

- 1 Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
- 2 Instituto de Química Medica – CSIC, Madrid, Spain
Inhibiting glycogen synthase kinase-3 (GSK-3) activity via pharmacological intervention has become an important strategy for treating neurodegenerative and psychiatric disorders. The known GSK-3 inhibitors are of diverse chemotypes and mechanisms of action and include compounds isolated from natural sources, cations, synthetic small-molecule ATP-competitive inhibitors, non-ATP-competitive inhibitors, and substrate–competitive inhibitors. Here we describe the variety of GSK-3 inhibitors with a specific emphasis on their biological activities in neurons and neurological disorders. We further highlight our current progress in the development of non-ATP-competitive inhibitors of GSK-3. The available data raise the hope that one or more of these drug design approaches will prove successful at stabilizing or even reversing the aberrant neuropathology and cognitive deficits of certain central nervous system disorders.
Introduction
The serine/threonine kinase GSK-3 is a conserved signaling molecule with essential roles in diverse biological processes. Aberrant GSK-3 activity has been linked with several human diseases including diabetes, inflammation, and neurodegenerative and psychiatric disorders (Eldar-Finkelman, 2002; Doble and Woodgett, 2003; Gould et al., 2004b; Jope et al., 2007; Hernandez and Avila, 2008; Hooper et al., 2008; Hur and Zhou, 2010). This supported the hypothesis that inhibition of GSK-3 will have therapeutic benefit and intensive efforts have been made in the search for and design of selective GSK-3 inhibitors. The reported GSK-3 inhibitors are of diverse chemotypes and mechanisms of action. These include inhibitors isolated from natural sources, cations, and synthetic small molecules. Regarding the mechanism of inhibition, we can find ATP-competitive inhibitors, non-ATP-competitive inhibitors, and substrate–competitive inhibitors. A major challenge in the field is achieving specificity, and advanced structure-based computational studies are conducted to improve GSK-3 inhibitor specificity and possibly to ensure targeting of specific GSK-3 isozymes. Here we review the state of the art of GSK-3 inhibitors with focus on their biological activities in neurons and neurological disorders. We further highlight our current progress in the development of non-ATP-competitive inhibitors of GSK-3 and their implications in CNS disorders.
Evolutionary, Structural, and Regulatory Features of GSK-3-Leadings in Drug Design
The importance of GSK-3 as a therapeutic target highlighted the need for in depth understanding of different features of GSK-3 with respect to sequence, structure, and regulation. GSK-3 is highly conserved in the animal kingdom. In mammals, GSK-3 is expressed as two isozymes: GSK-3α and GSK-3β. An alternative splice variant of GSK-3β, GSK-3β2, has also been reported (Mukai et al., 2002). GSK-3α and β share extensive similarities in their catalytic domains, but differ in their N- and C-terminal regions (Woodgett, 1990). In lower organisms such as choanoflagellates, sea squirts, and nematodes, a single gene encodes GSK-3 (Alon et al., 2011), whereas in vertebrates such as fish, amphibians, reptiles, and lizards the two genes coding for GSK-3α and β are identified; interestingly birds lack a copy of the GSK-3α gene (Alon et al., 2011). The existence of two GSK-3 isozymes suggested that at least one of the isozymes took on unique functions tied to the emergence of vertebrates, likely related to the development of highly ordered systems such as the central nervous system (CNS). Recent studies had found certain physiological differences between GSK-3 isozymes in functions related to embryonic development, brain structure, and behavior; although other studies clearly demonstrated redundant function for the two isozymes (Hoeflich et al., 2000; Hernandez et al., 2002; Prickaerts et al., 2006; Terwel et al., 2008; Kaidanovich-Beilin et al., 2009; Kim et al., 2009; Mines et al., 2010; Alon et al., 2011; Soutar et al., 2011). Our understanding of the distinct functions of GSK-3 isozymes in neuronal systems and, in particular, their relative contributions to neuropathologies is far from clear. This is of particular importance as we seek to determine the worthiness of development of isozyme-specific inhibitors.
Like other protein kinases, GSK-3 is composed of a conserved catalytic domain folded into a bi-lobal architecture with a smaller N-terminal lobe responsible for ATP binding and a larger, globular C-terminal domain that contains the conserved “activation loop” important for the kinase activity (Hanks and Hunter, 1995; Taylor et al., 1995). Tyrosine residue located within the activation loop is essential for full activation of GSK-3, and this process is a chaperone-dependent auto-phosphorylation event (Hughes et al., 1993; Cole et al., 2004; Lochhead et al., 2006). GSK-3 activity is further regulated by regions outside the catalytic domain. Its N-terminal end contains a highly conserved RPRTTSF motif that acts as an auto-inhibitory pseudosubstrate when phosphorylated at serine (Frame and Cohen, 2001; Ilouz et al., 2008). Another site located within the C-terminal region of GSK-3β (Thr 390) was recently identified as an inhibitory site (Thornton et al., 2008). In some instances, GSK-3 activity is also regulated via interactions with regulatory proteins. For example, GSK-3 interacts with presenilin proteins that, in turn, may regulate the production of the Alzheimer’s amyloid beta peptide (Aβ; Phiel et al., 2003). Interaction of GSK-3 with the scaffold protein Axin regulates the stability of the Wnt signaling effector β-catenin (Ikeda et al., 1998; Wu and Pan, 2011). FRAT/GBP competes with Axin and inhibits GSK-3 activity toward β-catenin (Yost et al., 1998). Axin and FRAT bind to GSK-3 via a similar hydrophobic patch located within the C-terminal region of GSK-3 (Fraser et al., 2002), this interaction has been exploited for inhibitor design (Hedgepeth et al., 1999; Thomas et al., 1999). Finally, the intracellular distribution of GSK-3 isozymes is differentially regulated (Diehl et al., 1998; Bijur and Jope, 2003; Meares and Jope, 2007; Caspi et al., 2008; Adachi et al., 2011; Azoulay-Alfaguter et al., 2011; Wu and Pan, 2011). Hence, GSK-3 function can be regulated at many levels, which in turn, may be exploited in development of selective GSK-3 inhibitors.
Unlike other protein kinases, GSK-3 is constitutively active in resting conditions and is inhibited in response to upstream signals. It can be inhibited or over-activated by diverse post-transductional modifications such as phosphorylation in response to upstream signals (Eldar-Finkelman, 2002). In addition, GSK-3 shows a unique preference in substrate recognition as it requires pre-phosphorylation of its substrates in the context of SXXXS(p) (Woodgett and Cohen, 1984; Fiol et al., 1994). Crystallographic studies of GSK-3β identified a phosphate binding pocket comprised of three basic residues, Arg 96, Lys 205, and Arg 189, that presumably binds the phosphorylated substrate (Dajani et al., 2001; ter Haar et al., 2001). Phosphorylation of GSK-3-downstream targets typically results in attenuation of the signaling pathway and/or inhibition of the substrate’s activity. In neurons, GSK-3 is intimately involved with control of apoptosis, synaptic plasticity, axon formation, and neurogenesis (Crowder and Freeman, 2000; Jiang et al., 2005; Yoshimura et al., 2005; Kim et al., 2006; Zhao et al., 2007; Muyllaert et al., 2008; Hur and Zhou, 2010). In vivo studies indicate that over-activity of GSK-3 results in adverse effects. This over-activity should be produced by an increase in GSK-3 expression or by an imbalance of its phosphorylation state leading to a super-active enzymatic state. Transgenic animals that overexpress GSK-3 display alterations in brain size, impaired long-term potentiation (LTP), and deficits in learning and memory (Lucas et al., 2001; Hernandez et al., 2002; Spittaels et al., 2002; Hooper et al., 2008). These animals also have characteristics typical of Alzheimer’s disease such as hyperphosphorylation of tau and enhanced production of Aβ peptide (Lucas et al., 2001; Phiel et al., 2003; Engel et al., 2006; Rockenstein et al., 2007). In addition, data from pharmacological and genetic models implicate GSK-3 activity in mood behavior and indicate that elevated GSK-3 activity is associated with manic and depressive behavior (Gould et al., 2004a; Kaidanovich-Beilin et al., 2004; O’Brien et al., 2004; Prickaerts et al., 2006; Beaulieu et al., 2008; Mines et al., 2010; Polter et al., 2010). Finally, abnormal regulation of GSK-3 activity was reported in patients with Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), major depression, schizophrenia, and bipolar disorder (Kozlovsky et al., 2002; Hu et al., 2003b; Hye et al., 2005; Karege et al., 2007; Lovestone et al., 2007; Pandey et al., 2010; Saus et al., 2010; Forlenza et al., 2011). Hence, increasing efforts are focused on development of selective GSK-3 inhibitors able to modulate this abnormal over-activity.
Small Metal Cations as GSK-3 Inhibitors
The cation lithium was the first “natural” GSK-3 inhibitor discovered (Klein and Melton, 1996; Stambolic et al., 1996). Lithium (meaning lithium salts) is a mood stabilizer long used in treatment of bipolar disorders. Lithium inhibits GSK-3 directly by competition with magnesium ions (Klein and Melton, 1996; Ryves and Harwood, 2001) and indirectly via enhanced serine phosphorylation and autoregulation (De Sarno et al., 2001; Zhang et al., 2003; Kirshennboim et al., 2004). Lithium has been widely used in many studies as a pharmacological inhibitor of GSK-3; these demonstrated that lithium produces similar biological consequences as inhibition of GSK-3 via other means. For example, treatment with lithium increases cellular β catenin levels (Stambolic et al., 1996; O’Brien and Klein, 2009), reduces tau phosphorylation at GSK-3 epitopes in neurons (Noble et al., 2005), activates glycogen synthase (Cheng et al., 1983), and promotes embryonic axis duplication (Klein and Melton, 1996). Lithium has striking morphological effects on neurons including a reduction in axon length, increase in growth cone area, and an increase in synapse formation (Burstein et al., 1985; Takahashi et al., 1999; Owens et al., 2003; Kim and Thayer, 2009). The therapeutic range of lithium is 0.5–1.5 mM, and its IC50 toward GSK-3 is 1–2 mM (Klein and Melton, 1996), suggested that lithium may clinically inhibit GSK-3. Indeed, numerous studies have evaluated the therapeutic activity of lithium in various neuronal systems, and verified a profound effect of lithium in neuroprotection against variety of insults in apoptotic and brain injury paradigms (Bijur et al., 2000; Hongisto et al., 2003; Perez et al., 2003; Williams et al., 2004; Jin et al., 2005; Wada et al., 2005; Brewster et al., 2006; Chuang and Manji, 2007; Mathew et al., 2008).
Lithium has been then tested in Alzheimer’s and related neurodegenerative models. These studies demonstrated that lithium blocks amyloid precursor protein (APP) deposits and reduces Aβ secretion in cells and transgenic mice overexpressing APP (Sun et al., 2002; Phiel et al., 2003; Rockenstein et al., 2007). Treatment with lithium also prevented Aβ neurotoxicity in rat brain (De Ferrari et al., 2003) and reduced tauopathy in transgenic mice overexpressing human mutant tau (Noble et al., 2005; Caccamo et al., 2007). Lithium was shown to provide therapeutic benefit in models of epileptic neurodegeneration (Busceti et al., 2007), motor performance in Huntington’s disease (Wood and Morton, 2003), and hippocampal neuropathology and neurological functions in spinocerebellar ataxia type 1 (SCA1; Watase et al., 2007). However, some studies reported that lithium had no effect on tau phosphorylation, Aβ loads, and neuroprotection (Ghribi et al., 2003; Song et al., 2004; Caccamo et al., 2007). These could be due to differences in the experimental sets (e.g., age, dose, time of treatment etc.) used in the different studies. Several clinical trials with lithium in AD and elderly patients have been conducted but results are not conclusive. Chronic treatment with lithium yielded positive results in dementia patients (Havens and Cole, 1982) and improved cognition and memory scores (MMSE) in patients receiving the drug as compared to non-treated patients (Terao et al., 2006). In AD patients, lithium reversed the reduction in brain-derived neurotrophic factor (BDNF) serum concentrations (Leyhe et al., 2009) and reduced the prevalence of AD in elderly patients with bipolar disorder (Nunes et al., 2007). A different clinical study found that treatment with lithium slowed the progression in ALS (Bedlack et al., 2008) via autophagy-induced degradation of aggregate-prone proteins (Bedlack et al., 2008). Other clinical trials, however, did not confirm the ability of lithium to prevent dementia or improve cognition or to reduce tau phosphorylation or Aβ levels in AD patients (Pachet and Wisniewski, 2003; Dunn et al., 2005; Hampel et al., 2009). In addition, lithium had toxic side effects in some elderly patients (Macdonald et al., 2008), and studies with lithium were thus discontinued (Tariot and Aisen, 2009).
Other metal ions such as beryllium, zinc, mercury, and copper are potent GSK-3 inhibitors (Ilouz et al., 2002; Ryves et al., 2002). Interestingly, these cations are more potent inhibitors of GSK-3 than lithium (IC50 in the micromolar concentration range as compared to the IC50 of lithium which is within the millimolar concentration range). Of particular interest is the trace element zinc, that unlike lithium, and other metal ions is naturally found in the body tissues. Zinc inhibits GSK-3 in the low micromolar range (IC50 = 15 μM) and elevates cellular β-catenin levels (Ilouz et al., 2002). It is noteworthy that zinc levels are linked with major depression and mental functions. In animal models, zinc deficiency results in increased depressive- and anxiety-like behaviors (Kroczka et al., 2001; Tassabehji et al., 2008), and treatment with zinc produces anti-depressive like activity in the mouse forced swimming test (FST) model (Kroczka et al., 2001). Hypozincemia is often detected in patients with major depression, and dietary supplementation of zinc improves symptoms of depression (Bodnar and Wisner, 2005; Nowak et al., 2005). Hence, it is tempting to speculate that the therapeutic activity of zinc in mood behavior and other cognitive symptoms is mediated by its ability to inhibit GSK-3. However, zinc is a co-factor of many enzymes and, like lithium, may initiate many cellular effects independently of GSK-3. Still, research with lithium and zinc may further our understanding of the biological functions of GSK-3 in man. Worth mentioning is also the indirect GSK-3 inhibition produced by the inorganic salt sodium tungstate (Gómez-Ramos et al., 2006). As a consequence of the GSK-3 inactivation, the phosphorylation of several GSK-3 dependent sites of the microtubule tau protein decreases (Gómez-Ramos et al., 2006). This fact points to a new potential drug for treatment of AD. Remarkably, this compound has a low toxicity profile and is currently in phase I of clinical trials as an antiobesity agent. Altogether, although mechanisms of action is not fully clear, these cations may serve as a stepping stone for development of new GSK-3 inhibitors that mimic their inhibitory paradigms.
Organic Molecules as GSK-3 Inhibitors
Much effort is done in the discovery and development of GSK-3 inhibitors in the last years being a very active field of research for academic centers and pharmaceutical companies. Nowadays, several chemical families have emerged as GSK-3 inhibitors, including great chemical diversity. Some of these GSK-3 inhibitors have synthetic origin but others have been derived directly or indirectly from small molecules of natural origin. Worth mentioning is the fact that the marine environment has been shown recently to provide a source of chemical structures with promising biological activities for CNS diseases. In fact, marine invertebrates have played a prominent role in the generation of novel GSK-3 inhibitors.
The number of small molecule GSK-3 inhibitors is continuously increasing with most in the early discovery phase. Here, we mainly focus on the GSK-3 inhibitors that have been tested in biological systems. Collectively, these studies provided compelling evidence of the specific roles of GSK-3 in neuronal functions under both normal and pathological conditions. Generally speaking, inhibition of GSK-3 has profound effects on neuroprotection, self renewal and pluripotency in embryonic stem (ES) cells, axonal morphogenesis, and mood behavior. As kinase selectivity is one of the main hazards in the development of this class of therapeutic agents, we will discuss GSK-3 inhibitors based on their ability to compete or not with ATP.
GSK-3 ATP-Competitive Inhibitors from Natural Resources
Many of this kind of GSK-3 inhibitors isolated from marine organisms were identified during the search for inhibitors for cyclin-dependent protein kinases (CDKs) with anti-tumor activity. The dual activity of these inhibitors (and others) toward GSK-3 and CDKs is a direct result of their structural similarity within the ATP-binding domain (about 86% sequence similarity).
The bis-indole indirubin isolated from the traditional Chinese medicine for treatment of myelocyte leukemia was initially characterized as a CDK inhibitor and then found to be a potent GSK-3 inhibitor (Hoessel et al., 1999; Leclerc et al., 2001). It inhibits both protein kinases within the nanomolar concentration range. The indirubin analogs that were synthesized and tested (collectively termed here “indirubins”) showed inhibitory activity toward both GSK-3 and CDKs (Leclerc et al., 2001; Meijer et al., 2003). The indirubin analog 6-bromoindirubin, isolated from a marine invertebrate, the mollusk known as “Tyrian purple,” showed a certain selectivity toward GSK-3 over CDKs (Meijer et al., 2003). Accordingly, a synthetic cell-permeable derivative, 6-bromoindirubin-3′-oxime (6BIO; Figure 1), was developed and was about 16-fold more selective for GSK-3 relative to CDKs (Meijer et al., 2003; Polychronopoulos et al., 2004). The biological activity of 6BIO has been evaluated in several neuronal systems. It reduced tau phosphorylation in cultured cortical neurons (Martin et al., 2009), and inhibited neurite outgrowth in cerebellar and embryonic or postnatal dorsal root ganglion (DRG) neurons (Kim et al., 2006; Alabed et al., 2011). This effect appeared to be dependent on the degree of GSK-3 inhibition, as a weak inhibition of GSK-3 promoted axon branching (Kim et al., 2006). 6BIO enhanced self renewal and pluripotency in human ES cells (Sato et al., 2004), via its ability to act as a Wnt mimetic (Sato et al., 2004). Additional studies with indirubins demonstrated their ability to provide neuroprotection against kainic acid, MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), and trophic deprivation (Hongisto et al., 2003; Jin et al., 2005; Wang et al., 2007; Magiatis et al., 2010). In vivo systemic administration of indirubin-3′-oxime to APP/Presenilin-1 transgenic mice, a established AD model, attenuated many AD symptoms including tau hyperphosphorylation, Aβ accumulation, inflammation, and spatial memory deficits (Ding et al., 2010). In contrast, treatment with indirubin-3′-oxime did not reduce tau phosphorylation in cultured neurons or in the rat brain (Selenica et al., 2007). These discrepancies could be due to the limited bioavailability of indirubin-3′-oxime, which, like other indirubins, has limited water solubility (Leclerc et al., 2001). Recent work reported the synthesis of additional indirubin derivatives with improved water solubility (Vougogiannopoulou et al., 2008). Their biological activity has not yet been reported.
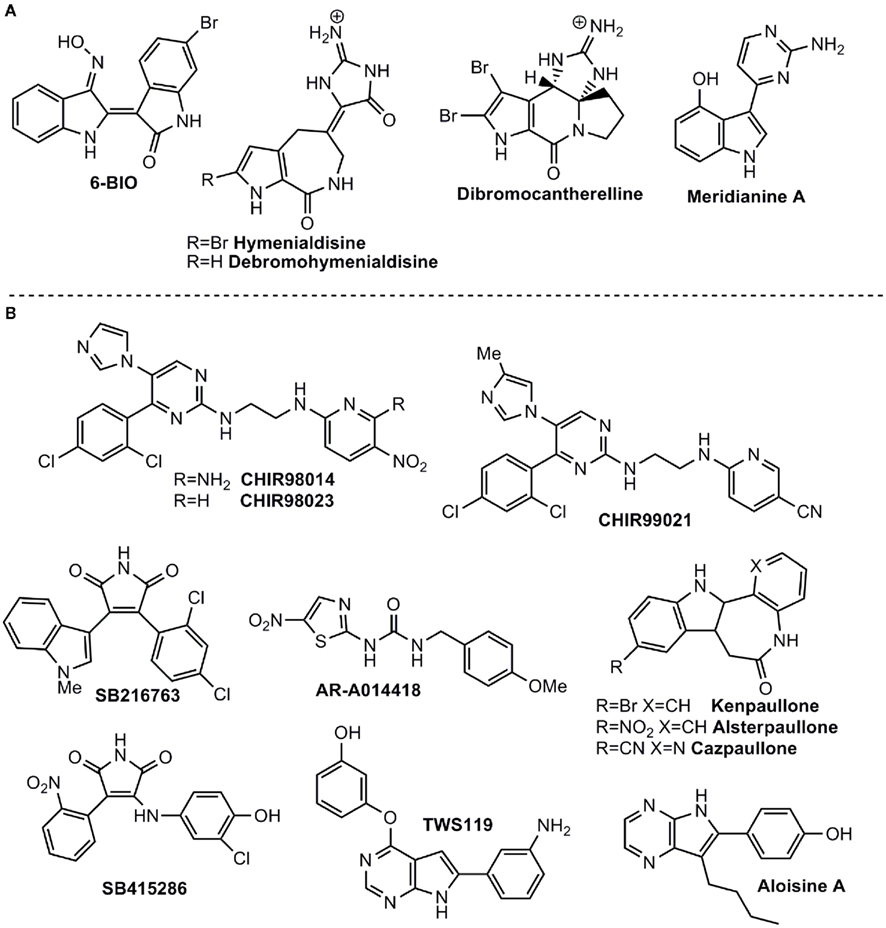
Figure 1. Some ATP-competitive GSK-3 inhibitors with potential for CNS disorders. (A) Isolated from marine organisms. (B) Small molecules from organic synthesis programs.
Sponges (Porifera), as the best known source of bioactive marine natural products in metazoans, play a significant role in marine drug discovery and development. The alkaloids debromohymenialdisine (DBH) and hymenialdisine (HD), which contain both bromopyrrole and guanidine groups (Williams and Faulkner, 1996; Figure 1) were originally isolated from sponges belonging to the Agelasidae, Axinellidae, and Halichondridae. HD structure was established using X-ray crystallography (Cimino et al., 1982) and it is a potent protein kinase inhibitor targeting GSK-3β, CDKs, MEK1, CK1, and Chk1 (Tasdemir et al., 2002; Sharma and Tepe, 2004) among others. In addition, HD has the ability to inhibit GSK-3β in vivo and also blocks the in vivo phosphorylation of the microtubule-binding protein tau at sites which are hyperphosphorylated by GSK-3 in AD (Meijer et al., 2000). As observed with other GSK-3 inhibitors, HD, and derivatives act in competition with ATP (Wan et al., 2004). Recently, it has been shown that HD and DBH are stored in spherulous cells from Axinella sp. (Song et al., 2011), which can help to define the bioactive production strategy in terms of sponge aquaculture.
The potent activity of HD as a competitive kinase inhibitor aroused interest in synthesizing a pyrrole-azepin-8-one ring system bonded to a glycocyamidine ring scaffold (Nguyen and Tepe, 2009). Crystallographic data of the HD complex with CDK2, led to the rational development of new analogs with increased potency and selectivity over GSK-3, CDK5, and CDK1. Moreover, using different docking methods and molecular dynamics simulation, the structural determinants that govern target selectivity on HD derivatives has been studied, explaining the significant inhibitory effect on GSK-3 of the related HD metabolite dibromocantharelline (Zhang et al., 2011). The specific residue Cys199 near the binding site of GSK-3 provides new clues for the design of potent and selective inhibitors.
Meridianins are brominated 3-(2-aminopyrimidine)-indoles, which are isolated from the tunicate Aplidium meridianum, an ascidian collected near the South Georgia Islands. These compounds inhibit various protein kinases such as CDKs, GSK-3, PKA, and CK1 (Gompel et al., 2004; Figure 1). The ability of Meridianins to prevent cell proliferation and to induce apoptosis, demonstrated their ability to enter cells and to interfere with the activity of kinases important for cell division and cell death (Gompel et al., 2004). Different medicinal chemistry approaches were employed to prepare more selective GSK-3 inhibitors (Akue-Gedu et al., 2009). However, in all cases, they were most potent toward CDKs, and inhibition of GSK-3 was marginal (Echalier et al., 2008).
Synthetic, ATP-Competitive GSK-3 Inhibitors
Among the first synthetic small molecule GSK-3 inhibitors reported were the purine analogs, the aminopyrimidines, developed by Chiron. The potent inhibitors CHIR98014 (CT98014), CHIR98023 (CT98023), CHIR99021 (CT99021) (collectively termed here the CHIRs) inhibit GSK-3 within the nanomolar concentration range (Ring et al., 2003; Figure 1). Systemic analysis that profiled protein kinase inhibitors also confirmed the high selectivity of CHIRs toward GSK-3 (Bain et al., 2003, 2007). A limited number of studies have tested CHIRs in neuronal systems (these compounds had been chiefly tested in diabetic models). They showed that CHIRs potently reduced tau phosphorylation in cultured neurons and the rat brain (Selenica et al., 2007). In addition, treatment with CHIRs inhibited neurite outgrowth in cerebellar and DRG neurons (Alabed et al., 2011) and blocked NMDA-mediated long-term depression (LTD) in hippocampus slices (Peineau et al., 2009), indicating an unexpected role of GSK-3 in LTD maintenance (Peineau et al., 2009). In agreement with the 6BIO studies, CHIRs enhanced self renewal and pluripotency in mouse ES cells mimicking the activation of Wnt signaling pathway (Ying et al., 2008; Li et al., 2011). Finally, CHIRs reduced neuronal death in a cerebral ischemia rat model (Kelly et al., 2004), and enhanced the levels of the survival motor neuron protein (SMN) in spinal muscular atrophy (SMA; Makhortova et al., 2011).
The arylindolemaleimides SB-216763 and SB-415286 are highly selective GSK-3 inhibitors developed by GlaxoSmithKline that inhibit GSK-3 within the low nanomolar concentration range (collectively termed here SBs; Coghlan et al., 2000; Figure 1). A Number of studies demonstrated the neuroprotective effects of SBs against variety of pro-apoptotic conditions including inhibition of the PI3 kinase/Akt survival pathway, trophic deprivation, Aβ toxicity, heat shock, ethanol, NMDA excitotoxicity, and polyglutamine toxicity caused by the Huntington’s disease protein (Bijur et al., 2000; Cross et al., 2001; Culbert et al., 2001; Carmichael et al., 2002; Facci et al., 2003; Hongisto et al., 2003, 2008; Takadera and Ohyashiki, 2004; Hu et al., 2009). In addition, SB-216763 was shown to inhibit axon growth in postnatal and embryonic DRG neurons (Owens et al., 2003; Alabed et al., 2011). On the other hand, different studies reported that SB-216763 induced the formation of multiple long axons in hippocampal, cerebellar granular (CG), and DRG neurons (Padilla et al., 1997; Jiang et al., 2005; Yoshimura et al., 2005; Seira et al., 2011). Furthermore, it improved axon regeneration in lesioned neurons (Seira et al., 2011). Apparently it appeared that axon fate is dependent on the degree of inhibition of GSK-3, namely, strong inhibition of GSK-3 with high concentration of inhibitor inhibited axon growth, while weak inhibition promoted axon branching (Kim et al., 2006). An alternative explanation for these discrepancies relies on different culture length (Jiang et al., 2005). In any event, it has been clearly demonstrated that GSK-3 regulates neurite polarity and neurite outgrowth. The therapeutic activity of SBs has been further tested in several in vivo models. SB-216763 reduced ischemic cerebral damage in mice subjected to middle cerebral artery occlusion (Valerio et al., 2011), and enhanced locomotor recovery after spinal cord injury (Padilla et al., 1997). Like CHIRs, treatment with SBs inhibited NMDA-induced LTD (Peineau et al., 2009). In an AD model of mice injected with Aβ peptide, SB-216763 reduced Aβ neurotoxic effects including reduction in tau phosphorylation, caspase-3, and the activity of the stress activated kinase JNK (c-Jun N-terminal kinase; Hu et al., 2009). Administration of SB-216763 to disrupted-in-schizophrenia-1 (DISC1) knockdown mice ameliorated schizophrenic symptoms such as depressive behavior and hyper-locomotion (Mao et al., 2009). In the postnatal rat model, administration of SB-216763 reduced tau phosphorylation in the hippocampus (Selenica et al., 2007). In one of these studies, however, SB-216763 produced neurodegenerative-like effects and behavior deficits in healthy mice (Hu et al., 2009). This demonstrates that over-inhibition of GSK-3 may result in conditions that prevent neurons from operating normally. Thus, GSK-3 inhibitors should be used preferentially in pathologies associated with elevated GSK-3 activity. Finally, additional maleimide derivatives were recently identified by phenotypic screen for defects in zebrafish embryogenesis (Zhang et al., 2011). These compounds were potent GSK-3 inhibitors (Zhang et al., 2011), but their biological activity remains to be tested in relevant systems.
The amino thiazole AR-A014418 developed by AstraZeneca was shown to be a selective inhibitor toward GSK-3 when compared to other protein kinases including CDKs (Bhat et al., 2003; Figure 1). In their initial study, Bhat et al. (2003 showed that AR-A014418 was neuroprotective in apoptotic conditions induced by inhibition of PI3 kinase and prevented neurodegeneration in hippocampal slices exposed to Aβ peptide. Consistent with studies using SBs and 6BIO (Owens et al., 2003; Kim et al., 2006; Alabed et al., 2011), treatment with AR-A014418 prevented axon elongation in hippocampal neuronal culture (Shi et al., 2004). AR-A014418 has also been examined in behavior models. It was shown to produce an anti-depressive like behavior in the mouse FST (Gould et al., 2004a; Silva et al., 2008) and, in a manic behavior model of amphetamine-induced hyperactivity, it reduced manic activity (Kalinichev and Dawson, 2011). These results suggest that inhibition of GSK-3 may be beneficial in both depressive and manic episodes. Finally, in JNPL3 transgenic mice overexpressing mutant human tau, AR-A014418 reduced levels of aggregated insoluble tau in the brainstem (Noble et al., 2005), and, in an ALS transgenic mouse model, AR-A014418 attenuated motor neuron death and improved cognition (Koh et al., 2007). Yet, unexpectedly, AR-A014418 showed no effect on tau phosphorylation in the cortex or hippocampus in the postnatal rat model (Selenica et al., 2007). A structurally closed related compound AZD-1080 entered into clinical trials phase I for AD in 2006, but unfortunately the development has been discontinued.1
The group of paullone compounds, in particular kenpaullone and alsterpaullone, are widely used in various experimental settings as GSK-3 inhibitors (Figure 1). Paullones are fused tetracyclic compounds that inhibit both GSK-3 and CDKs within the nanomolar concentration range (Schultz et al., 1999; Leost et al., 2000). A structurally similar compound, 1-azakenpaullone, is a more selective GSK-3 inhibitor (Kunick et al., 2004). Its derivative cazpaullone (9-cyano-1-azapaullone; Figure 1) has been recently characterized as a selective GSK-3 inhibitor (Stukenbrock et al., 2008). Both alsterpaullone and kenpaullone prevented neuron cell death in response to variety of insults including trophic deprivation, thapsigargin treatment, and mitochondrial stress (Takadera and Ohyashiki, 2004; Mishra et al., 2007; Petit-Paitel et al., 2009; Skardelly et al., 2011). Alsterpaullone was shown to reduce tau phosphorylation in cultured neurons (Leost et al., 2000; Selenica et al., 2007), and to block NMDA-induced LTD in hippocampal slices (Peineau et al., 2008). Kenpaullone decreased Aβ production in cells overexpressing APP (Phiel et al., 2003) and promoted differentiation of precursor cells into dopamine neurons (Castelo-Branco et al., 2004). This last supported the use of GSK-3 inhibition in treatment of Parkinson’s disease (Castelo-Branco et al., 2004). Recent work implicated a role for GSK-3 in SMA (Makhortova et al., 2011). Apparently, a search for small molecules that elevate the expression levels of SMN identified GSK-3 inhibitors as potential agents. Treatment with alsterpaullone verified this observation and showed that alsterpaullone slowed down the degradation of SMN in SMA human fibroblasts (Makhortova et al., 2011). Furthermore, the death of motor neurons induced by depletion of SMN was rescued by alsterpaullone (Makhortova et al., 2011). This study is a first indication of a therapeutic potential of inhibition of GSK-3 in SMA (Makhortova et al., 2011). Reports of alsterpaullone or kenpaullone in in vivo systems are limited. One study demonstrated the ability of alsterpaullone to reduce tau phosphorylation in the rat brain (Selenica et al., 2007).
Additional compounds were recently described as dual CDK/GSK-3 inhibitors. These include the purine derivatives pyrazolo [3,4-b] quinoxalines (Ortega et al., 2002), the pyrazolo[3,4-b]pyridine ring system (Chioua et al., 2009), the 9-oxo-thiazolo [5,4-f] quinazoline-2-carbonitrile derivatives (Loge et al., 2008), and the thiazolo[5,4-f]quinazolin-9-ones (Testard et al., 2006). Aloisines (6-phenyl[5H]pyrrolo[2,3-b]pyrazines) are a different class of dual CDK/GSK-3 inhibitors that inhibit the two kinases in the sub-micromolar range (Mettey et al., 2003). Aloisine A (Figure 1) is the most potent of those analogs tested and showed anti-proliferative effects in differentiated postmitotic neurons (Mettey et al., 2003). A series of bisindolylmaleimides were identified as potent GSK-3 inhibitors and were shown to enhance mouse ES cells self renewal in the presence of the leukemia inhibitory factor (LIF; Bone et al., 2009). Recent work identified TWS119, a 4,6-disubstituted pyrrolopyrimidine (Figure 1), from a phenotypic cellular screen for compounds with the ability to induce neuronal differentiation of pluripotent mouse ES cells; the compound proved to be a GSK-3 inhibitor (Ding et al., 2003). This result contrasts with earlier studies that demonstrated maintenance of pluripotency by other GSK-3 inhibitors (Sato et al., 2004; Ying et al., 2008). These differences could be due to differences between mouse and human ES cells, culture conditions, or differences in the developmental stages derivation (Welham et al., 2007). Finally, a novel series of macrocyclic polyoxygenated bis-7-azaindolylmaleimides were shown to be highly selective toward GSK-3 (Kuo et al., 2003; Shen et al., 2004). Their biological activity remains to be further elucidated.
Collectively, studies with ATP-competitive inhibitors verified that GSK-3 is essential for maintenance of normal neuronal activities, but also demonstrated an important role for GSK-3 in the etiological mechanisms of neurodegeneration and psychiatric disorders. Crystallographic data identified specific interactions within the ATP-binding pocket and with additional residues located at the surface of the C-lobe. These include Asp 133, Arg 141 Gln185, Asp200, and Arg220 and the conserved salt bridge of Lys 85/Glu 97 (Figure 3A). However, the limited specificity of ATP-competitive inhibitors is a serious drawback as demonstrated in series of studies that profiled many protein kinase inhibitors (Davies et al., 2000; Bain et al., 2003, 2007). It is possible that the high toxicity of this type of drugs prevented entering into the clinical phase or resulted in a failure in clinical treatments. It may be possible to improve specificity by exploiting unique features within the ATP-binding fold of GSK-3, but this strategy requires further validation. As a last caveat, the dual inhibition of GSK-3 and CDKs may be not disadvantageous. CDK5 which is largely restricted to neurons, is essential for regulation of many neuronal functions (Jessberger et al., 2009), and its aberrant activity has been implicated in variety of neurodegenerative conditions (Cruz and Tsai, 2004). CDK5 also serves as the “priming kinase” that phosphorylates prior its phosphorylation by GSK-3 (Li et al., 2006; Plattner et al., 2006). Thus, inhibition of both GSK-3 and CDK5 may be of therapeutic benefit, and the use of such dual activity inhibitors should not be discouraged until further evaluation in preclinical models.
Synthetic, Non-ATP-Competitive GSK-3 Inhibitors
As we have highlighted above, there are many compounds with different scaffolds able to inhibit GSK-3 in an ATP-competitive manner; these compounds may have adverse secondary effects if used in chronic treatment. The human kinome has more than 500 protein kinases that share a high degree of homology in the catalytic site, and in particular, within the ATP-binding pocket. Achieving kinase selectivity is one of the main challenges in the search and design of protein kinases inhibitors (Eglen and Reisine, 2009). ATP non-competitive GSK-3 inhibitors are likely to be more selective than those that inhibit ATP binding, since they should bind to unique regions within the kinase providing a more subtle modulation of kinase activity than simply blocking ATP entrance. This point is of utmost importance for GSK-3 modulation as a therapeutic approach, because only the aberrant GSK-3 activity should be inhibited.
There are different chemical families of organic compounds reported in the literature that do not compete with ATP in their GSK-3 inhibition and different binding modes to the enzyme have been described. The first reported family was the small heterocyclic thiadiazolidinones (TDZD) family (Martinez et al., 2002; Figure 2A). Although their mechanism of action has not yet been experimentally confirmed, a possible role has been postulated for an interaction with cysteine 199, a key residue located in the active site of GSK-3 (Mazanetz and Fischer, 2007). TDZD did not show inhibition over various kinases including PKA, PKC, CK-2, and CDK1/cyclin B. Treatment of primary culture neuronal cells with TDZD reduced tau phosphorylation, and treatment with diverse TDZDs such as NP00111 (Figure 2A), NP031112, NP03115, produced neuroprotective and antidepressant activities in several animals models (Luna-Medina et al., 2007; Rosa et al., 2008a,b).
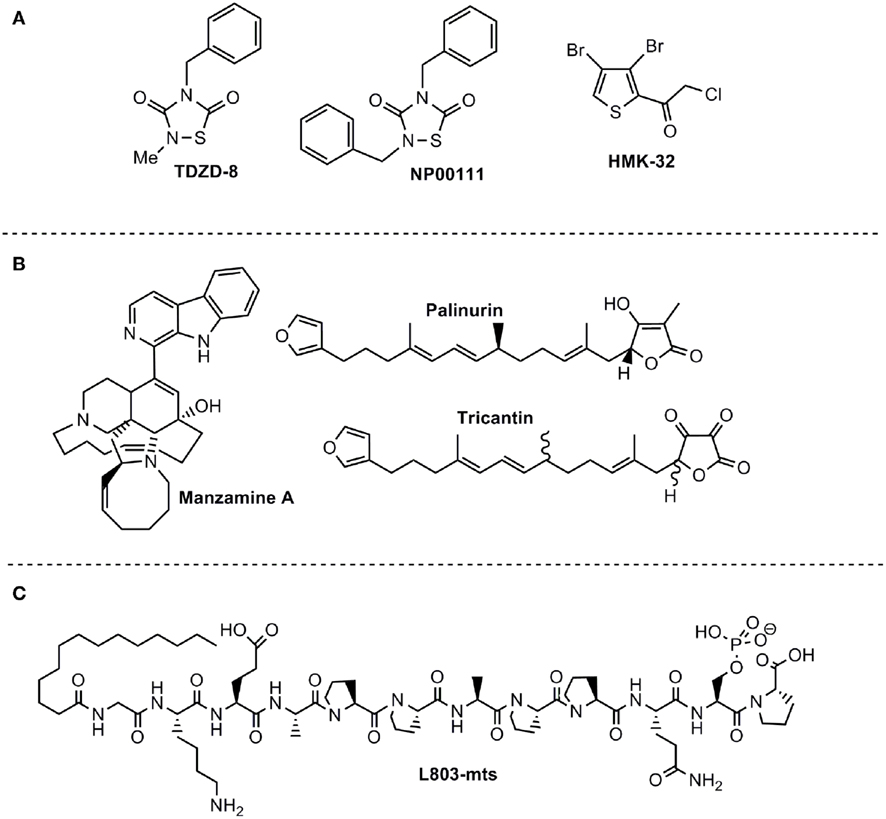
Figure 2. ATP non-competitive GSK-3 inhibitors with potential for CNS disorders. (A) Small molecules from organic synthesis programs. (B) Natural compounds isolated from marine organisms. (C) Peptide competitive with substrate.
With these others positive data, safety regulatory studies of one TDZD candidate was done with the aim to determinate the human dose. Very relevant for the translation from the bench to the treatment of AD patients are the results obtained in the chronic oral treatment for 3 months performed with TDZD compound named NP-12, to the double transgenic APP × tau rodent model (Ribe et al., 2005). Treatment with NP-12 improved cognitive performance in the Morris water maze test, while all histopathological features related to AD pathology such as beta-amyloid plaque load, tau hyperphosphorylation, gliosis, and neurons death were significantly reduced (Sereno et al., 2009). Of interest is the observation regarding the increase in insulin growth factor-1 (IGF-1) in mice brains of both on wild type and APP × PS1 mice, after an acute oral treatment with NP-12 for 5 days (Bolos et al., 2011). IGF-1 is a potent neurotrophic peptide with therapeutic value for many neurodegenerative diseases including AD pathology (Torres-Aleman, 2007). These increased IGF-1 levels in mice would mean that there is not only a direct action of GSK-3 inhibitors on the brain, but, there is also an effect on some peripheral signaling pathways (as activation of the IGF-1 transporter megalin), that could be modified with this innovative treatment improving the AD pathology.
Much of the work done in the hit-to-lead process, and lead optimization of TDZD heterocyclic family has been reported recently (Martinez et al., 2008). Two molecules have been well characterized. The first is TDZD-8, which is one of the pharmacological tools frequently used to study the biological and pathological roles of GSK-3 in cellular and animal models (Beaulieu et al., 2004; Cuzzocrea et al., 2006; Lipina et al., 2011). The second molecule is NP031112, also termed NP-12 or tideglusib. It is a brain permeable small molecule currently used in clinical trials phase II for AD and progressive supranuclear palsy (PSP).
Data from the phase IIa trial of tideglusib were recently reported and indicated a trend in improved cognitive abilities of the mild to moderate AD patients treated for 24 weeks (del Ser, 2010). The phase IIb trial for AD2 is ongoing with more than 20 centers involved. FDA and EMEA have approved the orphan drug status for the development of tideglusib in the rare tauopathy PSP3. The TAUROS study is ongoing and results are expected to be finalized by the end of 2011.
The second family of compounds known as ATP non-competitive GSK-3 inhibitors is the halomethylketone (HMK) derivatives (Conde et al., 2003) which have been recently described as the first irreversible inhibitors of this enzyme (Perez et al., 2009a; Figure 2A). In this case, inactivation of the enzyme is due to the formation of an irreversible covalent sulfur–carbon bond between the key cysteine 199 located at the entrance to the ATP site of GSK-3 and the HMK moiety (Perez et al., 2011).
HMKs are cell-permeable compounds. They are able to decrease tau hyperphosphorylation on primary neurons cell culture after 2 h of treatment (Perez et al., 2009a). They are rather selective in a wide protein kinase panel and its off-target activity were determined on different CNS receptors binding assays without any significant positive data (Perez et al., 2009a). There are a couple of HMKs commercially available from different commercial sources that confirms the importance of this series of compounds as pharmacological tools for the study of GSK-3 physiology and pathology in different cell models (Yasuda et al., 2009).
Non-ATP-Competitive GSK-3 Inhibitors from Natural Resources
Manzamines are complex β-carboline alkaloids isolated from Indo-Pacific sponges and characterized as having an intricate and novel polycyclic system (Hu et al., 2003a). Following a discovery program of GSK-3 inhibitors from marine sources, it was found that manzamine A (Figure 2B) inhibits human GSK-3β in vitro more than 70% at 25 μM (Rao et al., 2006). In order to identify the pharmacophore responsible for this new enzymatic inhibition, the potential GSK-3 inhibition of carboline and ircinal A, which can be considered the chemical precursors of manzamine A, were tested. Both moieties are inactive in their ability to bind to GSK-3, indicating the entire manzamine molecule is responsible for this activity. To further assess the potential of manzamine A in the treatment of AD, its ability to inhibit several different kinases (GSK-3β, GSK-3α, CDK1, PKA, MAPK, and CDK5) and decrease the hyperphosphorylation of tau protein mediated by GSK-3 in human neuroblastoma cell cultures was investigated (Hamann et al., 2007). Manzamine A specifically inhibits GSK-3β and CDK5 (the two key players in the hyperphosphorylation of tau protein in AD) with IC50s of 10 and 1.5 μM respectively; it is ineffective toward others kinases tested (Hamann et al., 2007). Kinetic studies indicated an ATP non-competitive inhibition regarding GSK-3 (Hamann et al., 2007) while susbstrate competitive inhibition has been recently proved experimentally (Palomo et al., 2011).
Treatment of SH-SY5Y cells in culture with manzamine A at different concentrations (5, 15, and 50 μM) resulted in a decrease in tau phosphorylation at the GSK-3 epitope Ser 396 (Hamann et al., 2007). as quantified by a specific ELISA sandwich methodology. Cell survival was determined in parallel by measuring LDH release. Manzamine A constitutes a promising scaffold from which more potent and selective GSK-3 inhibitors could be designed as potential therapeutic agents for the treatment of diseases mediated by GSK-3 such as the AD (Wahba and Hamann, 2011). Recently a potential binding site of manzamine A with GSK-3 was identified, and this will provide new directions in substrate competitive drug design (Peng et al., 2011).
Very recently extracts and compounds obtained from the marine organism Ircinia sp., and, more particularly, the furanoterpenoids isolated from the Mediterranean sponges Ircinia dendroides, Ircinia variabilis, and Ircinia oros, have been claimed as inhibitors of GSK-3 (Alonso et al., 2005). Fractionation and purification of active components from these extracts, guided by a GSK-3 inhibition assay, resulted in the isolation of furanosesquiterpenoids as new GSK-3 inhibitors with potential use as therapeutic agents. Palinurin and one unknown metabolite called tricantin were mainly isolated (Figure 2B). Kinetic analyses of isolated compounds were performed and showed that tricantin inhibits recombinant human GSK-3β with an IC50 value of 7.5 μM, whereas palinurin exhibited an IC50 value of 4.5 μM. They are cell-permeable inhibitors and described as ATP non-competitive GSK-3 inhibitor able to reduce tau phosphorylation in cell cultures (Alonso and Martinez, 2006). Total synthesis of palinurin has been recently described providing a new source for this lead compound (Perez et al., 2009b). Different studies have been performed to determine structure activity relationships among a series of congeneric molecules and the bioactive conformation has been proposed (Ermondi et al., 2011). However, the binding mode to GSK-3 remains unknown.
Peptides as Substrate–Competitive Inhibitors
Peptides have also been described as potential protein kinase inhibitors (Eldar-Finkelman and Eisenstein, 2009). The use of peptides, which copy natural motifs that specifically influence kinase activity and/or its intracellular interactions with associated partners, may be a promising approach for selective inhibition of protein kinases. Although substrate–competitive inhibitors have often been overlooked due their relative weak inhibition, they provide numerous advantages over the ATP-competitive inhibitors, mainly in selectivity. This is due to the fact that substrate recognition and types of interactions vary considerably among kinases, whereas ATP-binding domains are structurally conserved. In the case of GSK-3, the relatively weak affinities of substrate–competitive inhibitors, and, in general, ATP non-competitive inhibitors, may be advantageous. GSK-3 is essential for many aspects of neuronal function; hence, drastic inhibition may result in deleterious effects (as has been observed with some of GSK-3 inhibitors). Furthermore, pathological GSK-3 over-expression does not exceed two to threefold over normal levels. Thus, moderate-to-weak inhibition of GSK-3 (about 50% inhibition) is actually a desired approach for treatment of disorders associated with elevated activity of GSK-3. Finally because substrate–competitive inhibitors are more selective than ATP-competitive molecules, substrate–competitive inhibitors may be a favorable choice for clinical use.
The specific requirement of GSK-3 for pre-phosphorylated substrates supported the rational for the use of synthetic phosphorylated peptides as substrate–competitive inhibitors (Plotkin et al., 2003). The peptide L803-mts (11 residues) is a cell-permeable phosphorylated peptide derived from the GSK-3 substrate heat shock factor-1 (HSF-1; Figure 2C) that is very selective and inhibits cellular activity of GSK-3 within the low micromolar concentration range (Plotkin et al., 2003). L803-mts showed biological activity in diabetic models consisting with the paradigm of GSK-3 acting as a negative regulator of insulin signaling (Kaidanovich-Beilin and Eldar-Finkelman, 2006; Rao et al., 2007). Recent work further demonstrated the therapeutic activity of L803-mts in CNS models. Like other GSK-3 inhibitors, L803-mts promoted axon formation and elongation in hippocampal neurons (Kim et al., 2006). It was also shown to provide neuroprotection effects in neuron cultured cells exposed to 6-hydroxydopamine-induced cell death (Chen et al., 2004). In vivo treatment with L803-mts increased β-catenin levels in the mouse hippocampus and produced anti-depressive like activity in the FST (Kaidanovich-Beilin et al., 2004). Administration of L803-mts in a traumatic brain injury (TBI) model reversed depressive behavior in the injured animals (Shapira et al., 2007). Finally, L803-mts conferred low toxicity in neurons as compared with other GSK-3 inhibitors (Kim and Thayer, 2009).
Understanding of the mode of interaction of GSK-3 with its substrates is necessary for effective design and development of substrate–competitive inhibitors. A combined approach of mutagenesis and computational protein–protein docking analyses identified a novel substrate-binding site within the catalytic core of GSK-3β formed by Phe 67, Gln 89, Phe93, and Asn 95 (Ilouz et al., 2006; Licht-Murava et al., 2011), and Asp 181 as an additional binding site for the N-terminal pseudosubstrate (Ilouz et al., 2008; Figure 3B). These residues are spatially located near the ATP-binding site and the phosphate binding pocket that interacts with the phospho-serine moiety of the substrate (Dajani et al., 2001; ter Haar et al., 2001; Figure 3B). From studies of the binding mode of L803-mts with GSK-3 it was found that the inhibitor- and substrate-binding sites are not identical. Both substrate and L803-mts interact with the phosphate binding pocket, but the substrate interacts with the cavity bordered by Gln 89 and Asn 95, whereas L803-mts mainly interacts with Phe 93 and with a hydrophobic surface located away from the ATP-binding site (Licht-Murava et al., 2011; Figure 3B). This clarified our understanding of the different binding modes of substrates and inhibitors. Whereas the substrate requires appropriate alignment with the catalytic domain to allow catalysis, the inhibitor does not require an exact positioning within the catalytic cleft. Contacts other than those used by the substrate may be crucial for converting a substrate to an inhibitor. Based on this idea, new L803-mts variants were synthesized, and some were 3- to 10-fold better inhibitors than L803-mts (Licht-Murava et al., 2011). Taken together, substrate–competitive inhibitors of GSK-3 hold tremendous promise as potential therapeutics. An extensive understanding of molecular recognition of GSK-3 with its substrates and inhibitors should provide the basis for rational design and optimization of efficient and high affinity substrate–competitive inhibitors.
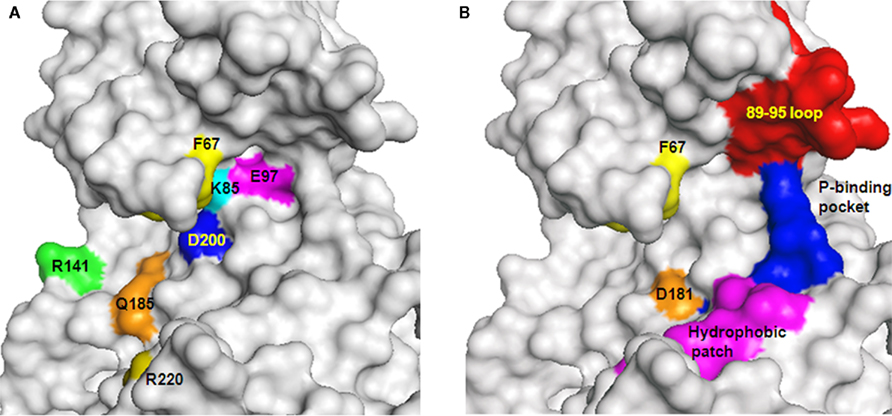
Figure 3. GSK-3-interacting sites with ATP-competitive inhibitors, substrates, and L803-mts. (A) Sites interacting with ATP or ATP-competitive inhibitors are indicated. These interacting sites are located at the interface within the N- and C-lobes of the catalytic domain. F67 (yellow) locates the P-loop interacting with ATP (B) Distinct and overlapping element in GSK-3 interaction with substrates and inhibitors. Sites interacting with substrates: F67 (yellow), 89–95 loop (red), phosphate binding pocket (P-binding pocket, blue). D181 (orange) interacts with the pseudosubstrate. Sites interacting with L803-mts: F93 (within the 89–95 loop), hydrophobic patch (V214, I217 Y216 magenta), and the phosphate binding pocket (blue). GSK-3 structure is based on PDB code 1gng and images were processed by PyMol software.
A different example for peptide inhibitors are two peptides derived from Frat (FRATide-39 residues) or Axin (Axin GID-25 residues) that compete with Axin binding to GSK-3, which in turn, leads to activation of Wnt signaling pathway (Hedgepeth et al., 1999; Thomas et al., 1999). Treatment with Axin GID induced multiple axon formation in hippocampal neurons (Jiang et al., 2005), and its exogenous expression in cerebellar granule neurons protected against trophic deprivation-induced cell death (Hongisto et al., 2003). These results could suggest that GSK-3 inhibition-mediated neuroprotection or axon formation involves activation of Wnt signaling and or an increase in Axin non-bound GSK-3 pool (Hongisto et al., 2003). However, from the therapeutic point of view, more relevant are small peptides.
Prospective and Challenges
Cumulative data now suggest a promising future for GSK-3 inhibitors (Table 1). However, some concerns had been raised regarding the potential toxicity of these compounds ranging from hypoglycemia to tumorigenesis and neuron deregulation. Furthermore, GSK-3 is essential for life, and there is a concern that its inhibition could prevent cells from operating normally. Nevertheless, it is worth mentioning that GSK-3 activity is elevated in pathological conditions, thus, a smooth inhibition of GSK-3 able to restore down levels of activity to physiological ones would be enough to produce an important therapeutic effects in unmet diseases, being that point crucial for not produce adverse effects.
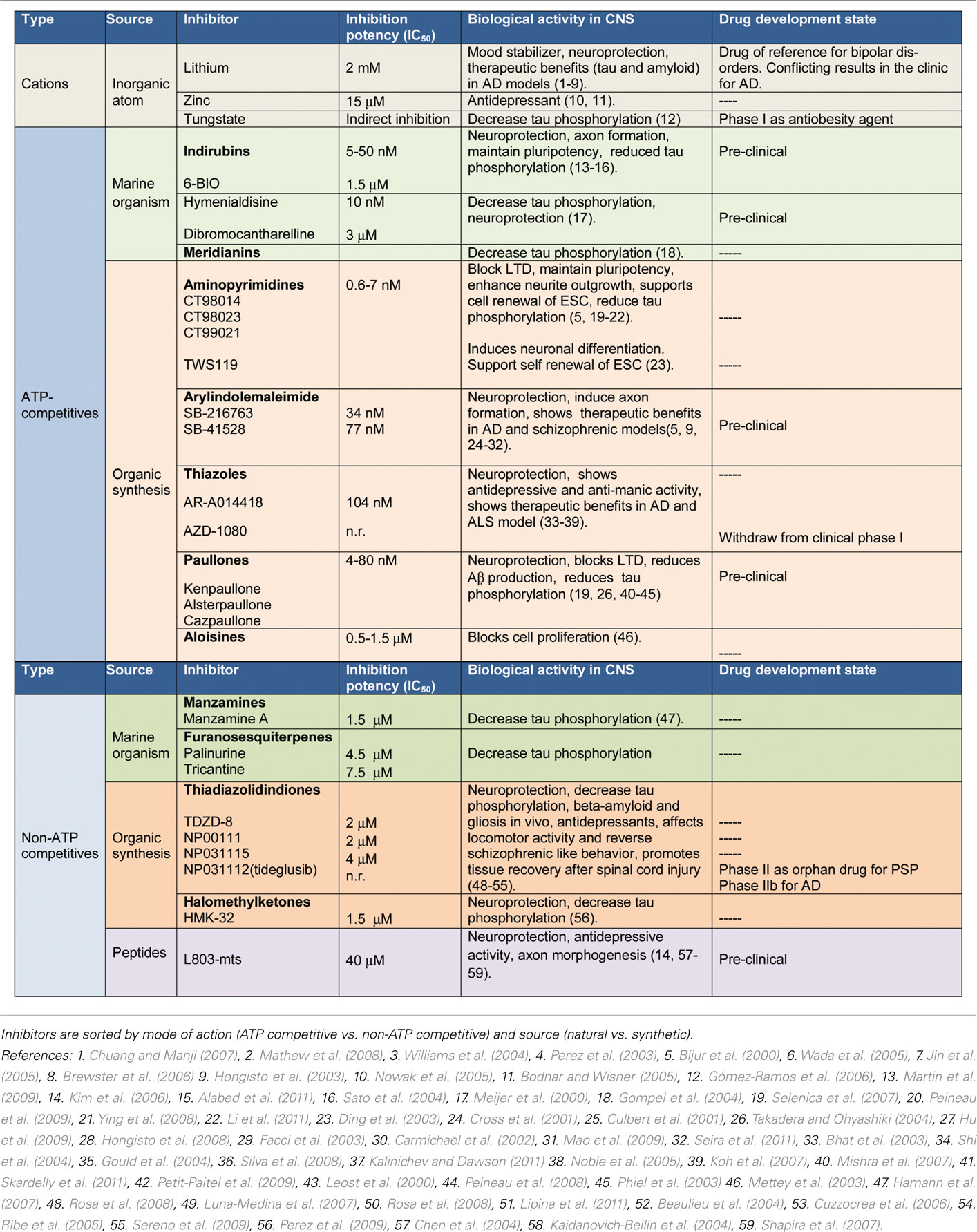
Table 1. GSK-3 inhibitors.
Evidently, treatment with GSK-3 inhibitors restored glucose homeostasis and did not provoke hypoglycemia or hyperinuslinemia in diabetic models. Activation of the proto-oncogenic molecule β-catenin by inhibition of GSK-3 is another major concern claiming that long-term inhibition of GSK-3 may promote cancer. However, no direct in vivo evidence has indicated tumorigenesis upon administration of GSK-3 inhibitors. On the contrary, in certain cancers GSK-3 inhibitors reduced cell proliferation and enhanced cell death upon irradiation treatment. Compelling evidence in this regard is the fact that treatment with the drug lithium which has been used as standard therapeutic for the treatment of bipolar disorder since the 1950s is not associated with increased levels of tumorigenesis or deaths from cancer. Certainly the degree of GSK-3 inhibition is a crucial element affecting toxicity, and a weak to moderate inhibition of GSK-3 is an optimal therapeutic approach.
GSK-3 inhibitors that are non-ATP-competitive provide important benefits in therapeutic use for several reasons. First of all because, better kinase selectivity may be expected from inhibitors that bind outside the ATP pocket, and secondly, because, this kind of kinase inhibitors should have lower values of IC50, which in the case of GSK-3 is not only beneficial but also necessary to avoid toxicity. Thus, non-ATP-competitive GSK-3 inhibitors, which comprise covalent inhibitors, substrate–competitive inhibitors and allosteric modulators, arise as the unique real potential drugs for the treatment of at least chronic diseases as AD. An interesting question in this regard is the feasibility in the design of selective inhibitors toward GSK-3α or GSK-3β. The fact that the catalytic domains of the two isozymes share more than 90% homology suggests that inhibitors targeting this domain may not discriminate between the two isozymes (as indeed is deduced in the in vitro kinase assays). A different strategic approach exploiting unique properties of GSK-3 isozymes such as protein–protein interactions, distinct cellular localization etc. should be used in development of such inhibitors. Hence, a better understanding of the distinct structure–function properties of GSK-3 isozymes is required for future design of isozymes-selective inhibitors.
Another important challenge to overcome for a GSK-3 inhibitor to be converted in an effective drug for AD treatment is its specific brain distribution. The drug needs to cross the blood brain barrier to exert its action in the regulation of exacerbated GSK-3 brain levels. Usually this is not an easy task for organic compounds and/or peptides, moreover when oral bioavailability is the preferred administration route for chronic AD treatment. It is very difficult to balance the equilibrium between molecular lipophilicity to enter into the brain and molecular hydrophilicity to be orally administrated. That reason has ruled out several promising GSK-3 inhibitors from the race to the market. Determination of potential brain penetration should be incorporated in the first stages of GSK-3 inhibitors development.
Finally, our knowledge regarding human clinical side effects of GSK-3 inhibitors is rather scarce since a limited number of compounds have reached the clinical phase. Moreover, these compounds are of distinct chemical structures; and thus differ in their bioclinical and pharmacological properties (absorption, distribution, metabolism, etc.). It is thus difficult to determine at this point what adverse events will be commonly associated with inhibition of GSK-3. Lithium is the only GSK-3 inhibitor that has been in clinical use for a significant time. However, lithium lacks target specificity, and its adverse side effects and high toxicity do not necessarily reflect events associated with inhibition of GSK-3 per se. AZD-1080 (AstraZeneca) and NP-12/Tideglusib (Noscria) reached the clinic in 2006. AZD-1080 was abandoned as a drug candidate due to nephrotoxicity observed in phase I clinical trials. NP-12 is currently in phase IIb trials for Alzheimer’s disease and paralysis supranuclear palsy, and no side effects/off targets effects have been described at this time. Their distinct chemical structures and/or different inhibition mode are most likely responsible for the different clinical impacts observed with these two compounds. Results from TAURUS and ARGO studies will reveal the safety and efficacy of Tideglusib in humans. Meanwhile, an increasing number of GSK-3 inhibitors are being tested in preclinical models, and it is anticipated that some will enter clinical trials.
Conclusion
As GSK-3 plays an important role in AD and some others unmet diseases, more of them related to CNS, many inhibitors have been discovered in the last years. Some of them have proven to be effective in specific cellular and animal models of different CNS pathologies. However, due to different hazards previously considered when GSK-3 is targeted, some risks should be avoid in a GSK-3 inhibitor development. High values for IC50 and ATP-competition when the compound binds to the enzyme, should not be present in a GSK-3 inhibitor if we want that the molecule become an effective drug. A mild inhibition of GSK-3 is indispensable to treat pathological states because it will be able to decrease the exacerbated GSK-3 function in the tissue affected by the disease but the simultaneous decrease of activity in other healthy tissues, will be compensate by alternative cellular mechanisms present in the human being.
ATP non-competitive GSK-3 inhibitors such allosteric modulators, substrate competitive or covalent inhibitors are emerging as an alternative and promising approach for a safer use in the clinic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Hagit Eldar-Finkelman acknowledges support from FP7 EU grant #223276 “NeuroGSK3” and the Israeli Academy of Sciences. Grant# 341/10. Ana Martinez acknowledge financial support from Spanish government through Ministry of Science and Innovation MICINN (Project SAF2009-13015-C02-01) and Instituto de Salud Carlos III ISCiii project no. RD07/0060/0015 (RETICS program).
Footnotes
References
Adachi, A., Kano, F., Tsuboi, T., Fujita, M., Maeda, Y., and Murata, M. (2011). Golgi-associated GSK3beta regulates the sorting process of post-Golgi membrane trafficking. J. Cell Sci. 123, 3215–3225.
Akue-Gedu, R., Debiton, E., Ferandin, Y., Meijer, L., Prudhomme, M., Anizon, F., and Moreau, P. (2009). Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins. Bioorg. Med. Chem. Lett. 17, 4420–4424.
Alabed, Y. Z., Pool, M., Ong Tone, S., Sutherland, C., and Fournier, A. E. (2011). GSK3 beta regulates myelin-dependent axon outgrowth inhibition through CRMP4. J. Neurosci. 30, 5635–5643.
Alon, L. T., Pietrokovski, S., Barkan, S., Avrahami, L., Kaidanovich-Beilin, O., Woodgett, J. R., Barnea, A., and Eldar-Finkelman, H. (2011). Selective loss of glycogen synthase kinase-3alpha in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 585, 1158–1162.
Alonso, D., Dorronsoro, I., Martinez, A., Panizo, G., Fuertes, A., Perez, J., Martin, E., Perez, D., and Medina, M. (2005). Glycogen synthase kinase-3 inhibitors isolated from marine organisms. WO 2005054221.
Alonso, D., and Martinez, A. (2006). “Marine compounds as a new source for glycogen synthase kinase 3 inhibitors,” in Glycogen Synthase Kinase 3 (GSK-3) and its Inhibitors, eds A. Martinez, A. Castro, and M. Medina (New Jersey, NJ: John Wiley & sons), 307–331.
Azoulay-Alfaguter, I., Yaffe, Y., Licht-Murava, A., Urbanska, M., Jaworski, J., Pietrokovski, S., Hirschberg, K., and Eldar-Finkelman, H. (2011). Distinct molecular regulation of glycogen synthase kinase-3{alpha} isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J. Biol. Chem. 286, 13470–13480.
Bain, J., McLauchlan, H., Elliott, M., and Cohen, P. (2003). The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204.
Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H., Klevernic, I., Arthur, J. S., Alessi, D. R., and Cohen, P. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Beaulieu, J. M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., Gainetdinov, R. R., and Caron, M. G. (2008). Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338.
Bedlack, R. S., Maragakis, N., and Heiman-Patterson, T. (2008). Lithium may slow progression of amyotrophic lateral sclerosis, but further study is needed. Proc. Natl. Acad. Sci. U.S.A. 105, E17; author reply E18.
Bhat, R., Xue, Y., Berg, S., Hellberg, S., Ormo, M., Nilsson, Y., Radesater, A. C., Jerning, E., Markgren, P. O., Borgegard, T., Nylöf, M., Giménez-Cassina, A., Hernández, F., Lucas, J. J., Díaz-Nido, J., and Avila, J. (2003). Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 278, 45937–45945.
Bijur, G. N., De Sarno, P., and Jope, R. S. (2000). Glycogen synthase kinase-3 beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J. Biol. Chem. 275, 7583–7590.
Bijur, G. N., and Jope, R. S. (2003). Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport 14, 2415–2419.
Bodnar, L. M., and Wisner, K. L. (2005). Nutrition and depression: implications for improving mental health among childbearing-aged women. Biol. Psychiatry 58, 679–685.
Bolos, M., Fernandez, S., and Torres-Aleman, I. (2011). Oral administration of a GSK3 inhibitor increases brain insulin-like growth factor I levels. J. Biol. Chem. 285, 17693–17700.
Bone, H. K., Damiano, T., Bartlett, S., Perry, A., Letchford, J., Ripoll, Y. S., Nelson, A. S., and Welham, M. J. (2009). Involvement of GSK-3 in regulation of murine embryonic stem cell self-renewal revealed by a series of bisindolylmaleimides. Chem. Biol. 16, 15–27.
Brewster, J. L., Linseman, D. A., Bouchard, R. J., Loucks, F. A., Precht, T. A., Esch, E. A., and Heidenreich, K. A. (2006). Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase-3 beta and a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol. Cell. Neurosci. 32, 242–253.
Burstein, D. E., Seeley, P. J., and Greene, L. A. (1985). Lithium ion inhibits nerve growth factor-induced neurite outgrowth and phosphorylation of nerve growth factor-modulated microtubule-associated proteins. J. Cell Biol. 101, 862–870.
Busceti, C. L., Biagioni, F., Aronica, E., Riozzi, B., Storto, M., Battaglia, G., Giorgi, F. S., Gradini, R., Fornai, F., Caricasole, A., Nicoletti, F., and Bruno, V. (2007). Induction of the Wnt inhibitor, Dickkopf-1, is associated with neurodegeneration related to temporal lobe epilepsy. Epilepsia 48, 694–705.
Caccamo, A., Oddo, S., Tran, L. X., and LaFerla, F. M. (2007). Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am. J. Pathol. 170, 1669–1675.
Carmichael, J., Sugars, K. L., Bao, Y. P., and Rubinsztein, D. C. (2002). Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 277, 33791–33798.
Caspi, M., Zilberberg, A., Eldar-Finkelman, H., and Rosin-Arbesfeld, R. (2008). Nuclear GSK-3beta inhibits the canonical Wnt signalling pathway in a beta-catenin phosphorylation-independent manner. Oncogene 27, 3546–3555.
Castelo-Branco, G., Rawal, N., and Arenas, E. (2004). GSK-3beta inhibition/beta-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J. Cell Sci. 117, 5731–5737.
Chen, G., Bower, K. A., Ma, C., Fang, S., Thiele, C. J., and Luo, J. (2004). Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 18, 1162–1164.
Cheng, K., Creacy, S., and Larner, J. (1983). “Insulin-like” effects of lithium ion on isolated rat adipocytes. II. Specific activation of glycogen synthase. Mol. Cell. Biochem. 56, 183–189.
Chioua, M., Samadi, A., Soriano, E., Lozach, O., Meijer, L., and Marco-Contelles, J. (2009). Synthesis and biological evaluation of 3,6-diamino-1H-pyrazolo[3,4-b]pyridine derivatives as protein kinase inhibitors. Bioorg. Med. Chem. Lett. 19, 4566–4569.
Chuang, D. M., and Manji, H. K. (2007). In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol. Psychiatry 62, 4–6.
Cimino, G., De Rosa, S., De Stefano, S., Mazzarella, L., Puliti, R., and Sodano, G. (1982). Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett. 23, 767–768.
Coghlan, M. P., Culbert, A. A., Cross, D. A., Corcoran, S. L., Yates, J. W., Pearce, N. J., Rausch, O. L., Murphy, G. J., Carter, P. S., Roxbee Cox, L., Mills, D., Brown, M. J., Haigh, D., Ward, R. W., Smith, D. G., Murray, K. J., Reith, A. D., and Holder, J. C. (2000). Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7, 793–803.
Cole, A., Frame, S., and Cohen, P. (2004). Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 377, 249–255.
Conde, S., Perez, D. I., Martinez, A., Perez, C., and Moreno, F. J. (2003). Thienyl and phenyl alpha-halomethyl ketones: new inhibitors of glycogen synthase kinase (GSK-3beta) from a library of compound searching. J. Med. Chem. 46, 4631–4633.
Cross, D. A., Culbert, A. A., Chalmers, K. A., Facci, L., Skaper, S. D., and Reith, A. D. (2001). Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 77, 94–102.
Crowder, R. J., and Freeman, R. S. (2000). Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J. Biol. Chem. 275, 34266–34271.
Cruz, J. C., and Tsai, L. H. (2004). Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends. Mol. Med. 10, 452–458.
Culbert, A. A., Brown, M. J., Frame, S., Hagen, T., Cross, D. A., Bax, B., and Reith, A. D. (2001). GSK-3 inhibition by adenoviral FRAT1 overexpression is neuroprotective and induces Tau dephosphorylation and beta-catenin stabilisation without elevation of glycogen synthase activity. FEBS Lett. 507, 288–294.
Cuzzocrea, S., Genovese, T., Mazzon, E., Crisafulli, C., Di Paola, R., Muia, C., Collin, M., Esposito, E., Bramanti, P., and Thiemermann, C. (2006). Glycogen synthase kinase-3 beta inhibition reduces secondary damage in experimental spinal cord trauma. J. Pharmacol. Exp. Ther. 318, 79–89.
Dajani, R., Fraser, E., Roe, S. M., Young, N., Good, V., Dale, T. C., and Pearl, L. H. (2001). Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105, 721–732.
Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105.
De Ferrari, G. V., Chacon, M. A., Barria, M. I., Garrido, J. L., Godoy, J. A., Olivares, G., Reyes, A. E., Alvarez, A., Bronfman, M., and Inestrosa, N. C. (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol. Psychiatry 8, 195–208.
De Sarno, P., Li, X., and Jope, R. S. (2001). Regulation of Akt and glycogen synthase kinase-3bet phosphorylation by sodium valproate and lithium. Neuropharmacology 43, 1158–1164.
del Ser, T. (2010). Phase IIa clinical trial on Alzheimer’s disease with NP12, a GSK-3 inhibitor. Alzheimers Dement. 6, S147.
Diehl, J. A., Cheng, M., Roussel, M. F., and Sherr, C. J. (1998). Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 12, 3499–3511.
Ding, S., Wu, T. Y., Brinker, A., Peters, E. C., Hur, W., Gray, N. S., and Schultz, P. G. (2003). Synthetic small molecules that control stem cell fate. Proc. Natl. Acad. Sci. U.S.A. 100, 7632–7637.
Ding, Y., Qiao, A., and Fan, G. H. (2010). Indirubin-3’-monoxime rescues spatial memory deficits and attenuates beta-amyloid-associated neuropathology in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 39, 156–168.
Doble, B. W., and Woodgett, J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186.
Dunn, N., Holmes, C., and Mullee, M. (2005). Does lithium therapy protect against the onset of dementia? Alzheimer Dis. Assoc. Disord. 19, 20–22.
Echalier, A., Bettayeb, K., Ferandin, Y., Lozach, O., Clement, M., Valette, A., Liger, F., Marquet, B., Morris, J. C., Endicott, J. A., Joseph, B., and Meijer, L. (2008). Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J. Med. Chem. 51, 737–751.
Eglen, R. M., and Reisine, T. (2009). The current status of drug discovery against the human kinome. Assay Drug Dev. Technol. 7, 22–43.
Eldar-Finkelman, H. (2002). Glycogen synthase kinase-3: an emerging therapeutic target. Trends Mol. Med. 8, 126–132.
Eldar-Finkelman, H., and Eisenstein, M. (2009). Peptide inhibitors targeting protein kinases. Curr. Pharm. Des. 15, 2463–2470.
Engel, T., Hernandez, F., Avila, J., and Lucas, J. J. (2006). Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 26, 5083–5090.
Ermondi, G., Caron, G., Pintos, I. G., Gerbaldo, M., Perez, M., Perez, D. I., Gandara, Z., Martinez, A., Gomez, G., and Fall, Y. (2011). An application of two MIFs-based tools (Volsurf+ and Pentacle) to binary QSAR: the case of a palinurin-related data set of non-ATP competitive glycogen synthase kinase 3beta (GSK-3beta) inhibitors. Eur. J. Med. Chem. 46, 860–869.
Facci, L., Stevens, D. A., and Skaper, S. D. (2003). Glycogen synthase kinase-3 inhibitors protect central neurons against excitotoxicity. Neuroreport 14, 1467–1470.
Fiol, C. J., Williams, J. S., Chou, C. H., Wang, Q. M., Roach, P. J., and Andrisani, O. M. (1994). A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J. Biol. Chem. 269, 32187–32193.
Forlenza, O. V., Torres, C. A., Talib, L. L., de Paula, V. J., Joaquim, H. P., Diniz, B. S., and Gattaz, W. F. (2011). Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer’s disease. J. Psychiatr. Res. 45, 220–224.
Frame, S., and Cohen, P. (2001). GSK-3 takes center stage more than 20 years after its discovery. Biochem. J. 359, 1–16.
Fraser, E., Young, N., Dajani, R., Franca-Koh, J., Ryves, J., Williams, R. S., Yeo, M., Webster, M. T., Richardson, C., Smalley, M. J., Pearl, L. H., Harwood, A., and Dale, T. C. (2002). Identification of the Axin and Frat binding region of glycogen synthase kinase-3. J. Biol. Chem. 277, 2176–2185.
Ghribi, O., Herman, M. M., and Savory, J. (2003). Lithium inhibits Abeta-induced stress in endoplasmic reticulum of rabbit hippocampus but does not prevent oxidative damage and tau phosphorylation. J. Neurosci. Res. 71, 853–862.
Gómez-Ramos, A., Domínguez, J., Zafra, D., Corominola, H., Gomis, R., Guinovart, J. J., and Avila, J. (2006). Sodium tungstate decreases the phosphorylation of tau through GSK3 inactivation. J. Neurosci. Res. 83, 264–273.
Gompel, M., Leost, M., De Kier Joffe, E. B., Puricelli, L., Franco, L. H., Palermo, J., and Meijer, L. (2004). Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 14, 1703–1707.
Gould, T. D., Einat, H., Bhat, R., and Manji, H. K. (2004a). AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 7, 387–390.
Gould, T. D., Zarate, C. A., and Manji, H. K. (2004b). Glycogen synthase kinase-3: a target for novel bipolar disorder treatments. J. Clin. Psychiatry 65, 10–21.
Hamann, M., Alonso, D., Martin-Aparicio, E., Fuertes, A., Perez-Puerto, M. J., Castro, A., Morales, S., Navarro, M. L., Del Monte-Millan, M., Medina, M., Pennaka, H., Balaiah, A., Peng, J., Cook, J., Wahyuono, S., and Martínez, A. (2007). Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure-activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod. 70, 1397–1405.
Hampel, H., Ewers, M., Burger, K., Annas, P., Mortberg, A., Bogstedt, A., Frolich, L., Schroder, J., Schonknecht, P., Riepe, M. W., Kraft, I., Gasser, T., Leyhe, T., Möller, H. J., Kurz, A., and Basun, H. (2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931.
Hanks, S. K., and Hunter, T. (1995). Protein kinases. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596.
Havens, W. W. II, and Cole, J. (1982). Successful treatment of dementia with lithium. J. Clin. Psychopharmacol. 2, 71–72.
Hedgepeth, C. M., Deardorff, M. A., Rankin, K., and Klein, P. S. (1999). Regulation of glycogen synthase kinase 3beta and downstream Wnt signaling by axin. Mol. Cell. Biol. 19, 7147–7157.
Hernandez, F., and Avila, J. (2008). The role of glycogen synthase kinase 3 in the early stages of Alzheimers’ disease. FEBS Lett. 582, 3848–3854.
Hernandez, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hoessel, R., Leclerc, S., Endicott, J. A., Nobel, M. E., Lawrie, A., Tunnah, P., Leost, M., Damiens, E., Marie, D., Marko, D., Niederberger, E., Tang, W., Eisenbrand, G., and Meijer, L. (1999). Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1, 60–67.
Hongisto, V., Smeds, N., Brecht, S., Herdegen, T., Courtney, M. J., and Coffey, E. T. (2003). Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 23, 6027–6036.
Hongisto, V., Vainio, J. C., Thompson, R., Courtney, M. J., and Coffey, E. T. (2008). The Wnt pool of glycogen synthase kinase 3beta is critical for trophic-deprivation-induced neuronal death. Mol. Cell. Biol. 28, 1515–1527.
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439.
Hu, J. F., Hamann, M. T., Hill, R., and Kelly, M. (2003a). The manzamine alkaloids. Alkaloids Chem. Biol. 60, 207–285.
Hu, J. H., Zhang, H., Wagey, R., Krieger, C., and Pelech, S. L. (2003b). Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 85, 432–442.
Hu, S., Begum, A. N., Jones, M. R., Oh, M. S., Beech, W. K., Beech, B. H., Yang, F., Chen, P., Ubeda, O. J., Kim, P. C., Davies, P., Ma, Q., Cole, G. M., and Frautschy, S. A. (2009). GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 33, 193–206.
Hughes, K., Nicolakaki, E., Plyte, S. E., Totty, N. F., and Woodgett, J. R. (1993). Modulation of glycogen synthase kinase-3 family by tyrosine kinase phosphorylation. EMBO J. 12, 803–808.
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551.
Hye, A., Kerr, F., Archer, N., Foy, C., Poppe, M., Brown, R., Hamilton, G., Powell, J., Anderton, B., and Lovestone, S. (2005). Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 373, 1–4.
Ikeda, S., Kishida, S., Yamamoto, H., Murai, H., Koyama, S., and Kikuchi, A. (1998). Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384.
Ilouz, R., Kaidanovich, O., Gurwitz, D., and Eldar-Finkelman, H. (2002). Inhibition of glycogen synthase kinase-3beta by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem. Biophys. Res. Commun. 295, 102–106.
Ilouz, R., Kowelsman, N., Eisenstein, M., and Eldar-Finkelman, H. (2006). Identification of novel glycogen synthase kinase-3beta substrate-interacting residues suggests a common mechanism for substrate recognition. J. Biol. Chem. 281, 30621–30630.
Ilouz, R., Pietrokovsky, S., Eisenstein, M., and Eldar-Finkelman, H. (2008). New insights into the autoinhibition mechanism of glycogen synthase kinase-3 beta. J. Mol. Biol. 385, 999–1007.
Jessberger, S., Gage, F. H., Eisch, A. J., and Lagace, D. C. (2009). Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci. 32, 575–582.
Jiang, H., Guo, W., Liang, X., and Rao, Y. (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120, 123–135.
Jin, N., Kovacs, A. D., Sui, Z., Dewhurst, S., and Maggirwar, S. B. (2005). Opposite effects of lithium and valproic acid on trophic factor deprivation-induced glycogen synthase kinase-3 activation, c-Jun expression and neuronal cell death. Neuropharmacology 48, 576–583.
Jope, R. S., Yuskaitis, C. J., and Beurel, E. (2007). Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 32, 577–595.
Kaidanovich-Beilin, O., and Eldar-Finkelman, H. (2006). Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: molecular characterization in liver and muscle. J. Pharmacol. Exp. Ther. 316, 17–24.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., van Eede, M., Hattori, S., Laliberté, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., MacAulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2, 1–23.
Kaidanovich-Beilin, O., Milman, A., Weizman, A., Pick, C., and Eldar-Finkelman, H. (2004). Rapid anti-depressive like activity of specific GSK-3 inhibitor, and its effect on -catenin in the mouse hippocampus. Biol. Psychiatry 55, 781–784.
Kalinichev, M., and Dawson, L. A. (2011). Evidence for antimanic efficacy of glycogen synthase kinase-3 (GSK3) inhibitors in a strain-specific model of acute mania. Int. J. Neuropsychopharmacol. 14, 1–17.
Karege, F., Perroud, N., Burkhardt, S., Schwald, M., Ballmann, E., La Harpe, R., and Malafosse, A. (2007). Alteration in kinase activity but not in protein levels of protein kinase B and glycogen synthase kinase-3beta in ventral prefrontal cortex of depressed suicide victims. Biol. Psychiatry 61, 240–245.
Kelly, S., Zhao, H., Hua Sun, G., Cheng, D., Qiao, Y., Luo, J., Martin, K., Steinberg, G. K., Harrison, S. D., and Yenari, M. A. (2004). Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp. Neurol. 188, 378–386.
Kim, H. J., and Thayer, S. A. (2009). Lithium increases synapse formation between hippocampal neurons by depleting phosphoinositides. Mol. Pharmacol. 75, 1021–1030.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Kim, W. Y., Zhou, F. Q., Zhou, J., Yokota, Y., Wang, Y. M., Yoshimura, T., Kaibuchi, K., Woodgett, J. R., Anton, E. S., and Snider, W. D. (2006). Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron 52, 981–996.
Kirshennboim, N., Plotkin, B., Ben Shlomo, S., Kaidanovich-Beilin, O., and Eldar-Finkelman, H. (2004). Lithium-mediated phosphorylation of glycogen synthase kinase-3 involves PI3 Kinase-dependent activation of protein kinase C- alpha. J. Mol. Neurosci. 24, 199–207.
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Koh, S. H., Kim, Y., Kim, H. Y., Hwang, S., Lee, C. H., and Kim, S. H. (2007). Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 205, 336–346.
Kozlovsky, N., Belmaker, R. H., and Agam, G. (2002). GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur. Neuropsychopharmacol. 12, 13–25.
Kroczka, B., Branski, P., Palucha, A., Pilc, A., and Nowak, G. (2001). Antidepressant-like properties of zinc in rodent forced swim test. Brain Res. Bull. 55, 297–300.
Kunick, C., Lauenroth, K., Leost, M., Meijer, L., and Lemcke, T. (2004). 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3 beta. Bioorg. Med. Chem. Lett. 14, 413–416.
Kuo, G. H., Prouty, C., DeAngelis, A., Shen, L., O’Neill, D. J., Shah, C., Connolly, P. J., Murray, W. V., Conway, B. R., Cheung, P., Westover, L., Xu, J. Z., Look, R. A., Demarest, K. T., Emanuel, S., Middleton, S. A., Jolliffe, L., Beavers, M. P., and Chen, X. (2003). Synthesis and discovery of macrocyclic polyoxygenated bis-7-azaindolylmaleimides as a novel series of potent and highly selective glycogen synthase kinase-3beta inhibitors. J. Med. Chem. 46, 4021–4031.
Leclerc, S., Garnier, M., Hoessel, R., Marko, D., Bibb, J. A., Snyder, G. L., Greengard, P., Biernat, J., Wu, Y. Z., Mandelkow, E. M., Eisenbrand, G., and Meijer, L. (2001). Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 276, 251–260.
Leost, M., Schultz, C., Link, A., Wu, Y. Z., Biernat, J., Mandelkow, E. M., Bibb, J. A., Snyder, G. L., Greengard, P., Zaharevitz, D. W., Gussio, R., Senderowicz, A. M., Sausville, E. A., Kunick, C., and Meijer, L. (2000). Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur. J. Biochem. 267, 5983–5994.
Leyhe, T., Eschweiler, G. W., Stransky, E., Gasser, T., Annas, P., Basun, H., and Laske, C. (2009). Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J. Alzheimers Dis. 16, 649–656.
Li, T., Hawkes, C., Qureshi, H. Y., Kar, S., and Paudel, H. K. (2006). Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 45, 3134–3145.
Li, W., Sun, W., Zhang, Y., Wei, W., Ambasudhan, R., Xia, P., Talantova, M., Lin, T., Kim, J., Wang, X., Kim, W. R., Lipton, S. A., Zhang, K., and Ding, S. (2011). Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 8299–8304.
Licht-Murava, A., Plotkin, B., Eisenstein, M., and Eldar-Finkelman, H. (2011). Elucidating substrate and inhibitor binding sites on the surface of GSK-3beta and the refinement of a competitive inhibitor. J. Mol. Biol. 408, 366–378.
Lipina, T. V., Kaidanovich-Beilin, O., Patel, S., Wang, M., Clapcote, S. J., Liu, F., Woodgett, J. R., and Roder, J. C. (2011). Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse 65, 234–248.
Lochhead, P. A., Kinstrie, R., Sibbet, G., Rawjee, T., Morrice, N., and Cleghon, V. (2006). A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol. Cell 24, 627–633.
Loge, C., Testard, A., Thiery, V., Lozach, O., Blairvacq, M., Robert, J. M., Meijer, L., and Besson, T. (2008). Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 43, 1469–1477.
Lovestone, S., Killick, R., Di Forti, M., and Murray, R. (2007). Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 30, 142–149.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39.
Luna-Medina, R., Cortes-Canteli, M., Sanchez-Galiano, S., Morales-Garcia, J. A., Martinez, A., Santos, A., and Perez-Castillo, A. (2007). NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: potential therapeutic role in brain disorders. J. Neurosci. 27, 5766–5776.
Macdonald, A., Briggs, K., Poppe, M., Higgins, A., Velayudhan, L., and Lovestone, S. (2008). A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 23, 704–711.
Magiatis, P., Polychronopoulos, P., Skaltsounis, A. L., Lozach, O., Meijer, L., Miller, D. B., and O’Callaghan, J. P. (2010). Indirubins deplete striatal monoamines in the Intact and MPTP-treated mouse brain and block kainate-induced striatal astrogliosis. Neurotoxicol. Teratol. 32, 212–219.
Makhortova, N. R., Hayhurst, M., Cerqueira, A., Sinor-Anderson, A. D., Zhao, W. N., Heiser, P. W., Arvanites, A. C., Davidow, L. S., Waldon, Z. O., Steen, J. A., Lam, K., Ngo, H. D., and Rubin, L. L. (2011). A screen for regulators of survival of motor neuron protein levels. Nat. Chem. Biol. 7, 544–552.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Martin, L., Magnaudeix, A., Esclaire, F., Yardin, C., and Terro, F. (2009). Inhibition of glycogen synthase kinase-3beta downregulates total tau proteins in cultured neurons and its reversal by the blockade of protein phosphatase-2A. Brain Res. 1252, 66–75.
Martinez, A., Alonso, A., Castro, A., and Dorronsoro, I. (2008). “GSK-3 inhibitors in Alzheimer’s disease: TDZDs, from the discovery to clinical trial,” in Medicinal Chemistry of Alzheimer Disease, ed. A. Martinez (Kerala: Transworld Research Network), 225–253.
Martinez, A., Alonso, M., Castro, A., Perez, C., and Moreno, F. J. (2002). First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 45, 1292–1299.
Mathew, S. J., Manji, H. K., and Charney, D. S. (2008). Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology 33, 2080–2092.
Mazanetz, M. P., and Fischer, P. M. (2007). Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 6, 464–479.
Meares, G. P., and Jope, R. S. (2007). Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J. Biol. Chem. 282, 16989–17001.
Meijer, L., Skaltsounis, A. L., Magiatis, P., Polychronopoulos, P., Knockaert, M., Leost, M., Ryan, X. P., Vonica, C. A., Brivanlou, A., Dajani, R., Crovace, C., Tarricone, C., Musacchio, A., Roe, S. M., Pearl, L., and Greengard, P. (2003). GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10, 1255–1266.
Meijer, L., Thunnissen, A. M., White, A. W., Garnier, M., Nikolic, M., Tsai, L. H., Walter, J., Cleverley, K. E., Salinas, P. C., Wu, Y. Z., Biernat, J., Mandelkow, E. M., Kim, S. H., and Pettit, G. R. (2000). Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 7, 51–63.
Mettey, Y., Gompel, M., Thomas, V., Garnier, M., Leost, M., Ceballos-Picot, I., Noble, M., Endicott, J., Vierfond, J. M., and Meijer, L. (2003). Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J. Med. Chem. 46, 222–236.
Mines, M. A., Yuskaitis, C. J., King, M. K., Beurel, E., and Jope, R. S. (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 5, e9706. doi: 10.1371/journal.pone.0009706
Mishra, R., Barthwal, M. K., Sondarva, G., Rana, B., Wong, L., Chatterjee, M., Woodgett, J. R., and Rana, A. (2007). Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J. Biol. Chem. 282, 30393–30405.
Mukai, F., Ishiguro, K., Sano, Y., and Fujita, S. C. (2002). Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 81, 1073–1083.
Muyllaert, D., Kremer, A., Jaworski, T., Borghgraef, P., Devijver, H., Croes, S., Dewachter, I., and Van Leuven, F. (2008). Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav. 7(Suppl. 1), 57–66.
Nguyen, T. N., and Tepe, J. J. (2009). Preparation of hymenialdisine, analogues and their evaluation as kinase inhibitors. Curr. Med. Chem. 16, 3122–3143.
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., Gaynor, K., Wang, L., LaFrancois, J., Feinstein, B., Burns, M., Krishnamurthy, P., Wen, Y., Bhat, R., Lewis, J., Dickson, D., and Duff, K. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995.
Nowak, G., Szewczyk, B., and Pilc, A. (2005). Zinc and depression. An update. Pharmacol. Rep. 57, 713–718.
Nunes, P. V., Forlenza, O. V., and Gattaz, W. F. (2007). Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br. J. Psychiatry 190, 359–360.
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
O’Brien, W. T., and Klein, P. S. (2009). Validating GSK3 as an in vivo target of lithium action. Biochem. Soc. Trans. 37, 1133–1138.
Ortega, M. A., Montoya, M. E., Zarranz, B., Jaso, A., Aldana, I., Leclerc, S., Meijer, L., and Monge, A. (2002). Pyrazolo[3,4-b]quinoxalines. A new class of cyclin-dependent kinases inhibitors. Bioorg. Med. Chem. 10, 2177–2184.
Owens, D. R., Zinman, B., and Bolli, G. (2003). Alternative routes of insulin delivery. Diabet. Med. 20, 886–898.
Pachet, A. K., and Wisniewski, A. M. (2003). The effects of lithium on cognition: an updated review. Psychopharmacology (Berl.) 170, 225–234.
Padilla, A., Hauer, J. A., Tsigelny, I., Parello, J., and Taylor, S. S. (1997). Solution structure of synthetic peptide inhibitor and substrate of cAMP-dependent protein kinase. A study by 2D H NMR and molecular dynamics. J. Pept. Res. 49, 210–220.
Palomo, V., Soteras, I., Perez, D. I., Perez, C., Gil, C., Campillo, N. E., Martinez, A. (2011). Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J. Med. Chem. (in press).
Pandey, G. N., Ren, X., Rizavi, H. S., and Dwivedi, Y. (2010). Glycogen synthase kinase-3beta in the platelets of patients with mood disorders: effect of treatment. J. Psychiatr. Res. 44, 143–148.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl. 1), S428–S437.
Peineau, S., Nicolas, C. S., Bortolotto, Z. A., Bhat, R. V., Ryves, W. J., Harwood, A. J., Dournaud, P., Fitzjohn, S. M., and Collingridge, G. L. (2009). A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Mol. Brain 2, 22.
Peng, J., Kudrimoti, S., Prasanna, S., Odde, S., Doerksen, R. J., Pennaka, H. K., Choo, Y. M., Rao, K. V., Tekwani, B. L., Madgula, V., Khan, S. I., Wang, B., Mayer, A. M., Jacob, M. R., Tu, L. C., Gertsch, J., and Hamann, M. T. (2011). Structure-activity relationship and mechanism of action studies of manzamine analogues for the control of neuroinflammation and cerebral infections. J. Med. Chem. 53, 61–76.
Perez, D. I., Conde, S., Perez, C., Gil, C., Simon, D., Wandosell, F., Moreno, F. J., Gelpi, J. L., Luque, F. J., and Martinez, A. (2009a). Thienylhalomethylketones: irreversible glycogen synthase kinase 3 inhibitors as useful pharmacological tools. Bioorg. Med. Chem. 17, 6914–6925.
Perez, M., Perez, D. I., Martinez, A., Castro, A., Gomez, G., and Fall, Y. (2009b). The first enantioselective synthesis of palinurin. Chem. Commun. (Camb.) 3252–3254.
Perez, D. I., Palomo, V., Perez, C., Gil, C., Dans, P. D., Luque, F. J., Conde, S., and Martinez, A. (2011). Switching reversibility to irreversibility in glycogen synthase kinase 3 inhibitors: clues for specific design of new compounds. J. Med. Chem. 54, 4042–4056.
Perez, M., Rojo, A. I., Wandosell, F., Diaz-Nido, J., and Avila, J. (2003). Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem. J. 372, 129–136.
Petit-Paitel, A., Brau, F., Cazareth, J., and Chabry, J. (2009). Involvment of cytosolic and mitochondrial GSK-3beta in mitochondrial dysfunction and neuronal cell death of MPTP/MPP-treated neurons. PLoS ONE 4, e5491. doi: 10.1371/journal.pone.0005491
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439.
Plattner, F., Angelo, M., and Giese, K. P. (2006). The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 281, 25457–25465.
Plotkin, B., Kaidanovich, O., Talior, I., and Eldar-Finkelman, H. (2003). Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 305, 974–980.
Polter, A., Beurel, E., Yang, S., Garner, R., Song, L., Miller, C. A., Sweatt, J. D., McMahon, L., Bartolucci, A. A., Li, X., and Jope, R. S. (2010). Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 35, 1761–1774.
Polychronopoulos, P., Magiatis, P., Skaltsounis, A. L., Myrianthopoulos, V., Mikros, E., Tarricone, A., Musacchio, A., Roe, S. M., Pearl, L., Leost, M., Greengard, P., and Meijer, L. (2004). Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 47, 935–946.
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., Daneels, G., Bouwknecht, J. A., and Steckler, T. (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029.
Rao, K. V., Donia, M. S., Peng, J., Garcia-Palomero, E., Alonso, D., Martinez, A., Medina, M., Franzblau, S. G., Tekwani, B. L., Khan, S. I., Wahyuono, S., Willett, K. L., and Hamann, M. T. (2006). Manzamine B and E and ircinal A related alkaloids from an Indonesian Acanthostrongylophora sponge and their activity against infectious, tropical parasitic, and Alzheimer’s diseases. J. Nat. Prod. 69, 1034–1040.
Rao, R., Hao, C. M., Redha, R., Wasserman, D. H., McGuinness, O. P., and Breyer, M. D. (2007). Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia 50, 452–460.
Ribe, E. M., Perez, M., Puig, B., Gich, I., Lim, F., Cuadrado, M., Sesma, T., Catena, S., Sanchez, B., Nieto, M., Gómez-Ramos, P., Morán, M. A., Cabodevilla, F., Samaranch, L., Ortiz, L., Pérez, A., Ferrer, I., Avila, J., and Gómez-Isla, T. (2005). Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis. 20, 814–822.
Ring, D. B., Johnson, K. W., Henriksen, E. J., Nuss, J. M., Goff, D., Kinnick, T. R., Ma, S. T., Reeder, J. W., Samuels, I., Slabiak, T., Wagman, A. S., Hammond, M. E., and Harrison, S. D. (2003). Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 52, 588–595.
Rockenstein, E., Torrance, M., Adame, A., Mante, M., Bar-on, P., Rose, J. B., Crews, L., and Masliah, E. (2007). Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27, 1981–1991.
Rosa, A., Egea, J., Martínez, A., García, A., and López, M. (2008a). Neuroprotective effect of the new thiadiazolidinone NP00111 against oxygen-glucose deprivation in rat hippocampal slices: implication of ERK1/2 and PPARgamma receptors. Exp. Neurol. 212, 93–99.
Rosa, A., Kaster, M., Binfaré, R., Morales, S., Martín-Aparicio, E., Navarro-Rico, M., Martinez, A., Medina, M., García, A., López, M., and Rodrigues, A. (2008b). Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1549–1456.
Ryves, W. J., Dajani, R., Pearl, L., and Harwood, A. J. (2002). Glycogen synthase kinase-3 inhibition by lithium and beryllium suggests the presence of two magnesium binding sites. Biochem. Biophys. Res. Commun. 290, 967–972.
Ryves, W. J., and Harwood, A. J. (2001). Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 280, 720–725.
Sato, N., Meijer, L., Skaltsounis, L., Greengard, P., and Brivanlou, A. H. (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 10, 55–63.
Saus, E., Soria, V., Escaramis, G., Crespo, J. M., Valero, J., Gutierrez-Zotes, A., Martorell, L., Vilella, E., Menchon, J. M., Estivill, X., Gratacòs, M., and Urretavizcaya, M. (2010). A haplotype of glycogen synthase kinase 3beta is associated with early onset of unipolar major depression. Genes Brain Behav. 9, 799–807.
Schultz, C., Link, A., Leost, M., Zaharevitz, D. W., Gussio, R., Sausville, E. A., Meijer, L., and Kunick, C. (1999). Paullones, a series of cyclin-dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity. J. Med. Chem. 42, 2909–2919.
Seira, O., Gavin, R., Gil, V., Llorens, F., Rangel, A., Soriano, E., and del Rio, J. A. (2011). Neurites regrowth of cortical neurons by GSK3beta inhibition independently of Nogo receptor 1. J. Neurochem. 113, 1644–1658.
Selenica, M. L., Jensen, H. S., Larsen, A. K., Pedersen, M. L., Helboe, L., Leist, M., and Lotharius, J. (2007). Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 152, 959–979.
Sereno, L., Coma, M., Rodriguez, M., Sanchez-Ferrer, P., Sanchez, M. B., Gich, I., Agullo, J. M., Perez, M., Avila, J., Guardia-Laguarta, C., Clarimón, J., Lleó, A., and Gómez-Isla, T. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367.
Shapira, M., Licht, A., Milman, A., Pick, C. G., Shohami, E., and Eldar-Finkelman, H. (2007). Role of glycogen synthase kinase-3beta in early depressive behavior induced by mild traumatic brain injury. Mol. Cell. Neurosci. 34, 571–577.
Sharma, V., and Tepe, J. J. (2004). Potent inhibition of checkpoint kinase activity by a hymenialdisine-derived indoloazepine. Bioorg. Med. Chem. Lett. 16, 4319–4321.
Shen, L., Prouty, C., Conway, B. R., Westover, L., Xu, J. Z., Look, R. A., Chen, X., Beavers, M. P., Roberts, J., Murray, W. V., Demarest, K. T., and Kuo, G. H. (2004). Synthesis and biological evaluation of novel macrocyclic bis-7-azaindolylmaleimides as potent and highly selective glycogen synthase kinase-3 beta (GSK-3 beta) inhibitors. Bioorg. Med. Chem. Lett. 12, 1239–1255.
Shi, S. H., Cheng, T., Jan, L. Y., and Jan, Y. N. (2004). APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 14, 2025–2032.
Silva, R., Mesquita, A. R., Bessa, J., Sousa, J. C., Sotiropoulos, I., Leao, P., Almeida, O. F., and Sousa, N. (2008). Lithium blocks stress-induced changes in depressive-like behavior and hippocampal cell fate: the role of glycogen-synthase-kinase-3beta. Neuroscience 152, 656–669.
Skardelly, M., Gaber, K., Schwarz, J., and Milosevic, J. (2011). Neuroprotective effects of the beta-catenin stabilization in an oxygen- and glucose-deprived human neural progenitor cell culture system. Int. J. Dev. Neurosci. 29, 543–547.
Song, L., Zhou, T., and Jope, R. S. (2004). Lithium facilitates apoptotic signaling induced by activation of the Fas death domain-containing receptor. BMC Neurosci. 5, 20. doi: 10.1186/1471-2202-5-20
Song, Y. F., Qu, Y., Cao, X. P., and Zhang, W. (2011). Cellular localization of debromohymenialdisine and hymenialdisine in the marine sponge Axinella sp. using a newly developed cell purification protocol. Mar. Biotechnol. 13, 868–882.
Soutar, M. P., Kim, W. Y., Williamson, R., Peggie, M., Hastie, C. J., McLauchlan, H., Snider, W. D., Gordon-Weeks, P. R., and Sutherland, C. (2011). Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 115, 974–983.
Spittaels, K., Van den Haute, C., Van Dorpe, J., Terwel, D., Vandezande, K., Lasrado, R., Bruynseels, K., Irizarry, M., Verhoye, M., Van Lint, J., Vandenheede, J. R., Ashton, D., Mercken, M., Loos, R., Hyman, B., Van der Linden, A., Geerts, H., and Van Leuven, F. (2002). Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 113, 797–808.
Stambolic, V., Ruel, L., and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668.
Stukenbrock, H., Mussmann, R., Geese, M., Ferandin, Y., Lozach, O., Lemcke, T., Kegel, S., Lomow, A., Burk, U., Dohrmann, C., Meijer, L., Austen, M., and Kunick, C. (2008). 9-cyano-1-azapaullone (cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic beta cell protection and replication. J. Med. Chem. 51, 2196–2207.
Sun, X., Sato, S., Murayama, O., Murayama, M., Park, J. M., Yamaguchi, H., and Takashima, A. (2002). Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci. Lett. 321, 61–64.
Takadera, T., and Ohyashiki, T. (2004). Glycogen synthase kinase-3 inhibitors prevent caspase-dependent apoptosis induced by ethanol in cultured rat cortical neurons. Eur. J. Pharmacol. 499, 239–245.
Takahashi, M., Yasutake, K., and Tomizawa, K. (1999). Lithium inhibits neurite growth and tau protein kinase I/glycogen synthase kinase-3beta-dependent phosphorylation of juvenile tau in cultured hippocampal neurons. J. Neurochem. 73, 2073–2083.
Tariot, P. N., and Aisen, P. S. (2009). Can lithium or valproate untie tangles in Alzheimer’s disease? J. Clin. Psychiatry 70, 919–921.
Tasdemir, D., Mallon, R., Greenstein, M., Feldberg, L. R., Kim, S. C., Collins, K., Wojciechowicz, D., Mangalindan, G., Concepción, G., Harper, M. K., and Ireland, C. M. (2002). Aldisine alkaloids from the Philippine sponge Stylissa massa are potent inhibitors of mitogen-activated protein kinase-1(MEK-1). J. Med. Chem. 45, 529–532.
Tassabehji, N. M., Corniola, R. S., Alshingiti, A., and Levenson, C. W. (2008). Zinc deficiency induces depression-like symptoms in adult rats. Physiol. Behav. 95, 365–369.
Taylor, S. S., Radzio-Andzelm, E., and Hunter, T. (1995). How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine kinase. FASEB J. 9, 1255–1266.
ter Haar, E., Coll, J. T., Austen, D. A., Hsiao, H. M., Swenson, L., and Jain, J. (2001). Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 8, 593–596.
Terao, T., Nakano, H., Inoue, Y., Okamoto, T., Nakamura, J., and Iwata, N. (2006). Lithium and dementia: a preliminary study. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 1125–1128.
Terwel, D., Muyllaert, D., Dewachter, I., Borghgraef, P., Croes, S., Devijver, H., and Van Leuven, F. (2008). Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 172, 786–798.
Testard, A., Loge, C., Leger, B., Robert, J. M., Lozach, O., Blairvacq, M., Meijer, L., Thiery, V., and Besson, T. (2006). Thiazolo[5,4-f]quinazolin-9-ones, inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 16, 3419–3423.
Thomas, G. M., Frame, S., Goedert, M., Nathke, I., Polakis, P., and Cohen, P. (1999). A GSK3-binding peptide from FRAT1 selectively inhibits the GSK3-catalysed phosphorylation of axin and beta-catenin. FEBS Lett. 458, 247–251.
Thornton, T. M., Pedraza-Alva, G., Deng, B., Wood, C. D., Aronshtam, A., Clements, J. L., Sabio, G., Davis, R. J., Matthews, D. E., Doble, B., and Rincon, M. (2008). Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320, 667–670.
Torres-Aleman, I. (2007). Targeting insulin-like growth factor-1 to treat Alzheimer’s disease. Expert Opin. Ther. Targets 11, 1535–1542.
Valerio, A., Bertolotti, P., Delbarba, A., Perego, C., Dossena, M., Ragni, M., Spano, P., Carruba, M. O., De Simoni, M. G., and Nisoli, E. (2011). Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J. Neurochem. 116, 1148–1159.
Vougogiannopoulou, K., Ferandin, Y., Bettayeb, K., Myrianthopoulos, V., Lozach, O., Fan, Y., Johnson, C. H., Magiatis, P., Skaltsounis, A. L., Mikros, E., and Meijer, L. (2008). Soluble 3′,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase -3 alter circadian period. J. Med. Chem. 51, 6421–6431.
Wada, A., Yokoo, H., Yanagita, T., and Kobayashi, H. (2005). Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J. Pharmacol. Sci. 99, 307–321.
Wahba, A. E., and Hamann, M. T. (2011). New one-pot methodologies for the modification or synthesis of alkaloid scaffolds. Mar. Drugs 8, 2395–2416.
Wan, Y., Hur, W., Cho, C. Y., Liu, Y., Adrian, F. J., Lozach, O., Bach, S., Mayer, T., Fabbro, D., Meijer, L., and Gray, N. S. (2004). Synthesis and target identification of hymenialdisine analogs. Chem. Biol. 11, 247–259.
Wang, W., Yang, Y., Ying, C., Li, W., Ruan, H., Zhu, X., You, Y., Han, Y., Chen, R., Wang, Y., and Li, M. (2007). Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 52, 1678–1684.
Watase, K., Gatchel, J. R., Sun, Y., Emamian, E., Atkinson, R., Richman, R., Mizusawa, H., Orr, H. T., Shaw, C., and Zoghbi, H. Y. (2007). Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 4, e182. doi: 10.1371/journal.pmed.0040182
Welham, M. J., Storm, M. P., Kingham, E., and Bone, H. K. (2007). Phosphoinositide 3-kinases and regulation of embryonic stem cell fate. Biochem. Soc. Trans. 35, 225–228.
Williams, D. H., and Faulkner, J. (1996). Isomers and tautomers of hymenialdisine and debromohymenialfisine. Nat. Prod. Lett. 9, 57–64.
Williams, R., Ryves, W. J., Dalton, E. C., Eickholt, B., Shaltiel, G., Agam, G., and Harwood, A. J. (2004). A molecular cell biology of lithium. Biochem. Soc. Trans. 32, 799–802.
Wood, N. I., and Morton, A. J. (2003). Chronic lithium chloride treatment has variable effects on motor behaviour and survival of mice transgenic for the Huntington’s disease mutation. Brain Res. Bull. 61, 375–383.
Woodgett, J. R. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factorA. EMBO J. 9, 2431–2438.
Woodgett, J. R., and Cohen, P. (1984). Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5). Biochim. Biophys. Acta 788, 339–347.
Wu, D., and Pan, W. (2011). GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 35, 161–168.
Yasuda, S., Liang, M., Marinova, Z., Yahyavi, A., and Chuang, D. (2009). The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 14, 51–59.
Ying, Q. L., Wray, J., Nichols, J., Batlle-Morera, L., Doble, B., Woodgett, J., Cohen, P., and Smith, A. (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523.
Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A., and Kaibuchi, K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149.
Yost, C., Farr, G. H. III, Pierce, S. B., Ferkey, D. M., Chen, M. M., and Kimelman, D. (1998). GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell 93, 1031–1041.
Zhang, F., Phiel, C. J., Spece, L., Gurvich, N., and Klein, P. S. (2003). Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 278, 33067–33077.
Zhang, N., Zhong, R., Yan, H., and Jiang, Y. (2011). Structural features underlying selective inhibition of GSK3beta by dibromocantharelline: implications for rational drug design. Chem. Biol. Drug Des. 77, 199–205.
Zhao, Y., Altman, B. J., Coloff, J. L., Herman, C. E., Jacobs, S. R., Wieman, H. L., Wofford, J. A., Dimascio, L. N., Ilkayeva, O., Kelekar, A., Reya, T., and Rathmell, J. C. (2007). Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol. Cell. Biol. 27, 4328–4339.
Keywords: protein kinases, GSK-3, GSK-3 inhibitors, CNS
Citation: Eldar-Finkelman H and Martinez A (2011) GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 4:32. doi: 10.3389/fnmol.2011.00032
Received: 08 August 2011; Accepted: 29 September 2011;
Published online: 31 October 2011.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaOksana Kaidanovich-Beilin, Samuel Lunenfeld Research Institute, Canada
Copyright: © 2011 Eldar-Finkelman and Martinez. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Hagit Eldar-Finkelman, Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University. e-mail:aGVsZGFyQHBvc3QudGF1LmFjLmls; Ana Martinez, Instituto de Química Medica – CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. e-mail:YW1hcnRpbmV6QGlxbS5jc2ljLmVz