

- School of Physiology and Pharmacology, University of Bristol, Bristol, UK
Calcium (Ca2+)/calmodulin (CaM)-dependent kinase II (CaMKII) activity plays a fundamental role in learning and memory. A key feature of CaMKII in memory formation is its ability to be regulated by autophosphorylation, which switches its activity on and off during synaptic plasticity. The synaptic scaffolding protein CASK (calcium (Ca2+)/calmodulin (CaM) associated serine kinase) is also important for learning and memory, as mutations in CASK result in intellectual disability and neurological defects in humans. We show that in Drosophila larvae, CASK interacts with CaMKII to control neuronal growth and calcium signaling. Furthermore, deletion of the CaMK-like and L27 domains of CASK (CASK β null) or expression of overactive CaMKII (T287D) produced similar effects on synaptic growth and Ca2+ signaling. CASK overexpression rescues the effects of CaMKII overactivity, consistent with the notion that CASK and CaMKII act in a common pathway that controls these neuronal processes. The reduction in Ca2+ signaling observed in the CASK β null mutant caused a decrease in vesicle trafficking at synapses. In addition, the decrease in Ca2+ signaling in CASK mutants was associated with an increase in Ether-à-go-go (EAG) potassium (K+) channel localization to synapses. Reducing EAG restored the decrease in Ca2+ signaling observed in CASK mutants to the level of wildtype, suggesting that CASK regulates Ca2+ signaling via EAG. CASK knockdown reduced both appetitive associative learning and odor evoked Ca2+ responses in Drosophila mushroom bodies, which are the learning centers of Drosophila. Expression of human CASK in Drosophila rescued the effect of CASK deletion on the activity state of CaMKII, suggesting that human CASK may also regulate CaMKII autophosphorylation.
Introduction
CaMKII has been proposed to act as a molecular switch during increased neuronal activity, when increased Ca2+ levels stimulate CaMKII activity to induce the changes in synaptic strength that underlie learning. The ability of CaMKII to induce long lasting changes in synaptic strength has been shown to be dependent on CaMKII autophosphorylation at T286 (T287 in Drosophila). T287 autophosphorylation occurs in response to prolonged increases in Ca2+, which result in constitutively active CaMKII that is independent of Ca2+. This constitutive CaMKII activity has been suggested to be important for long-term potentiation (LTP) and memory in rodents (Giese et al., 1998; Hardingham et al., 2003; Sanhueza et al., 2011) and Drosophila (Park et al., 2002; Mehren and Griffith, 2004; Hodge et al., 2006; Malik et al., 2013). In Drosophila, CaMKII autophosphorylation is regulated at synapses by CASK, a membrane-associated guanylate kinase (MAGUK) that is kinase dead in Drosophila (Lu et al., 2003; Hodge et al., 2006).
Interactions of CASK with CaMKII can lead to inhibition of CaMKII activity through CaMKII autophosphorylation at a second pair of sites, T305/T306. This process results in reduced binding of CaMKII to CaM, which decreases kinase activation by Ca2+ and thereby prevents T287 autophosphorylation. CaMKII autophosphorylation at this site is important for long-term depression (LTD) and behavioral plasticity in mice (Elgersma et al., 2002; Pi et al., 2010) and Drosophila (Lu et al., 2003; Malik et al., 2013).
The function of CASK has also been studied in mice, and while CASK knock-outs are lethal due to a cleft palate phenotype, neurons cultured from these animals show abnormalities in glutamatergic synaptic release (Atasoy et al., 2007). However, the early lethality of these mice prevents the modeling of CASK function in behavior and disease.
CASK has two isoforms, a full-length CASK β isoform that contains the CaMK-like and L27 domains and PDZ, SH3, and guanylate kinase domains. The other isoform, CASK α, is short and contains only the common PDZ, SH3 and guanylate kinase domains and forms a molecule with structural homology to vertebrate MPP (Slawson et al., 2011). Previous characterization of CASK has focused on a large chromosomal deficiency that removes both forms of CASK and genes on either side of CASK. These deficiency flies exhibit reduced synaptic currents, defects in neuromuscular junction (NMJ) growth, decreased neurotransmitter release, decreased vesicle cycling and a loss of interaction with neurexin (Zordan et al., 2005; Sun et al., 2009; Chen and Featherstone, 2011).
To investigate the true role of CASK in Drosophila, a deletion of the full length CASK β isoform that resulted in viable adults was used. This CASK β null allele deletes only the CaMK-like and L27 domains that are unique to CASK β and leaves the short CASK α isoform and all flanking genes intact (Slawson et al., 2011). The CASK β null has specific deficits in larval locomotor behavior (Slawson et al., 2011) and adult sleep and place preferences (Donelson et al., 2012). Furthermore, CASK β is required for 3 h and long-term memory as measured in an adult aversive olfactory conditioning assay; this assay revealed that CASK is required in the α′/β′ subset of mushroom body neurons during memory formation (Malik et al., 2013).
In this study, we used CASK β null flies to more accurately analyze CASK synaptic function, as flanking loci are not deleted in this flies, which is the case for flies with the large chromosomal deficiency of the CASK region, which has been phenotypically characterized previously (Lu et al., 2003; Zordan et al., 2005; Hodge et al., 2006; Sun et al., 2009; Chen and Featherstone, 2011; Slawson et al., 2011). CASK β is shown to regulate CaMKII autophosphorylation during synaptic bouton growth and Ca2+ signaling. We also demonstrate that CASK β controls vesicle trafficking and the localization of EAG K+ channels to synapses. In Drosophila larvae, CASK β is required for appetitive associative learning and olfactory evoked Ca2+ responses in the mushroom body. Finally, we provide evidence that the human form of CASK seems capable of compensating for the loss of Drosophila CASK in regulating CaMKII autophosphorylation, suggesting a high level of conservation.
Materials and Methods
Fly Strains and Genetics
All flies were grown at similar densities in bottles on standard medium at 22 ± 2°C. CASK β null, uas-CASK (10,20 MI), uas-CaMKII, uas-CaMKII-T287D, uas-CaMKII-T287A and uas-TrpA1 (Lu et al., 2003; Pulver et al., 2009; Slawson et al., 2011) were kind gifts from Dr. Leslie Griffith (Brandeis University, US). uas-EAG-RNAi (stock #9126) and uas-CASK-RNAi flies (stock #104793, Malik et al., 2013) were obtained from the Vienna Drosophila Stock Center (VDRC). MEF2-Gal4, 201Y-Gal4, and uas-cacophony-eGFP (Ranganayakulu et al., 1996; Kawasaki et al., 2004; Thum et al., 2007) were from the Bloomington Stock Center. Wildtype flies [Canton Sw-, (CSw-)] were a kind gift from Dr. Scott Waddell (Oxford University, UK). GCaMP3.1 flies (Tian et al., 2009) were a gift from Dr. Loren Looger (Janelia farm, VA, US). We are grateful to Dr. Hermann Aberle (Dusseldorf University, Germany) for OK371-Gal4 flies (Mahr and Aberle, 2006). The uas-human CASK line has been described previously (Malik et al., 2013). uas-GCaMP3 was meiotically recombined with 201Y-Gal4 flies on the 2nd chromosome and homozygosed. Similarly, uas-GCaMP3, uas-TrpA1 and OK371-Gal4, uas-GCaMP3 flies were generated by standard recombination crosses. All CASK mutants, Gal4, and UAS lines were outcrossed with the CSw- line for at least six generations prior to behavioral experiments.
Immunohistochemistry
Third-instar larvae were dissected in HL3.1 [70 mM NaCl, 5 mM KCl, 10 mM NaHCO3, 115 mM sucrose, 4 mM MgCl, 5 mM trehalose, 1.5 mM CaCl2, and 5 mM HEPES (pH 7.3)] using standard techniques that have been used previously for these antibodies (Hodge et al., 2006; Cavaliere et al., 2012). The dissected larvae were fixed in 4% paraformaldehyde in HL3.1, permeabilized in HL3.1 with 0.1% triton X (HL3.1-tx), and then blocked in HL3.1-tx, 0.1% BSA, and 2% normal donkey serum (HL3.1-tx-BSA-NGS). Primary antibody incubations were performed as indicted with anti-pT287 CaMKII (Santa Cruz, rabbit, 1:150), anti-Drosophila CaMKII (Cosmo, mouse, 1:100), anti-HRP-FITC (Jackson ImmunoResearch Laboratories, 1:200), anti-Drosophila DLG-PDZ2 [rabbit, 1:1000 (Sherwood et al., 2004)] anti-Drosophila EAG [rabbit, 1:50 (Sun et al., 2004)], at 4°C in HL3.1-tx-BSA-NGS. After washing three times in HL3.1-tx, incubations were performed with anti-rabbit 405 nm, anti-rabbit 488 nm, or anti-mouse 648 nm fluorescently conjugated secondary antibodies (Alexa, Invitrogen, 1:400) in HL3.1-tx-BSA for 2 h at room temperature, after which the larvae were washed and mounted in glycerol and Vectorshield (Vector Laboratories).
Imaging and Quantification
All preparations within each immunohistochemistry experiment were processed in parallel, and images were acquired with identical settings using a Leica SP5 confocal microscope. Care was taken to maintain all intensity readings within the linear range below saturation. Measurements of mean intensity, bouton area, and quantification were performed manually with Volocity software (PerkinElmer) by drawing round type Ib and Is boutons on muscle 12. Because bouton number increases with muscle area (Schuster et al., 1996), bouton number was normalized to muscle area. The muscle area was determined by measuring the length and width of muscle 12.
For live GCaMP3 experiments, larvae were dissected in HL3.1 and imaged essentially according to previously published protocols (Cavaliere et al., 2012). To prevent muscle contractions during NMJ imaging, HL3.1 was supplemented with 7 mM glutamate (HL3.1-glu). This concentration of glutamate desensitises the postsynaptic glutamate receptors without significantly affecting presynaptic Ca2+ signaling (Macleod et al., 2004). uas-TrpA1, uas-GCaMP3/OK371-Gal4 larvae were stimulated by bath application HL3.1-glutamate at 35°C. Images were captured through a 63 × water immersion lens (NA = 0.9) on a Zeiss widefield Axio Examiner microscope, and images were taken at 4 frames per second. The average of the first 10 frames during periods of neuronal inactivity was calculated after subtraction of background fluorescence. GCaMP3 intensity values were normalized to this baseline intensity. Ventral ganglion imaging was performed in similar manner using HL3.1 without glutamate on a Zeiss widefield Axio Examiner microscope (10 × water immersion lens NA = 0.3), and images were taken at 10 frames per second.
Similar to previous studies (Asahina et al., 2009), larval mushroom body imaging was performed by first cutting off the heads of the larvae in HL3.1. The salivary glands and digestive tract were removed to allow visualization of the brain. A small hole was made in a strip of PVDF membrane, which was then placed across a custom-made airflow chamber that allowed odor to be puffed onto the larval head. The dissected head of the larvae was placed into this hole so that the tip of the head was on one side of the PVDF membrane and the brain was on the other side. Low melt agarose (1%) in HL3.1 was applied over the top of the brain to prevent the media from covering the tip of the larvae's head and was allowed to set at 4°C for 1 min. HL3.1 was then added to the top of this agarose to allow for imaging of GCaMP3 in the mushroom body calyx through a 63× water immersion lens (NA = 0.9) on a Zeiss widefield Axio Examiner microscope. Images were acquired using identical exposure times at 10 frames per second. After recording for approximately 1 min, during which time a constant air flow was delivered via an aquarium pump, the air was the passed through a chamber containing a piece of tissue soaked in hexyl acetate (Sigma, 1:10 in mineral oil) for approximately 10 s before the odor-free air was applied. The part of the calyx specifically activated by the application of odor was selected as the region of interest, and the mean background-subtracted intensities for each time point were calculated for this region. The ratios of the mean intensity at each time point to the intensity at baseline were calculated.
FM1-43 Imaging
FM1-43 loading of motor neuron terminals at larval NMJs was performed as described previously (Sun et al., 2009). Wandering third-instar larvae were dissected in Ca2+-free HL3 as above. The larvae were then incubated with 1 μ M FM1-43 in 90 mM KCl and HL3 with 1.5 mM Ca2+ for 10 min. FM1-43 loading was terminated by washing the larvae five times for 10 min with Ca2+-free HL3. A stack of images of the labeled vesicles was acquired for quantification using the 63× objective (NA = 0.9) of a Zeiss widefield Axio Examiner microscope, and all settings remained constant between preparations. The mean labeling intensities of approximately 10 boutons per NMJ on muscle 12 were determined using Volocity.
Larval Appetitive Learning
Larval learning experiments were performed as previously described (Chen et al., 2011). Approximately 100 flies were allowed to lay eggs for 12 h in bottles containing standard media and maintained at 25°C. These eggs were then allowed to develop for 5 days, and the wandering third-instar larvae were then separated from the food by floating in a 15% sucrose solution prior to being washed in running tap water. These larvae were then used immediately in the larval learning experiments. Larval training was performed using 9 cm petri dishes containing approximately 10 ml 1% agarose dissolved in ddH2O with or without 2 M fructose. Odors were presented in an odor cup made from the top of a 500 μl thin-walled micro-centrifuge tube. The bottom of the tube (1 cm from the top) was cut away and replaced with the lid of another micro-centrifuge tube. Holes in the lid were then made using a pair of sharp forceps heated with a Bunsen burner. A small piece of blotting paper containing 20 μl of either hexyl acetate or benzaldehyde (Sigma) diluted 1:10 with mineral oil was placed in the odor cup. Odor cups containing the stimulus that had been conditioned with reinforcement (CS+) were placed 1 cm from the edge on either side of the petri dish containing fructose (unconditioned stimulus, US). The larvae were allowed to crawl in this dish for 5 min before being transferred to the petri dish that contained pure agarose and the stimulus that had been conditioned without reinforcement (CS−) for a further 5 min. This process was repeated three times. For testing, the larvae were then placed in a 7 mm wide stripe in the middle of a pure agarose plate with the CS+ and CS− odors on opposite sides of the dish. After 3 min the larvae on either side of dish and in the 7 mm wide middle strip were counted. A performance index (PI) was then calculated according to the following equation:
The odors representing the CS+ and CS− were then swapped, and the test was repeated to produce a second PI that was averaged with the first PI to yield an n of 1.
To measure fructose acuity, larvae were placed in the middle of a petri dish that contained pure agarose gel in one half and agarose gel containing 2 M fructose in the other half. Larvae were placed in the middle of the fructose acuity test plate and allowed to crawl for 5 min after which a PI was calculated according to the following equation:
Odor acuity control experiments were performed by placing odors on one side of a pure agarose petri dish and allowing larvae to crawl for 3 min after which the larvae on either side of dish and in the 7 mm wide middle strip were counted to calculate a preference index using the following equation:
Statistical Analysis
All statistical analyzes was performed in Prism (GraphPad). All data were analyzed with unpaired two-tailed Student's t-tests or One-Way ANOVAs with Bonferroni post-hoc testing where appropriate. We ensured that the variances did not differ significantly between groups. For each experiment, the details of the statistical analyzes and N's can be found in the figure legends. In all figures, error bars represent the SEM. No asterisk indicates p > 0.05, * indicates p < 0.05, ** indicates p < 0.01 and *** indicates p < 0.001.
Results
The CaMK-Like and L27 Domain-Containing Isoform of Cask Regulates Synaptic Terminal Growth
To determine the role of the long CASK isoform in control of synaptic morphology and the regulation of CaMKII autophosphorylation, we characterized the NMJs of homozygous CASK β wandering third-instar mutant larvae. Drosophila larval NMJs have a stereotyped pattern in which identified motor neurons form specific types of boutons (type Ib, Is, II and III boutons) on particular muscles (Hoang and Chiba, 2001). On muscle 12, motor neuron MN12-Ib forms type Ib boutons that are large (3–10 μm) and contain glutamatergic synapses. Motor neuron MNISNb/d-Is (RP5) terminates on muscle 12 and forms type Is boutons that are also glutamatergic, but these boutons are smaller in size (2–4 μm) (Hoang and Chiba, 2001). Larvae were dissected and co-stained with anti-HRP (green), which labels the presynaptic neuronal membrane (Jan and Jan, 1982), and anti-DLG-PDZ2 (magenta), a marker for the postsynaptic density (PSD) and sub-synaptic reticulum (Sherwood et al., 2004). Using pre- and post-synaptic markers, we quantified the effect of CASK and CaMKII on synaptic bouton morphology. Compared to controls (Figure 1A), CASK β null NMJs appeared to contain more boutons that were smaller in size (Figure 1B). Quantification of the number of type 1s boutons revealed similar numbers between control and CASK β null larvae (Figure 1C). However, the sizes of the CASK β null boutons were smaller than those of controls (Figure 1D). CASK β null NMJs contained more type 1b boutons (Figure 1E) that were also smaller in size than those of the controls (Figure 1F). To explore whether CASK was functioning pre- or post-synaptically to bring about these changes in synaptic terminal morphology, we used a transgene to overexpress CASK (Hodge et al., 2006) and an RNAi transgene specific for CASK (Malik et al., 2013) that were either expressed in motor neurons [OK371-Gal4, (Mahr and Aberle, 2006)] or muscle [MEF2-Gal4, (Ranganayakulu et al., 1996)]. The reduction in presynaptic CASK by RNAi knockdown was sufficient to cause the increase in type 1b boutons (Figure 1G) that was also observed in CASK β null larvae but had no effect of bouton area (Figure 1I). The change in bouton area observed in CASK β null larvae seemed to be mediated by a postsynaptic function of CASK, as muscle expression of CASK-RNAi reduced type Ib bouton area, and CASK overexpression increased type 1b bouton area (Figure 1J), but had no effect on bouton number (Figure 1H).
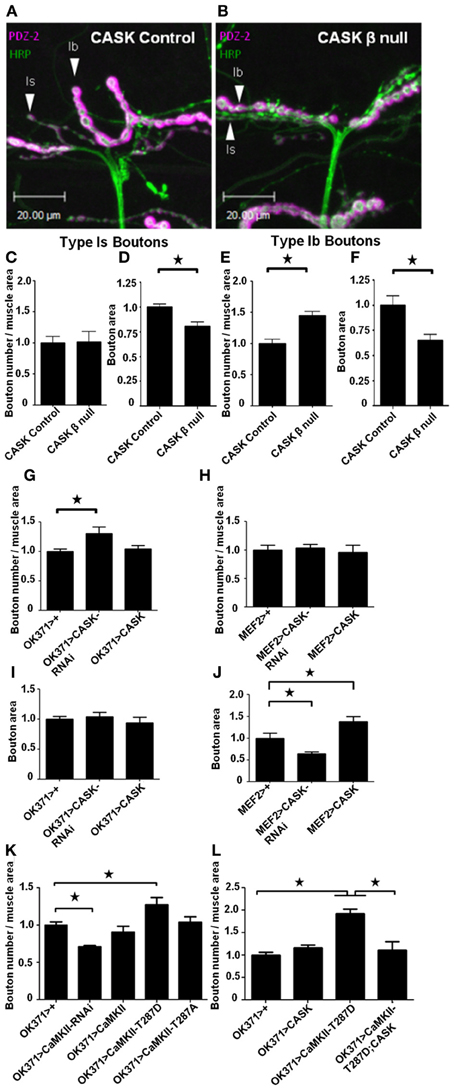
Figure 1. CASK and CaMKII regulate synaptic morphology. Representative images of muscle 12 of Drosophila third-instar larval neuromuscular junctions (NMJ) visualized with the presynaptic marker anti-HRP (green) and the postsynaptic marker anti-DLG (PDZ1-2, magenta). NMJs of larvae with precise (A) and imprecise (B) excisions of CASK (CASK β null) that remove the CaMK-like and L27 domain-containing version of CASK (Slawson et al., 2011). (C) Compared to controls, the total number of small (type 1s) boutons normalized to muscle area did not change (p > 0.05, n = 6). (D) However, the area of the 1s boutons was significantly (n = 6, p < 0.05) decreased in CASK β null larvae. Regarding, the larger (Type 1b) boutons, CASK β deletion caused an increase (p < 0.05, n = 6) in synapse number (E) and a decrease in size (F) (p < 0.05, n = 6). (G) Pre-synaptic (OK371-Gal4) reduction in CASK was sufficient to cause an increase (p < 0.05, n = 6) in 1b bouton number, but had no effect on bouton area (I). Postsynaptic (MEF2-Gal4) overexpression of CASK (J) caused an increase (p < 0.05, n = 6) in 1b bouton area (H) without affecting bouton number (p > 0.05, n = 6). Presynaptic overexpression of the constitutively active CaMKII phosphomimic (T287D) transgene (Park et al., 2002) caused an increase (p < 0.05, n = 6) in synapse number (K). Conversely, reducing presynaptic CaMKII activity (CaMKII-RNAi) reduced (p < 0.05, n = 4) 1b bouton number. (L) Presynaptic (OK371-Gal4) overexpression of T287D increased (p < 0.05, n = 6) type 1b bouton number, while CASK overexpression alone had little effect compared to controls. Co-expression of CaMKII-T287D with CASK (OK371-Gal4>uas-CASK/uas-CaMKII-T287D) returned type 1b bouton number to control levels (p < 0.05, n = 5). All data in (C–F) were analyzed with unpaired t-tests. All data in (G–L) were analyzed with One-Way ANOVA and Bonferroni post-hoc testing. In this and all subsequent figures, error bars represent the SEM. No asterisk indicates p > 0.05, * indicates p < 0.05, ** indicates p < 0.01 and *** indicates p < 0.001.
To address whether these CASK-dependent synaptic changes were brought about via CASK's regulation of CaMKII (Lu et al., 2003; Hodge et al., 2006), we tested whether transgenic manipulation of CaMKII would recapitulate these synaptic defects. Reducing CASK is known to result in increased CaMKII-T287 autophosphorylation (Hodge et al., 2006). Consistent with this, we found that expressing CaMKII-T287D presynaptically phenocopied the increase in type 1b boutons observed after deletion of CaMK-like and L27 domain-containing CASK or the presynaptic reduction of CASK. Conversely, RNAi mediated reduction in presynaptic CaMKII activity (Ashraf et al., 2006) caused a reduction in bouton number (Figure 1K).
Phosphorylation at TT306/7 is known to occur after T287 autophosphorylation and reduce the kinase activity of CaMKII by between 20 and 80% (Hanson and Schulman, 1992; Jama et al., 2009; Coultrap et al., 2010). Overexpression of CASK causes an increase in CaMKII-T306 T307 autophosphorylation (Hodge et al., 2006) that reduces kinase activity. Therefore, we co-expressed CASK concurrently with CaMKII-T287D presynaptically and found this rescued the increase in bouton number observed after presynaptic T287D expression alone, confirming that CASK and CaMKII act together in a common presynaptic pathway that controls synaptic growth (Figure 1L).
The CaMK-Like and L27 Domain-Containing Isoform of Cask Regulates the Switch of Synaptic CaMKII to Calcium Independence
While it has previously been shown that deletion of the CASK gene results in dysregulation of CaMKII autophosphorylation (Hodge et al., 2006), these mutant flies also have heterozygous deletions of genes on either side of the CASK locus. Furthermore, the shorter isoform of CASK (CASK α) was also deleted in these studies (Slawson et al., 2011). Therefore, to determine the role of the CaMK-like and L27 domain-containing isoform of CASK in the regulation of synaptic CaMKII autophosphorylation, we used a CaMKII-T287 phospho-specific antibody (Figures 2A,B, green). We found that the mean bouton pT287 CaMKII staining intensity was greater in CASK β null larvae for the type 1s (Figure 2C) and 1b boutons (Figure 2D) than in controls. While this result may be due to CASK β regulating CaMKII autophosphorylation, CASK could also conceivably have a scaffolding role that localizes CaMKII to synapses, or alternatively, CASK could increase the density of CaMKII present in the boutons by reducing bouton size. To address these questions, we co-stained NMJs with an antibody to total CaMKII (Figures 2A,B, magenta). The total level and spatial distribution of CaMKII was unchanged in CASK β null synapses compared to controls (Figures 2E,F), confirming that the amount of T287 autophosphorylation per synaptically localized CaMKII molecule had indeed increased (Figures 2G,H) and that CASK was not required for localizing CaMKII to synaptic bouton sites. To determine whether CASK was regulating CaMKII autophosphorylation by a pre- or post-synaptic mechanism, we knocked down CASK on either side of the synapse. The effect of these genetic manipulations on CASK levels were verified by immunohistochemistry, which showed that CASK knockdown in motor neurons reduced CASK expression at the NMJ by 52% compared to controls (Figures 2I,J). We found that either motor neuron (Figure 2K) or muscle (Figure 2L) expression of CASK-RNAi was sufficient to cause a switch to the Ca2+-independent constitutively active (pT287) state of CaMKII.
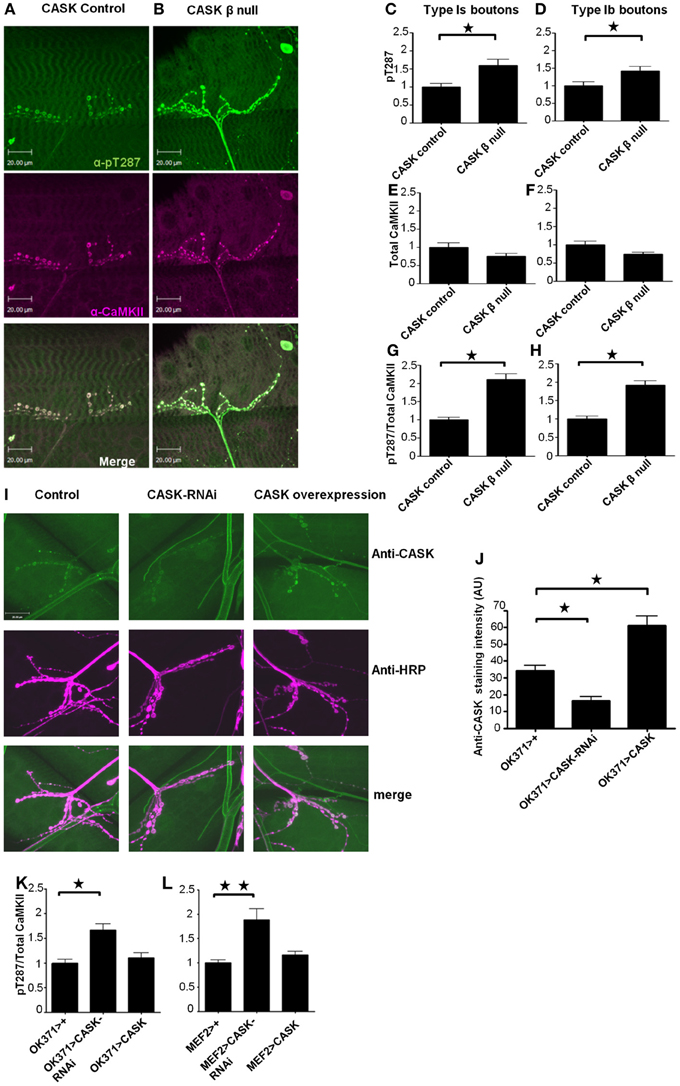
Figure 2. The CaMK-like and L27 domain-containing version of CASK regulates CaMKII autophosphorylation at synapses. Compared to control (A) CASK β null larvae (B) exhibited increased levels of synaptic CaMKII autophosphorylation at T287 [as visualized in green using a phospho-specific antibody to CaMKII-T287 (Hodge et al., 2006)] relative to total synaptic CaMKII [visualized in magenta using an antibody raised against CaMKII (Hodge et al., 2006)]. Overlapping expression is shown in white. Removal of the CaM-Kinase and L27 domains of CASK caused an increase (p < 0.05) in pT287 at 1s (C) and 1b (D) boutons. The total amount of CaMKII localized at synapses was not affected (p > 0.05) by the removal of CASK (E,F). The ratio of pT287 to total CaMKII (G,H) also showed a significant (p < 0.05) increase in the CASK β null larvae. (I) Representative images of CASK (green) and HRP (magenta) stained NMJs from control larvae, larvae overexpressing CASK-RNAi in their motor neurons and larvae overexpressing CASK in their motor neurons. (J) Quantification of the effect of CASK knockdown or overexpression on anti-CASK staining showed that genetic manipulation of CASK significantly changed the amount of CASK present at the NMJ (p < 0.05). Reductions in CASK either pre- (K) or post-synaptically (L) caused increases (p < 0.05) in CaMKII (T287) autophosphorylation. All data in (C–H) were analyzed by unpaired t-tests (n = 6). All data in (J–L) were analyzed by One-Way ANOVA with Bonferroni post-hoc tests (n = 6).
Cask and CaMKII Act in a Common Pathway Regulating Activity-Dependent Calcium Signaling
To further understand how CASK and CaMKII may be altering neuronal function in vivo, we expressed the genetically encoded calcium reporter GCaMP3 (Tian et al., 2009; Cavaliere et al., 2012) in these motor neurons to study activity-dependent changes in Ca2+ signaling (Figure 3). Transient increases in motor neuron Ca2+ are likely to control the movement of the larvae. Larvae crawl by peristaltic contractions of muscles in the tail segment that are followed by sequential posterior to anterior contraction of each segment (Cattaert and Birman, 2001). We reasoned that if there were endogenous peristaltic contractions of the larval preparation, then these contractions should be accompanied by increases in Ca2+ signaling at the NMJs of the contracting body wall segment (Figures 3A–C). As CASK β null larvae have a number of deficits in locomotion (Slawson et al., 2011), we sought to determine whether there were any changes in NMJ Ca2+ signaling that occurred during these peristaltic contractions. Although there was a reduction in this Ca2+ signaling, it was not significant (Figures 3A–C); this finding is likely due to the substantial movement artifacts, which increased the standard deviation of the recorded responses in this preparation. Therefore, we co-expressed the heat-activated ion channel TrpA1, which, when exposed to 30°C, causes a large depolarization and activation of these neurons (Pulver et al., 2009). Heat activation of TrpA1 robustly stimulated these motor neurons resulting in a large increase in synaptic Ca2+ signaling (Figures 3D–F), and while process induced some movement, the movement could be more easily controlled for than that during peristaltic contraction. The increase in peak Ca2+ induced by heat activation of TrpA1 was greatly reduced when the CaMK-like and L27 domain-containing version of CASK was absent in the CASK β null larvae (Figure 3F).
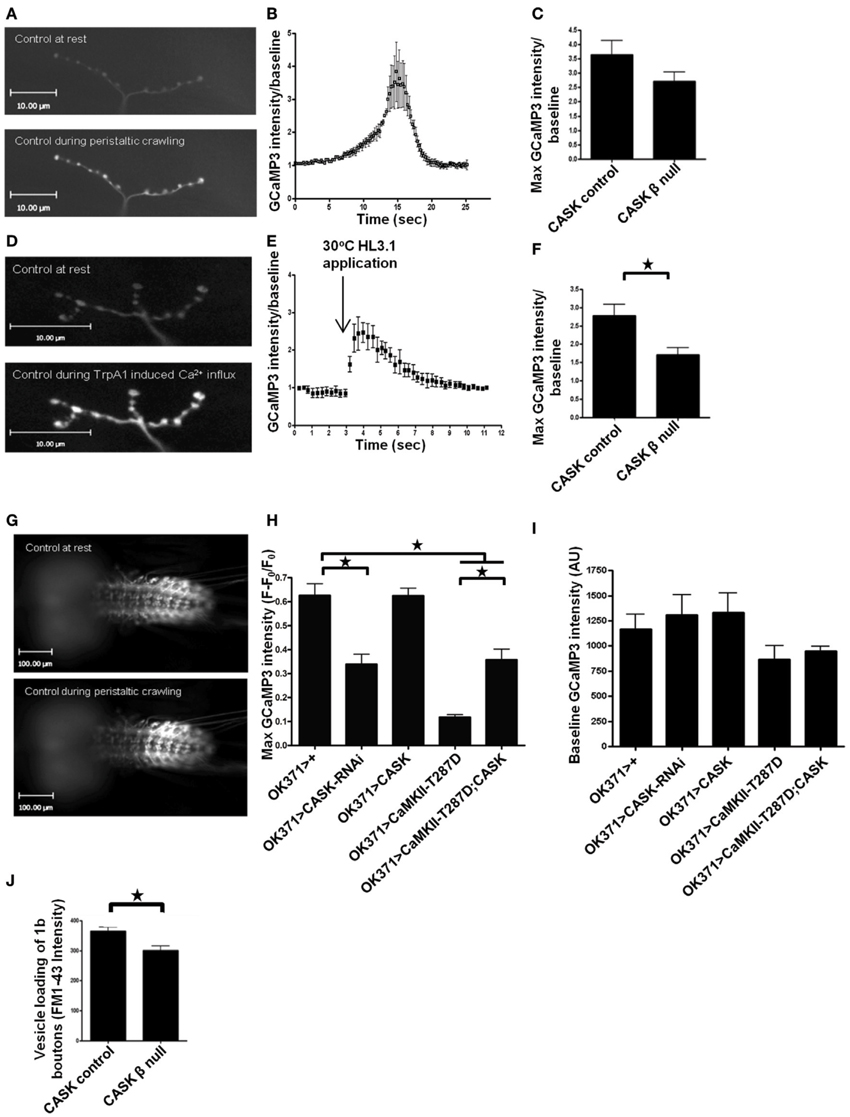
Figure 3. The CaMK-like and L27 domain-containing version of CASK regulates calcium signaling and vesicle cycling. Compared to resting synapses, there is an increase in Ca2+ signaling during peristaltic crawling, and this increase is reported by the genetically encoded Ca2+ indicator GCaMP3, which was expressed in motor neurons, as shown in representative images of larval muscle 12 NMJs (A) This increase can be observed as a Ca2+-dependent increase in GCaMP3 fluorescence intensity compared to baseline (B) CASK β deletion non-significantly reduced this signal (C) (p > 0.05, n = 3, unpaired t-test). Application of heated (30°C) HL3.1 saline to acutely heat-activate the synaptic terminals of motor neurons expressing TrpA1 (OK371-Gal4>uas-TrpA1/uas-GCaMP3) caused a robust Ca2+ influx at muscle 12 NMJs (D) that could also be observed on a time trace showing maximum GCaMP3 intensity/baseline (E). This increase in Ca2+ signaling was reduced (p < 0.05, n = 3, unpaired t-test) in the CASK β null background (F). The increase in Ca2+ signaling during peristaltic crawling also occurred in the segmentally arranged motor neuron cell bodies in the ventral ganglion (G,H). (H) Reduction in CASK (OK371-Gal4>uas-CASK-RNAi/uas-GCaMP3) caused a reduction (p < 0.05, n = 6, One-Way ANOVA with a Bonferroni post-hoc test) in peak GCaMP3 fluorescence relative to baseline compared to control. Similarly, increased expression of T287D reduced peak Ca2+ levels (p < 0.05, n = 4). CASK co-overexpression (OK371-Gal4>uas-GCaMP3/uas-CASK;uas-CaMKII-T287D) partially rescued the decrease in peak Ca2+ level compared to larvae with increased expression of T287D alone (p < 0.05, n = 4), but this peak level was still lower than observed in controls (p < 0.05, n = 4). (I) No significant differences were observed in baseline GCaMP3 levels between genotypes (p > 0.05, n = 4–6, One-Way ANOVA with a Bonferroni post-hoc test). (J) To investigate the synaptic consequences of the reduction in Ca2+ in presynaptic terminals, we measured vesicle recycling and found that there was a significant reduction (p < 0.05, n = 3, unpaired t-test) in loading of FM1-43 in CASK β null larvae compared to controls.
The cell bodies of the motor neuron NMJs that were imaged in the previous set of experiments are located in the ventral ganglion, where they are arranged in a segmentally repeated pattern such that motor neurons present posteriorly project to muscles located in the most posterior segments. Imaging of the ventral ganglion showed sequential activation of motor neurons characterized by waves of activation that moved from posterior to anterior segments and represented fictive crawling (Figure 3G). This increase in motor neuron cell body Ca2+ was quantified (Figure 3H). Reduction of CASK in these motor neurons reduced peak Ca2+ signaling but resulted in no significant change at baseline (Figure 3I). Similarly, increased CaMKII-T287D in these neurons also produced a similar reduction in peak Ca2+ level, and this deficit was partially reversed by co-expression of CASK (Figure 3H). This finding is again consistent with CASK and CaMKII autophosphorylation acting in the same pathway that regulates both activity-dependent Ca2+ signaling and synaptic bouton morphology.
We were interested in the functional consequences of the reduction in synaptic Ca2+ signaling observed in the CASK β null background. Previous work has shown that CASK is involved in neurotransmitter release and vesicle cycling in Drosophila synapses (Sun et al., 2009) because a chromosomal deficiency of CASK reduces vesicle trafficking. To determine whether this reduction was due to the CaMK-like and L27 domain-containing version of CASK, we measured activity-dependent vesicle recycling by loading FM1-43 dye into vesicles at the NMJ of control and CASK β null larvae and stimulating the synapses with 90 mM KCl for 10 min. We found that removal of CASK β was sufficient to reduce vesicle trafficking (Figure 3J).
Downstream Mechanisms of Cask that Regulate Calcium Signaling
CASK knockdown has previously been shown to reduce the localization of CaV2.2 Ca2+ channels to the plasma membrane in mouse hippocampal primary cell culture (Samuels et al., 2007). We investigated whether CASK knockdown in Drosophila larvae reduced the localization of a GFP-tagged CaV2 Ca2+ channel [uas-Cacophony-eGFP, (Kawasaki et al., 2004)] to the NMJ and found no significant effect (Figure 4A). CaMKII-T287D expression has previously been shown to phosphorylate the scaffolding protein DLG, which reduces the localization of this protein to synapses (Koh et al., 1999). This loss of synaptic DLG reduces FasII localization and results in an increase in bouton number. However, quantification of mean bouton DLG (Figure 4B) or FasII (Figure 4C) antibody-specific staining intensity at the NMJ showed no effect of CASK β deletion.
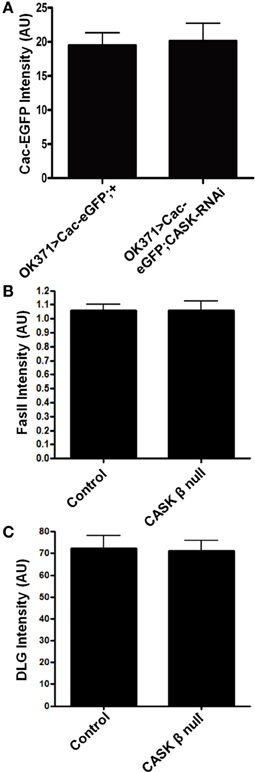
Figure 4. CASK does not regulate the localization of Cav2, FasII, or DLG to synapses. (A) CASK knockdown had no effect on the intensity of Cac-EGFP at the NMJ compared to control (p > 0.05, n = 6, unpaired t-test). (B) CASK β deletion did not significantly affect the intensity of FasII staining at the NMJ (p < 0.05, n = 6, unpaired t-test). (C) CASK β deletion did not significantly affect the intensity of DLG staining at the NMJ (p < 0.05, n = 6, unpaired t-test).
CaMKII-T287D is known to increase K+ conductance in larval Drosophila muscles and to cause an increase in action potential failures in motor neurons (Park et al., 2002). These effects may be mediated through CaMKII phosphorylation of the K+ channel EAG at T787, which increases EAG K+ current in oocytes (Wang et al., 2002). Mutation of this amino acid to a phospho-blocking residue also reduces localization of the channel to the plasma membrane (Marble et al., 2005). However, as CASK coexpression with EAG in oocytes increases EAG localization to the plasma membrane, this finding presents a divergence in the potential effects of CASK on EAG function; CASK may reduce EAG function via CaMKII inhibition or increasing EAG function via a direct interaction. To investigate whether CASK's regulation of EAG in Drosophila is involved with the decrease in Ca2+ signaling in motor neurons, NMJs were stained (Figure 5A) with an EAG-specific antibody (Sun et al., 2004). Compared to controls, there was an increase in EAG localization to the NMJ in CASK β null larvae (Figures 5B,C). This finding is consistent with the notion that CASK regulates Ca2+ signaling via an interaction with CaMKII to alter neuronal excitability via EAG. To explore whether this interpretation is correct, EAG-RNAi was co-expressed with CASK-RNAi to investigate whether EAG reduction would prevent the CASK knockdown-induced impairment of Ca2+ signaling. Compared to control, knock down of EAG was sufficient to rescue the effects of CASK knockdown (Figure 5D). This finding is consistent with the notion that CASK acts with EAG in a common pathway to regulate Ca2+ signaling.
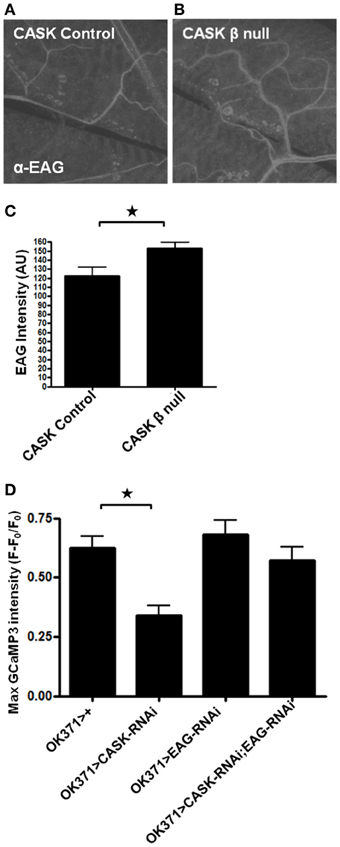
Figure 5. Signaling downstream of CASK in calcium signaling and synaptic growth. (A,B) Representative images of EAG staining at the NMJ in control and CASK β null larvae. (C) CASK β deletion significantly increased the intensity of EAG staining at the NMJ (p < 0.05, n = 6, unpaired t-test). (D) GCaMP3 imaging of the ventral ganglion during fictive crawling shows that the decrease in peak Ca2+ level induced by CASK knockdown was rescued by EAG knockdown. EAG knockdown alone did not significantly increase Ca2+ signaling (p > 0.05, n = 5–6, One-Way ANOVA with a Bonferroni post-hoc test).
Cask in the Mushroom Body is Required for Associative Memory Formation and Odor-Evoked Calcium Responses
These changes in activity-dependent neuronal Ca2+ signaling and synaptic morphology are consistent with a role of the CaMK-like and L27 domain-containing isoform of CASK in synaptic plasticity. To explore the functional consequence of CASK β-mediated changes in neural function, we tested CASK β null larvae for appetitive associative learning (Figure 6A) using adapted previously published protocols (Tully and Quinn, 1985; Chen et al., 2011). CASK β null larvae were deficient in the ability to associate an odor with a positive reward (fructose, Figure 6B). To determine which neurons mediated this deficit in memory caused by the loss of CASK function, we specifically knocked down CASK in the mushroom bodies using 201Y-Gal4 (Pauls et al., 2010). This Gal4 line allows for the expression of transgenes under uas control in the γ neurons of the larval mushroom body, the function of which is essential for larval appetitive learning (Pauls et al., 2010). Bidirectional changes in CASK expression in the γ neurons led to memory impairments (Figure 6C), indicating that the correct level of CASK in the mushroom body is required for memory formation. We expressed GCaMP3 under 201Y-Gal4 to investigate the responses of these olfactory memory neurons to odor application. CASK knockdown reduced odor-induced Ca2+ signaling (Figures 6D,E). The CASK mutant genotypes did not exhibit changes in odor acuity or the ability to sense fructose reward (Figure 7). This assay requires larvae to move toward these stimuli; thus, locomotor impairment does not prevent CASK mutant larvae from participating in this task.
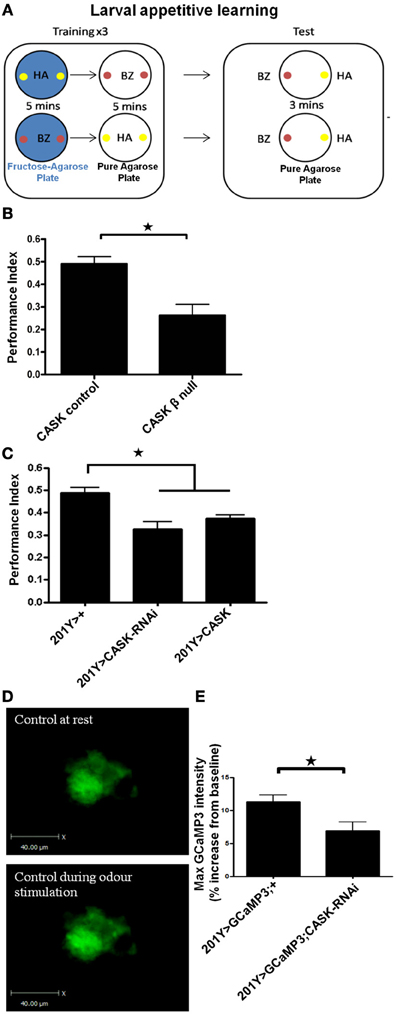
Figure 6. The CaMK-like and L27 domain-containing version of CASK is required for appetitive learning. (A) The role of CASK in associative memory was assessed by a larval olfactory-sugar reward conditioning protocol (Chen et al., 2011) described by the cartoon. The large rounded square on the left depicts the training phase of the protocol. Larvae were sequentially placed on petri dishes, represented by the large black circles, that contained either pure agarose (white filled circles) or 2 M fructose agarose (blue filled circles, the unconditioned stimulus, US). At the same time, the larvae were exposed to one of two neutral odors (the conditioned stimulus, CS). In the example given (top row) the larvae received 5 min of hexyl acetate on fructose-agarose (HA, small yellow filled circles, CS+) and then 5 min of benzaldehyde on agarose alone (BZ, small red filled circles, CS−). This training cycle was repeated 3 times before the test phase (depicted by the large rounded circle on the right) in which the larvae were allowed to move toward one of the two odors on pure agarose. The larvae showed learning by going toward HA (CS+). A performance index was calculated as the number of larvae that went to the CS+ minus the number of larvae that went to CS− divided by the total number of larvae. Bottom row, the odors representing the CS+ and CS− were swapped to produce a second performance index that was averaged with the reciprocal of the performance index to give an n of 1. (B) CASK β deletion caused a reduction (p < 0.05, n = 4, unpaired t-test) in learning compared to controls. (C) Compared to controls, learning was decreased (p < 0.05, n = 6, One-Way ANOVA with a Bonferroni post-hoc test) by both CASK knockdown and overexpression in the γ –lobe neurons of the mushroom body. (D) Representative images of GCaMP3 expressed in the γ –lobe neurons of the mushroom bodies at rest and during odor application. (E) Odor-evoked Ca2+ responses were reduced (p < 0.05, n = 6, unpaired t-test) by CASK knockdown in the mushroom body.
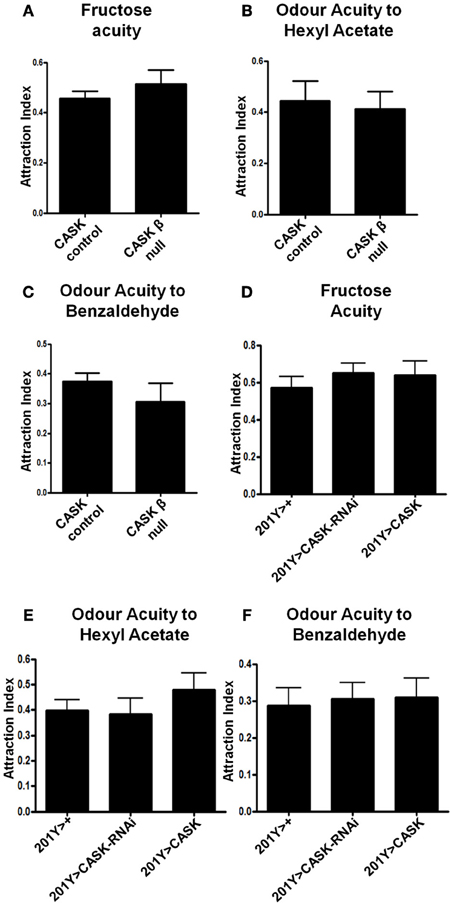
Figure 7. CASK does not regulate odor or fructose acuity. (A–C) CASK β deletion did not affect odor or fructose acuity compared to controls (p > 0.05, n = 3, unpaired t-test). (D–F) CASK knockdown or overexpression in the γ –lobe neurons of the mushroom body had no significant effects on odor or fructose acuity compared to controls (p > 0.05, n = 3, One Way ANOVA with a Bonferroni post-hoc test).
Human Cask Expression Rescues the Effect of Cask β Deletion on CaMKII Autophosphorylation
As the amino acids of Drosophila CASK and CaMKII are highly homologous with their human homologs [74 and 79%, respectively, (Cho et al., 1991; Hsueh, 2006)], it is possible that human CASK would also interact with CaMKII. To investigate if human CASK can regulate Drosophila CaMKII autophosphorylation, human CASK was expressed in Drosophila motor neurons in a CASK β null background. Immunohistochemistry was performed to examine changes in pT287 CaMKII at the NMJ and showed that the expression of human CASK rescued the dysregulation of CaMKII autophosphorylation induced by deletion of CASK β (Figures 8A–F).
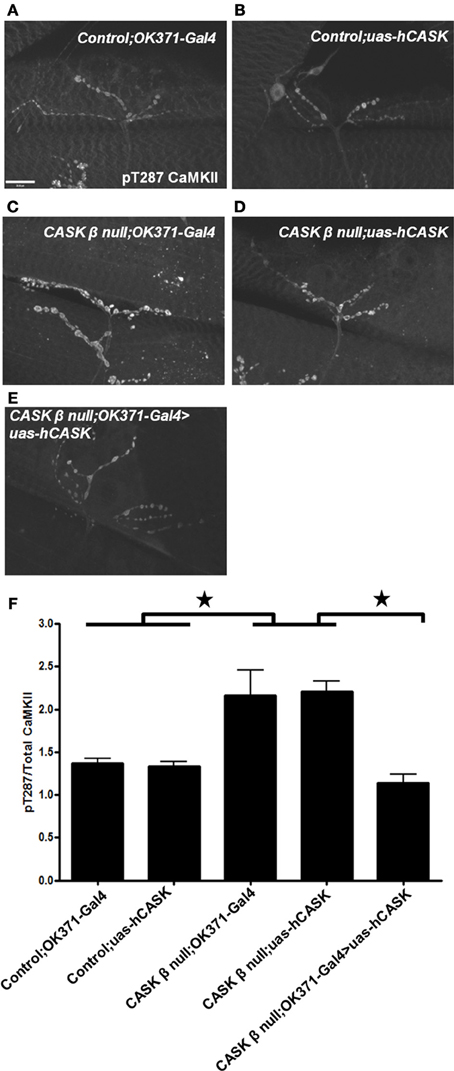
Figure 8. Human CASK expression rescues the effect of CASK β deletion on CaMKII autophosphorylation. Representation images of pT287 CaMKII-stained NMJs from (A) OK371-Gal4/+ control larvae (B) uas-human CASK/+ control larvae (C) CASK β null; OK371-Gal4 mutant larvae (D) CASK β null; uas-human CASK mutant larvae (E) CASK β null; OK371-Gal4>uas-human CASK rescue larvae. (F) CASK β null; uas-human CASK and CASK β null; OK371-Gal4 mutant larvae had significantly elevated pT287 CaMKII levels compared to uas-human CASK or OK371-Gal4 larvae (p < 0.05). Motor neuron expression of human CASK in a CASK β null background significantly reduced pT287 CaMKII levels to levels that were indistinguishable from wildtype (p > 0.05, n = 6, One Way ANOVA with a Bonferroni post-hoc test).
Discussion
CaMKII autophosphorylation is a central mechanism in synaptic plasticity and associative memory formation in mammals and Drosophila (Giese et al., 1998; Lisman et al., 2002; Park et al., 2002; Hardingham et al., 2003; Mehren and Griffith, 2004; Hodge et al., 2006; Sanhueza et al., 2011; Malik et al., 2013). Therefore, the finding that CASK can regulate CaMKII autophosphorylation suggests that this mechanism may have a role in the cognitive deficits induced by CASK mutation in humans (Froyen et al., 2007; Najm et al., 2008; Piluso et al., 2009; Tarpey et al., 2009). To explore this hypothesis, we investigated the physiological significance of the regulation of CaMKII autophosphorylation by CASK. We show for the first time that this interaction regulates synaptic growth, behaviorally induced Ca2+ signaling and appetitive learning. Furthermore, although it has previously been shown in Drosophila that CASK increases CaMKII TT306/7 phosphorylation to reduce T287 autophosphorylation (Lu et al., 2003; Hodge et al., 2006), we demonstrated that human CASK expression in Drosophila also regulates CaMKII autophosphorylation. These findings suggest that dysregulation of CaMKII autophosphorylation is potentially a mechanism that underlies neurological disorders resulting from CASK mutations.
Cask Regulates Synaptic Bouton Morphology and Rescues the Effects of CaMKII Overactivity
The role of CASK in regulating synaptic bouton growth at the NMJ has been studied, but the results are inconsistent. One study showed an increase in the total number of type 1 boutons at the NMJ of Drosophila larvae containing a large chromosomal deficiency that removed both CASK β and CASK α and produced heterozygous deletions of genes on either side of CASK (Sun et al., 2009). However, a second study that also used these CASK deficiency larvae showed that the total number of type 1 boutons did not change, although this study reported a reduction in the number of active zones (Chen and Featherstone, 2011). We have attempted to resolve these inconsistencies by studying larvae with a deletion that specifically removed the CaMK-like and L27 domain-containing isoform of CASK (Slawson et al., 2011). We show that deletion of the CASK β isoform alone caused an increase in type I synaptic bouton growth. Remodeling of presynaptic boutons is known to occur in the rat mossy fiber system after spatial learning, which suggests that control of presynaptic growth is likely to be important for the formation of long-lasting memories (Holahan et al., 2006). As we showed that CASK influences the control of synaptic growth by regulating CaMKII activity, this role of CASK may be a mechanism by which CASK dysfunction impairs learning and memory.
Cask Rescues the Effects of CaMKII Overactivity on Calcium Signaling
As Ca2+ signaling induced by neuronal activity is important in the control of synaptic strength (Unni et al., 2004), the reduction of Ca2+ signaling and synaptic growth resulting from CASK knockdown is consistent with the notion that CASK knock down causes deficits in synaptic plasticity and, hence, learning. These results suggest that CASK regulates Ca2+ signaling through CaMKII. At the Drosophila NMJ, CaMKII primarily decreases synaptic activity, as neuronal expression of CaMKII-T287D is known increase the probability of the failure of evoked responses (Park et al., 2002). Consistently, we found that CaMKII-T287D expression also reduced Ca2+ signaling. When both CASK and CaMKII-T287D were co-expressed, Ca2+ signaling was rescued compared to T287D expression alone, again suggesting that these molecules are interacting in a pathway that controls Ca2+ signaling. A similar reduction in GCaMP3 response has been observed with CASK knockdown or CaMKII-T287D expression in the adult mushroom body (Malik et al., 2013). As both reductions in CASK and CaMKII-T287D expression in motor neurons induce locomotor defects in larvae, the reduction of Ca2+ signaling induced by CASK knockdown or CaMKII-T287D expression may therefore, also be involved in this locomotor phenotype. We also showed that the reduction in presynaptic Ca2+ observed in the CASK β null terminals results in a reduction in activity-dependent vesicle trafficking. This finding is consistent with a role of CASK in control of synaptic vesicle release as suggested by studies in a range of other systems (Butz et al., 1998; Zordan et al., 2005; Hsueh, 2006; Spangler et al., 2013).
Cask is Required for Larval Appetitive Learning and Odor-Induced Calcium Signaling in the Mushroom Body
Using a larval appetitive olfactory learning assay, we demonstrated that CASK is required for associative learning and this function was localized to γ neurons in the mushroom body. This neuronal type is essential for larval appetitive learning (Pauls et al., 2010). We also showed that the Ca2+ signals induced by odor application are reduced by CASK knockdown in these neurons. As odor-induced Ca2+ signaling is likely to be critical for the induction of appetitive learning (Thum et al., 2007), this may be a mechanism by which CASK mutation impairs learning and memory.
One of the mechanisms through which CASK mutation may affect Ca2+ signaling is the regulation of EAG K+ channels. CaMKII and EAG act in a common pathway that is required for Drosophila synaptic and behavioral plasticity (Griffith et al., 1993, 1994). Overexpression of the EAG K+ channel BEC1 in the mouse forebrain impairs spatial working memory and tetanus-induced LTP, and the BEC1 heterozygous knockout exhibited significantly enhanced spatial working memory (Miyake et al., 2009). We provide evidence that CASK's regulation of EAG K+ channels, via CaMKII, regulates Ca2+ signaling, suggesting that this pathway may be important in learning and memory.
Expression of human CASK in the mushroom body has recently been shown to be able to completely restore the 3 h aversive memory deficit of CASK β null adults to wildtype levels (Malik et al., 2013). As human CASK was shown in the present study to regulate CaMKII autophosphorylation in Drosophila, this study, along with previous work, validates the use of Drosophila to study CASK and CaMKII in the healthy brain and in disease.
Mutations in human CASK can lead to a number of neurological diseases including FG syndrome 4, X-linked mental retardation with or without nystagmus and intellectual disability and microcephaly with pontine and cerebella hypoplasia (Froyen et al., 2007; Najm et al., 2008; Piluso et al., 2009; Tarpey et al., 2009). Therefore, investigating the mechanisms by which CaMKII autophosphorylation is regulated by CASK may be important for understanding the aetiologies of diseases involving these two proteins.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Hermann Aberle, Leslie Griffith, and Scott Waddell for flies and Dr. Vivian Budnik and Leslie Griffith for antibodies. Additional stocks were obtained from the Bloomington and Vienna RNAi stock centers. We acknowledge Drs. Michael Ashby, Matthias Landgraf, Brian McCabe, and Ralf Stanewsky for comments on the manuscript and the University of Bristol Wolfson bioimaging facility for the use of their microscopes. This work was supported by a Wellcome Trust 4-year Ph.D. studentship (Dynamic Cell Imaging) and BBSRC grants (BB/G008973/1 and BB/J017221/1).
References
Asahina, K., Louis, M., Piccinotti, S., and Vosshall, L. B. (2009). A circuit supporting concentration-invariant odor perception in Drosophila. J. Biol. 8, 9. doi: 10.1186/jbiol108
Ashraf, S. I., Mcloon, A. L., Sclarsic, S. M., and Kunes, S. (2006). Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell 124, 191–205. doi: 10.1016/j.cell.2005.12.017
Atasoy, D., Schoch, S., Ho, A., Nadasy, K. A., Liu, X., Zhang, W., et al. (2007). Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. U.S.A. 104, 2525–2530. doi: 10.1073/pnas.0611003104
Butz, S., Okamoto, M., and Sudhof, T. C. (1998). A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94, 773–782. doi: 10.1016/S0092-8674(00)81736-5
Cattaert, D., and Birman, S. (2001). Blockade of the central generator of locomotor rhythm by noncompetitive NMDA receptor antagonists in Drosophila larvae. J. Neurobiol. 48, 58–73. doi: 10.1002/neu.1042
Cavaliere, S., Gillespie, J. M., and Hodge, J. J. L. (2012). KCNQ channels show conserved ethanol block and function in ethanol behaviour. PLoS ONE 7:e50279. doi: 10.1371/journal.pone.0050279
Chen, K., and Featherstone, D. E. (2011). Pre and postsynaptic roles for Drosophila CASK. Mol. Cell. Neurosci. 48, 171–182. doi: 10.1016/j.mcn.2011.07.009
Chen, Y. C., Mishra, D., Schmitt, L., Schmuker, M., and Gerber, B. (2011). A behavioral odor similarity “space” in larval Drosophila. Chem. Senses 36, 237–249. doi: 10.1093/chemse/bjq123
Cho, K. O., Wall, J. B., Pugh, P. C., Ito, M., Mueller, S. A., and Kennedy, M. B. (1991). The alpha subunit of type II Ca2+/calmodulin-dependent protein kinase is highly conserved in Drosophila. Neuron 7, 439–450. doi: 10.1016/0896-6273(91)90296-C
Coultrap, S. J., Buard, I., Kulbe, J. R., Dell'acqua, M. L., and Bayer, K. U. (2010). CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J. Biol. Chem. 285, 17930–17937. doi: 10.1074/jbc.M109.069351
Donelson, N., Kim, E. Z., Slawson, J. B., Vecsey, C. G., Huber, R., and Griffith, L. C. (2012). High-resolution positional tracking for long-term analysis of Drosophila sleep and locomotion using the “tracker” program. PLoS ONE 7:e37250. doi: 10.1371/journal.pone.0037250
Elgersma, Y., Fedorov, N. B., Ikonen, S., Choi, E. S., Elgersma, M., Carvalho, O. M., et al. (2002). Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36, 493–505. doi: 10.1016/S0896-6273(02)01007-3
Froyen, G., Van Esch, H., Bauters, M., Hollanders, K., Frints, S. G., Vermeesch, J. R., et al. (2007). Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum. Mutat. 28, 1034–1042. doi: 10.1002/humu.20564
Giese, K. P., Fedorov, N. B., Filipkowski, R. K., and Silva, A. J. (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873. doi: 10.1126/science.279.5352.870
Griffith, L. C., Verselis, L. M., Aitken, K. M., Kyriacou, C. P., Danho, W., and Greenspan, R. J. (1993). Inhibition of calcium/calmodulin-dependent protein kinase in Drosophila disrupts behavioral plasticity. Neuron 10, 501–509. doi: 10.1016/0896-6273(93)90337-Q
Griffith, L. C., Wang, J., Zhong, Y., Wu, C. F., and Greenspan, R. J. (1994). Calcium/calmodulin-dependent pr-otein kinase II and potassium channel subunit eag similarly affect plasticity in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 91, 10044–10048. doi: 10.1073/pnas.91.21.10044
Hanson, P. I., and Schulman, H. (1992). Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analysed by site-directed mutagenesis. J. Biol. Chem. 267, 17216–17224.
Hardingham, N., Glazewski, S., Pakhotin, P., Mizuno, K., Chapman, P. F., Giese, K. P., et al. (2003). Neocortical long-term potentiation and experience-dependent synaptic plasticity require alpha-calcium/calmodulin-dependent protein kinase II autophosphorylation. J. Neurosci. 23, 4428–4436.
Hoang, B., and Chiba, A. (2001). Single-cell analysis of Drosophila larval neuromuscular synapses. Dev. Biol. 229, 55–70. doi: 10.1006/dbio.2000.9983
Hodge, J. J., Mullasseril, P., and Griffith, L. C. (2006). Activity-dependent gating of CaMKII autonomous activity by Drosophila CASK. Neuron 51, 327–337. doi: 10.1016/j.neuron.2006.06.020
Holahan, M. R., Rekart, J. L., Sandoval, J., and Routtenberg, A. (2006). Spatial learning induces presynaptic structural remodeling in the hippocampal mossy fiber system of two rat strains. Hippocampus 16, 560–570. doi: 10.1002/hipo.20185
Hsueh, Y. P. (2006). The role of the MAGUK protein CASK in neural development and synaptic function. Curr. Med. Chem. 13, 1915–1927. doi: 10.2174/092986706777585040
Jama, A. M., Fenton, J., Robertson, S. D., and Torok, K. (2009). Time-dependent autoinactivation of phospho-Thr286-alphaCa2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 284, 28146–28155. doi: 10.1074/jbc.M109.005900
Jan, L. Y., and Jan, Y. N. (1982). Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci. U.S.A. 79, 2700–2704. doi: 10.1073/pnas.79.8.2700
Kawasaki, F., Zou, B., Xu, X., and Ordway, R. W. (2004). Active zone localisation of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J. Neurosci. 24, 282–285. doi: 10.1523/JNEUROSCI.3553-03.2004
Koh, Y. H., Popova, E., Thomas, U., Griffith, L. C., and Budnik, V. (1999). Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell 98, 353–363. doi: 10.1016/S0092-8674(00)81964-9
Lisman, J., Schulman, H., and Cline, H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190. doi: 10.1038/nrn753
Lu, C. S., Hodge, J. J., Mehren, J., Sun, X. X., and Griffith, L. C. (2003). Regulation of the Ca2+/CaM-responsive pool of CaMKII by scaffold-dependent autophosphorylation. Neuron 40, 1185–1197. doi: 10.1016/S0896-6273(03)00786-4
Macleod, G. T., Marin, L., Charlton, M. P., and Atwood, H. L. (2004). Synaptic vesicles: test for a role in presynaptic calcium regulation. J. Neurosci. 24, 2496–2505. doi: 10.1523/JNEUROSCI.5372-03.2004
Mahr, A., and Aberle, H. (2006). The expression pattern of the Drosophila vesicular glutamate transporter: a marker protein for motoneurons and glutamatergic centers in the brain. Gene Expr. Patterns 6, 299–309. doi: 10.1016/j.modgep.2005.07.006
Malik, J. J., Gillespie, J. M., and Hodge, J. J. L. (2013). CASK and CaMKII function in the mushroom body α'/β' neurons during Drosophila memory formation. Front. Neural Cicuits 7:52. doi: 10.3389/fncir.2013.00052
Marble, D. D., Hegle, A. P., Snyder, E. D. 2nd., Dimitratos, S., Bryant, P. J., and Wilson, G. F. (2005). Camguk/CASK enhances Ether-a-go-go potassium current by a phosphorylation-dependent mechanism. J. Neurosci. 25, 4898–4907. doi: 10.1523/JNEUROSCI.4566-04.2005
Mehren, J. E., and Griffith, L. C. (2004). Calcium-independent calcium/calmodulin-dependent protein kinase II in the adult Drosophila CNS enhances the training of pheromonal cues. J. Neurosci. 24, 10584–10593. doi: 10.1523/JNEUROSCI.3560-04.2004
Miyake, A., Takahashi, S., Nakamura, Y., Inamura, K., Matsumoto, S., Mochizuki, S. et al. (2009). Disruption of the ether-a-go-go K+ channel gene BEC1/KCNH3 enhances cognitive function. J. Neurosci. 29, 14637–14645. doi: 10.1523/JNEUROSCI.0901-09.2009
Najm, J., Horn, D., Wimplinger, I., Golden, J. A., Chizhikov, V. V., Sudi, J., et al. (2008). Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 40, 1065–1067. doi: 10.1038/ng.194
Park, D., Coleman, M. J., Hodge, J. J., Budnik, V., and Griffith, L. C. (2002). Regulation of neuronal excitability in Drosophila by constitutively active CaMKII. J. Neurobiol. 52, 24–42. doi: 10.1002/neu.10066
Pauls, D., Selcho, M., Gendre, N., Stocker, R. F., and Thum, A. S. (2010). Drosophila larvae establish appetitive olfactory memories via mushroom body neurons of embryonic origin. J. Neurosci. 30, 10655–10666. doi: 10.1523/JNEUROSCI.1281-10.2010
Pi, H. J., Otmakhov, N., Lemelin, D., De Koninck, P., and Lisman, J. (2010). Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. J. Neurosci. 30, 8704–8709. doi: 10.1523/JNEUROSCI.0133-10.2010
Piluso, G., D'amico, F., Saccone, V., Bismuto, E., Rotundo, I. L., Di Domenico, M., et al. (2009). A missense mutation in CASK causes FG syndrome in an Italian family. Am. J. Hum. Genet. 84, 162–177. doi: 10.1016/j.ajhg.2008.12.018
Pulver, S. R., Pashkovski, S. L., Hornstein, N. J., Garrity, P. A., and Griffith, L. C. (2009). Temporal dynamics of neuronal activation by Channelrhodopsin-2 and TRPA1 determine behavioral output in Drosophila larvae. J. Neurophysiol. 101, 3075–3088. doi: 10.1152/jn.00071.2009
Ranganayakulu, G., Schulz, R. A., and Olson, E. N. (1996). Wingless signaling induces nautilus expression in the ventral mesoderm of the Drosophila embryo. Dev. Biol. 176, 143–148. doi: 10.1006/dbio.1996.9987
Samuels, B. A., Hsueh, Y. P., Shu, T., Liang, H., Tseng, H. C., Hong, C. J., et al. (2007). Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron 56, 823–837. doi: 10.1016/j.neuron.2007.09.035
Sanhueza, M., Fernandez-Villalobos, G., Stein, I. S., Kasumova, G., Zhang, P., Bayer, K. U., et al. (2011). Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J. Neurosci. 31, 9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011
Schuster, C. M., Davis, G. W., Fetter, R. D., and Goodman, C. S. (1996). Genetic dissection of structural and functional components of synaptic plasticity. II. Fasciclin II controls presynaptic structural plasticity. Neuron 17, 655–667. doi: 10.1016/S0896-6273(00)80198-1
Sherwood, N. T., Sun, Q., Xue, M., Zhang, B., and Zinn, K. (2004). Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2:e429. doi: 10.1371/journal.pbio.0020429
Slawson, J. B., Kuklin, E. A., Ejima, A., Mukherjee, K., Ostrovsky, L., and Griffith, L. C. (2011). Central regulation of locomotor behavior of Drosophila melanogaster depends on a CASK isoform containing CaMK-like and L27 domains. Genetics 187, 171–184. doi: 10.1534/genetics.110.123406
Spangler, S. A., Schmitz, S. K., Kevenaar, J. T., de Graaff, E., de Wit, H., Demmers, J., et al. (2013). Liprin-α2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J. Cell Biol. 201, 915–928. doi: 10.1083/jcb.201301011
Sun, M., Liu, L., Zeng, X., Xu, M., Fang, M., and Xie, W. (2009). Genetic interaction between Neurexin and CAKI/CMG is important for synaptic function in Drosophila neuromuscular junction. Neurosci. Res. 64, 362–371. doi: 10.1016/j.neures.2009.04.009
Sun, X. X., Hodge, J. J., Zhou, Y., Nguyen, M., and Griffith, L. C. (2004). The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 279, 10206–10214. doi: 10.1074/jbc.M310728200
Tarpey, P. S., Smith, R., Pleasance, E., Whibley, A., Edkins, S., Hardy, C., et al. (2009). A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543. doi: 10.1038/ng.367
Thum, A. S., Jenett, A., Ito, K., Heisenberg, M., and Tanimoto, H. (2007). Multiple memory traces for olfactory reward learning in Drosophila. J. Neurosci. 27, 11132–11138. doi: 10.1523/JNEUROSCI.2712-07.2007
Tian, L., Hires, S. A., Mao, T., Huber, D., Chiappe, M. E., Chalasani, S. H., et al. (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881. doi: 10.1038/nmeth.1398
Tully, T., and Quinn, W. G. (1985). Classical conditioning and retention in normal and mutant Drosophila melanogaster. J. Comp. Physiol. A 157, 263–277. doi: 10.1007/BF01350033
Unni, V. K., Zakharenko, S. S., Zablow, L., Decostanzo, A. J., and Siegelbaum, S. A. (2004). Calcium release from presynaptic ryanodine-sensitive stores is required for long-term depression at hippocampal CA3-CA3 pyramidal neuron synapses. J. Neurosci. 24, 9612–9622. doi: 10.1523/JNEUROSCI.5583-03.2004
Wang, Z., Wilson, G. F., and Griffith, L. C. (2002). Calcium/calmodulin-dependent protein kinase II phosphorylates and regulates the Drosophila eag potassium channel. J. Biol. Chem. 277, 24022–24029. doi: 10.1074/jbc.M201949200
Keywords: CASK, CaMKII, synaptic function, Drosophila, appetitive learning, calcium imaging, autophosphorylation
Citation: Gillespie JM and Hodge JJL (2013) CASK regulates CaMKII autophosphorylation in neuronal growth, calcium signaling, and learning. Front. Mol. Neurosci. 6:27. doi: 10.3389/fnmol.2013.00027
Received: 29 April 2013; Accepted: 24 August 2013;
Published online: 11 September 2013.
Edited by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelReviewed by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelHansen Wang, University of Toronto, Canada
Copyright © 2013 Gillespie and Hodge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James J. L. Hodge, School of Physiology and Pharmacology, University of Bristol, Medical Sciences Building, University Walk, Bristol, BS8 1TD, UK e-mail: james.hodge@bristol.ac.uk