 Tudor-Alexandru Popoiu1,2
Tudor-Alexandru Popoiu1,2 Edoardo Bertero
Edoardo Bertero
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Med, 23 August 2023
Sec. Molecular Medicine for Cardiology
Volume 3 - 2023 | https://doi.org/10.3389/fmmed.2023.1235188
This article is part of the Research TopicMitochondrial Dysfunction Affects Mechano-Energetic Coupling In Heart FailureView all 6 articles
The energy demand of cardiomyocytes changes continuously in response to variations in cardiac workload. Cardiac excitation-contraction coupling is fueled primarily by adenosine triphosphate (ATP) production by oxidative phosphorylation in mitochondria. The rate of mitochondrial oxidative metabolism is matched to the rate of ATP consumption in the cytosol by the parallel activation of oxidative phosphorylation by calcium (Ca2+) and adenosine diphosphate (ADP). During cardiac workload transitions, Ca2+ accumulates in the mitochondrial matrix, where it stimulates the activity of the tricarboxylic acid cycle. In this review, we describe how mitochondria internalize and extrude Ca2+, the relevance of this process for ATP production and redox homeostasis in the healthy heart, and how derangements in ion handling cause mitochondrial and cardiomyocyte dysfunction in heart failure.
Calcium (Ca2+) signaling plays a central role in the contractile, metabolic, and epigenetic functions of cardiac myocytes. Transient variations in cytosolic Ca2+ concentration ([Ca2+]c) trigger cardiac myocyte contraction and relaxation. During physiological elevations of cardiac workload, the accumulation of Ca2+ in the mitochondrial matrix adjusts the rate of mitochondrial oxidative metabolism to the energy requirements of the cell (McCormack and Denton, 1993). In cardiac disease, Ca2+ mishandling can wreak havoc on cellular functions: in heart failure (HF), defective mitochondrial Ca2+ accumulation can cause bioenergetic mismatch and oxidative stress (Kohlhaas et al., 2017); in ischemia/reperfusion (I/R) injury, mitochondrial Ca2+ overload leads to a sudden increase in mitochondrial membrane permeability that is one major driver of cell death (Xuxia et al., 2020). In addition to cardiac myocyte contraction, changes in [Ca2+]c modulate the activation of gene expression, a process coined excitation-transcription coupling. Here, we review the role of Ca2+ signaling as a regulator of mitochondrial oxidative metabolism and reactive oxygen species (ROS) emission from mitochondria, and how derangements in these processes contribute to the development of HF.
In cardiac myocytes, Ca2+ handling is mediated by voltage- and ligand-activated ion channels that are mainly found on the sarcolemma and on the sarcoplasmic reticulum (SR), a specialized endoplasmic reticulum that cyclically releases and removes Ca2+ ions from the cytosol during cardiac myocyte contraction and relaxation. During phase 2 of the action potential, opening of voltage-dependent L-type Ca2+ channels on the sarcolemma leads to Ca2+ entry into the cytosol, which in turn triggers the release of a larger amount of Ca2+ from the SR via type 2 ryanodine receptors (RyR2). This phenomenon, termed Ca2+-induced Ca2+ release, initiates contraction as Ca2+ binds the regulatory protein troponin C, thereby inducing a conformational change that enables the interaction of the myosin head with actin (Bers, 2002). During diastole, Ca2+ is removed from the cytosol by the SR Ca2+ ATPase (SERCA), which takes Ca2+ back into the SR, and to a lesser extent by the sarcolemmal sodium (Na+)/Ca2+ exchanger (NCX), which extrudes Ca2+ to the extracellular space; for each Ca2+ ion removed, the NCX imports 3 Na+ ions in the cytosol.
Ca2+ operates as a second messenger to adjust the rate of mitochondrial oxidative metabolism according to the energy requirements of cardiac myocytes (McCormack and Denton, 1993; Brandes and Bers, 1997). Increases in heart rate and contractility as seen during physical exercise increase the rate of adenosine triphosphate (ATP) turnover by cardiac myocytes up to 5- or 6-fold the baseline rate. The increase in adenosine diphosphate delivery to mitochondria via the creatine kinase system accelerates the rate of ATP production by oxidative phosphorylation, which needs to be sustained by an increased supply of reducing equivalents that are donated to complex I and complex II of the electron transport chain (ETC) by the reduced form of nicotinamide adenine dinucleotide (NADH) and succinate, respectively, derived from the tricarboxylic acid (TCA) cycle. Ca2+ is pivotal to this mechano-energetic coupling by stimulating the TCA cycle dehydrogenases (Figure 1).
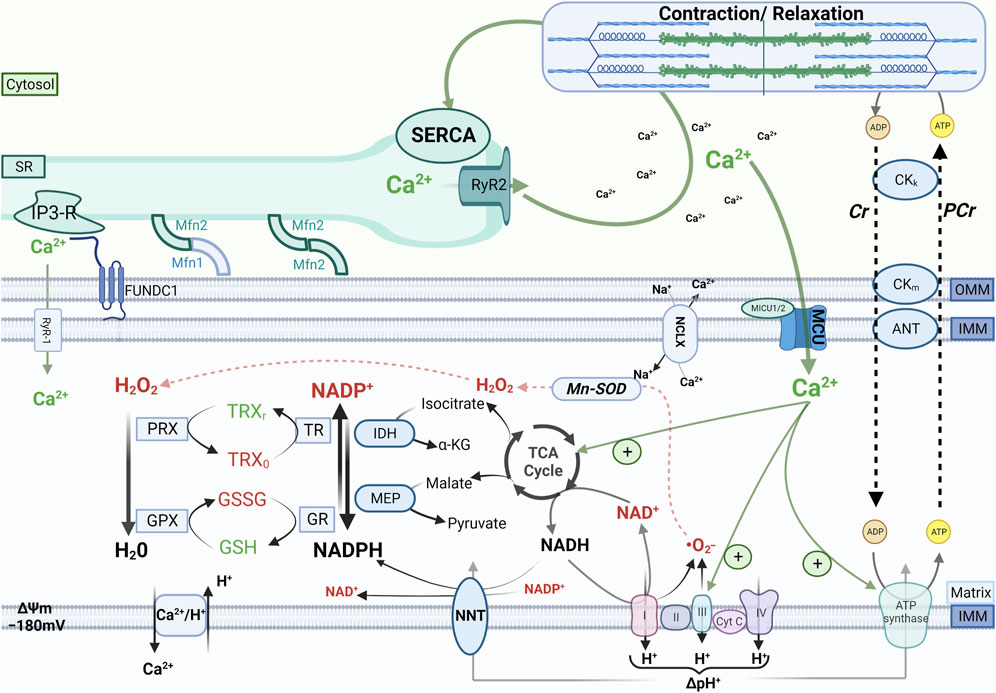
FIGURE 1. Cardiac mechano-energetic coupling. In the healthy heart, the tricarboxylic acid (TCA) cycle produces the reduced form of nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH). Electrons donated by NADH at complex I or by succinate at complex II of the respiratory chain are channeled through a series of electron acceptors that harness their energy to pump protons from the matrix to the intermembrane space. This process physiologically produces a certain amount of reactive oxygen species (ROS) mainly due to incomplete reduction of oxygen to superoxide (.O2−), which is rapidly converted to hydrogen peroxide (H2O2) by the manganese-dependent superoxide dismutase (Mn-SOD). In turn, H2O2 is reduced to H2O by peroxiredoxin (PRX) and glutathione peroxidase (GPX), which are regenerated in their active (reduced) form by a cascade of redox reactions fueled by NADPH. The main sources of mitochondrial NADPH are the TCA cycle enzymes isocitrate dehydrogenase and malate dehydrogenase and the nicotinamide nucleotide transhydrogenase (NNT). When adenosine triphosphate (ATP) consumption increases, increased flux of adenosine diphosphate (ADP) to mitochondria via the creatine kinase (CK) shuttle and Ca2+ accumulation in the mitochondrial matrix stimulate oxidative phosphorylation and the TCA cycle activity, respectively, thereby matching ATP supply and demand and providing reducing equivalents to maintain the redox state of mitochondrial antioxidant systems. Other abbreviations: SR, sarcoplasmic reticulum; RyR1 and RyR2, ryanodine receptors type 1 and type 2, respectively; MCU, mitochondrial calcium uniporter; NCLX, Na+/Ca2+ exchanger, IP3-R, inositol 1,4,5-trisphosphate receptors; Mfn1 and Mfn2, mitofusin 1 and mitofusin 2, respectively; IMM and OMM, inner and outer mitochondrial membrane, respectively; FUNDC1, FUN14 domain containing 1; Cr, creatine; PCr, phosphocreatine; TRX, thioredoxin; GSH and GSSG, reduced and oxidized glutathione, respectively; GR, glutathione reductase; TR, thioredoxin reductase; IDH, isocitrate dehydrogenase, MEP, malic enzyme; NNT, nicotinamide nucleotide transhydrogenase; SERCA, SR Ca2+ ATPase.
Furthermore, elevations in [Ca2+]c promote the shuttling of reducing equivalents from cytosolic NADH to the mitochondrial NADH pool by stimulating the activity of the two aspartate/glutamate carriers (AGC1/2, also known as SLC25A12 and SLC25A13 or aralar1 and citrin, respectively) that sense Ca2+ levels on the outer face of the IMM via their EF-hand motifs (Palmieri et al., 2001). The efflux of aspartate from the matrix in exchange for glutamate catalyzed by AGC1/2 is one of the steps of the malate-aspartate shuttle, a shuttle system whose net effect is that NADH in the cytosol is oxidized to NAD+, and NAD+ in the matrix is reduced to NADH. AGC1 is the predominant isoform found in cardiac mitochondria, and it responds to Ca2+ concentrations lower than those driving Ca2+ accumulation in the mitochondrial matrix. On these grounds, it has been proposed that the Ca2+-dependent activation of AGC1 might regenerate NADH to sustain oxidative phosphorylation before matrix Ca2+ levels rise (Contreras et al., 2007).
The reducing equivalents derived from oxidation of substrates via the TCA cycle are also used to sustain mitochondrial antioxidant systems. In fact, the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) serves as an electron donor for the redox reactions that convert hydrogen peroxide (H2O2) into water. Mitochondrial oxidative metabolism normally produces a certain quantity of ROS, which also serve physiological functions as signaling molecules, but can damage cellular components when produced in exaggerate amounts or not adequately scavenged. The best-characterized sites of ROS formation in mitochondria are complexes I and III of the ETC, where the superoxide radical (•O2−) can be generated by the incomplete reduction of O2 (Murphy, 2009). Superoxide is rapidly converted to H2O2 by the manganese-dependent superoxide dismutase (Mn-SOD). Another source of mitochondrial ROS is monoamine oxidases, a family of flavoprotein residing in the outer mitochondrial membrane that degrades endogenous monoamines using flavin adenine dinucleotide (FAD) as a cofactor and generating H2O2 as a product (Kaludercic et al., 2011). In turn, H2O2 is reduced to water by glutathione peroxidase or peroxiredoxin, which then need to be regenerated in their active (reduced) form by a cascade of redox reactions that are fueled by NADPH. Because the majority of mitochondrial NADPH is produced by reactions that use TCA cycle intermediates as substrates, the Ca2+-dependent stimulation of the TCA cycle is pivotal to maintain an adequate supply of reducing equivalents to both the ETC and H2O2-eliminating systems during elevations of cardiac workload (Figure 1).
Efficient Ca2+ uptake by mitochondria is made possible by the spatial proximity between mitochondria and the SR. Contact points between mitochondria and the SR create spatially localized microdomains where micromolar Ca2+ concentrations are transiently attained after Ca2+ is released from the SR. These local elevations of Ca2+ concentration are essential for efficient mitochondrial Ca2+ uptake (Giacomello et al., 2010). The regions of the SR that are tethered to mitochondria are also known as mitochondria-associated membranes (MAM), and the integrity of these contact sites is maintained by specialized proteins such as mitofusin 2 (Mfn2), a dynamin-related GTPase that is expressed on both the outer mitochondrial and SR membranes. Loss of Mfn2-mediated interorganelle tethering in the heart reduces the extent of mitochondria-SR contact sites, thereby impairing mitochondrial Ca2+ uptake and the Ca2+-dependent metabolic adaptation in response to β-adrenergic stimulation (Chen et al., 2012). Viceversa, chronic enhancement of cardiac mitochondria-SR tethering via expression of a cardiac myocyte-specific tether transgene potentiates mitochondrial-SR Ca2+ crosstalk, decreasing cardiac vulnerability to adrenergic stress and I/R injury (Nichtová et al., 2023).
Other proteins, such as PACS-2 (phosphofurin acidic cluster sorting protein 2), sigma-1 receptor, GRP75 (glucose-regulated protein 75), and FUNDC1 (FUN14 domain containing 1) were also identified as potential mitochondria-SR tethering proteins and shown to play a role in the communication between SR and mitochondria. Genetic deletion of these proteins results in cardiac dysfunction, ER stress, and mitochondrial fragmentation (Szabadkai et al., 2006; Hayashi and Su, 2007; Stoica et al., 2014; Wu et al., 2017). The ability of the SR to store large amounts of Ca2+ ions depends on the presence of Ca2+-binding proteins such as calsequestrin (CASQ) in its lumen. CASQ acts as a Ca2+ buffer that maintains the SR luminal free Ca2+ concentration between 100–500 μM. The expression of CASQ can be enhanced or suppressed at a gene level, resulting in an increase or decrease in SR Ca2+ load. In addition to acting as a major Ca2+ storage protein, CASQ also regulates the activity of the RyR2 channels (Fearnley et al., 2011).
Overall, there is a tight interplay between the ATP-consuming processes of excitation-contraction coupling and the ATP-producing processes of mitochondrial oxidative metabolism. Ca2+ plays a central role in matching cardiac energy supply to demand and sustaining mitochondrial antioxidative capacity during transitions of cardiac workload.
Mitochondrial Ca2+ uptake is mediated by a macromolecular complex embedded in the inner mitochondrial membrane (IMM) and coined the mitochondrial Ca2+ uniporter (MCU) complex. The MCU pore consists of a tetramer of 35-kDa MCUa subunits, which enclose a conserved DIME motif that accounts for the Ca2+ selectivity of the channel (Yoo et al., 2018; De Stefani et al., 2011). MCUa has a dominant negative homolog, coined MCUb, characterized by differences in the DIME motif that disrupt the Ca2+ permeability of the pore (Raffaello et al., 2013). The key difference between MCUa and MCUb is the replacement of Glu257 with Val242 in MCUb, which results in the loss of a negative charge and thus hinders the electrostatic attraction for Ca2+ (Colussi and Stathopulos, 2022). Therefore, MCUb operates as a negative regulator of mitochondrial Ca2+ uptake. Under physiological conditions, the MCU complex of cardiac myocytes does not comprise MCUb subunits, but these are incorporated in the MCU under pathological conditions, such as ischemic injury (Lambert et al., 2019). By reducing mitochondrial Ca2+ influx, MCUb renders mitochondria less sensitive to Ca2+ overload and consequent permeability transition. Indeed, MCUb overexpression reduces infarct size after I/R injury in mice (Lambert et al., 2019), and its upregulation after myocardial infarction might represent a cardioprotective mechanism to attenuate post-ischemic ventricular remodeling (Huo et al., 2020).
To protect mitochondria from the catastrophic effects of Ca2+ overload, MCU conductance is fine-tuned by several regulatory subunits, such as the EF-hand-containing proteins of the MICU family. MICU1 forms heterodimers with either MICU2 or MICU3 and modulates MCU flux depending on Ca2+ concentration in the intermembrane space (Wu et al., 2020). Although the mechanisms underlying the MICU-mediated regulation of MCU flux have not been completely elucidated, the prevailing view is that MICU1 occludes the cytoplasmic entry of the MCU pore at submicromolar [Ca2+]c, and moves away from the pore to permit Ca2+ penetration when [Ca2+]c increase to the micromolar range (Van Keuren et al., 2020). Therefore, the MICU proteins function as gatekeepers to prevent mitochondrial Ca2+ overload (Mallilankaraman et al., 2012).
The essential MCU regulatory element (EMRE) is a core component of the MCU complex. EMRE induces conformational changes in the pore domain that allow Ca2+ transit through the channel (Sancak et al., 2013). It was initially thought that the stoichiometry of MCUa and EMRE is in 1:1 ratio, but recent studies suggest that there are fewer EMRE subunits than MCUa subunits, with significant variability between different tissues (Watanabe et al., 2022). Upon EMRE binding to the MCU, the MCU-EMRE complex forms a V-shaped dimer, changing the structural organization of the MCU complex and allowing Ca2+ influx through the channel pore (Wang et al., 2019). EMRE downregulation might represent an endogenous mechanism to limit mitochondrial Ca2+ overload under conditions that chronically elevate MCU flux, such as in the MICU1-knockout mouse model (Liu et al., 2016).
MCU regulator 1 (MCUR1) was proposed as another putative component of the uniporter complex, but evidence in this regard is controversial: although MCUR1 modulates Ca2+ threshold for mitochondrial permeability transition and its deletion alters Ca2+ uptake and MCU current (Chaudhuri et al., 2016; Tomar et al., 2016), it was proposed that MCUR1 is in fact a cytochrome c oxidase (complex IV) assembly factor and not a regulator of the MCU (Paupe et al., 2015). Furthermore, a role in the regulation of the MCU current has been proposed for the solute carrier family 25 member 3 (SLC25A3), a metabolite transporter that interacts with MICU1 and MCU (Hoffman et al., 2014; Alevriadou et al., 2021).
The MCU complex has a low conductance and high Ca2+ selectivity. The complex is found in most tissues, but there are major tissue-specific differences in MCU fluxes that reflect the variability in current density and channel stoichiometry. In particular, the MICU1/MCU ratio is a key determinant of tissue-specific differences in the [Ca2+]c threshold for Ca2+ uptake and activation of oxidative metabolism (Paillard et al., 2017). MCU current density is the highest in skeletal muscle mitochondria and the lowest in cardiac mitochondria (Fieni et al., 2012; Boyman et al., 2021). Skeletal muscle mitochondria display the highest MCU current density, which is 30-fold higher than in cardiac mitochondria. This difference reflects the substantially lower volume occupied by mitochondria in cardiac vs. skeletal myocytes (37% vs. 5%). Moreover, the MCU current density varies also between different ages as it is lower in newborn that in adult mice (Fieni et al., 2012).
The main pathway for mitochondrial Ca2+ efflux is the mitochondrial Na+/Ca2+ exchanger, or NCLX (Palty et al., 2010). The existence of a Na+/Ca2+ exchanger as a primary Ca2+ extrusion mechanism from mitochondria implies that mitochondrial Ca2+ levels are controlled by cytosolic Na+, whereby elevations of intracellular Na+ concentration ([Na+]i) as seen in HF can hinder mitochondrial Ca2+ accumulation. Another relevant property of the NCLX is that its kinetics of Ca2+ extrusion are slower than Ca2+ uptake via the MCU, accounting for the accumulation of Ca2+ in the mitochondrial matrix during β-adrenergic stimulation.
The importance of NCLX in regulating mitochondrial Ca2+ levels is underscored by studies of genetic manipulation of NCLX expression. On the one hand, cardiac germline deletion of NCLX is embryonically lethal, and when the gene is silenced in adult mice, these develop severe cardiac dysfunction and sudden cardiac death secondary to mitochondrial Ca2+ overload (Luongo et al., 2017). Conversely, NCLX overexpression in cardiac myocytes is protective against cardiac maladaptive remodeling and prevents contractile disfunction in mice subjected to pressure overload by means of transverse aortic constriction (TAC) for 12 weeks (Garbincius et al., 2022).
The Ca2+-dependent stimulation of mitochondrial oxidative metabolism enables the heart to adjust cardiac inotropy to the circulatory demand, which is determined by neurohormonal activity. The physiological relevance of MCU-dependent mitochondrial Ca2+ uptake was initially investigated in studies using ruthenium derivatives to inhibit the MCU current. In a series of landmark studies conducted in rat cardiac trabeculae, Brandes and Bers demonstrated that elevations of cardiac workload simulated by simultaneous β-adrenergic stimulation and transient increases in pacing frequency transiently oxidize the redox state of the mitochondrial pyridine nucleotides NADH and FADH2 (Brandes and Bers, 1996; Brandes and Bers, 1997; Brandes and Bers, 1999; Brandes and Bers, 2002). The subsequent increase in mitochondrial Ca2+ restores the redox state of NADH and FADH2, but this is substantially blunted or abrogated by blocking the MCU current with the ruthenium derivative Ru360 or by elevating [Na+]i, which accelerates mitochondrial Ca2+ efflux via the NCLX (Maack et al., 2006; Liu and O’Rourke, 2008).
A long-standing debate is whether mitochondrial Ca2+ uptake happens on a beat-to-beat basis or more slowly, as a progressive accumulation over several cytosolic Ca2+ transients. In the beat-to-beat model, fluctuations in mitochondrial Ca2+ levels parallel changes in cytosolic Ca2+, and the increase in diastolic Ca2+ levels in the mitochondrial matrix induced by β-adrenergic stimulation stimulates the TCA cycle dehydrogenases (O’Rourke and Blatter, 2009; la Fuente and Sheu, 2019). Conversely, other studies argued against the existence of mitochondrial Ca2+ transients, suggesting that mitochondria accumulate Ca2+ by integrating cytosolic Ca2+ signals (Hüser et al., 2000). The controversial results supporting the two models might be explained by differences in experimental conditions, types of Ca2+ probes employed, and animal species (Bertero and Maack, 2018).
Following elucidation of the molecular identity of the MCU in the early 2010s, the role of the MCU current in cardiac metabolic adaptation has been further investigated in mouse models of genetic deletion of the MCUa subunit. These studies have shown that MCU deficiency has little or no consequence on cardiac function at rest, but hinders the cardiac chronotropic and inotropic response (Holmström et al., 2015; Kwong et al., 2015; Wu et al., 2015). Furthermore, it was recently discovered that in Barth syndrome, a rare X-linked mitochondrial disorder characterized by abnormal biosynthesis of the mitochondrial phospholipid cardiolipin, the altered composition of the IMM disrupts the structural and functional integrity of several macromolecular complexes embedded in the membrane, including the MCU (Ghosh et al., 2020; Bertero et al., 2021). We studied the functional consequences of MCU loss in a mouse model of Barth syndrome, and discovered that abrogation of mitochondrial Ca2+ uptake in cardiac myocytes abolished the cardiac inotropic response to β-adrenergic stimulation (Bertero et al., 2021). Noteworthy, Barth syndrome patients who do not develop overt systolic dysfunction early in life exhibit a blunted cardiac contractile reserve during physical exercise even in the presence near-normal left ventricular ejection fraction (Spencer et al., 2011). Therefore, Barth syndrome might represent the first example of a human disease in which loss of the MCU is a primary pathophysiological mechanism.
By regulating the rate of oxidative metabolism, mitochondrial Ca2+ levels induce epigenetic modifications that influence cellular differentiation in certain cell types. In mice, fibroblast-specific MCU deletion exacerbates fibrosis after myocardial infarction or angiotensin II administration (Lombardi et al., 2019). Mechanistically, pro-fibrotic signals such as transforming growth factor (TGF)-β reduce mitochondrial Ca2+ uptake by decreasing the MCU current in a MICU1-dependent manner. This leads to an induction of anabolic pathways feeding the TCA cycle that is associated with an increased availability of glutamine-derived α-ketoglutarate (α-KG). In turn, this activates the α-KG-dependent histone demethylases, thereby promoting changes in the structure of chromatin at locations related to the myofibroblast gene program, resulting in cellular differentiation. These findings indicate that the MCU plays a role in regulating the epigenome and influencing cellular differentiation beyond its role in metabolic regulation and cell death (Lombardi et al., 2019).
The existence and identity of alternative pathways of mitochondrial Ca2+ uptake and release is still a matter of debate (Lazaropoulos and Elrod, 2022). Mouse models of MCUa gene silencing showed that mitochondrial Ca2+ uptake is not completely abrogated in Mcua-knockout mice, although Ca2+ uptake kinetics are markedly slower compared with wild-type animals (Kwong et al., 2015; Luongo et al., 2015). It has been suggested that skeletal muscle-type ryanodine receptor type 1 (RyR1) is also found on the IMM of cardiac mitochondria and uptakes Ca2+ released from the SR upon stimulation with inositol triphosphate (Seidlmayer et al., 2016). Of note, RyR1 overexpression leads to mitochondrial fragmentation and increased ATP production (O-Uchi et al., 2013).
Furthermore, the existence of one or more Na+-independent mechanism(s) of mitochondrial Ca2+ efflux has been hypothesized since the 1970s (Carafoli et al., 1974). One recent study demonstrated that the transmembrane BAX inhibitor motif containing protein 5 (TMBIM5) mediates Na+-independent Ca2+ efflux from the matrix, indicating that it operates as a Ca2+/H+ exchanger (Austin et al., 2022). TMBIM5 was identified based on its interaction with the leucine zipper EF-hand containing transmembrane protein 1 (LETM1), a single transmembrane protein that was initially characterized as a potassium (K+)/H+ exchanger (Froschauer et al., 2005) and was subsequently proposed as a Ca2+/H+ exchanger (Jiang et al., 2009; Shao et al., 2016; Austin and Nowikovsky, 2019). However, its function, ion selectivity (Ca2+ vs. K+) and mode of operation as a ion transporter remain unclear.
The IMM is also host for the uncoupling proteins 2 and 3 (UCP2 and UCP3) that might operate as Ca2+ channels. Depending on the origin and mechanism of the elevation in cytosolic Ca2+, UCP2 and UCP3 might also mediate mitochondrial Ca2+ uptake. Under physiological conditions, UCP2 and UCP3 facilitate Ca2+ uptake, regulating mitochondrial Ca2+ levels and preventing Ca2+ overload and consequent mitochondrial dysfunction and cell death under pathological conditions (Waldeck-Weiermair et al., 2010; Zhang et al., 2022).
Mitochondrial Ca2+ uptake is driven by the large negative electrochemical potential (∆μH −180 mV) across the IMM that is maintained by the proton-pumping activity of the, ETC., complexes I, III, and IV (Vasington and Murphy, 1962). If the MCU flux was not modulated by its regulatory subunits, the negative IMM potential would drive large amounts of Ca2+ inside mitochondria, overwhelming the buffering capacity of inorganic phosphate contained in the mitochondrial matrix. Mitochondrial Ca2+ overload and uncontrolled ROS production lead to the opening of a large pore in the IMM denoted as the mitochondrial permeability transition pore (mPTP), which dissipates the proton-motive force and allows equilibration of solutes <1.5 kD, including proapoptotic factors such as cytochrome c (Rasola and Bernardi, 2011). The mPTP plays an important role in regulating cell death and mitophagy under physiological conditions (Bonora et al., 2022), and transient mPTP opening may be necessary to fine-tune mitochondrial Ca2+ levels, functioning like a pressure-release valve to prevent Ca2+ overload (Lu et al., 2016). Furthermore, mPTP opening is also a central mediator of cardiac myocyte death during ischemia-reperfusion injury (Morciano et al., 2017). Accordingly, MCU-knockout mice exhibit decreased infarct size and improved function after myocardial infarction, as defective Ca2+ uptake reduces cardiac myocyte death triggered by mitochondrial permeability transition (Luongo et al., 2015).
A major limitation of inhibiting the mPTP is that the molecular identity of the proteins that compose the pore is largely unknown. Multiple models were developed to explain the molecular architecture of the mPTP, and several proteins including the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT), and the F1-Fo ATP synthase (complex V) were proposed to be mPTP component (for a recent review, see (Bauer and Murphy, 2020)). Cyclosporine A inhibits permeability transition by targeting cyclophilin D, which is an activator of mPTP opening. Genetic deletion of Ppif worsens cardiac hypertrophy, fibrosis, and myocardial function in response to pressure overload. In Ppif-deficient mice, impaired mPTP opening causes an alteration in Ca2+ efflux, increasing mitochondrial Ca2+ and consequently activating Ca2+-dependent TCA cycle dehydrogenases. As a result, glucose oxidation is favored relative to fatty acids, reducing the metabolic flexibility of the heart during stress (Elrod et al., 2010). These findings suggest that the mPTP contributes to metabolic adaptation during changes in cardiac workload.
In the failing heart, altered Ca2+ handling is a major contributor to contractile dysfunction and arrhythmias (Luo and Anderson, 2013). A primary defect in HF is the reduced Ca2+ load of the SR (Lindner et al., 1998), which results from reduced Ca2+ reuptake via SERCA and Ca2+ release due to spontaneous RyR2 opening. Consequently, the amplitude and decay velocity of cytosolic Ca2+ transients are reduced, and [Ca2+]c during diastole is increased, thereby lowering systolic force development and increasing wall tension during diastole (Hasenfuss et al., 1996; O’Rourke et al., 2021). Another central alteration is the increase in [Na+]i, which results from increased late Na+ current (INa) (Valdivia et al., 2005), increased Na+/proton exchanger (NHE) activity (Baartscheer et al., 2003), and decreased Na+/K+ ATPase activity (Schwinger et al., 1999).
Alterations in cytosolic Na+ and Ca2+ handling and the resulting changes in mitochondrial Ca2+ signals have profound consequences on mitochondrial oxidative metabolism and antioxidant defense. In HF, reduced SR Ca2+ load and disruption of the tubular SR network (He et al., 2001) decrease Ca2+ concentration in the mitochondria-SR contact sites, thereby impairing mitochondrial Ca2+ uptake. In addition, mitoplasts (i.e., mitochondria stripped of the outer membrane) isolated from patients with HF exhibit decreased MCU activity compared with nonfailing controls (Michels et al., 2009), which might be explained by an increased expression of the regulatory subunits MICU1 and MICU2 (Paillard et al., 2022). Furthermore, elevated [Na+]i accelerates Ca2+ efflux from the mitochondrial matrix via the NCLX (Maack et al., 2006). Although elevated [Na+]i also favors Ca2+ influx in the cytosol via the reverse mode of the NCX, this is far less efficient in inducing mitochondrial Ca2+ uptake compared with SR Ca2+ release due to the slower NCX kinetics and the more remote location of the NCX flux compared with the RyR2 flux (Kohlhaas and Maack, 2010). Altogether, derangements in cellular Ca2+ and Na+ handling contribute to hamper mitochondrial Ca2+ signals, thereby hindering the Ca2+-mediated stimulation of oxidative metabolism that is required to adapt the production of reducing equivalents for the ETC to the ATP demand of cardiac myocytes (Figure 2).
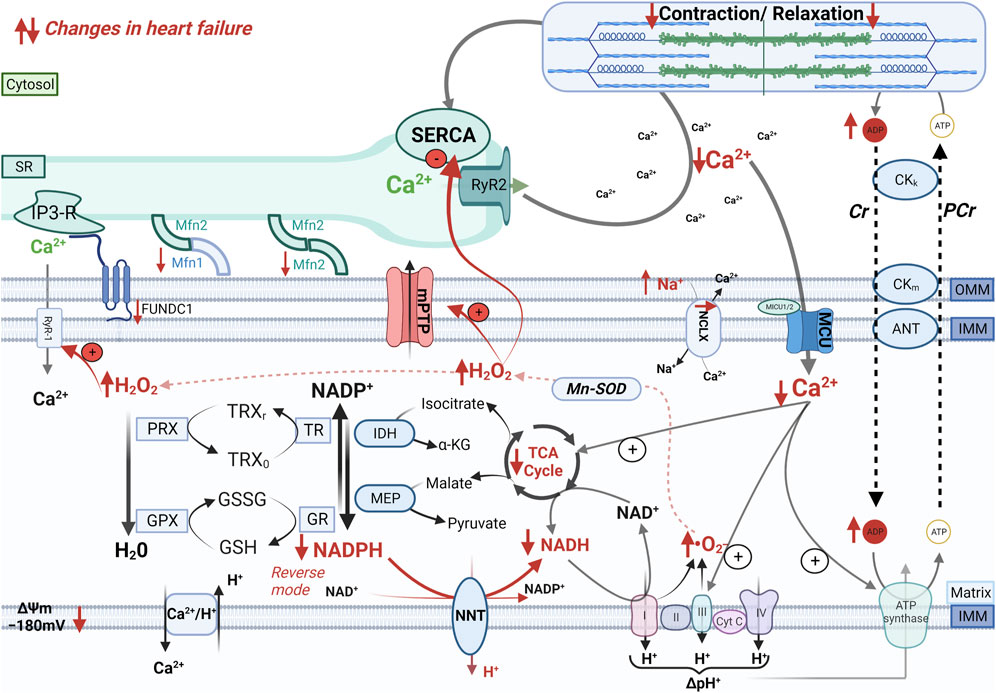
FIGURE 2. Mechano-energetic uncoupling in the failing heart. In the failing heart, altered mitochondria-SR communication, decreased MCU current, and elevation of cytosolic Na+ levels that accelerates Ca2+ extrusion via the mitochondrial Na+/Ca2+ exchanger (NCLX) impair Ca2+ accumulation in the mitochondrial matrix during elevations of cardiac workload. The insufficient stimulation of the TCA cycle dehydrogenases causes an oxidation of the mitochondrial pyridine nucleotides, thereby causes bioenergetic mismatch and oxidative stress, which contribute to the progression of heart failure. Furthermore, pathological increase in cardiac afterload can reverse the NNT reaction, which thereby regenerates NADH at the expense of the NADPH pool, thus further draining reducing equivalents from mitochondrial antioxidant systems.
At the same time, defective stimulation of the TCA cycle by mitochondrial Ca2+ leads to an inadequate supply of reducing equivalents required to maintain matrix NAD(P)H redox potential. In a guinea pig model of HF induced by combined chronic β-adrenergic stimulation and pressure overload, pharmacological inhibition of the NCLX could counteract the depletion of mitochondrial Ca2+ induced by elevated [Na+]i, thereby restoring NAD(P)H redox state and reducing ROS emission from mitochondria (Kohlhaas et al., 2010). This intervention ameliorated cardiac remodeling, improved cardiac function and decreased the risk of ventricular arrhythmias (Liu et al., 2014). In an analogous guinea pig model of HF, moderate overexpression of MCU by viral gene transfer reduced oxidative stress induced by increased cardiac workload, improved systolic function and decreased the number of premature ventricular complexes (Liu et al., 2021).
The mechanisms linking restoration of mitochondrial Ca2+ levels by NCLX inhibition or MCU overexpression and protection from ventricular arrhythmias and sudden cardiac death in this model are not completely resolved. One hypothesis is that the antiarrhythmic effect of correcting mitochondrial Ca2+ levels results from decreased ROS-mediated oxidation of RyR2, which decreases spontaneous SR Ca2+ release events that cause delayed afterdepolarizations, a known trigger of arrhythmias (Zima and Blatter, 2006). In mice, SR Ca2+ leaks feed a vicious cycle whereby mitochondrial Ca2+ mishandling leads to mitochondrial dysfunction, ROS emission, and RyR2 oxidation, further exacerbating proarrhythmic SR Ca2+ leaks (Santulli et al., 2015; Hamilton et al., 2020). One additional mechanism might be represented by ROS-induced activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), which prolongs action potential duration by phosphorylating Na+ channels (Erickson et al., 2008; Wagner et al., 2011). Therefore, oxidation of mitochondrial pyridine nucleotides and mitochondrial ROS production create both a trigger and a substrate for ventricular arrhythmias, but the underlying mechanisms have not been fully elucidated.
It is important to note that this mechanistic framework is supported by studies in guinea pigs exposed to combined chronic β-adrenergic stimulation and pressure overload, which might explain the apparently conflicting results obtained in smaller rodents subjected to myocardial infarction. In the latter model, it has been reported that mitochondrial Ca2+ levels are increased, rather than reduced (Santulli et al., 2015), and abrogating Ca2+ efflux by inhibiting the mPTP or the NCLX has beneficial, rather than detrimental effects (Elrod et al., 2010; Boyman et al., 2021). Importantly, guinea pigs more closely recapitulate human excitation-contraction coupling, whereas mice exhibit substantial differences including the higher [Na+]i (9–14 mmol/L vs. 4–8 mmol/L) and the smaller contractile reserve compared to humans that might partly explain the seemingly contradictory conclusions of these studies.
Pathological elevations of cardiac workload such as those induced by pressure overload also deplete mitochondrial antioxidative capacity and cause oxidative stress by reversing the reaction catalyzed by the mitochondrial nicotinamide nucleotide transhydrogenase (NNT) (Nickel et al., 2015). Under physiological conditions, the NNT transfers electrons from NADH to regenerate NADPH, thus bolstering mitochondrial antioxidative capacity (Figure 2). Pressure overload reverses the NNT reaction, thereby consuming NADPH to regenerate NADH required to sustain the ECT activity. The ensuing ROS production is a major driver of maladaptive remodeling after transverse aortic constriction in mice (Nickel et al., 2015). The yin/yang role of the NNT in the modulation of mitochondrial ROS emerged from the observation that C57BL/6J mice, an inbred mouse strain carrying a loss-of-function mutation of the Nnt gene, are protected from oxidative stress and heart failure induced by pressure overload (Nickel et al., 2015). Therefore, the use of this mouse strain in cardiovascular and/or metabolic research should be avoided.
Altogether, these studies indicate that alterations in cardiac myocyte Na+ and Ca2+ handling as seen in HF hinder cardiac metabolic adaptation and exacerbate mitochondrial ROS production by preventing Ca2+ accumulation in the mitochondrial matrix. In rodent models of HF, counteracting these changes with pharmacologic interventions or genetic manipulation had beneficial effects on cardiac remodeling and arrhythmias.
Despite the large body of evidence indicating that interventions aimed at ameliorating mitochondrial Ca2+ handling in cardiac myocytes have beneficial effects in cardiac disease, studies supporting this concept in humans remain limited. In the context of I/R injury, mitochondrial Ca2+ overload triggers mPTP opening, one central driver of cardiac myocyte loss. Accordingly, studies in rodent models of I/R injury demonstrated that genetic inhibition of the MCU reduced cardiac injury by inhibiting mPTP opening (Kwong et al., 2015; Wu et al., 2020). However, mPTP inhibition by cyclosporine infusion immediately prior to reperfusion did not improve cardiovascular outcomes nor attenuated maladaptive remodeling in patients with myocardial infarction in a phase 3 randomized controlled trial (Cung et al., 2015). One key translational challenge in this context is that mitochondrial Ca2+ overload ensues during ischemia, and mPTP opens as soon as the intracellular pH is restored upon reperfusion (Hartmann and Decking, 1999). Therefore, these detrimental processes are already at play when the patient arrives to the catheterization laboratory for percutaneous reperfusion. Viceversa, the therapeutic goal in chronic HF is to restore physiological levels of mitochondrial Ca2+, which can be achieved with two different approaches: boosting mitochondrial Ca2+ uptake with pharmacological agents or preventing mitochondrial Ca2+ efflux either by inhibiting the NCLX or by decreasing [Na+]i. Pharmacological agents that were tested in human HF an could potentially improve mitochondrial Ca2+ handling are discussed below.
An enhanced late Na+ inward current (INa) is one important contributor to high [Na+]i in HF (Wagner et al., 2006). The selective INa inhibitor ranolazine is approved for symptomatic treatment of stable angina (Silva et al., 2016). In animal models of HF, ranolazine effectively lowered [Na+]i, improved diastolic function, prevented arrhythmias, and pulmonary hypertension (Uran, 2021). On these grounds, ranolazine was tested in patients with HF with preserved ejection fraction, but did not affect ventricular relaxation in the RALI-DHF trial (Ranolazine in Diastolic HF) (Maier et al., 2013).
Increased activity of the NHE also contributes to the elevation in [Na+]i seen in failing cardiac myocytes (Baartscheer et al., 2003). Furthermore, abnormal activation of NHE can increase intracellular pH, thereby enhancing myofilament Ca2+ sensitivity and hindering myocardial relaxation, and activate Ca2+/calmodulin-dependent protein kinase II (CaMKII), which can further aggravate Ca2+ and Na+ mishandling in cardiac myocytes and contribute to oxidative stress (Deschaine et al., 2022). The NHE inhibitor cariproride had beneficial effects in rodent models of HF (Baartscheer et al., 2005; Baartscheer et al., 2008), but increased thromboembolic risk in clinical trials (Karmazyn, 2013).
Sodium-glucose cotransporter 2 inhibitors (SGLT2i) have demonstrated cardiovascular benefit in patients with acute or chronic HF, with or without diabetes, and independent of left ventricular ejection fraction (Packer et al., 2021a; Packer et al., 2021b; Kosiborod et al., 2022; Voors et al., 2022). The mechanisms underlying their cardioprotective activity are multifactorial and include beneficial effects on systemic metabolism, renal function, hemodynamics, and most likely, direct cardiac effects. It is still unclear whether SGLT2i lower [Na+]i and consequently increase mitochondrial Ca2+ levels by inhibiting the NHE expressed on cardiac myocytes. Although this model found experimental confirmation in one study performed in isolated ventricular myocytes from rats and rabbits (Baartscheer et al., 2017) and in human atrial cardiac myocytes (Trum et al., 2020), subsequent studies challenged these results (Chung et al., 2021). Another potential mechanism how SGLT2i could reduce [Na+]i is inhibition of late INa, which was observed in cardiac myocytes from mouse models of HF (Philippaert et al., 2021). Even if [Na+]i was not affected, reduction in late INa might have beneficial effects also by reverting action potential prolongation due to increased activity of this current in HF (Philippaert et al., 2021).
Alterations in Na+ and Ca2+ handling contribute to contractile dysfunction and arrhythmias in HF. In spite of our detailed mechanistic understanding of these derangements derived from animal models of HF, therapeutic strategies aimed at ameliorating cardiac myocyte ion handling and restoring mitochondrial Ca2+ levels have not provided clinical benefit in HF patients thus far. Further studies are needed to understand whether the cardioprotective effects of SGLT2i are mediated, at least in part, by their beneficial activity on mitochondrial Ca2+ and cardiac energetics.
T-AP, CM, EB drafted the manuscript and gave substantial intellectual contribution. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alevriadou, B. R., Patel, A., Noble, M., Ghosh, S., Gohil, V. M., Stathopulos, P. B., et al. (2021). Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. - Cell Physiol. 320 (4), C465–C482. doi:10.1152/ajpcell.00502.2020
Austin, S., Mekis, R., Mohammed, S. E. M., Scalise, M., Wang, W-A., Galluccio, M., et al. (2022). TMBIM5 is the Ca2+/H+ antiporter of mammalian mitochondria. EMBO Rep. 23 (12), e54978. doi:10.15252/embr.202254978
Austin, S., and Nowikovsky, K. (2019). LETM1: essential for mitochondrial biology and cation homeostasis? Trends Biochem. Sci. 44 (8), 648–658. doi:10.1016/j.tibs.2019.04.002
Baartscheer, A., Hardziyenka, M., Schumacher, C. A., Belterman, C. N. W., Van Borren, M. M. G. J., Verkerk, A. O., et al. (2008). Chronic inhibition of the Na +/H +- exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodelling. Br. J. Pharmacol. 154 (6), 1266–1275. doi:10.1038/bjp.2008.189
Baartscheer, A., Schumacher, C. A., Van Borren, M., Belterman, C. N. W., Coronel, R., and Fiolet, J. W. T. (2003). Increased Na+/H+-exchange activity is the cause of increased [Na+] i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 57 (4), 1015–1024. doi:10.1016/s0008-6363(02)00809-x
Baartscheer, A., Schumacher, C. A., van Borren, M. M. G. J., Belterman, C. N. W., Coronel, R., Opthof, T., et al. (2005). Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res. 65 (1), 83–92. doi:10.1016/j.cardiores.2004.09.024
Baartscheer, A., Schumacher, C. A., Wüst, R. C. I., Fiolet, J. W. T., Stienen, G. J. M., Coronel, R., et al. (2017). Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 60 (3), 568–573. doi:10.1007/s00125-016-4134-x
Bauer, T. M., and Murphy, E. (2020). Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 126 (2), 280–293. doi:10.1161/CIRCRESAHA.119.316306
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi:10.1038/415198a
Bertero, E., and Maack, C. (2018). Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 122 (10), 1460–1478. doi:10.1161/CIRCRESAHA.118.310082
Bertero, E., Nickel, A., Kohlhaas, M., Hohl, M., Sequeira, V., Brune, C., et al. (2021). Loss of mitochondrial Ca2+ uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth syndrome cardiomyopathy. Circulation 144 (21), 1694–1713. doi:10.1161/CIRCULATIONAHA.121.053755
Bonora, M., Giorgi, C., and Pinton, P. (2022). Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 23 (4), 266–285. doi:10.1038/s41580-021-00433-y
Boyman, L., Greiser, M., and Lederer, W. J. (2021). Calcium influx through the mitochondrial calcium uniporter holocomplex, MCUcx. MCUcx. J. Mol. Cell Cardiol. 151, 145–154. doi:10.1016/j.yjmcc.2020.10.015
Brandes, R., and Bers, D. M. (1999). Analysis of the mechanisms of mitochondrial NADH regulation in cardiac trabeculae. Biophys. J. 77 (3), 1666–1682. doi:10.1016/S0006-3495(99)77014-1
Brandes, R., and Bers, D. M. (1996). Increased work in cardiac trabeculae causes decreased mitochondrial NADH fluorescence followed by slow recovery. Biophys. J. 71 (2), 1024–1035. doi:10.1016/S0006-3495(96)79303-7
Brandes, R., and Bers, D. M. (1997). Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ. Res. 80 (1), 82–87. doi:10.1161/01.res.80.1.82
Brandes, R., and Bers, D. M. (2002). Simultaneous measurements of mitochondrial NADH and Ca2+ during increased work in intact rat heart trabeculae. Biophys. J. 83 (2), 587–604. doi:10.1016/S0006-3495(02)75194-1
Carafoli, E., Tiozzo, R., Lugli, G., Crovetti, F., and Kratzing, C. (1974). The release of calcium from heart mitochondria by sodium. J. Mol. Cell Cardiol. 6 (4), 361–371. doi:10.1016/0022-2828(74)90077-7
Chaudhuri, D., Artiga, D. J., Abiria, S. A., and Clapham, D. E. (2016). Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. U. S. A. 113 (13), E1872–E1880. doi:10.1073/pnas.1602264113
Chen, Y., Csordás, G., Jowdy, C., Schneider, T. G., Csordás, N., Wang, W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 111 (7), 863–875. doi:10.1161/CIRCRESAHA.112.266585
Chung, Y. J., Park, K. C., Tokar, S., Eykyn, T. R., Fuller, W., Pavlovic, D., et al. (2021). Off-target effects of sodium-glucose co-transporter 2 blockers: empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+] i in the heart. Cardiovasc Res. 117 (14), 2794–2806. doi:10.1093/cvr/cvaa323
Colussi, D. M., and Stathopulos, P. B. (2022). From passage to inhibition: uncovering the structural and physiological inhibitory mechanisms of MCUb in mitochondrial calcium regulation. FASEB J. 37, 1–16. doi:10.1096/fj.202201080r
Contreras, L., Gomez-Puertas, P., Iijima, M., Kobayashi, K., Saheki, T., and Satrustegui, J. (2007). Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 282 (10), 7098–7106. doi:10.1074/jbc.M610491200
Cung, T-T., Morel, O., Cayla, G., Rioufol, G., Garcia-Dorado, D., Angoulvant, D., et al. (2015). Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 373 (11), 1021–1031. doi:10.1056/NEJMoa1505489
Deschaine, B., Verma, S., and Rayatzadeh, H. (2022). Clinical evidence and proposed mechanisms of sodium–glucose cotransporter 2 inhibitors in heart failure with preserved ejection fraction: a class effect? Card. Fail Rev. 8, e23. doi:10.15420/cfr.2022.11
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi:10.1038/nature10230
Elrod, J. W., Wong, R., Mishra, S., Vagnozzi, R. J., Sakthievel, B., Goonasekera, S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca 2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120 (10), 3680–3687. doi:10.1172/JCI43171
Erickson, J. R., Mei-ling, A. J., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133 (3), 462–474. doi:10.1016/j.cell.2008.02.048
Fearnley, C. J., Roderick, H. L., and Bootman, M. D. (2011). Calcium signaling in cardiac myocytes. Cold Spring Harb. Perspect. Biol. 3 (11), a004242. doi:10.1101/cshperspect.a004242
Fieni, F., Lee, S. B., Jan, Y. N., and Kirichok, Y. (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 3, 1317. doi:10.1038/ncomms2325
Froschauer, E., Nowikovsky, K., and Schweyen, R. J. (2005). Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim. Biophys. Acta 1711 (1), 41–48. doi:10.1016/j.bbamem.2005.02.018
Garbincius, J. F., Luongo, T. S., Jadiya, P., Hildebrand, A. N., Kolmetzky, D. W., Mangold, A. S., et al. (2022). Enhanced NCLX-dependent mitochondrial Ca2+ efflux attenuates pathological remodeling in heart failure. J. Mol. Cell Cardiol. 167, 52–66. doi:10.1016/j.yjmcc.2022.03.001
Ghosh, S., Basu Ball, W., Madaris, T. R., Srikantan, S., Madesh, M., Mootha, V. K., et al. (2020). An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. 117 (28), 16383–16390. doi:10.1073/pnas.2000640117
Giacomello, M., Drago, I., Bortolozzi, M., Scorzeto, M., Gianelle, A., Pizzo, P., et al. (2010). Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 38 (2), 280–290. doi:10.1016/j.molcel.2010.04.003
Hamilton, S., Terentyeva, R., Martin, B., Perger, F., Li, J., Stepanov, A., et al. (2020). Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res. Cardiol. 115 (4), 38–20. doi:10.1007/s00395-020-0797-z
Hartmann, M., and Decking, U. K. M. (1999). Blocking Na+--H+ exchange by cariporide reduces Na+-overload in ischemia and is cardioprotective. J. Mol. Cell Cardiol. 31 (11), 1985–1995. doi:10.1006/jmcc.1999.1029
Hasenfuss, G., Reinecke, H., Studer, R., Pieske, B., Meyer, M., Drexler, H., et al. (1996). Calcium cycling proteins and forcefrequency relationship in heart failure. Basic Res. Cardiol. 91 (1), 17–22. doi:10.1007/BF00795357
Hayashi, T., and Su, T-P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131 (3), 596–610. doi:10.1016/j.cell.2007.08.036
He, J-Q., Conklin, M. W., Foell, J. D., Wolff, M. R., Haworth, R. A., Coronado, R., et al. (2001). Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 49 (2), 298–307. doi:10.1016/s0008-6363(00)00256-x
Hoffman, N. E., Chandramoorthy, H. C., Shanmughapriya, S., Zhang, X. Q., Vallem, S., Doonan, P. J., et al. (2014). SLC25A23 augments mitochondrial Ca²⁺ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 25 (6), 936–947. doi:10.1091/mbc.E13-08-0502
Holmström, K. M., Pan, X., Liu, J. C., Menazza, S., Liu, J., Nguyen, T. T., et al. (2015). Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell Cardiol. 85, 178–182. doi:10.1016/j.yjmcc.2015.05.022
Huo, J., Lu, S., Kwong, J. Q., Bround, M. J., Grimes, K. M., Sargent, M. A., et al. (2020). MCUb induction protects the heart from postischemic remodeling. Circ. Res. 127 (3), 379–390. doi:10.1161/CIRCRESAHA.119.316369
Hüser, J., Blatter, L. A., and Sheu, S-S. (2000). Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J. Bioenerg. Biomembr. 32, 27–33. doi:10.1023/a:1005556227425
Jiang, D., Zhao, L., and Clapham, D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326 (5949), 144–147. doi:10.1126/science.1175145
Kaludercic, N., Carpi, A., Menabò, R., Di Lisa, F., and Paolocci, N. (2011). Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta (BBA)-Molecular Cell Res. 1813 (7), 1323–1332. doi:10.1016/j.bbamcr.2010.09.010
Karmazyn, M. (2013). NHE-1: still a viable therapeutic target. J. Mol. Cell Cardiol. 61, 77–82. doi:10.1016/j.yjmcc.2013.02.006
Kohlhaas, M., Liu, T., Knopp, A., Zeller, T., Ong, M. F., Bohm, M., et al. (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121 (14), 1606–1613. doi:10.1161/CIRCULATIONAHA.109.914911
Kohlhaas, M., and Maack, C. (2010). Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation 122 (22), 2273–2280. doi:10.1161/CIRCULATIONAHA.110.968057
Kohlhaas, M., Nickel, A. G., and Maack, C. (2017). Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 595 (12), 3753–3763. doi:10.1113/JP273609
Kosiborod, M. N., Angermann, C. E., Collins, S. P., Teerlink, J. R., Ponikowski, P., Biegus, J., et al. (2022). Effects of empagliflozin on symptoms, physical limitations, and quality of life in patients hospitalized for acute heart failure: results from the EMPULSE trial. Circulation 146 (4), 279–288. doi:10.1161/CIRCULATIONAHA.122.059725
Kwong, J. Q., Lu, X., Correll, R. N., Schwanekamp, J. A., Vagnozzi, R. J., Sargent, M. A., et al. (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12 (1), 15–22. doi:10.1016/j.celrep.2015.06.002
la Fuente, S., and Sheu, S-S. (2019). SR-Mitochondria communication in adult cardiomyocytes: a close relationship where the Ca2+ has a lot to say. Arch. Biochem. Biophys. 663, 259–268. doi:10.1016/j.abb.2019.01.026
Lambert, J. P., Luongo, T. S., Tomar, D., Jadiya, P., Gao, E., Zhang, X., et al. (2019). MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 140 (21), 1720–1733. doi:10.1161/CIRCULATIONAHA.118.037968
Lazaropoulos, M. P., and Elrod, J. W. (2022). Mitochondria in pathological cardiac remodeling. Curr. Opin. Physiol. 25, 100489. doi:10.1016/j.cophys.2022.100489
Lindner, M., Erdmann, E., and Beuckelmann, D. J. (1998). Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J. Mol. Cell Cardiol. 30 (4), 743–749. doi:10.1006/jmcc.1997.0626
Liu, J. C., Liu, J., Holmström, K. M., Menazza, S., Parks, R. J., Fergusson, M. M., et al. (2016). MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16 (6), 1561–1573. doi:10.1016/j.celrep.2016.07.011
Liu, T., and O’Rourke, B. (2008). Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 103 (3), 279–288. doi:10.1161/CIRCRESAHA.108.175919
Liu, T., Takimoto, E., Dimaano, V. L., DeMazumder, D., Kettlewell, S., Smith, G., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 115 (1), 44–54. doi:10.1161/CIRCRESAHA.115.303062
Liu, T., Yang, N., Sidor, A., O’Rourke, B., Zhang, H. H., Li, Y., et al. (2021). MCU overexpression rescues inotropy and reverses heart failure by reducing SR Ca2+Leak. Circ. Res. 49, 1191–1204. doi:10.1161/circresaha.120.318562
Lombardi, A. A., Gibb, A. A., Arif, E., Kolmetzky, D. W., Tomar, D., Luongo, T. S., et al. (2019). Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun. 10 (1), 4509. doi:10.1038/s41467-019-12103-x
Lu, X., Kwong, J. Q., Molkentin, J. D., and Bers, D. M. (2016). Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ. Res. 118 (5), 834–841. doi:10.1161/CIRCRESAHA.115.308093
Luo, M., and Anderson, M. E. (2013). Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 113 (6), 690–708. doi:10.1161/CIRCRESAHA.113.301651
Luongo, T. S., Lambert, J. P., Gross, P., Nwokedi, M., Lombardi, A. A., Shanmughapriya, S., et al. (2017). The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545 (7652), 93–97. doi:10.1038/nature22082
Luongo, T. S., Lambert, J. P., Yuan, A., Zhang, X., Gross, P., Song, J., et al. (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12 (1), 23–34. doi:10.1016/j.celrep.2015.06.017
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O’Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99 (2), 172–182. doi:10.1161/01.RES.0000232546.92777.05
Maier, L. S., Layug, B., Karwatowska-Prokopczuk, E., Belardinelli, L., Lee, S., Sander, J., et al. (2013). RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: the RALI-DHF proof-of-concept study. JACC Hear Fail 1 (2), 115–122. doi:10.1016/j.jchf.2012.12.002
Mallilankaraman, K., Doonan, P., Cárdenas, C., Chandramoorthy, H. C., Müller, M., Miller, R., et al. (2012). MICU1 is an essential gatekeeper for mcu-mediated mitochondrial Ca 2+ uptake that regulates cell survival. Cell 151 (3), 630–644. doi:10.1016/j.cell.2012.10.011
McCormack, J. G., and Denton, R. M. (1993). The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc Trans. 21 (3), 793–799.
Michels, G., Khan, I. F., Endres-Becker, J., Rottlaender, D., Herzig, S., Ruhparwar, A., et al. (2009). Regulation of the human cardiac mitochondrial Ca 2+ uptake by 2 different voltage-gated Ca 2+ channels. Circulation 119 (18), 2435–2443. doi:10.1161/CIRCULATIONAHA.108.835389
Morciano, G., Bonora, M., Campo, G., Aquila, G., Rizzo, P., Giorgi, C., et al. (2017). Mechanistic role of mPTP in ischemia-reperfusion injury. Mitochondrial Dyn. Cardiovasc Med. 982, 169–189. doi:10.1007/978-3-319-55330-6_9
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem J. 417, 1–13. doi:10.1042/BJ20081386
Nichtová, Z., Fernandez-Sanz, C., De La Fuente, S., Yuan, Y., Hurst, S., Lanvermann, S., et al. (2023). Enhanced mitochondria-SR tethering triggers adaptive cardiac muscle remodeling. Circ. Res. 132 (11), e171–e187. doi:10.1161/CIRCRESAHA.122.321833
Nickel, A. G., Von Hardenberg, A., Hohl, M., Löffler, J. R., Kohlhaas, M., Becker, J., et al. (2015). Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 22 (3), 472–484. doi:10.1016/j.cmet.2015.07.008
O-Uchi, J., Jhun, B. S., Hurst, S., Bisetto, S., Gross, P., Chen, M., et al. (2013). Overexpression of ryanodine receptor type 1 enhances mitochondrial fragmentation and Ca2+-induced ATP production in cardiac H9c2 myoblasts. Am. J. Physiol. - Hear Circ. Physiol. 305 (12), 1736–1751. doi:10.1152/ajpheart.00094.2013
O’Rourke, B., Ashok, D., and Liu, T. (2021). Mitochondrial Ca2+ in heart failure: not enough or too much? J. Mol. Cell Cardiol. 151, 126–134. doi:10.1016/j.yjmcc.2020.11.014
O’Rourke, B., and Blatter, L. A. (2009). Mitochondrial Ca2+ uptake: tortoise or hare? J. Mol. Cell Cardiol. 46 (6), 767–774. doi:10.1016/j.yjmcc.2008.12.011
Packer, M., Anker, S. D., Butler, J., Filippatos, G., Ferreira, J. P., Pocock, S. J., et al. (2021a). Effect of empagliflozin on the clinical stability of patients with heart failure and a reduced ejection fraction: the EMPEROR-reduced trial. Circulation 143 (4), 326–336. doi:10.1161/CIRCULATIONAHA.120.051783
Packer, M., Butler, J., Zannad, F., Filippatos, G., Ferreira, J. P., Pocock, S. J., et al. (2021b). Effect of empagliflozin on worsening heart failure events in patients with heart failure and preserved ejection fraction: EMPEROR-preserved trial. Circulation 144 (16), 1284–1294. doi:10.1161/CIRCULATIONAHA.121.056824
Paillard, M., Csordás, G., Szanda, G., Golenár, T., Debattisti, V., Bartok, A., et al. (2017). Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 18 (10), 2291–2300. doi:10.1016/j.celrep.2017.02.032
Paillard, M., Huang, K-T., Weaver, D., Lambert, J. P., Elrod, J. W., and Hajnóczky, G. (2022). Altered composition of the mitochondrial Ca2+ uniporter in the failing human heart. Cell Calcium 105, 102618. doi:10.1016/j.ceca.2022.102618
Palmieri, L., Pardo, B., Lasorsa, F. M., Del Arco, A., Kobayashi, K., Iijima, M., et al. (2001). Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 20 (18), 5060–5069. doi:10.1093/emboj/20.18.5060
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., and Parnis, J. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 107 (2), 436–441. doi:10.1073/pnas.0908099107
Paupe, V., Prudent, J., Dassa, E. P., Rendon, O. Z., and Shoubridge, E. A. (2015). CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 21 (1), 109–116. doi:10.1016/j.cmet.2014.12.004
Philippaert, K., Kalyaanamoorthy, S., Fatehi, M., Long, W., Soni, S., Byrne, N. J., et al. (2021). Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation 143 (22), 2188–2204. doi:10.1161/CIRCULATIONAHA.121.053350
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32 (17), 2362–2376. doi:10.1038/emboj.2013.157
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50 (3), 222–233. doi:10.1016/j.ceca.2011.04.007
Sancak, Y., Markhard, A. L., Kitami, T., Kovács-Bogdán, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342 (6164), 1379–1382. doi:10.1126/science.1242993
Santulli, G., Xie, W., Reiken, S. R., and Marks, A. R. (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. 112 (36), 11389–11394. doi:10.1073/pnas.1513047112
Schwinger, R. H. G., Wang, J., Frank, K., Muller-Ehmsen, J., Brixius, K., McDonough, A. A., et al. (1999). Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 99 (16), 2105–2112. doi:10.1161/01.cir.99.16.2105
Seidlmayer, L. K., Kuhn, J., Berbner, A., Arias-Loza, P-A., Williams, T., Kaspar, M., et al. (2016). Inositol 1, 4, 5-trisphosphate-mediated sarcoplasmic reticulum-mitochondrial crosstalk influences adenosine triphosphate production via mitochondrial Ca 2+ uptake through the mitochondrial ryanodine receptor in cardiac myocytes. Cardiovasc Res. 112 (1), 491–501. doi:10.1093/cvr/cvw185
Shao, J., Fu, Z., Ji, Y., Guan, X., Guo, S., Ding, Z., et al. (2016). Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca2+/H+ antiporter. Sci. Rep. 6, 34174–34179. doi:10.1038/srep34174
Silva, S. G. F., F Simoes, R., Couto, R., and J Oliveira, P. (2016). Targeting mitochondria in cardiovascular diseases. Curr. Pharm. Des. 22 (37), 5698–5717. doi:10.2174/1381612822666160822150243
Spencer, C. T., Byrne, B. J., Bryant, R. M., Margossian, R., Maisenbacher, M., Breitenger, P., et al. (2011). Impaired cardiac reserve and severely diminished skeletal muscle O- utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Circ. Physiol. 301 (5), H2122–H2129. doi:10.1152/ajpheart.00479.2010
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K-F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5 (1), 3996–4012. doi:10.1038/ncomms4996
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175 (6), 901–911. doi:10.1083/jcb.200608073
Teiger, E., Than, V. D., Richard, L., Wisnewsky, C., Tea, B-S., Gaboury, L., et al. (1996). Apoptosis in pressure overload-induced heart hypertrophy in the rat. J. Clin. Invest. 97 (12), 2891–2897. doi:10.1172/JCI118747
Tomar, D., Dong, Z., Shanmughapriya, S., Koch, D. A., Thomas, T., Hoffman, N. E., et al. (2016). MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 15 (8), 1673–1685. doi:10.1016/j.celrep.2016.04.050
Trum, M., Riechel, J., Lebek, S., Pabel, S., Sossalla, S. T., Hirt, S., et al. (2020). Empagliflozin inhibits Na+/H+ exchanger activity in human atrial cardiomyocytes. ESC Hear Fail 7 (6), 4429–4437. doi:10.1002/ehf2.13024
Uran, C. (2021). Through the heart and beyond: a review on ranolazine. Monaldi Arch. chest Dis. 92 (1). 1806. doi:10.4081/monaldi.2021.1806
Valdivia, C. R., Chu, W. W., Pu, J., Foell, J. D., Haworth, R. A., Wolff, M. R., et al. (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell Cardiol. 38 (3), 475–483. doi:10.1016/j.yjmcc.2004.12.012
Van Keuren, A. M., Tsai, C. W., Balderas, E., Rodriguez, M. X., Chaudhuri, D., and Tsai, M. F. (2020). Mechanisms of EMRE-dependent MCU opening in the mitochondrial calcium uniporter complex. Cell Rep. 33 (10), 108486. doi:10.1016/j.celrep.2020.108486
Vasington, F. D., and Murphy, J. V. (1962). Ca++ uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 237 (8), 2670–2677. doi:10.1016/s0021-9258(19)73805-8
Voglhuber, J., Holzer, M., Radulović, S., Thai, P. N., Djalinac, N., Matzer, I., et al. (2022). Functional remodelling of perinuclear mitochondria alters nucleoplasmic Ca 2+ signalling in heart failure. Philos. Trans. R. Soc. B Biol. Sci. 377 (1864), 20210320. doi:10.1098/rstb.2021.0320
Voors, A. A., Angermann, C. E., Teerlink, J. R., Collins, S. P., Kosiborod, M., Biegus, J., et al. (2022). The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: a multinational randomized trial. Nat. Med. 28 (3), 568–574. doi:10.1038/s41591-021-01659-1
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca 2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116 (12), 3127–3138. doi:10.1172/JCI26620
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108 (5), 555–565. doi:10.1161/CIRCRESAHA.110.221911
Waldeck-Weiermair, M., Malli, R., Naghdi, S., Trenker, M., Kahn, M. J., and Graier, W. F. (2010). The contribution of UCP2 and UCP3 to mitochondrial Ca2+ uptake is differentially determined by the source of supplied Ca2+. Cell Calcium 47 (5), 433–440. doi:10.1016/j.ceca.2010.03.004
Wang, Y., Nguyen, N. X., She, J., Zeng, W., Yang, Y., Chen, B. X., et al. (2019). Structural mechanism of EMRE-dependent gating of the human mitochondrial calcium uniporter. Cell 177 (5), 1252–1261. doi:10.1016/j.cell.2019.03.050
Watanabe, A., Maeda, K., Nara, A., Hashida, M., Ozono, M., Nakao, A., et al. (2022). Quantitative analysis of mitochondrial calcium uniporter (MCU) and essential MCU regulator (EMRE) in mitochondria from mouse tissues and HeLa cells. FEBS Open Bio 12 (4), 811–826. doi:10.1002/2211-5463.13371
Wu, S., Lu, Q., Wang, Q., Ding, Y., Ma, Z., Mao, X., et al. (2017). Binding of FUN14 domain containing 1 with inositol 1, 4, 5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136 (23), 2248–2266. doi:10.1161/CIRCULATIONAHA.117.030235
Wu, W., Shen, Q., Zhang, R., Qiu, Z., Wang, Y., Zheng, J., et al. (2020). The structure of the MICU 1- MICU 2 complex unveils the regulation of the mitochondrial calcium uniporter. EMBO J. 39 (19), 1042855–e104314. doi:10.15252/embj.2019104285
Wu, Y., Rasmussen, T. P., Koval, O. M., Joiner, M. A., Hall, D. D., Chen, B., et al. (2015). The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 6 (1), 6081. doi:10.1038/ncomms7081
Xuxia, H., Cuimei, S., Zhangmei, Y., and Ren, J. (2020). Mitochondrial Ca2+ regulation in the etiology of heart failure: physiological and pathophysiological implications. Acta Pharmacol. Sin. 41 (10), 1301–1309. doi:10.1038/s41401-020-0476-5
Yoo, J. (2022). Structural basis of Ca 2 + uptake by mitochondrial calcium uniporter in mitochondria: a brief review. BMB Rep. 55 (11), 528–534. doi:10.5483/BMBRep.2022.55.11.134
Yoo, J., Wu, M., Yin, Y., Herzik, M. A., Lander, G. C., and Lee, S. Y. (2018). Cryo-EM structure of a mitochondrial calcium uniporter. Science 361 (6401), 506–511. doi:10.1126/science.aar4056
Zhang, D., Wang, F., Li, P., and Gao, Y. (2022). Mitochondrial Ca2+ homeostasis: emerging roles and clinical significance in cardiac remodeling. Int. J. Mol. Sci. 23 (6), 3025. doi:10.3390/ijms23063025
Keywords: mitochondria, cardiomyocyte, calcium, redox homeostasis, heart failure, reactive oxygen species
Citation: Popoiu T-A, Maack C and Bertero E (2023) Mitochondrial calcium signaling and redox homeostasis in cardiac health and disease. Front. Mol. Med. 3:1235188. doi: 10.3389/fmmed.2023.1235188
Received: 05 June 2023; Accepted: 10 August 2023;
Published: 23 August 2023.
Edited by:
Julia Ritterhoff, University of Washington, United StatesReviewed by:
Jonathan Lambert, University of Padua, ItalyCopyright © 2023 Popoiu, Maack and Bertero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edoardo Bertero, ZWRvLmJlcnRlcm9AZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.