 Lynette M. Bustos
Lynette M. Bustos Rita Sattler
Rita Sattler
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Med , 16 February 2023
Sec. Molecular Mechanisms of Neurodegeneration
Volume 3 - 2023 | https://doi.org/10.3389/fmmed.2023.1075805
This article is part of the Research Topic The Role of Glial Cells in Neurodegeneration View all 9 articles
Many neurodegenerative diseases fall under the class of diseases known as proteinopathies, whereby the structure and localization of specific proteins become abnormal. These aberrant proteins often aggregate within cells which disrupts vital homeostatic and physiological cellular functions, ultimately contributing to cell death. Although neurodegenerative disease research is typically neurocentric, there is evidence supporting the role of non-neuronal cells in the pathogenesis of these diseases. Specifically, the role of astrocytes in neurodegenerative diseases has been an ever-growing area of research. Astrocytes are one of the most abundant cell types in the central nervous system (CNS) and provide an array of essential homeostatic functions that are disrupted in neurodegenerative diseases. Astrocytes can exhibit a reactive phenotype that is characterized by molecular changes, as well as changes in morphology and function. In neurodegenerative diseases, there is potential for reactive astrocytes to assume a loss-of-function phenotype in homeostatic operations such as synapse maintenance, neuronal metabolic support, and facilitating cell-cell communication between glia and neurons. They are also able to concurrently exhibit gain-of-function phenotypes that can be destructive to neural networks and the astrocytes themselves. Additionally, astrocytes have been shown to internalize disease related proteins and reflect similar or exacerbated pathology that has been observed in neurons. Here, we review several major neurodegenerative disease-specific proteinopathies and what is known about their presence in astrocytes and the potential consequences regarding cell and non-cell autonomous neurodegeneration.
The concept of astrocytes was first introduced in the 1800’s by Rudolf Virchow, where he described “neuroglia” as connective material between neurons (Kettenmann and Verkhratsky, 2008). In the same century, Michael von Lenhossek branded these cells as “astrocyten” or “star cells” describing their star-like morphology. Astrocytes are one of the most abundant cell types in the central nervous system (CNS) and undergo a morphological and functional maturation process based on environmental cues from the surrounding microenvironment (Molofsky and Deneen, 2015; Khakh and Deneen, 2019). This maturation coincides with an increase in formation, density, and elimination of synapses that aids in neuronal circuitry maturation (Molofsky and Deneen, 2015). This intricate relationship between astrocytes and neurons was first proposed by Carl Ludwig Schleich in 1899 (Kettenmann and Verkhratsky, 2008). In addition to aiding in the formation and maintenance of synapses, astrocytes contribute to the blood-brain barrier, provide trophic support to neurons, and facilitate neuron-glia and glia-glia communication (Khakh and Deneen, 2019).
In the human cortex, astrocytes have been characterized by their distinct morphological properties. The four subtypes are interlaminar (located in layers I and II), protoplasmic (located in layers III and IV), varicose projections (located in layers V and VI), and fibrous (located in the white matter) (Oberheim et al., 2009; Vasile et al., 2017). Although studies use morphology as one defining characteristic of an astrocyte’s function within the brain, they have been recently categorized molecularly in the murine brain using single-cell transcriptomics; thus, defining specialized astrocyte subtypes between brain regions (Batiuk et al., 2020). Cortical astrocytes express, at higher levels, genes that encode proteins required for metabolism, gliotransmission and neurotransmitter uptake and recycling (Oberheim et al., 2009; Zhang et al., 2016; Vasile et al., 2017; Peteri et al., 2019).
In the same way astrocyte function is heterogeneous, so too is their activation and reactivity to physiological conditions and disease pathologies, respectively. Much work has demonstrated that astrocytes can take on neurotoxic and neuroprotective phenotypes in response to a CNS insult (Faulkner et al., 2004; Okada et al., 2006; Herrmann et al., 2008; Zamanian et al., 2012; Liddelow et al., 2017; Clarke et al., 2018). However, as technologies used in the field of astrocyte biology have advanced, the dualistic view of “A1 vs. A2” reactive astrocytes has blurred. The term “reactive astrogliosis” has recently been redefined as a process induced in a pathological context-specific manner that results in a loss or upregulation of homeostatic functions and/or the gain of new function(s) (Escartin et al., 2021). Therefore, the term “astrocyte reactivity” is the ability of astrocytes to adopt a discrete state in response to a specific disease-related pathology, and the term “astrocyte activation” is to be associated with a change to physiological conditions not due to a pathological source (Escartin et al., 2021). The consensus of the field is that astrocyte reactivity is most likely a secondary event triggered by an external signal at a specific timepoint in the pathological progression, within some contexts where the phenotypes are reversible (Escartin et al., 2021).
There is mounting evidence for the important role of astrocytes in disease pathogenesis of various neurodegenerative diseases. There is a critical loss of supportive astrocytic functions and in some contexts a toxic gain-of-function that causes astrocytes to not only affect the health of vulnerable neurons, but to develop a disease specific reactive phenotype. Recently, a study assessed astrocyte-derived toxic factors in an induced reactive state that has been previously shown to be detrimental to the health and survival of neurons and other glia (Guttenplan et al., 2021). The authors screened reactive and quiescent astrocytes and conditioned media (ACM) using mass spectrometry to assess for changes in protein abundance following activation via addition of pro-inflammatory cytokines, IL-1α, TNFα and C1q. There was an increase in C3 and other reactivity factors in both cells and ACM, but none facilitated cell death in their in vitro systems. In reactive ACM, other than known reactivity markers, lipoparticle proteins such as APOE and APOJ were upregulated. By immunodepleting reactive ACM of APOE and APOJ, the author’s observed a decrease in toxicity of reactive ACM. Furthermore, they described an upregulation of long-chain saturated free fatty acids in reactive ACM. Through conditional knockout of the metabolic enzyme ELOVL3, they were able to reduce long-chain saturated free fatty acid production and reduce reactive ACM toxicity towards neurons and oligodendrocytes in both in vivo and in vitro conditions, respectively. Demonstrating that reactive astrocytes, through the secretion of toxic lipids, contributes to neuronal death. However, clear direct evidence of this altered lipid metabolism occurring in neurodegenerative diseases is missing.
Astrocytes have a role in mediating or exacerbating pathological conditions, especially in the context of neurodegenerative diseases. In many neurodegenerative diseases, astrocytes have also shown evidence of pathological inclusions. It is not certain whether this is initiated by the disease itself directly affecting astrocytes and causing protein inclusions, or if it is a compensatory mechanism where astrocytes are internalizing pathological proteins released by or crowding around neurons. In this review, we will summarize what is currently known about how the distribution of these aberrant proteins causes disease-specific alterations in astrocytes, and how this might contribute to neuronal cell death (see Table 1). We will discuss the presence of astrocytic proteinopathies in the spectrum disease of Amyotrophic lateral sclerosis and Frontotemporal Dementia (ALS/FTD), as well as Alzheimer’s disease, Huntington’s disease and Parkinson’s disease.
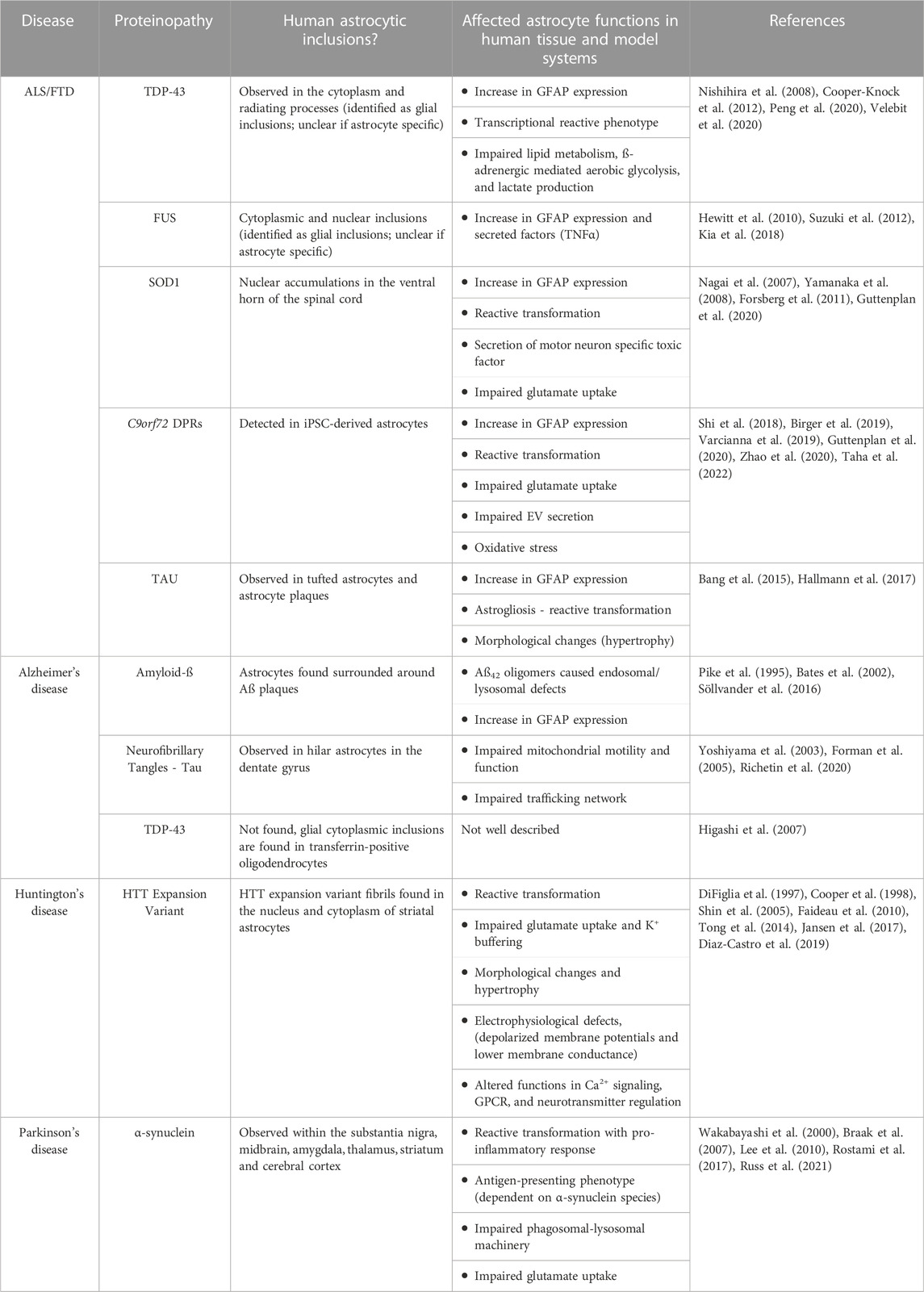
Table 1. Neurodegenerative disease pathologies affecting astrocytes.
Amyotrophic lateral sclerosis (ALS) is a fatal progressive neurodegenerative disorder that causes the loss of upper and lower motor neurons, leading to muscle atrophy and loss of muscle control, and ultimately the inability to walk and breath (Hardiman et al., 2017; Balendra and Isaacs, 2018). The onset of disease can occur between 40 and 70 years of age. The majority of ALS cases are categorized as sporadic (sALS), and just over 10% of sALS cases are associated with a genetic variant (Yun and Ha, 2020). Only a small subset, about 10% of ALS cases, are classified as familial (fALS), where about 70% of fALS cases are associated with a genetic variant (Yun and Ha, 2020). Over 50 potential genes have been associated with ALS, and the two most common genetic abnormalities are those found in the C9orf72 and SOD1 genes (Mejzini et al., 2019). The GGGGCC (G4C2) hexanucleotide repeat expansion (HRE) in the first intron of the gene C9orf72, is the most common genetic abnormality associated with both ALS and frontal temporal dementia (FTD) (DeJesus-Hernandez et al., 2011; Renton et al., 2011). ALS and FTD patient brains can exhibit multiple proteinopathies, dependent on the specific patient subgroup. Here, we will discuss ALS-associated proteinopathy, and then further discuss FTD-tau associated proteinopathy and several overlapping ALS/FTD proteinopathies.
Non-cell autonomous disease mechanisms in ALS have been well-accepted for over 2 decades, suggesting a significant contribution of the observed neurodegeneration is exacerbated by non-neuronal cells (Rothstein et al., 1996; Clement et al., 2003). A common pathogenic feature of ALS astrocytes is the loss-of-function mechanism that leads to neuronal glutamate excitotoxicity. In ALS patients, there is significant evidence demonstrating a decrease in glutamate transport within the cortex and spinal cord (Rothstein et al., 1992). The reduced expression of astrocytic glutamate transporter, EAAT2, is responsible for this phenomenon (Rothstein et al., 1996). Consequently, ALS astrocytes have a reduced ability to uptake glutamate at the synapse, leading already vulnerable motor neurons to also undergo glutamate excitotoxicity. More recently, a study proposed the astrocytic release of inorganic polyphosphate (polyP) as a motor neuron specific vulnerability in ALS/FTD (Arredondo et al., 2022). In this study, they utilized in vitro and in vivo models with SOD1, TARDBP and C9orf72 ALS/FTD-related genetic variants to describe an excessive and ultimately toxic release of inorganic polyP by astrocytes. They further observed enrichment of polyP in astrocyte-like cells in postmortem spinal cord tissue from SOD1, C9orf72 and sALS patients. Ultimately, astrocytes have been shown to elicit both a cell autonomous and non-cell autonomous disease pathogenesis in both sporadic and familial forms of ALS leading to the death of motor neurons.
Early works have primarily studied spinal cord astrocytes in the context of the first genetic abnormality associated with ALS—missense point mutations in the Superoxide Dismutase 1 (SOD1) gene. SOD1-ALS accounts for ∼12% of familial and ∼1% of sporadic cases (Renton et al., 2014). Although this genetic variation of ALS presents with progressive motor neuron degeneration, there is no TDP-43 pathology present. In SOD1-ALS, SOD1 variants undergo misfolding that causes structural and functional defects in catalytic activity and metal binding leading to a toxic gain-of-function (Huai and Zhang, 2019). Misfolded SOD1 variants have been observed to localize not only in motor neurons of the spinal cord, but also in the nuclei of astrocytes (see Figure 1), microglia and oligodendrocytes in the ventral horn (Forsberg et al., 2011). Early studies demonstrated that when SOD1 variants are expressed only in motor neurons of a chimeric mouse model, this is not sufficient to initiate neurodegeneration in the spinal cord (Clement et al., 2003). However, primary mouse astrocytes expressing SOD1G93A variant reduced the survival of both wild type and SOD1G93A motor neurons in co-culture conditions, eliciting a direct effect of neuronal degeneration (Di Giorgio et al., 2007). Additionally, primary mouse motor neuron cultures treated with preconditioned media from primary astrocytes from SOD1G93A mice displayed a selective toxic effect on neuronal morphometry and survival (Nagai et al., 2007). When conditioned media from SOD1G93A cortical neurons, microglia, myocytes and fibroblasts were added to the primary motor neurons, there was no effect on survival (Nagai et al., 2007). However, when SOD1G37R variant was selectively deleted in mouse astrocytes in vivo, disease onset was not altered, but instead astrocyte-specific deletion of SOD1G37R delayed microglial activation and significantly slowed later disease progression (Yamanaka et al., 2008).
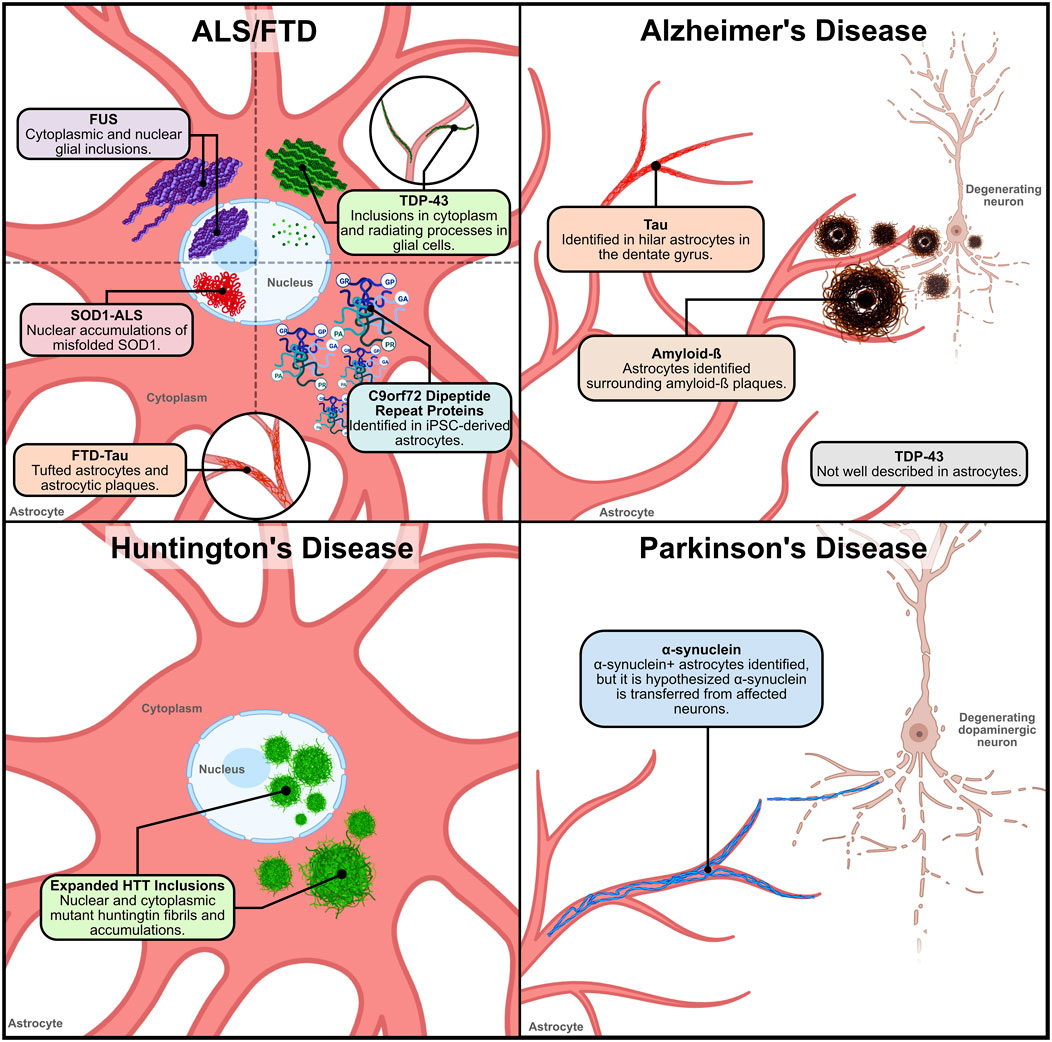
Figure 1. Location and distribution of neurodegenerative disease proteinopathies within and around astrocytes. 1) ALS/FTD – Various proteinaceous inclusions occur in ALS and FTD astrocytes. In ALS and FTD, TDP-43 inclusions have been found in the cytoplasm and radiating processes of glial cells. In ALS and FTD cases with FUS variants, cytoplasmic and nuclear glial inclusions have been identified. However, the identification of TDP-43 and FUS inclusions specifically in astrocytes has not been clarified. Nuclear accumulations of misfolded SOD1 caused by ALS-linked genetic variants have been identified in ventral horn astrocytes. In FTD-TAU, tufted astrocytes and astrocytic plaques have been observed in patient tissue. This pathology is similar to what has also been observed in AD tauopathy. and in C9orf72-ALS iPSC-derived spinal cord-like astrocytes the presence of DPRs were detected. 2) Alzheimer’s disease–In Alzheimer’s disease, astrocytes have been described to surround and internalize extracellular amyloid-ß (Aß) plaques. Reactive astrocytes found in postmortem AD tissue have been observed to have a significant Aß load. Tau is not expressed in astrocytes explicitly, so the source of tau in astrocytic accumulations in AD is unknown. However, tau accumulation in hilar astrocytes has been described. AD TDP-43 pathology has not been well characterized in astrocytes. 3) Huntington’s disease – In Huntington’s disease, expanded huntingtin (HTT) fibrils in the nucleus and cytoplasm of cortical and striatal astrocytes have been observed. Expanded HTT inclusions in astrocytes occurs early in the disease pathogenesis, suggesting that astrocytic disruptions may contribute to disease progression. 4) Parkinson’s disease – In Parkinson’s disease, α-synuclein+ inclusions in astrocytes of PD patient tissue found within the SN, midbrain, amygdala, thalamus, striatum and cerebral cortex. In early stages of disease pathogenesis, α-synuclein+ astrocytes are found in the amygdala, striatum, and thalamic nuclei projecting to the cortex. In the cortex, astrocytic α-synuclein+ inclusions are found in layers V and VI, with limited prevalence in layers III and IV. However, studies hypothesize that the α-synuclein found in astrocytes is possibly transferred from affected dopaminergic neurons. Figure created with Biorender.com.
More recently, the knockout of pro-inflammatory cytokines, IL-1α, TNFα and C1q, was found to significantly delay the death of motor neurons in the SOD1G93A mouse model (Guttenplan et al., 2020). In these IL-1α−/− TNFα−/− C1q−/− SOD1G93A mice, lower levels of the microglia-mediated reactive astrocyte marker C3, identified in Liddelow et al. (2017), was observed (Guttenplan et al., 2020). Additionally, these triple knockout SOD1G93A mice had a 50% lifespan extension compared to strain matched SOD1G93A mice (Guttenplan et al., 2020). Together these studies provide evidence that astrocytes expressing ALS-linked SOD1 variants have the ability to elicit direct and indirect toxic effects selective to motor neurons, while exacerbating microglial activation and disease progression.
Frontotemporal dementia is the clinical term used to encompass a heterogeneous group of disorders associated with progressive behavioral and personality changes, and/or impairments in language and executive function (Bang et al., 2015; Pottier et al., 2016; Woollacott and Rohrer, 2016). FTD is the clinical syndrome resulting from frontotemporal lobar degeneration (FTLD) which can be caused by several different molecular pathologies. There are both familial and sporadic forms of FTD, with the most common variants discovered in genes encoding for the microtubule-associated protein Tau (MAPT; FTLD-TAU), progranulin (GRN; FTLD-TDP), and C9orf72 (FTLD-TDP) (Mackenzie and Neumann, 2016).
Tau is encoded by the gene MAPT and due to alternative splicing in exons 2, 3, and 10, six isoforms of the protein are produced. These isoforms make-up various combinations of three or four microtubule-binding repeat-regions in the C-terminal, and zero to two inserts in the N-terminal; resulting in 0N3R, 1N3R, 2N3R, 0N4R, 1N4R and 2N4R tau species (Buée et al., 2000). It is expected that the isoforms have various specific functions, but the primary function of tau is to promote the assembly of microtubules (Buée et al., 2000). Although tau isoforms are expressed differentially among neuronal subpopulations, tau is not expressed in astrocytes under physiological conditions (Muller et al., 1997).
Neurons are the primary cell type affected by FTLD-TAU, but astrocytes have been identified as a contributor of disease pathology. There is evidence indicating that astrocytic degeneration is associated with the degree of tissue atrophy in the frontal and temporal lobes in FTLD (Broe et al., 2004). FTLD-TAU accounts for 36%–50% of all FTLD cases with Pick’s disease, corticobasal degeneration, and progressive supranuclear palsy as the most common subtypes (Bang et al., 2015). Tufted astrocytes and astrocytic plaques (see Figure 1) are common pathologies in FTLD-TAU (Bang et al., 2015). When iPSC-derived astrocytes expressing the MAPTN279K variant were co-cultured with control neurons, there was a direct toxic effect decreasing neuronal survival during a rotenone-induced oxidative stress assay (Hallmann et al., 2017). Furthermore, these MAPTN279K FTD astrocytes displayed increased cell death and astrogliosis, defined by hypertrophy and increased glial fibrillary acidic protein (GFAP) levels (Hallmann et al., 2017). These results suggest that the MAPTN279K variant elicited a detrimental reactive astrocyte transformation that mediated an astrocyte cell-autonomous and non-cell autonomous neuron pathology. It is important to note that tau-related inclusions overlap with Alzheimer’s disease and enact similar astrocyte pathology, which is further discussed below.
The C9orf72 HRE variant is responsible for 30%–50% of familial ALS cases, 10% of sALS cases, and up to 50% of FTD cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Cooper-Knock et al., 2015). The discovery of this variant provided the strongest genetic evidence to date of the significant overlap between ALS and FTD. In addition to TDP-43 pathology (see below), there are three primary non-exclusive mechanisms hypothesized to cause C9orf72-ALS/FTD that necessitate the cooperation of both toxic loss and gain-of-functions. The repeat expansion suppresses the production of C9orf72 protein by inhibiting transcription, causing haploinsufficiency and loss-of function of the C9orf72 protein (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The toxic gain-of-function from sense and antisense C9orf72 repeat RNA, generated by bidirectional transcription, form sense and antisense HRE-RNA foci (DeJesus-Hernandez et al., 2011; Donnelly et al., 2013). The secondary structures of the repeat RNA, G-quadruplexes and DNA/RNA hairpins, bind and sequester RNA-binding proteins and cause DNA damage, respectively (Haeusler et al., 2014). Protein products of the repeat RNA, dipeptide repeat proteins (DPRs), are generated via repeat-associated non-ATG (RAN) translation forming five species: poly-GA, poly-GP, poly-GR, poly-PA and poly-PR (Ash et al., 2013; Tran et al., 2015). DPRs are aggregation prone and are thought to have some level of pathogenic effect. However, DPR pathology does not coincide with TDP-43 pathology and neurodegeneration observed in these patients. Nucleocytoplasmic trafficking deficits have also been associated with C9orf72 pathogenesis (Zhang et al., 2015; Boeynaems et al., 2016; Moore et al., 2020). These pathological features of the disease have been observed in spinal cord, motor, and frontal cortex neurons of C9orf72-ALS/FTD patient postmortem tissue (Murray et al., 2011; Mackenzie et al., 2015; DeJesus-Hernandez et al., 2017).
In C9orf72-ALS, few studies have described a cell-autonomous astrocyte pathology and an astrocyte-mediated non-cell autonomous motor neuron pathology (Madill et al., 2017; Allen et al., 2019; Birger et al., 2019; Taga et al., 2019; Varcianna et al., 2019; Zhao et al., 2020). One study demonstrated that C9orf72-ALS human induced pluripotent stem cell (iPSC)-derived spinal cord astrocytes develop RNA foci and DPR pathology (Zhao et al., 2020). DPRs have led to mis-splicing of the SLC1A2 gene causing dysfunction of EAAT2 (Lin et al., 1998; Kwon et al., 2014). Consequently, DPR pathology in astrocytes reduces their ability to uptake glutamate from their surroundings, leading to glutamate excitotoxicity in motor neurons already made vulnerable by C9orf72 haploinsufficiency (Shi et al., 2018). In co-culture, C9orf72-ALS astrocytes with confirmed RNA foci and poly-GP presence, triggered adverse effects on the electrophysiological properties of motor neurons (Zhao et al., 2020). Additionally, it has been found that DPRs can be transmitted from neurons to astrocytes through both contact dependent and independent mechanisms (Westergard et al., 2016). The transmission of different DPR species leads to nuclear and cytoplasmic aggregates in astrocytes co-cultured with DPR transfected neurons (Westergard et al., 2016).
Furthermore, when exposed to C9orf72-ALS astrocyte derived extracellular vesicles (EVs) and conditioned media, motor neurons experienced a 50%–70% increase in death compared to control astrocyte EVs and conditioned media (Varcianna et al., 2019). In this same study, C9orf72-ALS astrocytes were observed to secrete fewer EVs compared to controls (Varcianna et al., 2019). This contrasts studies describing astrocytes expressing SOD1 variants having an increase in EV secretion, therefore suggesting a characteristic difference between SOD1 and C9orf72 ALS astrocytes (Basso et al., 2013; Varcianna et al., 2019). Together, these studies indicate a role for C9orf72 astrocytes in the neurodegeneration of motor neurons observed in C9orf72-ALS/FTD; however, there is little to no information on the role of astrocytes in the neurodegeneration of cortical neurons leading to dementia symptoms in this disease spectrum.
The hallmark pathology of approximately 97% of ALS cases and 50% of FTD cases is the simultaneous nuclear depletion and cytoplasmic accumulation of TDP-43 (TAR-DNA binding protein 43), encoded by the TARDBP gene (Arai et al., 2006; Neumann et al., 2006). TDP-43 is an RNA and DNA binding protein, serving multiple functions in the regulation of gene expression at both the transcription and translation levels (Ratti and Buratti, 2016). Similar to other heterogeneous nuclear ribonucleoproteins (hnRNPs), TDP-43 consists of an N-terminus region followed by two RNA recognition motifs (RRMs) that bind RNA and DNA, and a prion-like domain containing C-terminus that aids in its ability to participate in protein-protein binding, phase separation and aggregation. Predominantly a nuclear protein, TDP-43 can shuttle to and from the cytoplasm via active and passive transport under physiological conditions. However, the aberrant nuclear depletion and cytoplasmic mislocalization of TDP-43 causes two toxic mechanisms—a toxic loss-of-function in the nucleus and toxic gain-of-function in the cytoplasm that work in conjunction (Tziortzouda et al., 2021). The resulting pathology in affected neurons and glia are cytoplasmic inclusions containing TDP-43, phosphorylated TDP-43 (pTDP-43), and cleaved C-terminal fragments. This pathology primarily occurs in neurons in the spinal cord, frontal, temporal and primary cortices, as well as subcortical regions, such as the hippocampus, basal ganglia, amygdala, thalamus and regions within the brainstem (Arai et al., 2006; Neumann et al., 2006; Murray et al., 2011; Mackenzie et al., 2015). TDP-43 inclusions are found in the cytoplasm and radiating processes of astrocytes (see Figure 1), but are more common in oligodendrocytes in the spinal cord and some cortical regions (Nishihira et al., 2008; Cooper-Knock et al., 2012; Heo et al., 2022).
The achievement of TDP-43 pathology in vitro and in vivo models most often requires either biological stressors or overexpression of specific TDP-43 variants. Astrocytic models require TDP-43 (full length, phosphorylated or the C-terminal fragment) to be selectively overexpressed or seeded into astrocytes (Serio et al., 2013; Hebron et al., 2014; Heyburn et al., 2016; Peng et al., 2020; Velebit et al., 2020). In a recent study investigating TDP-43 proteinopathy in sALS, hiPSC-derived motor neurons and astrocytes were exposed to seeded pathological TDP-43 aggregates from serially passaged sALS postmortem tissue (Smethurst et al., 2020). Through this seeding model, iPSC-motor neurons were observed to readily transfer TDP-43 aggregates to iPSC-astrocytes and vice versa. However, TDP-43 aggregates in iPSC-astrocytes were cleared more readily and there were less aggregates transferred from astrocytes to motor neurons (Smethurst et al., 2020). This study suggests a cell-type specific vulnerability and protein clearance of TDP-43, whereby astrocytes are less vulnerable and more efficient at clearing TDP-43 than motor neurons.
Another study, using a conditional TDP-43 knockdown mouse model to investigate the effects of nuclear depletion of TDP-43 in spinal cord astrocytes, revealed enhanced GFAP immunoreactivity and a molecular signature resembling that of the “A1-astrocytes” identified in Liddelow et al. (2017) (Peng et al., 2020). These TDP-43 depleted astrocyte mice presented with motor deficits, upregulation of C1 complement expression in microglia, and reduction of mature oligodendrocytes without directly affecting motor neuron survival (Peng et al., 2020). This indicates that nuclear loss-of-function of TDP-43 in affected astrocytes has both a cell autonomous effect and a non-cell autonomous effect on other glial types. Furthermore, a study that transfected primary rat cortical astrocytes with the C-terminal fragment of TDP-43, found that affected cells developed cytoplasmic TDP-43 inclusions and nuclear depletion of endogenous TDP-43 (Velebit et al., 2020). This is consistent with findings in neurons and cell lines where expression of the C-terminal fragment affects endogenous TDP-43 nucleocytoplasmic trafficking (Geser et al., 2008; Winton et al., 2008). Transfected astrocytes with TDP-43 cytoplasmic inclusions also presented with a stress state that involved an increase in lipid droplets, signifying an impaired lipid metabolism and disruptions in ß-adrenergic mediated aerobic glycolysis and lactate production (Velebit et al., 2020). This study further supports the idea that astrocytes affected by TDP-43 pathology in ALS and FTD may undergo a reactive transformation that disables an astrocyte’s ability to metabolically support surrounding neurons.
Given the dominant role of TDP-43 in ALS pathogenesis, ALS-linked TDP-43 variants have also been studied in astrocytes. In iPSC-derived astrocytes, carrying the TDP-43M337V variant, cells displayed cytoplasmic mislocalization of TDP-43, but not simultaneous nuclear depletion (Serio et al., 2013). Motor neurons with the same TDP-43M337V variant presented with high levels of insoluble TDP-43 (Bilican et al., 2012), whereas astrocytes did not (Serio et al., 2013). Notably, this suggests a cell-type specific function of TDP-43 between neurons and astrocytes. In vivo, the same TDP-43M337V variant expressed only in rat astrocytes led to the progressive loss of spinal motor neurons, skeletal muscle atrophy, and eventual paralysis (Tong et al., 2013). However, a separate study where the TDP-43A315T variant was overexpressed in astrocytes, found no motor neuron death in either an in vitro or transplant mouse model (Haidet-Phillips et al., 2013). The lack of consensus among these studies regarding whether astrocytes harboring a TDP-43 disease related-variant can cause motor neuron death may be due to variant-specific astrocyte-associated TDP-43 dysfunction. Additionally, variations in protein processing responses between cell types and models could also account for these differences.
Another RNA binding protein commonly associated with both ALS and FTD is the fused in sarcoma (FUS) protein. This predominantly nuclear protein also has a large role in regulating RNA metabolism events (Lagier-Tourenne and Cleveland, 2009; Dormann et al., 2010). Similar to TDP-43 pathology, FUS pathology is characterized by a simultaneous nuclear depletion and cytoplasmic mislocalization (Deng et al., 2010; Mackenzie et al., 2011). However, ALS and FTD cases with FUS inclusions do not present with TDP-43 inclusions; vice versa for cases with TDP-43 inclusions (Ling et al., 2013). ALS and FTD related variants in FUS lead to a dysfunctional protein. The ubiquitous knock down and conditional knock out of FUS in mice does not lead to ALS related motor deficits, but rather behavioral impairments (Kino et al., 2015; Udagawa et al., 2015). FUS inclusions are observed in neurons and glia in human and model tissue (see Figure 1), with a specific set of variants causing wide-spread FUS pathology triggering cell death (Kwiatkowski et al., 2009; Blair et al., 2010; Hewitt et al., 2010; Suzuki et al., 2012). Primary astrocytes transduced with FUSR521G variant, presented with cytoplasmic mislocalization in 90% of the cultured cells and a substantial increase in GFAP protein expression (Kia et al., 2018). Additionally, motor neuron death occurred in cultures treated with conditioned media from the FUSR521G astrocytes (Kia et al., 2018). This study indicated that the presence of FUSR521G in astrocytes was not detrimental to astrocytes, rather it caused a toxic phenotype that triggered motor neuron death through secreted factors like TNFα (Kia et al., 2018).
The RNA binding protein adenosine deaminase acting on RNA 2 (ADAR2), was recently found to also undergo nucleocytoplasmic mislocalization (Moore et al., 2019). ADAR2 is a member of the ADAR family whose primary role is to deaminate adenosine residues, specifically in double-stranded RNA (Melcher et al., 1995; O'Connell et al., 1995). This catalytic process converts a single adenosine to an inosine resulting in A-to-I editing (Melcher et al., 1995; Hwang et al., 2016). In C9orf72-ALS/FTD human tissue from the motor cortex and spinal cord, ADAR2 was found to mislocalize and form cytoplasmic accumulations, and co-localize with TDP-43 inclusions in the spinal cord (Moore et al., 2019). Nucleocytoplasmic mislocalization of ADAR2 was also observed in C9orf72-ALS/FTD hiPSC-motor neurons and in an AAV9-(G4C2)149-transduced WT mouse model (Moore et al., 2019). Substantial RNA editing aberrations associated with A-to-I editing were identified in spinal cord, cerebellum, motor and frontal cortices from C9orf72-ALS/FTD patients (Moore et al., 2019). Although a rigorous characterization of ADAR2 mislocalization and its transcriptional consequences in motor neurons were performed, there is no evidence yet supporting the mislocalization of ADAR2 in astrocytes or any other glial cells.
Collectively, these studies suggest astrocytes have a critical involvement in the degeneration of neurons through the loss of supportive and/or gain of toxic functions in various genetic subtypes of ALS and FTD. It is important to note that a C3+ reactive astrocyte transformation has been observed in C9orf72, SOD1 and sALS human tissue samples in the spinal cord and motor cortex, indicating a neuroinflammatory astrogliosis as a common pathology in ALS (Guttenplan et al., 2020). This is further supported by several studies utilizing RNA-sequencing data from ALS mouse models and iPSC-derived astrocytes, indicating that ALS astrocytes undergo a cell autonomous reactive transformation and pro-inflammatory profile in early disease stages (Ziff et al., 2021; Taha et al., 2022). However, the notion that astrocytes are directly the cause of motor neuron death is unlikely, instead it seems that diseased astrocytes exacerbate the vulnerable condition of diseased neurons. Whether this aggravation is due to a compensatory mechanism initiated by a loss of homeostatic functions and/or a toxic reactivity state is unclear. What is clear is that astrocytes do develop similar protein accumulations to neurons such as TDP-43 cytoplasmic inclusions, SOD1 nuclear accumulations, and DPRs. Nevertheless, the incidence of these protein accumulations in astrocytes is reduced compared to neurons, which suggests astrocytic clearance is either more efficient or not completely affected by underlying disease mechanisms (Yoshiyama et al., 2003; Forman et al., 2005; Serio et al., 2013; Westergard et al., 2016; Kia et al., 2018; Shi et al., 2018; Wang and Ye, 2021).
Alzheimer’s disease (AD) is the most common neurodegenerative disease-causing dementia. The onset of disease for a majority of patients is after the age of 65 (late-onset AD; LOAD), whereas a smaller population of patients will experience clinical symptoms before the age of 65 (early onset AD; EOAD) (Long and Holtzman, 2019). Pathogenesis of disease can be inherited and caused by specific genetic variants in genes APP, PSEN1 and PSEN2 (St George-Hyslop et al., 1987; Schellenberg et al., 1992; Goate et al., 1991; Sherrington et al., 1995), and with higher risk of disease development in carriers of the apolipoprotein E (APOE) ε4 allele (Pericak-Vance et al., 1988; Corder et al., 1993; Strittmatter et al., 1993). These are usually associated with faster progressing patients compared to sporadic AD cases. AD patients suffer from a set of heterogenous clinical and preclinical symptoms such as deficits in learning and memory, attention, executive function, language, visuospatial function, and behavioral impairments (Jack et al., 2018). The hallmark pathologies of AD are the extracellular deposits of amyloid-ß that form diffuse and neuritic plaques, and the intracytoplasmic neurofibrillary tangles (NFTs) that consist of aggregated hyperphosphorylated tau protein. Additionally, TDP-43 pathology, similar to what is observed in ALS and FTD, has also been found to occur in various brain regions of AD patients. There is substantial evidence indicating a localized role of reactive astrocytes in AD pathogenesis (Liddelow et al., 2017). Whereby these cells undergo both gains and losses of functions that are dependent on their given subtype and regional pathogenic load (Sadick et al., 2022). Here, we discuss AD-associated amyloid-ß plaques and neurofibrillary tau tangles and their proximity and distribution within astrocytes. Then, we will discuss AD-associated TDP-43 pathology and potential astrocytic occurrence, briefly review how it compares to FTD-tau related pathology, and incidence with other AD proteinopathies.
Amyloid-ß (Aß) is the product of the proteolytic cleavage of the amyloid precursor protein (APP). Cleaved sequentially by ß-secretase and γ-secretase, the Aß protein ranges from 40–42 amino acids in length (Hardy and Allsop, 1991). Identification of genetic variants in the APP, PSEN1 and PSEN2 genes associated with familial AD, were found to favor the longer Aß42 peptide and enhance its production (Chartier-Harlin et al., 1991; Goate et al., 1991; Murrell et al., 1991; Citron et al., 1992; Hendriks et al., 1992; Mullan et al., 1992; Cai et al., 1993; Scheuner et al., 1996). These findings resulted in the conception of the controversial “Amyloid Cascade Hypothesis” that asserts the imbalance of Aß42 production and clearance is the cause of AD (Beyreuther and Masters, 1991; Hardy and Allsop, 1991; Hardy and Higgins, 1992). The pathological distribution of Aß begins in pre-clinical stages, where deposits can be found in neocortical areas of high metabolic demand, then extending into the allocortex and brainstem whereby patients have transitioned into clinical stages (Thal et al., 2000; Buckner et al., 2005). However, failures in therapeutics against Aß have resulted in a refocus from improving the clearance of Aß plaques to also improving the clearance of NFTs and other misfolded aggregate prone proteins, and alleviating neuroinflammation—all of which are believed to contribute to neuronal death and the clinical manifestations of AD.
Surrounding Aß plaques are activated microglia and astrocytes (see Figure 1) (Pike et al., 1995; Bates et al., 2002). Astrocytes have been shown to be important mediators of amyloid-ß clearance (Thal et al., 2000). Specifically, astrocytes can internalize Aß1-42 in vitro, with aggregate size and conformation limiting astrocytic Aß uptake (Nielsen et al., 2010). In a 2010 study, primary adult human astrocytes from the temporal lobe were shown to internalize smaller size Aß1-42 oligomers more efficiently than fibrillar Aß1-42 species (Nielsen et al., 2010). Furthermore, amyloid associated proteins, like ApoE, significantly altered the astrocytic uptake of oligomeric Aß, suggesting a mechanism that could ultimately effect clearance and deposition at different disease stages (Nielsen et al., 2010). Reactive astrocytes found in postmortem AD tissue of the entorhinal cortex have a significant Aß load (Nagele et al., 2003), however astrocytes with large phagocytic burden become inefficient at degradation of cell debris (Loov et al., 2012). One study found that Aß accumulation in astrocytes resulted in enlarged endosomes, where engulfed Aß42 was stored instead of being actively degraded (Söllvander et al., 2016). These intracellular Aß42 accumulations further caused endosomal/lysosomal defects, which led to microvesicle-induced apoptosis of neurons (Söllvander et al., 2016).
A recent study aimed at characterizing activation states within AD pathogenesis used single-nuclei RNA-sequencing to identify differences in astrocyte states in the hippocampus of the 5xFAD transgenic AD mouse model (Habib et al., 2020). This mouse model expresses five familial AD-linked variants in the human APP and PS1 transgenes, causing severe extracellular amyloid deposition, synaptic and cortical neuron loss in layer V and the subiculum, and impaired spatial and working memory deficits (Oakley et al., 2006). A continuous range of astrocyte profiles were identified and divided into six transcriptional sub-clusters. Both AD and WT groups had clusters ranging from a homeostatic Gfap-low state, to an intermediate state, into a Gfap-high state (Habib et al., 2020). The AD group had an additional Gfap-high cluster that was designated as the diseased-associated astrocyte (DAA) cluster and an intermediate cluster between the Gfap-high and DAA clusters, described as a transitional DAA state. The DAAs were associated with different molecular pathways involved in amyloid accumulation, metabolism and clearance, and the proteolytic processing of APP. The authors determined that the DAAs were only present in the hippocampus of AD mice using co-staining of GFAP+SERPINA3N+VIM+ astrocytes. DAAs were also found to be adjacent to Aß plaques, appearing before the onset of cognitive decline and continuing to develop in parallel with disease progression in the AD model. The DAAs were found to develop from homeostatic Gfap-cells in WT mice, who acquired these with age, suggesting a similar phenotypic DAA-like stage that also occurs in normal aging. In human postmortem tissue, DAA-like cells were identified with higher frequency in AD patients along a spectrum with the intermediate transitional DAA cluster, which suggests a dynamic activation process in AD. This study supports the notion that astrocytes take on pathogenic changes with a role in early stages of AD disease pathogenesis.
In the pathological context of AD, the 3R and 4R ratio of tau isoforms is disturbed, leading to aggregation and NFT formation. Tau pathology begins in the entorhinal cortex, spreading to limbic regions and then finally to the neocortex (Braak and Braak, 1991; Braak et al., 2011). Because tau is not expressed in astrocytes (Muller et al., 1997), the source of tau in astrocytic accumulations in AD is unknown. Nevertheless, a recent study described tau accumulation in hilar astrocytes (see Figure 1) of the dentate gyrus in patients (Richetin et al., 2020). The presence of 3R tau+ inclusions were more prevalent in astrocytes in the hippocampus and dentate gyrus compared to controls, with incidence increasing with disease state (Richetin et al., 2020). The occurrence of 3R tau+ inclusions were exacerbated when hyperphosphorylated tau and Aß plaques were observed, which correlated with synaptic alterations. Astrocytic 4R tau+ inclusions were present, but only in cases without hyperphosphorylated tau and Aß plaques. Furthermore, when 1N3R tau was overexpressed in astrocytes in vivo, these cells experienced impaired mitochondrial motility and function, and decreased adult neurogenesis and inhibitory synapses, ultimately contributing to impaired spatial memory in the animals (Richetin et al., 2020). In a recent study examining the transmission of tau aggregates released by neurons and its effects on receptive astrocytes, it was observed that filamentous tau aggregates bind to the integrin receptor complex ITG⍺V/ß1 located on immune-panned primary mouse astrocytes (Wang and Ye, 2021). It was observed that this integrin complex facilitated the uptake of tau fibrils while also regulating tau fibril-induced inflammation and conversion to a reactive astrocyte state similar to that of an “A1 astrocyte” (Wang and Ye, 2021). The authors proposed that astrocytic integrin receptors act as a sensing mechanism for extracellular misfolded proteins that help with clearance. However, when overburdened by these extracellular misfolded proteins astrocytes then shift towards an inflammatory reactivity state. Providing further evidence that the overexpression or presence of tau in astrocytes may lead to disruptions in their trafficking network of cellular cargo leading to degeneration of not only neurons, but also astrocytes themselves (Yoshiyama et al., 2003; Forman et al., 2005). The role of astrocytes in tauopathies, other than AD, has been extensively reviewed in Kovacs (2020).
In 57% of AD cases there is evidence of TDP-43 pathology similar to what is observed in ALS and FTD (McAleese et al., 2017). Recently updated staging schemes describe TDP-43 pathology occurring first within limbic brain regions such as the amygdala, hippocampal formation and entorhinal cortex, with early distribution in the occipitotemporal cortex and ventral striatum (stages 1–3), and finally extending into neocortical areas such as the mid-temporal and mid-frontal cortex, as well as the basal ganglia (stages 4 and 5/6) (Josephs et al., 2016; Nag et al., 2018). TDP-43 inclusions are predominantly found in neurons, but there are reports of glial cytoplasmic inclusions (GCI) of TDP-43 in AD tissue (see Figure 1). However, GCIs are found only in transferrin-positive oligodendrocytes (Higashi et al., 2007).
A recent study examined whether TDP-43 in AD represents a coexistence of AD and FTD-TDP or if the two TDP-43 pathologies are discrete. They identified distinct molecular patterns of TDP-43 species in AD human tissue of hippocampus, entorhinal, frontal, temporal and occipital cortex, amygdala (Tome et al., 2020). They found a range of TDP-43 inclusion patterns from the absence of inclusions to inclusions only positive for pTDP-43 (serine 409/410), and inclusions positive for pTDP-43 (serines 403/404 and 409/410) and full length TDP-43 (N and C-terminal domains), similar to that of FTLD-TDP. About 20% of AD cases with only pTDP-43 inclusions, presented with language deficits, and about 36% of AD cases positive for both pTDP-43 and full length TDP-43 experienced FTD-like behavioral or language problems. This further supported the hypothesis that distinct phosphorylation sites have an impact on the type of TDP-43 pathology and its relation to FTD-like symptoms observed in AD. However, glial TDP-43 inclusions were not individually assessed, but were considered in the classification of samples.
Limbic-predominant age-associated TDP-43 encephalopathy-related neuropathological changes (LATE-NC), is a newly defined TDP-43 related proteinopathy with inclusions primarily in limbic regions of the brain (Nelson et al., 2019). LATE-NC affects up to 50% of individuals past the age of 80 years old and individuals with or without AD (Nelson et al., 2019). LATE-NC clinically manifests with amnestic cognitive syndrome affecting emotional lability and global cognitive function, which is independent of co-existing pathologies (Nelson et al., 2019). TDP-43 proteinopathy extends from the amygdala to the hippocampus, then to the middle frontal gyrus; this distribution can occur with or without hippocampal sclerosis (Nelson et al., 2019). Individuals with LOAD with TDP-43 pathology in limbic regions are now recognized as having simultaneous LATE-NC. A diagnosis of ALS and/or FTLD-TDP excludes a diagnosis of LATE-NC. Unlike ALS and FTD, there are rare mentions of glial TDP-43 inclusions associated with LATE, however there is no clarification on glial cell type (Hunter et al., 2020).
Finally, there are a handful of studies implicating TDP-43 interactions with Aß and tau. Studies investigating the relationship between Aß and TDP-43 pathology in AD have found that Aß42 expression in vivo upregulated endogenous TDP-43 and caused cytosolic pTDP-43 inclusions (Herman et al., 2011; Herman et al., 2012). Using an AD mouse harboring APP and PS1 variants (APP/PS1 model), the injection of TDP-43 oligomers increased Aß plaques and further impaired spatial memory (Shih et al., 2020). The relationship between tau and TDP-43 has indicated that TDP-43 has a role in assisting with the MAPT mRNA splicing leading to a decrease of 4R isoforms by exclusion of exon 10 (Gu et al., 2017). Furthermore, ALS/FTD related TDP-43 variants led to inclusion of exon 10, resulting in an increase and disruption of the 3R:4R ratio (Gu et al., 2017). Specificity of phosphorylation sites on TDP-43 dictate its ability to suppress exon 10 in MAPT mRNA (Gu et al., 2018). Additionally, TDP-43 and tau have been shown to have a collaborative seeding process in the brain whereby TDP-43 oligomers could serve as a nucleation site for tau aggregates (Montalbano et al., 2020). However, these interactions have not been identified or studied in astrocytes or any other glia.
The role of astrocytes in AD is ultimately important to maintain the integrity of neurons; however, there is evidence that AD pathology has an early and eventual impact on astrocytic integrity caused by a reactive transformation. Both Aß and tau pathology have been observed in relation to astrocytes in both models of AD and AD human tissue. In a recent study investigating how the characteristic AD proteinopathies influence astrocytes, TRAP-seq data from mouse models of tauopathy and ß-amyloidopathy revealed that neither of these proteinopathies triggered “A1” or “A2” reactivity profiles in these mice, but rather model specific deleterious and adaptive-protective profiles (Jiwaji et al., 2022). Specifically, these pathologies induced activation of inflammatory pathways, alongside protein degradation pathways, all the while exhibiting an induction of a set of genes normally elevated in aged mouse brains (Jiwaji et al., 2022). Thus, furthering the argument that astrocyte reactivity is context and disease dependent. TDP-43 pathology has also been observed in glia, but the identification of TDP-43 in an AD context in astrocytes has yet to be properly described. There is much more research needed in describing AD-related pathology in astrocytes and identifying astrocyte specific perturbations that occur due to the accruement of Aß, tau, and TDP-43 pathologies.
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by the expansion of the CAG repeat in the huntingtin (HTT) gene, resulting in a poly-glutamine tract in the translated protein. HD onset commonly occurs around 45 years of age, with aggressive progression of involuntary choreiform movements (repetitive, rapid, jerky movements); in addition, patients can experience behavioral, psychiatric, and dementia like symptoms (Ghosh and Tabrizi, 2015). However, there are cases where motor symptom onset can begin in childhood (Ghosh and Tabrizi, 2015). Patients generally have 36 or more CAG repeat units, with longer CAG expansions associated with earlier disease onset (Andrew et al., 1993). Much of the neurodegeneration in HD occurs in striatal medium spiny neurons (MSNs), as these are more vulnerable to the HTT expansion variant’s cellular disruptions. However, there is also substantial cellular disruption and death in the cortex, accompanied by progressive reactive astrogliosis (Ghosh and Tabrizi, 2015). HD neuropathology is measured on a graded scale of 0–4 that is dependent on the degree of striatal atrophy and astrocyte reactivity (Rüb et al., 2015; Khakh et al., 2017). The major pathological hallmark of HD are large inclusions of expanded HTT fibrils in the nucleus and cytoplasm of cells (see Figure 1) (DiFiglia et al., 1997; Cooper et al., 1998). Additionally, there have been occurrences of a co-pathology with TDP-43 inclusions in neurons (Schwab et al., 2008). The large expanded HTT inclusions are not only found in neurons, but also at comparable levels in cortical and striatal astrocytes of HD patients and models (Shin et al., 2005; Faideau et al., 2010; Tong et al., 2014; Jansen et al., 2017). Expanded HTT in astrocytes occurs in grade 0 of HD pathogenesis, suggesting that astrocytes may contribute to disease progression (Faideau et al., 2010). The physiological function of the huntingtin protein is still relatively undetermined. Information about the protein sequence, posttranslational modifications and regional expression patterns implicate the protein in CNS developmental processes, cell survival, vesicle transport, and synaptic maintenance (De Souza and Leavitt, 2015).
Evidence from human and HD murine model tissue display expanded HTT accumulations in striatal astrocytes, indicative of an astrocytic role in HD pathogenesis (Shin et al., 2005; Faideau et al., 2010; Tong et al., 2014). It has been observed that astrocytic expression of expanded HTT has the ability to reduce the expression of both Glt-1, the murine homolog of the astrocyte specific glutamate receptor EAAT2, and Kir4.1, a potassium ion channel that aids in extracellular K+ buffering (Lievens et al., 2001; Behrens et al., 2002; Shin et al., 2005; Faideau et al., 2010; Tong et al., 2014; Jiang et al., 2016). This reduction allows for an increase in extracellular glutamate and K+, increasing the excitability of MSNs and leaving them susceptible to excitotoxicity. In HD affected regions, astrocytes exhibit a reactive state defined by an increase in GFAP expression (Selkoe et al., 1982). When expanded HTT is expressed selectively in astrocytes, these cells undergo morphological changes and hypertrophy with these changes being greater in the putamen compared to the caudate nucleus (Faideau et al., 2010), suggestive of regional differences of astrocyte HD pathology. When striatal astrocytes expressing expanded HTT were co-cultured with striatal neurons there was an increase in neuron death (Aizawa et al., 2016). However, when expanded HTT is expressed only in cortical neurons in vivo, these mice do not develop neuropathology (Gu et al., 2005).
Two HD mice models are widely used, the transgenic R6/2, a model of aggressive disease progression mimicking juvenile HD (Mangiarini et al., 1996), and the knock-in Q175, a model with less aggressive progression mimicking adult-onset HD (Menalled et al., 2003). The amount of nuclear expanded HTT inclusions in astrocytes increases as both models develop symptoms and pathology (Tong et al., 2014). The astrocytes in the striatum have electrophysiological defects, such as depolarized membrane potentials and lower membrane conductance, attributed to reduced expression of Kir4.1 (Tong et al., 2014). By increasing astrocytic Kir4.1 the motor deficits and longevity of both models improved, and even more interestingly the expression of the glutamate transporter Glt-1 increased (Tong et al., 2014).
In a recent study examining the molecular signatures of astrocytes in HD, again the two HD mice models, R6/2 and Q175, were used in conjunction with human HD postmortem tissue to assess astrocyte specific gene and protein expression (Diaz-Castro et al., 2019). There were a core set of gene expression changes in both HD models and human tissue samples that confirmed altered functions in Ca2+ signaling, G-protein coupled receptors (GPCRs), and neurotransmitter regulation. The mice were assessed at three different ages, and the functions of the common dysregulated genes were similar at early and late stages. To further support that data, astrocyte reactivity was identified in human tissue samples from patients at grade 3 and altered astrocyte gene expression was observed as early as grade 0. This is indicative of early astrocyte dysfunction in HD that persists throughout disease progression. Additionally, there was little evidence indicating an “A1 neurotoxic” astrocyte signature, further supporting the idea that astrocyte reactivity is context specific.
To explore the notion of context-specific and region-specific astrocyte differences, 14 experimental perturbations relevant to the contribution of striatal astrocytes in HD were selected and executed in the R6/2 model (Yu et al., 2020). At the level of genes, pathways and upstream transcriptional signaling regulators, the molecular responses of striatal astrocytes were found to be context dependent. With this data, the authors identified that activation of GPCR signaling in striatal astrocytes could correct HD related astrocyte alterations that effect Ca2+ signaling and astrocyte territory size, in addition to an enhancement of neuroimmune responses. Striatal astrocyte GPCR activation corrected behavioral deficits in the R6/2 model, effecting anxiety-related behaviors and motor function. Interestingly, there was also an observed increased expression of thrombospondin 1, a synaptogenic astrocyte-secreted protein important for the formation of synapses.
Together these studies suggest the disruption of essential functions of astrocytes and astrocyte-neuron interactions having a large role in HD pathogenesis. A recent study employing single-nucleus RNA-sequencing of cingulate cortex from grade III/IV HD patients found three distinct reactivity states among the astrocyte cluster (Al-Dalahmah et al., 2020). These reactivity states ranged from low, intermediate and high GFAP expression, while all had decreased expression of glutamate transporters SLC1A2 and SLC1A3 (Al-Dalahmah et al., 2020). The authors report HTT aggregates in a small population of astrocytes in the cingulate cortex from cases used for the snRNA-sequencing, and further suggest that reactive transformation of the astrocytes is secondary to neuronal loss, but exacerbated by the reactive nature of the surrounding astrocytes (Al-Dalahmah et al., 2020). Further contributing to the evidence of astrocytic expanded HTT inclusions and a reactive astrocyte transformation in HD, ultimately exacerbating disease pathogenesis.
Parkinson’s disease (PD) is the second-most common age-related neurodegenerative disease after AD (Wright Willis et al., 2010). Most PD cases are idiopathic, but there are various monogenic mutations that have been linked with disease development. The hallmark pathology of PD is the degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the depletion of dopamine levels in the striatum (Kordower et al., 2013). In addition to the loss of DA neurons, there are intracellular accumulations of misfolded proteins known as Lewy bodies. These inclusions consist of α-synuclein and other associated proteins such as phosphorylated tau and amyloid-ß (Maiti et al., 2017). Diagnosis of PD is based on the presence of at least two of the following clinical symptoms: resting tremor, bradykinesia, muscle rigidity, and postural imbalance or impaired gait (de Lau and Breteler, 2006; Dickson, 2012; Maiti et al., 2017). Additionally, asymmetric symptom onset and good response to levodopa further supports a parkinsonian diagnosis (de Lau and Breteler, 2006). In about half of PD patients there are also executive function deficits mediated by the fronto-striatal pathways, such as attention, mental processing speed, and impairments in language, working memory and impulsivity (Dickson, 2012; Maiti et al., 2017). Here we will discuss what is known about the distribution of α-synuclein inclusions in astrocytes and their effects on astrocyte function. Then we will briefly discuss monogenic mutations that cause parkinsonian phenotypes and their effects on astrocyte function.
The role of astrocytes in PD is still unclear, however there is substantial evidence implicating these cells in disease progression. The gene SNCA, encoding the protein α-synuclein is lowly expressed in astrocytes compared to neurons. However, there are studies showing α-synuclein+ inclusions in astrocytes of PD patient tissue (Wakabayashi et al., 2000; Braak et al., 2007). Specifically in sporadic PD cases, these α-synuclein+ astrocytes are found within the SN, midbrain, amygdala, thalamus, striatum and cerebral cortex (Wakabayashi et al., 2000; Braak et al., 2007). In early stages of disease pathogenesis, these α-synuclein+ astrocytes are primarily found in the amygdala, striatum, and thalamic nuclei projecting to the cortex (Braak et al., 2007). In the cortex, these astrocytic inclusions are observed in layers V and VI, with limited prevalence in layers III and IV (Braak et al., 2007). Previous studies suggest that the α-synuclein found in astrocytes are possibly transferred from neurons (see Figure 1) and may be an exogenous pro-inflammatory activator of astrocytes (Lee et al., 2010; Fellner et al., 2013; Rannikko et al., 2015).
In a 2010 study by Lee et al., they observed that SH-SY5Y derived α-synuclein was endocytosed by rodent astrocytes and further concentrated in the astrocytic lysosomes. This direct transmission of “neuronal” α-synuclein to astrocytes also caused an astrocytic inflammatory response and build-up of glial cytoplasmic inclusions (Lee et al., 2010). Further exploring the effect of α-synuclein on astrocytes, a recent study tested different α-synuclein species and how they would induce an astrocytic inflammatory response (Russ et al., 2021). iPSC-derived astrocytes from healthy individuals were treated with TNFα or various α-synuclein species (monomers, oligomers, high molecular weight fibrils). Astrocytes treated with TNFα experienced a pro-inflammatory phenotype characterized by an increase in cytokine release, nuclear localization of NF-κB and altered mitochondrial function, whereas α-synuclein treatment did not. Instead α-synuclein treated astrocytes took on an antigen-presenting phenotype, similar to oligodendrocyte precursor cells in an in vitro model of multiple sclerosis (Kirby et al., 2019). Furthermore, only α-synuclein fibrils caused alterations in mitochondrial function, similar to that of TNFα treatment (Russ et al., 2021). Treatment of PD iPSC-astrocytes with α-synuclein resulted in an exacerbated pro-inflammatory response with an increase in secretion of pro-inflammatory cytokines. Co-treatment of TNFα and α-synuclein resulted in a decrease in the effects resulting from TNFα or α-synuclein treatment alone. This suggested to the authors that the astrocytic responses to α-synuclein can be positively altered in the presence of pro-inflammatory cytokines, but negatively altered by degradation resistant α-synuclein aggregate species.
It has been further hypothesized that an increase in α-synuclein concentration can overload the astrocytic lysosomal system, causing α-synuclein accumulations and aggregates that may lead to further deficits in physiological astrocyte function. In a recent study, astrocytes derived from embryonic stem cells were shown to engulf large amounts of α-synuclein (Rostami et al., 2017). These large oligomers were observed to be stored in the trans-golgi region that led to defects in the phagosomal-lysosomal machinery within the astrocytes. Ultimately these astrocytes produced tunneling nanotubes that allowed physical connection to neighboring cells, mediating α-synuclein cell-cell transfer. Additionally, there are reports of deficits in astrocytic glutamate uptake resulting in excitotoxicity similar to what is observed in ALS, which has been thoroughly reviewed (Hindeya Gebreyesus and Gebrehiwot Gebremichael, 2020).
A small percentage of PD cases are attributed to monogenic mutations in LRRK2, Parkin, PINK1 and increased risk of PD with GBA1 variants (Klein and Westenberger, 2012). The leucine-rich repeat kinase 2 is encoded by the LRRK2 gene and causes autosomal dominant PD through multiple pathogenic variants with varying penetrance (Roosen and Cookson, 2016; Tolosa et al., 2020). Depending on the mutation, LRRK2-PD can present differently neuropathologically. About 50% of individuals have nigral degeneration, and 65%–80% will present with typical Lewy body pathology (Kalia et al., 2015). Other pathological changes can include non-specific cell loss without signs of proteinopathy, and, interestingly, tau and TDP-43 pathology (Vilas et al., 2018). LRRK2 has been implicated in regulating the autophagy-lysosome pathway. In primary mouse astrocytes, PD-associated mutations within LRRK2 compromised lysosomal enzyme function by lowering lysosomal pH (Henry et al., 2015). Furthermore, mutant LRRK2 astrocytes exhibited lysosomes with an enlarged morphology and reduced the capacity of lysosomes in the cell, however these deficits were rescued upon selective inhibition of LRRK2 kinase activity (Henry et al., 2015). In addition to showing localization with lysosomes, LRRK2 has also shown an association with endosomes and other vesicular bodies (Roosen and Cookson, 2016). Furthermore, iPSC-astrocytes harboring LRRK2 or GBA mutations exhibited increased expression of α -synuclein caused a disruption in calcium signaling and altered metabolic activity (Sonninen et al., 2020). Whereas, primary PINK1-knock out mouse astrocytes exhibited mitochondrial dysfunction resulting in proliferation defects (Choi et al., 2013).
These studies are suggestive of a reactive astrocyte transformation in PD, whereby astrocytes not only suffer phagosomal-lysosomal defects, but contribute to disease pathogenesis through potential transfer of α-synuclein and pro-inflammatory processes. In addition, various PD-related mutations have a detrimental effect on homeostatic astrocyte functions within the CNS.
In summary, there is mounting evidence for the important role of astrocytes in disease pathogenesis of various neurodegenerative diseases. There is a critical loss of supportive astrocytic functions and in some contexts a toxic gain-of-function that cause astrocytes to not only affect the health of vulnerable neurons, but to develop a disease-specific astrocytic reactive phenotype. Overall, the presence of proteinopathies in astrocytes is significantly less well described and studied as compared to proteinopathies in a neuronal context, although there is evidence that in some diseases astrocytes not only develop disease-specific proteinopathies, but further propagate it (Lee et al., 2010; Fellner et al., 2013; Rannikko et al., 2015; Söllvander et al., 2016; Westergard et al., 2016; Smethurst et al., 2020; Wang and Ye, 2021). Therefore, it is unlikely that astrocytes are the direct cause of neurodegeneration, but rather active participants of disease. Moreover, these studies show that reactive transformations of astrocytes do have commonalities between diseases (e.g., pro-inflammatory activations, increased GFAP expression, trafficking and homeostasis disruptions). With more studies investigating transcriptional, biochemical, morphological, and metabolic variables, we are expected to find more defined disease and CNS region-specific astrocytic phenotypes. Thus, research on the role of astrocytes in specific disease contexts and regions of interest is essential to further understand the selective neurodegeneration and cell death leading to disease pathogenesis in these debilitating and fatal diseases.
LMB wrote the manuscript in consultation with RS. RS performed the critical revision of the paper.
This work was supported by the NIH/NINDS R21NS125861 (RS), 3R21NS125861-01S1 (RS) and the Barrow Neurological Foundation (RS and LMB).
The authors thank Lauren Gittings, Stephen Moore, Ileana Lorenzini and Ryan Pevey from the Department of Translational Neuroscience, Barrow Neurological Institute, and Joyce Bustos for feedback and comments during manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmmed.2023.1075805/full#supplementary-material
Aizawa, H., Hideyama, T., Yamashita, T., Kimura, T., Suzuki, N., Aoki, M., et al. (2016). Deficient RNA-editing enzyme ADAR2 in an amyotrophic lateral sclerosis patient with a FUS(P525L) mutation. J. Clin. Neurosci. 32, 128–129. doi:10.1016/j.jocn.2015.12.039
Al-Dalahmah, O., Sosunov, A. A., Shaik, A., Ofori, K., Liu, Y., Vonsattel, J. P., et al. (2020). Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 8, 19. doi:10.1186/s40478-020-0880-6
Allen, S. P., Hall, B., Woof, R., Francis, L., Gatto, N., Shaw, A. C., et al. (2019). C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142 (12), 3771–3790. doi:10.1093/brain/awz302
Andrew, S. E., Goldberg, Y. P., Kremer, B., Telenius, H., Theilmann, J., Adam, S., et al. (1993). The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat. Genet. 4 (4), 398–403. doi:10.1038/ng0893-398
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351 (3), 602–611. doi:10.1016/j.bbrc.2006.10.093
Arredondo, C., Cefaliello, C., Dyrda, A., Jury, N., Martinez, P., Diaz, I., et al. (2022). Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons. Neuron 110 (10), 1656–1670 e1612. doi:10.1016/j.neuron.2022.02.010
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77 (4), 639–646. doi:10.1016/j.neuron.2013.02.004
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14 (9), 544–558. doi:10.1038/s41582-018-0047-2
Bang, J., Spina, S., and Miller, B. L. (2015). Frontotemporal dementia. Lancet 386 (10004), 1672–1682. doi:10.1016/S0140-6736(15)00461-4
Basso, M., Pozzi, S., Tortarolo, M., Fiordaliso, F., Bisighini, C., Pasetto, L., et al. (2013). Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 288 (22), 15699–15711. doi:10.1074/jbc.M112.425066
Bates, K. A., Fonte, J., Robertson, T. A., Martins, R. N., and Harvey, A. R. (2002). Chronic gliosis triggers Alzheimer’s disease-like processing of amyloid precursor protein. Neuroscience 113 (4), 785–796. doi:10.1016/s0306-4522(02)00230-0
Batiuk, M. Y., Martirosyan, A., Wahis, J., de Vin, F., Marneffe, C., Kusserow, C., et al. (2020). Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 11 (1), 1220. doi:10.1038/s41467-019-14198-8
Behrens, P. F., Franz, P., Woodman, B., Lindenberg, K. S., and Landwehrmeyer, G. B. (2002). Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125, 1908–1922. doi:10.1093/brain/awf180
Beyreuther, K., and Masters, C. L. (1991). Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of alzheimer's disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1 (4), 241–251. doi:10.1111/j.1750-3639.1991.tb00667.x
Bilican, B., Serio, A., Barmada, S. J., Nishimura, A. L., Sullivan, G. J., Carrasco, M., et al. (2012). Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. U. S. A. 109 (15), 5803–5808. doi:10.1073/pnas.1202922109
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50, 274–289. doi:10.1016/j.ebiom.2019.11.026
Blair, I. P., Williams, K. L., Warraich, S. T., Durnall, J. C., Thoeng, A. D., Manavis, J., et al. (2010). FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 81 (6), 639–645. doi:10.1136/jnnp.2009.194399
Boeynaems, S., Bogaert, E., Michiels, E., Gijselinck, I., Sieben, A., Jovicic, A., et al. (2016). Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6, 20877. doi:10.1038/srep20877
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82 (4), 239–259. doi:10.1007/BF00308809
Braak, H., Sastre, M., and Del Tredici, K. (2007). Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol. 114 (3), 231–241. doi:10.1007/s00401-007-0244-3
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70 (11), 960–969. doi:10.1097/NEN.0b013e318232a379
Broe, M., Kril, J., and Halliday, G. M. (2004). Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain 127, 2214–2220. doi:10.1093/brain/awh250
Buckner, R. L., Snyder, A. Z., Shannon, B. J., LaRossa, G., Sachs, R., Fotenos, A. F., et al. (2005). Molecular, structural, and functional characterization of alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 25 (34), 7709–7717. doi:10.1523/JNEUROSCI.2177-05.2005
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 33 (1), 95–130. doi:10.1016/s0165-0173(00)00019-9
Cai, X. D., Golde, T. E., and Younkin, S. G. (1993). Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science 259 (5094), 514–516. doi:10.1126/science.8424174
Chartier-Harlin, M. C., Crawford, F., Houlden, H., Warren, A., Hughes, D., Fidani, L., et al. (1991). Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353 (6347), 844–846. doi:10.1038/353844a0
Choi, I., Kim, J., Jeong, H. K., Kim, B., Jou, I., Park, S. M., et al. (2013). PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia 61 (5), 800–812. doi:10.1002/glia.22475
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature 360 (6405), 672–674. doi:10.1038/360672a0
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Munch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U. S. A. 115 (8), E1896–E1905. doi:10.1073/pnas.1800165115
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillée, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302 (5642), 113–117. doi:10.1126/science.1086071
Cooper, J. K., Schilling, G., Peters, M. F., Herring, W. J., Sharp, A. H., Kaminsky, Z., et al. (1998). Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 7 (5), 783–790. doi:10.1093/hmg/7.5.783
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764. doi:10.1093/brain/awr365
Cooper-Knock, J., Kirby, J., Highley, R., and Shaw, P. J. (2015). The spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics 12 (2), 326–339. doi:10.1007/s13311-015-0342-1
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261 (5123), 921–923. doi:10.1126/science.8346443
de Lau, L. M. L., and Breteler, M. M. B. (2006). Epidemiology of Parkinson's disease. Lancet Neurology 5 (6), 525–535. doi:10.1016/s1474-4422(06)70471-9
De Souza, R. A., and Leavitt, B. R. (2015). Neurobiology of huntington's disease. Curr. Top. Behav. Neurosci. 22, 81–100. doi:10.1007/7854_2014_353
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72 (2), 245–256. doi:10.1016/j.neuron.2011.09.011
DeJesus-Hernandez, M., Finch, N. C. A., Wang, X., Gendron, T. F., Bieniek, K. F., Heckman, M. G., et al. (2017). In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. 134 (2), 255–269. doi:10.1007/s00401-017-1725-7
Deng, H. X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., et al. (2010). FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67 (6), 739–748. doi:10.1002/ana.22051
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10 (5), 608–614. doi:10.1038/nn1885
Diaz-Castro, B., Gangwani, M. R., Yu, X., Coppola, G., and Khakh, B. S. (2019). Astrocyte molecular signatures in Huntington's disease. Sci. Transl. Med. 11 (514), eaaw8546. doi:10.1126/scitranslmed.aaw8546
Dickson, D. W. (2012). Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2 (8), a009258. doi:10.1101/cshperspect.a009258
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277 (5334), 1990–1993. doi:10.1126/science.277.5334.1990
Donnelly, C. J., Zhang, P. W., Pham, J. T., Heusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80 (2), 415–428. doi:10.1016/j.neuron.2013.10.015
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29 (16), 2841–2857. doi:10.1038/emboj.2010.143
Escartin, C., Galea, E., Lakatos, A., O'Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24 (3), 312–325. doi:10.1038/s41593-020-00783-4
Faideau, M., Kim, J., Cormier, K., Gilmore, R., Welch, M., Auregan, G., et al. (2010). In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with huntington's disease subjects. Hum. Mol. Genet. 19 (15), 3053–3067. doi:10.1093/hmg/ddq212
Faulkner, J. R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B., and Sofroniew, M. V. (2004). Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24 (9), 2143–2155. doi:10.1523/JNEUROSCI.3547-03.2004
Fellner, L., Irschick, R., Schanda, K., Reindl, M., Klimaschewski, L., Poewe, W., et al. (2013). Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 61 (3), 349–360. doi:10.1002/glia.22437
Forman, M. S., Lal, D., Zhang, B., Dabir, D. V., Swanson, E., Lee, V. M., et al. (2005). Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J. Neurosci. 25 (14), 3539–3550. doi:10.1523/JNEUROSCI.0081-05.2005
Forsberg, K., Andersen, P. M., Marklund, S. L., and Brannstrom, T. (2011). Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 121 (5), 623–634. doi:10.1007/s00401-011-0805-3
Geser, F., Brandmeir, N. J., Kwong, L. K., Martinez-Lage, M., Elman, L., McCluskey, L., et al. (2008). Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol. 65 (5), 636–641. doi:10.1001/archneur.65.5.636
Ghosh, R., and Tabrizi, S. J. (2015). Clinical aspects of huntington's disease. Curr. Top. Behav. Neurosci. 22, 3–31. doi:10.1007/7854_2013_238
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349 (6311), 704–706. doi:10.1038/349704a0
Gu, X., Li, C., Wei, W., Lo, V., Gong, S., Li, S. H., et al. (2005). Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46 (3), 433–444. doi:10.1016/j.neuron.2005.03.025
Gu, J., Chen, F., Iqbal, K., Gong, C. X., Wang, X., and Liu, F. (2017). Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J. Biol. Chem. 292 (25), 10600–10612. doi:10.1074/jbc.M117.783498
Gu, J., Wang, W., Miao, S., Chen, F., Wu, F., Hu, W., et al. (2018). Protein Phosphatase 1 dephosphorylates TDP-43 and suppresses its function in tau exon 10 inclusion. FEBS Lett. 592 (3), 402–410. doi:10.1002/1873-3468.12976
Guttenplan, K. A., Weigel, M. K., Adler, D. I., Couthouis, J., Liddelow, S. A., Gitler, A. D., et al. (2020). Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 11 (1), 3753. doi:10.1038/s41467-020-17514-9
Guttenplan, K. A., Weigel, M. K., Prakash, P., Wijewardhane, P. R., Hasel, P., Rufen-Blanchette, U., et al. (2021). Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 599 (7883), 102–107. doi:10.1038/s41586-021-03960-y
Habib, N., McCabe, C., Medina, S., Varshavsky, M., Kitsberg, D., Dvir-Szternfeld, R., et al. (2020). Disease-associated astrocytes in Alzheimer's disease and aging. Nat. Neurosci. 23 (6), 701–706. doi:10.1038/s41593-020-0624-8
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A., Shaw, P. G., Kim, M. S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507 (7491), 195–200. doi:10.1038/nature13124
Haidet-Phillips, A. M., Gross, S. K., Williams, T., Tuteja, A., Sherman, A., Ko, M., et al. (2013). Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp. Neurol. 250, 250–259. doi:10.1016/j.expneurol.2013.10.004
Hallmann, A. L., Arauzo-Bravo, M. J., Mavrommatis, L., Ehrlich, M., Ropke, A., Brockhaus, J., et al. (2017). Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 7, 42991. doi:10.1038/srep42991
Hardiman, O., Al-chalabi, A., Chio, A., Corr, E. M., Robberecht, W., Shaw, P. J., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. 3, 17071–17119. doi:10.1038/nrdp.2017.71
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388. doi:10.1016/0165-6147(91)90609-v
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: The amyloid cascade hypothesis. Science 256 (5054), 184–185. doi:10.1126/science.1566067
Hebron, M., Chen, W., Miessau, M. J., Lonskaya, I., and Moussa, C. E. (2014). Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J. Neurochem. 129 (2), 350–361. doi:10.1111/jnc.12630
Hendriks, L., van Duijn, C. M., Cras, P., Cruts, M., Van Hul, W., van Harskamp, F., et al. (1992). Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat. Genet. 1 (3), 218–221. doi:10.1038/ng0692-218
Henry, A. G., Aghamohammadzadeh, S., Samaroo, H., Chen, Y., Mou, K., Needle, E., et al. (2015). Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 24 (21), 6013–6028. doi:10.1093/hmg/ddv314
Heo, D., Ling, J. P., Molina-Castro, G. C., Langseth, A. J., Waisman, A., Nave, K. A., et al. (2022). Stage-specific control of oligodendrocyte survival and morphogenesis by TDP-43. Elife 11, e75230. doi:10.7554/eLife.75230
Herman, A. M., Khandelwal, P. J., Stanczyk, B. B., Rebeck, G. W., and Moussa, C. E. (2011). β-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 1386, 191–199. doi:10.1016/j.brainres.2011.02.052
Herman, A. M., Khandelwal, P. J., Rebeck, G. W., and Moussa, C. E. (2012). Wild type TDP-43 induces neuro-inflammation and alters APP metabolism in lentiviral gene transfer models. Exp. Neurol. 235 (1), 297–305. doi:10.1016/j.expneurol.2012.02.011
Herrmann, J. E., Imura, T., Song, B., Qi, J., Ao, Y., Nguyen, T. K., et al. (2008). STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 28 (28), 7231–7243. doi:10.1523/JNEUROSCI.1709-08.2008
Hewitt, C., Kirby, J., Highley, J. R., Hartley, J. A., Hibberd, R., Hollinger, H. C., et al. (2010). Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 67 (4), 455–461. doi:10.1001/archneurol.2010.52
Heyburn, L., Hebron, M. L., Smith, J., Winston, C., Bechara, J., Li, Z., et al. (2016). Tyrosine kinase inhibition reverses TDP-43 effects on synaptic protein expression, astrocytic function and amino acid dis-homeostasis. J. Neurochem. 139 (4), 610–623. doi:10.1111/jnc.13763
Higashi, S., Iseki, E., Yamamoto, R., Minegishi, M., Hino, H., Fujisawa, K., et al. (2007). Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 1184, 284–294. doi:10.1016/j.brainres.2007.09.048
Hindeya Gebreyesus, H., and Gebrehiwot Gebremichael, T. (2020). The potential role of astrocytes in Parkinson's disease (PD). Med. Sci. (Basel) 8 (1), 7. doi:10.3390/medsci8010007
Huai, J., and Zhang, Z. (2019). Structural properties and interaction partners of familial ALS-Associated SOD1 mutants. Front. Neurol. 10, 527. doi:10.3389/fneur.2019.00527
Hunter, S., Hokkanen, S. R. K., Keage, H. A. D., Fleming, J., Minett, T., Polvikoski, T., et al. (2020). TDP-43 related neuropathologies and phosphorylation state: Associations with age and clinical dementia in the cambridge city over-75s cohort. J. Alzheimers Dis. 75 (1), 337–350. doi:10.3233/JAD-191093
Hwang, T., Park, C. K., Leung, A. K., Gao, Y., Hyde, T. M., Kleinman, J. E., et al. (2016). Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 19 (8), 1093–1099. doi:10.1038/nn.4337
Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 14 (4), 535–562. doi:10.1016/j.jalz.2018.02.018
Jansen, A. H., van Hal, M., Op den Kelder, I. C., Meier, R. T., de Ruiter, A. A., Schut, M. H., et al. (2017). Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 65 (1), 50–61. doi:10.1002/glia.23050
Jiang, R., Diaz-Castro, B., Looger, L. L., and Khakh, B. S. (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from huntington's disease model mice. J. Neurosci. 36 (12), 3453–3470. doi:10.1523/JNEUROSCI.3693-15.2016
Jiwaji, Z., Tiwari, S. S., Avilés-Reyes, R. X., Hooley, M., Hampton, D., Torvell, M., et al. (2022). Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 10 (1), 135. Springer US. doi:10.1038/s41467-021-27702-w
Josephs, K. A., Murray, M. E., Whitwell, J. L., Tosakulwong, N., Weigand, S. D., Petrucelli, L., et al. (2016). Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol. 131 (4), 571–585. doi:10.1007/s00401-016-1537-1
Kalia, L. V., Lang, A. E., Hazrati, L. N., Fujioka, S., Wszolek, Z. K., Dickson, D. W., et al. (2015). Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 72 (1), 100–105. doi:10.1001/jamaneurol.2014.2704
Kettenmann, H., and Verkhratsky, A. (2008). Neuroglia: The 150 years after. Trends Neurosci. 31 (12), 653–659. doi:10.1016/j.tins.2008.09.003
Khakh, B. S., and Deneen, B. (2019). The emerging nature of astrocyte diversity. Annu. Rev. Neurosci. 42, 187–207. doi:10.1146/annurev-neuro-070918-050443
Khakh, B. S., Beaumont, V., Cachope, R., Munoz-Sanjuan, I., Goldman, S. A., and Grantyn, R. (2017). Unravelling and exploiting astrocyte dysfunction in huntington's disease. Trends Neurosci. 40 (7), 422–437. doi:10.1016/j.tins.2017.05.002
Kia, A., McAvoy, K., Krishnamurthy, K., Trotti, D., and Pasinelli, P. (2018). Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 66 (5), 1016–1033. doi:10.1002/glia.23298
Kino, Y., Washizu, C., Kurosawa, M., Yamada, M., Miyazaki, H., Akagi, T., et al. (2015). FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 3, 24. doi:10.1186/s40478-015-0202-6
Kirby, L., Jin, J., Cardona, J. G., Smith, M. D., Martin, K. A., Wang, J., et al. (2019). Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 10 (1), 3887. doi:10.1038/s41467-019-11638-3
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson's disease. Cold Spring Harb. Perspect. Med. 2 (1), a008888. doi:10.1101/cshperspect.a008888
Kordower, J. H., Olanow, C. W., Dodiya, H. B., Chu, Y., Beach, T. G., Adler, C. H., et al. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136, 2419–2431. doi:10.1093/brain/awt192
Kovacs, G. G. (2020). Astroglia and tau: New perspectives. Front. Aging Neurosci. 12, 96. doi:10.3389/fnagi.2020.00096
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323 (5918), 1205–1208. doi:10.1126/science.1166066
Kwon, I., Xiang, S., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., et al. (2014). Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345 (6201), 1139–1145. doi:10.1126/science.1254917
Lagier-Tourenne, C., and Cleveland, D. W. (2009). Rethinking ALS: The FUS about TDP-43. Cell 136 (6), 1001–1004. doi:10.1016/j.cell.2009.03.006
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285 (12), 9262–9272. doi:10.1074/jbc.M109.081125
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541 (7638), 481–487. doi:10.1038/nature21029
Lievens, J. C., Woodman, B., Mahal, A., Spasic-Boscovic, O., Samuel, D., Kerkerian-Le Goff, L., et al. (2001). Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol. Dis. 8 (5), 807–821. doi:10.1006/nbdi.2001.0430
Lin, C.-L. G., Bristol, L. A., Jin, L., Dykes-Hoberg, M., Crawford, T., Clawson, L., et al. (1998). Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20 (3), 589–602. doi:10.1016/s0896-6273(00)80997-6
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79 (3), 416–438. doi:10.1016/j.neuron.2013.07.033
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179 (2), 312–339. doi:10.1016/j.cell.2019.09.001
Loov, C., Hillered, L., Ebendal, T., and Erlandsson, A. (2012). Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS One 7 (3), e33090. doi:10.1371/journal.pone.0033090
Mackenzie, I. R. A., and Neumann, M. (2016). Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J. Neurochem. 138, 54–70. doi:10.1111/jnc.13588
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122 (1), 111–113. doi:10.1007/s00401-011-0845-8
Mackenzie, I. R. A., Frick, P., Grässer, F. A., Gendron, T. F., Petrucelli, L., Cashman, N. R., et al. (2015). Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 130 (6), 845–861. doi:10.1007/s00401-015-1476-2
Madill, M., McDonagh, K., Ma, J., Vajda, A., McLoughlin, P., O'Brien, T., et al. (2017). Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 10 (1), 22. doi:10.1186/s13041-017-0300-4
Maiti, P., Manna, J., and Dunbar, G. L. (2017). Current understanding of the molecular mechanisms in Parkinson's disease: Targets for potential treatments. Transl. Neurodegener. 6, 28. doi:10.1186/s40035-017-0099-z
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87 (3), 493–506. doi:10.1016/s0092-8674(00)81369-0
McAleese, K. E., Walker, L., Erskine, D., Thomas, A. J., McKeith, I. G., and Attems, J. (2017). TDP-43 pathology in Alzheimer's disease, dementia with Lewy bodies and ageing. Brain Pathol. 27 (4), 472–479. doi:10.1111/bpa.12424
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 13, 1310. doi:10.3389/fnins.2019.01310
Melcher, T., Maas, S., Higuchi, M., Keller, W., and Seeburg, P. H. (1995). Editing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR-B pre-mRNA in vitro reveals site-selective adenosine to inosine conversion. J. Biol. Chem. 270 (15), 8566–8570. doi:10.1074/jbc.270.15.8566
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S., and Chesselet, M. F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J. Comp. Neurol. 465 (1), 11–26. doi:10.1002/cne.10776
Molofsky, A. V., and Deneen, B. (2015). Astrocyte development: A guide for the perplexed. Glia 63 (8), 1320–1329. doi:10.1002/glia.22836
Montalbano, M., McAllen, S., Cascio, F. L., Sengupta, U., Garcia, S., Bhatt, N., et al. (2020). TDP-43 and tau oligomers in alzheimer's disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol. Dis. 146, 105130. doi:10.1016/j.nbd.2020.105130
Moore, S., Alsop, E., Lorenzini, I., Starr, A., Rabichow, B. E., Mendez, E., et al. (2019). ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol. 138 (1), 49–65. doi:10.1007/s00401-019-01999-w
Moore, S., Rabichow, B. E., and Sattler, R. (2020). The hitchhiker's guide to nucleocytoplasmic trafficking in neurodegeneration. Neurochem. Res. 45 (6), 1306–1327. doi:10.1007/s11064-020-02989-1
Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B., et al. (1992). A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1 (5), 345–347. doi:10.1038/ng0892-345
Muller, R., Heinrich, M., Heck, S., Blohm, D., and Richter-Landsberg, C. (1997). Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 288 (2), 239–249. doi:10.1007/s004410050809
Murray, M. E., DeJesus-Hernandez, M., Rutherford, N. J., Baker, M., Duara, R., Graff-Radford, N. R., et al. (2011). Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 122 (6), 673–690. doi:10.1007/s00401-011-0907-y
Murrell, J., Farlow, M., Ghetti, B., and Benson, M. D. (1991). A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 254 (5028), 97–99. doi:10.1126/science.1925564
Nag, S., Yu, L., Boyle, P. A., Leurgans, S. E., Bennett, D. A., and Schneider, J. A. (2018). TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer's disease. Acta Neuropathol. Commun. 6 (1), 33. doi:10.1186/s40478-018-0531-3
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10 (5), 615–622. doi:10.1038/nn1876
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., and Wang, H.-Y. (2003). Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971 (2), 197–209. doi:10.1016/s0006-8993(03)02361-8
Nelson, P. T., Dickson, D. W., Trojanowski, J. Q., Jack, C. R., Boyle, P. A., Arfanakis, K., et al. (2019). Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142 (6), 1503–1527. doi:10.1093/brain/awz099
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 (5796), 130–133. doi:10.1126/science.1134108
Nielsen, H. M., Mulder, S. D., Belien, J. A., Musters, R. J., Eikelenboom, P., and Veerhuis, R. (2010). Astrocytic A beta 1-42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins. Glia 58 (10), 1235–1246. doi:10.1002/glia.21004
Nishihira, Y., Tan, C. F., Onodera, O., Toyoshima, Y., Yamada, M., Morita, T., et al. (2008). Sporadic amyotrophic lateral sclerosis: Two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 116 (2), 169–182. doi:10.1007/s00401-008-0385-z
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26 (40), 10129–10140. doi:10.1523/JNEUROSCI.1202-06.2006
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29 (10), 3276–3287. doi:10.1523/JNEUROSCI.4707-08.2009
O'Connell, M. A., Krause, S., Higuchi, M., Hsuan, J. J., Totty, N. F., Jenny, A., et al. (1995). Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol. Cell Biol. 15 (3), 1389–1397. doi:10.1128/MCB.15.3.1389
Okada, S., Nakamura, M., Katoh, H., Miyao, T., Shimazaki, T., Ishii, K., et al. (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 12 (7), 829–834. doi:10.1038/nm1425
Peng, A. Y. T., Agrawal, I., Ho, W. Y., Yen, Y. C., Pinter, A. J., Liu, J., et al. (2020). Loss of TDP-43 in astrocytes leads to motor deficits by triggering A1-like reactive phenotype and triglial dysfunction. Proc. Natl. Acad. Sci. U. S. A. 117 (46), 29101–29112. doi:10.1073/pnas.2007806117
Pericak-Vance, M. A., Yamaoka, L. H., Haynes, C. S., Speer, M. C., Haines, J. L., Gaskell, P. C., et al. (1988). Genetic linkage studies in Alzheimer's disease families. Exp. Neurol. 102 (3), 271–279. doi:10.1016/0014-4886(88)90220-8
Peteri, U. K., Niukkanen, M., and Castren, M. L. (2019). Astrocytes in neuropathologies affecting the frontal cortex. Front. Cell Neurosci. 13, 44. doi:10.3389/fncel.2019.00044
Pike, C. J., Cummings, B. J., and Cotman, C. W. (1995). Early association of reactive astrocytes with senile plaques in Alzheimer's disease. Exp. Neurol. 132 (2), 172–179. doi:10.1016/0014-4886(95)90022-5
Pottier, C., Ravenscroft, T. A., Sanchez-Contreras, M., and Rademakers, R. (2016). Genetics of FTLD: overview and what else we can expect from genetic studies. J. Neurochem. 138, 32–53. doi:10.1111/jnc.13622
Rannikko, E. H., Weber, S. S., and Kahle, P. J. (2015). Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 16, 57. doi:10.1186/s12868-015-0192-0
Ratti, A., and Buratti, E. (2016). Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 138, 95–111. doi:10.1111/jnc.13625
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72 (2), 257–268. doi:10.1016/j.neuron.2011.09.010
Renton, A. E., Chio, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17 (1), 17–23. doi:10.1038/nn.3584
Richetin, K., Steullet, P., Pachoud, M., Perbet, R., Parietti, E., Maheswaran, M., et al. (2020). Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer's disease. Nat. Neurosci. 23 (12), 1567–1579. doi:10.1038/s41593-020-00728-x
Roosen, D. A., and Cookson, M. R. (2016). LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 11 (1), 73. doi:10.1186/s13024-016-0140-1
Rostami, J., Holmqvist, S., Lindstrom, V., Sigvardson, J., Westermark, G. T., Ingelsson, M., et al. (2017). Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 37 (49), 11835–11853. doi:10.1523/JNEUROSCI.0983-17.2017
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326 (22), 1464–1468. doi:10.1056/NEJM199205283262204
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16 (3), 675–686. doi:10.1016/s0896-6273(00)80086-0
Rüb, U., Vonsattel, J. P. G., Heinsen, H., and Korf, H.-W. (2015). “The neuropathological grading of Huntington’s disease (HD),” in The neuropathology of Huntington’s disease: Classical findings, recent developments and correlation to functional neuroanatomy, 7–23.
Russ, K., Teku, G., Bousset, L., Redeker, V., Piel, S., Savchenko, E., et al. (2021). TNF-alpha and alpha-synuclein fibrils differently regulate human astrocyte immune reactivity and impair mitochondrial respiration. Cell Rep. 34 (12), 108895. doi:10.1016/j.celrep.2021.108895
Sadick, J. S., O'Dea, M. R., Hasel, P., Dykstra, T., Faustin, A., and Liddelow, S. A. (2022). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer's disease. Neuron 110 (11), 1788–1805.e10. Elsevier Inc. doi:10.1016/j.neuron.2022.03.008
Schellenberg, G. D., Bird, T. D., Wijsman, E. M., Orr, H. T., Anderson, L., Nemens, E., et al. (1992). Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 258 (5082), 668–671. doi:10.1126/science.1411576
Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., et al. (1996). Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 2 (8), 864–870. doi:10.1038/nm0896-864
Schwab, C., Arai, T., Hasegawa, M., Yu, S., and McGeer, P. L. (2008). Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 67 (12), 1159–1165. doi:10.1097/NEN.0b013e31818e8951
Selkoe, D. J., Salazar, F. J., Abraham, C., and Kosik, K. S. (1982). Huntington's disease: changes in striatal proteins reflect astrocytic gliosis. Brain Res. 245 (1), 117–125. doi:10.1016/0006-8993(82)90344-4
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U. S. A. 110 (12), 4697–4702. doi:10.1073/pnas.1300398110
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375 (6534), 754–760. doi:10.1038/375754a0
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24 (3), 313–325. doi:10.1038/nm.4490
Shih, Y. H., Tu, L. H., Chang, T. Y., Ganesan, K., Chang, W. W., Chang, P. S., et al. (2020). TDP-43 interacts with amyloid-beta, inhibits fibrillization, and worsens pathology in a model of Alzheimer's disease. Nat. Commun. 11 (1), 5950. doi:10.1038/s41467-020-19786-7
Shin, J. Y., Fang, Z. H., Yu, Z. X., Wang, C. E., Li, S. H., and Li, X. J. (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 171 (6), 1001–1012. doi:10.1083/jcb.200508072
Smethurst, P., Risse, E., Tyzack, G. E., Mitchell, J. S., Taha, D. M., Chen, Y. R., et al. (2020). Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 143 (2), 430–440. doi:10.1093/brain/awz419
Söllvander, S., Nikitidou, E., Brolin, R., Söderberg, L., Sehlin, D., Lannfelt, L., et al. (2016). Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 11 (1), 38. doi:10.1186/s13024-016-0098-z
Sonninen, T. M., Hamalainen, R. H., Koskuvi, M., Oksanen, M., Shakirzyanova, A., Wojciechowski, S., et al. (2020). Metabolic alterations in Parkinson's disease astrocytes. Sci. Rep. 10 (1), 14474. doi:10.1038/s41598-020-71329-8
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 90 (5), 1977–1981. doi:10.1073/pnas.90.5.1977
St George-Hyslop, P. H., Tanzi, R. E., Polinsky, R. J., Haines, J. L., Nee, L., Watkins, P. C., et al. (1987). The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 235 (4791), 885–890. doi:10.1126/science.2880399
Suzuki, N., Kato, S., Kato, M., Warita, H., Mizuno, H., Kato, M., et al. (2012). FUS/TLS-immunoreactive neuronal and glial cell inclusions increase with disease duration in familial amyotrophic lateral sclerosis with an R521C FUS/TLS mutation. J. Neuropathol. Exp. Neurol. 71 (9), 779–788. doi:10.1097/NEN.0b013e318264f164
Taga, A., Dastgheyb, R., Habela, C., Joseph, J., Richard, J. P., Gross, S. K., et al. (2019). Role of human-induced pluripotent stem cell-derived spinal cord astrocytes in the functional maturation of motor neurons in a multielectrode array system. Stem Cells Transl. Med. 8 (12), 1272–1285. doi:10.1002/sctm.19-0147
Taha, D. M., Clarke, B. E., Hall, C. E., Tyzack, G. E., Ziff, O. J., Greensmith, L., et al. (2022). Astrocytes display cell autonomous and diverse early reactive states in familial amyotrophic lateral sclerosis. Brain 145 (2), 481–489. doi:10.1093/brain/awab328
Thal, D. R., Schultz, C., Dehghani, F., Yamaguchi, H., Braak, H., and Braak, E. (2000). Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol. 100 (6), 608–617. doi:10.1007/s004010000242
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16 (2), 97–107. doi:10.1038/s41582-019-0301-2
Tome, S. O., Vandenberghe, R., Ospitalieri, S., Van Schoor, E., Tousseyn, T., Otto, M., et al. (2020). Distinct molecular patterns of TDP-43 pathology in alzheimer's disease: relationship with clinical phenotypes. Acta Neuropathol. Commun. 8 (1), 61. doi:10.1186/s40478-020-00934-5
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., Liu, X., et al. (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 32 (13), 1917–1926. doi:10.1038/emboj.2013.122
Tong, X., Ao, Y., Faas, G. C., Nwaobi, S. E., Xu, J., Haustein, M. D., et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat. Neurosci. 17 (5), 694–703. doi:10.1038/nn.3691
Tran, H., Almeida, S., Moore, J., Gendron, T. F., Chalasani, U., Lu, Y., et al. (2015). Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron 87 (6), 1207–1214. doi:10.1016/j.neuron.2015.09.015
Tziortzouda, P., Van Den Bosch, L., and Hirth, F. (2021). Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat. Rev. Neurosci. 22 (4), 197–208. doi:10.1038/s41583-021-00431-1
Udagawa, T., Fujioka, Y., Tanaka, M., Honda, D., Yokoi, S., Riku, Y., et al. (2015). FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 6, 7098. doi:10.1038/ncomms8098
Varcianna, A., Myszczynska, M. A., Castelli, L. M., O'Neill, B., Kim, Y., Talbot, J., et al. (2019). Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40, 626–635. doi:10.1016/j.ebiom.2018.11.067
Vasile, F., Dossi, E., and Rouach, N. (2017). Human astrocytes: structure and functions in the healthy brain. Brain Struct. Funct. 222 (5), 2017–2029. doi:10.1007/s00429-017-1383-5
Velebit, J., Horvat, A., Smolic, T., Prpar Mihevc, S., Rogelj, B., Zorec, R., et al. (2020). Astrocytes with TDP-43 inclusions exhibit reduced noradrenergic cAMP and Ca(2+) signaling and dysregulated cell metabolism. Sci. Rep. 10 (1), 6003. doi:10.1038/s41598-020-62864-5
Vilas, D., Sharp, M., Gelpi, E., Genis, D., Marder, K. S., Cortes, E., et al. (2018). Clinical and neuropathological features of progressive supranuclear palsy in Leucine rich repeat kinase (LRRK2) G2019S mutation carriers. Mov. Disord. 33 (2), 335–338. doi:10.1002/mds.27225
Wakabayashi, K., Hayashi, S., Yoshimoto, M., Kudo, H., and Takahashi, H. (2000). NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson's disease brains. Acta Neuropathol. 99 (1), 14–20. doi:10.1007/pl00007400
Wang, P., and Ye, Y. (2021). Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 12 (1), 95. Springer US. doi:10.1038/s41467-020-20322-w
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-Cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17 (3), 645–652. doi:10.1016/j.celrep.2016.09.032
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283 (19), 13302–13309. doi:10.1074/jbc.M800342200
Woollacott, I. O., and Rohrer, J. D. (2016). The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J. Neurochem. 138, 6–31. doi:10.1111/jnc.13654
Wright Willis, A., Evanoff, B. A., Lian, M., Criswell, S. R., and Racette, B. A. (2010). Geographic and ethnic variation in Parkinson disease: a population-based study of US medicare beneficiaries. Neuroepidemiology 34 (3), 143–151. doi:10.1159/000275491
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11 (3), 251–253. doi:10.1038/nn2047
Yoshiyama, Y., Zhang, B., Bruce, J., Trojanowski, J. Q., and Lee, V. M. Y. (2003). Reduction of detyrosinated microtubules and golgi fragmentation are linked to tau-induced degeneration in astrocytes. J. Neurosci. 23 (33), 10662–10671. doi:10.1523/jneurosci.23-33-10662.2003
Yu, X., Nagai, J., Marti-Solano, M., Soto, J. S., Coppola, G., Babu, M. M., et al. (2020). Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron 108 (6), 1146–1162.e10. doi:10.1016/j.neuron.2020.09.021
Yun, Y., and Ha, Y. (2020). CRISPR/Cas9-Mediated gene correction to understand ALS. Int. J. Mol. Sci. 21 (11), 3801. doi:10.3390/ijms21113801
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32 (18), 6391–6410. doi:10.1523/JNEUROSCI.6221-11.2012
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525 (7567), 56–61. doi:10.1038/nature14973
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89 (1), 37–53. doi:10.1016/j.neuron.2015.11.013
Zhao, C., Devlin, A. C., Chouhan, A. K., Selvaraj, B. T., Stavrou, M., Burr, K., et al. (2020). Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 68 (5), 1046–1064. doi:10.1002/glia.23761
Keywords: astrocytes, neurodegenaration, proteinopathies, ALS, FTD, Alzheimer's disease, Huntingtons’s disease, Parkinson’s disease
Citation: Bustos LM and Sattler R (2023) The Fault in Our Astrocytes - cause or casualties of proteinopathies of ALS/FTD and other neurodegenerative diseases?. Front. Mol. Med. 3:1075805. doi: 10.3389/fmmed.2023.1075805
Received: 20 October 2022; Accepted: 08 February 2023;
Published: 16 February 2023.
Edited by:
Marija Cvetanovic, University of Minnesota Twin Cities, United StatesReviewed by:
José Julio Rodríguez Arellano, University of the Basque Country, SpainCopyright © 2023 Bustos and Sattler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rita Sattler, Uml0YS5TYXR0bGVyQEJhcnJvd25ldXJvLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.