
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 20 March 2025
Sec. Molecular Diagnostics and Therapeutics
Volume 12 - 2025 | https://doi.org/10.3389/fmolb.2025.1547152
This article is part of the Research Topic Distinct phenotype but same genotype: Hints for the diversity of phenotypes in ciliopathies View all articles
 Mine Yuksel Kalyoncu1*†
Mine Yuksel Kalyoncu1*† Rim Hjeij2*†
Rim Hjeij2*† Muruvvet Yanaz3Aynur Gulieva3Merve Selcuk Balcı3Şeyda Karabulut3Neval Metin Cakar3†Almala Pınar Ergenekon3
Muruvvet Yanaz3Aynur Gulieva3Merve Selcuk Balcı3Şeyda Karabulut3Neval Metin Cakar3†Almala Pınar Ergenekon3 Ela Erdem Eralp3Yasemin Gokdemir3
Ela Erdem Eralp3Yasemin Gokdemir3 Heymut Omran2
Heymut Omran2 Bülent Taner Karadag3
Bülent Taner Karadag3Introduction: Primary ciliary dyskinesia (PCD) is an autosomal recessive rare disease caused by alterations in ciliary structure and function. Without a unique gold standard diagnostic test, the European Respiratory Society and the American Thoracic Society recommend using various diagnostic techniques to improve accuracy. This study aimed to demonstrate the effectiveness of immunofluorescence (IF) analysis in the diagnosis of PCD cases with uncertain genetic results and to demonstrate the importance of international collaboration in the diagnosis of PCD.
Methods: In collaboration with IF specialists at the University of Münster, individuals with inconclusive results in the Marmara University PCD panel consisting of the 22 most common genes and clinically suggestive of PCD were included in the study. IF imaging determined the subcellular localization of DNAH5 and GAS8 in respiratory epithelial cells. Nasal nitric oxide measurements, high-speed video microscopy (HSVM) analysis, and genetic analyses were performed.
Results: 19 patients were evaluated. The median age (25–75p) was 15 years (10–20 years) with 12 (63.2%) males. Three cases (15.7%) showed an absence of DNAH5, and one (5.3%) had a proximal distribution of DNAH5 in the ciliary axoneme. One case (5.3%) had cells without cilia, indicating a possible ciliogenesis defect. All individuals with abnormal IF analysis had a PICADAR score of 6 or above, and their cilia were immotile in HSVM.
Discussion: Consistent with the IF finding suggesting a ciliogenesis defect, further genetic analysis revealed biallelic pathogenic variants in CCNO in the affected individual. The absence of DNAH5 in the respiratory epithelial cells of an individual carrying heterozygous pathogenic splice variants in DNAH5 suggests the need for further genetic analysis. This study underscores the importance of international collaboration in diagnosing rare diseases like PCD.
Primary ciliary dyskinesia (PCD) (ORPHA:244) is a rare autosomal recessive genetic disorder characterized by abnormal movement of motile cilia (Frommer et al., 2015). It is a complex, heterogeneous, and multisystemic disease presenting with a range of clinical symptoms, including rhinosinusitis, middle ear disease, bronchiectasis (BE), and subfertility. Approximately half of the PCD-affected individuals exhibit situs abnormalities, and a subset of these individuals may also have associated congenital heart diseases or other organ abnormalities, particularly those with situs ambiguous or heterotaxy (Kennedy and Plant, 2014).
Diagnosing PCD can be challenging because there is no single gold standard test or method (O'Connor et al., 2021). The American Thoracic Society (ATS) and European Respiratory Society (ERS) recommend using a combination of diagnostic techniques to increase sensitivity and specificity (Shapiro et al., 2018; Lucas et al., 2017). Another barrier to accurate diagnosis is the complexity of the diagnostic methods, which require specialized equipment and trained staff (Leigh et al., 2011). Additionally, the lack of diagnostic devices in low-income countries further complicates the diagnosis process (Surdut et al., 2023).
In recent years, immunofluorescence (IF) staining has gained popularity as a diagnostic tool for PCD (Omran and Loges, 2009). This technique involves detecting ciliary proteins in respiratory cells using fluorescence or confocal microscopy and labeling them with antibodies targeting proteins such as outer (ODA) and inner (IDA) dynein arms, radial spokes (RS), dynein regulatory complex proteins (N-DRC), and central pair (CP) (Shapiro et al., 2018; Lucas et al., 2017). IF staining helps in recognizing the structure of the ciliary axoneme and has proven effective in confirming specific variants as pathogenic by identifying the location of protein compounds within the ciliary axoneme (Bhatt and Hogg, 2020). IF analysis is cheaper, faster, and more convenient than other ultrastructural analysis methods like transmission electron microscopy (TEM) (Shoemark et al., 2017). ERS Task Force experts agree that IF can be helpful in clinical settings (Lucas et al., 2017).
Our diagnostic approach for PCD primarily includes genetic analysis and nasal nitric oxide (NO) measurements. While nasal NO measurement is a valuable screening tool, its diagnostic utility is limited (Knowles et al., 2014; Raidt et al., 2022). Although the majority of PCD cases present with low nasal NO levels, certain genetic variants have been reported to exhibit normal or even elevated values (Knowles et al., 2014; Yiallouros et al., 2019; Collins et al., 2014). Consequently, this test cannot be relied upon as a conclusive diagnostic tool for PCD as it may not accurately identify all cases. To address this limitation, we introduced high-speed video microscopy (HSVM) in January 2023. A potential drawback of our diagnostic approach is that our PCD genetic panel comprises only 22 PCD-associated genes, making it challenging to diagnose patients with rare or novel variants. Consequently, we often require panels with more comprehensive genes or ultrastructural tests such as TEM and IF. Some patients exhibit highly suspicious clinical findings, but their genetic results show heterogeneous variations in PCD genes, necessitating ultrastructural tests like IF to confirm the presence of disease variants.
This study aims to diagnose individuals with highly suspicious clinical findings and unclear genetic results, emphasize the significance of IF analysis in diagnosing PCD, and highlight the benefits of collaborating with specialized centers in the diagnostic process.
Individuals with clinical symptoms and medical history suggestive of PCD and no pathological variants detected in PCD genetics performed using a targeted genetic panel, including the 22 most frequently identified PCD-associated genes (Table 1), were included in the study. Written informed consent was obtained from patients aged over 18 years and from the parents of patients under 18 years. Clinical and demographic data were collected from medical records. This study was approved by the Clinical Research Ethics Committee of Marmara University (Protocol No: 09.2023.111). Nasal NO measurements were conducted according to ERS recommendations using a CLD 88sp NO analyzer (ECO MEDICS, AG, Duerten, Switzerland) (Omran and Loges, 2009).
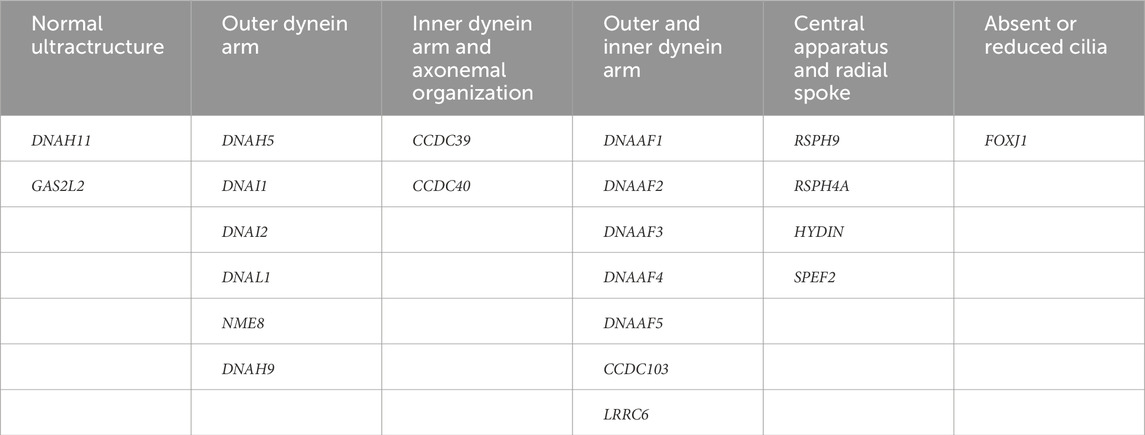
Table 1. Targeted genetic panel used for PCD in Marmara University.
To obtain respiratory epithelial cells, a transnasal brush biopsy (Cytobrush Plus; Medscand Medical, Malmö, Sweden) was performed (Omran and Loges, 2009). The cells were suspended in cell culture medium (RPMI), spread onto glass slides, and air-dried at the outpatient clinics of Marmara University. These samples were then sent to the IF laboratories at the University of Münster between November and December 2022. IF analysis was performed in a cohort of 19 PCD-suspected individuals and five healthy controls at the University of Münster. The cells were treated with 4% paraformaldehyde, 0.2% Triton X-100, and 1% skim milk before incubation with primary antibodies (3–4 h) and secondary antibodies (30 min) at room temperature (Omran and Loges, 2009). Individuals were analyzed using antibodies against component proteins for the ODAs (DNAH5) and the nexin-dynein regulatory complex (GAS8) of the ciliary axoneme (Omran and Loges, 2009). Monoclonal Mouse anti-DNAH5 and polyclonal rabbit anti-GAS8 (HPA041311) primary antibodies were used for double labeling at a 1:500 dilution (Omran and Loges, 2009). Mouse monoclonal anti-DNAH5 antibody was generated as previously described (Omran et al., 2008). Goat Anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 546 secondary antibodies were used as 1:1000 dilution. To visualize cell nuclei, DNA was stained with Hoechst 33342 (Sigma). High-resolution fluorescence images were were taken using a Zeiss Axiovert 200 equiped with ApoTome.2 using a PlanApo 63X/1.4NA oil objective. Images were taken using a AxionCam712 mono and processed with ZEN2 Blue software. Figures were prepared with Adobe Creative Suite 4 (Omran and Loges, 2009; Dougherty et al., 2016).
After IF staining was completed in Münster, HSVM analyses were performed as an additional diagnostic tool at Marmara University between January 2023 and March 2023. Nasal epithelial cells were obtained by using the nasal brushing technique from patients (Dougherty et al., 2016). Participants were recruited if they had not taken nasal steroids or decongestants for at least 4 weeks and had not developed symptoms of acute respiratory tract infection for at least 4 weeks. Ciliated cells placed in a pre-warmed culture medium (RPMI 1640-Medium) at a temperature of 37°C17. The cells were subsequently equilibrated to the optimal temperature of 37°C on a heater plate (Tpi-TSX, Tokai, Japan). The frequency of ciliary movement was measured using HSVM with the Sisson-Ammons Video Analysis software (SAVA, MI, United States). The measurements were taken with an inverted phase-contrast Nikon Eclipse TS100 microscope (Nikon, Japan) equipped with a ×40 objective and linked to a digital high-speed video camera (Basler acA1300-200um, Germany). The digital image sampling was set at 640 × 480 pixels and a frame rate of 120–150 frames per second (fps) for a duration of one minute, with intervals of 15 s. The ciliary beat was analyzed from both top and side views using real-time and slow-motion replay (Raidt et al., 2014). Ciliary beat patterns (CBP) were defined as “normal, virtually immotile, stiff beating with a reduced amplitude, circular gyrating motion” and ciliary beat frequency were measured (Raidt et al., 2014). The results obtained in HSVM could not be confirmed through air–liquid interface (ALI) culture as it is not available at our center.
Statistical analysis was conducted using IBM® SPSS® Statistics Version 20 software. For descriptive analysis, normally distributed data were expressed as mean ± SD, while non-normally distributed data were expressed as median [interquartile range (IQR)]. Statistical significance was set at p < 0.05.
Nineteen PCD-affected individuals were included in this study. The median age (25th-75th percentile) was 15 years (10–20 years) with 12 (63.2%) individuals being male (Table 2).
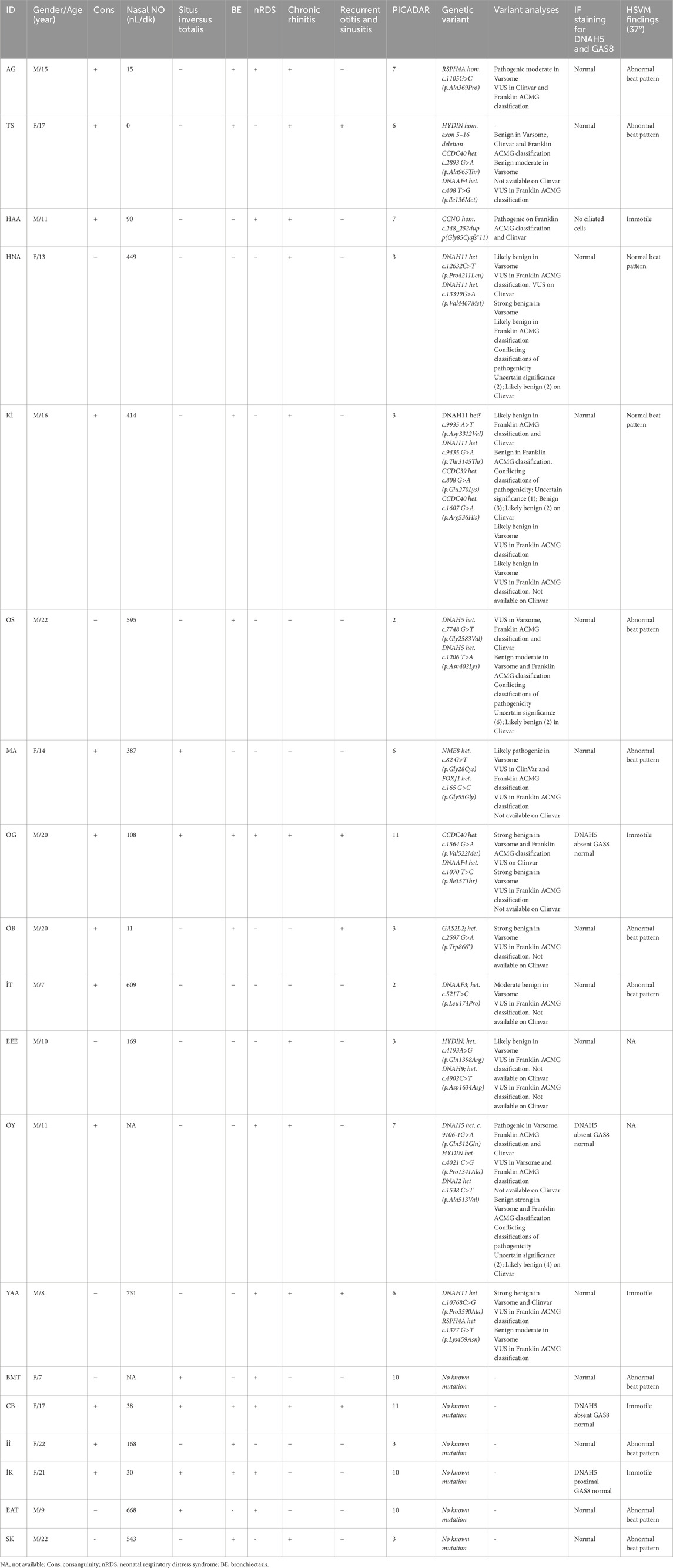
Table 2. Characteristics of PCD-suspected individuals (n:19).
We performed IF analysis in controls and PCD-affected individuals targeting DNAH5 (structural component of ODAs) and GAS8 (structural component of N-DRCs). In unaffected controls, DNAH5 and GAS8 localize to the axonemal length. However, we detected an abnormal localization of DNAH5 in five cases (Table 2). DNAH5 was completely absent in three cases (15.7%) (CB, ÖG, and ÖY) who shared clinical characteristics: neonatal respiratory distress, chronic rhinitis, recurrent otitis and sinusitis. All had immotile cilia on HSVM. CB and ÖG presented with situs inversus totalis and BE, while ÖY lacked these. Genetic analysis revealed no pathogenic variants in CB and ÖG, whereas ÖY carried a heterozygous DNAH5 splice variant (c.9106-1G>A). Nasal NO levels varied, with CB showing lower values (38.94) compared to ÖG (108.23 nL/min).
In only one case (IK) (5.3%), IF staining showed proximal distribution of DNAH5 in the ciliary axoneme. This patient, in contrast to numerous PCD cases, did not exhibit chronic rhinitis, recurrent otitis, sinusitis, or situs inversus totalis; however, the patient presented with a history of neonatal respiratory distress and BE. Nasal NO levels were low (30.68 nL/min), genetic analysis did not identify a pathogenic variant, and HSVM findings demonstrated immotile cilia.
One individual (HAA) (5.3%) showed cells without cilia in IF staining. This patient presented with a history of neonatal respiratory distress, chronic rhinitis, and recurrent otitis and sinusitis. In contrast to typical PCD cases, the patient did not exhibit situs inversus totalis or BE, and nasal NO levels were relatively elevated (90.27 nL/min). Initial genetic analysis did not detect any pathogenic variants. Subsequently, whole exome sequencing (WES) was performed. However, after the patient submitted a sample for IF staining, WES analysis was completed, revealing a biallelic pathogenic CCNO variant (c.248_252dup p (Gly85Cysfs*11). Figure 1 illustrates examples of the absence or abnormal localization of DNAH5 in four individuals with confirmed PCD.
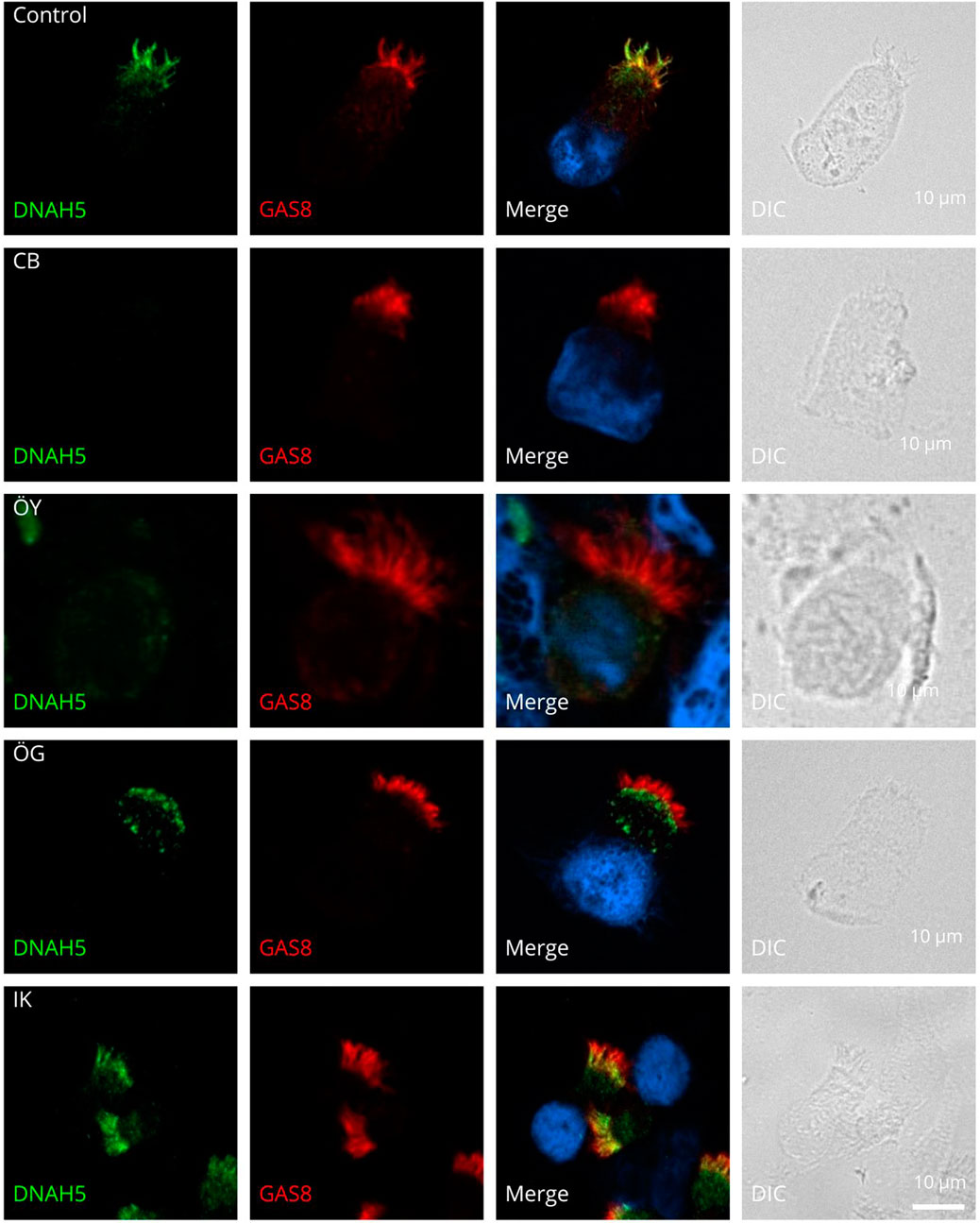
Figure 1. Outer dynein arm defects by IF. Respiratory cilia double-labeled with antibodies directed against DNAH5 (green) and GAS8 (red) show colocalization of DNAH5 with GAS8 along the cilia from healthy controls. In cells of three PCD-affected individuals CB, OY and OG, DNAH5 is absent from the ciliary axonemes and is proximal in one individual IK. Nuclei were stained with Hoechst33342 (blue). Scale bars represent 10 μm.
In two individuals (AG and TS), IF results were normal despite low nasal NO and an abnormal beat pattern observed through HSVM (Table 2). One individual with situs inversus (BMT), a PICADAR score of 10, and an abnormal beat pattern detected by HSVM also had normal IF results (Table 2). WES analysis was performed at the time of IF sampling for AG; however, the results were not yet available. Following IF analysis, a homozygous RSPH4A c.1105G>C (p.Ala369Pro) pathogenic variant was identified.
After the implementation of HSVM in our center in collaboration with Münster, the remaining patients were recalled for HSVM assessment. WES analysis were planned for cases with abnormal HSVM results, low nasal NO measurements, and a PICADAR score above 5.
All individuals with abnormal IF analysis had a PICADAR score greater than 7. The cilia of all individuals with abnormal IF were immotile according to HSVM.
Parental consanguinity was observed in 100% of the individuals, and all had neonatal respiratory distress. Table 2 summarizes the clinical and demographic features, along with HSVM findings, of the patients with abnormal IF results.
In this study, we successfully diagnosed PCD using IF analysis in individuals with inconclusive results from multiple diagnostic tools, including genetic testing, HSVM, and nasal NO measurement. The addition of IF analysis significantly enhanced our diagnostic capabilities. Furthermore, through international collaboration, HSVM has been introduced at our institution, resulting in notable advancements in the diagnosis of PCD.
At our center, current practice involves using nasal NO measurements and genetic testing to evaluate suspected PCD cases. Genetic panels now offer broader gene coverage and are more cost-effective than WES (Platt et al., 2021). Our panel includes the 22 most frequently observed PCD-related genes; however, limitations persist, such as incomplete genetic diagnoses due to unknown genes, variants of uncertain significance, or single heterozygous variants not included in the panel. As a result, many individuals with highly suggestive clinical features remain without genetic confirmation of PCD.
Diagnosing PCD remains complex and challenging due to the absence of disease-specific symptoms, screening tests, and a single gold-standard diagnostic method. Additionally, PCD centers are rare, and the lack of a standardized diagnostic algorithm further complicates diagnosis and awareness efforts (Behan et al., 2016a; Rubbo and Lucas, 2017). Current guidelines recommend a combination of diagnostic tools, but algorithms vary significantly between regions (Shapiro et al., 2018; Lucas et al., 2017). For instance, the ERS recommends HSVM as the first diagnostic test, while the ATS suggests extended genetic panel testing (Shapiro et al., 2018; Lucas et al., 2017). In our country, limited diagnostic resources have resulted in an approach similar to the ATS algorithm, where genetic testing follows nasal NO screening. However, genetic analysis is often time-consuming and limited in scope. To address these limitations, we introduced HSVM into our diagnostic repertoire through international collaboration with the University of Münster.
International collaborations have demonstrated effectiveness in diagnosing and managing rare or complex conditions in resource-limited settings. Such partnerships have facilitated the diagnosis of inherited thrombocytopenias in Argentina and improved the understanding of barriers to pediatric cancer diagnosis in Western Kenya (Glembotsky et al., 2012; Severance et al., 2022). However, examples of collaboration for PCD diagnosis remain limited (Rumman et al., 2023). Our study aimed to address diagnostic uncertainties for individuals with inconclusive results through IF analysis, enabling identification of underlying causes in a subset of cases. Moreover, our institution has successfully integrated HSVM into its diagnostic protocol. Similar to our findings, Rumman et al. reported that 68 of 464 clinically suspected PCD patients were diagnosed through international collaboration (Rumman et al., 2023).
Previous studies have reported cases of PCD where patients exhibited normal IF results despite a definitive diagnosis (Biebach et al., 2022; Shapiro and Leigh, 2017). Additionally, IF analysis may be insufficient for diagnosing PCD individuals with central pair (CP) defects, as the absence of the CP-associated SPEF2 protein has been noted in HYDIN mutant cells (Cindric et al., 2020). In our study, three individuals with high clinical suspicion of PCD had abnormal nasal NO levels and HSVM findings but normal IF results. This discrepancy may arise from genes associated with normal ultrastructure or the need for additional staining with specific IF antibodies. Although IF analysis is widely used to confirm the absence of proteins caused by genetic mutations, its diagnostic efficacy is constrained by the availability of validated antibodies and its reported low sensitivity in PCD diagnosis (Shoemark et al., 2017; Baz-Redon et al., 2020). Furthermore, as novel genes and proteins associated with PCD continue to be identified, the IF antibody panel will require expansion (Knowles and Leigh, 2017).
To determine the likelihood of PCD, researchers have proposed various predictive tools (Martinu et al., 2021). Among these, the PICADAR score has emerged as a widely utilized tool, particularly in low-resource settings (Behan et al., 2016b; Rubin, 1988). A PICADAR score of 10 corresponds to a probability of 92.6% for PCD, while a score >5 demonstrates good sensitivity and specificity for clinically suspected cases (Behan et al., 2016b). However, the optimal cut-off may vary; one study suggested a score of 2 as the best discriminative value (Rademacher et al., 2017). In a Japanese cohort, most patients had PICADAR scores ≤5, indicating that individuals with chronic wet cough, even with low scores, should still be evaluated for PCD (Chiyonobu et al., 2022). In our cohort, all five individuals had PICADAR scores ≥7. Notably, all three patients with normal IF results had PICADAR scores of ≥6 and low nasal NO levels. Further studies involving larger populations are needed to better assess the sensitivity and specificity of the PICADAR score.
The primary limitation of this study is its small sample size. Additionally, the absence of a reliable reference standard for assessing IF accuracy presents a significant challenge. For benchmarking purposes, we utilized the ERS Task Force criteria to classify confirmed and highly probable PCD cases (Lucas et al., 2017).
In conclusion, this study underscores the critical role of international collaboration in diagnosing rare diseases like PCD, which often require expertise available only in specialized centers. IF analysis is not routinely available at our institution; however, collaboration with the University of Münster enabled us to diagnose individuals in whom conventional methods were inconclusive. Such partnerships between resource-limited settings and specialized centers can significantly enhance diagnostic capabilities, particularly when disease prevalence is lower than anticipated. Furthermore, establishing international registry systems that include data from developing countries will contribute to improved diagnosis and management of PCD. Collaboration and shared expertise are key to addressing the diagnostic challenges associated with rare diseases.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Clinical Research Ethics Committee of Marmara University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
MY: Data curation, Formal Analysis, Software, Writing–original draft. RH: Conceptualization, Formal Analysis, Methodology, Software, Writing–original draft. MY: Methodology, Resources, Writing–review and editing. AG: Data curation, Writing–review and editing. MS: Resources, Visualization, Writing–review and editing. ŞK: Methodology, Resources, Writing–review and editing. NM: Resources, Writing–review and editing. AE: Investigation, Methodology, Resources, Writing–review and editing. EE: Supervision, Visualization, Writing–review and editing. YG: Project administration, Supervision, Visualization, Writing–review and editing. HO: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing–review and editing. BK: Supervision, Visualization, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Deutsche Forschungsgemeinschaft DFG OM6/7, OM6/8, OM6/10 and OM6/11 to HO and HJ 7/1-1, HJ 7/1-3 to RH. This work is generated within the European Reference Network for rare respiratory diseases ERN-LUNG.
We thank the Turkish Pediatric Association (Özdemir İlter Grant) for supporting PCD research and all PCD-affected individuals and their families who participated in this study. We also acknowledge Ataşehir and Caddebostan Rotary Clubs for providing a nasal NO analyzer (ECO MEDICS, AG, Duerten, Switzerland) and HSVM. We appreciate the technical assistance provided by members of Prof. Dr. Heymut Omran’s laboratory.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Baz-Redon, N., Rovira-Amigo, S., Fernandez-Cancio, M., Castillo-Corullon, S., Cols, M., Caballero-Rabasco, M. A., et al. (2020). Immunofluorescence analysis as a diagnostic tool in a Spanish cohort of patients with suspected primary ciliary dyskinesia. J. Clin. Med. 9 (11), 3603. doi:10.3390/jcm9113603
Behan, L., Dimitrov, B. D., Kuehni, C. E., Hogg, C., Carroll, M., Evans, H. J., et al. (2016b). Picadar: a diagnostic predictive tool for primary ciliary dyskinesia. Eur. Respir. J. 47 (4), 1103–1112. doi:10.1183/13993003.01551-2015
Behan, L., Dunn Galvin, A., Rubbo, B., Masefield, S., Copeland, F., Manion, M., et al. (2016a). Diagnosing primary ciliary dyskinesia: an international patient perspective. Eur. Respir. J. 48 (4), 1096–1107. doi:10.1183/13993003.02018-2015
Bhatt, R., and Hogg, C. (2020). Primary ciliary dyskinesia: a major player in a bigger game. Breathe (Sheff). 16 (2), 200047. doi:10.1183/20734735.0047-2020
Biebach, L., Cindric, S., Koenig, J., Aprea, I., Dougherty, G. W., Raidt, J., et al. (2022). Recessive mutations in cfap74 cause primary ciliary dyskinesia with normal ciliary ultrastructure. Am. J. Respir. Cell Mol. Biol. 67 (3), 409–413. doi:10.1165/rcmb.2022-0032LE
Chiyonobu, K., Xu, Y., Feng, G., Saso, S., Ogawa, S., Ikejiri, M., et al. (2022). Analysis of the clinical features of Japanese patients with primary ciliary dyskinesia. Auris Nasus Larynx 49 (2), 248–257. doi:10.1016/j.anl.2021.08.003
Cindric, S., Dougherty, G. W., Olbrich, H., Hjeij, R., Loges, N. T., Amirav, I., et al. (2020). Spef2-and hydin-mutant cilia lack the central pair-associated protein spef2, aiding primary ciliary dyskinesia diagnostics. Am. J. Respir. Cell Mol. Biol. 62 (3), 382–396. doi:10.1165/rcmb.2019-0086OC
Collins, S. A., Gove, K., Walker, W., and Lucas, J. S. (2014). Nasal nitric oxide screening for primary ciliary dyskinesia: systematic review and meta-analysis. Eur. Respir. J. 44 (6), 1589–1599. doi:10.1183/09031936.00088614
Dougherty, G. W., Loges, N. T., Klinkenbusch, J. A., Olbrich, H., Pennekamp, P., Menchen, T., et al. (2016). Dnah11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am. J. Respir. Cell Mol. Biol. 55 (2), 213–224. doi:10.1165/rcmb.2015-0353OC
Frommer, A., Hjeij, R., Loges, N. T., Edelbusch, C., Jahnke, C., Raidt, J., et al. (2015). Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects. Am. J. Respir. Cell Mol. Biol. 53 (4), 563–573. doi:10.1165/rcmb.2014-0483OC
Glembotsky, A. C., Marta, R. F., Pecci, A., De Rocco, D., Gnan, C., Espasandin, Y. R., et al. (2012). International collaboration as a tool for diagnosis of patients with inherited thrombocytopenia in the setting of a developing country. J. Thromb. Haemost. 10 (8), 1653–1661. doi:10.1111/j.1538-7836.2012.04805.x
Kennedy, M. P., and Plant, B. J. (2014). Primary ciliary dyskinesia and the heart: cilia breaking symmetry. Chest 146 (5), 1136–1138. doi:10.1378/chest.14-0722
Knowles, M. R., and Leigh, M. W. (2017). Primary ciliary dyskinesia diagnosis. Is color better than black and white? Am. J. Respir. Crit. Care Med. 196 (1), 9–10. doi:10.1164/rccm.201702-0426ED
Knowles, M. R., Ostrowski, L. E., Leigh, M. W., Sears, P. R., Davis, S. D., Wolf, W. E., et al. (2014). Mutations in rsph1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am. J. Respir. Crit. Care Med. 189 (6), 707–717. doi:10.1164/rccm.201311-2047OC
Leigh, M. W., O'Callaghan, C., and Knowles, M. R. (2011). The challenges of diagnosing primary ciliary dyskinesia. Proc. Am. Thorac. Soc. 8 (5), 434–437. doi:10.1513/pats.201103-028SD
Lucas, J. S., Barbato, A., Collins, S. A., Goutaki, M., Behan, L., Caudri, D., et al. (2017). European respiratory society guidelines for the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 49 (1), 1601090. doi:10.1183/13993003.01090-2016
Martinu, V., Borek-Dohalska, L., Varenyiova, Z., Uhlik, J., Capek, V., Pohunek, P., et al. (2021). Evaluation of a clinical index as a predictive tool for primary ciliary dyskinesia. Diagn. (Basel) 11 (6), 1088. doi:10.3390/diagnostics11061088
O'Connor, M. G., Horani, A., and Shapiro, A. J. (2021). Progress in diagnosing primary ciliary dyskinesia: the north american perspective. Diagn. (Basel) 11 (7), 1278. doi:10.3390/diagnostics11071278
Omran, H., Kobayashi, D., Olbrich, H., Tsukahara, T., Loges, N. T., Hagiwara, H., et al. (2008). Ktu/pf13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 456 (7222), 611–616. doi:10.1038/nature07471
Omran, H., and Loges, N. T. (2009). Immunofluorescence staining of ciliated respiratory epithelial cells. Methods Cell Biol. 91, 123–133. doi:10.1016/S0091-679X(08)91007-4
Platt, C. D., Zaman, F., Bainter, W., Stafstrom, K., Almutairi, A., Reigle, M., et al. (2021). Efficacy and economics of targeted panel versus whole-exome sequencing in 878 patients with suspected primary immunodeficiency. J. Allergy Clin. Immunol. 147 (2), 723–726. doi:10.1016/j.jaci.2020.08.022
Rademacher, J., Buck, A., Schwerk, N., Price, M., Fuge, J., Welte, T., et al. (2017). Nasal nitric oxide measurement and a modified picadar score for the screening of primary ciliary dyskinesia in adults with bronchiectasis. Pneumologie 71 (8), 543–548. doi:10.1055/s-0043-111909
Raidt, J., Krenz, H., Tebbe, J., Grosse-Onnebrink, J., Olbrich, H., Loges, N. T., et al. (2022). Limitations of nasal nitric oxide measurement for diagnosis of primary ciliary dyskinesia with normal ultrastructure. Ann. Am. Thorac. Soc. 19 (8), 1275–1284. doi:10.1513/AnnalsATS.202106-728OC
Raidt, J., Wallmeier, J., Hjeij, R., Onnebrink, J. G., Pennekamp, P., Loges, N. T., et al. (2014). Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur. Respir. J. 44 (6), 1579–1588. doi:10.1183/09031936.00052014
Rubbo, B., and Lucas, J. S. (2017). Clinical care for primary ciliary dyskinesia: current challenges and future directions. Eur. Respir. Rev. 26 (145), 170023. doi:10.1183/16000617.0023-2017
Rubin, B. K. (1988). Immotile cilia syndrome (primary ciliary dyskinesia) and inflammatory lung disease. Clin. Chest Med. 9 (4), 657–668. doi:10.1016/s0272-5231(21)00590-6
Rumman, N., Fassad, M. R., Driessens, C., Goggin, P., Abdelrahman, N., Adwan, A., et al. (2023). The palestinian primary ciliary dyskinesia population: first results of the diagnostic and genetic spectrum. ERJ Open Res. 9 (2), 00714. doi:10.1183/23120541.00714-2022
Severance, T. S., Njuguna, F., Olbara, G., Kugo, M., Langat, S., Mostert, S., et al. (2022). An evaluation of the disparities affecting the underdiagnosis of pediatric cancer in western Kenya. Pediatr. Blood Cancer 69 (10), e29768. doi:10.1002/pbc.29768
Shapiro, A. J., Davis, S. D., Polineni, D., Manion, M., Rosenfeld, M., Dell, S. D., et al. (2018). Diagnosis of primary ciliary dyskinesia. An official american thoracic society clinical practice guideline. Am. J. Respir. Crit. Care Med. 197 (12), e24–e39. doi:10.1164/rccm.201805-0819ST
Shapiro, A. J., and Leigh, M. W. (2017). Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: genetic defects with normal and non-diagnostic ciliary ultrastructure. Ultrastruct. Pathol. 41 (6), 373–385. doi:10.1080/01913123.2017.1362088
Shoemark, A., Frost, E., Dixon, M., Ollosson, S., Kilpin, K., Patel, M., et al. (2017). Accuracy of immunofluorescence in the diagnosis of primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 196 (1), 94–101. doi:10.1164/rccm.201607-1351OC
Surdut, S. P., van der Merwe, E., Goussard, P., and Urban, M. F. (2023). Which side are they on? Diagnosing primary ciliary dyskinesias in low- or middle-income countries: a review and case series. Afr. J. Thorac. Crit. Care Med. 29 (3), 131–138. doi:10.7196/AJTCCM.2023.v29i3.425
Keywords: primary ciliary dyskinesia (PCD), international collaboration, low income countries, high speed video microscopy, rare disease, diagnostic challenges
Citation: Yuksel Kalyoncu M, Hjeij R, Yanaz M, Gulieva A, Selcuk Balcı M, Karabulut Ş, Metin Cakar N, Ergenekon AP, Erdem Eralp E, Gokdemir Y, Omran H and Karadag BT (2025) Empowering limited-resource countries: collaborating with expert centers for diagnosis of primary ciliary dyskinesia. Front. Mol. Biosci. 12:1547152. doi: 10.3389/fmolb.2025.1547152
Received: 17 December 2024; Accepted: 19 February 2025;
Published: 20 March 2025.
Edited by:
Alessandro Palma, Sapienza University of Rome, ItalyReviewed by:
Sara Carvalhal, University of Algarve, PortugalCopyright © 2025 Yuksel Kalyoncu, Hjeij, Yanaz, Gulieva, Selcuk Balcı, Karabulut, Metin Cakar, Ergenekon, Erdem Eralp, Gokdemir, Omran and Karadag. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mine Yüksel Kalyoncu, bWluZWV5dWtzZWxsQGdtYWlsLmNvbQ==; Rim Hjeij, cmltLmhqZWlqQHVrbXVlbnN0ZXIuZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.