
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 03 April 2025
Sec. Molecular Biophysics
Volume 12 - 2025 | https://doi.org/10.3389/fmolb.2025.1524951
This article is part of the Research Topic Computational and Experimental Techniques to Battle Viral Infectious Diseases: Identifying Antiviral Agents for Global Health Security View all articles
 R. P. Yadav
R. P. Yadav N. R. Jena*
N. R. Jena*Introduction: The flavivirus infections caused by the Zika virus (ZIKV), Dengue virus (DENV), and West Nile virus (WNV) cause mild to serious pathological conditions, such as fever, joint pain, shock, internal bleeding, organ failure, nausea, breathlessness, brain tissue damage, neurodegenerative diseases, and deaths. As currently no efficient vaccine or drug is available to prevent or treat these diseases in humans, it is essential to identify potential drug-like molecules to treat these diseases. For these reasons, several known anti-viral drugs are repurposed against the proteases of ZIKV, WNV, and DENV to inhibit their activities.
Methods: The GOLD 5.0 molecular docking program was used to dock 20 HIV and HCV drugs against the ZIKV protease. Based on docking scores, 5 drugs were found to bind to the ZIKV protease with high affinities. Subsequently, the AMBER ff14SB force field was employed to simulate these drug-bound complexes of ZIKV protease. The MM/PBSA free energy method was utilized to compute the binding free energies of these complexes. Consequently, the two best ZIKV protease inhibitors were repurposed against the proteases of DENV and WNV.
Results and Discussion: It is found that out of the 5 drugs, Ritonavir and Paritaprevir bind to the NS2B-NS3 protease of the ZIKV strongly with the Gibbs binding free energies (∆Gbind) of −17.44±3.18 kcal/mol and −14.25±3.11 kcal/mol respectively. Remarkably, Ritonavir binds to the ZIKV Protease about 12 kcal/mol more strongly compared to its binding to the HIV protease. It is further found that Paritaprevir binds to DENV and WNV proteases as strongly as it binds to the ZIKV protease. Hence it is proposed that Paritaprevir may act as a potent pan-antiviral against the Zika, West Nile, and Dengue viral diseases.
The flavivirus infections caused by the Zika virus (ZIKV), Dengue virus (DENV), and West Nile virus (WNV) cause mild to serious pathological conditions, such as fever, joint pain, shock, internal bleeding, organ failure, nausea, breathlessness, brain tissue damage, neurodegenerative diseases, and deaths (Broutet et al., 2016; Carod-Artal, 2018; de Araújo et al., 2018; Hersh et al., 2017; Kong et al., 2018; Mlakar et al., 2016; Moghadam et al., 2016; Musso and Gubler, 2016; Nash et al., 2001; Paixao et al., 2022). Unfortunately, no efficient vaccine or drug is available to date to prevent or treat these diseases in humans (Poland et al., 2018). Therefore, it is imperative to identify potential drug-like candidates that can be used to treat these diseases.
The ZIKV, DENV, and WNV possess structurally and functionally identical proteins (Nitsche, 2019; Wahaab et al., 2021; El Sahili et al., 2019; Chang et al., 2017). For example, the NS2B-NS3 proteases of these viruses are about 65% similar in amino acid sequence. The 3-dimensional structures of these proteins are also identical (Nitsche, 2019; Wahaab et al., 2021; El Sahili et al., 2019; Chang et al., 2017). Further, the main function of the NS2B-NS3 protease across these viruses is to cleave the polyprotein chain of these viruses to produce three structural (sp) and seven non-structural proteins (nsp) (Kuno et al., 1998; Falgout et al., 1991; Phoo et al., 2016; Zhang et al., 2016; Li and Kang, 2021). Therefore, it is necessary to inhibit the protease activities of these viruses to control their spread and subsequent deadly effects. It is now established that the NS2B-NS3 protease of these viruses contains identical folds and subdomains that directly or indirectly facilitate substrate binding and catalysis (Nitsche, 2019; Wahaab et al., 2021; El Sahili et al., 2019; Chang et al., 2017; Kuno et al., 1998; Falgout et al., 1991; Phoo et al., 2016; Zhang et al., 2016; Li and Kang, 2021). The substrate binding site consists of various sub-sites, namely, S1′, S2′, S3′, S1, S2, S3, and S4 (Zhang et al., 2016; Li and Kang, 2021; Robin et al., 2009; Li et al., 2018; Li et al., 2017; Pant and Jena, 2022). The primed sites facilitate catalysis, whereas the non-primed sites help accurately place the substrates into the active site and hold the substrates firmly till the completion of the catalysis (Pant and Jena, 2022). Therefore, identifying suitable inhibitors that can block the substrate binding sites of ZIKV, DENV, and WNV proteases will help inhibit the protease activities of these viruses. These inhibitors may further be developed as drug candidates for treating these viral diseases.
Although several attempts were made to identify potent inhibitors against these viral proteases individually (Zhang et al., 2016; Li and Kang, 2021; Robin et al., 2009; Li et al., 2018; Li et al., 2017; Pant and Jena, 2022; Phoo et al., 2018; Knox et al., 2006; Roy et al., 2017; Schüller et al., 2011; Behnam et al., 2015; Li et al., 2015; Grazia Martina et al., 2021; Song et al., 2021; Pushpakom et al., 2019; Pant et al., 2023; Pant and Jena, 2023), not a single drug has cleared the clinical trial. As the drug designing process is financially expensive and takes several years to discover a potent drug, drug repurposing is an efficient, less expensive, and prompt way of identifying existing drugs against these viral diseases (Talevi and Bellera, 2020; V Kleandrova and Speck-Planche, 2021; Cregar-Hernandez et al., 2011). Further, identifying common drugs against ZIKV, DENV, and WNV will substantially reduce the financial burden of designing new drugs and expedite the drug discovery process (Talevi and Bellera, 2020; V Kleandrova and Speck-Planche, 2021). For these reasons, the repurposing of 20 antiviral drugs (Table 1) recently proposed against proteases of the human immunodeficiency virus (HIV) (Wang et al., 2011; Tie et al., 2007; Walmsley et al., 2002; Klibanov et al., 2015; Koudriakova et al., 1998; Kempf and Sham, 1996) and hepatitis C virus (HCV) (Keating, 2016; Lam and Salazar, 2016; Shen et al., 2016; Kiang et al., 2013) is studied herein to evaluate their ability to bind to the substrate binding site of the NS2B-NS3 protease of the ZIKV. The two best drugs that have stable interactions with the ZIKV protease are repurposed against the WNV and DENV proteases (Yuan et al., 2017). Some of these drugs were also repurposed against ZIKV and SARS-COV-2 (Bolcato et al., 2020; Gurung et al., 2021; Jena, 2021). As these drugs were originally shown to inhibit the protease activities of HIV (Wang et al., 2011; Tie et al., 2007; Walmsley et al., 2002; Klibanov et al., 2015; Koudriakova et al., 1998; Kempf and Sham, 1996) and HCV (Keating, 2016; Lam and Salazar, 2016; Shen et al., 2016; Kiang et al., 2013), it is believed that these drugs may also inhibit proteases of other viral diseases due to their structural and functional similarities. For example, the catalytic residues of HCV (S139, H57, D81) are conserved in ZIKV (S135, H51, D75), WNV (S135, H51, D75), and DENV (S135, H51, D75) (Li and Kang, 2021; Robin et al., 2009; Li et al., 2018; Li et al., 2017; Pant and Jena, 2022; Phoo et al., 2018; Knox et al., 2006; Roy et al., 2017; Schüller et al., 2011; Behnam et al., 2015) and all of these proteases cleave the polypeptide chain to generate non-structural and structural proteins (Falgout et al., 1991; Phoo et al., 2016).

Table 1. Docking scores of different antiviral drugs bound to NS2B-NS3 of the ZIKV.
To examine if these drugs can inhibit the ZIKV, WNV, and DENV proteases more effectively than their parent viral proteins (say, Ritonavir for HIV protease), the stability of Ritonavir bound to the HIV protease (PDB ID 1HXW) (Kempf et al., 1995) were compared with different complexes where Ritonavir is bound to ZIKV, WNV, and DENV proteases. Ritonavir was chosen because it effectively inhibits the HIV protease with good oral bioavailability in humans (Kempf et al., 1995).
Initially, the high-resolution (2.0 Å) crystal structure of the NS2B-NS3 protease of the ZIKV bound to benzimidazol-1-yl-methanol (7HQ) (PDB ID 5H4I) (Zhang et al., 2016) was downloaded from the protein databank (https://www.rcsb.org/). Later, 7HQ was removed from the complex structure (PDB ID 5H4I) (Zhang et al., 2016) to generate the isolated protease structure. Subsequently, the missing ends of the protease were capped by NME (C-terminal) and ACE (N-terminal) molecules, and hydrogen atoms were added to the protease by using the GOLD 5.0 program (Jones et al., 1997; Li et al., 2014; Verdonk et al., 2003; Liebeschuetz et al., 2012). Consequently, 20 HIV and HCV drugs as summarized in Table 1 and Figure 1 were docked into the active site of the ZIKV protease by using the GOLD 5.0 program (Jones et al., 1997; Li et al., 2014; Verdonk et al., 2003; Liebeschuetz et al., 2012). The 3-dimensional structures of these drugs were downloaded from the DrugBank database (Wishart et al., 2018) and were energy-minimized before docking.
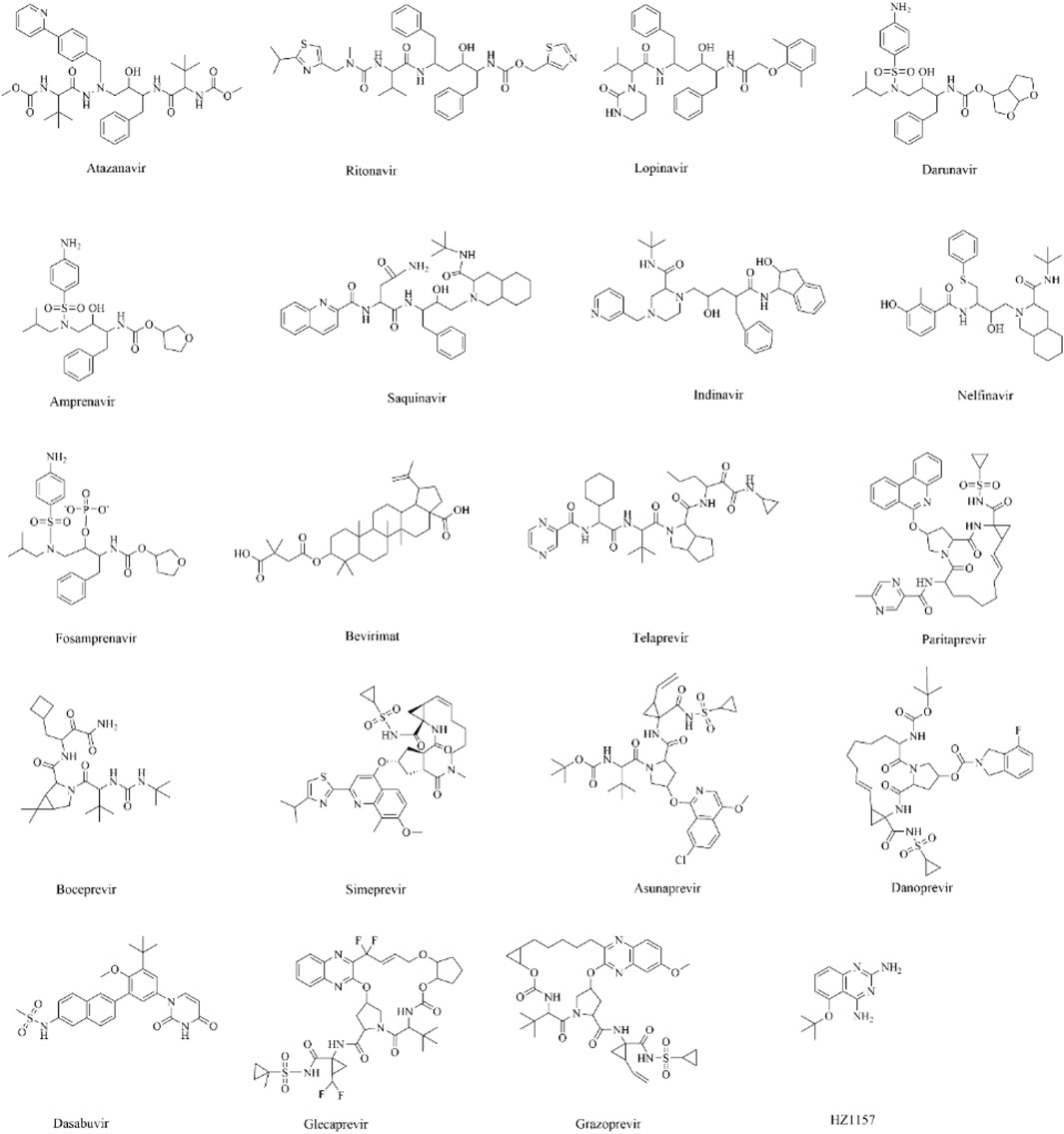
Figure 1. The two-dimensional structures of different drugs considered herein for molecular docking.
To ensure that the docking program can accurately generate the protease-drug complex structures, the 7HQ was docked into the active site of NS2B-NS3 protease of the ZIKV to reproduce the experimental structure of the NS2B-NS3-7HQ complex (PDB ID 5H4I) (Zhang et al., 2016). The following docking protocols were used for this purpose. (1) The binding site was considered to be situated within a radius of 10 Å from the conserved Tyr161. (2) The genetic algorithm was used to create 10 different poses of each drug by using the ChemScore of GOLD 5.0 program (Jones et al., 1997; Li et al., 2014; Verdonk et al., 2003; Liebeschuetz et al., 2012). 100,000 genetic operations were performed to generate accurate poses. (3) The ChemPLP function was used to rank these 10 conformations based on different energy terms. (4) Although the drug molecules could adopt different conformations during docking, the protease was held rigid. The torsion angles of amino acids containing hydroxyl groups (Ser, Thr, and Tyr) were allowed to rotate to optimize their hydrogen bonding interactions. For the same reason, the Lysine NH3+ groups were allowed to rotate during docking. The ligand conformations were varied by not altering their bond lengths and angles. The stereochemistry of ligands was also not changed during docking. The atomic charges (formal and partial) were ignored during docking. Whether an atom is charged was decided by counting the bond orders and comparing the results with the atom’s normal valency (Jones et al., 1997; Li et al., 2014; Verdonk et al., 2003; Liebeschuetz et al., 2012). It should be mentioned that the ChemScore function was derived empirically from a set of 82 protein-ligand complexes for which measured binding affinities were available (Jones et al., 1997; Li et al., 2014; Verdonk et al., 2003; Liebeschuetz et al., 2012). However, the ChemPLP function uses Piecewise Linear Potential to model the steric complementarity between protein and ligand. Both functions calculate distance- and angle-dependent hydrogen bonding terms, lipophilic, and rotational energy terms. They also contain a clash penalty and internal torsion terms, which militate against close contacts in docking and poor internal conformations (Korb et al., 2009; Baxter et al., 1998) Similar docking protocols were used elsewhere (Saini et al., 2021; Saini et al., 2019).
Remarkably, the rank-1 pose of 7HQ was found to match its crystallographic conformation with a RMSD of 0.85 Å. The only difference between the docked and experimental complex structures arose from the adoption of different rotameric conformations of the C-OH bond of 7HQ (Pant and Jena, 2023). Therefore, the above docking protocols were used to produce different protease-drug complexes. Out of the 10 different conformations of each complex, the one that makes the maximum interactions with the protease and possesses a binding mode similar to that of 7HQ was short-listed for further analysis. This docking protocol was shown earlier to produce accurate docking poses (Pant and Jena, 2022; Pant et al., 2023; Pant and Jena, 2023; Pant and Jena, 2024).
It was found that among these 20 protease-drug complexes, 5 complexes possess the highest docking scores (Table 1). These complexes include drugs like Ritonavir, Saquinavir, Indinavir, Paritaprevir, and Lopinavir (Table 1). As the protease was held rigid and no solvent effects were considered during docking, it is expected that in the presence of water molecules and under the influence of protein dynamics, protease-drug complexes may undergo further conformational changes to make optimal interactions. Due to these reasons, the above 5 short-listed protein-drug complexes were subjected to molecular dynamics (MD) simulations. It should be mentioned that in an earlier in vitro structure-based drug discovery study (Yuan et al., 2017), the combination of Lopinavir-Ritonavir was found to inhibit the ZIKV replication by neutralizing the NS2B-NS3 protease. As the Lopinavir-Ritonavir combination is commercially available and commonly used clinically, the inhibition abilities of these individual drugs to the ZIKV protease were not evaluated (Yuan et al., 2017). Moreover, the binding mechanisms of these drugs to the ZIKV protease are also not known.
Before MD simulations, the short-listed docked complexes containing the ZIKV protease were solvated in a water box of size 10 Å. The TIP3P method (Mark and Nilsson, 2001; Onufriev and Izadi, 2018) was used to treat water molecules. Sufficient ions (Na+ and Cl−) were added to make the solvated complexes neutral. The GAFF method and AM1-BCC charge model were used to generate force fields for the drug molecules (Huang and Roux, 2013; He et al., 2020). The AMBER ff14SB force field (Maier et al., 2015) as implemented in the AMBER 14 program (Maier et al., 2015) was used to model the protease. Subsequently, each solvated neutral complex was energy minimized by 500 steps by using the steepest descent algorithm (Meza, 2010) followed by 1,000 steps by using the conjugate gradient algorithm (Štich et al., 1989). The minimizations were performed in three steps. In the first step, the water molecules were minimized by restraining the protease-drug complexes with a force constant of 50 kcal mol-1 Å−2. In the second step, the drug and water molecules were energy minimized by restraining the protease by a force constant of 50 kcal mol-1 Å−2. In the last step, all molecules were energy minimized without any restraints. Subsequently, each system was heated slowly to reach a temperature (T) of 300 K throughout 20 ps in the NVT ensemble. During this process, a force constant of 20 kcal mol-1 Å−2 was used to restrain the protease and drug molecules (except for their hydrogen (H) atoms). The temperature was regulated by using the weak-coupling algorithm (Berendsen et al., 1984). Later, a 100 ps of MD simulation was performed to equilibrate the system in the NPT ensemble while applying a harmonic restraint of 5 kcal mol-1 Å−2 at T = 300 K and a pressure (P) of 1.0 atm. The constant pressure of 1.0 atm was maintained using the Barendsen barostat (Ryckaert et al., 1977). Subsequently, each complex was equilibrated for 1,000 ps without restraining any molecules. During these processes, H atoms were constrained by using the SHAKE algorithm (Kräutler et al., 2001). The time step of MD simulations was set to 2 fs. A cutoff of 10 Å was set for non-bonded intermolecular interactions and long-range electrostatic interactions were treated by using the particle-mesh Ewald method (Darden et al., 1993). Consequently, each complex was subjected to a production run for 100 ns by using the NPT ensemble (T = 300 K and P = 1 atm).
As discussed later, among these drugs, Ritonavir and Paritaprevir made the most stable complexes with the ZIKA virus protease (Table 2). Therefore, to understand if these drugs can also bind strongly to the DENV and WNV proteases, the average simulated structures of the ZIKV protease-Ritonavir and ZIKV protease-Paritaprevir complexes were superimposed onto the crystal structures of WNV (PDB ID 2FP7) (Erbel et al., 2006) and DENV (PDB ID 3U1I) (Noble et al., 2012) proteases after removing crystallographic ligands bound to them. It should be mentioned that the amino acid sequences of PDB ID 3UI1 correspond to the DENV serotype 3 (DENV3) (Noble et al., 2012). Subsequently, the coordinates of Ritonavir and Paritaprevir were saved to generate the three-dimensional structures of the WNV protease-Ritonavir, WNV protease-Paritaprevir, DENV protease-Ritonavir, and DENV-Paritaprevir complexes. As the proteases of ZIKV, DENV, and WNV are structurally similar, it is believed that these drugs may bind to them by adopting a similar mechanism. However, the protease dynamics may induce conformational changes in the drugs, which can be captured by using the MD simulation techniques. Therefore, the generated protease-drug complexes were initially energy minimized, heated to 300 K, equilibrated for 1000 ps, and subjected to production run for 100 ns by employing all the steps mentioned above for the ZIKV.
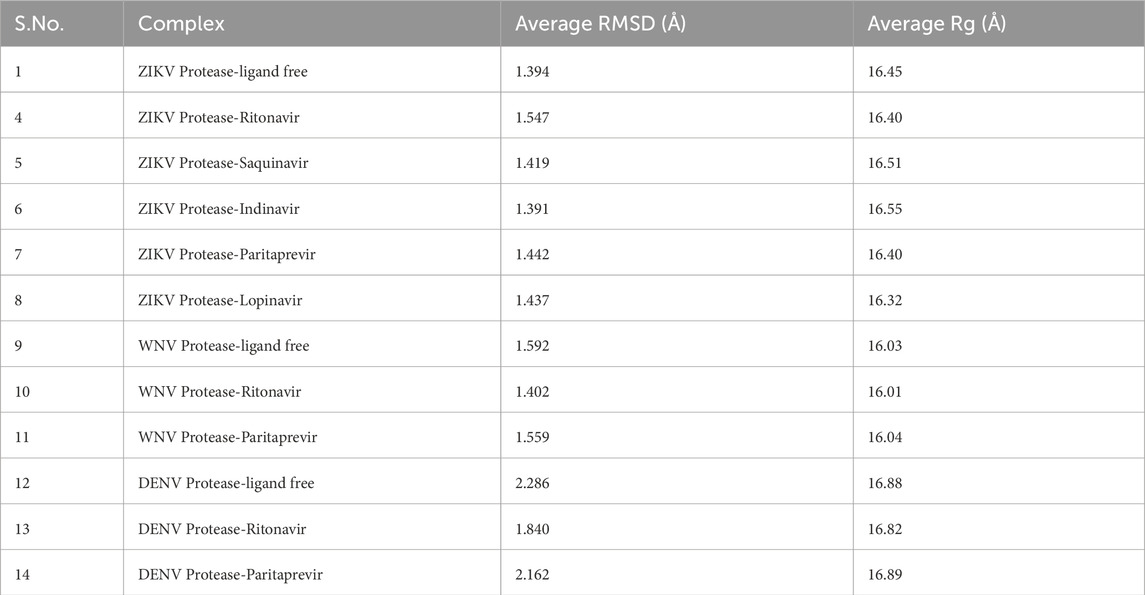
Table 2. The average root mean square deviations (RMSD) of the Cα atoms and the average radius of gyration (Rg) of the NS2B-NS3 protease in ligand-free and ligand-bound conformations.
To compare the structures of the NS2B-NS3-drug complexes with the ligand-free structures of the NS2B-NS3 proteases, the active conformations of ZIKV (PDB ID 5H4I) (Zhang et al., 2016), WNV (PDB ID 2FP7) (Erbel et al., 2006), and DENV (PDB ID 3UI1) (Noble et al., 2012) proteases were simulated for 100ns each after removing their crystallographic ligands from their binding sites. As in the apo (inactive) conformations of WNV (PDB ID 2GGV) (Aleshin et al., 2007) and DENV proteases (PDB ID 2FOM) (Erbel et al., 2006), the NS2B is protruding to the solvent and is placed away from the active site of the NS3 (Supplementary Figure S4), these structures cannot facilitate the substrate binding. Therefore, the comparison of MD-simulated ligand-bound conformations of the ZIKV, WNV, and DENV proteases with their apo structures would not yield meaningful results. For this reason, the ligand-free structures of these viral proteases were simulated to compare with the simulated drug-bound conformations of different viral proteases. The HIV protease-Ritonavir complex (PDB ID 1HXW) (Kempf et al., 1995) was also simulated for 100ns to compare its binding free-energy data with the ZIKV-Ritonavir, WNV-Ritonavir, and DENV-Ritonavir complexes.
The Gibb’s binding free energy
where
The root mean square deviations (RMSD in Å) of Cα atoms of the ZIKV, WNV, and DENV proteases and the root mean square fluctuations (RMSF in Å) of different residues of these proteases are illustrated in Figure 2. Their radius of gyration are illustrated in Figure 3. The average RMSD of the of Cα atoms and the average radius of gyration of these virals proteases are presented in Table 2. From Figures 2, 3 and Table 2, it is clear that the average RMSD is less than 2.0 Å for all proteases studied herein and hence the ligand binding to them would stabilize their structure. Although RMSD of ZIKV protease-Lopinavir complex slightly increased from the average value between 60–80 ns, it decreased afterward. It could be due to conformational changes occurring in the ZIKV-protease due to Lopinavir binding. The computed radius of gyration of all complexes suggests that the binding of different drugs to the active site of the viral proteases would help the protease to acquire a compact structure (Figure 3). It should be mentioned that the ligand-free structures of the viral proteases possess the similar Rg values as computed for the protease-drug complexes (Table 2; Figure 3). This is because the ligand-free structures were derived from the ligand-bound complex structures after removing crystallographic ligands, where the protease had already acquired a compact structure (Zhang et al., 2016; Erbel et al., 2006; Noble et al., 2012).
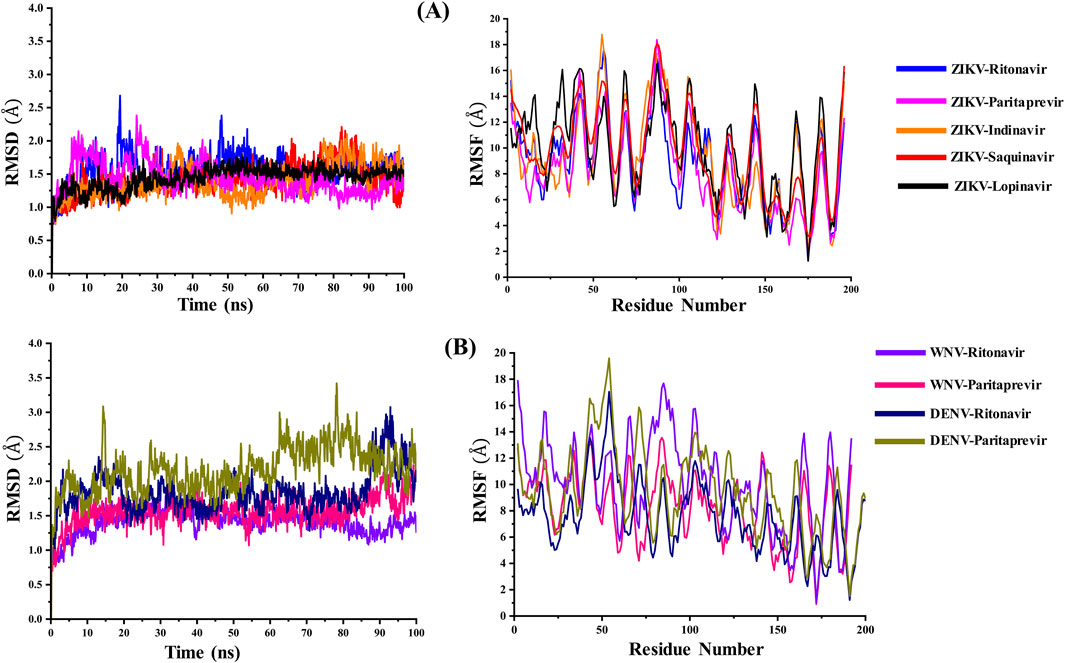
Figure 2. (A) The variations in the root mean square deviations (RMSD in Å) of the Cα atoms and the root mean square fluctuations (RMSF in Å) of amino acid residues of the ZIKV NS2B-NS3 protease bound to different ligands with simulation time. (B) The variations in the RMSD (Å) of the Cα atoms and the RMSF (Å) of amino acid residues of the WNV and DENV NS2B-NS3 proteases bound to Ritonavir and Paritaprevir with simulation time. These values were calculated by considering the minimized structure as the reference.

Figure 3. The variations in the radius of gyration (Rg in Å) of the (A) ZIKV and (B) WNV, and DENV NS2B-NS3 proteases in ligand-free and ligand-bound conformations with simulation time.
The average MD-simulated structures of different complexes are shown in Figures 4–6 and Supplementary Figures S1–S3. Important interactions made between the drug molecules and different residues of these proteases are also depicted in these Figures (Figures 4–6; Supplementary Figures S1–S3). From these figures, it is evident that the short-listed drug molecules are well placed in the active site of the protease and are making stable interactions with them. The stability of these complexes is also evident from the negative ΔGbind values (Table 3; Supplementary Table S1). It should be mentioned that the hydrogen bonding interactions that lasted for >80% (occupancy >80%), 50%–79% (occupancy 50%–79%), and <50% of the simulation time (occupancy <50%) are considered to be strong, moderate, and weak respectively (Pant and Jena, 2022; Pant et al., 2023). The π-π stacking interaction between two aromatic rings is considered to be strong if the distance between them is <3.5 Å (Riwar et al., 2017).
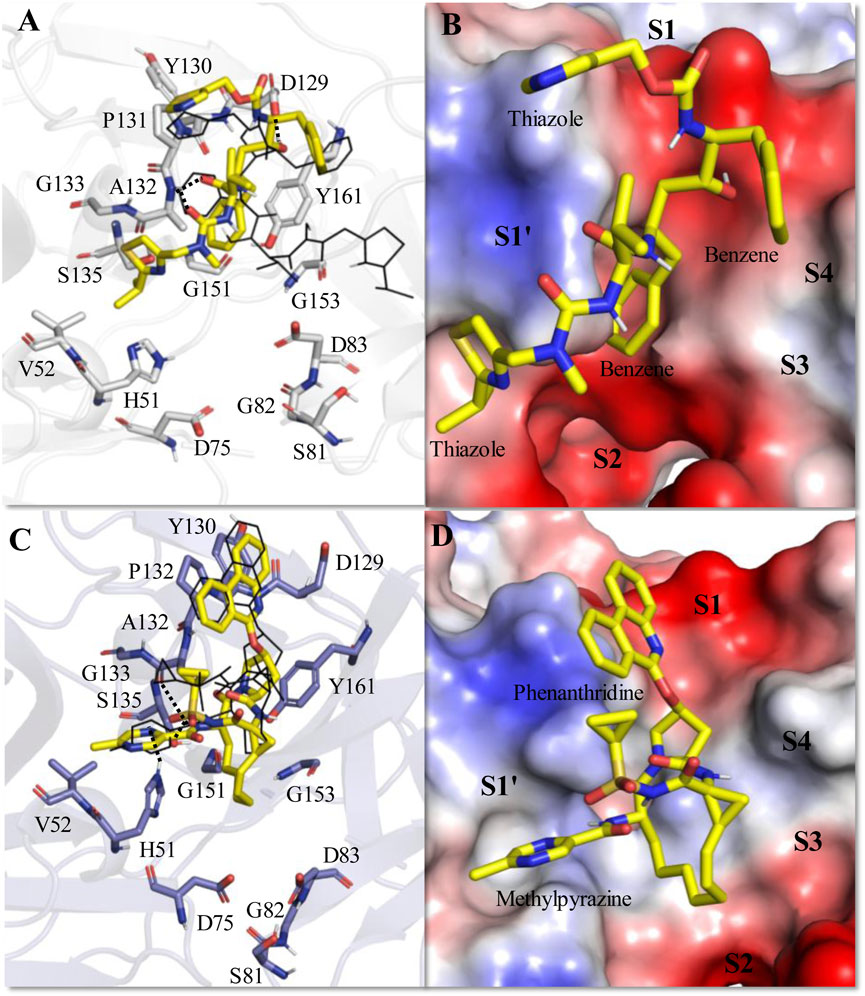
Figure 4. Average MD-simulated structures of (A, B) ZIKV NS2B-NS3-Ritonavir and (C, D) ZIKV NS2B-NS3-Paritaprevir complexes. The initial docking conformations of Ritonavir and Paritaprevir are shown in lines (in black color). These drugs bound to the electrostatic potential surface of the NS2B-NS3 protease and different substrate sites (S1–S4 and S1′) of the protease are also shown (B, D). The red and blue colors refer to the negative and positive electrostatic potential regions respectively. Important rings of Ritonavir and Paritaprevir are mentioned. Hydrogen bonding interactions are shown by dotted lines.
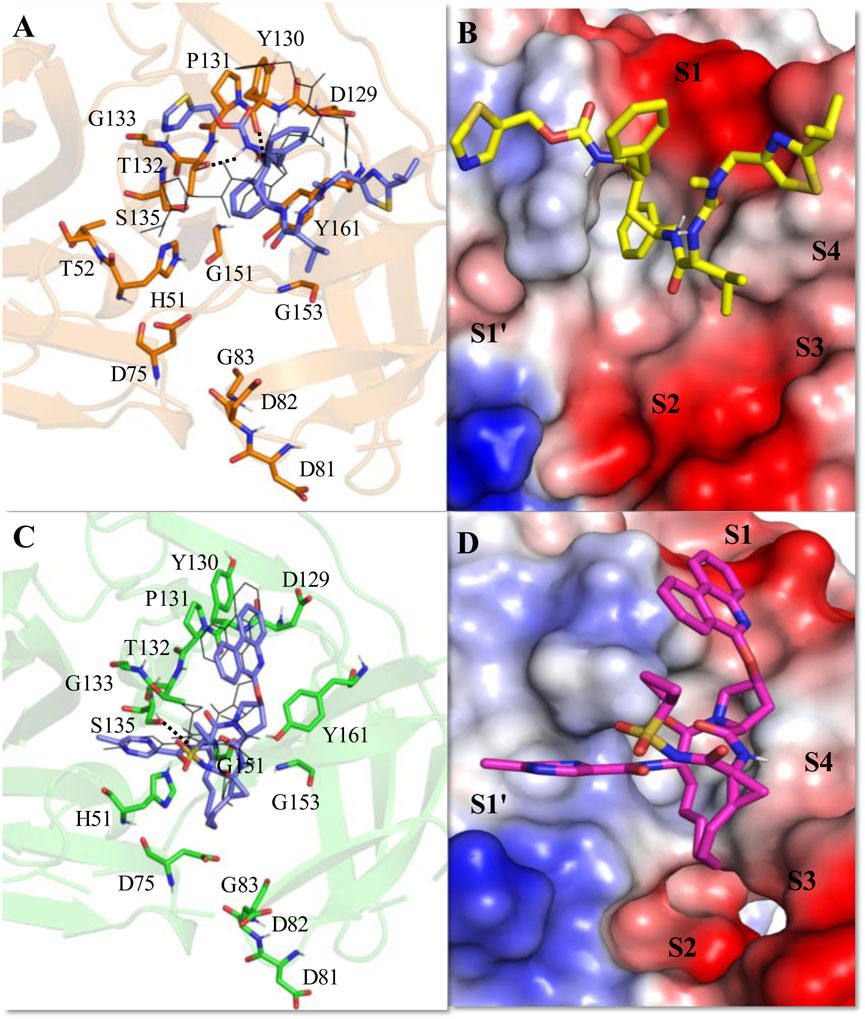
Figure 5. Average MD-simulated structures of (A, B) WNV NS2B-NS3-Ritonavir and (C, D) WNV NS2B-NS3-Paritaprevir complexes. The initial conformations of Ritonavir and Paritaprevir (generated after superimpositions of ZIKV protease-Ritonavir and ZIKV protease-Paritaprevir complexes onto the crystal structure of WNV protease (PDB ID (2FP7)) are shown in lines (in black color). Hydrogen bonding interactions are shown by dotted lines. These drugs bound to the electrostatic potential surface of the NS2B-NS3 protease and different substrate sites (S1–S4 and S1′) of the protease are also shown (B, D). The red and blue colors refer to the negative and positive electrostatic potential regions respectively.
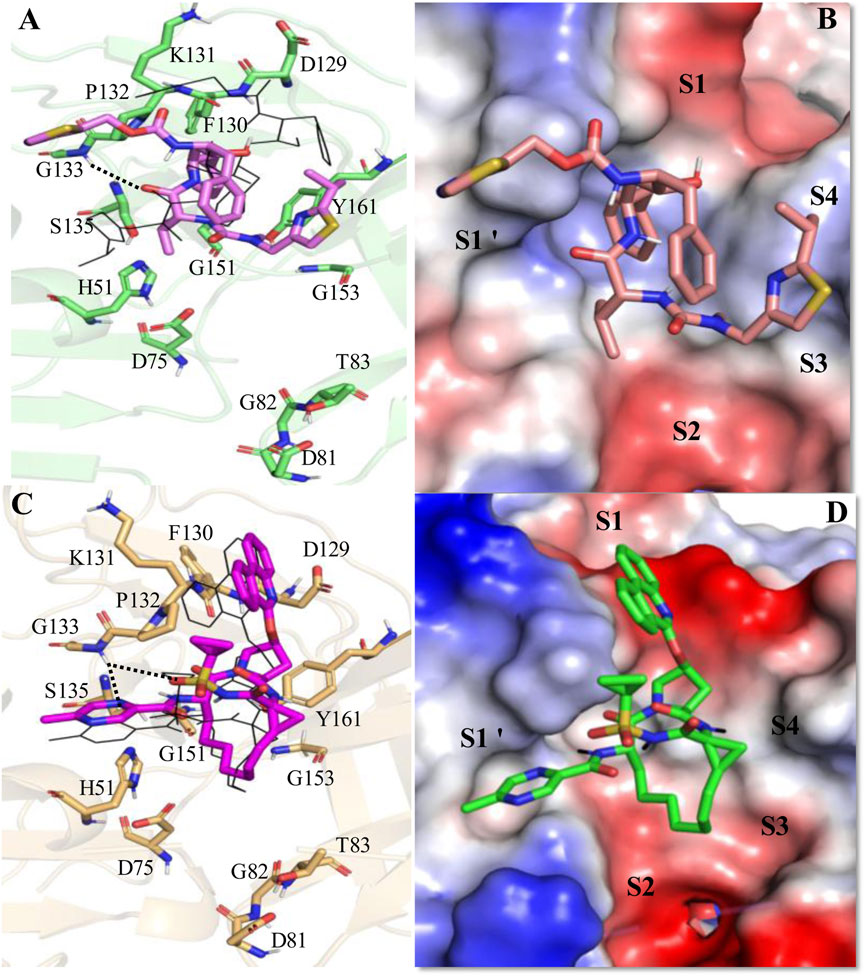
Figure 6. Average MD-simulated structures of (A, B) DENV NS2B-NS3-Ritonavir and (C, D) DENV NS2B-NS3-Paritaprevir complexes. The initial conformations of Ritonavir and Paritaprevir (generated after superimposition of the ZIKV protease-Ritonavir and ZIKV protease-Paritaprevir complexes onto the crystal structure of DENV protease (PDB ID 3U1I)) are shown in lines (in black color). Dotted lines show hydrogen bonding interactions. These drugs bound to the electrostatic potential surface of the NS2B-NS3 protease and different substrate sites (S1–S4 and S1′) of the protease are also shown (B, D). The red and blue colors refer to the negative and positive electrostatic potential regions respectively.

Table 3. Gibb’s binding free energies (ΔGbind) of different protease-drug complexes obtained by using the MM/PBSA method.
If we compare the ΔGbind values of different drugs bound to the ZIKV protease, they follow the order Ritonavir > Paritaprevir > Lopinavir > Saquinavir > Indinavir (Table 3). The contributions of different energies to ΔGbind indicate that the higher stabilities of Ritonavir and Paritaprevir arise mainly due to the favorable van der Wall and non-polar interactions with the protease (Supplementary Table S1). Therefore, Ritonavir and Paritaprevir may act as potent inhibitors of the NS2B-NS3 protease of the ZIKV.
The average MD simulated structure of the ZIKV protease-Ritonavir complex indicates that the tail thiazole ring of Ritonavir is quite mobile and adopts a different conformation than its original docked conformation (Figures 4A, B). During the MD simulation, the tail thiazole ring of Ritonavir moved from the S3 site and was placed in between the S2 and S1′ sites. The head thiazole ring of Ritonavir is also slightly moved away from the S1 pocket (Figure 4B). In this conformation, the head and tail thiazole rings of Ritonavir make a T-shaped stacking interaction with P131 and V52 respectively. It also makes two weak hydrogen bonds (<50% occupancy) with D129, and A132 of the protease (Figure 4A). One of its benzene rings makes a stable π-π stacking interaction with Y161 and the tail thiazole ring makes hydrophobic interactions with A132 and G133. This indicates that Ritonavir is tightly bound to the protease, which is evident from its ΔGbind of −17.44 ± 3.18 kcal/mol (Table 3). This complex is about 12 kcal/mol more stable than the HIV-Ritonavir complex (Table 3). This suggests that Ritonavir would bind to the ZIKV protease more strongly than that of HIV protease, and therefore, it may act as a better drug to inhibit the ZIKV protease activities. Interestingly, the docked structure of Paritaprevir did not change much in the MD simulation. Its phenanthridine ring is placed in the S1 site and the methylpyrazine ring is placed in the S1′ site as was obtained in the docked conformation (Figures 4C, D). In this conformation, it makes two strong π-π interactions with P132, and Y161 and several hydrophobic interactions with different residues of the protease (Figure 4C). Its tail methyl pyrazine ring can make a transient hydrogen bond with H51 and the NH group can make two hydrogen bonds with S135 and A132 (Figure 4C). For these reasons, its ΔGbind is −14.25 ± 3.11 kcal/mol, which is only about 3 kcal/mol less stable than that of the ZIKV protease-Ritonavir complex (Table 3).
During MD-simulation, Lopinavir slightly moved away from its initial docking conformation mainly because of the rotation of its dimethylphenoxy group away from the S1 site (Supplementary Figure S1). However, its tetrahydropyrimidine ring remained intact in its initial position. Because of this, the latter group made one moderate hydrogen bond with Y161 (62% occupancy), and two weak hydrogen bonds with G151 (34% occupation), and G153 (30% occupation). Because of these interactions, its ΔGbind is calculated to be −7.34 ± 2.56 kcal/mol, which is about 10 kcal/mol less negative than that of the ZIKV protease-Ritonavir complex (Table 3). The MD-simulated structure of Saquinavir is similar to its docking structure (Supplementary Figure S2). Although it remains within the binding pocket of the protease, it does not make any stable hydrogen bond with the protease (Supplementary Figure S2). Its stability is mainly governed by two weak π-π interactions with 161 of the protease. For this reason, its ΔGbind is computed to be −5.03 ± 2.98 kcal/mol, which is about 12 kcal/mol less stable than that of the ZIKV-protease-Ritonavir complex (Table 3). Interestingly, during MD simulation, Indinavir adopted a folded confirmation (Supplementary Figure S3), wherein it makes two weak hydrogen bonds (<50% occupancy) with S135 and H51. However, it failed to make the π-π stacking interaction with Y161 (Supplementary Figure S3). It is also unable to make any hydrophobic interactions with the protease. For this reason, its binding free energy is the lowest (−2.95 ± 2.30 kcal/mol) among all drugs studied herein (Table 3).
It should be mentioned that in an earlier experimental study (Li et al., 2018), a small molecule ligand (5-amino-1-((4-methoxyphenyl)sulphonyl)-1H-pyrazol-3-yl benzoate) was found to covalently bound to the ZIKV protease. Although the complete structure of the ligand could not be crystallized, the X-ray study (Li et al., 2018) resolved a fragment of the ligand (benzoyl moiety), occupying the S1 site of the protease. In this conformation, the benzoyl moiety was found to make a π-π -stacking interaction with Y161 in similar manner as obtained for Paritaprevir and other ligands. Another small molecule ligand (1H-benzo[d]imidazole-1-yl methanol) was also observed to be stabilized by making a π-π -stacking interaction with Y161 (Zhang et al., 2016).
In the active site of the WNV protease, Ritonavir adopted a new conformation (Figures 5A, B), which is significantly different from its conformation adopted in the active site of ZIKV protease (Figures 4A, B). This happened because of the rotation of the head thiazole group of Ritonavir away from the S1 site toward the S1′ site (Figures 5 A, B). Similarly, the tail thiazole ring moved toward the S4 site. Because of these, it failed to interact with P131 and T52 of WNV protease as noticed in the case of ZIKV protease (Figure 4A). This conformation is mainly stabilized by a strong π-π stacking interaction with Tyr161 and transient hydrogen bonds with T132 and Y130 (Figure 5A). For these reasons, a ΔGbind of −7.43 ± 2.16 kcal/mol is obtained for the WNV-protease-Ritonavir complex (Table 3). Therefore, Ritonavir is loosely bound to the WNV protease compared to that of ZIKV protease. However, its binding to WNV protease is about 2 kcal/mol more stable than its binding of HIV protease (Table 3). Interestingly, Paritaprevir is bound to the WNV protease in a similar manner as it binds to the ZIKV protease (Figures 5 C, D). Its binding to the WNV protease is mainly stabilized by strong π-π stacking interactions with Tyr161 and Pro131 (Figures 5 C, D). Its NH group makes a transient hydrogen bond with S135. It also makes several hydrophobic interactions with the protease. Its phenanthridine ring is placed in the S1 site and the methyl pyrazine ring is bound to the S1′ site. As the catalytic reactions are induced by the residues of the S1′ site, the placement of the methyl pyrazine ring of Paritaprevir in this site would affect the protease activities severely. Interestingly, a ΔGbind of −17.3 ± 0.25 kcal/mol is obtained for the WNV-Paritaprevir complex (Table 3), which is about 3 kcal/mol more stable than that of the ZIKV-Paritaprevir complex. This suggests that Paritaprevir binds to the WNV protease more strongly compared to the ZIKV protease.
In the substrate binding site of DENV protease (Figure 6), Ritonavir undergoes a similar conformational change as noticed in the case of WNV protease (Figures 5A, B). However, it adopted a somewhat compact and folded structure compared to the extended conformation obtained in the case of ZIKV (Figure 4A) and WNV proteases (Figure 5A). It is mainly stabilized by making a weak hydrogen bond with G133 and π-π stacking interactions with P132, Y161, and V155 (Figures 6A, B). For these reasons, a ΔGbind of −11.51 ± 2.82 kcal/mol is obtained for the DENV-Ritonavir complex, which is about 3 kcal/mol more stable than that of the WNV protease-Ritonavir complex and about 6 kcal/mol more stable than the HIV-Ritonavir complex (Table 3). This indicates that Ritonavir would act as a better inhibitor of DENV protease compared to WNV and HIV proteases. However, as the ZIKV protease-Ritonavir complex is about 6 kcal/mol more stable than that of the Ritonavir-DENV protease complex (Table 3), Ritonavir would be more potent against the ZIKV protease. Interestingly, the binding mode of Paritaprevir (Figures 6C, D) is identical to its binding mode obtained in the case of ZIKV and WNV proteases. As Paritaprevir contains more hydrophobic groups, is heavier compared to Ritonavir, and contains a cyclic ring, it does not move much during the MD simulations. Further, due to strong π-π stacking interactions, it remains intact within the protease binding site. It is found that in the DENV protease active site, the head phenanthridine ring of Paritaprevir maintains its π-π stacking interaction with Y161 (Figures 6C, D). However, as DENV protease contains K131, (instead of P131 of ZIKV) its Phenanthridine ring failed to make another strong π-π stacking interaction with K131. Despite this, it made a relatively weak stacking interaction with P132 (Figure 6C). The tail methyl pyrazine ring and the NH group of Paritaprevir can also make weak hydrogen bonds with G133 (Figure 6C). As a result, a ΔGbind of −12.76 ± 2.91 kcal/mol is obtained for this complex, which is about 1 kcal/mol less stable than the ZIKV protease-Paritaprevir complex and about 4 kcal/mol less stable than that of the WNV protease-Paritaprevir complex (Table 3). These results indicate that Paritaprevir binds most strongly to the WNV protease and moderately strongly to the ZIKV and DENV proteases.
If we compare the structures of Ritonavir bound to different proteases, it appears that Ritonavir is quite flexible and can adopt different conformations within the active site of ZIKV, WNV, and DENV proteases (Figure 7). However, its binding to the ZIKV protease is stronger compared to other viral proteases considered here. Although the S1 site residues of ZIKV (D129, Y130, P131, A132) and WNV (D129, Y130, P131, T132) possess identical sequences and structures (Figures 7A, B), the less binding affinity of Ritonavir toward the WNV protease is presumably because of the inward movements of its S2 (S81-G83) and S4 site residues (V153-Y161) that push Ritonavir to move away from the active site (Figures 7 A, B). However, in the case of DENV, the S2 and S4 site residues moved away from the active site, thereby, providing Ritonavir more space to undergo further conformational changes (folding conformation). Further, as the S1 site (D129, F130, K131, P132) and S2 site residues (D81, G82, T83, and M84) of the DENV are significantly different from those of WNV in sequence and structure (Figures 7 C, D), Ritonavir faces different protein dynamics in the active site of these proteases. This ultimately produces different stabilities for Ritonavir (Table 3). However, interestingly, except for the S4 site residues, other binding site residues of ZIKV and WNV proteases do not move much during the binding of Paritaprevir to them (Figures 7 E, F). This is also true for the DENV protease (Figures 7G, H). We also noted that the binding of Paritaprevir to WNV and DENV proteases stabilizes the S2 site residues (Figures 7G, H) unlike in the case of the binding of Ritonavir (Figures 7C, D).
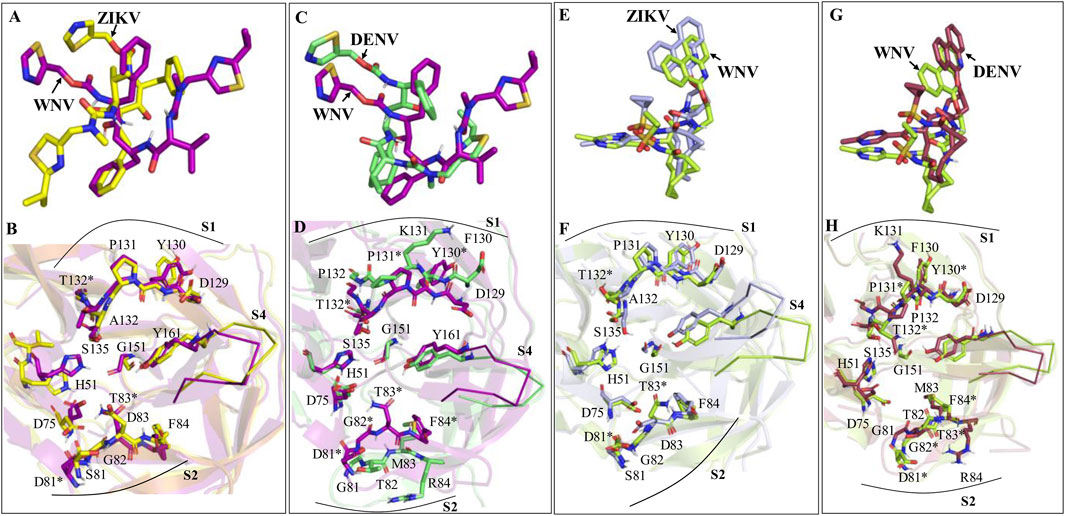
Figure 7. (A, B) Superimpositions of the WNV-Ritonavir complex onto the ZIKV-Ritonavir complex. (C, D) Superimpositions of the DENV-Ritonavir complex onto the WNV-Ritonavir complex. (E, F) Superimpositions of the WNV-Paritaprevir complex onto the ZIKV-Paritaprevir complex. (G, H) Superimpositions of the DENV-Paritaprevir complex onto the WNV-Paritaprevir complex. Some of the residues of ZIKV, WNV, and DENV are common. However, distinct residues of WNV (not present in ZIKV and DENV) are marked by *. Residues of S1 and S2 sites are shown by sticks and residues of S4 sites are shown by ribbons.
To further understand the role of protein dynamics in the ligand binding to the proteases of ZIKV, WNV, and DENV, the simulated complex structures of Paritaprevir bound to these proteases were superimposed to the crystal structures of ZIKV-7HQ (PDB ID 5H4I) (Zhang et al., 2016), WNV-benzoyl-Nle-K-R-R-H (PDB ID 2FP7) (Erbel et al., 2006) and DENV-tripeptide (PDB ID 3U1I) (Noble et al., 2012) complexes (Figures 8A–C). It is found that mainly S2 and S4 site residues of the ZIKV and WNV proteases undergo conformational changes during the ligand binding (Figures 8A, B) and a minor conformational change occurs in the S4 site residues of the DENV protease (Figure 8C). Moreover, important catalytic residues, such as S135, H51, and D75 are found to adopt different conformations depending on the nature of the ligand entering into the active site of the ZIKV and WNV proteases. These results indicate that active site residues of DENV are more rigid compared to those of ZIKV and WNV, and therefore, DENV would respond equally to different ligands irrespective of their nature (small molecules or peptides or peptidomimetics) (Zhang et al., 2016; Li and Kang, 2021; Robin et al., 2009; Li et al., 2018; Li et al., 2017; Pant and Jena, 2022; Phoo et al., 2018; Knox et al., 2006; Roy et al., 2017; Schüller et al., 2011; Behnam et al., 2015; Li et al., 2015; Grazia Martina et al., 2021; Song et al., 2021; Pushpakom et al., 2019; Pant et al., 2023; Pant and Jena, 2023).
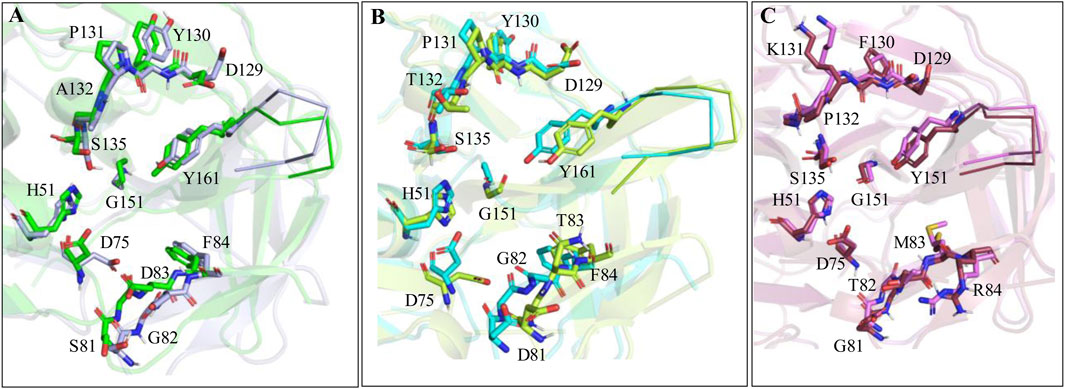
Figure 8. (A) Superimposition of ZIKV protease-Paritaprevir complex structure onto the crystal structure of ZIKV protease (PDB ID 5H4I). (B) Superimposition of WNV-protease-Paritaprevir complex onto the crystal structure of the WNV protease (PDB ID 2PF7). (C) Superimposition of DENV-Paritaprevir complex onto the crystal structure of DENV protease (PDB ID 3U1I).
We also noticed that Paritaprevir binds to the active site of ZIKV, WNV, and DENV proteases similarly as observed for the 7HQ and peptide ligands (Supplementary Figure S4). In an earlier study (Viswanathan et al., 2014) about 2211 different DENV strains from four different DENV sterotypes were compared. It was found that about 40% of residues of DENV are invariant, while about 66% of active site amino acid sequences are identical (Viswanathan et al., 2014). Therefore, Paritaprevir may bind to all strains and sterotypes of DENV similarly as obtained here for DENV3 protease. Therefore, it may inhibit the activities of all DENV stereotypes. It should be mentioned that as the substrate binding site of these proteases is acidic in nature, peptide or peptidomimetic inhibitors containing basic residues were thought to be effective against these proteases (Viswanathan et al., 2014). However, the highly negative ΔGbind values obtained here (Table 2) suggest that neutral and small molecule ligands like Ritonavir and Paritaprevir may also inhibit the protease activities of ZIKV, WNV, and DENV effectively. Among the two, Paritaprevir may act as a pan-antiviral against the Zika, Dengue, and West Nile viral diseases due to its strong binding with their NS2B-NS3 proteases. Interestingly, as Paritaprevir in combination with other drugs has already been approved for its use against different hepatitis C virus genotypes (Klibanov et al., 2015; Keating, 2016; Hussaini, 2016; Menon et al., 2016), it would be safe for humans and can be repurposed against ZIKV, DENV, and WNV viral diseases. However, as the computational results obtained herein are force-field dependent and no wet-lab experiments were performed to validate the computational findings, it is necessary to verify the efficacy of Paritaprevir against ZIKV, DENV, and WNV proteases by conducting in vitro enzymatic assays or viral replication studies.
Among the five screened drugs, such as Ritonavir, Saquinavir, Indinavir, Paritaprevir, and Lopinavir, the anti-HIV drug Ritonavir (ΔGbind = −17.44 ± 3.18) and the anti-HCV drug Paritaprevir (ΔGbind = −14.25 ± 3.11) are found to bind to the ZIKV NS2B-NS3 protease quite strongly. Remarkably, Ritonavir is found to bind to ZIKV protease significantly more strongly than that of the HIV Protease. Therefore, it is proposed that the repurposing of these drugs against the ZIKV may potentially decrease the virus loads in patients. It is further found that Paritaprevir binds to WNV protease even more strongly than ZIKV protease with a ΔGbind = −17.3 ± 2.55 kcal/mol. Hence, Paritaprevir may also act as a potent drug to reduce the virus loads in patients suffering from West Nile Viral disease. However, Ritonavir may not be a good drug candidate against the WNV protease due to its relatively loose binding to the WNV protease (ΔGbind = −7.43 ± 2.16). Due to the moderately strong binding of Ritonavir (ΔGbind = −11.51 ± 2.82) and Paritaprevir (ΔGbind = −12.76 ± 2.91) to the DENV protease, they may be effective for patients suffering from Dengue viral disease. These results also indicate that Paritaprevir, due to its better binding with proteases of ZIKV, WNV, and DENV, may act as a pan-antiviral against these viral diseases. However, experimental biochemical studies evaluating the binding and toxicological effects of these drugs are necessary before confirming their use against these viral diseases. Nevertheless, this study is expected to aid in the further design and analysis of drugs against these viral diseases considering the main scaffolds of Ritonavir and Paritaprevir.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
RY: Formal Analysis, Investigation, Writing–review and editing. NJ: Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. Funding was received from the Council of Scientific and Industrial Research (CSIR, India) via grant 01/3061/21/EMR II.
NJ is thankful to the Council of Scientific and Industrial Research (CSIR, India) for financial support (Grant No-01-3061/21/EMR II).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2025.1524951/full#supplementary-material
Aleshin, A. E., Shiryaev, S. A., Strongin, A. Y., and Liddington, R. C. (2007). Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 16, 795–806. doi:10.1110/ps.072753207
Baxter, C. A., Murray, C. W., Clark, D. E., Westhead, D. R., and Eldridge, M. D. (1998). Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins Struct. Funct. Bioinforma. 33, 367–382. doi:10.1002/(SICI)1097-0134(19981115)33:3<367::AID-PROT6>3.0.CO;2-W
Behnam, M. A., Graf, D., Bartenschlager, R., Zlotos, D. P., and Klein, C. D. (2015). Discovery of nanomolar dengue and West Nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue. J. Med. Chem. 58, 9354–9370. doi:10.1021/acs.jmedchem.5b01441
Berendsen, H. J., Postma, J. van, Van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi:10.1063/1.448118
Bolcato, G., Bissaro, M., Pavan, M., Sturlese, M., and Moro, S. (2020). Targeting the coronavirus SARS-CoV-2: computational insights into the mechanism of action of the protease inhibitors lopinavir, ritonavir and nelfinavir. Sci. Rep. 10, 20927. doi:10.1038/s41598-020-77700-z
Broutet, N., Krauer, F., Riesen, M., Khalakdina, A., Almiron, M., Aldighieri, S., et al. (2016). Zika virus as a cause of neurologic disorders. N. Engl. J. Med. 374, 1506–1509. doi:10.1056/NEJMp1602708
Carod-Artal, F. J. (2018). Neurological complications of Zika virus infection. Expert Rev. Anti-infective Ther. 16, 399–410. doi:10.1080/14787210.2018.1466702
Case, D. A., Babin, V., Berryman, J. T., Betz, R. M., Cai, Q., Cerutti, D. S., et al. (2014). Amber 14. San Francisco: University of California, 1–826.
Chang, H.-H., Huber, R. G., Bond, P. J., Grad, Y. H., Camerini, D., Maurer-Stroh, S., et al. (2017). Systematic analysis of protein identity between Zika virus and other arthropod-borne viruses. Bull. World Health Organ. 95, 517–525I. doi:10.2471/blt.16.182105
Cregar-Hernandez, L., Jiao, G.-S., Johnson, A. T., Lehrer, A. T., Wong, T. A. S., and Margosiak, S. A. (2011). Small molecule pan-dengue and West Nile virus NS3 protease inhibitors. Antivir. Chem. Chemother. 21, 209–217. doi:10.3851/imp1767
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: an N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
de Araújo, T. V. B., de Alencar Ximenes, R. A., de Barros Miranda-Filho, D., Souza, W. V., Montarroyos, U. R., de Melo, A. P. L., et al. (2018). Association between microcephaly, Zika virus infection, and other risk factors in Brazil: final report of a case-control study. Lancet Infect. Dis. 18, 328–336. doi:10.1016/S1473-3099(17)30727-2
El Sahili, A., Soh, T. S., Schiltz, J., Gharbi-Ayachi, A., Seh, C. C., Shi, P. Y., et al. (2019). NS5 from dengue virus serotype 2 can adopt a conformation analogous to that of its Zika virus and Japanese encephalitis virus homologues. J. virology 94, 012944-19–e11128. doi:10.1128/jvi.01294-19
Erbel, P., Schiering, N., D’Arcy, A., Renatus, M., Kroemer, M., Lim, S. P., et al. (2006). Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. and Mol. Biol. 13, 372–373. doi:10.1038/nsmb1073
Falgout, B., Pethel, M., Zhang, Y.-M., and Lai, C.-J. (1991). Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. virology 65, 2467–2475. doi:10.1128/jvi.65.5.2467-2475.1991
Grazia Martina, M., Vicenti, I., Bauer, L., Crespan, E., Rango, E., Boccuto, A., et al. (2021). Bithiazole inhibitors of phosphatidylinositol 4 kinase (PI4KIIIβ) as broad spectrum antivirals blocking the replication of SARS CoV 2, Zika virus, and human rhinoviruses. ChemMedChem 16, 3548–3552. doi:10.1002/cmdc.202100483
Gurung, A. B., Ali, M. A., Lee, J., Farah, M. A., and Al-Anazi, K. M. (2021). The potential of paritaprevir and emetine as inhibitors of SARS-CoV-2 RdRp. Saudi J. Biol. Sci. 28, 1426–1432. doi:10.1016/j.sjbs.2020.11.078
He, X., Man, V. H., Yang, W., Lee, T. S., and Wang, J. (2020). A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 114502. doi:10.1063/5.0019056
Hersh, A. M., Gundacker, N. D., and Boltax, J. (2017). Zika-associated shock and multi-organ dysfunction. Ann. Am. Thorac. Soc. 14, 1706–1708. doi:10.1513/AnnalsATS.201612-988CC
Homeyer, N., and Gohlke, H. (2012). Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Mol. Inf. 31, 114–122. doi:10.1002/minf.201100135
Hou, T., Wang, J., Li, Y., and Wang, W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82. doi:10.1021/ci100275a
Huang, L., and Roux, B. (2013). Automated force field parameterization for nonpolarizable and polarizable atomic models based on ab initio target data. J. Chem. Theory Comput. 9, 3543–3556. doi:10.1021/ct4003477
Hussaini, T. (2016). Paritaprevir/ritonavir-ombitasvir and dasabuvir, the 3D regimen for the treatment of chronic hepatitis C virus infection: a concise review. Hepat. Med. 8 (8), 61–68. doi:10.2147/HMER.S72429
Jena, N. R. (2021). Drug targets, mechanisms of drug action, and therapeutics against SARS-CoV-2. Chem. Phys. Impact 2, 100011. doi:10.1016/j.chphi.2021.100011
Jones, G., Willett, P., Glen, R. C., Leach, A. R., and Taylor, R. (1997). Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748. doi:10.1006/jmbi.1996.0897
Keating, G. M. (2016). Ombitasvir/paritaprevir/ritonavir: a review in chronic HCV genotype 4 infection. Drugs 76, 1203–1211. doi:10.1007/s40265-016-0612-1
Kempf, D., and Sham, H. (1996). HIV protease inhibitors. Curr. Pharm. Des. 2, 225–246. doi:10.2174/1381612802666220921175941
Kempf, D. J., Marsh, K. C., Denissen, J. F., McDonald, E., Vasavanonda, S., Flentge, C. A., et al. (1995). ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. 92, 2484–2488. doi:10.1073/pnas.92.7.2484
Kiang, T. K., Wilby, K. J., and Ensom, M. H. (2013). Telaprevir: clinical pharmacokinetics, pharmacodynamics, and drug–drug interactions. Clin. Pharmacokinet. 52, 487–510. doi:10.1007/s40262-013-0053-x
Klibanov, O. M., Gale, S. E., and Santevecchi, B. (2015). Ombitasvir/paritaprevir/ritonavir and dasabuvir tablets for hepatitis C virus genotype 1 infection. Ann. Pharmacother. 49, 566–581. doi:10.1177/1060028015570729
Knox, J. E., Ma, N. L., Yin, Z., Patel, S. J., Wang, W. L., Chan, W. L., et al. (2006). Peptide inhibitors of West Nile NS3 protease: SAR study of tetrapeptide aldehyde inhibitors. J. Med. Chem. 49, 6585–6590. doi:10.1021/jm0607606
Kong, W., Li, H., and Zhu, J. (2018). Zika virus: the transboundary pathogen from mosquito and updates. Microb. Pathog. 114, 476–482. doi:10.1016/j.micpath.2017.12.031
Korb, O., Stutzle, T., and Exner, T. E. (2009). Empirical scoring functions for advanced protein−ligand docking with PLANTS. J. Chem. Inf. Model. 49, 84–96. doi:10.1021/ci800298z
Koudriakova, T., Iatsimirskaia, E., Utkin, I., Gangl, E., Vouros, P., Storozhuk, E., et al. (1998). Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metabolism Dispos. 26, 552–561. doi:10.3390/2Fijms23179866
Kräutler, V., Van Gunsteren, W. F., and Hünenberger, P. H. (2001). A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508. doi:10.1002/1096-987x(20010415)22:5<501::aid-jcc1021>3.0.co;2-v
Kuno, G., Chang, G.-J. J., Tsuchiya, K. R., Karabatsos, N., and Cropp, C. B. (1998). Phylogeny of the genus Flavivirus. J. virology 72, 73–83. doi:10.1128/jvi.72.1.73-83.1998
Lam, J. T., and Salazar, L. (2016). New combination antiviral for the treatment of hepatitis C. Am. J. Health-System Pharm. 73, 1042–1050. doi:10.2146/ajhp150163
Li, L., Basavannacharya, C., Chan, K. W. K., Shang, L., Vasudevan, S. G., and Yin, Z. (2015). Structure guided discovery of a Novel non peptide inhibitor of dengue virus NS2B–NS3 protease. Chem. Biol. and drug Des. 86, 255–264. doi:10.1111/cbdd.12500
Li, Q., and Kang, C. (2021). Structure and dynamics of Zika virus protease and its insights into inhibitor design. Biomedicines 9, 1044. doi:10.3390/biomedicines9081044
Li, Y., Han, L., Liu, Z., and Wang, R. (2014). Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 54, 1717–1736. doi:10.1021/ci500081m
Li, Y., Zhang, Z., Phoo, W. W., Loh, Y. R., Wang, W., Liu, S., et al. (2017). Structural dynamics of Zika virus NS2B-NS3 protease binding to dipeptide inhibitors. Structure 25, 1242–1250. doi:10.1016/j.str.2017.06.006
Li, Y., Zhang, Z., Phoo, W. W., Loh, Y. R., Yang, H. Y., et al. (2018). Structural insights into the inhibition of Zika virus NS2B-NS3 protease by a small-molecule inhibitor. Structure 26, 555–564. doi:10.1016/j.str.2018.02.005
Liebeschuetz, J. W., Cole, J. C., and Korb, O. (2012). Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Computer-aided Mol. Des. 26, 737–748. doi:10.1007/s10822-012-9551-4
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi:10.1021/acs.jctc.5b00255
Mark, P., and Nilsson, L. (2001). Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105, 9954–9960. doi:10.1021/jp003020w
Menon, R. M., Klein, C. E., Podsadecki, T. J., Chiu, Y.-L., Dutta, S., and Awni, W. M. (2016). Br. J. Clin. Pharmacol. 24 (5), 929–940. doi:10.1111/bcp.12873
Meza, J. C. (2010). Steepest descent. Wiley Interdiscip. Rev. Comput. Stat. 2, 719–722. doi:10.1002/wics.117
Mlakar, J., Korva, M., Tul, N., Popović, M., Poljšak-Prijatelj, M., Mraz, J., et al. (2016). Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958. doi:10.1056/nejmoa1600651
Moghadam, S. R. J., Bayrami, S., Moghadam, S. J., Golrokhi, R., Golsoorat Pahlaviani, F., and SeyedAlinaghi, S. (2016). Zika virus: A review of literature. Asian Pac. J. Trop. Biomed. 6, 989–994. doi:10.1016/j.apjtb.2016.09.007
Musso, D., and Gubler, D. J. (2016). Zika virus. Clin. Microbiol. Rev. 29, 487–524. doi:10.1128/cmr.00072-15
Nash, D., Mostashari, F., Fine, A., Miller, J., O'Leary, D., Murray, K., et al. (2001). The outbreak of West Nile virus infection in the New York City area in 1999. N. Engl. J. Med. 344, 1807–1814. doi:10.1056/nejm200106143442401
Nitsche, C. (2019). Proteases from dengue, West Nile and Zika viruses as drug targets. Biophys. Rev. 11, 157–165. doi:10.1007/s12551-019-00508-3
Noble, C. G., Seh, C. C., Chao, A. T., and Shi, P. Y. (2012). Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virology 86, 438–446. doi:10.1128/jvi.06225-11
Onufriev, A. V., and Izadi, S. (2018). Water models for biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1347. doi:10.1002/wcms.1347
Paixao, E. S., Cardim, L. L., Costa, M. C., Brickley, E. B., de Carvalho-Sauer, R. C. O., Carmo, E. H., et al. (2022). Mortality from congenital Zika syndrome—nationwide cohort study in Brazil. N. Engl. J. Med. 386, 757–767. doi:10.1056/nejmoa2101195
Pant, S., Bhattacharya, G., and Jena, N. R. (2023). Structures and dynamics of peptide and peptidomimetic inhibitors bound to the NS2B-NS3 protease of the ZIKA virus. J. Biomol. Struct. Dyn. 41, 3076–3088. doi:10.1080/07391102.2022.2045223
Pant, S., and Jena, N. R. (2022). C-terminal extended hexapeptides as potent inhibitors of the NS2B-NS3 protease of the ZIKA virus. Front. Med. 9, 921060. doi:10.3389/fmed.2022.921060
Pant, S., and Jena, N. R. (2023). Repurposing of antiparasitic drugs against the NS2B-NS3 protease of the Zika virus. J. Biomol. Struct. Dyn. 42, 10101–10113. doi:10.1080/07391102.2023.2255648
Pant, S., and Jena, N. R. (2024). Binding of nucleotide inhibitors to the NS5 RdRp of the ZIKA virus in the replication initiation state. Curr. Med. Chem. 32, 2460–2476. doi:10.2174/0109298673259914231213052438
Phoo, W. W., Li, Y., Zhang, Z., Lee, M. Y., Loh, Y. R., Tan, Y. B., et al. (2016). Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 7, 13410. doi:10.1038/ncomms13410
Phoo, W. W., Zhang, Z., Wirawan, M., Chew, E. J. C., Chew, A. B. L., Kouretova, J., et al. (2018). Structures of Zika virus NS2B-NS3 protease in complex with peptidomimetic inhibitors. Antivir. Res. 160, 17–24. doi:10.1016/j.antiviral.2018.10.006
Poland, G. A., Kennedy, R. B., Ovsyannikova, I. G., Palacios, R., Ho, P. L., and Kalil, J. (2018). Development of vaccines against Zika virus. Lancet Infect. Dis. 18, e211–e219. doi:10.1016/s1473-3099(18)30063-x
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58. doi:10.1038/nrd.2018.168
Riwar, L., Trapp, N., Kuhn, B., and Diederich, F. (2017). Substituent effects in parallel displaced π–π stacking interactions: distance matters. Angew. Chem. Int. Ed. 56, 11252–11257. doi:10.1002/anie.201703744
Robin, G., Chappell, K., Stoermer, M. J., Hu, S. H., Young, P. R., Fairlie, D. P., et al. (2009). Structure of West Nile virus NS3 protease: ligand stabilization of the catalytic conformation. J. Mol. Biol. 385, 1568–1577. doi:10.1016/j.jmb.2008.11.026
Roy, A., Lim, L., Srivastava, S., Lu, Y., and Song, J. (2017). Solution conformations of Zika NS2B-NS3pro and its inhibition by natural products from edible plants. PloS one 12, e0180632. doi:10.1371/journal.pone.0180632
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. doi:10.1016/0021-9991(77)90098-5
Saini, G., Dalal, V., Gupta, D. N., Sharma, N., Kumar, P., and Sharma, A. K. (2021). A molecular docking and dynamic approach to screen inhibitors against ZnuA1 of Candidatus Liberibacter asiaticus. Mol. Simul. 47, 510–525. doi:10.1080/08927022.2021.1888948
Saini, G., Dalal, V., Savita, B. K., Sharma, N., Kumar, P., and Sharma, A. K. (2019). Molecular docking and dynamic approach to virtual screen inhibitors against Esbp of Candidatus Liberibacter asiaticus. J. Mol. Graph. Model. 92, 329–340. doi:10.1016/j.jmgm.2019.08.012
Schüller, A., Yin, Z., Chia, C. B., Doan, D. N. P., Kim, H. K., Shang, L., et al. (2011). Tripeptide inhibitors of dengue and West Nile virus NS2B–NS3 protease. Antivir. Res. 92, 96–101. doi:10.1016/j.antiviral.2011.07.002
Shen, J., Serby, M., Reed, A., Lee, A. J., Menon, R., Zhang, X., et al. (2016). Metabolism and disposition of hepatitis C polymerase inhibitor dasabuvir in humans. Drug Metabolism Dispos. 44, 1139–1147. doi:10.1124/dmd.115.067512
Song, W., Zhang, H., Zhang, Y., Li, R., Han, Y., Lin, Y., et al. (2021). Repurposing clinical drugs is a promising strategy to discover drugs against Zika virus infection. Front. Med. 15, 404–415. doi:10.1007/s11684-021-0834-9
Štich, I., Car, R., Parrinello, M., and Baroni, S. (1989). Conjugate gradient minimization of the energy functional: a new method for electronic structure calculation. Phys. Rev. B 39, 4997–5004. doi:10.1103/physrevb.39.4997
Talevi, A., and Bellera, C. L. (2020). Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 15, 397–401. doi:10.1080/17460441.2020.1704729
Tie, Y., Kovalevsky, A. Y., Boross, P., Wang, Y. F., Ghosh, A. K., Tozser, J., et al. (2007). Atomic resolution crystal structures of HIV 1 protease and mutants V82A and I84V with saquinavir. Proteins Struct. Funct. Bioinforma. 67, 232–242. doi:10.1002/prot.21304
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., and Taylor, R. D. (2003). Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinforma. 52, 609–623. doi:10.1002/prot.10465
Viswanathan, U., Tomlinson, S. M., Fonner, J. M., Mock, S. A., and Watowich, S. J. (2014). Identification of a novel inhibitor of dengue virus protease through use of a virtual screening drug discovery Web portal. J. Chem. Inf. Model. 54, 2816–2825. doi:10.1021/ci500531r
V Kleandrova, V., and Speck-Planche, A. (2021). The urgent need for pan-antiviral agents: from multitarget discovery to multiscale design. Future Med. Chem. 13, 5–8. doi:10.4155/fmc-2020-0134
Wahaab, A., Mustafa, B. E., Hameed, M., Stevenson, N. J., Anwar, M. N., Liu, K., et al. (2021). Potential role of flavivirus NS2B-NS3 proteases in viral pathogenesis and anti-flavivirus drug discovery employing animal cells and models: a review. Viruses 14, 44. doi:10.3390/v14010044
Walmsley, S., Bernstein, B., King, M., Arribas, J., Beall, G., Ruane, P., et al. (2002). Lopinavir–ritonavir versus nelfinavir for the initial treatment of HIV infection. N. Engl. J. Med. 346, 2039–2046. doi:10.1056/nejmoa012354
Wang, E., Sun, H., Wang, J., Wang, Z., Liu, H., Zhang, J. Z. H., et al. (2019). End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 119, 9478–9508. doi:10.1021/acs.chemrev.9b00055
Wang, Y., Liu, Z., Brunzelle, J. S., Kovari, I. A., Dewdney, T. G., Reiter, S. J., et al. (2011). The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochem. Biophysical Res. Commun. 412, 737–742. doi:10.1016/j.bbrc.2011.08.045
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082. doi:10.1093/nar/gkx1037
Yuan, S., Chan, J. F.-W., den-Haan, H., Chik, K. K. H., Zhang, A. J., Chan, C. C. S., et al. (2017). Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antivir. Res. 145, 33–43. doi:10.1016/j.antiviral.2017.07.007
Keywords: Zika virus, Dengue virus, West Nile virus, NS2B-NS3 protease, pan antiviral, docking, MD-simulations
Citation: Yadav RP and Jena NR (2025) Paritaprevir as a pan-antiviral against different flaviviruses. Front. Mol. Biosci. 12:1524951. doi: 10.3389/fmolb.2025.1524951
Received: 08 November 2024; Accepted: 07 March 2025;
Published: 03 April 2025.
Edited by:
Poonam Dhankhar, Cornell University, United StatesReviewed by:
Nitin Sharma, Washington University in St. Louis, United StatesCopyright © 2025 Yadav and Jena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: N.R. Jena, bnJqZW5hQGlpaXRkbWouYWMuaW4=, bnJqZW5hQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.