
 Eva Baquedano1,2,3,4
Eva Baquedano1,2,3,4

- 1 Department of Pediatrics, Universidad Autónoma de Madrid, Madrid, Spain
- 2 Department of Endocrinology, Hospital Infantil Universitario Niño Jesús, Madrid, Spain
- 3 CIBER Fisiopatología de Obesidad y Nutrición, Madrid, Spain
- 4 Instituto de Investigación Sanitaria La Princesa, Instituto de Salud Carlos III, Madrid, Spain
The brain incorporates and coordinates information based on the hormonal environment, receiving information from peripheral tissues through the circulation. Although it was initially thought that hormones only acted on the hypothalamus to perform endocrine functions, it is now known that they in fact exert diverse actions on many different brain regions including the hypothalamus. Ghrelin is a gastric hormone that stimulates growth hormone secretion and food intake to regulate energy homeostasis and body weight by binding to its receptor, growth hormone secretagogues–GH secretagogue-receptor, which is most highly expressed in the pituitary and hypothalamus. In addition, ghrelin has effects on learning and memory, reward and motivation, anxiety, and depression, and could be a potential therapeutic agent in neurodegenerative disorders where excitotoxic neuronal cell death and inflammatory processes are involved.
Ghrelin and Growth Hormone Secretagogues
Ghrelin is a 28-amino acid peptide that is esterified with octanoic acid on serine 3 and is mainly released from the oxyntic cells of the stomach mucosa. It was discovered based on its ability to stimulate growth hormone (GH) release by activating the GH secretagogue-receptor (GHSR-1a) in the pituitary (Kojima et al., 1999; Date et al., 2000). Ghrelin has many other peripheral actions including direct effects on exocrine and endocrine pancreatic functions, carbohydrate metabolism, the cardiovascular system, gastric secretion, stomach motility, and sleep (Van der Lely et al., 2004; Ghigo et al., 2005; Kojima and Kangawa, 2005).
One of the most studied functions of ghrelin is its orexigenic properties, which has prompted the investigation of this hormone as a target for the treatment of obesity (Foster-Schubert and Cummings, 2006; Zorrilla et al., 2006). Ghrelin promotes feeding, weight gain, and adiposity by acting both at central and peripheral targets (Tschop et al., 2001a; Wren et al., 2001) with an inverse correlation between circulating ghrelin levels and body weight (Tschop et al., 2000). Moreover, lack of ghrelin in KO mice (Wortley et al., 2005) or ghrelin signaling in growth hormone secretagogues (GHS)–R1a KO mice (Zigman et al., 2005) protects against diet-induced obesity (DIO). Indeed, a vast number of studies point to ghrelin as an important hormonal signal promoting feeding and regulating metabolism in humans and rodents (Cummings et al., 2001; Tschop et al., 2001b; Drazen et al., 2006).
Ghrelin originates from the proteolytic cleavage of pre-pro-ghrelin and pro-ghrelin. Posttranslational acylation of pro-ghrelin by the ghrelin O-acyltransferase (GOAT) enzyme is required for its activation of GHS–R1a (Figure 1). GOAT is found mostly in the stomach (Yang et al., 2008), acylating newly synthesized pro-ghrelin, with ghrelin exists as two forms in the plasma, acylated ghrelin and des-acylated ghrelin. Although des-acyl ghrelin is found at high concentrations in the plasma, it neither activates GHSR-1a nor causes GH release in vivo or in vitro (Kojima et al., 1999; Bednarek et al., 2000; Hosoda et al., 2000; Toshinai, 2006). However, it is now thought that des-acylated ghrelin may exert physiologically relevant effects through as yet unidentified receptors.
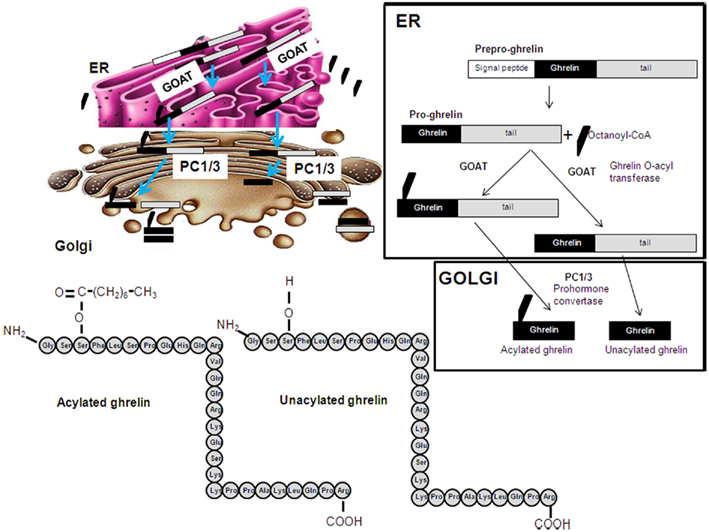
Figure 1. Schematic representation of the posttranslational processing of ghrelin. A signal peptide peptidase cleaves the signal sequence. Acylation of pro-ghrelin occurs by means of ghrelin O-acyl transferase (GOAT), which is located in the ER compartment and mediates the translocation of octanoyl-CoA. Once the pro-ghrelin precursor reaches the trans-Golgi compartment, it is cleaved by PC1/3 prohormone convertase. Different forms of ghrelin are released to the circulation: acylated, unacylated, and other shorter forms.
Growth hormone secretagogues (GHS) are metenkephalin-derived synthetic oligopeptides and non-peptidyl molecules that activate the GHS–R1a (Davenport et al., 2005). Multiple peptidyl and several non-peptidyl analogs of ghrelin have been developed and functionally assessed over the last three decades. Unlike ghrelin, they do not require acylation to activate GHS–R1a and are not known to be GOAT substrates. These synthetic analogs can exhibit partial agonism (e.g., stimulation of appetite, but not GH release and vice versa), antagonism, and inverse agonism despite similar GHS–R1a binding affinities (Holst et al., 2004, 2006; Halem et al., 2005; Veldhuis and Bowers, 2010).
Ghrelin Receptor
The ghrelin receptor, GHS–R1a, is a G protein-coupled 7-transmembrane receptor that was first cloned from the pituitary and hypothalamus. It belongs to a family of receptors operating via the Gq-phospholipase C signaling pathways (Howard et al., 1996). Other signaling pathways involved are ERK1/2, the PLC–PKC pathway and Raf–MEK–MAPK. Ghrelin acts on GHS–R1a to increase intracellular Ca2+ via the guanine nucleotide binding protein q phospholipase C (Gq-PLC) pathway (Kojima and Kangawa, 2005). Through the α-subunit of GHS–R1a, activation of the PLC–protein kinase C pathway and Raf–MEK–MAPK occurs (Chung et al., 2007). Ghrelin also transactivates the tyrosine kinase receptor via the β- and Γ-subunits, leading to activation of MAPK via the Ras–Raf–MEK pathway (Nanzer et al., 2004). Furthermore, ghrelin and des-acylated ghrelin have been shown to exert their effects through stimulation of c-AMP-mediated PKA pathways (Granata et al., 2007). The anti-apoptotic effects of ghrelin and des-acylated ghrelin are mediated via the MAPK and PI3K signaling pathways, with Akt downstream effectors being involved in many of these effects. Akt can phosphorylate effector proteins at the membrane or cytoplasmic levels, including for example GSK-3β. It can also act as a transcription factor or phosphorylate other transcription factor at the nuclear level (Song et al., 2005).
This receptor is found at highest concentrations in the pituitary and hypothalamus (Howard et al., 1996) and acts primarily at these sites to stimulate GH release, induce a positive energy balance by stimulating food intake and decrease adipose usage through GH-independent mechanisms (Date et al., 2000). The abundant expression of GHS–R1a in the hypothalamus highlights its important role in energy metabolism. This receptor is highly expressed in agouti-related peptide/neuropeptide Y neurons in the arcuate nucleus and neurons in the ventromedial nucleus that express fatty acid synthase (Bennet et al., 1997; Guan et al., 1997; Tannenbaum et al., 1998; Willesen et al., 1999; Mitchell et al., 2001; Nogueiras et al., 2004; Smith et al., 2005; Zigman et al., 2006; López et al., 2008; Lage et al., 2010). There is also abundant GHSR-1a expression in other neuronal populations (Kohno et al., 2003) including the dentate gyrus of the hippocampus, the CA2 and CA3 regions of the hippocampus, the substantia nigra (SN), the ventral tegmental area (VTA), and various thalamic and brainstem nuclei, including the dorsal raphe nucleus (DRN; Shiiya et al., 2002). GHSR-1a has recently been identified in the thoracic, lumbar and sacral regions of the spinal cord (Ferens et al., 2010), and expressed in sympathetic and autonomic preganglionic neurons.
Anti-Apoptotic Actions of Ghrelin, Des-Acylated Ghrelin and GH Secretagogues
Ghrelin protects several cell types such as adipocytes (Kim et al., 2004) osteoblasts (Kim et al., 2005), cardiomyocytes, and endothelial cells (Baldanzi et al., 2002) by inhibiting apoptotic stimuli. Ghrelin, even at rather low doses (10−13 M), protects hypothalamic neuronal cells from cell death by inhibiting apoptosis (Chung et al., 2007). It has been reported to have protective effects against a variety of stimuli including ischemia/reperfusion (Chang et al., 2004; Konturek et al., 2006), alendronate (Iseri et al., 2005), serum deprivation (Kim et al., 2004), doxorubicin (Baldanzi et al., 2002), and TNF-α (Kim et al., 2005). At least some of the neuroprotective effects of ghrelin appear to be mediated through activation of GHS–R1a, as the specific receptor antagonist D-Lys-3-GH-releasing peptide (GHRP-6) completely blocks the protective effects of ghrelin against oxygen–glucose deprivation (OGD) insult. In contrast, Baldanzi et al. (2002) reported that in cardiomyocytes ghrelin exhibits an anti-apoptotic effect through binding to a novel, unidentified receptor that is distinct from GHS–R1a.
Multiple signaling pathways are involved in ghrelin-induced ERK1/2 activation, and the anti-apoptotic effects of ghrelin are mediated via the PI3K, PKC, and PKA signaling pathways. Ghrelin activates ERK1/2 in 3T3-L1 adipocytes (Kim et al., 2004), osteoblasts (Kim et al., 2005), cardiomyocytes, and endothelial cells (Baldanzi et al., 2002). This activation is believed to be an important mechanism to limit ischemic damage in hypothalamic neuronal cells (Jiang et al., 2002). A selective inhibitor of ERK1/2, PD98059, inhibits ghrelin-induced phosphorylation of ERK1/2 and the anti-apoptotic activities of ghrelin. On the other hand, ghrelin has been shown to exert its effects in various cells through stimulation of c-AMP-mediated PKA pathways (Kohno et al., 2003). Pretreatment with a PI3K inhibitor (wortmannin), PKC inhibitor (GF109203X), or PKA inhibitor (H89) significantly attenuates ghrelin-induced phosphorylation of ERK1/2 and the anti-apoptotic effects of ghrelin (Chung et al., 2007).
Des-acylated ghrelin protects cortical neurons from the apoptotic stimuli induced by OGD insult. Moreover, both types of ghrelin inhibit OGD-induced apoptosis even when administered 4 h after an OGD insult. This suggests that these peptides may have the ability to attenuate disease progression through activation of MAPK and PI3K/Akt signaling pathways even when administered sometime after the insult has occurred (Chung et al., 2011). Des-acylated ghrelin also stimulates PI3K/Akt pathways, with both ghrelin and des-acylated ghrelin capable of altering the status of the Bcl-2 family of proteins, inhibiting cytochrome c release and caspase-3 activity and promoting the survival of cortical neurons (Chung et al., 2008). Some studies have also reported the protective effects of des-acylated ghrelin on systemic tissues (Cassoni et al., 2001; Baldanzi et al., 2002; Nanzer et al., 2004; Maccarinelli et al., 2005; Delhanty et al., 2006; Filigheddu et al., 2007; Granata et al., 2007).
The neuroprotective effects of des-acylated ghrelin do not appear to be mediated through activation of GHS–R1a, as antagonism of this receptor fails to block the protective effect of des-acylated ghrelin against OGD insult. This is to be expected as des-acylated ghrelin is reported to neither activate nor bind GHS–R1a (Kojima et al., 1999); thus, the existence of a separate specific receptor for des-acylated ghrelin is suspected. This hypothesis is supported by the observation by Toshinai et al. (2006) in which des-acylated ghrelin, but not acylated ghrelin stimulated food intake in GHS–R1a-deficient mice, as well as by other studies (Baldanzi et al., 2002; Muccioli et al., 2004; Gauna et al., 2006; Filigheddu et al., 2007; Granata et al., 2007). Taken together, the findings provide evidence that ghrelin, regardless of its acylation, may function as a survival factor for neuronal cells and offer a new perspective on the potential role of these peptides in neuronal injury.
The Neuroprotective Actions of Ghrelin and GHS
As explained above, ghrelin stimulates the ERK1/2 and PI3K/Akt pathways through activation of GHS–R1a, which has been implicated in the regulation of cell survival (Figure 2; Datta et al., 1999; Pearson et al., 2001; Chung et al., 2007, 2008). Systemic administration of GHRP-6, a GHS, increases expression of proteins involved in cell survival or neuroprotection (Frago et al., 2002, 2005; Figure 3). Treatment of adult male rats with GHRP-6 for 1 week significantly increases insulin-like growth factor (IGF)-I mRNA levels in the hypothalamus, cerebellum, and hippocampus and this is associated with increased phosphorylation of Akt and Bad, with no change in MAPK or GSK-3. This suggests that GHRP-6 activates phosphatidylinositol kinase intracellular pathways involved in cell survival in response to growth factors and that this could be mediated through stimulation of local IGF-I production. Indeed, the anti-apoptotic protein Bcl-2 was augmented in these same areas, with no change in the pro-apoptotic protein Bax. Moreover, GHRP-6, reduces cerebellar cell death in aged rats and this phenomenon appears to be mediated via the stimulation of IGF-I production, which in turn inhibits the activation of caspases 9 and 3 (Pañeda et al., 2003). Other studies show that ghrelin exerts its neuroprotective effects through stimulation of the protein kinase A and C pathways (Chung et al., 2007). Taken together, these findings suggest that multiple signaling pathways are involved in ghrelin-mediated protection.
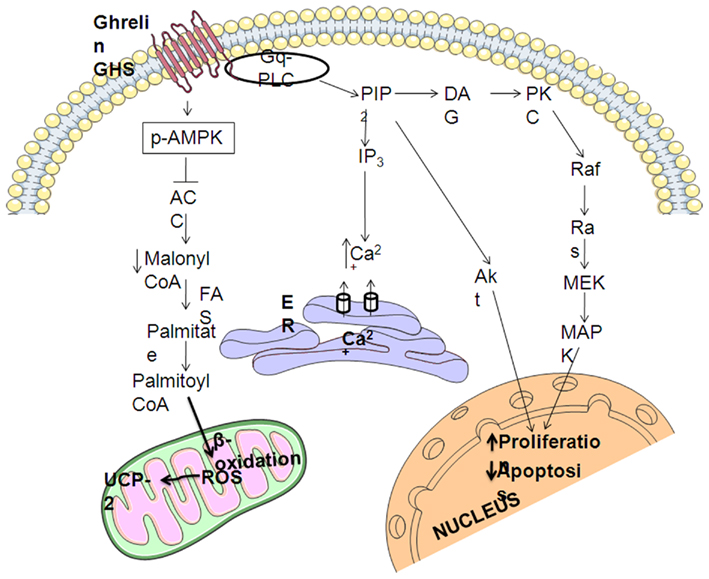
Figure 2. Summary of intracellular mechanisms mediating the neuroprotective effects of ghrelin. GHS–R1a activation result in release of intracellular calcium and protein kinase C (PKC) activation that leads to the stimulation of mitogen-activated protein kinases (MAPKs) pathway. The generation of phosphatidylinositol phosphates PIP3 and PIP2 induces the protein inositol 3 kinase (PI3K)/Akt pathway. MAPK and Akt stimulate cell proliferation and inhibit apoptosis. Ghrelin also regulates hypothalamic AMP-activated protein kinase (AMPK), phosphorylating (pAMPK), and activating it, which in turn phosphorylates and inactivates acetyl-CoA carboxylase (ACC), decreasing the cytoplasmic pool of malonyl-CoA, which promotes the generation of reactive oxygen species (ROS), which are buffered by uncoupling protein 2 (UCP-2).
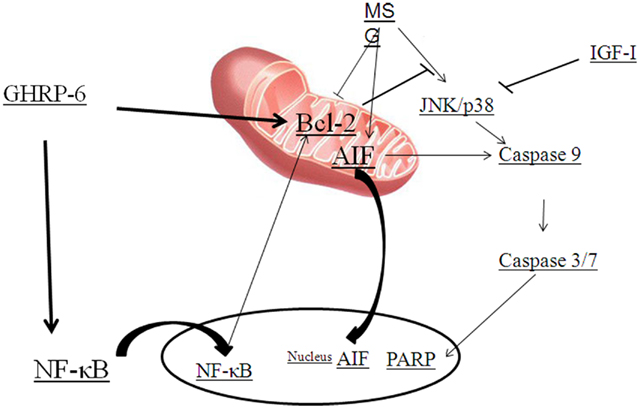
Figure 3. Diagram summarizing the GHRP-6 survival actions proposed against monosodium glutamate (MSG) excitotoxicity. MSG activates JNK or p38, caspases and stimulates the translocation of apoptosis inducing factor (AIF). Growth hormone-releasing peptide (GHRP)-6 prevents cell death by inducing Bcl-2 and nuclear factor-kappa B (NF-κB) that results in the blockage of AIF translocation and caspase and PARP activation. Insulin-like growth factor (IGF)-I prevents cell death by blocking caspase activation.
Table 1 shows the effect of ghrelin, des-acyl ghrelin, GHS, and antagonists in different diseases.
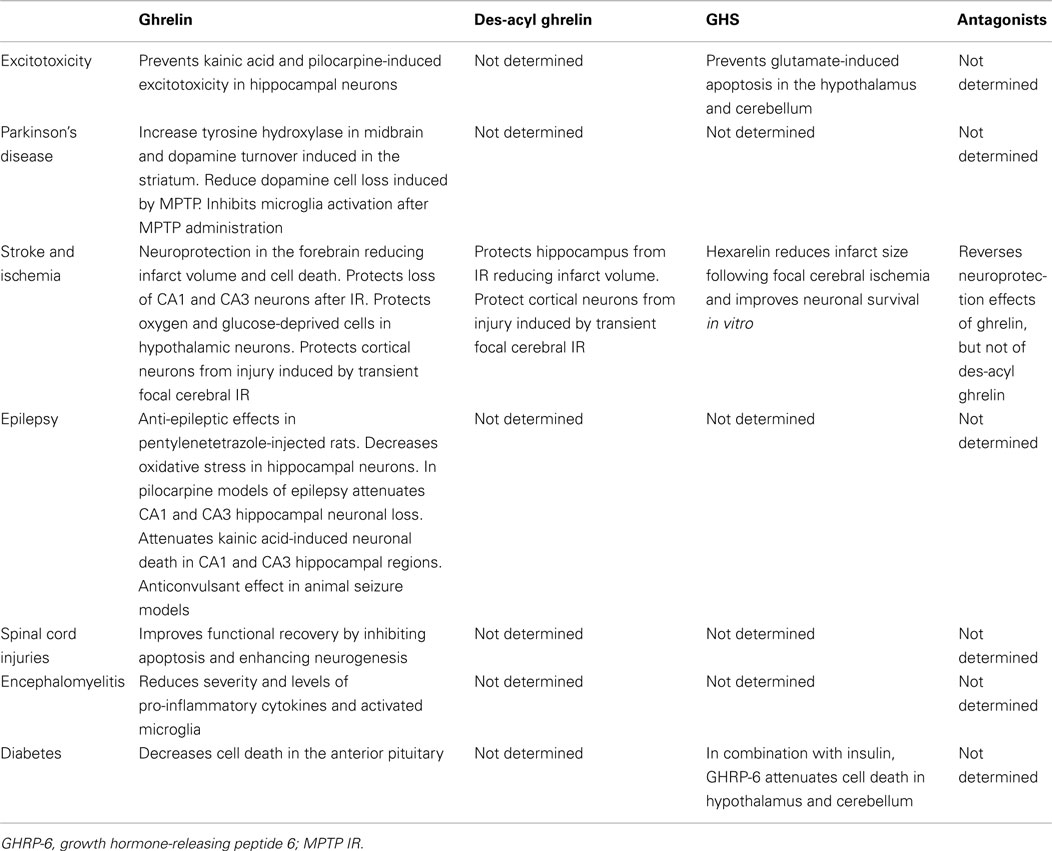
Table 1. Summary of the neuroprotective effects of ghrelin, des-acyl ghrelin, and synthetic antagonists and agonists of the ghrelin receptor.
Excitotoxicity
Excitotoxic brain damage is one of the major mechanisms by which neurons die in the adult central nervous system (CNS). L-Glutamate is the principal excitatory neurotransmitter in the mammalian CNS and under pathological conditions such as trauma, stroke, epilepsy, or hypoglycemia, excess release of L-Glutamic acid and other excitatory amino acids can lead to excitotoxic lesions in the brain due to over-excitation of nerve cells (Gill and Pulido, 2001). Over-activation of two major subtypes of ionotropic glutamate receptors, AMPA and NMDA, initiates a cascade of events involving protease activation and mitochondrial dysfunction that can result in cell death (Seeburg, 1993; Glazner and Mattson, 2000). Systemic administration of monosodium glutamate (MSG) induces apoptosis of hypothalamic and cerebellar neurons, which is associated with activation of caspases 9 and 7. GHRP-6 is capable of preventing this glutamate-induced cell death in both the hypothalamus and cerebellum (Delgado-Rubín de Célix et al., 2006). Moreover, MSG also induces cell death in the neuronal cell line RCA-6 by activating the intrinsic pathway of apoptosis through caspase-9 and -3/7 (Delgado-Rubín et al., 2009), which is in agreement with previous studies indicating that neurons and the neuroblastoma cell lines B50 and SH-SY5Y use the intrinsic pathway to undergo apoptotic cell death in response to excitotoxicity (Henshall et al., 2001; Mattson, 2003). However, although GHRP-6 partially inhibits MSG-induced cell death in RCA-6 neurons, it does not modify caspase-9 or -3/7 activities (Delgado-Rubín et al., 2009). Indeed, MSG also activates a caspase-independent pathway in these cells. Similar results showing two mechanisms of MSG-induced cell death have been reported in primary cortical neurons where MSG activates calpain, caspase-3 and the translocation of apoptosis-inducing factor (AIF) from the mitochondria to the cytosol and nuclei (Zhang and Bhavnani, 2006). Indeed, AIF plays an important role in caspase-independent mechanisms of cell death and is reported to be a key factor in neuronal death during excitotoxicity (Cheung et al., 2005). In RCA-6 neurons MSG promotes a perinuclear aggregation of AIF and this change in cellular localization is impeded by the presence of GHRP-6. As the insertion of Bax in the inner mitochondrial membrane is necessary for AIF release (Selznick et al., 2000; Arnoult et al., 2003), the GHRP-6-induced increment of Bcl-2, and hence its ability to complex with Bax, could block the release of AIF to the cytosol and its translocation to the nucleus (Susin et al., 1999).
Administration of kainic acid (KA) induces a sequence of altered behavioral events characterized by epileptiform seizures (Ben-Ari et al., 1980; Sperk, 1994), which are followed by neurodegeneration in specific brain regions, including the hippocampus, piriform cortex, thalamus, and amygdala. In the hippocampus, the CA3 pyramidal cells and interneurons in the hilus of the dentate gyrus are the most vulnerable, followed by CA1 pyramidal cells (Coyle, 1983; Sperk et al., 1985). Ghrelin could have a neuroprotective role in hippocampal neurons against KA-induced excitotoxicity, as it protects hippocampal neurons against pilocarpine-induced seizures (Xu et al., 2009) via activation of the PI3K/AKT pathway and inhibition of the mitochondrial apoptotic pathway.
Excitotoxic neuronal damage evoked by excessive or prolonged activation of excitatory amino acid receptors is recognized as an important mechanism in several neurodegenerative disorders such as stroke, traumatic brain injury, amyotrophic lateral sclerosis, Parkinson’s disease (PD), Huntington’s disease, and Alzheimer’s disease (AD; Doble, 1999; Salinska et al., 2005).
Parkinson’s Disease
Parkinson’s disease is characterized by the progressive degeneration of dopaminergic neurons that project from the SN to the dorsal striatum. The GHS–R1a is abundantly expressed in dopaminergic neurons in the SN (Guan et al., 1997; Zigman et al., 2006) with ghrelin increasing tyrosine hydroxylase expression in the midbrain and dopamine turnover in the dorsal striatum (Andrews et al., 2009). Sub-acute MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) administration, which selectively kills dopaminergic neurons in the SN, is suggested to be a model for PD (Przedborski et al., 2000). MPTP is taken up by dopaminergic neurons and leads to impairment in mitochondrial function. The subsequent energy failure with ATP depletion increases the formation of free radicals (Schmidt and Ferger, 2001) and cytochrome c release (Gómez et al., 2001; Singh and Dikshit, 2007). Intraperitoneal injection of ghrelin reduces dopamine cell loss in the SN in mice treated with MPTP (Andrews et al., 2009), most likely through attenuation of MPTP-induced caspase-3 activation by regulating intracellular apoptotic signaling molecules, such as Bcl-2 and Bax (Jiang et al., 2008). MPTP is known to decrease Bcl-2 and increase Bax in the striatum (Youdim and Arraf, 2004), thereby tilting the balance toward apoptosis. Ghrelin-treated cells have an increased Bcl-2/Bax ratio, decreased cytochrome c release, and inhibition of caspase-3 activation; thus, rescuing SN pars compacta (SNpc) neurons from MPTP-induced toxicity (Youdim and Arraf, 2004).
Increased microglia activation, which participates in the pathogenesis of PD, is inhibited by ghrelin injection in the SN after MPTP administration (Jiang et al., 2008). The neuroprotective effects of ghrelin on MPTP-induced nigrostriatal dopamine dysfunction involve the activation of uncoupling protein (UCP)-2-dependent mitochondrial respiration, suppression of reactive oxygen species (ROS) production, and mitochondrial biogenesis (Andrews, 2011), but only if mice remained fasted after ghrelin injection. Considering that matrix metalloproteinase-3 (MMP-3) plays a pivotal role in dopaminergic neurodegeneration in PD (Kim et al., 2005, 2007) and ghrelin suppresses MPTP-induced MMP-3 expression (Andrews, 2011), ghrelin may mediate the inhibition of MMP-3 expression.
In conclusion, ghrelin can antagonize MPTP-induced neurotoxicity of nigral dopaminergic neurons in vivo through a GHS-R 1a mediated anti-apoptotic effect. If ghrelin can induce regeneration or prevent degeneration of dopaminergic neurons it could be a potential important tool in the therapeutic strategy for treatment of PD, but this possibility requires further basic and clinical research.
Stroke and Ischemia
Recent studies highlight a neuroprotective action of ghrelin in ischemic models of stroke, both in vivo and in vitro. In vivo rat ischemia–reperfusion models show that ghrelin-administered ip or intravenously (iv) elicits significant neuroprotection in the forebrain by reducing infarct volume and cell death (Chung et al., 2007, 2008; Hwang et al., 2009). Both ischemic preconditioning and ghrelin administration protect the hippocampus from ischemia/reperfusion and up-regulate UCP-2, with acylated and des-acylated ghrelin reported to be equally effective in reducing infarct volume (Hwang et al., 2009). Interestingly, pre-administration of a GHSR-1a antagonist completely reverse the neuroprotective effect of ghrelin, but not des-acylated ghrelin, suggesting that des-acylated ghrelin has neuroprotective effects in ischemia independent of GHSR-1a (Chung et al., 2008; Hwang et al., 2009). The neuroprotective mechanisms involved include reduced apoptosis and increased mitochondrial function. Pretreatment of cells with acylated ghrelin prevents the induction of apoptosis by activating ERK1/2, as well as preventing caspase activation, cytochrome C (Cyt) release and increasing the Bcl-2:Bax ratio (Miao et al., 2007; Chung et al., 2008; Hwang et al., 2009).
Ghrelin protects OGD cells by decreasing the generation of ROS and stabilizing the mitochondrial membrane potential (Chung et al., 2007), with this being dependent on the MAPK, PI3K, PKC, and PKA signaling pathways. Consistent with an antioxidant effect, ghrelin has been shown to increase UCP-2 in the hippocampus and prevent the loss of CA1 neurons after ischemia/reperfusion (Liu et al., 2006). Therefore, it appears that a common neuroprotective or neuromodulatory role of ghrelin in the brain could involve UCP-2-dependent mitochondrial adaptation.
The protective effects of ghrelin could also involve the GH–IGF-I axis, as IGF-I is an important neuroprotective factor in stroke and local IGF-I production is induced by this hormone (Frago et al., 2002, 2005). However, some in vitro studies suggest that a direct effect of ghrelin is most likely, given that IGF-I was not increased in vitro (Miao et al., 2007; Chung et al., 2008; Hwang et al., 2009). In hypothalamic neuronal cells, ghrelin treatment also prevents OGD-induced ROS generation involving Bcl-2 preventing ROS accumulation (Sidoti-De et al., 1998) and/or shifting the cellular redox potential to a more reduced state (Ellerby et al., 1996). The increased levels of Bcl-2 protein in ghrelin-treated cells may both promote cell survival and protect against ischemic oxidative stress. Ghrelin also prevents the OGD-induced collapse of mitochondrial transmembrane potential by regulating Bcl-2 family proteins during ischemic injury (Chung et al., 2007).
Ghrelin mRNA and protein have been detected in cortical neurons, suggesting a possible autocrine/paracrine mode of action of ghrelin in the inhibition of apoptosis. The suppression of ghrelin expression by transfecting cells with siRNA against pre-pro-ghrelin significantly increased apoptosis during an OGD insult and even in normoxic conditions (Chung et al., 2008). This observation is comparable with a report by Granata et al. (2007), in which an antibody against ghrelin significantly inhibited apoptosis in pancreatic β-cells. It should be noted that the survival effect of endogenous ghrelin and des-acylated ghrelin could not be distinguished because the siRNA used in this study was directed against pre-pro-ghrelin. Chemical inhibition of both ghrelin and des-acylated ghrelin-induced phosphorylation of Akt and ERK1/2 completely blocked the ghrelin-induced anti-apoptotic effects, indicating that these peptides suppress OGD-induced apoptosis in cortical neuronal cells through PI3K/Akt and ERK1/2.
The GHS hexarelin increases GSK-3β phosphorylation in post-hypoxic-ischemic animals (Brywe et al., 2005). GSK-3β is a pro-apoptotic protein (Eldar-Finkelman, 2002) and inhibitors of GSK-3β reduce infarct size following focal cerebral ischemia in vivo (Kelly et al., 2004) and improve neuronal survival in vitro (Cross et al., 2001). Thus the PI3K/Akt-mediated inactivation of GSK-3β is most likely at least partly responsible for the anti-apoptotic effects of ghrelin and des-acylated ghrelin. Several transcription factors, such as cAMP-response element-binding protein (D’Amico et al., 2000), nuclear factor-κB (Madrid et al., 2000; Sanchez et al., 2003), and β-catenin (Haq et al., 2003) can be regulated by GSK-3β. Ghrelin and des-acylated ghrelin-induced Akt signaling is associated with downstream attenuation of GSK-3β and nuclear translocation of β-catenin, targeting the Bcl-2 protein family, inhibiting cytochrome c release and caspase-3 activity, thus inhibiting the apoptotic cascade and favoring cell survival. Indeed, cytosolic Bcl-2 protein levels are decreased by OGD insult, whereas treatment with des-acylated ghrelin increases Bcl-2 levels and inhibits the OGD-induced rise in Bax levels in mitochondria, resulting in complete normalization of the Bcl-2/Bax ratio.
The change in the status of Bcl-2 and the Bax proteins caused by either ghrelin or des-acylated ghrelin treatment inhibits apoptosis and favors cell survival. It is known that the Bcl-2 protein family tightly regulates cytochrome c release from the mitochondria into the cytosol (Merry and Korsmeyer, 1997). After release from the mitochondrial intermembrane space, cytochrome c forms the apoptosome together with apoptosis-activating factor Apaf-1 and procaspase-9, leading to activation of the initiator caspase-9 (Li et al., 1997). Subsequent activation of downstream members of the caspase family, including the effector caspase-3, leads to apoptosis (Slee et al., 1999). Cytochrome c is translocated from the mitochondria to the cytosolic compartment after OGD insult (Pérez-Pinzón et al., 1999) and both ghrelin and des-acylated ghrelin prevent this translocation and the subsequent activation of caspase-3, thus inhibiting activation of the apoptotic cascade (Chung et al., 2007).
Intraperitoneally administered des-acylated ghrelin or ghrelin, protects cortical neurons from injury induced by transient focal cerebral ischemia and reperfusion in vivo, significantly reducing infarct volumes after initiation of ischemia, at least in part through suppression of Par-4 expression (Miao et al., 2007). Ghrelin injected intravenously also has neuroprotective affects in transient focal ischemia/reperfusion in rats by inhibiting apoptotic molecules of the mitochondrial pathway and activating endogenous protective molecules (Miao et al., 2007). Given that ghrelin can pass through the blood–brain barrier (Banks et al., 2002), it is possible that these effects, at least in part, are directly due to ghrelin and not through other systemic changes induced by the treatment.
Epilepsy
Anti-epileptic effects of ghrelin have been reported in an acute experimental epilepsy model of pentylenetetrazole-injected rats (Obay et al., 2007). The time of onset of pentylenetetrazole-induced seizures in rats was delayed following previous treatment with ghrelin (Obay et al., 2007), with protein markers for oxidative stress being decreased in hippocampal neurons of ghrelin-administered rats (Obay et al., 2008). Ghrelin has also been tested in the pilocarpine-model of epilepsy, where it attenuated CA1 and CA3 hippocampal neuronal loss by inhibiting caspase-3 activation and maintaining the Bcl-2:Bax ratio (Xu et al., 2009). Lee et al. (2010) demonstrated that ip injection of ghrelin significantly reduces hippocampal neuronal death, the number of TUNEL-positive cells and caspase-3 expression in association with decreased seizures after KA injection, with ghrelin significantly attenuating KA-induced neuronal cell death in CA1 and CA3 hippocampal regions (Lee et al., 2010). In this case, the neuroprotective effect involves suppression of microglia and astrocyte activation, as well as suppression of inflammatory mediators, such as TNF-α, interleukin-1b, and cyclooxygenase-2 (Lee et al., 2010).
Neuropeptide Y (NPY) and Γ-aminobutyric acid (GABA) may be involved in the anticonvulsant effects of ghrelin as NPY and GABA exert anti-epileptic effects in animal seizure models (Czapinski et al., 2005) and ghrelin not only enhances NPY synthesis, but also increases GABA-ergic activity in the brain (Cowley et al., 2003). It is well known that a majority of epileptic seizures are due to an imbalance between the activities of inhibitory and excitatory neurotransmitters (Sharma et al., 2007). Therefore, the stimulatory effect of ghrelin on NPY and GABA activities may contribute to the anti-epileptic properties of ghrelin.
Other Neurological Disease Models
GH secretagogue-receptor is present in the spinal cord (Ferens et al., 2010) and recent studies demonstrate that ghrelin improves functional recovery after spinal cord injury by inhibiting apoptosis and potentially enhancing neurogenesis (Lee et al., 2010). Ghrelin also restricts the development of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (Theil et al., 2009). Daily ip injections of ghrelin are reported to reduce the severity of EAE and decrease levels of pro-inflammatory cytokines and activated microglia, thus further consolidating the anti-inflammatory properties of ghrelin.
Ghrelin is also reported to protect against apoptotic cell death in cortical neurons exposed to endoplasmic reticulum (ER) stress by activating the PI3K/Akt pathway (Chung et al., 2011). Ghrelin targets CHOP protein via the inhibition of the PERK/eIF2α/ATF4 pathway. It is assumed that ghrelin-mediated suppression of CHOP is associated with the exclusion of FOXO 1 from the nucleus. Moreover, increased Akt signaling by ghrelin is associated with downstream attenuation of GSK-3β, BAD, and FOXO1. These findings suggest that ghrelin could also function as a neuroprotective agent in treatment of neurodegenerative diseases where ER stress responses are involved.
Diabetes
Diabetes mellitus results in diverse complications when poorly controlled over an extended period of time. Indeed, poor glycemic control is not only associated with metabolic and hormonal imbalances (Bestetti et al., 1985), but also with an increased risk of disorders in the CNS as a result of changes in brain metabolism, vascular reactivity, blood–brain barrier integrity and increased oxidative stress (Fouyas et al., 2003; Valko et al., 2007). Some of these alterations could be due, at least in part, to increased apoptosis of both neurons and glial cells, as chronic hyperglycemia has been reported to induce cell death of cortical, hippocampal and hypothalamic neurons (Klein et al., 2004), as well as to induce death and decrease cell proliferation of astrocytes both in vivo and in vitro (Rungger-Brandle et al., 2000; Lechuga-Sancho et al., 2006a,b; García-Cáceres et al., 2008).
In poorly controlled diabetes increased cell death occurs in different tissues and organs (Arroba et al., 2003; García-Cáceres et al., 2008; Granado et al., 2011), with this cell loss being involved in many of the secondary complications of diabetes. In the anterior pituitary, lactotrophs appear to be more susceptible to diabetes-induced death than other cell types (Arroba et al., 2003, 2005, 2006), with this process involving activation of caspase-8, the prototypical initiator caspase of the extrinsic cell death pathway. Ghrelin treatment decreases cell death and activation of caspase-8 and increases Bcl-2, Hsp70, and iNOS levels in the anterior pituitary (Granado et al., 2009).
Poorly controlled diabetes also increases cell death in the hypothalamus and cerebellum (Lechuga-Sancho et al., 2006a,b; García-Cáceres et al., 2008; Granado et al., 2009). Although treatment with GHRP-6 affected cell death in these areas, it was more effective in combination with insulin treatment. This combined treatment reduced the diabetes-induced-decrease in glial fibrillary acidic protein (GFAP) levels, suggesting an effect on glial cells. Thus, GHRP-6, and possibly ghrelin, may work in concert with other hormones such as insulin to prevent disease processes.
Future Perspectives
Numerous clinical trials are underway employing both ghrelin agonists and antagonists in diverse diseases. Simulation of appetite and fat accumulation by ghrelin agonists in wasting diseases or cachexia is a prime area of investigation. This hormone is currently a therapeutical target for the development of obesity treatment, as ghrelin antagonist should decrease appetite. In addition, the above findings indicate that ghrelin, as well as des-acylated ghrelin, or their analogs could function as neuroprotective agents. Further understanding of this facet is of great importance as.
However, a number of systems may also be affected with these treatments, as suggested by the wide distribution of the known ghrelin receptor and with a number of tissues and cell types known to respond to this hormone through a yet unidentified mechanism. Thus, it is imperative that we understand the diverse functions of this hormone and their mechanisms of action, including the receptors involved, in order to develop specific and safe agonists or antagonists targeting the mechanism of interest. This will most likely result in the identification of new receptors for both ghrelin and des-acyl ghrelin in the new future.
Involvement of the ghrelin system in diseases of the CNS is unknown. However, alterations in cell metabolism are well known to be involved in pathological processes and it is possible that this hormone may have both direct and indirect roles in these processes. Thus, detection of early disturbances in the ghrelin system might possibly be of future diagnostic value, in addition to being a therapeutical target.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by grant BFU200802950C033 from the Ministerio de Ciencia e Innovación and a grant from Fondo de Investigación Sanitaria (PI100747), CIBER Fisiopatologíade Obesidad y Nutrición (CIBEROBN) Instituto de Salud Carlos III, and Fundación de Endocrinología y Nutrición.
References
Andrews, Z. B. (2011). The extra-hypothalamic actions ghrelin on neuronal function. Trends Neurosci. 34, 31–40.
Andrews, Z. B., Erion, D., Beiler, R., Liu, Z. W., Abizaid, A., Zigman, J., Elsworth, J. D., Savitt, J. M., DiMarchi, R., Tschoep, M., Roth, R. H., Gao, X. B., and Horvath, T. L. (2009). Ghrelin promotes and protects nigrostriatal dopamine function via UCP2-dependent mitochondrial mechanism. J. Neurosci. 29, 14057–14065.
Arnoult, D., Karbowski, M., and Youle, R. J. (2003). Caspase inhibition prevents the mitochondrial release of apoptosis-inducing factor. Cell Death Differ. 10, 845–849.
Arroba, A. I., Frago, L. M., Argente, J., and Chowen, J. A. (2005). Activation of caspase 8 in the pituitaries of streptozotocin-induced diabetic rats: implication in increased apoptosis of lactotrophs. Endocrinology 146, 4417–4424.
Arroba, A. I., Frago, L. M., Pañeda, C., Argente, J., and Chowen, J. A. (2003). The number of lactotrophs is reduced in the anterior pituitary of streptozotocin-induced diabetic rats. Diabetologia 46, 634–638.
Arroba, A. I., Lechuga-Sancho, A. M., Frago, L. M., Argente, J., and Chowen, J. A. (2006). Increased apoptosis of lactotrophs in streptozotocin-induced diabetic rats is followed by increased proliferation. J. Endocrinol. 191, 55–63.
Baldanzi, G., Filigheddu, N., Cutrupi, S., Catapano, F., Bonissoni, S., Fubini, A., Malan, D., Baj, G., Granata, R., Broglio, F., Papotti, M., Surico, N., Bussolino, F., Isgaard, J., Deghenghi, R., Sinigaglia, F., Prat, M., Muccioli, G., Ghigo, E., and Graziani, A. (2002). Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 159, 1029–1037.
Banks, W. A., Tschop, M., Robinson, S. M., and Heiman, M. L. (2002). Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 302, 822–827.
Bednarek, M. A., Freighner, S. D., Pong, S. S., McKee, K. K., Hreniuk, D. L., Silva, M. V., Warren, V. A., Howard, A. D., Van der Ploeg, L. H., and Heck, J. V. (2000). Structure–function studies on the new growth hormone-releasing peptide, ghrelin: minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J. Med. Chem. 43, 4370–4376.
Ben-Ari, Y., Tremblaym, E., Ottersenm, O. P., and Meldrum, B. S. (1980). The role of epileptic activity in hippocampal and “remote” cerebral lesions induced by kainic acid. Brain Res. 191, 79–97.
Bennet, P. A., Thomas, G. B., Howard, A. D., Feighner, S. D., Van der Ploeg, L. H., Smith, R. G., and Robinson, I. C. (1997). Hypothalamic growth hormone secretagogue-receptor (GHS-R) expression is regulated by growth hormone in the rat. Endocrinology 138, 4552–4557.
Bestetti, G., Locatelli, V., Tirone, F., Rossi, G. L., and Muller, E. E. (1985). One month of streptozotocin-diabetes induces different neuroendocrine and morphological alterations in the hypothalamo-pituitary axis of male and female rats. Endocrinology 117, 208–216.
Brywe, K. G., Leverin, A. L., Gustavsson, M., Mallard, C., Granata, R., Destefanis, S., Volante, M., Hagberg, H., Ghigo, E., and Isgaard, J. (2005). Growth hormone-releasing peptide hexarelin reduces neonatal brain injury and alters Akt/glycogen synthase kinase-3beta phosphorylation. Endocrinology 146, 4665–4672.
Cassoni, P., Papotti, M., Ghe, C., Catapano, F., Sapino, A., Graziani, A., Deghenghi, R., Reissmann, T., Ghigo, E., and Muccioli, G. (2001). Identification, characterization, and biological activity of specific receptors for natural (ghrelin) and synthetic growth hormone secretagogues and analogs in human breast carcinomas and cell lines. J. Clin. Endocrinol. Metab. 86, 1738–1745.
Chang, L., Ren, Y., Liu, X., Li, W. G., Yang, J., Geng, B., Weintraub, N. L., and Tang, C. (2004). Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 43, 165–170.
Cheung, E. C., Melanson-Drapeau, L., Cregan, S. P., Vanderluit, J. L., Ferguson, K. L., McIntosh, W. C., Park, D. S., Bennett, S. A., and Slack, R. S. (2005). Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J. Neurosci. 25, 1324–1334.
Chung, H., Chung, H. Y., Bae, C. W., Kim, C. J., and Park, S. (2011). Ghrelin suppresses tunicamycin- or thapsigargin-triggered endoplasmic reticulum stress-mediated apoptosis in primary cultured rat cortical neuronal cells. Endocrinol. J. 58, 409–420.
Chung, H., Kim, E., Lee, D. H., Seo, S., Ju, S., Lee, D., Kim, H., and Park, S. (2007). Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology 148, 148–159.
Chung, H., Seo, S., Moon, M., and Park, S. (2008). Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3b and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen–glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J. Endocrinol. 198, 511–521.
Cowley, M. A., Smith, R. G., Diano, S., Tschöp, M., Pronchuk, N., Grove, K. L., Strasburger, C. J., Bidlingmaier, M., Esterman, M., Heiman, M. L., Garcia-Segura, L. M., Nillni, E. A., Mendez, P., Low, M. J., Sotonyi, P., Friedman, J. M., Liu, H., Pinto, S., Colmers, W. F., Cone, R. D., and Horvath, T. L. (2003). The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661.
Cross, D. A., Culbert, A. A., Chalmers, K. A., Facci, L., Skaper, S. D., and Reith, A. D. (2001). Selective small-molecules inhibitors of glycogen synthase kinase-3 activity protect primary neurons from death. J. Neurochem. 77, 94–102.
Cummings, D. E., Purnell, J. Q., Frayo, R. S., Schimdova, K., Wisse, B. E., and Weigle, D. S. (2001). Prepandrial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719.
Czapinski, P., Blaszczyk, B., and Czuczwar, S. J. (2005) Mechanisms of action of antiepileptic drugs. Curr. Top. Med. Chem. 5, 3–14.
D’Amico, M., Hulit, J., Amanatullah, D. F., Zafonte, B. T., Albanese, C., Bouzahzah, B., Fu, M., Augenlicht, L. H, Donehower, L. A., Takemaru, K., Moon, R. T., Davis, R., Lisanti, M. P., Shtutman, M., Zhurinsky, J., Ben-Ze’ev, A., Troussard, A. A., Dedhar, S., and Pestell, R. G. (2000). The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. J. Biol. Chem. 275, 32649–32657.
Date, Y., Kojima, M., Hosoda, H., Sawaguchi, A., Mondal, M. S., Suganuma, T., Matsukura, S., Kangawa, K., and Nakazato, M. (2000). Ghrelin, a novel growth hormone-releasing peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141, 4255–4261.
Datta, S. R., Brunet, A., and Greenberg, M. E. (1999). Cellular survival: a play in three Akts. Genes Dev. 13, 2905–2927.
Davenport, A. P., Bonner, T. I., Foord, S. M., Harmar, A. J., Neubiq, R. R, Pin, J. P., Spedding, M., Kojima, M., and Kangawa, K. (2005). International union of pharmacology. LVI: ghrelin receptor nomenclature, distribution and function . Pharmacol. Rev. 57, 541–546.
Delgado-Rubín, A., Chowen, J. A., Argente, J., and Frago, L. M. (2009). Growth hormone-releasing peptide 6 protection of hypothalamic neurons from glutamate excitotoxicity is caspase independent and not mediated by insulin-like growth factor I. Eur. J. Neurosci. 29, 2115–2124.
Delgado-Rubín de Célix, A., Chowen, J. A., Argente, J., and Frago, L. M. (2006). Growth hormone releasing peptide-6 acts as a survival factor in glutamate-induced excitotoxicity. J. Neurochem. 99, 839–849.
Delhanty, P. J., Van der Eerden, B. C., Van der Velde, M., Gauna, C., Pols, H. A., Jahr, H., Chiba, H., Van der Lely, A. J., and Van Leeuwen, J. P. (2006). Ghrelin and unacylated ghrelin stimulate human osteoblast growth via mitogen-activated protein kinase (MAPK)/phosphoinositide 3-kinase (PI3K) pathways in the absence of GHS–R1a. J. Endocrinol. 188, 37–47.
Doble, A. (1999). The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther. 81, 163–221.
Drazen, D. L., Vahl, T. P., D’Alesio, D. A., Seeley, R. J., and Woods, S. C. (2006). Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology 147, 23–30.
Eldar-Finkelman, H. (2002) Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol. Med. 8, 126–132.
Ellerby, L. M., Ellerby, H. M., Park, S. M., Holleran, A. L., Murphy, A. N., Fiskum, G., Kane, D. J., Testa, M. P., Kayalar, C., and Bredesen, D. E. (1996). Shift of the cellular oxidation- reduction potential in neural cells expressing Bcl-2. J. Neurochem. 67, 1259–1267.
Ferens, D. M., Yin, L., Bron, R., Hunne, B., Ohashi-Doi, K., Kitchener, P. D., Sanger, G. J., Witherington, J., Shimizu, Y., and Furness, J. B. (2010). Functional and in situ hybridization evidence that preganglionic sympathetic vasoconstrictor neurons express ghrelin receptors. Neuroscience 166, 671–679.
Filigheddu, N., Gnocchi, V. F., Coscia, M., Cappelli, M., Porporato, P. E., Taulli, R., Traini, S., Baldanzi, G., Chianale, F., Cutrupi, S., Arnoletti, E., Ghé, C., Fubini, A., Surico, N., Siniqaglia, F., Ponzetto, C., Mucciolli, G., Crepaldi, T., and Graziani, A. (2007). Ghrelin and des-acyl ghrelin promote differentiation and fusion of C2C12 skeletal muscle cells. Mol. Biol. Cell 18, 986–994.
Foster-Schubert, K. E., and Cummings, D. E. (2006). Emerging therapeutic strategies for obesity. Endocrinol. Rev. 27, 779–793.
Fouyas, I. P., Kelly, P. A., Ritchie, I. M., Lammie, G. A., and Whittle, I. R. (2003). Cerebrovascular responses to pathophysiological insult in diabetic rats. J. Clin. Neurosci. 10, 88–91.
Frago, L. M., Pañeda, C., Argente, J., and Chowen, J. A. (2005). Growth hormone-releasing peptide-6 increases insulin-like growth factor-I mRNA levels and activates Akt in RCA-6 cells as a model of neuropeptide Y neurons. J. Neuroendocrinol. 17, 701–710.
Frago, L. M., Pañeda, C., Dickson, S. L., Hewson, A. K., Argente, J., and Chowen, J. A. (2002). Growth hormone (GH) and GH-releasing peptide-6 increase brain insulin-like growth factor-I expression and activate intracellular signaling pathways involved in neuroprotection. Endocrinology 143, 4113–4122.
García-Cáceres, C., Lechuga-Sancho, A. M., Argente, J., Frago, L. M., and Chowen, J. A. (2008). Death of hypothalamic astrocytes in poorly controlled diabetic rats is associated with nuclear translocation of apoptosis inducing factor. J. Neuroendocrinol. 20, 1348–1360.
Gauna, C., Delhanty, P. J., van Aken, M. O., Janssen, J. A., Themmen, A. P., Hofland, L. J., Culler, M., Broglio, F., Ghigo, E., and Van der Lely, A. J. (2006). Unacylated ghrelin is active on the INS-1E rat insulinoma cell line independently of the growth hormone secretagogue receptor type 1a and the corticotropin releasing factor 2 receptor. Mol. Cell. Endocrinol. 251, 103–111.
Ghigo, E., Broglio, F., Arvat, E., Maccariom, M., Papotti, M., and Muccioli, G. (2005). Ghrelin: more than a natural GH secretagogue and/or an orexigenic factor. Clin. Endocrinol. (Oxf) 62, 1–17.
Gill, S. S., and Pulido, O. M. (2001). Glutamate receptors in peripheral tissues: current knowledge, future research and implications for toxicology. Toxicol. Pathol. 29, 208–223.
Glazner, G. W., and Mattson, M. P. (2000). Differential effects of BDNF, ADNF9, and TNF alpha on levels of NMDA receptor subunits, calcium homeostasis, and neuronal vulnerability to excitotoxicity. Exp. Neurol. 161, 442–452.
Gómez, C., Reiriz, J., Piqué, M., Gil, J., Ferrer, I., and Ambrosio, S. (2001). Low concentrations of 1-methyl-4-phenylpyridinium ion induce caspase-mediated apoptosis in human SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 63, 421–428.
Granado, M., Chowen, J. A., García-Cáceres, C., Delgado-Rubín, A., Barrios, V., Castillero, E., Argente, J., and Frago, L. M. (2009). Ghrelin treatment protects lactotrophs from apoptosis in the pituitary of diabetic rats. Mol. Cell. Endocrinol. 309, 67–75.
Granado, M., García-Cáceres, C., Tuda, M., Frago, L. M., Chowen, J. A., and Argente, J. (2011). Insulin and growth hormone-releasing peptide-6 (GHRP-6) have differential beneficial effects on cell turnover in the pituitary, hypothalamus and cerebellum of streptozotocin (STZ)-induced diabetic rats. Mol. Cell. Endocrinol. 337, 101–113.
Granata, R., Settanni, F., Biancone, L., Trovato, L., Nano, R., Bertuzzi, F., Destefanis, S., Annunziata, M., Martinetti, M., Catapano, F., Ghè, C., Isgaard, J., Papotti, M., Ghigo, E., and Muccioli, G. (2007). Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-Kinase/Akt signaling. Endocrinology 148, 512–529.
Guan, X. M., Yu, H., Palyha, O. C., McKee, K. K., Feighner, S. D., Sirinathsinghji, D. J., Smith, R. G., Van der Ploeg, L. H., and Howard, A. D. (1997). Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res. Mol. Brain Res. 48, 23–29.
Halem, H. A., Taylor, J. E., Dong, J. Z., Shen, Y., Datta, R., Abizaid, A., Diano, S., Horvath, T. L., and Culler, M. D. (2005). A novel growth hormone secretagogue-1a receptor antagonist that blocks ghrelin-induced growth hormone secretion but induces increased body weight gain. Neuroendocrinology 81, 339–349.
Haq, S., Michael, A., Andreucci, M., Bhattacharya, K., Dotto, P., Walters, B., Woodgett, J., Kilter, H., and Force, T. (2003). Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc. Natl. Acad. Sci. U.S.A 100, 4610–4615.
Henshall, D. C., Bonislawski, D. P., Skradski, S. L., Araki, T., Lan, J. Q., Schindler, C. K., Meller, R., and Simon, R. P. (2001). Formation of the Apaf-1/cytochrome C complex precedes activation of caspase-9 during seizure-induced neuronal death. Cell Death Differ. 8, 1169–1181.
Holst, B., Holliday, N. D., Bach, A., Elling, C. E., Cox, H. M., and Schwartz, T. W. (2004). Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 279, 53806–53817.
Holst, B., Lang, M., Brandt, E., Bach, A., Howard, A., Frimurer, T. M., Beck-Sickinger, A., and Schwartz, T. W. (2006). Ghrelin receptor inverse agonists: identification of an active peptide core and its interactions epitopes on the receptor. Mol. Pharmacol. 70, 936–946.
Hosoda, H., Kojima, M., Matsuo, H., and Kangawa, K. (2000). Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem. Biophys. Res. Commun. 279, 909–913.
Howard, A. D., Feighner, S. D, Cully, D. F., Arena, J. P., Liberator, P. A., Rosenblum, C. I., Hamelin, M., Hreniuk, D. L., Palyha, O. C., Anderson, J., Paress, P. S., Diaz, C., Chou, M., Liu, K. K., McKee, K. K., Pong, S. S., Chaung, L. Y., Elbrecht, A., Dashkevicz, M., Heavens, R., Rigby, M., Sirinathsinghji, D. J., Dean, D. C., Melillo, D. G., Patchett, A. A., Nargund, R., Griffin, P. R., DeMartino, J. A., Gupta, S. K., Schaeffer, J. M., Smith, R. G., and Van der Ploeg, L. H. (1996). A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273, 974–977.
Hwang, S., Moon, M., Kim, S., Hwang, L., Ahn, K. J., and Park, S. (2009). Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response-4. Endocrinol. J. 56, 609–617.
Iseri, S. O., Sener, G., Yuksel, M., Contukm, G., Cetinelm, S., Gedik, N., and Yegen, B. C. (2005). Ghrelin against alendronate-induced gastric damage in rats. J. Endocrinol. 187, 399–406.
Jiang, H., Li, L. J., Wang, J., and Xie, J. X. (2008). Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 212, 532–537.
Jiang, Z., Zhang, Y., Chen, X., Lam, P. Y., Yang, H., Xu, Q., and Yu, A. C. (2002). Activation of Erk1/2 and Akt in astrocytes under ischemia. Biochem. Biophys. Res. Commun. 294, 726–733.
Kelly, S., Zhao, H., Hua-Sun, G., Cheng, D., Qiao, Y., Luo, J., Martin, K., Steinberg, G. K., Harrison, S. D., and Yenari, M. A. (2004). Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp. Neurol. 188, 378–386.
Kim, M. S., Yoon, C. Y., Jang, P. G., Park, Y. J., Shin, C. S., Park, H. S., Ryu, J. W., Pak, Y. K., Park, J. Y., Lee, K. U., Kim, S. Y., Lee, H. K., Kim, Y. B., and Park, K. S. (2004). The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol. Endocrinol. 18, 2291–2301.
Kim, S. W., Her, S. J., Park, S. J., Kim, D., Park, K. S., Lee, H. K., Han, B. H., Kim, M. S., Shin, C. S., and Kim, S. Y. (2005). Ghrelin stimulates proliferation and differentiation and inhibits apoptosis in osteoblastic MC3T3-E1 cells. Bone 37, 359–369.
Kim, Y. S., Choi, D. H., Block, M. L., Lorenzl, S., Yang, L., Kim, Y. J., Sugama, S., Cho, B. P., Hwang, O., Browne, S. E., Kim, S. Y., Hong, J. S., Beal, M. F., and Joh, T. H. (2007). A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J 21, 179–187.
Klein, J. P., Hains, B. C., Craner, M. J., Black, J. A., and Waxman, S. G. (2004). Apoptosis of vasopressinergic hypothalamic neurons in chronic diabetes mellitus. Neurobiol. Dis. 15, 221–228.
Kohno, D., Gao, H. Z., Muroya, S., Kikuyama, S., and Yada, T. (2003). Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2_ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes 52, 948–956.
Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H., and Kangawa, K. (1999). Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660.
Konturek, P. C., Brzozowski, T., Walter, B., Burnat, G., Hess, T., Hahn, E. G., and Konturek, S. J. (2006). Ghrelin-induced gastroprotection against ischemia-reperfusion injury involves an activation of sensory afferent nerves and hyperemia mediated by nitric oxide. Eur. J. Pharmacol. 536, 171–181.
Lage, R., Vázquez, M. J., Varela, L., Saha, A. K., Vidal-Puig, A., Nogueiras, R., Diéguez, C., and López, M. (2010). Ghrelin effects on neuropeptides in the rat hypothalamus dependo n fatty acid metabolism actions on BSX but not on gender. FASEB J. 24, 2670–2679.
Lechuga-Sancho, A. M., Arroba, A. I., Frago, L. M., García-Cáceres, C., Delgado-Rubín, A., Argente, J., and Chowen, J. A. (2006a). Reduction in the number of astrocytes and their projections is associated with increased synaptic protein density in the hypothalamus of poorly controlled diabetic rats. Endocrinology 147, 5314–5324.
Lechuga-Sancho, A. M., Arroba, A. I., Frago, L. M., Pañeda, C., García-Cáceres, C., Delgado-Rubín, A., Argente, J., and Chowen, J. A. (2006b). Activation of the intrinsic cell death pathway, increased apoptosis and modulation of astrocytes in the cerebellum of diabetic rats. Neurobiol. Dis. 23, 290–299.
Lee, J., Lim, E., Kim, Y., Li, E., and Park, S. (2010). Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse. J. Endocrinol. 205, 263–720.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., and Wang, X. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489.
Liu, Y., Wang, P. S., Xie, D., Liu, K., and Chen, L. (2006). Ghrelin reduces injury of hippocampal neurons in a rat model of cerebral ischemia/reperfusion. Chin. J. Physiol. 49, 244–250.
López, M., Lage, R., Saha, A. K., Pérez-Tilve, D., Vázquez, M. J., Varela, L., Sangiao-Alvarellos, S., Tovar, S., Raghay, K., Rodríguez-Cuenca, S., Deoliveira, R. M., Castañeda, T., Datta, R., Dong, J. Z., Culler, M., Sleeman, M. W., Alvarez, C. V., Gallego, R., Lelliottm, C. J., Carling, D., Tschöp, M. H., Diéguez, C., and Vidal-Puig, A. (2008). Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 7, 389–399.
Maccarinelli, G., Sibilia, V., Torsello, A., Raimondo, F., Pitto, M., Giustina, A., Netti, C., and Cocchi, D. (2005). Ghrelin regulates proliferation and differentiation of osteoblastic cells. J. Endocrinol. 184, 249–256.
Madrid, L. V., Wang, C. Y., Guttridge, D. C., Schottelius, A. J., Baldwin, A. S. Jr., and Mayo, M. W. (2000). Mol. Cell Biol. 20, 1626–1638.
Mattson, M. P. (2003). Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 3, 65–94.
Merry, D. E., and Korsmeyer, S. J. (1997). Bcl-2 gene family in the nervous system. Annu. Rev. Neurosci. 20, 245–267.
Miao, Y., Xia, Q., Hou, Z., Zheng, Y., Pan, H., and Zhu, S. (2007). Ghrelin protects cortical neuron against focal ischemia/reperfusion in rats. Biochem. Biophys. Res. Commun. 359, 795–800.
Mitchell, V., Bouret, S., Beauvillain, J. C., Schilling, A., Perret, M., Kordon, C., and Epelbaum, J. (2001). Comparative distribution of mRNA encoding the growth hormones secretagogue-receptor (GHS-R) in Microcebus murinus (Primate, lemurian) and rat forebrain and pituitary. J. Comp. Neurol. 429, 469–489.
Muccioli, G., Pons, N., Ghe, C., Catapano, F., Granata, R., and Ghigo, E. (2004). Ghrelin and des-acyl ghrelin both inhibit isoproterenol-induced lipolysis in rat adipocytes via a non-type 1a growth hormone secretagogue receptor. Eur. J. Pharmacol. 498, 27–35.
Nanzer, A. M., Khalaf, S., Mozid, A. M., Fowkes, R. C., Patel, M. V., Burrin, J. M., Grossman, A. B., and Korbonits, M. (2004). Ghrelin exerts a proliferative effect on a rat pituitary somatotroph cell line via the mitogen-activated protein kinase pathway. Eur. J. Endocrinol. 151, 233–240.
Nogueiras, R., Tovar, S., Mitchell, S. E., Rayner, D. V., Archer, Z. A., Dieguez, C., and Williams, L. M. (2004). Regulation of growth hormone secretagogue receptor gene expression in the arcuate nuclei of the rat by leptin and ghrelin. Diabetes 53, 2552–2558.
Obay, B. D., Tasdemir, E., Tümer, C., Bilgin, H. M., and Atmaca, M. (2008). Dose dependent effects of ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptide 29, 448–455.
Obay, B. D., Tasdemir, E., Tumer, C., Bilgin, H. M., and Sermet, A. (2007). Antiepileptic effects of ghrelin on pentylenetetrazole-induced seizures in rats. Peptides 28, 1214–1219.
Pañeda, C., Arroba, A. I., Frago, L. M., Holm, A. M., Rømer, J., Argente, J., and Chowen, J. A. (2003). Growth hormone-releasing peptide-6 inhibits cerebellar cell death in aged rats. Neuroreport 14, 1633–1635.
Pearson, G., Robinson, F., Beers, G. T., Xu, B. E., Karandikar, M., Berman, K., and Cobb, M. H. (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrinol. Rev. 22, 153–183.
Pérez-Pinzón, M. A., Xu, G. P., Born, J., Lorenzo, J., Busto, R., Rosenthal, M., and Sick, T. J. (1999). Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J. Cereb. Blood Flow Metab. 19, 39–43.
Przedborski, S., Jackson-Lewis, V., Djaldetti, R., Liberatore, G., Vila, M., Vukosavic, S., and Almer, G. (2000). The parkinsonian toxin MPTP: action and mechanism. Restor. Neurol. Neurosci. 16, 135–142.
Rungger-Brandle, E., Dosso, A. A., and Leuenberger, P. M. (2000). Glial reactivity, an early feature of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 41, 1971–1980.
Salinska, E., Danysz, W., and Lazarewicz, J. W. (2005). The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 43, 322–339.
Sanchez, J. F., Sniderhan, L. F., Williamson, A. L., Fan, S., Chakraborty-Sett, S., and Maggirwar, S. B. (2003). Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol. Cell. Biol. 23, 4649–4662.
Schmidt, N., and Ferger, B. (2001). Neurochemical findings in the MPTP model of Parkinson’s disease. J. Neural Transm. 108, 1263–1282.
Seeburg, P. H. (1993). The TIS/TiPS Lecture. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 16, 359–365.
Selznick, L. A., Zheng, T. S., Flavell, R. A., Rakic, P., and Roth, K. A. (2000). Amyloid beta-induced neuronal death is bax-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 59, 271–279.
Sharma, A. K., Reams, R. Y., Jordan, W. H., Miller, M. A., Thacker, H. L., and Snyder, P. W. (2007). Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol. Pathol. 35, 984–999.
Shiiya, T., Nakazato, M., Mizuta, M., Date, Y., Mondal, M. S., Tanaka, M., Nozoe, S., Hosoda, H., Kangawa, K., and Matsukura, S. (2002). Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J. Clin. Endocrinol. Metab. 87, 240–244
Sidoti-De, F. C., Rincheval, V., Risler, Y., Mignotte, B., and Vayssiere, J. L. (1998). TNF-alpha activates at least two apoptotic signaling cascades. Oncogene 17, 1639–1651.
Singh, S., and Dikshit, M. (2007). Apoptotic neuronal death in Parkinson’s disease: involvement of nitric oxide. Brain Res. Rev. 54, 233–250.
Slee, E. A., Adrain, C., and Martin, S. J. (1999). Serial killers: ordering caspase activation events in apoptosis. Cell Death Differ. 6, 1067–1074.
Smith, R. G., Jiang, H., and Sun, Y. (2005). Developments in ghrelin biology and potential clinical relevance. Trends Endocrinol. Metab. 16, 436–442.
Song, G., Ouyang, G., and Bao, S. (2005) The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 9, 59–71.
Sperk, G., Lassmann, H., Baran, H., Seteilberger, F., and Hornykiewicz, O. (1985). Kainic acid-induced seizures: dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 338, 289–295.
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Brenner, C., Larochette, N., Prévost, M. C., Alzari, P. M., and Kroemer, G. (1999) Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 189, 381–394.
Tannenbaum, G. S., Lapointe, M., Beaudet, A., and Howard, A. D. (1998). Expression of growth hormone secretagogue-receptors by growth hormone-releasing hormone neurons in the mediobasal hypothalamus. Endocrinology 139, 4420–4423.
Theil, M. M., Miyake, S., Mizuno, M., Tomi, C., Croxford, J. L., Hosoda, H., Theil, J., von Hörsten, S., Yokote, H., Chiba, A., Lin, Y., Oki, S., Akamizu, T., Kangawa, K., and Yamamura, T. (2009). Suppression of experimental autoimmune encephalomyelitis by ghrelin. J. Immunol. 183, 2859–2866.
Toshinai, K. (2006). Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology 147, 2306–2314.
Toshinai, K., Yamaguchi, H., Sun, Y., Smith, R. G., Yamanaka, A., Sakurai, T., Date, Y., Mondal, M. S., Shimbara, T., Kawagoe, T., Murakami, N., Miyazato, M., Kangawa, K., and Nakazato, M. (2006). Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology 147, 2306–2314.
Tschop, M., Smiley, D. L., and Heiman, M. L. (2000). Ghrelin induces adiposity in rodents. Nature 407, 908–913.
Tschop, M., Weyer, C., Tataranni, P. A., Devanarayan, V., Ravussin, E., and Heiman, M. L. (2001a). Circulating ghrelin levels are decreased in human obesity. Diabetes 50, 707–709.
Tschop, M., Wawarta, R., Riepl, R. L., Friedrich, S., Bidlingmaier, M., Landgraf, R., and Folwaczny, C. (2001b). Post-prandrial decrease of circulating human ghrelin levels. J. Endocrinol. Invest. 24, 19–21.
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 49, 6958–6971.
Van der Lely, A. J., Tschop, M., Heiman, M. L., and Ghigo, E. (2004). Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocrinol. Rev. 25, 426–457.
Veldhuis, J. D., and Bowers, C. Y. (2010). Integrating GHS into the Ghrelin system. Int. J. Pept. doi: 10.1155/2010/879503
Willesen, M. G., Kristensen, P., and Romer, J. (1999). Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 70, 306–316.
Wortley, K. E., Del Rincón, J. P., Murray, J. D., García, K., Iida, K., Thorner, M. O., and Sleeman, W. (2005). Absence of ghrelin protects against early-onset obesity. 115, 3573–3578.
Wren, A. M., Seal, L. J., Cohen, M. A., Brynes, A. E., Frost, G. S., Murphy, K. G., Dhillo, W. S., and Bloom, S. R. (2001). J. Clin. Endocrinol. Metab. 86, 5992–5995.
Xu, J., Wang, S., Lin, Y., Cao, L., Wang, R., and Chi, Z. (2009). Ghrelin protects against cell death of hippocampal neurons in pilocarpine-induced seizures in rats. Neurosci. Lett. 453, 58–61.
Yang, J., Brown, M. S., Liang, G., Grishin, N. V., and Goldstein, J. L. (2008). Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132, 387–396.
Youdim, M. B., and Arraf, Z. (2004). Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology 46, 1130–1140.
Zhang, Y., and Bhavnani, B. R. (2006). Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 7, 49. doi: 10.1186/1471-2202-7-49
Zigman, J. M., Jones, J. E., Lee, C. E., Saper, C. B., and Elmquist, J. K. (2006). Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 494, 528–548.
Zigman, J. M., Nakano, Y., Coppari, R., Balthasar, N., Marcus, J. N., Lee, C. E., Jones, J. E., Deysher, A. E., Waxman, A. R., White, R. D., Williams, T. D., Lachey, J. L., Seeley, R. J., Lowell, B. B., and Elmquist, J. K. (2005). Mice lacking ghrelin receptors resist the development of diet-induced obesity. J. Clin. Invest. 115, 3564–3572.
Keywords: Ghrelin, growth hormone secretagogues, neuroprotection, apoptosis, cell survival, signaling pathways
Citation: Frago LM, Baquedano E, Argente J and Chowen JA (2011) Neuroprotective actions of ghrelin and growth hormone secretagogues. Front. Mol. Neurosci. 4:23. doi: 10.3389/fnmol.2011.00023
Received: 08 July 2011; Paper pending published: 19 July 2011;
Accepted: 29 August 2011; Published online: 28 September 2011.
Edited by:
Isabel Varela-Nieto, Consejo Superior Investigaciones Científicas, SpainReviewed by:
Isabel Varela-Nieto, Consejo Superior Investigaciones Científicas, SpainZane Andrews, Monash University, Australia
Copyright: © 2011 Frago, Baquedano, Argente and Chowen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Julie A. Chowen, Department of Endocrinology, Hospital Infantil Universitario Niño Jesús, Avenida Menéndez Pelayo 65, 28009 Madrid, Spain. e-mail: jachowen@telefonica.net