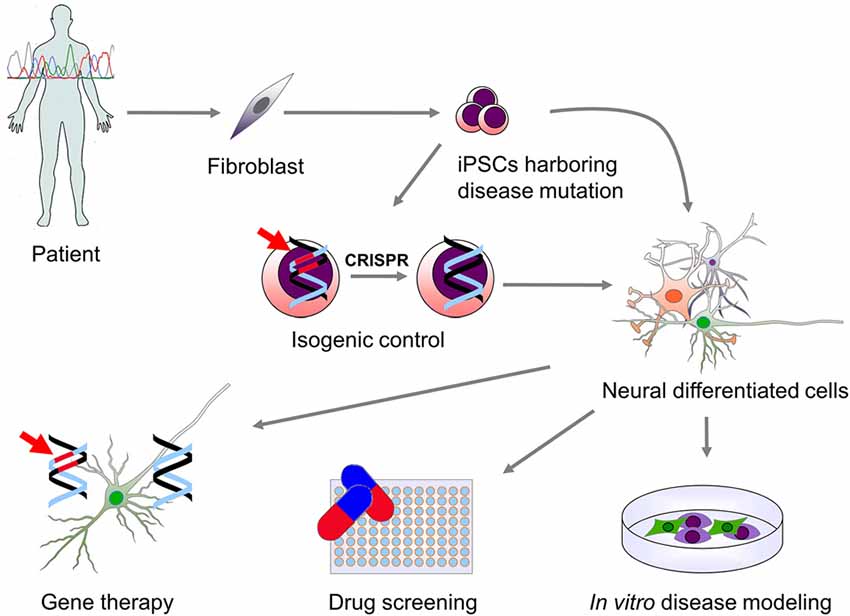
Figure 1. Use of patient-derived induced pluripotent stem cells (iPSCs) for modeling genetic neurological diseases. Genetic screening of patients affected by a neurological disorder may lead to the identification of specific mutations causing disease. Patient-derived somatic cells (fibroblast and other cell types) can be then reprogrammed to a pluripotent state. iPSCs carrying the disease-related mutation (indicated in red) can be then differentiated into the neural cells type (neurons and glial cells) which are affected in the disease. This allows for the in vitro study of the molecular mechanisms downstream the genetic mutation. In order to overcome genetic background variability and to validate the effect of the genetic mutation on phenotype observed in vitro, isogenic control iPSCs can be generated via genomic correction of the mutation. Moreover, in vitro differentiated cells can be used for high-throughput screening of drugs or the validation of specific genetic therapies that can then be translated into clinical practice.
When approaching the study of neurological diseases using human induced pluripotent stem cell (iPSC)-derived neurons as an in vitro model system, several considerations need to be taken into account, including the accurate generation of truly pluripotent cells, the relative efficiency of neuronal differentiation, and the strengths, utility and limitations of generated neurons. In this review, we provide a brief overview of what we consider to be the most important advantages and disadvantages of using human iPSCs to model neurological diseases, and their translational utility at a clinical level. Our main focus will be to evaluate this model system for early-onset genetic neurological disorders (Table 1), although, where relevant and appropriate, we will use examples from other later-onset neurological diseases.

Table 1. Utility of induced pluripotent stem cells (iPSC) in childhood-onset neurodevelopmental and neurological disorders.
Differentiation of iPSCs into neural cells is based on recapitulating embryonic development and relies on the use of specific factors that can promote or inhibit specific signaling pathways. All methods published so far can guarantee high purity of NSCs, but it is more challenging to obtain decent percentages of specific subtypes of desired mature neural cells. To date, it is possible to derive a wide variety of neuronal cell types from PSCs, including forebrain neuronal neurons (Espuny-Camacho et al., 2013; Lancaster et al., 2013); motor neurons (Wada et al., 2009; Nizzardo et al., 2010); dopaminergic neurons (Kriks et al., 2011; Kirkeby et al., 2012); GABAergic neurons (Maroof et al., 2013; Nicholas et al., 2013); medium spiny neurons (Delli Carri et al., 2013); forebrain cholinergic neurons (Wicklund et al., 2010; Hu et al., 2016); serotonergic neurons (Erceg et al., 2008); caudal neurons (Kirkeby et al., 2012); cerebellar neurons (Erceg et al., 2010); astrocytes (Emdad et al., 2012; Juopperi et al., 2012); oligodendrocytes (Nistor et al., 2005; Ogawa et al., 2011). Neuronal populations generated are typically heterogeneous, presenting both mature and immature cells, and thus need further technologies to achieve a high level of purity. Sorting techniques are often useful, but there are few neuronal subtype-specific surface markers available to select desired neural subpopulations (Pruszak et al., 2009; Yuan et al., 2011; Doi et al., 2014). To overcome this issue, sorting can sometimes be achieved by the expression of a selectable marker included as a reporter under expression of specific transcription factors or proteins (DeRosa et al., 2015; Toli et al., 2015).
During reprogramming into iPSCs, somatic cells return to a developmental stage similar to that of ESCs, independent of their original age. Indeed, age-related characteristics of the original cells, (such as nuclear abnormalities, telomere length, and mitochondrial activity) are lost during this re-set to an embryonic stage (Marion et al., 2009; Suhr et al., 2010). Differentiation protocols for the generation of neuronal subtypes from PSCs require a significant amount of time spanning from weeks to months (Srikanth and Young-Pearse, 2014) to produce neurons that show a relatively mature morphological, molecular and electrophysiological phenotype. Despite this maturation process, generated neurons are still reminiscent of human fetal neurons (Mariani et al., 2012; Lancaster et al., 2013; Miller et al., 2013; Vera and Studer, 2015). It is therefore conceivable that such in vitro model systems could fail to recapitulate the disease phenotype especially for late-onset disorders.
Studer and colleagues (Miller et al., 2013) have addressed this issue, by developing a genetic strategy for introducing aging-related features in iPSC-derived neurons, specifically studying Parkinson disease (PD). Specifically, PD patient iPSC-derived midbrain dopaminergic neurons (mDA) recapitulate some PD disease features, including α-synuclein (αSYN) accumulation, oxidative stress, defects in neural outgrowth and mitochondrial dysfunction (Byers et al., 2011; Jiang et al., 2012; Reinhardt et al., 2013). However key late disease features of PD, such as neural degeneration, were only evident in model systems exposed to external stressors (Byers et al., 2011; Nguyen et al., 2011). Importantly, the accumulation of Lewy bodies and appearance of neuromelanin, a distinct feature of adult mDA neurons, have not been observed in iPSC-derived neurons. In this case, Studer and colleagues transiently overexpressed progerin, and showed restoration of aging features in both fibroblasts and mDA neurons derived from iPSCs. In particular, in iPSC-derived mDA neurons, they observed features of normal neural aging, with degeneration of dendrites in vitro and neuromelanin accumulation after grafting in vivo (Miller et al., 2013).
The relatively immature characteristics of iPSC-derived neurons should therefore always be a major consideration when modeling postnatal-, childhood- and adult-onset neurological diseases, where pathological features only manifest during postnatal development or the aging process.
Using patient derived iPSCs guarantees a unique opportunity to study the phenotype associated with a specific mutation in the context of the genetic background in which the mutation leads to disease. However, genetic background and potential genetic modulators of disease could conceivably affect the phenotype of both healthy control and patient lines. This is a definite limiting factor in the study of both Mendelian and more complex multigenic/multifactorial disorders. One solution is to use iPSCs derived from un-affected relatives for comparison, or to compare several control and several different patient lines in the same study. However, both these methods can be costly and time consuming. In more recent times, gene-editing technologies have become a more robust method by which the effect of a specific genotype on the iPSC model system can be unequivocally validated. Indeed, for monogenic disorders, correction of the single mutation in an iPSC cell line allows development of a unique “isogenic” control which harbors the same genetic background of the patient, thereby decreasing any “background noise” that could mask or affect cell phenotype. Furthermore, the insertion of a specific mutation into a control line can be utilized to show that such mutagenesis can induce disease phenotypes into a control line, akin to those seen in patient lines. Such concepts have been elegantly illustrated by Reinhardt et al. (2013) in their study of LRRK2-related PD, where they generated and compared several control lines, from healthy age- and gender-matched individuals, vs. isogenic controls using gene editing tools. No significant difference in αSYN levels was observed when comparing mDA neurons from wild type to LRRK2-mutated patient derived iPSC lines. However, αSYN levels were markedly reduced in patient lines when compared to the corresponding corrected isogenic lines. Gene expression profiles after 30 days of differentiation revealed significant differences in the age- and gender-matched iPSC lines when compared to both patient and isogenic lines. It is therefore clear that genetic background can have effects on gene expression, and the comparison of patient lines with isogenic controls can help overcome such genetic variability.
A number of different techniques can be used to generate isogenic controls. Reinhardt et al. (2013) used Zinc Finger Nuclease (ZFNs) technology. Both ZFN and the similar Transcription Activator-Like Effector Nuclease (TALENs) have been used successfully for gene editing in iPSCs (Hockemeyer et al., 2009, 2011). Both methods rely on the generation of costumed DNA binding domains conjugated to Fok1 nuclease, which can induce double strand breaks in a non-specific manner. The landmark discovery of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9 system has added a highly efficient tool to manipulate the iPSCs genome (Ding et al., 2013) using endonuclease Cas9 and a guide RNA, targeting a specific region. Due to the efficient, relatively fast and targeted approach of the CRISPR-Cas9 technique compared to ZFNs and TALENs, this newer technique appears to be the preferred genome editing methodology for many researchers. Furthermore, the different commercial companies now offer generation of custom isogenic control lines using CRISPR-Cas9 technology (Baker, 2014).
Although iPSCs represent an advantageous tool to study molecular phenotypes in neurological disease, their two dimensional nature means that some of the environmental factors, regional identities and complex neural circuits are absent when compared to either an animal disease model or human patient. Different studies have shown the intrinsic ability of NSCs to spontaneously self-organize in 3-dimensional (3D) structures resembling whole organs (Lancaster and Knoblich, 2014). Lancaster et al. (2013) showed how simple cultivation of control iPSCs in suspension can give rise to organoids which display several brain regions along the rostro-caudal and dorso-ventral pathways of mainly the forebrain and mid-hindbrain areas. The degree of cellular organization was maximal in the dorsal cortical region of the generated organoid where they observed a layered organization typical of the developing human forebrain. The same group then generated organoids from iPSCs derived from patients with microcephaly. The patient-derived cortical organoids mimicked a number of the features seen in disease, including smaller neural tissues with few progenitors regions, and radial glial maturation and orientation abnormalities, which were not previously recapitulated in a murine model. Organoids therefore represent a powerful tool to study both human fetal neocortical development (Camp et al., 2015) and developmental disorders. The generation of organoids representing more caudal fetal regions, (such as the midbrain and hindbrain) is also no doubt useful for studying neurological disorders affecting these areas of the central nervous system. Indeed, midbrain-like organoids (MLOs) have recently been derived from human ESCs (Jo et al., 2016). Neural cells of such MLOs not only expressed midbrain markers and displayed characteristic dopaminergic electrical activity, but also produced neuromelanin, a characteristic not observed in bi-dimensional systems.
The development of new drugs to treat human disorders is a challenging field. Indeed, many drugs tested in animal models have failed in human clinical trials due to lack of efficacy or intolerability (Scannell et al., 2012). Overall, new drugs for pharmacoresistant disorders are an unmet need, and constitute a priority area for research. The development of new drugs is hindered by the lack of appropriate models. Human iPSCs offer a unique opportunity for high-throughput drug screening in patient derived cells to assess drug efficacy and toxicity. Despite being in the early stages, several studies have demonstrated the feasibility and usefulness of this approach for childhood neurological disorders. For example, motor neurons derived from patients affected by Spinal Muscular Atrophy (SMA) have been used to test specific drugs (Xu et al., 2016). SMA is characterized by mutations in SMN1, which leads to degeneration of spinal motor neurons associated with mitochondrial dysfunction. Treatment of SMA human iPSCs derived motor neurons with N-acetylcysteine (NAC) improved mitochondrial functionality, thereby rescuing motor neuron degeneration in vitro.
In Rett syndrome (RTT) patient-derived iPSCs have been utilized to test the effect of IGF1 and gentamicin in vitro (Marchetto et al., 2010). RTT iPSCs derived glutamatergic neurons showed a decreased level of glutamatergic synapses when compared to controls, which was increased by IGF1 treatment. Gentamicin, administrated at high dose acted as a suppressor of nonsense mutations which cause impaired function of MeCP2 in RTT. Furthermore, IGF1 has been used to rescue the phenotype observed in neurons differentiated from Phelan–McDermid syndrome (PMDS)-derived iPSCs (Shcheglovitov et al., 2013). PMDS neurons showed impairment in excitatory synaptic transmission and reduced number of excitatory synapses, which were restored after treatment with IGF1.
These and other studies (Table 1), have mainly tested small number of compounds on iPSC differentiated cells, thereby demonstrating the feasibility of using iPSCs for drug testing. In the future, high-throughput technologies allowing the screening of an extensive library of compounds will be useful. High-content imaging and analysis are likely to be helpful for such high-throughput approaches (Sirenko et al., 2014). High-content high-throughput assays have already been undertaken on iPSC-derived neural cells in 348-well assays, with analysis focusing on neurite outgrowth, cell number and viability, mitochondrial integrity and membrane potential.
Two studies have used high-throughput assays to screen large numbers of candidate drugs for Fragile X syndrome, a neurodevelopmental disorder characterized by learning problems, autism, and anxiety. This syndrome is associated with CGG repeat expansion in the 5′-untranslated region of FMR1, which leads to the absence of FMR protein. Both studies used patient derived iPSCs and differentiated them either in NSCs or neural precursors. In the first study 5000 compounds (both novel and approved drugs) were tested (Kumari et al., 2015), while the second study expanded drug screening to over 50,000 compounds (Kaufmann et al., 2015). In both studies FMR1-neural cells, treated with compounds as decanehydroxamate, deserpidine or tibrofan, showed increased mRNA levels, though not to clinically significant levels. Nevertheless, both studies show how promising high-throughput techniques can be in expanding the potential of drug testing using iPSCs derived cells.
In addition to the screening of new drugs, human iPSCs also offer other therapeutic possibilities that can be translated into clinical practice, as evident in the Phase I SMA study (Chiriboga et al., 2016). The antisense oligonucleotide nusinersen was designed to alter splicing of SMN2 mRNA. SMN1 motor neurons compensate with the paralogous gene SMN2. SMN2 shows a high sequence homology to SMN1, the only difference resides in the C-to-T base change inside exon 7. This mutation leads to abnormal SMN2 splicing and to the generation of truncated highly unstable proteins that trigger neural degeneration of motor neurons. SMN2 splicing correction with the use of oligonucleotides resulted in the production of a greater amount of full-length SMN2. This strategy has been tested on patient-derived iPSCs differentiated motor neurons (Corti et al., 2012) and showed the ability to convert SMA-differentiated motor neurons to a normal phenotype, both in vitro and after grafting into a mouse model of disease.
iPSC-derived neural cells can also be a patient-derived platform to test genetic therapies. Juvenile neuronal ceroid lipofuscinosis (NCL) disorder is caused by loss-of-function mutations in CLN3. Patient-derived iPSCs neurons showed abnormal lysosomal storage with abnormalities observed in mitochondria, Golgi apparatus and endoplasmic reticulum. After restoring the function of CLN3 via AAVrh.10 virus bearing wild-type human CLN3, in vitro differentiated neurons showed a rescued phenotype, without excess accumulation of storage material (Lojewski et al., 2014). Similarly, a lentiviral approach was used for a genetic early onset form of PD. iPSC-derived mDA harboring mutations in PINK1, a gene encoding a mitochondrial kinase, showed dysfunctional mitochondrial function. Expression of the non-mutated PINK1 via lentivirus in patient-derived mDA neurons restored normal recruitment of PINK1 upon mitochondrial depolarization, and normalized mitochondrial number and biogenesis (Seibler et al., 2011).
The combination of genetic engineering techniques and the promise of human PSCs differentiated cells as a donor source for cell replacement therapies, could in the future lead to the generation of patient-derived “corrected” cells that could potentially be used in autologous transplantation to replace affected disease cells. Such use of patient derived, genetically corrected neural cells may also potentially overcome immune-mediated responses that might be triggered by using allogenic neural cells. Overall, even though such approaches are extremely promising and although PSCs are already used in clinical trials (Kimbrel and Lanza, 2015), there remain many issues regarding the use of PSC as cell replacement therapy, particularly concerning cell identity, purity, safety and long term risks. For this reason, even though it remains an extremely promising approach, clinical translation of such a therapeutic approach is likely to take some time.
Patient derived iPSCs represent a unique and increasingly utilized tool for the study of human genetic neurological diseases of childhood. iPSCs are an extraordinary model that can facilitate new insight into the molecular basis of disease and aid the development of new therapies, especially for pharmacoresistant diseases where human tissue is inaccessible for research purposes. Like all other laboratory models, human iPSCs have some limitations, namely that the model can be time consuming and costly to establish, shows clonal variability and genetic background can influence phenotype. Despite this, the ever-growing number of studies using human iPSCs to both model genetic disease and discover new therapies, render them an extremely promising tool, capturing the attention of researchers worldwide.
SB: conceptional design and writing of the manuscript. MAK: conceptional design and writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with the authors SB, MAK and states that the process nevertheless met the standards of a fair and objective review.
SB is funded by the Wellcome Trust (158065). MAK is funded by a Wellcome Trust Intermediate Clinical Fellowship and receives funding from GOSHCC, the Gracious Heart Charity Foundation, Rosetrees Trust, the AADC Research Trust, and supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
Amenduni, M., De Filippis, R., Cheung, A. Y., Disciglio, V., Epistolato, M. C., Ariani, F., et al. (2011). iPS cells to model CDKL5-related disorders. Eur. J. Hum. Genet. 19, 1246–1255. doi: 10.1038/ejhg.2011.131
An, M. C., Zhang, N., Scott, G., Montoro, D., Wittkop, T., Mooney, S., et al. (2012). Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11, 253–263. doi: 10.1016/j.stem.2012.04.026
Ananiev, G., Williams, E. C., Li, H., and Chang, Q. (2011). Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One 6:e25255. doi: 10.1371/journal.pone.0025255
Andoh-Noda, T., Akamatsu, W., Miyake, K., Matsumoto, T., Yamaguchi, R., Sanosaka, T., et al. (2015). Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain 8:31. doi: 10.1186/s13041-015-0121-2
Awad, O., Sarkar, C., Panicker, L. M., Miller, D., Zeng, X., Sgambato, J. A., et al. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 24, 5775–5788. doi: 10.1093/hmg/ddv297
Baarine, M., Khan, M., Singh, A., and Singh, I. (2015). Functional characterization of IPSC-derived brain cells as a model for X-linked Adrenoleukodystrophy. PLoS One 10:e0143238. doi: 10.1371/journal.pone.0143238
Bar-Nur, O., Brumbaugh, J., Verheul, C., Apostolou, E., Pruteanu-Malinici, I., Walsh, R. M., et al. (2014). Small molecules facilitate rapid and synchronous iPSC generation. Nat. Methods 11, 1170–1176. doi: 10.1038/nmeth.3142
Bird, M. J., Needham, K., Frazier, A. E., van Rooijen, J., Leung, J., Hough, S., et al. (2014). Functional characterization of Friedreich ataxia iPS-derived neuronal progenitors and their integration in the adult brain. PLoS One 9:e101718. doi: 10.1371/journal.pone.0101718
Boza-Morán, M. G., Martínez-Hernández, R., Bernal, S., Wanisch, K., Also-Rallo, E., Le Heron, A., et al. (2015). Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC-derived human motor neurons. Sci. Rep. 5:11696. doi: 10.1038/srep11696
Byers, B., Cord, B., Nguyen, H. N., Schüle, B., Fenno, L., Lee, P. C., et al. (2011). SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS One 6:e26159. doi: 10.1371/journal.pone.0026159
Camnasio, S., Delli Carri, A., Lombardo, A., Grad, I., Mariotti, C., Castucci, A., et al. (2012). The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 46, 41–51. doi: 10.1016/j.nbd.2011.12.042
Camp, J. G., Badsha, F., Florio, M., Kanton, S., Gerber, T., Wilsch-Bräuninger, M., et al. (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. U S A 112, 15672–15677. doi: 10.1073/pnas.1520760112
Carlessi, L., Fusar Poli, E., Bechi, G., Mantegazza, M., Pascucci, B., Narciso, L., et al. (2014). Functional and molecular defects of hiPSC-derived neurons from patients with ATM deficiency. Cell Death Dis. 5:e1342. doi: 10.1038/cddis.2014.310
Chae, J. I., Kim, D. W., Lee, N., Jeon, Y. J., Jeon, I., Kwon, J., et al. (2012). Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem. J. 446, 359–371. doi: 10.1042/bj20111495
Chamberlain, S. J., Chen, P. F., Ng, K. Y., Bourgois-Rocha, F., Lemtiri-Chlieh, F., Levine, E. S., et al. (2010). Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. U S A 107, 17668–17673. doi: 10.1073/pnas.1004487107
Chang, T., Zheng, W., Tsark, W., Bates, S., Huang, H., Lin, R. J., et al. (2011). Brief report: phenotypic rescue of induced pluripotent stem cell-derived motoneurons of a spinal muscular atrophy patient. Stem Cells 29, 2090–2093. doi: 10.1002/stem.749
Cheng, P. H., Li, C. L., Chang, Y. F., Tsai, S. J., Lai, Y. Y., Chan, A. W., et al. (2013). miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse and induced pluripotent stem cell models. Am. J. Hum. Genet. 93, 306–312. doi: 10.1016/j.ajhg.2013.05.025
Cheung, A. Y., Horvath, L. M., Grafodatskaya, D., Pasceri, P., Weksberg, R., Hotta, A., et al. (2011). Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 20, 2103–2115. doi: 10.1093/hmg/ddr093
Chiriboga, C. A., Swoboda, K. J., Darras, B. T., Iannaccone, S. T., Montes, J., De Vivo, D. C., et al. (2016). Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 86, 890–897. doi: 10.1212/wnl.0000000000002445
Corti, S., Nizzardo, M., Simone, C., Falcone, M., Nardini, M., Ronchi, D., et al. (2012). Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 4:165ra162. doi: 10.1126/scitranslmed.3004108
Delli Carri, A., Onorati, M., Lelos, M. J., Castiglioni, V., Faedo, A., Menon, R., et al. (2013). Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32+ medium-sized spiny neurons. Development 140, 301–312. doi: 10.1242/dev.084608
Demestre, M., Orth, M., Föhr, K. J., Achberger, K., Ludolph, A. C., Liebau, S., et al. (2015). Formation and characterisation of neuromuscular junctions between hiPSC derived motoneurons and myotubes. Stem Cell Res. 15, 328–336. doi: 10.1016/j.scr.2015.07.005
Denton, K. R., Lei, L., Grenier, J., Rodionov, V., Blackstone, C., and Li, X. J. (2014). Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 32, 414–423. doi: 10.1002/stem.1569
DeRosa, B. A., Belle, K. C., Thomas, B. J., Cukier, H. N., Pericak-Vance, M. A., Vance, J. M., et al. (2015). hVGAT-mCherry: a novel molecular tool for analysis of GABAergic neurons derived from human pluripotent stem cells. Mol. Cell. Neurosci. 68, 244–257. doi: 10.1016/j.mcn.2015.08.007
Ding, Q., Regan, S. N., Xia, Y., Oostrom, L. A., Cowan, C. A., and Musunuru, K. (2013). Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394. doi: 10.1016/j.stem.2013.03.006
Djuric, U., Cheung, A. Y., Zhang, W., Mok, R. S., Lai, W., Piekna, A., et al. (2015). MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 76, 37–45. doi: 10.1016/j.nbd.2015.01.001
Doerr, J., Böckenhoff, A., Ewald, B., Ladewig, J., Eckhardt, M., Gieselmann, V., et al. (2015). Arylsulfatase a overexpressing Human iPSC-derived neural cells reduce CNS sulfatide storage in a mouse model of Metachromatic Leukodystrophy. Mol. Ther. 23, 1519–1531. doi: 10.1038/mt.2015.106
Doers, M. E., Musser, M. T., Nichol, R., Berndt, E. R., Baker, M., Gomez, T. M., et al. (2014). iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 23, 1777–1787. doi: 10.1089/scd.2014.0030
Doi, D., Samata, B., Katsukawa, M., Kikuchi, T., Morizane, A., Ono, Y., et al. (2014). Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Reports 2, 337–350. doi: 10.1016/j.stemcr.2014.01.013
Ebert, A. D., Yu, J., Rose, F. F. Jr., Mattis, V. B., Lorson, C. L., Thomson, J. A., et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280. doi: 10.1038/nature07677
Efthymiou, A. G., Steiner, J., Pavan, W. J., Wincovitch, S., Larson, D. M., Porter, F. D., et al. (2015). Rescue of an in vitro neuron phenotype identified in Niemann-Pick disease, type C1 induced pluripotent stem cell-derived neurons by modulating the WNT pathway and calcium signaling. Stem Cells Transl. Med. 4, 230–238. doi: 10.5966/sctm.2014-0127
Eigentler, A., Boesch, S., Schneider, R., Dechant, G., and Nat, R. (2013). Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev. 22, 3271–3282. doi: 10.1089/scd.2013.0126
Emdad, L., D’Souza, S. L., Kothari, H. P., Qadeer, Z. A., and Germano, I. M. (2012). Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev. 21, 404–410. doi: 10.1089/scd.2010.0560
Erceg, S., Laínez, S., Ronaghi, M., Stojkovic, P., Pérez-Aragó, M. A., Moreno-Manzano, V., et al. (2008). Differentiation of human embryonic stem cells to regional specific neural precursors in chemically defined medium conditions. PLoS One 3:e2122. doi: 10.1371/journal.pone.0002122
Erceg, S., Ronaghi, M., Zipancic, I., Lainez, S., Roselló, M. G., Xiong, C., et al. (2010). Efficient differentiation of human embryonic stem cells into functional cerebellar-like cells. Stem Cells Dev. 19, 1745–1756. doi: 10.1089/scd.2009.0498
Espuny-Camacho, I., Michelsen, K. A., Gall, D., Linaro, D., Hasche, A., Bonnefont, J., et al. (2013). Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 77, 440–456. doi: 10.1016/j.neuron.2012.12.011
Farra, N., Zhang, W. B., Pasceri, P., Eubanks, J. H., Salter, M. W., and Ellis, J. (2012). Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 17, 1261–1271. doi: 10.1038/mp.2011.180
Fernandes, T. G., Duarte, S. T., Ghazvini, M., Gaspar, C., Santos, D. C., Porteira, A. R., et al. (2015). Neural commitment of human pluripotent stem cells under defined conditions recapitulates neural development and generates patient-specific neural cells. Biotechnol. J. 10, 1578–1588. doi: 10.1002/biot.201400751
Fuller, H. R., Mandefro, B., Shirran, S. L., Gross, A. R., Kaus, A. S., Botting, C. H., et al. (2016). Spinal muscular atrophy patient iPSC-derived motor neurons have reduced expression of proteins important in neuronal development. Front. Cell. Neurosci. 9:506. doi: 10.3389/fncel.2015.00506
Fusaki, N., Ban, H., Nishiyama, A., Saeki, K., and Hasegawa, M. (2009). Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 85, 348–362. doi: 10.2183/pjab.85.348
Guo, X., Disatnik, M. H., Monbureau, M., Shamloo, M., Mochly-Rosen, D., and Qi, X. (2013). Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Invest. 123, 5371–5388. doi: 10.1172/jci70911
Halevy, T., Czech, C., and Benvenisty, N. (2015). Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Reports 4, 37–46. doi: 10.1016/j.stemcr.2014.10.015
Havlicek, S., Kohl, Z., Mishra, H. K., Prots, I., Eberhardt, E., Denguir, N., et al. (2014). Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 23, 2527–2541. doi: 10.1093/hmg/ddt644
HD iPSC Consortium. (2012). Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11, 264–278. doi: 10.1016/j.stem.2012.04.027
Heesen, L., Peitz, M., Torres-Benito, L., Hölker, I., Hupperich, K., Dobrindt, K., et al. (2016). Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell. Mol. Life Sci. 73, 2089–2104. doi: 10.1007/s00018-015-2084-y
Hick, A., Wattenhofer-Donzé, M., Chintawar, S., Tropel, P., Simard, J. P., Vaucamps, N., et al. (2013). Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Model. Mech. 6, 608–621. doi: 10.1242/dmm.010900
Higurashi, N., Uchida, T., Lossin, C., Misumi, Y., Okada, Y., Akamatsu, W., et al. (2013). A human Dravet syndrome model from patient induced pluripotent stem cells. Mol. Brain 6:19. doi: 10.1186/1756-6606-6-19
Hockemeyer, D., Soldner, F., Beard, C., Gao, Q., Mitalipova, M., DeKelver, R. C., et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857. doi: 10.1038/nbt.1562
Hockemeyer, D., Wang, H., Kiani, S., Lai, C. S., Gao, Q., Cassady, J. P., et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731–744. doi: 10.1038/nbt.1927
Hsiao, H. Y., Chiu, F. L., Chen, C. M., Wu, Y. R., Chen, H. M., Chen, Y. C., et al. (2014). Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 23, 4328–4344. doi: 10.1093/hmg/ddu151
Hu, Y., Qu, Z. Y., Cao, S. Y., Li, Q., Ma, L., Krencik, R., et al. (2016). Directed differentiation of basal forebrain cholinergic neurons from human pluripotent stem cells. J. Neurosci. Methods 266, 42–49. doi: 10.1016/j.jneumeth.2016.03.017
Igoillo-Esteve, M., Gurgul-Convey, E., Hu, A., Romagueira Bichara Dos Santos, L., Abdulkarim, B., Chintawar, S., et al. (2015). Unveiling a common mechanism of apoptosis in β-cells and neurons in Friedreich’s ataxia. Hum. Mol. Genet. 24, 2274–2286. doi: 10.1093/hmg/ddu745
Jang, J., Kang, H. C., Kim, H. S., Kim, J. Y., Huh, Y. J., Kim, D. S., et al. (2011). Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann. Neurol. 70, 402–409. doi: 10.1002/ana.22486
Jeon, I., Lee, N., Li, J. Y., Park, I. H., Park, K. S., Moon, J., et al. (2012). Neuronal properties, in vivo effects and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 30, 2054–2062. doi: 10.1002/stem.1245
Jiang, H., Ren, Y., Yuen, E. Y., Zhong, P., Ghaedi, M., Hu, Z., et al. (2012). Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 3:668. doi: 10.1038/ncomms1669
Jiao, J., Yang, Y., Shi, Y., Chen, J., Gao, R., Fan, Y., et al. (2013). Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum. Mol. Genet. 22, 4241–4252. doi: 10.1093/hmg/ddt275
Jo, J., Xiao, Y., Sun, A. X., Cukuroglu, E., Tran, H. D., Göke, J., et al. (2016). Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19, 248–257. doi: 10.1016/j.stem.2016.07.005
Juopperi, T. A., Kim, W. R., Chiang, C. H., Yu, H., Margolis, R. L., Ross, C. A., et al. (2012). Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 5:17. doi: 10.1186/1756-6606-5-17
Kaufmann, M., Schuffenhauer, A., Fruh, I., Klein, J., Thiemeyer, A., Rigo, P., et al. (2015). High-throughput screening using iPSC-derived neuronal progenitors to identify compounds counteracting epigenetic gene silencing in Fragile X syndrome. J. Biomol. Screen. 20, 1101–1111. doi: 10.1177/1087057115588287
Kim, K. Y., Hysolli, E., and Park, I. H. (2011). Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. U S A 108, 14169–14174. doi: 10.1073/pnas.1018979108
Kim, D., Kim, C. H., Moon, J. I., Chung, Y. G., Chang, M. Y., Han, B. S., et al. (2009). Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4, 472–476. doi: 10.1016/j.stem.2009.05.005
Kimbrel, E. A., and Lanza, R. (2015). Current status of pluripotent stem cells: moving the first therapies to the clinic. Nat. Rev. Drug Discov. 14, 681–692. doi: 10.1038/nrd4738
Kirkeby, A., Grealish, S., Wolf, D. A., Nelander, J., Wood, J., Lundblad, M., et al. (2012). Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 1, 703–714. doi: 10.1016/j.celrep.2012.04.009
Krey, J. F., Paşca, S. P., Shcheglovitov, A., Yazawa, M., Schwemberger, R., Rasmusson, R., et al. (2013). Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209. doi: 10.1038/nn.3307
Kriks, S., Shim, J. W., Piao, J., Ganat, Y. M., Wakeman, D. R., Xie, Z., et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. doi: 10.1038/nature10648
Kumari, D., Swaroop, M., Southall, N., Huang, W., Zheng, W., and Usdin, K. (2015). High-throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl. Med. 4, 800–808. doi: 10.5966/sctm.2014-0278
Lancaster, M. A., and Knoblich, J. A. (2014). Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345:1247125. doi: 10.1126/science.1247125
Lancaster, M. A., Renner, M., Martin, C. A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi: 10.1038/nature12517
Larimore, J., Ryder, P. V., Kim, K. Y., Ambrose, L. A., Chapleau, C., Calfa, G., et al. (2013). MeCP2 regulates the synaptic expression of a Dysbindin-BLOC-1 network component in mouse brain and human induced pluripotent stem cell-derived neurons. PLoS One 8:e65069. doi: 10.1371/journal.pone.0065069
Lee, P., Martin, N. T., Nakamura, K., Azghadi, S., Amiri, M., Ben-David, U., et al. (2013). SMRT compounds abrogate cellular phenotypes of ataxia telangiectasia in neural derivatives of patient-specific hiPSCs. Nat. Commun. 4:1824. doi: 10.1038/ncomms2824
Li, Y., Polak, U., Bhalla, A. D., Rozwadowska, N., Butler, J. S., Lynch, D. R., et al. (2015). Excision of expanded GAA repeats alleviates the molecular phenotype of Friedreich’s ataxia. Mol. Ther. 23, 1055–1065. doi: 10.1038/mt.2015.41
Liu, J., Gao, C., Chen, W., Ma, W., Li, X., Shi, Y., et al. (2016). CRISPR/Cas9 facilitates investigation of neural circuit disease using human iPSCs: mechanism of epilepsy caused by an SCN1A loss-of-function mutation. Transl. Psychiatry 6:e703. doi: 10.1038/tp.2015.203
Liu, Y., Lopez-Santiago, L. F., Yuan, Y., Jones, J. M., Zhang, H., O’Malley, H. A., et al. (2013). Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol. 74, 128–139. doi: 10.1002/ana.23897
Liu, H., Lu, J., Chen, H., Du, Z., Li, X. J., and Zhang, S. C. (2015). Spinal muscular atrophy patient-derived motor neurons exhibit hyperexcitability. Sci. Rep. 5:12189. doi: 10.1038/srep12189
Liu, J., Verma, P. J., Evans-Galea, M. V., Delatycki, M. B., Michalska, A., Leung, J., et al. (2011). Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev. 7, 703–713. doi: 10.1007/s12015-010-9210-x
Livide, G., Patriarchi, T., Amenduni, M., Amabile, S., Yasui, D., Calcagno, E., et al. (2015). GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur. J. Hum. Genet. 23, 195–201. doi: 10.1038/ejhg.2014.81
Lojewski, X., Staropoli, J. F., Biswas-Legrand, S., Simas, A. M., Haliw, L., Selig, M. K., et al. (2014). Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 23, 2005–2022. doi: 10.1093/hmg/ddt596
Lu, X. H., Mattis, V. B., Wang, N., Al-Ramahi, I., Van den Berg, N., Fratantoni, S. A., et al. (2014). Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 6:268ra178. doi: 10.1126/scitranslmed.3010523
Maeda, H., Chiyonobu, T., Yoshida, M., Yamashita, S., Zuiki, M., Kidowaki, S., et al. (2016). Establishment of isogenic iPSCs from an individual with SCN1A mutation mosaicism as a model for investigating neurocognitive impairment in Dravet syndrome. J. Hum. Genet. 61, 565–569. doi: 10.1038/jhg.2016.5
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Mariani, J., Simonini, M. V., Palejev, D., Tomasini, L., Coppola, G., Szekely, A. M., et al. (2012). Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. U S A 109, 12770–12775. doi: 10.1073/pnas.1202944109
Marion, R. M., Strati, K., Li, H., Tejera, A., Schoeftner, S., Ortega, S., et al. (2009). Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141–154. doi: 10.1016/j.stem.2008.12.010
Maroof, A. M., Keros, S., Tyson, J. A., Ying, S. W., Ganat, Y. M., Merkle, F. T., et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. doi: 10.1016/j.stem.2013.04.008
Mastrangelo, L., Kim, J. E., Miyanohara, A., Kang, T. H., and Friedmann, T. (2012). Purinergic signaling in human pluripotent stem cells is regulated by the housekeeping gene encoding hypoxanthine guanine phosphoribosyltransferase. Proc. Natl. Acad. Sci. U S A 109, 3377–3382. doi: 10.1073/pnas.1118067109
Mattis, V. B., Tom, C., Akimov, S., Saeedian, J., Østergaard, M. E., Southwell, A. L., et al. (2015). HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet. 24, 3257–3271. doi: 10.1093/hmg/ddv080
McGivern, J. V., Patitucci, T. N., Nord, J. A., Barabas, M. E., Stucky, C. L., and Ebert, A. D. (2013). Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 61, 1418–1428. doi: 10.1002/glia.22522
Mekhoubad, S., Bock, C., de Boer, A. S., Kiskinis, E., Meissner, A., and Eggan, K. (2012). Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10, 595–609. doi: 10.1016/j.stem.2012.02.014
Miller, J. D., Ganat, Y. M., Kishinevsky, S., Bowman, R. L., Liu, B., Tu, E. Y., et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. doi: 10.1016/j.stem.2013.11.006
Mishra, H. K., Prots, I., Havlicek, S., Kohl, Z., Perez-Branguli, F., Boerstler, T., et al. (2016). GSK3ß-dependent dysregulation of neurodevelopment in SPG11-patient iPSC model. Ann. Neurol. doi: 10.1002/ana.24633 [Epub ahead of print].
Muotri, A. R., Marchetto, M. C., Coufal, N. G., Oefner, R., Yeo, G., Nakashima, K., et al. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446. doi: 10.1038/nature09544
Nayler, S., Gatei, M., Kozlov, S., Gatti, R., Mar, J. C., Wells, C. A., et al. (2012). Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl. Med. 1, 523–535. doi: 10.5966/sctm.2012-0024
Ng, S. Y., Soh, B. S., Rodriguez-Muela, N., Hendrickson, D. G., Price, F., Rinn, J. L., et al. (2015). Genome-wide RNA-seq of human motor neurons implicates selective ER stress activation in spinal muscular atrophy. Cell Stem Cell 17, 569–584. doi: 10.1016/j.stem.2015.08.003
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D., et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Nistor, G. I., Totoiu, M. O., Haque, N., Carpenter, M. K., and Keirstead, H. S. (2005). Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 49, 385–396. doi: 10.1002/glia.20127
Nizzardo, M., Simone, C., Dametti, S., Salani, S., Ulzi, G., Pagliarani, S., et al. (2015). Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci. Rep. 5:11746. doi: 10.1038/srep11746
Nizzardo, M., Simone, C., Falcone, M., Locatelli, F., Riboldi, G., Comi, G. P., et al. (2010). Human motor neuron generation from embryonic stem cells and induced pluripotent stem cells. Cell. Mol. Life Sci. 67, 3837–3847. doi: 10.1007/s00018-010-0463-y
Ogawa, S., Tokumoto, Y., Miyake, J., and Nagamune, T. (2011). Induction of oligodendrocyte differentiation from adult human fibroblast-derived induced pluripotent stem cells. In Vitro Cell. Dev. Biol. Anim. 47, 464–469. doi: 10.1007/s11626-011-9435-2
Ohuchi, K., Funato, M., Kato, Z., Seki, J., Kawase, C., Tamai, Y., et al. (2016). Established stem cell model of spinal muscular atrophy is applicable in the evaluation of the efficacy of thyrotropin-releasing hormone analog. Stem Cells Transl. Med. 5, 152–163. doi: 10.5966/sctm.2015-0059
Okita, K., Matsumura, Y., Sato, Y., Okada, A., Morizane, A., Okamoto, S., et al. (2011). A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412. doi: 10.1038/nmeth.1591
Park, C. Y., Halevy, T., Lee, D. R., Sung, J. J., Lee, J. S., Yanuka, O., et al. (2015). Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived. Cell Rep. 13, 234–241. doi: 10.1016/j.celrep.2015.08.084
Paşca, S. P., Portmann, T., Voineagu, I., Yazawa, M., Shcheglovitov, A., Paşca, A. M., et al. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662. doi: 10.1038/nm.2576
Patitucci, T. N., and Ebert, A. D. (2016). SMN deficiency does not induce oxidative stress in SMA iPSC-derived astrocytes or motor neurons. Hum. Mol. Genet. 25, 514–523. doi: 10.1093/hmg/ddv489
Pruszak, J., Ludwig, W., Blak, A., Alavian, K., and Isacson, O. (2009). CD15, CD24 and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells 27, 2928–2940. doi: 10.1002/stem.211
Reinhardt, P., Schmid, B., Burbulla, L. F., Schöndorf, D. C., Wagner, L., Glatza, M., et al. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. doi: 10.1016/j.stem.2013.01.008
Sareen, D., Ebert, A. D., Heins, B. M., McGivern, J. V., Ornelas, L., and Svendsen, C. N. (2012). Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7:e39113. doi: 10.1371/journal.pone.0039113
Scannell, J. W., Blanckley, A., Boldon, H., and Warrington, B. (2012). Diagnosing the decline in pharmaceutical RandD efficiency. Nat. Rev. Drug Discov. 11, 191–200. doi: 10.1038/nrd3681
Schwab, A. J., and Ebert, A. D. (2014). Sensory neurons do not induce motor neuron loss in a human stem cell model of spinal muscular atrophy. PLoS One 9:e103112. doi: 10.1371/journal.pone.0103112
Seibler, P., Graziotto, J., Jeong, H., Simunovic, F., Klein, C., and Krainc, D. (2011). Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976. doi: 10.1523/JNEUROSCI.4441-10.2011
Shan, B., Xu, C., Zhang, Y., Xu, T., Gottesfeld, J. M., and Yates, J. R. (2014). Quantitative proteomic analysis identifies targets and pathways of a 2-aminobenzamide HDAC inhibitor in Friedreich’s ataxia patient iPSC-derived neural stem cells. J. Proteome Res. 13, 4558–4566. doi: 10.1021/pr500514r
Shcheglovitov, A., Shcheglovitova, O., Yazawa, M., Portmann, T., Shu, R., Sebastiano, V., et al. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. doi: 10.1038/nature12618
Sheridan, S. D., Theriault, K. M., Reis, S. A., Zhou, F., Madison, J. M., Daheron, L., et al. (2011). Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 6:e26203. doi: 10.1371/journal.pone.0026203
Sirenko, O., Hesley, J., Rusyn, I., and Cromwell, E. F. (2014). High-content high-throughput assays for characterizing the viability and morphology of human iPSC-derived neuronal cultures. Assay Drug Dev. Technol. 12, 536–547. doi: 10.1089/adt.2014.592
Soragni, E., Miao, W., Iudicello, M., Jacoby, D., De Mercanti, S., Clerico, M., et al. (2014). Epigenetic therapy for Friedreich ataxia. Ann. Neurol. 76, 489–508. doi: 10.1002/ana.24260
Srikanth, P., and Young-Pearse, T. L. (2014). Stem cells on the brain: modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J. Neurogenet. 28, 5–29. doi: 10.3109/01677063.2014.881358
Suhr, S. T., Chang, E. A., Tjong, J., Alcasid, N., Perkins, G. A., Goissis, M. D., et al. (2010). Mitochondrial rejuvenation after induced pluripotency. PLoS One 5:e14095. doi: 10.1371/journal.pone.0014095
Sun, Y., Florer, J., Mayhew, C. N., Jia, Z., Zhao, Z., Xu, K., et al. (2015). Properties of neurons derived from induced pluripotent stem cells of Gaucher disease type 2 patient fibroblasts: potential role in neuropathology. PLoS One 10:e0118771. doi: 10.1371/journal.pone.0118771
Szlachcic, W. J., Switonski, P. M., Krzyzosiak, W. J., Figlerowicz, M., and Figiel, M. (2015). Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis. Model. Mech. 8, 1047–1057. doi: 10.1242/dmm.019406
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takahashi, K., and Yamanaka, S. (2016). A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 17, 183–193. doi: 10.1038/nrm.2016.8
Tang, X., Kim, J., Zhou, L., Wengert, E., Zhang, L., Wu, Z., et al. (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U S A 113, 751–756. doi: 10.1073/pnas.1524013113
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1126/science.282.5391.1145
Tian, Y., Voineagu, I., Paşca, S. P., Won, H., Chandran, V., Horvath, S., et al. (2014). Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 6:75. doi: 10.1186/s13073-014-0075-5
Tiscornia, G., Vivas, E. L., Matalonga, L., Berniakovich, I., Barragán Monasterio, M., Eguizábal, C., et al. (2013). Neuronopathic Gaucher’s disease: induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum. Mol. Genet. 22, 633–645. doi: 10.1093/hmg/dds471
Toli, D., Buttigieg, D., Blanchard, S., Lemonnier, T., Lamotte d’Incamps, B., Bellouze, S., et al. (2015). Modeling amyotrophic lateral sclerosis in pure human iPSc-derived motor neurons isolated by a novel FACS double selection technique. Neurobiol. Dis. 82, 269–280. doi: 10.1016/j.nbd.2015.06.011
Trilck, M., Hübner, R., and Frech, M. J. (2016). Generation and neuronal differentiation of patient-specific induced pluripotent stem cells derived from niemann-pick type C1 fibroblasts. Methods Mol. Biol. 1353, 233–259. doi: 10.1007/7651_2014_166
Trilck, M., Hübner, R., Seibler, P., Klein, C., Rolfs, A., and Frech, M. J. (2013). Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 8:144. doi: 10.1186/1750-1172-8-144
Urbach, A., Bar-Nur, O., Daley, G. Q., and Benvenisty, N. (2010). Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6, 407–411. doi: 10.1016/j.stem.2010.04.005
Vera, E., and Studer, L. (2015). When rejuvenation is a problem: challenges of modeling late-onset neurodegenerative disease. Development 142, 3085–3089. doi: 10.1242/dev.120667
Vessoni, A. T., Herai, R. H., Karpiak, J. V., Leal, A. M., Trujillo, C. A., Quinet, A., et al. (2016). Cockayne syndrome-derived neurons display reduced synapse density and altered neural network synchrony. Hum. Mol. Genet. 25, 1271–1280. doi: 10.1093/hmg/ddw008
Wada, T., Honda, M., Minami, I., Tooi, N., Amagai, Y., Nakatsuji, N., et al. (2009). Highly efficient differentiation and enrichment of spinal motor neurons derived from human and monkey embryonic stem cells. PLoS One 4:e6722. doi: 10.1371/journal.pone.0006722
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y. H., Li, H., Lau, F., et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630. doi: 10.1016/j.stem.2010.08.012
Wicklund, L., Leão, R. N., Strömberg, A. M., Mousavi, M., Hovatta, O., Nordberg, A., et al. (2010). β-amyloid 1–42 oligomers impair function of human embryonic stem cell-derived forebrain cholinergic neurons. PLoS One 5:e15600. doi: 10.1371/journal.pone.0015600
Williams, E. C., Zhong, X., Mohamed, A., Li, R., Liu, Y., Dong, Q., et al. (2014). Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum. Mol. Genet. 23, 2968–2980. doi: 10.1093/hmg/ddu008
Xu, C. C., Denton, K. R., Wang, Z. B., Zhang, X., and Li, X. J. (2016). Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis. Model. Mech. 9, 39–49. doi: 10.1242/dmm.021766
Yamashita, S., Chiyonobu, T., Yoshida, M., Maeda, H., Zuiki, M., Kidowaki, S., et al. (2016). Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia 57, e81–e86. doi: 10.1111/epi.13338
Yoshida, M., Kitaoka, S., Egawa, N., Yamane, M., Ikeda, R., Tsukita, K., et al. (2015). Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs. Stem Cell Reports 4, 561–568. doi: 10.1016/j.stemcr.2015.02.010
Yuan, S. H., Martin, J., Elia, J., Flippin, J., Paramban, R. I., Hefferan, M. P., et al. (2011). Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One 6:e17540. doi: 10.1371/journal.pone.0017540
Zhang, Z. N., Freitas, B. C., Qian, H., Lux, J., Acab, A., Trujillo, C. A., et al. (2016). Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc. Natl. Acad. Sci. U S A 113, 3185–3190. doi: 10.1073/pnas.1521255113
Zhou, W., and Freed, C. R. (2009). Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27, 2667–2674. doi: 10.1002/stem.201
Keywords: iPSCs, childhood neurological disorders, in vitro disease modeling, gene therapies, drug screening, isogenic control
Citation: Barral S and Kurian MA (2016) Utility of Induced Pluripotent Stem Cells for the Study and Treatment of Genetic Diseases: Focus on Childhood Neurological Disorders. Front. Mol. Neurosci. 9:78. doi: 10.3389/fnmol.2016.00078
Received: 02 June 2016; Accepted: 15 August 2016;
Published: 06 September 2016.
Edited by:
Kirsten Harvey, University College London, UKCopyright © 2016 Barral and Kurian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manju A. Kurian, bWFuanUua3VyaWFuQHVjbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.