
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 16 July 2012
Sec. Methods and Model Organisms
Volume 5 - 2012 | https://doi.org/10.3389/fnmol.2012.00080
 Norelle C. Wildburger1,2,3
Norelle C. Wildburger1,2,3 Fernanda Laezza1,4,5*
Fernanda Laezza1,4,5*Glycogen synthase kinase 3 (GSK-3) is an evolutionarily conserved multifaceted ubiquitous enzyme. In the central nervous system (CNS), GSK-3 acts through an intricate network of intracellular signaling pathways culminating in a highly divergent cascade of phosphorylations that control neuronal function during development and adulthood. Accumulated evidence indicates that altered levels of GSK-3 correlate with maladaptive plasticity of neuronal circuitries in psychiatric disorders, addictive behaviors, and neurodegenerative diseases, and pharmacological interventions known to limit GSK-3 can counteract some of these deficits. Thus, targeting the GSK-3 cascade for therapeutic interventions against this broad spectrum of brain diseases has raised a tremendous interest. Yet, the multitude of GSK-3 downstream effectors poses a substantial challenge in the development of selective and potent medications that could efficiently block or modulate the activity of this enzyme. Although the full range of GSK-3 molecular targets are far from resolved, exciting new evidence indicates that ion channels regulating excitability, neurotransmitter release, and synaptic transmission, which ultimately contribute to the mechanisms underling brain plasticity and higher level cognitive and emotional processing, are new promising targets of this enzyme. Here, we will revise this new emerging role of GSK-3 in controling the activity of voltage-gated Na+, K+, Ca2+ channels and ligand-gated glutamate receptors with the goal of highlighting new relevant endpoints of the neuronal GSK-3 cascade that could provide a platform for a better understanding of the mechanisms underlying the dysfunction of this kinase in the CNS and serve as a guidance for medication development against the broad range of GSK-3-linked human diseases.
Glycogen synthase kinase 3 (GSK-3) is a highly evolutionarily conserved multifaceted ubiquitous enzyme (Plyte et al., 1992; Kaidanovich-Beilin and Woodgett, 2011), which was first identified as a regulator of glycogen metabolism through phosphorylation and inactivation of glycogen synthase in skeletal muscle (Embi et al., 1980; Rylatt et al., 1980). Since then, GSK-3 has been identified in a wide spectrum of cellular processes, such as cell proliferation and differentiation (Frame and Cohen, 2001; Grimes and Jope, 2001a), cell survival (Takashima et al., 1993; Pap and Cooper, 1998), and cell motility (Wagner et al., 1997; Lucas et al., 1998; Sanchez et al., 2000a,b). GSK-3 is a proline-directed serine/threonine kinase, belonging to the CMCG [cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), CDK-like kinases and GSKs], that has been implicated as either a downstream or upstream effector in a number of intracellular signaling pathways and transcription factor activity, including but not limited to: Wnt/β-catenin (Dominguez et al., 1995; Cadigan and Nusse, 1997; Patapoutian and Reichardt, 2000; Seidensticker and Behrens, 2000; Chen et al., 2006; Ataman et al., 2008; Wan et al., 2012), brain-derived neurotrophic factor (BDNF) (Namekata et al., 2012), insulin (Garcia-Segura et al., 2010) and Hedgehog signaling (Jia et al., 2002; Price and Kalderon, 2002), and the translation initiation factor eIF-2B (Coghlan et al., 2000; Frame and Cohen, 2001; Doble and Woodgett, 2003).
The consensus sequence for GSK-3 kinase targets—at least 40 putative so far identified (Grimes and Jope, 2001b; Doble and Woodgett, 2003; Jope and Johnson, 2004)—typically consists of S/T-X-X-X-S/T in which the first S/T residue is the GSK-3 phosphorylation site and the downstream S/T residue is usually phosphorylated by casein kinase I or casein kinase II (Fiol et al., 1987). When phosphorylated, this downstream residue acts as a “priming” site enhancing phosphorylation by GSK-3 at the active site (Dajani et al., 2001). This unique property of GSK-3 suggests the existence of a hierarchical regulation of this enzyme prone to fine-tuning regulation of cell signaling through multiple kinase pathways (Fiol et al., 1987; Roach, 1990). GSK-3 is constitutively active in cells (Grimes and Jope, 2001a; Doble and Woodgett, 2003) and negatively regulated by the PI-3K/Akt pathway through phosphorylation of Ser-9 and Ser-21 (GSK-3β and GSK-3α, respectively) located at the N-terminus (Plyte et al., 1992; Cross et al., 1995; Grimes and Jope, 2001a). Conversely, phosphorylation at Tyr-216 (GSK-3β) or Tyr-279 (GSK-3α) serves as an activator (Hughes et al., 1993), possibly through autophosphorylation (Frame and Cohen, 2001). These positive and negative intramolecular mechanisms at the basis of the enzyme activity have been resolved through crystal structure resolution (Dajani et al., 2001) illustrated in Figure 1.
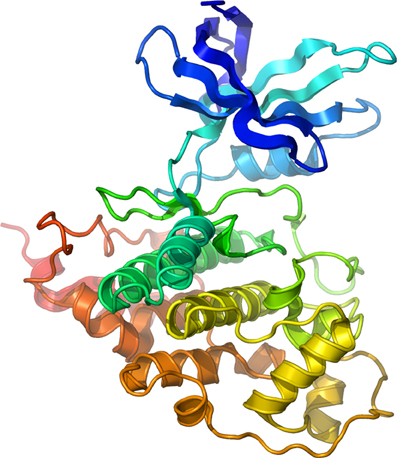
Figure 1. Structure of Human GSK-3β. Secondary structure model of human GSK-3β rainbow colored from the N-terminus (blue) to the C-terminus (red). Courtesy of Dr. Lawrence Pearl.
While GSK-3 is found in nearly all tissues (Woodgett, 1990), its highest expression and activity levels are in the brain (Leroy and Brion, 1999). Interestingly, in animal models over-expression of GSK-3 induces increased vulnerability to mood-related behavioral disturbances and impaired socialization behaviors (Mines et al., 2010; Polter et al., 2010), whereas GSK-3β haploinsufficiency leads to anti-depressant-like behavioral phenotypes (O'Brien et al., 2004; Kaidanovich-Beilin et al., 2009; Kaidanovich-Beilin and Woodgett, 2011). Furthermore, in clinical studies changes in the expression and activity of GSK-3 are found in schizophrenia (Kozlovsky et al., 2001, 2002; Jope, 2003; Lovestone et al., 2007; Emamian, 2012), mood disorders (Eldar-Finkelman, 2002; Jope, 2011), addictive behaviors (Miller et al., 2009, 2010) and Alzheimer's disease (Balaraman et al., 2006; Hooper et al., 2008; Kremer et al., 2011), indicating a prominent role of this enzyme for higher level cognitive and emotional processing. Yet, a complete picture of GSK-3 activity and of its molecular targets in the CNS is still unresolved. Here, we will discuss the emerging role of GSK-3 in the regulation of neuronal ion channel function and its implications for intrinsic excitability, synaptic transmission, and neuronal plasticity as a step forward to the comprehension of GSK-3 activity in the human brain. We hope our effort will provide a new template to identify relevant endpoints of the neuronal GSK-3 cascade that could help elucidate disease mechanisms and serve as optimal targets for drug development against the broad spectrum of neurological and psychiatric disorders linked to GSK-3 dysfunction.
Of the two mammalian GSK-3 isoforms, GSK-3α and GSK-3β (Woodgett, 1990; Boyle et al., 1991), GSK-3β is the most abundant in the brain. Recent studies indicate the existence of two splice variants of GSK-3β, which generate a short form, GSK-3β1, and a long form containing an additional 13 amino acids in the catalytic domain, GSK-3β2 (Mukai et al., 2002). Both of these isoforms are highly expressed in the CNS during development and adulthood, but GSK-3β1 remains the dominant splice variant; GSK-3β2 is more abundant in the brain compared to other tissues like liver, heart, and skeletal muscle, but is expressed at lower levels than GSK-3β1, and its expression tends to decrease after birth (Mukai et al., 2002). At the cellular level, the two different splice variants of GSK-3β show a distinct pattern distribution with GSK-3β1 predominantly in the cell body and processes and GSK-3β2 primarily in the soma, possibly hinting at isoform-specific functions (Mukai et al., 2002), though a more recent study suggests that both splice variant are found in the soma and processes (Wood-Kaczmar et al., 2009). Even so, the existence of two splice variant of GSK-3β raises the question of the differential functions. Lastly, mechanisms that control the expression levels of GSK-3 itself are of great interest, as they could be part of homeostatic loops, which, if abrogated, could lead to the insurgence of the psychiatric and neurological disorders discussed above. Among potential mechanisms, activity-regulated Wnt signaling is a strong candidate (Chen et al., 2006; Tang, 2007; Ataman et al., 2008; Wan et al., 2012).
In the next section, we will review what is known about the direct or indirect effects of GSK-3 on voltage-gated ion channels, and discuss the potential implications of these findings for intrinsic excitability and neurotransmitter release.
Nav channels are heteromeric transmembrane proteins consisting of a pore-forming α-subunit (Nav1.1–Nav1.9 and Nax) and accessory β-subunits (β1−β4) (Catterall et al., 2005). These channels are activated by membrane depolarization giving rise to action potentials in neurons and other excitable cells, and thus play a critical role in synaptic transmission, signal integration, and neuronal plasticity. The intracellular portion of Nav channels contains multiple phosphorylation motifs known to regulate the channel biophysical properties, its subcellular distribution, and trafficking (Shao et al., 2009; Baek et al., 2011). Some of the target kinases for these sites include protein kinase A (PKA) (Zhou et al., 2002), protein kinase C (PKC) (Vijayaragavan et al., 2004), and Ca2+/calmodulin kinase II (CaMKII) (Deschenes et al., 2002).
A series of studies on the Nav1.7 isoform indicate that the Nav channel is regulated by GSK-3. In bovine adrenal chromaffin cells, treatment with lithium, a therapeutic agent that inhibits GSK-3 activity (Beaulieu et al., 2009; Hernandez et al., 2009), increases the cell surface [3H] saxitoxin binding of Nav1.7 and augments veratridine-induced Na+ currents by a mechanism that is prevented by more specific GSK-3 inhibitors such as SB216763 and SB415286 (Coghlan et al., 2000; Yanagita et al., 2009), although some additional GSK-3-independent mechanisms might also be mediating the effects of lithium (Yanagita et al., 2007). Likewise, stimulation of the insulin pathway, which is an upstream negative regulator of GSK-3, increases surface expression of Nav1.7 channels in the same cell type, supposedly through activation of the PI-3K/Akt pathway and subsequent decrease in active GSK-3 levels (Yamamoto et al., 1996; Nemoto et al., 2009). Overall, these data suggest that both constitutive and regulated GSK-3 might exert control on the surface level expression of Nav channels (Yamamoto et al., 1996; Yanagita et al., 2009). Regulation of the channel trafficking might be one underlying mechanism of the GSK-3 pathway, but more global effects on the Nav α-subunit mRNA expression level may also account for some of the observed phenotypes (Wada et al., 2005; Yanagita et al., 2009), as treatment with lithium or insulin increases Nav α-subunit mRNA gene transcription (Yamamoto et al., 1996; Yanagita et al., 2009). Whether these results are reproducible in the CNS and to what extent they can be extended to other Nav channel isoforms should be investigated.
Interestingly, although these studies suggest Nav channel regulation by GSK-3, to the best of our knowledge, no consensus sites for GSK-3 have been reported for any Nav α subunits or β accessory subunits (Berendt et al., 2010; Scheuer, 2011), suggesting indirect Nav channel regulation by GSK-3. The protein–protein interaction network that composes the macromolecular complex of Nav channels in the CNS is rich in key determinants for Nav channel function and excitability (Shao et al., 2009). The intracellular fibroblast growth factor 14 (FGF14), a member of the intracellular FGF family (iFGF) (Itoh and Ornitz, 2008), is a critical accessory protein of the Nav channel that binds to the intracellular C-tail of the Nav α subunit in an Nav-isoform specific manner, and controls biophysical properties and channel sub-cellular targeting to the axonal initial segment (AIS) (Laezza et al., 2007, 2009; Shavkunov et al., 2012). Loss of FGF14 function down-regulates Na+ currents, reduces expression of Nav channels at the AIS, and impairs excitability in the hippocampus and cerebellum (Goldfarb et al., 2007; Laezza et al., 2007), indicating that, under normal conditions, FGF14 is required for proper activity of Nav channels acting as a multivalent stimulator of excitability. Through a chemical screening of kinase inhibitors, we recently identified the FGF14:Nav1.6 C-tail complex as a potential new GSK-3 target (Laezza et al., 2011). In these studies, GSK-3 inhibitors reduce the association of the FGF14:Nav1.6 C-tail complex in heterologous cells, (Laezza et al., 2011). If this mechanism occurs in neurons, then by controling the assembly of the FGF14 with Nav channels, active GSK-3 might stimulate intrinsic excitability. Notably, the primary pharmacotherapeutic strategy in bipolar disorder consists of limiting neuronal hyperexcitability by blocking Nav channels with anticonvulsants and reducing the activity of GSK-3 with lithium (Rogawski and Loscher, 2004; Rowe et al., 2007). Whether the Nav channel macromolecular complex is a relevant end point of the GSK-3 cascade and a common molecular target of mood stabilizers (Harwood and Agam, 2003; Bazinet, 2009) would be an attractive hypothesis to test.
The Kv channel family includes the most heterogeneous and abundant group of ion channels in excitable cells, comprising more than 40 subunit genes divided into separate families based on structural and functional properties (Gutman et al., 2003). A functional channel typically requires association of four α subunits, usually within the same family, and may include β subunits or other accessory proteins that regulate channel trafficking and biophysical properties (Norris et al., 2010; Vacher and Trimmer, 2011). Kv channels are critical players in the repolarization phase of the action potential, controling neuronal firing patterns (Pongs, 1999), and setting the resting membrane potential. These channels also contribute to the action potential back-propagation with broad implications for dendritic signal integration and synaptic plasticity (Johnston et al., 2003; Thompson, 2007). Kv channels and their accessory proteins are directly phosphorylated by a number of kinases which affect the channel biophysical properties, trafficking, and subcellular targeting (Cerda and Trimmer, 2010; Baek et al., 2011; Cerda et al., 2011; Cerda and Trimmer, 2011).
Interestingly, of all different Kv channel subtypes, the Kv channel encoded by the gene KCNQ2 (Singh et al., 1998), which corresponds to Kv7.2 (Cooper, 2010), is the only one so far identified as a GSK-3β substrate (though GSK-3α cannot be discounted). KCNQ2 mediates M-currents (Jentsch, 2000), which exert an overall dampening effect on excitability by promoting firing accommodation (Otto et al., 2006; Kapfhamer et al., 2010). Loss-of function or dominant negative mutations in the KCNQ2 gene are found in epilepsy and bipolar disorder (Singh et al., 1998; Cooper et al., 2000; Mulley et al., 2003), and inhibition of M-channel activity has been linked to schizophrenia (Fedorenko et al., 2008), suggesting that neuronal hyperexcitability resulting from impaired M-channel function may be a common denominator in a number of neurological and psychiatric illnesses in which GSK-3 activity is dysfunctional (Li et al., 2002). Recent studies have shown that both the KCNQ2 channel mutants—associated with bipolar disorder—and wild-type KCNQ2 channels are phosphorylated by GSK-3β in vitro, at a site that requires PKA priming and is dephosphorylated by protein phosphatase 2A (PP2A; also known as calcineurin) (Borsotto et al., 2007). Phosphorylation of the KCNQ2 channel typically results in inhibition of the channel activity (Schroeder et al., 1998; Hoshi et al., 2003) and expression of PP2A increases KCNQ2-mediated current (Borsotto et al., 2007), suggesting that GSK-3 might be part of a regulatory mechanism that suppress M-currents. At cellular level, GSK-3β and KCNQ2 colocalize in the apical dendrites of pyramidal neurons in the medial prefrontal cortex (mPFC) (Kapfhamer et al., 2010). Furthermore, pharmacological inhibition of GSK-3 in the mPFC mimics and occludes the effect of the M-channel inhibitor linopirdine, resulting in sensorimotor disinhibition (measured by prepulse inhibition), reduced M-type-dependent firing accommodation, and an overall increase in excitability (Kapfhamer et al., 2010). In both bipolar disorder and schizophrenia sensorimotor gating is often impaired (Braff et al., 1995), and inhibition of KCNQ2 channels induced by GSK-3-dependent phosphorylation might be a potential mechanism underlying hyperexcitability and circuit dysfunction that accompany some of the clinical symptoms observed in these psychiatric disorders (Sharp and Hendren, 2007). Given the high degree of homology between different Kv channels and their complex and heterogeneous role in excitability, determining whether other Kv channels are GSK-3 targets should be a priority.
Cav channels are composed of four or five distinct subunits, which are encoded by multiple genes (Catterall, 2000). These include the Cav1 subfamily (Cav1.1–1.4), which mediates L-type Ca2+ currents, the Cav2 subfamily (Cav2.1–2.3), which mediates P/Q-, N-, and R-type Ca2+ currents, respectively, and the Cav3 subfamily (Cav3.1–Cav3.3), which mediates T-type Ca2+ currents (Carr et al., 2003). Found in all excitable cells, Cav channels are closed at resting membrane potential and opened by depolarizing potentials, allowing Ca2+ influx into the cell, which serves as a potent and highly versatile intracellular signaling molecule both pre- and postsynaptically (Catterall, 2011). Cav channels are highly regulated by phosphorylation (Jiang et al., 2008) both directly and indirectly through their associated multiprotein complexes (Catterall, 2010). Ca2+ flux through Cav channels regulates neurotransmitter release presynaptically (Sudhof, 2012), signal transduction (Xie, 2004), and contribute to the induction of synaptic plasticity (Kochlamazashvili et al., 2010).
Evidence exists in the hippocampus for direct GSK-3β phosphorylation of the intracellular loop-connecting domains II and III (LII−III) of the P/Q-type Ca2+ channels at a threonine site (predicted Thr915 residue) of the synprint site (Zhu et al., 2010). The synprint site on the P/Q and N-type channels mediates the binding to the soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) complex, a protein complex required for neurotransmitter release (Kim and Catterall, 1997). Neurotransmitter release occurs through a series of steps including: (1) binding of the synprint sites on N-type or P/Q-type Ca2+ channels with the presynaptic membrane proteins synaptotagmin and t-SNARE (i.e., SNAP25 and syntaxin); (2) dissociation of synaptobrevin (Syb) (also called VAMP2, vesicle associated membrane protein 2), a vesicular SNARE protein responsible for synaptic vesicle fusion, from synaptophysin I (SypI); (3) association of Syb with t-SNARE (Pennuto et al., 2003). All of these molecular events are tightly regulated by intracellular Ca2+ concentration ([Ca2+]i) and by the phosphorylation of the synprint sites on N-type and P/Q-type Cav channels (Yokoyama et al., 1997). By phosphorylation of the synprint site, GSK-3 decreases Ca2+ currents and Ca2+ influx in hippocampal neurons (Zhu et al., 2010), preventing the channel's association with SNAP25, syntaxin (Kim and Catterall, 1997; Yokoyama et al., 1997, 2005), and synaptotagmin blocking presynaptic vesicle release (Zhu et al., 2010). This effect is remarkably similar to previously reported Cdk5/p25-dependent phosphorylation of the same II and III (LII−III) loop of P/Q-type Cav channels, which also resulted in inhibition of the interaction of the channel with the SNARE complex required for neurotransmitter release (Tomizawa et al., 2002). Whether competition or convergence exist between these two kinases on Cav channels remains to be determined.
In addition to the effects on Cav channels, a number of reports have indicated a role of GSK-3 in presynaptic vesicle recycling and in the regulation of the expression of other relevant presynaptic proteins (Smillie and Cousin, 2011). GSK-3β phosphorylation of Ser-774 at the C-terminal proline-rich domain (PRD) of dynamin I (Clayton et al., 2010), a protein involved in vesicle endocytosis (Newton et al., 2006; Clayton et al., 2009; Zhu et al., 2009), is required during activity-dependent bulk endocytosis (ADBE) (Clayton et al., 2008, 2010). Furthermore, in hippocampal neurons activation of GSK-3β decreases presynaptic glutamate release following high-frequency stimulation and diminishes the expression of synapsin I (SynI) (Zhu et al., 2007), another critical component of the SNARE complex that regulates synaptic vesicle mobilization (Nichols et al., 1992; Rosahl et al., 1995; Terada et al., 1999; Chi et al., 2001), and is upregulated during long-term potentiation (LTP) (Sato et al., 2000). Pre-treatment with lithium or other GSK-3 inhibitors opposes this effect (Welch et al., 2007). The cadherin/β-catenin complex also has been shown to play a role in the clustering of synaptic vesicles at synapses (Bamji et al., 2003) and β-catenin itself is a phosphorylation substrate of GSK-3β (Hinoi et al., 2000; Liu et al., 2002). Thus, through a concerted modulation of protein–protein interactions at the level of Cav channels and SNARE complex, the GSK-3 signaling pathway controls presynaptic transmitter release exerting an overall suppressive effect on synaptic vesicle fusion and neurotransmitter release.
Presynaptic Cav channels include also the N-type (Cav2.2) which similarly to the P/Q channels impact transmitter release (Sudhof, 2012) through phosphorylation-dependent binding to the SNARE complex (Yokoyama et al., 1997). A novel component of the Cav2.2 macromolecular complex is the collapsin response mediator protein 2 (CRMP-2), a member of a family of five proteins implicated in signal transduction, axonal growth, and guidance (Goshima et al., 1999; Uchida et al., 2005). Cav2.2 associates with CRMP-2 at both the I–II intracellular loop and the distal C-terminus in the presynaptic terminals of dorsal root ganglion (DRG) and hippocampal neurons (Brittain et al., 2009; Chi et al., 2009; Wang et al., 2010). Functional coupling of synaptic CRMP-2 with Cav2.2 is enhanced by depolarization and overexpression of CRMP-2 increases surface expression of Cav2.2, leading to up-regulation of presynaptic Ca2+ flux, augmented glutamate release, and increase in synapse size (Chi et al., 2009; Wang et al., 2010). The molecular machinery leading to these phenotypes includes the interaction of CRMP-2 with the tubulin-heterodimer and stimulation of microtubule assembly (Fukata et al., 2002; Schmidt and Strittmatter, 2007). Upon extracellular activation of the semaphorin3A/Neuropilin-1/PlexinA receptors pathway, CRMP-2 is sequentially phosphorylated by Cdk5 and GSK-3 (Uchida et al., 2005). Phosphorylated CRMPs have a reduced affinity for tubulin and, as such, they lose their stimulatory effect on axon elongation, promoting growth cone collapse (Arimura et al., 2000, 2005). Although it is still unclear whether Cav2.2 binds to the phosphorylated or non-phosphorylated state of CRMP-2, one might speculate that, like tubulin, Cav2.2 binds to the non-phosphorylated, active form of CRMP-2 (Wang et al., 2010). A potential working model could be that GSK-3 phosphorylation of CRMP-2 through the semaphoring 3A signaling could serve as a detector of presynaptic inactivity (reduction in Ca2+ flux and presynaptic release) leading to suppression of axonal growth with critical implications for plasticity, Wnt-dependent control of neuronal polarity (Kim and Snider, 2011; Nishiyama et al., 2011; Shelly et al., 2011; Yang and Luo, 2011; Yamashita et al., 2012; Yamashita and Goshima, 2012), with functional consequences for schizophrenia (Singh et al., 2011) and neurodegeneration (Williamson et al., 2011). A simple working model of the role of GSK-3 at presynaptic terminals is depicted in Figure 2.
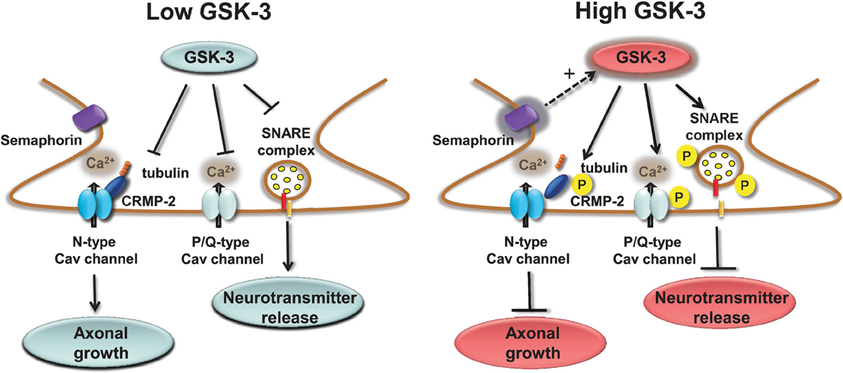
Figure 2. Regulation of presynaptic Cav channels by GSK-3. Under low GSK-3 conditions the interaction of P/Q Cav channels with the SNARE complex is favored and Ca2+ flux and neurotransmitter release are enhanced; likewise, N-type Cav channel interaction with CPMP-2 is stabilized and axonal growth is stimulated. Opposite effects are expected upon high levels of GSK-3 induced, for example, by stimulation of semaphorin 3A signaling pathway.
Interesting results indicate that α1C-subunit of Cav1.2b channels, which mediate L-type currents, associates with and is a substrate of GSK-3β in colonic smooth muscle cells (Li and Sarna, 2011). This complex is downstream of the norepinephrine-mediated signaling pathway. Stimulation of norepinephrine inactivates GSK-3β via the PI-3K/Akt pathway, decreasing phosphorylation of the α1C-subunit, and concurrently enhances α1C-protein translation and blocks its polyubiquitination and proteasomal degradation (Li and Sarna, 2011). Whether these data are applicable to the L-type channels in the CNS it remains to be determined. Finally, modulation of T- and R-type calcium channels either directly or indirectly by GSK-3 remains unknown and presently to the best of our knowledge, an unexplored possibility.
Glutamate receptors, broadly divided into two ionotropic and metabotropic families, mediate fast and slow G-protein coupled mediated signaling, respectively. Ionotropic glutamate receptors are the most abundant ligand-gated ion channels in the CNS, mediate virtually all excitatory neuronal transmission and play critical roles for induction and expression of activity-dependent synaptic remodeling and neuronal plasticity. Here, we will discuss what is currently known on GSK-3 phosphorylation of ligand-gated glutamate receptors.
The α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors are heterotetramers formed by the heterologous combination of GluA1-GluA4 subunits. These subunits determine ionic permeability and contribute to the biophysical properties of the channel (Nakagawa, 2010; Traynelis et al., 2010), its trafficking routes (Henley et al., 2011), and dictate its roles in induction and expression of synaptic plasticity (Shi et al., 2001). Upon binding to glutamate, Na+ and/or Ca2+ permeability of the AMPA receptors increases, leading to an excitatory postsynaptic potential. If AMPA receptors are GluA2-lacking, the channels are permeable to Ca2+ and can participate in the induction of LTP and long-term depression (LTD) in both principal cells (Plant et al., 2006; Rozov et al., 2012) and interneurons (Laezza et al., 1999; Laezza and Dingledine, 2004, 2011). In hippocampal pyramidal neurons most of the functional AMPA receptors, though, contain the GluA2 subunit, either combined with GluA1 or in a small percentage associated with GluA3 (Lu et al., 2009). In CA1 pyramidal neurons evidence exists, although controversial (Adesnik and Nicoll, 2007), for a GluA1 homomeric species that appears to be inserted into the membrane following LTP induction (Plant et al., 2006). The GluA1/GluA2 receptors are delivered and inserted into the plasma membrane by activity-dependent mechanisms that require protein-protein interactions at the GluA1 C-terminus, whereas the GluA3/GluA2 heterodimer is part of a constitutive pathway controlled by proteins binding to the C-terminus of GluA2 (Shi et al., 2001). Extensive studies have addressed the role of these different heterodimers or of GluA1 homomers (Plant et al., 2006) in the expression mechanisms of LTP and LTD (Adesnik and Nicoll, 2007; Shepherd and Huganir, 2007; Kessels and Malinow, 2009).
Early studies showed that in vivo chronic treatment with lithium decreased surface expression of GluA1 in the rat hippocampus (Du et al., 2004). The mechanism appears mediated by a decrease in GluR1 phosphorylation at a specific PKA site (GluR1p845), which is responsible for GluA1 insertion into the plasma membrane (Lee et al., 2000; Malinow and Malenka, 2002) and controls the channel open probability (Banke et al., 2000). More recent studies have shown that active GSK-3 forms a complex with both GluA1 and GluA2 subunits in the CA1 region of the hippocampus, as determined by reciprocal co-immunoprecipitation and that this interaction is relevant for synaptic plasticity (Peineau et al., 2007; Bradley et al., 2012). In these studies the activity of GSK-3 was enhanced during LTD via activation of PP1. Conversely, following chemically-induced LTP (Lu et al., 2001; Man et al., 2003) the association of AMPA receptors (GluA1 and GluA2) with GSK-3 is reduced (Peineau et al., 2007; Bradley et al., 2012), suggesting a GSK-3-dependent pathway that controls AMPA receptor surface levels during LTD (Peineau et al., 2008; Bradley et al., 2012).
The molecular mechanisms underlying GSK-3-dependent trafficking of AMPA receptors have been addressed in other studies. Pharmacological inhibition or knockdown of GSK-3 in cortical neurons decrease AMPA receptor-mediated mEPSC amplitude and occlude the effect of insulin, a known upstream negative effector of GSK-3, through an effect on AMPA receptor trafficking (Wei et al., 2010). The decrease in mEPSCs amplitude that accompanies GSK-3 inhibition correlates with a loss of surface GluA1 and GluA2 resulting in a marked decrease in the number and size of PSD95 positive synaptic clusters (Wei et al., 2010). The molecular machinery underlying GSK-3-dependent trafficking of AMPA receptor includes the guanyl nucleotide dissociation inhibitor (GDI):Rab5 complex (Zerial and McBride, 2001; Huang et al., 2004). If GSK-3 phosphorylation at S45 is impaired, GDI loses its affinity for Rab5, one of the small GTPase that controls receptor trafficking from the plasma membrane to early endosomes (Brown et al., 2005). When free from GDI, Rab5 can recruit surface GluA1/GluA2 complexes into early endosomes, promoting AMPA receptor internalization and leading to decreased amplitude of mEPSCs (Wei et al., 2010), as illustrated in Figure 3. Stimulation of AMPA receptors by bath application of agonists attenuates GSK-3 activity (Nishimoto et al., 2008) in cortical neurons, suggesting that GSK-3 might be part of a regulatory loop that controls internalization of AMPA receptors in response to synaptic activity. Overall, these results indicate the role of both constitutive and regulated (insulin) GSK-3 activity in the maintenance of synaptic AMPA receptors (Wei et al., 2010). Given that the complex endocytic machinery regulating AMPA receptor trafficking is one of the key postsynaptic mechanisms underlying activity-dependent synaptic plasticity (Carroll et al., 1999, 2001; Luscher et al., 1999; Beattie et al., 2000), these studies open new avenues and broaden the role of this kinase to critical postsynaptic functions.
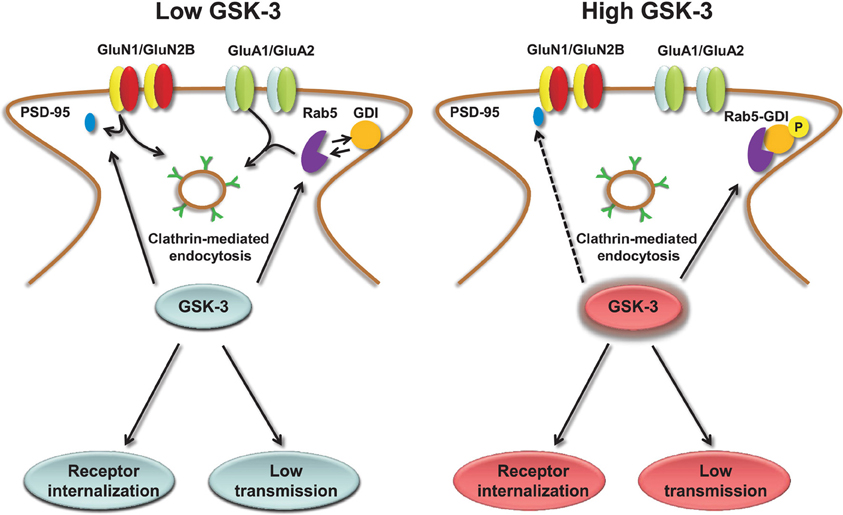
Figure 3. Regulation of AMPA and NMDA receptors by GSK-3. Under low levels of GSK-3, internalization of AMPA and GluN2B-containing NMDA receptors through clathrin-mediated endocytosis is facilitated. GSK-3-dependent AMPA receptor internalization requires dissociation of the GDI:Rab5 complex, induced by GSK-3 phosphorylation of GDI at S45; GSK-3-dependent NMDA receptor internalization is limited to GluN2B containing receptors and requires dissociation of PSD-95 from the GluN2B C-terminal tail through a series of steps that are induced by GSK-3 inhibitors. Conversely, under high levels of GSK-3, AMPA and NMDA receptors are highly expressed at the plasma membrane, and receptor internalization is prevented.
N-methyl-D-aspartate (NMDA) receptors are ligand-gated glutamate receptors that mediate fast synaptic transmission in the brain. The NMDA receptor's unique properties as a ligand-gated and voltage-sensitive receptor modulated by extracellular Mg2+ enables the channel to act as a coincidence detector of high-frequency signals that trigger plasticity at glutamatergic synapses in development and during adulthood (Seeburg et al., 1995). NMDA receptors are formed by the assembly of different subunits including GluN1 subunit, four different GluN2 subunits (GluN2A–D), and two GluN3 subunits (GluN3A, B) (Traynelis et al., 2010). A functional NMDA receptor requires two GluN1 subunits and two GluN2 subunits, which are activated by simultaneous binding of glycine and glutamate to GluN1 and GluN2 subunits (Hedegaard et al., 2011). The four GluN2 subunits play different roles during neuronal development and in synaptic plasticity (Liu et al., 2004; Yang et al., 2011). For example, the GluN2A-containing NMDA receptors mediate Ca2+ currents leading to LTP, while GluN2B-containing receptors are prominently expressed during development and critical for LTD induction (Liu et al., 2004).
The earliest studies indicating a role of GSK-3 in regulating NMDA receptors showed that pharmacological inhibition or silencing of GSK-3 causes a long-lasting reduction of NMDA receptor-mediated ionic synaptic current in cortical pyramidal neurons with no effects on the receptor desensitization (Chen et al., 2007). In the same studies GSK-3 inhibitors were shown to mimic and occlude the effect of insulin, indicating that both constitutive and regulated GSK-3 activity controls NMDA receptor (Chen et al., 2007). Silencing of GSK-3 in neuronal cultures reduces NMDA receptor currents and prevents its regulation by GSK-3 inhibitors (Chen et al., 2007).
Similarly to AMPA receptors, the down-regulation of NMDA receptor-mediated currents is mediated by increased Rab5-dependent internalization (Chen et al., 2007) through a mechanism that requires disruption of GluN2B interaction with the scaffolding PDZ (Luscher et al., 2000) domain protein PSD95 and depends upon clathrin/dynamin-dependent endocytosis (Chen et al., 2007). Inhibition of dynamin (the GTPase responsible for “pinching” the vesicle off of the plasma membrane) prevents endocytosis and occludes the effect of GSK-3 inhibitors on surface NMDA receptors (Chen et al., 2007). The effect of GSK-3 appears specific for GluN1/GluN2B versus GluN1/GluN2A receptors, and does not involve any other intracellular transport mechanisms that are involved in NMDA receptor trafficking, including the ones mediated by kinesins (Setou et al., 2000; Morfini et al., 2002), microtubules (Yuen et al., 2005a,b; Zhou and Snider, 2005) or by F-actin (Eickholt et al., 2002), suggesting that GSK-3 regulates fairly specific pools of NMDA receptors (Chen et al., 2007). A simple scheme of the role of GSK-3 in regulating NMDA receptors is shown in Figure 3.
In other studies treatment of cortical neurons with AMPA was shown to decrease cell surface NMDA receptors in a GSK-3 activity-dependent manner, and to reduce glutamate-induced intracellular Ca2+ influx (Nishimoto et al., 2009), suggesting a functional cross talk between AMPA and NMDA receptor activity through GSK-3. This mechanism may contribute to neuroprotection in cortical and in cerebellar granule cells and explain some of the neuroprotective effects of therapeutic dosages of lithium (Nonaka et al., 1998). In addition, data in cortical neurons suggests that NMDA receptor activation, key for LTP induction, increases the levels of active phospho-Akt, a negative regulator of GSK-3 (Sutton and Chandler, 2002), providing further evidence for a role of the GSK-3 pathway in NMDA dependent synaptic remodeling. At present there are no existing data as to the effects of GSK-3 on metabotropic glutamate receptors (mGluRs), but scaffolding proteins such as Shank and Homer might functionally link mGluRs to the NMDA receptors (Berridge et al., 2003) and provide a molecular basis for a reciprocal cross-talk between these two receptors (Liu et al., 2005).
Extensive studies have explored the functional interaction between dopaminergic signaling and NMDA receptors (Wang and Goldman-Rakic, 2004; Cepeda and Levine, 2006) and investigated the role of GSK-3 in this context. In the mPFC, for example, high concentration of dopamine activates dopamine 2 (D2) receptors and induces NMDA-mediated EPSCs down-regulation through activation of GSK-3 (Li et al., 2009) via its upstream regulator PP2A (Beaulieu et al., 2009). D2-dependent activation of GSK-3 stimulates two separate pathways. One triggers endocytosis of surface NMDA receptors through increased phosphorylation of GluN2B at S1480; the other one promotes phosphorylation of β-catenin (Ser33/37/Tyr41) and its subsequent degradation, leading to inhibition of GluN2B gene transcription (Li et al., 2009). The overall result is a decrease in both surface and intracellular pools of NMDA receptors via a rapid cytosolic mechanism and gene transcription. The mechanism that triggers phosphorylation of GluN2B at Ser1480 depends upon dynamin and is independent from Gq11 or PLC (phospholipase C) (Li et al., 2009). In the same study it was shown that inhibition of GSK-3 in hyperdopamine conditions reverses internalization of cortical GluN2B thereby restoring NMDA receptor-mediated EPSCs (Li et al., 2009). As a further support of the role of GSK-3 in regulating NMDA receptors, the use of wortmannin, an inhibitor of the PI-3K/Akt pathway was shown to cause a significant decrease in surface expression of both GluN2A and GluN2B in the hippocampus, which GSK-3 inhibitors such as lithium or SB216763, are able to restore to control levels (Zhu et al., 2007). Overall, there appear to be a common control mechanism for AMPA and NMDA receptors by GSK-3 (Figure 3) that has potential critical implications for synaptic maintenance of glutamate receptors and for dopamine receptor-mediated psychostimulant effects and hyperdopamine-dependent behaviors in the brain (Beaulieu, 2012).
Changes in pre- and postsynaptic function are critical components of neuronal plasticity underlying memory formation (Hernandez et al., 2002; Hooper et al., 2007; Zhu et al., 2007; Dewachter et al., 2009; Lee et al., 2010). Compelling evidence indicates that modulation of constitutively active and/or regulated GSK-3 has a direct impact on the induction and expression of neuronal plasticity (Lucas et al., 1998; Kelly and Lynch, 2000; Lin et al., 2001; Daw et al., 2002; Sanna et al., 2002; Liu et al., 2003; Peineau et al., 2007; Kimura et al., 2008; Peineau et al., 2008; Bradley et al., 2012) and that, conversely, synaptic stimuli that induce plasticity, affect GSK-3 activity (Szatmari et al., 2005; Peineau et al., 2007, 2008). Furthermore, evidence indicates a role of GSK-3 in setting the threshold for the induction of LTP and LTD, a phenomenon called metaplasticity (Abraham and Tate, 1997; Peineau et al., 2007; Bradley et al., 2012). The roles of GSK-3 in regulating pre- and postsynaptic ion channels, discussed above, might provide new molecular insights into the mechanisms underlying neuronal plasticity and metaplasticity. The internalization of AMPA and NMDA receptors induced by GSK-3 inhibition (Chen et al., 2007; Wei et al., 2010), for example, might contribute to the mechanisms underling LTD expression (Oliet et al., 1996; Beattie et al., 2000). Furthermore, structural proteins that comprise the postsynaptic density (PSD) decrease upon GSK-3 stimulation opposing the effect of high-frequency LTP-inducing stimuli (Zhu et al., 2007). At the presynaptic site, GSK-3 has been shown to prevent the expression of Syn1 associated with LTP induction (Rosahl et al., 1995; Terada et al., 1999; Sato et al., 2000; Zhu et al., 2007) and together with a suppressing effect on presynaptic Cav channels, the GSK-3 pathway might decode signals that oppose LTP expression, suppressing the probability of neurotransmitter release.
Although a comprehensive model of the role of GSK-3 in synaptic plasticity is still lacking, high levels of active GSK-3 in response to neuronal activity might predispose synapses to LTD, repress LTP, and/or shift the threshold of LTP induction, shaping the mechanisms of memory acquisition, formation, and retention (Peineau et al., 2007, 2008; Bradley et al., 2012) through a concerted activity on pre- and postsynaptic ion channel trafficking.
Collectively these results indicate an emerging and significant role of the GSK-3 pathway in regulating neuronal voltage-gated and ligand-gated ion channels (Figure 4), revealing novel mechanisms underlying the complex effects of this kinase pathway in the brain. One of the most remarkable features emerging from these studies is the great deal of diversity in GSK-3 signaling, despite the high degree of target specificity and the precise functional outcome resulting from targeted phosphorylation. Some of the factors that might contribute to target specificity in the GSK-3 signaling pathway are the stringent consensus sequence for primary phosphorylation and the requirement for priming phosphorylation by other kinases (Frame et al., 2001). Priming phosphorylation events may act through combinatorial mechanisms, either sequentially or concurrently, and, as such, serve as powerful converging points of multiple intracellular signaling cascades, increasing the level of signal specificity. Other post-translational modifications may also play a role in target specificity. For example, O-glycosylation (Butkinaree et al., 2010), nitrosylation, or palmitoylation (Salaun et al., 2010) could compete with or facilitate phosphorylation and may provide additional fine-tuning, increasing signal specificity. At the broader cellular level, transient and stable interactions of GSK-3 with macromolecular scaffolds can provide a mechanism for subcellular compartmentalization of this kinase allowing spatial and temporal segregation of signaling (Frame et al., 2001).
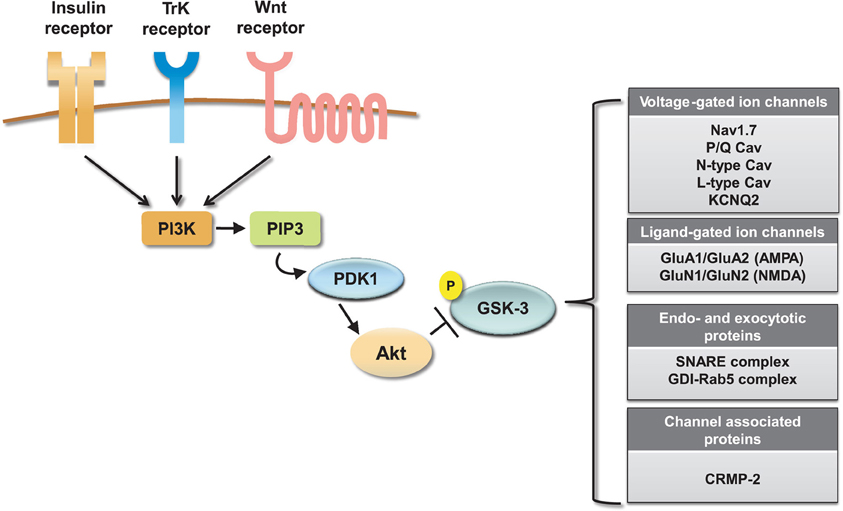
Figure 4. Summary of ion channel targets of the GSK-3 pathway. The activity of GSK-3 is controlled by activation of insulin, TrK, and Wnt signaling through a cascade of positive downstream effectors including phosphatidylinositol 3-kinase (PI-3K), phosphatidylinositol (3,4,5)-triphosphate (PIP3), and 3-phosphoinositide-dependent protein kinase (PDK1). This cascade activates the major GKS-3 negative regulator, Akt. Activation of Akt results in inhibition of GSK-3 by phosphorylation at S9 (for GSK-3β). On the side, a summary table shows relevant ion channels, endo- and exocytotic proteins and channel associated proteins that have been identified as GSK-3 targets.
GSK-3 specificity in the presynaptic regulation of P/Q and N-type Cav channels is achieved within distinct functional domains. As illustrated in Figure 2, phosphorylation of P/Q Cav channels by GSK-3 inhibits the channel interaction with the SNARE complex, suppresses Ca2+ currents and blocks presynaptic release. Through a separate indirect mechanism, induced by semaphorin 3A signaling, GSK-3 might decrease the affinity of CRMP-2, an accessory protein of N-type Cav channels, presumably inhibiting Cav channels insertion to the cell membrane. Signal specificity and conservation coexist for AMPA and NMDA receptors. GSK-3 regulation of these receptors is mediated in both cases through the small GTPase Rab-5, clathrin and dynamin-dependent endocytosis, and involves either the dissociation of the GDI:Rab-5 complex (for AMPA receptors) or the weakening of GluN2B interaction with the scaffolding protein PSD95 (Figure 3).
Thus far it seems that GSK-3 has both direct and indirect effects on voltage-gated ion channels. GSK-3 acts by either directly phosphorylating the channel as in the cases of the Kv7.2 and P/Q-type Cav channels or indirectly in the case of the Nav and N-type Cav channels through modulation of protein–protein interactions of the channel macromolecular complex. Both direct and indirect mechanisms can result in the same outcome—inhibition—as in the case of Cav channels, or opposite outcomes. For example, GSK-3 through indirect means is critical for Nav channel functionality, while direct phosphorylation inhibits Kv7.2 function. These complex bi-directional regulations allow a very fine-tune modulation of ion channel function by the GSK-3 signaling that could serve as a point of cross-talk with other transduction systems, such as G-protein receptors (Beaulieu et al., 2011). The net effect of these functional interactions will ultimately be the result of signal integration from multiple signaling modalities with GSK-3 being the master player.
In the ligand-gated receptors examined to-date, it appears that GSK-3 promotes internalization of AMPA and NMDA receptors at the postsynaptic sites through indirect means, which is in keeping with its role in LTD (Peineau et al., 2007). From these examples, it emerges that GSK-3 signaling might be part of a network of globally conserved, yet targeted regulation of the presynaptic and postsynaptic function that converges on either presynaptic suppression and postsynaptic maintenance of synaptic transmission (Figure 5). Furthermore, given the extensive evidence for dysfunctional GSK-3 signaling in psychiatric disorders, neurodegenerative diseases, and addictive behaviors (Grimes and Jope, 2001b; Kozlovsky et al., 2001, 2002; Eldar-Finkelman, 2002; Li et al., 2002; Hooper et al., 2007; Lovestone et al., 2007; Kremer et al., 2011; Emamian, 2012), any new molecular discoveries that could link GSK-3 with synaptic function and/or neuronal excitability are likely to provide useful platforms for elucidating the mechanisms underlying cognitive and emotional processing in the human brain and for developing a more targeted therapeutic approach against this multifaceted, divergent intracellular pathway.
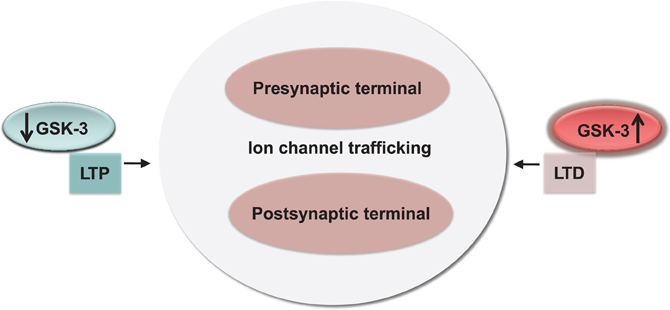
Figure 5. Conservation and functional relevance of the GSK-3 signaling. High and low-levels of GSK-3 correlate with bidirectional changes in synaptic strength (LTP and LTD) through a concerted effect on channel trafficking and internalization at both pre- and postsynaptic locations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the PhRMA Foundation (Fernanda Laezza). The authors would like to thank Dr. Myoung-Goo Kang and Dr. Shao-Jun Tang for their helpful comments, Dr. Laurence H. Pearl for the crystal structure of GSK-3β and Marcy Bubar Jordan for proofreading the manuscript.
Abraham, W. C., and Tate, W. P. (1997). Metaplasticity: a new vista across the field of synaptic plasticity. Prog. Neurobiol. 52, 303–323.
Adesnik, H., and Nicoll, R. A. (2007). Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602.
Arimura, N., Inagaki, N., Chihara, K., Menager, C., Nakamura, N., Amano, M., Iwamatsu, A., Goshima, Y., and Kaibuchi, K. (2000). Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 275, 23973–23980.
Arimura, N., Menager, C., Kawano, Y., Yoshimura, T., Kawabata, S., Hattori, A., Fukata, Y., Amano, M., Goshima, Y., Inagaki, M., Morone, N., Usukura, J., and Kaibuchi, K. (2005). Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell. Biol. 25, 9973–9984.
Ataman, B., Ashley, J., Gorczyca, M., Ramachandran, P., Fouquet, W., Sigrist, S. J., and Budnik, V. (2008). Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron 57, 705–718.
Baek, J. H., Cerda, O., and Trimmer, J. S. (2011). Mass spectrometry-based phosphoproteomics reveals multisite phosphorylation on mammalian brain voltage-gated sodium and potassium channels. Semin. Cell Dev. Biol. 22, 153–159.
Balaraman, Y., Limaye, A. R., Levey, A. I., and Srinivasan, S. (2006). Glycogen synthase kinase 3beta and Alzheimer's disease: pathophysiological and therapeutic significance. Cell. Mol. Life Sci. 63, 1226–1235.
Bamji, S. X., Shimazu, K., Kimes, N., Huelsken, J., Birchmeier, W., Lu, B., and Reichardt, L. F. (2003). Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40, 719–731.
Banke, T. G., Bowie, D., Lee, H., Huganir, R. L., Schousboe, A., and Traynelis, S. F. (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20, 89–102.
Bazinet, R. P. (2009). Is the brain arachidonic acid cascade a common target of drugs used to manage bipolar disorder? Biochem. Soc. Trans. 37, 1104–1109.
Beattie, E. C., Carroll, R. C., Yu, X., Morishita, W., Yasuda, H., Von Zastrow, M., and Malenka, R. C. (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 3, 1291–1300.
Beaulieu, J. M. (2012). A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 37, 7–16.
Beaulieu, J. M., Del'guidice, T., Sotnikova, T. D., Lemasson, M., and Gainetdinov, R. R. (2011). Beyond cAMP: the regulation of Akt and GSK3 by dopamine receptors. Front. Mol. Neurosci. 4:38. doi: 10.3389/fnmol.2011.00038
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347.
Berendt, F. J., Park, K. S., and Trimmer, J. S. (2010). Multisite phosphorylation of voltage-gated sodium channel alpha subunits from rat brain. J. Proteome Res. 9, 1976–1984.
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 4, 517–529.
Borsotto, M., Cavarec, L., Bouillot, M., Romey, G., Macciardi, F., Delaye, A., Nasroune, M., Bastucci, M., Sambucy, J. L., Luan, J. J., Charpagne, A., Jouet, V., Leger, R., Lazdunski, M., Cohen, D., and Chumakov, I. (2007). PP2A-Bgamma subunit and KCNQ2 K+ channels in bipolar disorder. Pharmacogenomics J. 7, 123–132.
Boyle, W. J., Smeal, T., Defize, L. H., Angel, P., Woodgett, J. R., Karin, M., and Hunter, T. (1991). Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 64, 573–584.
Bradley, C. A., Peineau, S., Taghibiglou, C., Nicolas, C. S., Whitcomb, D. J., Bortolotto, Z. A., Kaang, B. K., Cho, K., Wang, Y. T., and Collingridge, G. L. (2012). A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 5:13. doi: 10.3389/fnmol.2012.00013
Braff, D. L., Swerdlow, N. R., and Geyer, M. A. (1995). Gating and habituation deficits in the schizophrenia disorders. Clin. Neurosci. 3, 131–139.
Brittain, J. M., Piekarz, A. D., Wang, Y., Kondo, T., Cummins, T. R., and Khanna, R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390.
Brown, T. C., Tran, I. C., Backos, D. S., and Esteban, J. A. (2005). NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron 45, 81–94.
Butkinaree, C., Park, K., and Hart, G. W. (2010). O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta 1800, 96–106.
Cadigan, K. M., and Nusse, R. (1997). Wnt signaling: a common theme in animal development. Genes Dev. 11, 3286–3305.
Carr, D. B., Day, M., Cantrell, A. R., Held, J., Scheuer, T., Catterall, W. A., and Surmeier, D. J. (2003). Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron 39, 793–806.
Carroll, R. C., Beattie, E. C., Von Zastrow, M., and Malenka, R. C. (2001). Role of AMPA receptor endocytosis in synaptic plasticity. Nat. Rev. Neurosci. 2, 315–324.
Carroll, R. C., Beattie, E. C., Xia, H., Luscher, C., Altschuler, Y., Nicoll, R. A., Malenka, R. C., and Von Zastrow, M. (1999). Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 96, 14112–14117.
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555.
Catterall, W. A. (2010). Signaling complexes of voltage-gated sodium and calcium channels. Neurosci. Lett. 486, 107–116.
Catterall, W. A. (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3, a003947.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Cepeda, C., and Levine, M. S. (2006). Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE 2006, pe20.
Cerda, O., Baek, J. H., and Trimmer, J. S. (2011). Mining recent brain proteomic databases for ion channel phosphosite nuggets. J. Gen. Physiol. 137, 3–16.
Cerda, O., and Trimmer, J. S. (2010). Analysis and functional implications of phosphorylation of neuronal voltage-gated potassium channels. Neurosci. Lett. 486, 60–67.
Cerda, O., and Trimmer, J. S. (2011). Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J. Biol. Chem. 286, 28738–28748.
Chen, J., Park, C. S., and Tang, S. J. (2006). Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J. Biol. Chem. 281, 11910–11916.
Chen, P., Gu, Z., Liu, W., and Yan, Z. (2007). Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 72, 40–51.
Chi, P., Greengard, P., and Ryan, T. A. (2001). Synapsin dispersion and reclustering during synaptic activity. Nat. Neurosci. 4, 1187–1193.
Chi, X. X., Schmutzler, B. S., Brittain, J. M., Wang, Y., Hingtgen, C. M., Nicol, G. D., and Khanna, R. (2009). Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 122, 4351–4362.
Clayton, E. L., Anggono, V., Smillie, K. J., Chau, N., Robinson, P. J., and Cousin, M. A. (2009). The phospho-dependent dynamin-syndapin interaction triggers activity-dependent bulk endocytosis of synaptic vesicles. J. Neurosci. 29, 7706–7717.
Clayton, E. L., Evans, G. J., and Cousin, M. A. (2008). Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J. Neurosci. 28, 6627–6632.
Clayton, E. L., Sue, N., Smillie, K. J., O'Leary, T., Bache, N., Cheung, G., Cole, A. R., Wyllie, D. J., Sutherland, C., Robinson, P. J., and Cousin, M. A. (2010). Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat. Neurosci. 13, 845–851.
Coghlan, M. P., Culbert, A. A., Cross, D. A., Corcoran, S. L., Yates, J. W., Pearce, N. J., Rausch, O. L., Murphy, G. J., Carter, P. S., Roxbee Cox, L., Mills, D., Brown, M. J., Haigh, D., Ward, R. W., Smith, D. G., Murray, K. J., Reith, A. D., and Holder, J. C. (2000). Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7, 793–803.
Cooper, E. C. (2010). Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels, the modulation of neuronal excitability in vertebrate axons. Semin. Cell Dev. Biol. 22, 185–192.
Cooper, E. C., Aldape, K. D., Abosch, A., Barbaro, N. M., Berger, M. S., Peacock, W. S., Jan, Y. N., and Jan, L. Y. (2000). Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc. Natl. Acad. Sci. U.S.A. 97, 4914–4919.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789.
Dajani, R., Fraser, E., Roe, S. M., Young, N., Good, V., Dale, T. C., and Pearl, L. H. (2001). Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105, 721–732.
Daw, M. I., Bortolotto, Z. A., Saulle, E., Zaman, S., Collingridge, G. L., and Isaac, J. T. (2002). Phosphatidylinositol 3 kinase regulates synapse specificity of hippocampal long-term depression. Nat. Neurosci. 5, 835–836.
Deschenes, I., Neyroud, N., Disilvestre, D., Marban, E., Yue, D. T., and Tomaselli, G. F. (2002). Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ. Res. 90, E49–E57.
Dewachter, I., Ris, L., Jaworski, T., Seymour, C. M., Kremer, A., Borghgraef, P., De Vijver, H., Godaux, E., and Van Leuven, F. (2009). GSK3beta, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol. Dis. 35, 193–200.
Doble, B. W., and Woodgett, J. R. (2003). GSK-3, tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186.
Dominguez, I., Itoh, K., and Sokol, S. Y. (1995). Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proc. Natl. Acad. Sci. U.S.A. 92, 8498–8502.
Du, J., Gray, N. A., Falke, C. A., Chen, W., Yuan, P., Szabo, S. T., Einat, H., and Manji, H. K. (2004). Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J. Neurosci. 24, 6578–6589.
Eickholt, B. J., Walsh, F. S., and Doherty, P. (2002). An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J. Cell Biol. 157, 211–217.
Eldar-Finkelman, H. (2002). Glycogen synthase kinase 3, an emerging therapeutic target. Trends Mol. Med. 8, 126–132.
Emamian, E. S. (2012). AKT/GSK3 signaling pathway and schizophrenia. Front Mol Neurosci 5:33. doi: 10.3389/fnmol.2012.00033
Embi, N., Rylatt, D. B., and Cohen, P. (1980). Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527.
Fedorenko, O., Strutz-Seebohm, N., Henrion, U., Ureche, O. N., Lang, F., Seebohm, G., and Lang, U. E. (2008). A schizophrenia-linked mutation in PIP5K2A fails to activate neuronal M channels. Psychopharmacology (Berl.) 199, 47–54.
Fiol, C. J., Mahrenholz, A. M., Wang, Y., Roeske, R. W., and Roach, P. J. (1987). Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem. 262, 14042–14048.
Frame, S., and Cohen, P. (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16.
Frame, S., Cohen, P., and Biondi, R. M. (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 7, 1321–1327.
Fukata, Y., Itoh, T. J., Kimura, T., Menager, C., Nishimura, T., Shiromizu, T., Watanabe, H., Inagaki, N., Iwamatsu, A., Hotani, H., and Kaibuchi, K. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583–591.
Garcia-Segura, L. M., Arevalo, M. A., and Azcoitia, I. (2010). Interactions of estradiol and insulin-like growth factor-I signalling in the nervous system: new advances. Prog. Brain Res. 181, 251–272.
Goldfarb, M., Schoorlemmer, J., Williams, A., Diwakar, S., Wang, Q., Huang, X., Giza, J., Tchetchik, D., Kelley, K., Vega, A., Matthews, G., Rossi, P., Ornitz, D. M., and D'angelo, E. (2007). Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron 55, 449–463.
Goshima, Y., Hori, H., Sasaki, Y., Yang, T., Kagoshima-Maezono, M., Li, C., Takenaka, T., Nakamura, F., Takahashi, T., Strittmatter, S. M., Misu, Y., and Kawakami, T. (1999). Growth cone neuropilin-1 mediates collapsin-1/Sema III facilitation of antero- and retrograde axoplasmic transport. J. Neurobiol. 39, 579–589.
Grimes, C. A., and Jope, R. S. (2001a). CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. 78, 1219–1232.
Grimes, C. A., and Jope, R. S. (2001b). The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65, 391–426.
Gutman, G. A., Chandy, K. G., Adelman, J. P., Aiyar, J., Bayliss, D. A., Clapham, D. E., Covarriubias, M., Desir, G. V., Furuichi, K., Ganetzky, B., Garcia, M. L., Grissmer, S., Jan, L. Y., Karschin, A., Kim, D., Kuperschmidt, S., Kurachi, Y., Lazdunski, M., Lesage, F., Lester, H. A., McKinnon, D., Nichols, C. G., O'Kelly, I., Robbins, J., Robertson, G. A., Rudy, B., Sanguinetti, M., Seino, S., Stuehmer, W., Tamkun, M. M., Vandenberg, C. A., Wei, A., Wulff, H., and Wymore, R. S. (2003). International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol. Rev. 55, 583–586.
Harwood, A. J., and Agam, G. (2003). Search for a common mechanism of mood stabilizers. Biochem. Pharmacol. 66, 179–189.
Hedegaard, M., Hansen, K. B., Andersen, K. T., Brauner-Osborne, H., and Traynelis, S. F. (2011). Molecular pharmacology of human NMDA receptors. Neurochem. Int. doi: 10.1016/j.neuint.2011.11.016. [Epub ahead of print].
Henley, J. M., Barker, E. A., and Glebov, O. O. (2011). Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 34, 258–268.
Hernandez, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533.
Hernandez, F., Nido, J. D., Avila, J., and Villanueva, N. (2009). GSK3 inhibitors and disease. Mini Rev. Med. Chem. 9, 1024–1029.
Hinoi, T., Yamamoto, H., Kishida, M., Takada, S., Kishida, S., and Kikuchi, A. (2000). Complex formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3 beta-dependent phosphorylation of beta-catenin and down-regulates beta-catenin. J. Biol. Chem. 275, 34399–34406.
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer's disease. J. Neurochem. 104, 1433–1439.
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., Hernandez, F., Anderton, B., Rosenblum, K., Bliss, T., Cooke, S. F., Avila, J., Lucas, J. J., Giese, K. P., Stephenson, J., and Lovestone, S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86.
Hoshi, N., Zhang, J. S., Omaki, M., Takeuchi, T., Yokoyama, S., Wanaverbecq, N., Langeberg, L. K., Yoneda, Y., Scott, J. D., Brown, D. A., and Higashida, H. (2003). AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 6, 564–571.
Huang, C. C., You, J. L., Wu, M. Y., and Hsu, K. S. (2004). Rap1-induced p38 mitogen-activated protein kinase activation facilitates AMPA receptor trafficking via the GDI.Rab5 complex. Potential role in (S)-3, 5-dihydroxyphenylglycene-induced long term depression. J. Biol. Chem. 279, 12286–12292.
Hughes, K., Nikolakaki, E., Plyte, S. E., Totty, N. F., and Woodgett, J. R. (1993). Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 12, 803–808.
Itoh, N., and Ornitz, D. M. (2008). Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 237, 18–27.
Jentsch, T. J. (2000). Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21–30.
Jia, J., Amanai, K., Wang, G., Tang, J., Wang, B., and Jiang, J. (2002). Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416, 548–552.
Jiang, X., Lautermilch, N. J., Watari, H., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2008). Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 105, 341–346.
Johnston, D., Christie, B. R., Frick, A., Gray, R., Hoffman, D. A., Schexnayder, L. K., Watanabe, S., and Yuan, L. L. (2003). Active dendrites, potassium channels and synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 667–674.
Jope, R. S. (2003). Lithium and GSK-3, one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 24, 441–443.
Jope, R. S. (2011). Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front. Mol. Neurosci. 4:16. doi: 10.3389/fnmol.2011.00016
Jope, R. S., and Johnson, G. V. (2004). The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., Van Eede, M., Hattori, S., Laliberte, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., Macaulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2, 35.
Kaidanovich-Beilin, O., and Woodgett, J. R. (2011). GSK-3, Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 4:40. doi: 10.3389/fnmol.2011.00040
Kapfhamer, D., Berger, K. H., Hopf, F. W., Seif, T., Kharazia, V., Bonci, A., and Heberlein, U. (2010). Protein Phosphatase 2a and glycogen synthase kinase 3 signaling modulate prepulse inhibition of the acoustic startle response by altering cortical M-Type potassium channel activity. J. Neurosci. 30, 8830–8840.
Kelly, A., and Lynch, M. A. (2000). Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology 39, 643–651.
Kessels, H. W., and Malinow, R. (2009). Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350.
Kim, D. K., and Catterall, W. A. (1997). Ca2+-dependent and -independent interactions of the isoforms of the alpha1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc. Natl. Acad. Sci. U.S.A. 94, 14782–14786.
Kim, W. Y., and Snider, W. D. (2011). Functions of GSK-3 Signaling in Development of the Nervous System. Front. Mol. Neurosci. 4:44. doi: 10.3389/fnmol.2011.00044
Kimura, T., Yamashita, S., Nakao, S., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2008). GSK-3beta is required for memory reconsolidation in adult brain. PLoS ONE 3:e3540. doi: 10.1371/journal.pone.0003540
Kochlamazashvili, G., Henneberger, C., Bukalo, O., Dvoretskova, E., Senkov, O., Lievens, P. M., Westenbroek, R., Engel, A. K., Catterall, W. A., Rusakov, D. A., Schachner, M., and Dityatev, A. (2010). The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca(2+) channels. Neuron 67, 116–128.
Kozlovsky, N., Belmaker, R. H., and Agam, G. (2001). Low GSK-3 activity in frontal cortex of schizophrenic patients. Schizophr. Res. 52, 101–105.
Kozlovsky, N., Belmaker, R. H., and Agam, G. (2002). GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur. Neuropsychopharmacol. 12, 13–25.
Kremer, A., Louis, J. V., Jaworski, T., and Van Leuven, F. (2011). GSK3 and Alzheimer's Disease: facts and fiction. Front. Mol. Neurosci. 4:17. doi: 10.3389/fnmol.2011.00017
Laezza, F., and Dingledine, R. (2004). Voltage-controlled plasticity at GluR2-deficient synapses onto hippocampal interneurons. J. Neurophysiol. 92, 3575–3581.
Laezza, F., and Dingledine, R. (2011). Induction and expression rules of synaptic plasticity in hippocampal interneurons. Neuropharmacology 60, 720–729.
Laezza, F., Doherty, J. J., and Dingledine, R. (1999). Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science 285, 1411–1414.
Laezza, F., Gerber, B. R., Lou, J. Y., Kozel, M. A., Hartman, H., Craig, A. M., Ornitz, D. M., and Nerbonne, J. M. (2007). The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J. Neurosci. 27, 12033–12044.
Laezza, F., Lampert, A., Kozel, M. A., Gerber, B. R., Rush, A. M., Nerbonne, J. M., Waxman, S. G., Dib-Hajj, S. D., and Ornitz, D. M. (2009). FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol. Cell. Neurosci. 42, 90–101.
Laezza, F., Shavkunov, A., Buzhdygan, T., and Nenov, M. (2011). The FGF14, Nav channel complex is a new target of the Akt/GSK3 signaling pathway. Neuropsychopharmacology 36, S89–S90.
Lee, H. K., Barbarosie, M., Kameyama, K., Bear, M. F., and Huganir, R. L. (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959.
Lee, M. C., Yasuda, R., and Ehlers, M. D. (2010). Metaplasticity at single glutamatergic synapses. Neuron 66, 859–870.
Leroy, K., and Brion, J. P. (1999). Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J. Chem. Neuroanat. 16, 279–293.
Li, Q., and Sarna, S. K. (2011). Chronic stress targets posttranscriptional mechanisms to rapidly upregulate alpha1C-subunit of Cav1.2b calcium channels in colonic smooth muscle cells. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G154–G163.
Li, X., Bijur, G. N., and Jope, R. S. (2002). Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 4, 137–144.
Li, Y. C., Xi, D., Roman, J., Huang, Y. Q., and Gao, W. J. (2009). Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J. Neurosci. 29, 15551–15563.
Lin, C. H., Yeh, S. H., Lin, C. H., Lu, K. T., Leu, T. H., Chang, W. C., and Gean, P. W. (2001). A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 31, 841–851.
Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G. H., Tan, Y., Zhang, Z., Lin, X., and He, X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847.
Liu, F., Gong, X., Zhang, G., Marquis, K., Reinhart, P., and Andree, T. H. (2005). The inhibition of glycogen synthase kinase 3beta by a metabotropic glutamate receptor 5 mediated pathway confers neuroprotection to Abeta peptides. J. Neurochem. 95, 1363–1372.
Liu, S. J., Zhang, A. H., Li, H. L., Wang, Q., Deng, H. M., Netzer, W. J., Xu, H., and Wang, J. Z. (2003). Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J. Neurochem. 87, 1333–1344.
Liu, X. B., Murray, K. D., and Jones, E. G. (2004). Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J. Neurosci. 24, 8885–8895.
Lovestone, S., Killick, R., Di Forti, M., and Murray, R. (2007). Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 30, 142–149.
Lu, W., Man, H., Ju, W., Trimble, W. S., Macdonald, J. F., and Wang, Y. T. (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254.
Lu, W., Shi, Y., Jackson, A. C., Bjorgan, K., During, M. J., Sprengel, R., Seeburg, P. H., and Nicoll, R. A. (2009). Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268.
Lucas, F. R., Goold, R. G., Gordon-Weeks, P. R., and Salinas, P. C. (1998). Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J. Cell Sci. 111(Pt 10), 1351–1361.
Luscher, C., Nicoll, R. A., Malenka, R. C., and Muller, D. (2000). Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat. Neurosci. 3, 545–550.
Luscher, C., Xia, H., Beattie, E. C., Carroll, R. C., Von Zastrow, M., Malenka, R. C., and Nicoll, R. A. (1999). Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 24, 649–658.
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Man, H. Y., Wang, Q., Lu, W. Y., Ju, W., Ahmadian, G., Liu, L., D'Souza, S., Wong, T. P., Taghibiglou, C., Lu, J., Becker, L. E., Pei, L., Liu, F., Wymann, M. P., Macdonald, J. F., and Wang, Y. T. (2003). Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron 38, 611–624.
Miller, J. S., Tallarida, R. J., and Unterwald, E. M. (2009). Cocaine-induced hyperactivity and sensitization are dependent on GSK3. Neuropharmacology 56, 1116–1123.
Miller, J. S., Tallarida, R. J., and Unterwald, E. M. (2010). Inhibition of GSK3 attenuates dopamine D1 receptor agonist-induced hyperactivity in mice. Brain Res. Bull. 82, 184–187.
Mines, M. A., Yuskaitis, C. J., King, M. K., Beurel, E., and Jope, R. S. (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 5:e9706. doi: 10.1371/journal.pone.0009706
Morfini, G., Szebenyi, G., Elluru, R., Ratner, N., and Brady, S. T. (2002). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293.
Mukai, F., Ishiguro, K., Sano, Y., and Fujita, S. C. (2002). Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 81, 1073–1083.
Mulley, J. C., Scheffer, I. E., Petrou, S., and Berkovic, S. F. (2003). Channelopathies as a genetic cause of epilepsy. Curr. Opin. Neurol. 16, 171–176.
Nakagawa, T. (2010). The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol. Neurobiol. 42, 161–184.
Namekata, K., Harada, C., Guo, X., Kimura, A., Kittaka, D., Watanabe, H., and Harada, T. (2012). Dock3 stimulates axonal outgrowth via GSK-3beta-mediated microtubule assembly. J. Neurosci. 32, 264–274.
Nemoto, T., Yanagita, T., Kanai, T., and Wada, A. (2009). Drug development targeting the glycogen synthase kinase-3beta (GSK-3beta)-mediated signal transduction pathway: the role of GSK-3beta in the maintenance of steady-state levels of insulin receptor signaling molecules and Na(v)1.7 sodium channel in adrenal chromaffin cells. J. Pharmacol. Sci. 109, 157–161.
Newton, A. J., Kirchhausen, T., and Murthy, V. N. (2006). Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc. Natl. Acad. Sci. U.S.A. 103, 17955–17960.
Nichols, R. A., Chilcote, T. J., Czernik, A. J., and Greengard, P. (1992). Synapsin I regulates glutamate release from rat brain synaptosomes. J. Neurochem. 58, 783–785.
Nishimoto, T., Kihara, T., Akaike, A., Niidome, T., and Sugimoto, H. (2008). alpha-Amino-3-hydroxy-5-methyl-4-isoxazole propionate attenuates glutamate-induced caspase-3 cleavage via regulation of glycogen synthase kinase 3beta. J. Neurosci. Res. 86, 1096–1105.
Nishimoto, T., Kihara, T., Akaike, A., Niidome, T., and Sugimoto, H. (2009). AMPA reduces surface expression of NR1 through regulation of GSK3beta. Neuroreport 20, 161–165.
Nishiyama, M., Togashi, K., Von Schimmelmann, M. J., Lim, C. S., Maeda, S., Yamashita, N., Goshima, Y., Ishii, S., and Hong, K. (2011). Semaphorin 3A induces CaV2.3 channel-dependent conversion of axons to dendrites. Nat. Cell Biol. 13, 676–685.
Nonaka, S., Hough, C. J., and Chuang, D. M. (1998). Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. U.S.A. 95, 2642–2647.
Norris, A. J., Foeger, N. C., and Nerbonne, J. M. (2010). Neuronal voltage-gated K+ (Kv) channels function in macromolecular complexes. Neurosci. Lett. 486, 73–77.
O'Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
Oliet, S. H., Malenka, R. C., and Nicoll, R. A. (1996). Bidirectional control of quantal size by synaptic activity in the hippocampus. Science 271, 1294–1297.
Otto, J. F., Yang, Y., Frankel, W. N., White, H. S., and Wilcox, K. S. (2006). A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J. Neurosci. 26, 2053–2059.
Pap, M., and Cooper, G. M. (1998). Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932.
Patapoutian, A., and Reichardt, L. F. (2000). Roles of Wnt proteins in neural development and maintenance. Curr. Opin. Neurobiol. 10, 392–399.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl. 1), S428–S437.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Pennuto, M., Bonanomi, D., Benfenati, F., and Valtorta, F. (2003). Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles. Mol. Biol. Cell 14, 4909–4919.
Plant, K., Pelkey, K. A., Bortolotto, Z. A., Morita, D., Terashima, A., McBain, C. J., Collingridge, G. L., and Isaac, J. T. (2006). Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Plyte, S. E., Hughes, K., Nikolakaki, E., Pulverer, B. J., and Woodgett, J. R. (1992). Glycogen synthase kinase-3, functions in oncogenesis and development. Biochim. Biophys. Acta 1114, 147–162.
Polter, A., Beurel, E., Yang, S., Garner, R., Song, L., Miller, C. A., Sweatt, J. D., McMahon, L., Bartolucci, A. A., Li, X., and Jope, R. S. (2010). Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 35, 1761–1774.
Pongs, O. (1999). Voltage-gated potassium channels: from hyperexcitability to excitement. FEBS Lett. 452, 31–35.
Price, M. A., and Kalderon, D. (2002). Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 108, 823–835.
Roach, P. J. (1990). Control of glycogen synthase by hierarchal protein phosphorylation. FASEB J. 4, 2961–2968.
Rogawski, M. A., and Loscher, W. (2004). The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat. Med. 10, 685–692.
Rosahl, T. W., Spillane, D., Missler, M., Herz, J., Selig, D. K., Wolff, J. R., Hammer, R. E., Malenka, R. C., and Sudhof, T. C. (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493.
Rowe, M. K., Wiest, C., and Chuang, D. M. (2007). GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci. Biobehav. Rev. 31, 920–931.
Rozov, A., Sprengel, R., and Seeburg, P. H. (2012). GluA2-lacking AMPA receptors in hippocampal CA1 cell synapses: evidence from gene-targeted mice. Front. Mol. Neurosci. 5:22. doi: 10.3389/fnmol.2012.00022
Rylatt, D. B., Aitken, A., Bilham, T., Condon, G. D., Embi, N., and Cohen, P. (1980). Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur. J. Biochem. 107, 529–537.
Salaun, C., Greaves, J., and Chamberlain, L. H. (2010). The intracellular dynamic of protein palmitoylation. J. Cell Biol. 191, 1229–1238.
Sanchez, C., Diaz-Nido, J., and Avila, J. (2000a). Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog. Neurobiol. 61, 133–168.
Sanchez, C., Perez, M., and Avila, J. (2000b). GSK3beta-mediated phosphorylation of the microtubule-associated protein 2C (MAP2C) prevents microtubule bundling. Eur. J. Cell Biol. 79, 252–260.
Sanna, P. P., Cammalleri, M., Berton, F., Simpson, C., Lutjens, R., Bloom, F. E., and Francesconi, W. (2002). Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J. Neurosci. 22, 3359–3365.
Sato, K., Morimoto, K., Suemaru, S., Sato, T., and Yamada, N. (2000). Increased synapsin I immunoreactivity during long-term potentiation in rat hippocampus. Brain Res. 872, 219–222.
Scheuer, T. (2011). Regulation of sodium channel activity by phosphorylation. Semin. Cell Dev. Biol. 22, 160–165.
Schmidt, E. F., and Strittmatter, S. M. (2007). The CRMP family of proteins and their role in Sema3A signaling. Adv. Exp. Med. Biol. 600, 1–11.
Schroeder, B. C., Kubisch, C., Stein, V., and Jentsch, T. J. (1998). Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690.
Seeburg, P. H., Burnashev, N., Kohr, G., Kuner, T., Sprengel, R., and Monyer, H. (1995). The NMDA receptor channel: molecular design of a coincidence detector. Recent Prog. Horm. Res. 50, 19–34.
Seidensticker, M. J., and Behrens, J. (2000). Biochemical interactions in the wnt pathway. Biochim. Biophys. Acta 1495, 168–182.
Setou, M., Nakagawa, T., Seog, D. H., and Hirokawa, N. (2000). Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science 288, 1796–1802.
Shao, D., Okuse, K., and Djamgoz, M. B. (2009). Protein-protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. Int. J. Biochem. Cell Biol. 41, 1471–1481.
Sharp, F. R., and Hendren, R. L. (2007). Psychosis: atypical limbic epilepsy versus limbic hyperexcitability with onset at puberty? Epilepsy Behav. 10, 515–520.
Shavkunov, A., Panova, N., Prasai, A., Veselenak, R., Bourne, N., Stoilova-McPhie, S., and Laezza, F. (2012). Bioluminescence methodology for the detection of protein-protein interactions within the voltage-gated sodium channel macromolecular complex. Assay Drug Dev. Technol. 10, 148–160.
Shelly, M., Cancedda, L., Lim, B. K., Popescu, A. T., Cheng, P. L., Gao, H., and Poo, M. M. (2011). Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71, 433–446.
Shepherd, J. D., and Huganir, R. L. (2007). The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643.
Shi, S., Hayashi, Y., Esteban, J. A., and Malinow, R. (2001). Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105, 331–343.
Singh, K. K., De Rienzo, G., Drane, L., Mao, Y., Flood, Z., Madison, J., Ferreira, M., Bergen, S., King, C., Sklar, P., Sive, H., and Tsai, L. H. (2011). Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron 72, 545–558.
Singh, N. A., Charlier, C., Stauffer, D., Dupont, B. R., Leach, R. J., Melis, R., Ronen, G. M., Bjerre, I., Quattlebaum, T., Murphy, J. V., McHarg, M. L., Gagnon, D., Rosales, T. O., Peiffer, A., Anderson, V. E., and Leppert, M. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29.
Smillie, K. J., and Cousin, M. A. (2011). The Role of GSK3 in Presynaptic Function. Int. J. Alzheimers Dis. 2011, 263673.
Sudhof, T. C. (2012). Calcium control of neurotransmitter release. Cold Spring Harb. Perspect. Biol. 4, a011353.
Sutton, G., and Chandler, L. J. (2002). Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J. Neurochem. 82, 1097–1105.
Szatmari, E., Habas, A., Yang, P., Zheng, J. J., Hagg, T., and Hetman, M. (2005). A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J. Biol. Chem. 280, 37526–37535.
Takashima, A., Noguchi, K., Sato, K., Hoshino, T., and Imahori, K. (1993). Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 7789–7793.
Terada, S., Tsujimoto, T., Takei, Y., Takahashi, T., and Hirokawa, N. (1999). Impairment of inhibitory synaptic transmission in mice lacking synapsin I. J. Cell Biol. 145, 1039–1048.
Tomizawa, K., Ohta, J., Matsushita, M., Moriwaki, A., Li, S. T., Takei, K., and Matsui, H. (2002). Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J. Neurosci. 22, 2590–2597.
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., Hansen, K. B., Yuan, H., Myers, S. J., Dingledine, R., and Sibley, D. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Uchida, Y., Ohshima, T., Sasaki, Y., Suzuki, H., Yanai, S., Yamashita, N., Nakamura, F., Takei, K., Ihara, Y., Mikoshiba, K., Kolattukudy, P., Honnorat, J., and Goshima, Y. (2005). Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2, implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells 10, 165–179.
Vacher, H., and Trimmer, J. S. (2011). Diverse roles for auxiliary subunits in phosphorylation-dependent regulation of mammalian brain voltage-gated potassium channels. Pflugers Arch. 462, 631–643.
Vijayaragavan, K., Boutjdir, M., and Chahine, M. (2004). Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J. Neurophysiol. 91, 1556–1569.
Wada, A., Yokoo, H., Yanagita, T., and Kobayashi, H. (2005). Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J. Pharmacol. Sci. 99, 307–321.
Wagner, U., Brownlees, J., Irving, N. G., Lucas, F. R., Salinas, P. C., and Miller, C. C. (1997). Overexpression of the mouse dishevelled-1 protein inhibits GSK-3beta-mediated phosphorylation of tau in transfected mammalian cells. FEBS Lett. 411, 369–372.
Wan, X. Z., Li, B., Li, Y. C., Yang, X. L., Zhang, W., Zhong, L., and Tang, S. J. (2012). Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J. Neurosci. 32, 3910–3916.
Wang, Y., Brittain, J. M., Wilson, S. M., and Khanna, R. (2010). Emerging roles of collapsin response mediator proteins (CRMPs) as regulators of voltage-gated calcium channels and synaptic transmission. Commun. Integr. Biol. 3, 172–175.
Wang, Y., and Goldman-Rakic, P. S. (2004). D2 receptor regulation of synaptic burst firing in prefrontal cortical pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 101, 5093–5098.
Wei, J., Liu, W., and Yan, Z. (2010). Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 285, 26369–26376.
Welch, E. M., Barton, E. R., Zhuo, J., Tomizawa, Y., Friesen, W. J., Trifillis, P., Paushkin, S., Patel, M., Trotta, C. R., Hwang, S., Wilde, R. G., Karp, G., Takasugi, J., Chen, G., Jones, S., Ren, H., Moon, Y. C., Corson, D., Turpoff, A. A., Campbell, J. A., Conn, M. M., Khan, A., Almstead, N. G., Hedrick, J., Mollin, A., Risher, N., Weetall, M., Yeh, S., Branstrom, A. A., Colacino, J. M., Babiak, J., Ju, W. D., Hirawat, S., Northcutt, V. J., Miller, L. L., Spatrick, P., He, F., Kawana, M., Feng, H., Jacobson, A., Peltz, S. W., and Sweeney, H. L. (2007). PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91.
Williamson, R., Van Aalten, L., Mann, D. M., Platt, B., Plattner, F., Bedford, L., Mayer, J., Howlett, D., Usardi, A., Sutherland, C., and Cole, A. R. (2011). CRMP2 hyperphosphorylation is characteristic of Alzheimer's disease and not a feature common to other neurodegenerative diseases. J. Alzheimers Dis. 27, 615–625.
Wood-Kaczmar, A., Kraus, M., Ishiguro, K., Philpott, K. L., and Gordon-Weeks, P. R. (2009). An alternatively spliced form of glycogen synthase kinase-3beta is targeted to growing neurites and growth cones. Mol. Cell. Neurosci. 42, 184–194.
Woodgett, J. R. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438.
Xie, C. W. (2004). Calcium-regulated signaling pathways: role in amyloid beta-induced synaptic dysfunction. Neuromolecular Med. 6, 53–64.
Yamamoto, R., Yanagita, T., Kobayashi, H., Yuhi, T., Yokoo, H., and Wada, A. (1996). Up-regulation of functional voltage-dependent sodium channels by insulin in cultured bovine adrenal chromaffin cells. J. Neurochem. 67, 1401–1408.
Yamashita, N., and Goshima, Y. (2012). Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol. Neurobiol. 45, 234–246.
Yamashita, N., Ohshima, T., Nakamura, F., Kolattukudy, P., Honnorat, J., Mikoshiba, K., and Goshima, Y. (2012). Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J. Neurosci. 32, 1360–1365.
Yanagita, T., Maruta, T., Nemoto, T., Uezono, Y., Matsuo, K., Satoh, S., Yoshikawa, N., Kanai, T., Kobayashi, H., and Wada, A. (2009). Chronic lithium treatment up-regulates cell surface Na(V)1.7 sodium channels via inhibition of glycogen synthase kinase-3 in adrenal chromaffin cells: enhancement of Na(+) influx, Ca(2+) influx and catecholamine secretion after lithium withdrawal. Neuropharmacology 57, 311–321.
Yanagita, T., Maruta, T., Uezono, Y., Satoh, S., Yoshikawa, N., Nemoto, T., Kobayashi, H., and Wada, A. (2007). Lithium inhibits function of voltage-dependent sodium channels and catecholamine secretion independent of glycogen synthase kinase-3 in adrenal chromaffin cells. Neuropharmacology 53, 881–889.
Yang, G. Y., and Luo, Z. G. (2011). Implication of Wnt signaling in neuronal polarization. Dev. Neurobiol. 71, 495–507.
Yang, Q., Liao, Z. H., Xiao, Y. X., Lin, Q. S., Zhu, Y. S., and Li, S. T. (2011). Hippocampal synaptic metaplasticity requires the activation of NR2B-containing NMDA receptors. Brain Res. Bull. 84, 137–143.
Yokoyama, C. T., Myers, S. J., Fu, J., Mockus, S. M., Scheuer, T., and Catterall, W. A. (2005). Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol. Cell. Neurosci. 28, 1–17.
Yokoyama, C. T., Sheng, Z. H., and Catterall, W. A. (1997). Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J. Neurosci. 17, 6929–6938.
Yuen, E. Y., Jiang, Q., Chen, P., Gu, Z., Feng, J., and Yan, Z. (2005a). Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J. Neurosci. 25, 5488–5501.
Yuen, E. Y., Jiang, Q., Feng, J., and Yan, Z. (2005b). Microtubule regulation of N-methyl-D-aspartate receptor channels in neurons. J. Biol. Chem. 280, 29420–29427.
Zerial, M., and McBride, H. (2001). Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117.
Zhou, F. Q., and Snider, W. D. (2005). Cell biology. GSK-3beta and microtubule assembly in axons. Science 308, 211–214.
Zhou, J., Shin, H. G., Yi, J., Shen, W., Williams, C. P., and Murray, K. T. (2002). Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ. Res. 91, 540–546.
Zhu, L. Q., Liu, D., Hu, J., Cheng, J., Wang, S. H., Wang, Q., Wang, F., Chen, J. G., and Wang, J. Z. (2010). GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J. Neurosci. 30, 3624–3633.
Zhu, L. Q., Wang, S. H., Liu, D., Yin, Y. Y., Tian, Q., Wang, X. C., Wang, Q., Chen, J. G., and Wang, J. Z. (2007). Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J. Neurosci. 27, 12211–12220.
Keywords: voltage-gated ion channels, glutamate receptors, synaptic transmission, neurotransmitter release, excitability
Citation: Wildburger NC and Laezza F (2012) Control of neuronal ion channel function by glycogen synthase kinase-3: new prospective for an old kinase. Front. Mol. Neurosci. 5:80. doi: 10.3389/fnmol.2012.00080
Received: 17 April 2012; Accepted: 20 June 2012;
Published online: 16 July 2012.
Edited by:
Carmen Villmann, Julius-Maximilians-University Würzburg, GermanyReviewed by:
Erhard Wischmeyer, University of Wuerzburg, GermanyCopyright © 2012 Wildburger and Laezza. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Fernanda Laezza, Department of Pharmacology and Toxicology, University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA. e-mail:ZmVsYWV6emFAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.