

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 15 March 2012
Sec. Neuroplasticity and Development
Volume 5 - 2012 | https://doi.org/10.3389/fnmol.2012.00025
This article is part of the Research Topic The neuronal functions of EF-hand Ca(2+)-binding proteins View all 22 articles
Many neurons of the vertebrate central nervous system (CNS) express the Ca2+ binding protein calbindin D-28k (CB), including important projection neurons like cerebellar Purkinje cells but also neocortical interneurons. CB has moderate cytoplasmic mobility and comprises at least four EF-hands that function in Ca2+ binding with rapid to intermediate kinetics and affinity. Classically it was viewed as a pure Ca2+ buffer important for neuronal survival. This view was extended by showing that CB is a critical determinant in the control of synaptic Ca2+ dynamics, presumably with strong impact on plasticity and information processing. Already 30 years ago, in vitro studies suggested that CB could have an additional Ca2+ sensor function, like its prominent acquaintance calmodulin (CaM). More recent work substantiated this hypothesis, revealing direct CB interactions with several target proteins. Different from a classical sensor, however, CB appears to interact with its targets both, in its Ca2+-loaded and Ca2+-free forms. Finally, CB has been shown to be involved in buffered transport of Ca2+, in neurons but also in kidney. Thus, CB serves a threefold function as buffer, transporter and likely as a non-canonical sensor.
Despite the wealth of information on expression patterns of Ca2+ binding proteins (CaBPs), their functional significance is only slowly emerging. In particular, this is due to their complex interplay with other Ca2+ controlling mechanisms and the inherent technical difficulties in studying biophysical properties of individual proteins (Neher, 2000), including the differentiation between Ca2+-buffer and Ca2+-sensor (da Silva and Reinach, 1991). Buffers are characterized by more or less specific binding/chelating of Ca2+ ions without further Ca2+-dependent target interactions. Their function is in the control of the spatio-temporal extent of Ca2+ signaling domains (Augustine et al., 2003; Eggermann et al., 2012). Sensors, on the other hand, undergo additional characteristic conformational changes upon Ca2+-binding, resulting in exposure of hydrophobic surfaces necessary for binding and subsequent regulation of downstream effectors (Ikura, 1996; Schwaller, 2008, 2010). Their functional significance lies in both, the control of intracellular free Ca2+ ([Ca2+]i) and in triggering Ca2+-dependent downstream signaling.
In consequence, characterization of a CaBP requires determination of several biophysical parameters (Table 1), including affinity and kinetics of Ca2+-binding, intracellular mobility, structural and conformational analysis, and the identification of binding partners. Following some general remarks on buffering, I will review advances in gathering biophysical parameters of CB that allowed deducing its functional facets, with emphasis on its neuronal function.
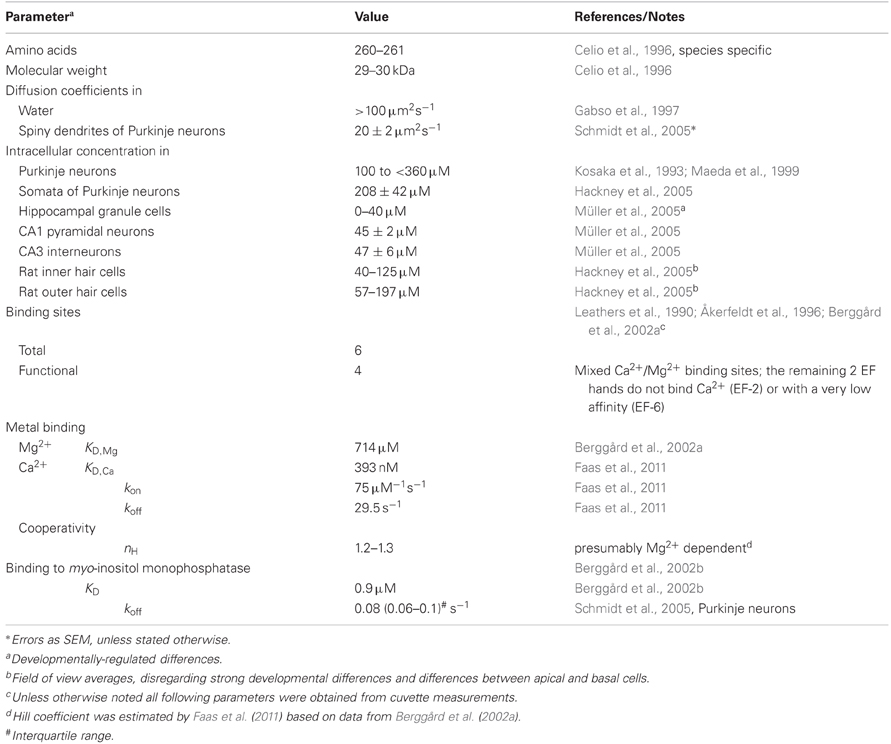
Table 1. Properties of calbindin D-28k.
Dissociation constants (KD) of proton buffers are optimized to clamp pH at 7–7.4 in living tissue by bidirectional buffering of free protons to a concentration of ∼100 nM. [Ca2+]i in resting cells is similar, however, contrasting to pH buffers, KD values of most CaBPs are well above [Ca2+]i; a notable exception is parvalbumin (PV, KD,Ca ∼9 nM; Lee et al., 2000b). Thus, under resting conditions most binding sites are unoccupied by Ca2+, such that CaBPs limit increases in [Ca2+]i from the resting level rather than clamping [Ca2+]i at a given level, i.e., they act as unidirectional buffers. This can even be augmented by an additional Mg2+ affinity, which reduces the effective affinity for Ca2+ due to competition or a necessity for preceding Mg2+ unbinding (Figure 1).
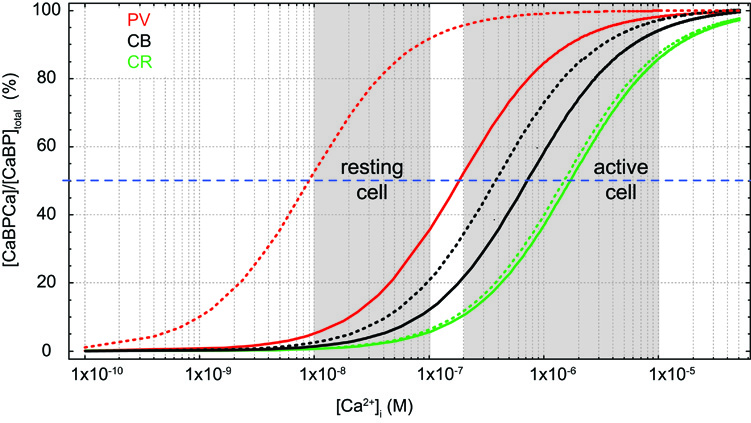
Figure 1. Effects of Mg2+ on Ca2+ binding. Simulated steady-state Ca2+-binding curves of the Ca2+ binding proteins (CaBP) parvalbumin (PV, red, KD,Ca 9 nM, KD,Mg 31 μM), calbindin (CB, black, average KD,Ca 393 nM, KD,Mg 714 μM; cf. Table 1), and calretinin (CR, green, average KD,Ca 1.5 μM, KD,Mg 4.5 mM) in the absence of Mg2+ (dotted lines) and in the presence of 600 μM Mg2+ (solid lines).
Neuronal Ca2+ signals are typically short lived and the amount of Ca2+ bound to a specific buffer can substantially deviate from the steady state value (Markram et al., 1998). Non-equilibrium conditions require a kinetic description of Ca2+ binding by forward (kon) and backward (koff) rates, values that could be quantified only recently due to a notable technical advance (Nägerl et al., 2000; Faas et al., 2007, 2011).
I will close these general remarks with a note on the ambiguous term “saturation.” In biochemistry, saturation refers to the fraction of total binding sites occupied at a given time. In chemistry, it often marks the 100% occupancy, probably the most intuitive meaning. In descriptions of Ca2+ dynamics saturation is often used to mark the deviation from linearity. A linear signaling process has the property that the combined effects of two or more elementary events result in a response which is given by the sum of the individual responses. This is the case if increases in [Ca2+]i are much smaller than KD of the buffer(s). Ca2+ kinetics become more complex if the increase in [Ca2+]i approaches KD (Neher, 1998a), i.e., saturation of a CaBP occurs around its half-occupancy.
CB has six EF-hands, of which one or two are non-functioning in metal binding (Leathers et al., 1990; Åkerfeldt et al., 1996; Berggård et al., 2002a; Cedervall et al., 2005; Kojetin et al., 2006). EF-2 is consistently viewed as non-metal binding while EF-6 may have a very low Ca2+ affinity (Åkerfeldt et al., 1996; Cedervall et al., 2005). EF-1, 3, 4, 5 are mixed metal binding sites, however, with much higher affinity for Ca2+ (393 nM; Faas et al., 2011) than for Mg2+ (714 μM; Berggård et al., 2002a). Thus, under physiological conditions ([Mg2+]i ∼600 μM) CB will function predominantly as a Ca2+ buffer. In resting neurons ([Ca2+]i ∼50 nM, [Mg2+]i ∼600 μM), it will be loaded by approx. ten percent with Ca2+ and 45% with Mg2+ (Figure 1; Berggård et al., 2002a). Compared to the related PV, the presence of Mg2+ produces only a minor shift in the apparent KD,Ca of CB (Figure 1), while it appears to increase cooperativity in Ca2+ binding (Berggård et al., 2002a); however, cooperativity is still negligible, with an estimated Hill coefficient (nH) of ∼1.25 (Faas et al., 2011).
Disregarding cooperativity, CB binds Ca2+ with rapid to intermediate kinetics (kon 75 μM−1s−1) and medium (393 nM) affinity (Faas et al., 2011; Table 1). Its on-rates are between those of EGTA (kon 10 μM−1s−1, KD 70 nM; Nägerl et al., 2000; Meinrenken et al., 2002) and BAPTA (kon 400 μM−1s−1, KD 220 nM; Naraghi, 1997; Naraghi and Neher, 1997; Meinrenken et al., 2002), while its affinity is closer to BAPTA. Typically, these properties endow CB to function as a major determinant of neuronal Ca2+ kinetics (Airaksinen et al., 1997; Barski et al., 2003; Schmidt et al., 2003).
The diffusion of CB has been quantified in spiny dendrites of cerebellar Purkinje neurons (PNs). Apart from a smaller immobilized fraction (see below), CB diffused with an apparent diffusion coefficient (D) of 20 μm2/s between spines and parent dendrites (Schmidt et al., 2005). This is ∼2-fold slower than the diffusional mobility of PV in the same cellular compartments, but identical to the D of a mobile calmodulin (CaM) fraction in HEK293 cells (Kim et al., 2004) and in spiny dendrites of PNs (Schmidt et al., 2007). To my knowledge there is no quantification of aqueous CB diffusion, but it might be estimated to be >100 μm2/s (Gabso et al., 1997).
In vitro studies from the 80th and 90th already suggested that CB might have an additional Ca2+ sensor function. It was shown to activate isolated erythrocyte membrane Ca2+-Mg2+ -ATPases (Morgan et al., 1986) and cyclic nucleotide phosphodiesterases (Reisner et al., 1992); in centrifugation studies of different tissues CB was found not only in the cytoplasmatic fractions but also in membrane/organelle containing fractions (Hubbard and McHugh, 1995; Winsky and Kuźnicki, 1995). The CB content in the membrane fractions was decreased in samples prepared or incubated in low Ca2+ (Winsky and Kuźnicki, 1995).
Three studies from the group of S. Linse (Berggård et al., 2000, 2002a,b) laid the foundation for substantiating the sensor hypothesis: They found that CB underwent substantial conformational rearrangements upon Ca2+-binding and protonation, likely exposing EF-2, but not upon Mg2+-binding. These changes went beyond the moderate redistributions in Mg2+-induced cooperativity in Ca2+-binding (see above). Unlike classical sensors CB had exposed hydrophobic regions also in its Ca2+-free (apo-) conformation, which is thermodynamically unfavorable under aqueous conditions, thus, suggesting additional Ca2+-independent interactions with target proteins. Subsequently, CB was shown to interact with myo-inositol monophosphatase-1 (IMPase), a key enzyme in the IP3 second messenger pathway (see below) both, in its apo- and Ca2+-bound form with low affinity (KD ∼0.9 μM). Finally, they identified a 12 amino-acid motive as the putative CB binding domain of IMPase.
Another CB ligand identified is Ran-binding-protein-M (RanBPM), a small GTPase involved in nuclear transport processes and microtubule formation (Lutz et al., 2003). Using NMR, it was shown that RanBPM interacted with Ca2+ loaded CB. Finally, CB was found to inhibit caspase-3 in osteoblastic cells independent of Ca2+, thereby, suppressing apoptosis (Bellido et al., 2000). Caspases are important enzymes in apoptosis, with activation of caspase-3 triggering the common executive pathway of cell death (Grütter, 2000; Yuan and Yankner, 2000; Yan and Shi, 2005). In central nervous system (CNS), incorrect execution of death pathways is thought to be associated with severe disorders including Chorea Huntington and Alzheimer disease. The finding that CB markedly reduces caspase-3 activity is particularly noteworthy in this context, since it might act as a neuroprotective agent.
So far, only in vitro studies showed a sensor function of CB. The CB-IMPase interaction was demonstrated in situ in PNs in acute slices (Schmidt et al., 2005). In multi-photon fluorescence recovery after photobleaching experiments a fraction of dye-labeled CB (∼20–30%) was found to be immobilized for >1 s in spines and dendrites, but not in smooth parts of axons. Use of the above peptide sequence from IMPase in a competition assay led to significant relief from immobilization, suggesting that CB indeed interacted with IMPase in PNs. Further experiments showed that the interaction was influenced by synaptic activation associated with increased [Ca2+]i.
A detailed NMR analysis of CB structure (Kojetin et al., 2006), completing earlier work (Klaus et al., 1999; Berggård et al., 2000, 2002a,b; Venters et al., 2003; Venyaminov et al., 2004; Vanbelle et al., 2005), confirmed that upon Ca2+ binding CB adopts discrete hydrophobic states but also has exposed hydrophobic surfaces in its apo-form. In addition, the regions mediating the interactions with RanBPM, IMPase, Caspase-3 and also its pro-domain were mapped.
Finally, in kidney CB was found to associate with TRPV5 channels which are involved in Ca2+ reabsorption from the urea (Lambers et al., 2006). The family of TRP channels is extraordinarily large with several members being abundantly expressed in the CNS. While it is temping to speculate on further direct CB interactions with Ca2+-conductances in neurons, experimental evidence is missing.
Taken together, there is growing evidence that CB not only functions as Ca2+-buffer but also binds to and regulates a variety of target proteins, including membrane ATPases, IMPase, RanBPM, procaspase-3, caspase-3, and TRPV5. Unlike canonical Ca2+-sensors, however, CB likely interacts in Ca2+-free and Ca2+-occupied form with its targets.
CB appears to fulfill three functions: First, it functions as a mobile or partly immobilized Ca2+-buffer with medium kinetics and affinity—buffer function. Second, it functions in buffered Ca2+-diffusion—transport function. Finally, it interacts with target proteins likely both, in its apo- and Ca2+-loaded form—sensor-like function.
In neurons expressing CB, it makes a major contribution to the total buffer capacitance (Fierro and Llano, 1996; Jackson and Redman, 2003; but see Faas et al. (2011) for an alternative view on CA1 spines in the presumed presence of large amounts of CaM). In dendrites and spines, CB clips the peak amplitude of synaptically induced Ca2+ transients, speeds their initial decay kinetics and prolongs their later phase (Airaksinen et al., 1997; Schmidt et al., 2003, 2007), while the rise time of the Ca2+ transients remained essentially unaffected (Koster et al., 1995; Schmidt et al., 2003). Thus, in postsynaptic structures CB induces characteristic biphasic decay kinetics of volume averaged Ca2+ transients and controls their amplitude.
Long lasting alterations of synaptic weight, like long-term potentiation (LTP) or depression (LTD) comprise strong Ca2+-dependent postsynaptic components. LTP in CA1 pyramidal neurons with reduced CB content could be induced normally but its maintenance was affected, leading to impaired spatial learning (Molinari et al., 1996). Whether this is attributable to altered Ca2+-signaling in the absence of CB or a direct target modulation by CB remained unclear. Lack of CB results also in deficits in motor coordination (Airaksinen et al., 1997; Barski et al., 2003), which is consistent with the strong CB expression in cerebellar cortex and its impact on synaptically mediated Ca2+ transients. However, parallel-fiber (PF) LTD in PNs lacking CB was normal and also Ca2+-signals mediated via activation of metabotropic glutamate receptors (mGluRs), known to be required in LTD induction (Daniel et al., 1998; Ito, 2001), were unaltered compared to the WT (Barski et al., 2003). It remained elusive, however, why rapid Ca2+ transients mediated by climbing-fiber and PF inputs were affected by lack of CB, whereas longer lasting, mGluR mediated Ca2+-signals were not (cf. discussion in Barski et al., 2003 for a possible explanation). Given that the conditional knock-outs used in the study are not affected by compensations for lack of CB (Vecellio et al., 2000; Kreiner et al., 2010), the answer may involve the as yet not further characterized CB-IMPase interaction (Schmidt et al., 2005; cf. below).
In presynaptic terminals, relevant Ca2+-signaling domains and their topographical relationships to Ca2+ dependent processes are well defined (Neher, 1998b; Augustine et al., 2003; Eggermann et al., 2012). However, the function of individual CaBPs has been rarely specified, although they are generally believed to be crucial in regulating transmitter release and short-term plasticity. Specifically, it has been postulated that saturation of CB underlies a form of paired pulse facilitation (PPF) at neocortical interneuron to pyramidal neuron synapses, hippocampal mossy-fiber to CA-3 synapses (Blatow et al., 2003), and recurrent PN synapses (Orduz and Llano, 2007). This form of PPF has been termed “pseudofacilitation” (Rozov et al., 2001) in order to distinguish it from more classical mechanisms like residual Ca2+ (Zucker and Stockbridge, 1983; Connor et al., 1986), Ca2+ remaining bound to the release sensor or a facilitation sensor (“active Ca2+”; Katz and Miledi, 1968; Yamada and Zucker, 1992; Atluri and Regehr, 1996) or Ca2+ dependent facilitation of Ca2+-currents (CDF; Lee et al., 1999, 2000a; Tsujimoto et al., 2002; for more detail see, e.g., Neher, 1998b; Zucker and Regehr, 2002; Stevens, 2003). In pseudofacilitation a substantial amount of Ca2+ entering the presynapse during the first action potential (AP) is thought to be buffered by CB, thereby, reducing the initial release probability but also saturating CB, which in turn results in increased [Ca2+]i during the second AP and potentiated release (Neher, 1998b; Rozov et al., 2001; Blatow et al., 2003; Felmy et al., 2003). Albeit direct evidence for CB saturation is scarce and Ca2+ imaging experiments from CB containing versus CB deficient presynaptic terminals are lacking, the hypothesis is consistent with conclusions drawn from experiments with BAPTA (Rozov et al., 2001). However, Ca2+ binding by endogenous CaBPs can be more complex than binding by exogenous buffers, as exemplified recently for PV (Caillard et al., 2000; Eggermann et al., 2012; Eggermann and Jonas, 2012). Thus, more direct evidence for saturation is desirable.
CDF and its counterpart Ca2+-dependent inactivation (CDI) are Ca2+ driven feedback mechanisms regulating voltage operated Cav2.1 (P/Q type) Ca2+-channels. CDF is mediated by Ca2+ loaded CaM or NCS-1 (Lee et al., 1999, 2000a; Tsujimoto et al., 2002), while CB only affected CDI but not CDF. Different from the CaM and NCS-1 effects on CDF, CB effects on CDI were essentially consistent with its buffering action and did not require the assumption of a Ca2+-sensor function (Kreiner and Lee, 2006).
Beyond Ca2+ transporting epithelia (Bronner and Stein, 1988; Bronner, 1989; Koster et al., 1995; Lambers et al., 2006), buffered Ca2+ transport by CB has recently also been suggested between activated spines and their parent dendritic shafts (Schmidt et al., 2007; Schmidt and Eilers, 2009). Although spill-over of Ca2+ from spines into dendrites had been reported before by several groups (Majewska et al., 2000; Holthoff et al., 2002; Schmidt et al., 2003), these experimental observations were significantly influenced by the action of Ca2+ dyes (Sabatini et al., 2002; Schmidt et al., 2003) and in the majority view spine necks remained substantial diffusion barriers for second messengers, making spines biochemically isolated compartments (Gamble and Koch, 1987; Zador et al., 1990; Müller and Connor, 1991; Svoboda et al., 1996; Sabatini et al., 2002). An analysis of spine Ca2+ dynamics under minimally perturbed conditions, however, confirmed that indeed most spines of pyramidal neurons allow a sizeable Ca2+ efflux that was tightly regulated by the geometry of the spine neck (Noguchi et al., 2005). Consecutively, it was shown that in PNs the majority of Ca2+ left the spine bound to CB (Schmidt et al., 2007), with the buffered efflux again being tightly controlled by the spine neck geometry (Schmidt and Eilers, 2009). This diffusional coupling drove a spatial summation process in which coincident activity of neighboring spines was integrated in the dendrite with the potential to activate dendritic CaM. This biochemical summation might exist in parallel to the classical summation of electrical signals in dendrites, possibly with reciprocal interaction (Nemri and Ghisovan, 2007).
Despite buffered Ca2+ transport, even neurons expressing large amounts of CB can retain Ca2+ signals that are spatially restricted to activated dendritic branches (Eilers et al., 1995, 1997) Although significant Ca2+ transport out of the active branches indeed occurred, it was outweighed by Ca2+ extrusion along the dendrite. This close interplay between diffusion and extrusion defined the capability of Ca2+ to spread between dendritic branches (Schmidt et al., 2011).
In this final function-section, I will focus on the neuronal CB-IMPase interaction. IMPase catalyzes the hydrolysis of myo-inositol-1(or 4)-monophosphate to form free myo-inositol, the resource for IP3 and DAG second messengers. CB was shown to bind IMPase in vitro (Berggård et al., 2002b) and in PNs (Schmidt et al., 2005) with a KD of ∼0.9 μM and an off-rate of ∼0.08 s−1 (Table 1). In vitro, apo- and Ca2+-bound CB activated IMPase similarity up to 250-fold. The activation was most pronounced under conditions that otherwise were associated with very low IMPase activity, precisely at reduced pH and at low substrate concentration. In spiny dendrites, binding of CB to IMPase was apparent at resting Ca2+ levels but further amplified by synaptic activation associated with increases in intracellular Ca2+ in a frequency dependent way. Thus, while the in vitro interaction appeared essentially independent of Ca2+, in dendrites it is likely boosted by increasing Ca2+ levels.
Still unresolved is the impact of the CB-IMPase interaction on dendritic IP3 mediated Ca2+-signaling. Two scenarios would be conceivable: First, the increased IMPase activity speeds the degradation of IP and could, in consequence, result in accelerated IP3 degradation and in reduced Ca2+-signals. Second, increased IMPase activity could result in an accelerated source substance supply for IP3 production and consequently in increased IP3 levels and amplified Ca2+-signaling. Considering that IP3 mediated Ca2+-signals in dendrites of PN-specific CB knock-outs were, despite the absence of a major buffer, unaltered compared to the WT (Barski et al., 2003) this argues in favor of the second scenario.
I reviewed evidence for a threefold function of CB, consisting of a sensor-like and a transport function in addition to buffering of Ca2+. Different from a canonical Ca2+-sensor, CB appears to bind its targets in Ca2+-occupied and Ca2+-free conformation. Spino-dendritic Ca2+-coupling and its regulation by the geometry of the spine neck were shown for pyramidal as well as PNs. In the latter the coupling was essentially mediated by CB. The control of this coupling via the spine neck, which itself undergoes use-dependent regulation, will increase the computational capacitance of dendrites. Despite growing evidence for these two additional functions, decades of investigation on CB mainly underlined its importance as a Ca2+-buffer.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I thank Oliver Arendt for critical reading of the manuscript. This work was supported by the DFG (EI 341/4–2).
Airaksinen, M. S., Eilers, J., Garaschuk, O., Thoenen, H., Konnerth, A., and Meyer, M. (1997). Ataxia and altered dendritic calcium signalling in mice carrying a targeted nullmutation of the calbindin D28k gene. Proc. Natl. Acad. Sci. U.S.A. 94, 1488–1493.
Åkerfeldt, K. S., Coyne, A. N., Wilk, R. R., Thulin, E., and Linse, S. (1996). Ca2+-binding stoichiometry of calbindin D28k as assessed by spectroscopic analyses of synthetic peptide fragments. Biochemistry 35, 3662–3669.
Atluri, P. P., and Regehr, W. G. (1996). Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J. Neurosci. 16, 5661–5671.
Augustine, G. J., Santamaria, F., and Tanaka, K. (2003). Local calcium signaling in neurons. Neuron 40, 331–346.
Barski, J. J., Hartmann, J., Rose, C. R., Hoebeek, F., Mörl, K., Noll-Hussong, M., De Zeeuw, C. I., Konnerth, A., and Meyer, M. (2003). Calbindin in cerebellar Purkinje cells is a critical determinant of the precision of motor coordination. J. Neurosci. 23, 3469–3477.
Bellido, T., Huening, M., Raval-Pandya, M., Manolagas, S. C., and Christakos, S. (2000). Calbindin-D28k is expressed in osteoblastic cells and suppresses their apoptosis by inhibiting caspase-3 activity. J. Biol. Chem. 275, 26328–26332.
Berggård, T., Miron, S., Önnerfjord, P., Thulin, E., Åkerfeldt, K. S., Enghild, J. J., Akke, M., and Linse, S. (2002a). Calbindin D28k exhibits properties characteristic of a Ca2+ sensor. J. Biol. Chem. 277, 16662–16672.
Berggård, T., Szczepankiewicz, O., Thulin, E., and Linse, S. (2002b). Myo-inositol monophosphatase is an activated target of calbindin D28k. J. Biol. Chem. 277, 41954–41959.
Berggård, T., Silow, M., Thulin, E., and Linse, S. (2000). Ca2+- and H+-dependent conformational changes of calbindin D28k. Biochemistry 39, 6864–6873.
Blatow, M., Caputi, A., Burnashev, N., Monyer, H., and Rozov, A. (2003). Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-d28k-containing terminals. Neuron 38, 79–88.
Bronner, F. (1989). Renal calcium transport: mechanisms and regulation-an overview. Am. J. Physiol. 257, F707–F711.
Bronner, F., and Stein, W. D. (1988). CaBPr facilitates intracellular diffusion for Ca pumping in distal convoluted tubule. Am. J. Physiol. 255, F558–F562.
Caillard, O., Moreno, H., Schwaller, B., Llano, I., Celio, M. R., and Marty, A. (2000). Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 97, 13372–13377.
Cedervall, T., Andre, I., Selah, C., Robblee, J. P., Krecioch, P. C., Fairman, R., Linse, S., and Akerfeldt, K. S. (2005). Calbindin D28k EF-hand ligand binding and oligomerization: four high-affinity sites–three modes of action. Biochemistry 44, 13522–13532.
Celio, M. R., Pauls, T., and Schwaller, B. (1996). Guidebook to the Calcium-Binding Proteins. Oxford: Sambrook & Tooze Publication at Oxford University Press.
Connor, J. A., Kretz, R., and Shapiro, E. (1986). Calcium levels measured in a presynaptic neurone of Aplysia under conditions that modulate transmitter release. J. Physiol. 375, 625–642.
da Silva, A. C., and Reinach, F. C. (1991). Calcium binding induces conformational changes in muscle regulatory proteins. Trends Biochem. Sci. 16, 53–57.
Daniel, H., Levenes, C., and Crepél, F. (1998). Cellular mechanisms of cerebellar LTD. Trends Neurosci. 21, 401–407.
Eggermann, E., Bucurenciu, I., Goswami, S. P., and Jonas, P. (2012). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 13, 7–21.
Eggermann, E., and Jonas, P. (2012). How the ‘slow’ Ca2+ buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat. Neurosci. 15, 20–22.
Eilers, J., Augustine, G. J., and Konnerth, A. (1995). Subthreshold synaptic Ca2+ signalling in fine dendrites and spines of cerebellar Purkinje neurons. Nature 373, 155–158.
Eilers, J., Takechi, H., Finch, E. A., Augustine, G. J., and Konnerth, A. (1997). Local dendritic Ca2+ signaling induces cerebellar LTD. Learn. Mem. 3, 159–168.
Faas, G. C., Raghavachari, S., Lisman, J. E., and Mody, I. (2011). Calmodulin as a direct detector of Ca2+ signals. Nat. Neurosci. 14, 301–306.
Faas, G. C., Schwaller, B., Vergara, J. L., and Mody, I. (2007). Resolving the fast kinetics of cooperative binding: Ca2+ buffering by calretinin. PLoS Biol. 5, 2646–2660. doi: 10.1371/journal.pbio.0050311
Felmy, F., Neher, E., and Schneggenburger, R. (2003). Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron 37, 801–811.
Fierro, L., and Llano, I. (1996). High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J. Physiol. 496, 617–625.
Gabso, M., Neher, E., and Spira, M. E. (1997). Low mobility of the Ca2+ buffers in axons of cultured Aplysia neurons. Neuron 18, 473–481.
Gamble, E., and Koch, C. (1987). The dynamics of free calcium in dendritic spines in response to repetitive synaptic input. Science 236, 1311–1315.
Grütter, M. G. (2000). Caspases: key players in programmed cell death. Curr. Opin. Struct. Biol. 10, 649–655.
Hackney, C. M., Mahendrasingam, S., Penn, A., and Fettiplace, R. (2005). The concentrations of calcium buffering proteins in mammalian cochlear hair cells. J. Neurosci. 25, 7867–7875.
Holthoff, K., Tsay, D., and Yuste, R. (2002). Calcium dynamics of spines depend on their dendritic location. Neuron 33, 425–437.
Hubbard, M. J., and McHugh, N. J. (1995). Calbindin28kDa and calbindin30kDa (calretinin) are substantially localised in the particulate fraction of rat brain. FEBS Lett. 374, 333–337.
Ikura, M. (1996). Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 21, 14–17.
Ito, M. (2001). Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol. Rev. 81, 1143–1195.
Jackson, M. B., and Redman, S. J. (2003). Calcium dynamics, buffering, and buffer saturation in the boutons of dentate granule-cell axons in the hilus. J. Neurosci. 23, 1612–1621.
Katz, B., and Miledi, R. (1968). The role of calcium in neuromuscular facilitation. J. Physiol. 195, 481–492.
Kim, S. A., Heinze, K. G., Waxham, M. N., and Schwille, P. (2004). Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl. Acad. Sci. U.S.A. 101, 105–110.
Klaus, W., Grzesiek, S., Labhardt, A. M., Buchwald, P., Hunziker, W., Gross, M. D., and Kallick, D. A. (1999). NMR investigation and secondary structure of domains I and II of rat brain calbindin D28k (1–93). Eur. J. Biochem. 262, 933–938.
Kojetin, D. J., Venters, R. A., Kordys, D. R., Thompson, R. J., Kumar, R., and Cavanagh, J. (2006). Structure, binding interface and hydrophobic transitions of Ca2+-loaded calbindin-D28K. Nat. Struct. Mol. Biol. 13, 641–647.
Kosaka, T., Kosaka, K., Nakayama, T., Hunziker, W., and Heizmann, C. W. (1993). Axons and axon terminals of cerebellar Purkinje cells and basket cells have higher levels of parvalbumin immunoreactivity than somata and dendrites: quantitative analysis by immunogold labeling. Exp. Brain Res. 93, 483–491.
Koster, H. P., Hartog, A., Van Os, C. H., and Bindels, R. J. (1995). Calbindin-D28K facilitates cytosolic calcium diffusion without interfering with calcium signaling. Cell Calcium 18, 187–196.
Kreiner, L., Christel, C. J., Benveniste, M., Schwaller, B., and Lee, A. (2010). Compensatory regulation of Cav2.1 Ca2+ channels in cerebellar Purkinje neurons lacking parvalbumin and calbindin D-28k. J. Neurophysiol. 103, 371–381.
Kreiner, L., and Lee, A. (2006). Endogenous and exogenous Ca2+ buffers differentially modulate Ca2+-dependent inactivation of Cav2.1 Ca2+ channels. J. Biol. Chem. 281, 4691–4698.
Lambers, T. T., Mahieu, F., Oancea, E., Hoofd, L., De Lange, F., Mensenkamp, A. R., Voets, T., Nilius, B., Clapham, D. E., Hoenderop, J. G., and Bindels, R. J. (2006). Calbindin-D28k dynamically controls TRPV5-mediated Ca2+ transport. Embo J. 25, 2978–2988.
Leathers, V. L., Linse, S., Forsen, S., and Norman, A. W. (1990). Calbindin-D28k, a 1 alpha,25-dihydroxyvitamin D3-induced calcium-binding protein, binds five or six Ca2+ ions with high affinity. J. Biol. Chem. 265, 9838–9841.
Lee, A., Scheuer, T., and Catterall, W. A. (2000a). Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J. Neurosci. 20, 6830–6838.
Lee, S-H., Schwaller, B., and Neher, E. (2000b). Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for Ca2+ transients of neuronal dendrites. J. Physiol. 525, 419–432.
Lee, A., Wong, S. T., Gallagher, D., Li, B., Storm, D. R., Scheuer, T., and Catterall, W. A. (1999). Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399, 155–159.
Lutz, W., Frank, E. M., Craig, T. A., Thompson, R., Venters, R. A., Kojetin, D., Cavanagh, J., and Kumar, R. (2003). Calbindin D28K interacts with Ran-binding protein M: identification of interacting domains by NMR spectroscopy. Biochem. Biophys. Res. Commun. 303, 1186–1192.
Maeda, H., Ellis-Davies, G. C., Ito, K., Miyashita, Y., and Kasai, H. (1999). Supralinear Ca2+ signaling by cooperative and mobile Ca2+ buffering in Purkinje neurons. Neuron 24, 989–1002.
Majewska, A., Brown, E., Ross, J., and Yuste, R. (2000). Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J. Neurosci. 20, 1722–1734.
Markram, H., Roth, A., and Helmchen, F. (1998). Competitive calcium binding: implications for dendritic calcium signaling. J. Comput. Neurosci. 5, 331–348.
Meinrenken, C. J., Borst, J. G., and Sakmann, B. (2002). Calcium secretion coupling at calyx of held governed by nonuniform channel-vesicle topography. J. Neurosci. 22, 1648–1667.
Molinari, S., Battini, R., Ferrari, S., Pozzi, L., Killcross, A. S., Robbins, T. W., Jouvenceau, A., Billard, J. M., Dutar, P., Lamour, Y., Baker, W. A., Cox, H., and Emson, P. C. (1996). Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc. Natl. Acad. Sci. U.S.A. 93, 8028–8033.
Morgan, D. W., Welton, A. F., Heick, A. E., and Christakos, S. (1986). Specific in vitro activation of Ca, Mg-ATPase by vitamin D-dependent rat renal calcium binding protein (calbindin D28K). Biochem. Biophys. Res. Commun. 138, 547–553.
Müller, W., and Connor, J. A. (1991). Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature 354, 73–76.
Müller, A., Kukley, M., Stausberg, P., Beck, H., Müller, W., and Dietrich, D. (2005). Endogenous Ca2+ buffer concentration and Ca2+ microdomains in hippocampal neurons. J. Neurosci. 25, 558–565.
Nägerl, U. V., Novo, D., Mody, I., and Vergara, J. L. (2000). Binding kinetics of calbindin-D28k determined by flash photolysis of caged Ca2+. Biophys. J. 79, 3009–3018.
Naraghi, M. (1997). T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium 22, 255–268.
Naraghi, M., and Neher, E. (1997). Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of Ca2+ at the mouth of a calcium channel. J. Neurosci. 17, 6961–6973.
Neher, E. (1998a). Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium 24, 345–357.
Neher, E. (1998b). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399.
Nemri, A., and Ghisovan, N. (2007). Dendritic spines: it takes two to make an impression. J. Physiol. 582, 15–16.
Noguchi, J., Matsuzaki, M., Ellis-Davies, G. C., and Kasai, H. (2005). Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron 46, 609–622.
Orduz, D., and Llano, I. (2007). Recurrent axon collaterals underlie facilitating synapses between cerebellar Purkinje cells. Proc. Natl. Acad. Sci. U.S.A. 104, 17831–17836.
Reisner, P. D., Christakos, S., and Vanaman, T. C. (1992). In vitro enzyme activation with calbindin-D28k, the vitamin D-dependent 28 kDa calcium binding protein. FEBS Lett. 297, 127–131.
Rozov, A., Burnashev, N., Sakmann, B., and Neher, E. (2001). Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol. 531, 807–826.
Sabatini, B. L., Oertner, T. G., and Svoboda, K. (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33, 439–452.
Schmidt, H., Arendt, O., and Eilers, J. (2011). Diffusion and extrusion shape standing calcium gradients during ongoing parallel fiber activity in dendrites of Purkinje neurons. Cerebellum doi: 10.1007/s12311-010-0246-x. [Epub ahead of print].
Schmidt, H., and Eilers, J. (2009). Spine neck geometry determines spino-dendritic cross-talk in the presence of mobile endogenous calcium binding proteins. J. Comput. Neurosci. 27, 229–243.
Schmidt, H., Kunerth, S., Wilms, C., Strotmann, R., and Eilers, J. (2007). Spino-dendritic cross-talk in rodent Purkinje neurons mediated by endogenous Ca2+-binding proteins. J. Physiol. 581, 619–629.
Schmidt, H., Schwaller, B., and Eilers, J. (2005). Calbindin D28k targets myo-inositol monophosphatase in spines and dendrites of cerebellar Purkinje neurons. Proc. Natl. Acad. Sci. U.S.A. 102, 5850–5855.
Schmidt, H., Stiefel, K., Racay, P., Schwaller, B., and Eilers, J. (2003). Mutational analysis of dendritic Ca2+ kinetics in rodent Purkinje cells: role of parvalbumin and calbindin D28k. J. Physiol. 551, 13–32.
Schwaller, B. (2008). The continuing disappearance of “pure” Ca2+ buffers. Cell. Mol. Life Sci. 66, 275–300.
Svoboda, K., Tank, D. W., and Denk, W. (1996). Direct measurement of coupling between dendritic spines and shafts. Science 272, 716–719.
Tsujimoto, T., Jeromin, A., Saitoh, N., Roder, J. C., and Takahashi, T. (2002). Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science 295, 2276–2279.
Vanbelle, C., Halgand, F., Cedervall, T., Thulin, E., Akerfeldt, K. S., Laprevote, O., and Linse, S. (2005). Deamidation and disulfide bridge formation in human calbindin D28k with effects on calcium binding. Protein Sci. 14, 968–979.
Vecellio, M., Schwaller, B., Meyer, M., Hunziker, W., and Celio, M. R. (2000). Alterations in Purkinje cell spines of calbindin D-28 k and parvalbumin knock-out mice. Eur. J. Neurosci. 12, 945–954.
Venters, R. A., Benson, L. M., Craig, T. A., Paul, K. H., Kordys, D. R., Thompson, R., Naylor, S., Kumar, R., and Cavanagh, J. (2003). The effects of Ca2+ binding on the conformation of calbindin D28K: a nuclear magnetic resonance and microelectrospray mass spectrometry study. Anal. Biochem. 317, 59–66.
Venyaminov, S. Y., Klimtchuk, E. S., Bajzer, Z., and Craig, T. A. (2004). Changes in structure and stability of calbindin-D(28K) upon calcium binding. Anal. Biochem. 334, 97–105.
Winsky, L., and Kuźnicki, J. (1995). Distribution of calretinin, calbindin D28k, and parvalbumin in subcellular fractions of rat cerebellum: effects of calcium. J. Neurochem. 65, 381–388.
Yamada, W. M., and Zucker, R. S. (1992). Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys. J. 61, 671–682.
Yan, N., and Shi, Y. (2005). Mechanisms of apoptosis through structural biology. Annu. Rev. Cell Dev. Biol. 21, 35–56.
Zador, A., Koch, C., and Brown, T. H. (1990). Biophysical model of a Hebbian synapse. Proc. Natl. Acad. Sci. U.S.A. 87, 6718–6722.
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405.
Keywords: calcium, sensor, transporter, buffer, synaptic plasticity, neurons, transmitter release
Citation: Schmidt H (2012) Three functional facets of calbindin D-28k. Front. Mol. Neurosci. 5:25. doi: 10.3389/fnmol.2012.00025
Received: 12 January 2012; Paper pending published: 30 January 2012;
Accepted: 14 February 2012;Published online: 15 March 2012.
Edited by:
Beat Schwaller, University of Fribourg, SwitzerlandReviewed by:
Guido C. Faas, University of California, USACopyright: © 2012 Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hartmut Schmidt, Medical Faculty, Carl-Ludwig Institute for Physiology, University of Leipzig, Liebigstr. 27, 04103 Leipzig, Germany. e-mail:aGFydG11dC5zY2htaWR0QG1lZGl6aW4udW5pLWxlaXB6aWcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.