 Hyunju Cho
Hyunju Cho- Center for Biomolecular and Cellular Structure, Institute for Basic Science, Daejeon, Republic of Korea
Huntington’s disease (HD) is primarily caused by the aberrant aggregation of the N-terminal exon 1 fragment of mutant huntingtin protein (mHttex1) with expanded polyglutamine (polyQ) repeats in neurons. The first 17 amino acids of the N-terminus of Httex1 (N17 domain) immediately preceding the polyQ repeat domain are evolutionarily conserved across vertebrates and play multifaceted roles in the pathogenesis of HD. Due to its amphipathic helical properties, the N17 domain, both alone and when membrane-associated, promotes mHttEx1 aggregation. Diverse post-translational modifications (PTMs) in the N17 domain alter the aggregation state, thus modulating the cellular toxicity of mHttex1. Furthermore, the N17 domain serves as a nuclear export signal (NES) and mediates the cytoplasmic localization of mHttex1. This review summarizes the four main roles of the N17 domain in regulating HD pathology and discusses potential therapeutic approaches targeting this N17 domain to mitigate HD progression.
Introduction
Huntington’s disease (HD) is the most common dominantly inherited neurological disorder, and is characterized by progressive involuntary chorea, cognitive dysfunction, psychiatric disturbances, and premature death (Bates et al., 2015). HD is caused by abnormal expansion of CAG (polyQ) repeats in exon 1 of the huntingtin gene. PolyQ repeats longer than 36 are pathogenic and positively correlate with an increased propensity to form intracellular aggregates and increased disease severity (Scherzinger et al., 1997; Gusella and MacDonald, 2000). The intracellular accumulation of pathological huntingtin aggregates impairs the overall proteostasis network and disrupts the structure and dynamics of the endoplasmic reticulum (ER) and mitochondrial membranes (Gidalevitz et al., 2006; Kim et al., 2016; Bäuerlein et al., 2017; Riguet et al., 2021). This eventually leads to the dysregulation of various cellular processes, including transcription, mitochondrial respiration, ER homeostasis, vesicular trafficking, and axonal transport (Kim et al., 2016; Riguet et al., 2021).
The full-length huntingtin protein (Htt) with 23Q contains a total of 3,144 amino acids (348 kDa). Huntingtin plays diverse functional roles in nervous system development, the transport of vesicles containing brain-derived neurotrophic factor (BDNF), and selective autophagy (Saudou and Humbert, 2016). N-terminal fragments containing exon 1 of huntingtin (Httex1) with expanded polyQ repeats, generated by either aberrant splicing or proteolytic cleavage, have been observed in human postmortem brains and mouse HD models (DiFiglia et al., 1997; Lunkes et al., 2002; Sathasivam et al., 2013; Neueder et al., 2017). Moreover, mutant huntingtin exon 1 carrying a polyQ expansion (mHttex1) is sufficient to recapitulate HD-associated phenotypes in animal models (Yu et al., 2003; Southwell et al., 2016; Yang et al., 2020), and is thus widely used as a relevant model for HD biology and pathology.
Httex1 comprises three main domains: the N-terminal N17 domain, the PolyQ repeat domain, and the C-terminal proline-rich domain (PRD) (Figure 1A). The 17 amino acid residues in the N17 domain are highly conserved across diverse vertebrate species (Atwal et al., 2007) (Figure 1A). Consistent with this, the N17 domain plays several essential roles in HD pathogenesis by stimulating mHttex1 aggregation and altering its post-translational modifications (PTMs) and cellular localization (Figure 2). In this review, I will focus on these functional roles of the N17 domain and further discuss the implications of N17-targeted therapeutic development for HD.
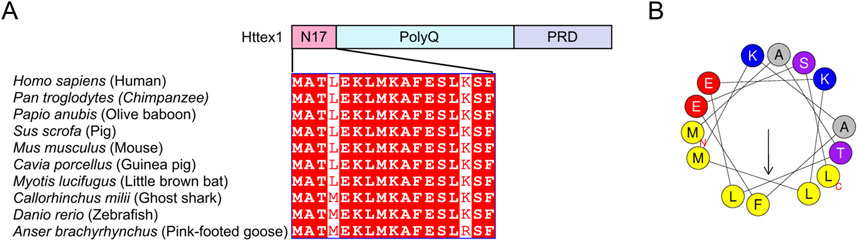
Figure 1. Sequences and amphipathic helical property of the N17 domain of Httex1 (A) Sequence alignment of the N17 domain of Httex1. Httex1 is composed of three main domains: N17 domain, polyQ repeat domain, and proline-rich domain (PRD). The sequences of the N17 domain are highly conserved among tested species (Atwal et al., 2007). (B) The helical wheel illustrates the distribution of hydrophobic and hydrophilic amino acids in the amphipathic helix of the N17 domain.
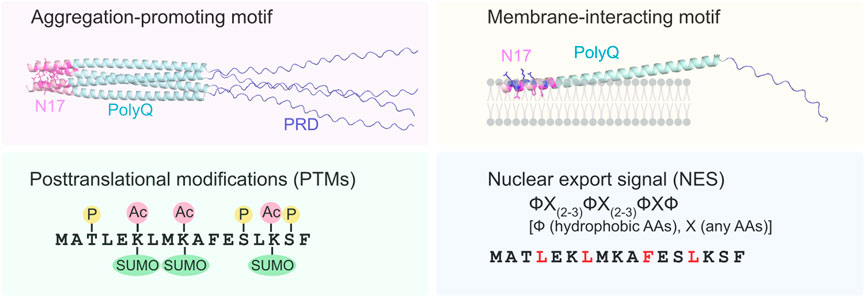
Figure 2. Multi-functional roles of the N17 domain in modulating aggregation and cellular localization of mHttex1. As an aggregation-promoting motif, the hydrophobic residues (highlighted in magenta) in the N17 domain (light pink) form a core hydrophobic interface that may further stabilize polyQ aggregation. The amphipathic N17 domain also interacts with the lipid bilayers, thereby enhancing the local concentration and aggregation of mHttex1. Hydrophobic (magenta) and charged (blue) residues in the N17 domain are located on the interior and exterior surfaces of the membrane, respectively. Three types of PTMs (phosphorylation, acetylation, and SUMOylation) in the N17 domain are known to alter the aggregation state of mHttex1. Furthermore, the N17 domain contains NES-like consensus sequences, ФX(2–3) ФX(2–3)ФXФ, where Ф and X are hydrophobic amino acids (highlighted in red) and random amino acids, respectively. In the absence of the N17 domain, toxic mHttex1 aggregates accumulate in the nucleus.
N17 domain as an aggregation-promoting motif
The N17 domain is known to enhance the aggregation kinetics of various huntingtin model proteins (Tam et al., 2009; Thakur et al., 2009; Sivanandam et al., 2011; Crick et al., 2013; Vieweg et al., 2021). The N17 domain has an amphipathic helical property (Atwal et al., 2007; Sivanandam et al., 2011) (Figure 1B), and the hydrophobic surface of the amphipathic N17 domain is crucial for the rapid aggregation of mHttex1 (Tam et al., 2009). Consistent with this, the AlphaFold (Jumper et al., 2021) prediction of the tetrameric structure of Httex1-51Q suggests that the hydrophobic residues of the N17 domain interact with each other to form a core hydrophobic interface on the tetramer, thereby potentially stabilizing the self-assembly of mHttex1 (Figure 2). The isolated N17 peptide exhibits the ability to interact with both the N17 and polyQ repeat domains in mHttex1 (Tam et al., 2009). In this proposed mechanism, the N17-N17 and N17-polyQ intermolecular interactions lower the kinetic barrier between the oligomeric and fibril states, thus promoting amyloid fibril aggregation (Shen et al., 2016).
An alternative mechanism is that the hydrogen bonds between the residues in the N17 domain and the polyQ repeat domain enable the stabilization of a long α-helix constituting both domains (Urbanek et al., 2020; Elena-Real et al., 2023). The bifurcated hydrogen bonds between the peptide backbone of F17 or S16 at the i-4 position and side chains of Q21 or Q20 at the i position strengthen the structural coupling between the N17 and polyQ repeat domains, thus stably extending the N17 α-helical content to the polyQ repeat domain through the hydrogen bond network (Elena-Real et al., 2023). The propagation of α-helical content from N17 to polyQ repeat domains increases with the increasing length of the polyQ repeat domain (Elena-Real et al., 2023). Since the N17 domain alone is not sufficient to form aggregation (Thakur et al., 2009), this structural coupling between two adjacent domains could be crucial for accelerating Httex1 aggregation.
N17 domain as a membrane-interacting motif
The membrane interaction of mutant huntingtin (mHtt) with expanded polyQ repeats appears to be disease-relevant. Both endogenous Htt and the exogenously expressed full-length and truncated N-terminal fragments (∼90 kDa) are present in the membrane fraction of neuron-like clonal striatal cells (Kegel et al., 2000). The N-terminal mHtt fragments are also associated with the brain membranes of both human HD patients and R6/2 mice (Kim et al., 2001; Suopanki et al., 2006). Moreover, the inclusions and fibrils of mHttex1 are known to disrupt various organellar membranes, including nuclear, ER, and mitochondrial membranes. Perinuclear inclusions of mHttex1 interact with the nuclear membrane, destroying nuclear membrane integrity (Waelter et al., 2001; Liu et al., 2015; Riguet et al., 2021). Httex1-97Q-GFP fibrils impinge on the ER membrane and alter the membrane curvature and dynamics (Bäuerlein et al., 2017). N-terminal mHtt is also associated with mitochondrial membranes, potentially contributing to mitochondrial dysfunction in HD (Panov et al., 2002; Orr et al., 2008). Collectively all these results suggest that large fibrillar inclusions impair the membrane integrity of various organelles, thereby contributing to HD pathology.
The N17 domain forms an amphipathic helix in both phospholipid bilayers and DPC micelles (Michalek et al., 2013a; Michalek et al., 2013b; Tao et al., 2019). Similar to other amphipathic helices, hydrophobic residues (L4, L7, F11, L14, and F17) are embedded in the lipid bilayer, and charged residues are located on the membrane surface (Michalek et al., 2013a; Michalek et al., 2013b; Tao et al., 2019) (Figure 2). The N17-anchoring in the membrane is thought to increase the local concentrations of mHttex1, thereby inducing polyQ aggregation on the membrane surface (Michalek et al., 2013b; Tao et al., 2019). Consistent with this, large unilamellar vesicles (LUVs) with a diameter of 100 nm (25% POPS and 75% POPC) enhance the aggregation kinetics of mHttex1 (Pandey et al., 2018). The aggregation-enhancing effects of the N17 domain appear to depend on lipid head charges, saturation levels of phospholipid acyl tails, and membrane curvature (Tao et al., 2019; Beasley et al., 2021; Beasley et al., 2022). Small unilamellar vesicles (SUVs) with anionic lipids (POPG and POPS) dramatically increase Httex1-46Q fibrils, whereas the zwitterionic lipid SUVs with POPC and POPE do not alter the Httex1-46Q fibril content at a 1:10 protein-to-lipid ratio (Beasley et al., 2021). Furthermore, LUVs with saturated DMPC lipids significantly increase Httex1-46Q fibril formation, whereas LUVs with unsaturated DOPC lipids reduce Httex1-46Q fibrils (Beasley et al., 2022). Several studies have suggested that Httex1 preferentially interacts with highly curved membrane surfaces (Chaibva et al., 2014; Tao et al., 2019). Despite the growing evidence of membrane composition-specific effects via the N17 domain, since different organellar membranes have various lipid compositions (van Meer et al., 2008), how the in vitro findings can be extended to the organellar-specific membranes remains to be addressed.
Posttranslational modifications on the N17 domain
PTMs, including phosphorylation, acetylation, and SUMOylation, in the N17 domain alter the hydrophobicity, charge, and secondary structure of the amphipathic helix. These altered physicochemical properties and conformation of the N17 helix affect N17-induced aggregation and membrane interactions, ultimately modulating the cellular toxicity of mHttex1. Thus, PTMs in the N17 domain have been suggested to act as molecular switches in HD pathogenesis, providing an important target for the therapeutic development of HD (Bowie et al., 2018).
Among known PTMs, phosphorylation is the most well-characterized modification. The phosphorylation of the T3, S13, and S16 residues of the N17 domain is generally associated with reduced mHtt toxicity. Consistent with this, decreased levels of mHtt T3 phosphorylation were observed in cellular and mouse HD models, as well as in human HD patient samples (Aiken et al., 2009; Cariulo et al., 2017). Phosphorylation at T3 stabilizes the N17 helical conformation and decreases SDS-insoluble aggregation and fibril formation of Httex1 in vitro (Cariulo et al., 2017; Chiki et al., 2017). Despite the relatively lower inhibitory effect of T3D compared to phosphorylated T3 (Chiki et al., 2017), the phosphomimetic T3D mutation is still able to abolish the formation of large inclusions of Httex1-97Q in human H4 glioma cells (Branco-Santos et al., 2017). In contrast, the phosphorylation-deficient T3A mutant was not significantly different from the unmodified Httex1-97Q (Branco-Santos et al., 2017). Furthermore, full-length Htt with phosphomimetic S13D/S16D mutations, but not phosphoresistant S13A/S16A mutations, is known to prevent progressive neuronal dysfunction, mHtt aggregation, and late-onset neurodegenerative pathology in vivo (Gu et al., 2009). Similar to phosphorylated S13/S16, the phosphomimetic S13D/S16D mutant delayed the aggregation kinetics of mHttex1 in vitro (Mishra et al., 2012). Interestingly, the S13D/S16D mutant showed decreased binding affinity to the membrane, thereby preventing membrane-mediated aggregation (Tao et al., 2019).
Consistent with the notion that increased phosphorylation levels at the N17 domain reduce the toxicity of mHttex1, overexpression of several kinases has been shown to decrease the aggregation of mHttex1 or promote the degradation of mHttex1. Overexpression of the inflammatory kinase IKKβ increases phosphorylation levels at S13 of mHttex1 in ST14A cells, thereby inducing mHttex1 clearance via autophagy (Thompson et al., 2009). Similarly, TANK-binding kinase 1 (TBK1)-mediated phosphorylation at S13 reduces mHttex1 aggregation in an autophagy-dependent manner, leading to decreased neurotoxicity in several HD models (Hegde et al., 2020). Overexpression of the nuclear factor kappa B kinase subunit beta (IKBKB) increases endogenous phosphorylated S13 levels of mHttex1 and reduces Httex1 aggregation in HEK293T cells (Cariulo et al., 2023). Taken together, all previous studies suggest that modulating the expression levels of these specific kinases could be a potential therapeutic strategy to reduce toxic mHttex1 proteins.
Both lysine acetylation at the K6, K9, and K15 residues and N-terminal acetylation have been suggested to modulate the aggregation of mHttex1. A previous mass spectrometry study identified the K9 acetylation of Htt-23Q (1–612) in HEK293T cells (Cong et al., 2011). Single lysine acetylation at K6, K9, or K15 does not play a critical role in the regulation of Httex1-43Q aggregation in vitro (Chiki et al., 2017), whereas non-specific acetylation of Httex1-51Q at K6, K9, and K15 with sulfo-N-hydroxysuccinimide (NHSA) prevents fibril formation (Chaibva et al., 2016). In this study, since the in vitro aggregation reactions were coupled with acetylation reactions involving NHSA, it remains unclear whether multiple lysine acetylations regulate the aggregation of mHttex1 in vitro (Chaibva et al., 2016).
SUMOylation at K6, K9, or K15 of the N17 domain alters the aggregation propensities and toxicity of mHttex1 (Steffan et al., 2004; Subramaniam et al., 2009; O’Rourke et al., 2013). Overexpression of small ubiquitin-like modifier 1 (SUMO1) predominantly SUMOylates K6 and K9 residues of mHttex1 (Steffan et al., 2004). N-terminal SUMO-fused Httex1-97Q also reduced SDS-insoluble aggregation in striatal cells (Steffan et al., 2004). Consistent with this, incubation of Httex1-46Q with SUMO1 significantly decreased the formation of SDS-insoluble aggregates and fibrils in vitro (Sedighi et al., 2020). In addition, overexpression of both the Ras homolog enriched in the striatum (Rhes) and SUMO1 increased SUMOylation at K9 and K15 residues and reduced SDS-insoluble aggregates of Httex1-82Q in HEK293 cells (Subramaniam et al., 2009). Given that Rhes interacts more effectively with mHttex1 than with Httex1-WT in both HD cellular and mouse models, and that SUMO modification of mHtt suppresses general transcription, accumulation of SUMOylated mHttex1 has been suggested to cause cytotoxicity despite its reduced aggregation (Steffan et al., 2004; Subramaniam et al., 2009). Reduction of SUMO activity and deletion of SUMO1 ameliorates neurodegeneration in HD fly and mouse models (Steffan et al., 2004; Ramírez-Jarquín et al., 2022).
In addition to SUMO1, overexpression of SUMO2 SUMOylates mHttex1 but enhances SDS-insoluble mHttex1 in HeLa cells (O’Rourke et al., 2013). Furthermore, SDS-insoluble SUMO2-modified proteins largely accumulate in human HD brains compared to controls, suggesting that SUMO2 modification of various proteins could occur during HD progression (O’Rourke et al., 2013). Since most of these studies used the overexpression of SUMO1 and SUMO2 in mammalian cells, the molecular mechanism by which purified SUMOylated mHttex1 directly alters aggregation states remains unclear.
N17 as a nuclear export signal (NES)
The N17 domain serves as a nuclear export signal (NES) (Maiuri et al., 2013; Zheng et al., 2013). The amino acid sequence of the N17 domain displays a NES consensus-like sequence (ФХ(2–3)ФX(2–3)ФXФ, where Ф are hydrophobic amino acids (L4, L7, F11, L14 for the N17 domain) and X are random amino acids) (Kutay and Güttinger, 2005; Maiuri et al., 2013; Zheng et al., 2013) (Figure 2). Single mutations in these hydrophobic amino acids or deletion of partial and full-length N17 increased the nuclear localization of Httex1-WT and mHttex1 in various cellular HD models (Cornett et al., 2005; Rockabrand et al., 2007; Zheng et al., 2013). Furthermore, L7S and F11G mutants induce nuclear inclusions of Httex1-72Q in primary mouse cortical neurons (Zheng et al., 2013). Deletion of N17 in full-length mHtt leads to a dramatic acceleration of nuclear aggregation of small mHtt N-terminal fragments in BACHD-ΔN17 (97Q) mouse brains (Gu et al., 2015). BACHD-ΔN17 (97Q) mice exhibit early disease onset and more severe motor and behavioral deficits than BACHD-WT (97Q) mice (Gu et al., 2015). Consistent with this, the induced nuclear accumulation of toxic mHttex1 aggregates in Httex1-ΔN17 (97Q) zebrafish leads to an accelerated HD-like phenotype (Veldman et al., 2015). Therefore, the N17 domain plays an important role in mediating the cytoplasmic targeting of mHtt and in mitigating nuclear toxicity.
N17-mediated nuclear export is regulated by both the CRM1/Exportin 1 receptor and the nucleoporin translocated promoter region (TPR), nuclear basket protein (Cornett et al., 2005; Maiuri et al., 2013; Zheng et al., 2013). The N17 domain alone is shown to be associated with CRM1 and TPR (Cornett et al., 2005; Maiuri et al., 2013). Phosphorylation mimetic mutations (S13D/S16D) and helix-disruption mutations (M8P) reduce the cytoplasmic localization of Httex1 and disrupt the interaction between the N17 domain and CRM1, suggesting that the secondary structure and PTM of N17 could also modulate the cellular localization of Httex1 (Maiuri et al., 2013; Zheng et al., 2013). Furthermore, mHtt interferes with its interaction with nucleoporin TPR, thereby diminishing nuclear export and enhancing mHtt nuclear aggregation (Cornett et al., 2005).
Potential therapeutic approaches and further directions
The highly conserved N17 domain of huntingtin plays a versatile role in modulating the aggregation and cellular localization of mHtt, thereby contributing to the pathology of HD. Given that N17 acts as an aggregation-promoting motif, direct targeting of the N17 domain could be a promising therapeutic approach for inhibiting mHtt aggregation. A few chaperones, such as TRiC chaperonin and the Hsc70 chaperone, are known to interact with the N17 domain of mHttex1, leading to the suppression of mHttex1 aggregation (Tam et al., 2009; Monsellier et al., 2015). Hsp70s bind their substrates promiscuously and use co-chaperones to enhance substrate selectivity (Mayer and Gierasch, 2019), while TriC requires a complex subunit assembly step (Shahmoradian et al., 2013). However, these requirements may complicate the therapeutic application for HD, with unwanted side effects. Thus, engineering artificial chaperones with enhanced activity and high substrate specificity for N17-mediated mHtt aggregation could be a feasible approach for alleviating the cellular toxicity of mHtt. In addition, a previous study discovered a human antibody that specifically binds to the N17 domain and confirmed its capacity to counteract the aggregation of mHttex1 in cells (Lecerf et al., 2001). Therefore, designing antibodies, short peptides, or small-molecule drugs that directly bind to the N17 domain could be an alternative approach to prevent N17-mediated mHtt aggregation.
In addition to the direct targeting of the N17 domain, indirect modulation of PTM states on the N17 domain of mHttex1 could provide therapeutic potential in HD. Since decreased phosphorylation levels of N17 have been observed in human HD patient samples, and several kinases are known to phosphorylate T3, S13, or S16 residues at the N17 domain, high-throughput screening of compound libraries could enable the identification of small-molecule drugs that restore the phosphorylation levels of the N17 domain (Atwal et al., 2011). However, despite the overall substrate specificity of S/T kinases and tyrosine kinases (Johnson et al., 2023; Yaron-Barir et al., 2024), assessing whether potential kinase activators specifically upregulate the phosphorylation levels of the N17 domain of mHtt could be critical in eliminating any side effects associated with HD therapeutics. In contrast to phosphorylation, the mechanistic understanding of how other PTMs directly regulate N17-induced toxicity lags behind, mainly due to the lack of in vitro assays with modified mHttex1 proteins. Therefore, further mechanistic studies on PTMs of the N17 domain could provide therapeutic opportunities to mitigate HD progression by altering PTMs of the N17 domain.
Author contributions
HC: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from the Institute for Basic Science (IBS-R030-Y1 to HC).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aiken, C. T., Steffan, J. S., Guerrero, C. M., Khashwji, H., Lukacsovich, T., Simmons, D., et al. (2009). Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284, 29427–29436. doi:10.1074/jbc.M109.013193
Atwal, R. S., Desmond, C. R., Caron, N., Maiuri, T., Xia, J., Sipione, S., et al. (2011). Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460. doi:10.1038/nchembio.582
Atwal, R. S., Xia, J., Pinchev, D., Taylor, J., Epand, R. M., and Truant, R. (2007). Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 16, 2600–2615. doi:10.1093/hmg/ddm217
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Prim. 1, 15005. doi:10.1038/nrdp.2015.5
Bäuerlein, F. J. B., Saha, I., Mishra, A., Kalemanov, M., Martínez-Sánchez, A., Klein, R., et al. (2017). In situ architecture and cellular interactions of PolyQ inclusions. Cell. 171, 179–187. doi:10.1016/j.cell.2017.08.009
Beasley, M., Frazee, N., Groover, S., Valentine, S. J., Mertz, B., and Legleiter, J. (2022). Physicochemical properties altered by the tail group of lipid membranes influence huntingtin aggregation and lipid binding. J. Phys. Chem. B 126, 3067–3081. doi:10.1021/acs.jpcb.1c10254
Beasley, M., Groover, S., Valentine, S. J., and Legleiter, J. (2021). Lipid headgroups alter huntingtin aggregation on membranes. Biochim. Biophys. Acta Biomembr. 1863, 183497. doi:10.1016/j.bbamem.2020.183497
Bowie, L. E., Maiuri, T., Alpaugh, M., Gabriel, M., Arbez, N., Galleguillos, D., et al. (2018). N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 115, E7081-E7090–E7090. doi:10.1073/pnas.1801772115
Branco-Santos, J., Herrera, F., Poças, G. M., Pires-Afonso, Y., Giorgini, F., Domingos, P. M., et al. (2017). Protein phosphatase 1 regulates huntingtin exon 1 aggregation and toxicity. Hum. Mol. Genet. 26, 3763–3775. doi:10.1093/hmg/ddx260
Cariulo, C., Azzollini, L., Verani, M., Martufi, P., Boggio, R., Chiki, A., et al. (2017). Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc. Natl. Acad. Sci. U. S. A. 114, E10809-E10818–E10818. doi:10.1073/pnas.1705372114
Cariulo, C., Martufi, P., Verani, M., Toledo-Sherman, L., Lee, R., Dominguez, C., et al. (2023). IKBKB reduces huntingtin aggregation by phosphorylating serine 13 via a non-canonical IKK pathway. Life Sci. Alliance 6, e202302006. doi:10.26508/lsa.202302006
Chaibva, M., Burke, K. A., and Legleiter, J. (2014). Curvature enhances binding and aggregation of huntingtin at lipid membranes. Biochemistry 53, 2355–2365. doi:10.1021/bi401619q
Chaibva, M., Jawahery, S., Pilkington, A. W., Arndt, J. R., Sarver, O., Valentine, S., et al. (2016). Acetylation within the first 17 residues of huntingtin exon 1 alters aggregation and lipid binding. Biophys. J. 111, 349–362. doi:10.1016/j.bpj.2016.06.018
Chiki, A., DeGuire, S. M., Ruggeri, F. S., Sanfelice, D., Ansaloni, A., Wang, Z.-M., et al. (2017). Mutant Exon1 huntingtin aggregation is regulated by T3 phosphorylation-induced structural changes and crosstalk between T3 phosphorylation and acetylation at K6. Angew. Chem. Int. Ed. Engl. 56, 5202–5207. doi:10.1002/anie.201611750
Cong, X., Held, J. M., DeGiacomo, F., Bonner, A., Chen, J. M., Schilling, B., et al. (2011). Mass spectrometric identification of novel lysine acetylation sites in huntingtin. Mol. and Cell. Proteomics 10, M111.009829. doi:10.1074/mcp.M111.009829
Cornett, J., Cao, F., Wang, C.-E., Ross, C. A., Bates, G. P., Li, S.-H., et al. (2005). Polyglutamine expansion of huntingtin impairs its nuclear export. Nat. Genet. 37, 198–204. doi:10.1038/ng1503
Crick, S. L., Ruff, K. M., Garai, K., Frieden, C., and Pappu, R. V. (2013). Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U. S. A. 110, 20075–20080. doi:10.1073/pnas.1320626110
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi:10.1126/science.277.5334.1990
Elena-Real, C. A., Sagar, A., Urbanek, A., Popovic, M., Morató, A., Estaña, A., et al. (2023). The structure of pathogenic huntingtin exon 1 defines the bases of its aggregation propensity. Nat. Struct. Mol. Biol. 30, 309–320. doi:10.1038/s41594-023-00920-0
Gidalevitz, T., Ben-Zvi, A., Ho, K. H., Brignull, H. R., and Morimoto, R. I. (2006). Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471–1474. doi:10.1126/science.1124514
Gu, X., Cantle, J. P., Greiner, E. R., Lee, C. Y. D., Barth, A. M., Gao, F., et al. (2015). N17 Modifies mutant Huntingtin nuclear pathogenesis and severity of disease in HD BAC transgenic mice. Neuron 85, 726–741. doi:10.1016/j.neuron.2015.01.008
Gu, X., Greiner, E. R., Mishra, R., Kodali, R., Osmand, A., Finkbeiner, S., et al. (2009). Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840. doi:10.1016/j.neuron.2009.11.020
Gusella, J. F., and MacDonald, M. E. (2000). Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 1, 109–115. doi:10.1038/35039051
Hegde, R. N., Chiki, A., Petricca, L., Martufi, P., Arbez, N., Mouchiroud, L., et al. (2020). TBK1 phosphorylates mutant Huntingtin and suppresses its aggregation and toxicity in Huntington’s disease models. EMBO J. 39, e104671. doi:10.15252/embj.2020104671
Johnson, J. L., Yaron, T. M., Huntsman, E. M., Kerelsky, A., Song, J., Regev, A., et al. (2023). An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766. doi:10.1038/s41586-022-05575-3
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kegel, K. B., Kim, M., Sapp, E., McIntyre, C., Castaño, J. G., Aronin, N., et al. (2000). Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20, 7268–7278. doi:10.1523/JNEUROSCI.20-19-07268.2000
Kim, Y. E., Hosp, F., Frottin, F., Ge, H., Mann, M., Hayer-Hartl, M., et al. (2016). Soluble oligomers of PolyQ-expanded huntingtin target a multiplicity of key cellular factors. Mol. Cell. 63, 951–964. doi:10.1016/j.molcel.2016.07.022
Kim, Y. J., Yi, Y., Sapp, E., Wang, Y., Cuiffo, B., Kegel, K. B., et al. (2001). Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U. S. A. 98, 12784–12789. doi:10.1073/pnas.221451398
Kutay, U., and Güttinger, S. (2005). Leucine-rich nuclear-export signals: born to be weak. Trends Cell. Biol. 15, 121–124. doi:10.1016/j.tcb.2005.01.005
Lecerf, J.-M., Shirley, T. L., Zhu, Q., Kazantsev, A., Amersdorfer, P., Housman, D. E., et al. (2001). Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. 98, 4764–4769. doi:10.1073/pnas.071058398
Liu, K.-Y., Shyu, Y.-C., Barbaro, B. A., Lin, Y.-T., Chern, Y., Thompson, L. M., et al. (2015). Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington’s disease. Hum. Mol. Genet. 24, 1602–1616. doi:10.1093/hmg/ddu574
Lunkes, A., Lindenberg, K. S., Ben-Haïem, L., Weber, C., Devys, D., Landwehrmeyer, G. B., et al. (2002). Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell. 10, 259–269. doi:10.1016/s1097-2765(02)00602-0
Maiuri, T., Woloshansky, T., Xia, J., and Truant, R. (2013). The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum. Mol. Genet. 22, 1383–1394. doi:10.1093/hmg/dds554
Mayer, M. P., and Gierasch, L. M. (2019). Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 294, 2085–2097. doi:10.1074/jbc.REV118.002810
Michalek, M., Salnikov, E. S., and Bechinger, B. (2013a). Structure and topology of the huntingtin 1-17 membrane anchor by a combined solution and solid-state NMR approach. Biophys. J. 105, 699–710. doi:10.1016/j.bpj.2013.06.030
Michalek, M., Salnikov, E. S., Werten, S., and Bechinger, B. (2013b). Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry 52, 847–858. doi:10.1021/bi301325q
Mishra, R., Hoop, C. L., Kodali, R., Sahoo, B., van der Wel, P. C. A., and Wetzel, R. (2012). Serine phosphorylation suppresses huntingtin amyloid accumulation by altering protein aggregation properties. J. Mol. Biol. 424, 1–14. doi:10.1016/j.jmb.2012.09.011
Monsellier, E., Redeker, V., Ruiz-Arlandis, G., Bousset, L., and Melki, R. (2015). Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576. doi:10.1074/jbc.M114.603332
Neueder, A., Landles, C., Ghosh, R., Howland, D., Myers, R. H., Faull, R. L. M., et al. (2017). The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 7, 1307. doi:10.1038/s41598-017-01510-z
O’Rourke, J. G., Gareau, J. R., Ochaba, J., Song, W., Raskó, T., Reverter, D., et al. (2013). SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell. Rep. 4, 362–375. doi:10.1016/j.celrep.2013.06.034
Orr, A. L., Li, S., Wang, C.-E., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 28, 2783–2792. doi:10.1523/JNEUROSCI.0106-08.2008
Pandey, N. K., Isas, J. M., Rawat, A., Lee, R. V., Langen, J., Pandey, P., et al. (2018). The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem. 293, 2597–2605. doi:10.1074/jbc.M117.813667
Panov, A. V., Gutekunst, C.-A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736. doi:10.1038/nn884
Ramírez-Jarquín, U. N., Sharma, M., Zhou, W., Shahani, N., and Subramaniam, S. (2022). Deletion of SUMO1 attenuates behavioral and anatomical deficits by regulating autophagic activities in Huntington disease. Proc. Natl. Acad. Sci. U. S. A. 119, e2107187119. doi:10.1073/pnas.2107187119
Riguet, N., Mahul-Mellier, A.-L., Maharjan, N., Burtscher, J., Croisier, M., Knott, G., et al. (2021). Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties. Nat. Commun. 12, 6579. doi:10.1038/s41467-021-26684-z
Rockabrand, E., Slepko, N., Pantalone, A., Nukala, V. N., Kazantsev, A., Marsh, J. L., et al. (2007). The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77. doi:10.1093/hmg/ddl440
Sathasivam, K., Neueder, A., Gipson, T. A., Landles, C., Benjamin, A. C., Bondulich, M. K., et al. (2013). Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. 110, 2366–2370. doi:10.1073/pnas.1221891110
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi:10.1016/j.neuron.2016.02.003
Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., et al. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 90, 549–558. doi:10.1016/s0092-8674(00)80514-0
Sedighi, F., Adegbuyiro, A., and Legleiter, J. (2020). SUMOylation prevents huntingtin fibrillization and localization onto lipid membranes. ACS Chem. Neurosci. 11, 328–343. doi:10.1021/acschemneuro.9b00509
Shahmoradian, S. H., Galaz-Montoya, J. G., Schmid, M. F., Cong, Y., Ma, B., Spiess, C., et al. (2013). TRiC’s tricks inhibit huntingtin aggregation. eLife 2, e00710. doi:10.7554/eLife.00710
Shen, K., Calamini, B., Fauerbach, J. A., Ma, B., Shahmoradian, S. H., Serrano Lachapel, I. L., et al. (2016). Control of the structural landscape and neuronal proteotoxicity of mutant Huntingtin by domains flanking the polyQ tract. Elife 5, e18065. doi:10.7554/eLife.18065
Sivanandam, V. N., Jayaraman, M., Hoop, C. L., Kodali, R., Wetzel, R., and van der Wel, P. C. A. (2011). The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566. doi:10.1021/ja110715f
Southwell, A. L., Smith-Dijak, A., Kay, C., Sepers, M., Villanueva, E. B., Parsons, M. P., et al. (2016). An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum. Mol. Genet. 25, 3654–3675. doi:10.1093/hmg/ddw212
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304, 100–104. doi:10.1126/science.1092194
Subramaniam, S., Sixt, K. M., Barrow, R., and Snyder, S. H. (2009). Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330. doi:10.1126/science.1172871
Suopanki, J., Götz, C., Lutsch, G., Schiller, J., Harjes, P., Herrmann, A., et al. (2006). Interaction of huntingtin fragments with brain membranes--clues to early dysfunction in Huntington’s disease. J. Neurochem. 96, 870–884. doi:10.1111/j.1471-4159.2005.03620.x
Tam, S., Spiess, C., Auyeung, W., Joachimiak, L., Chen, B., Poirier, M. A., et al. (2009). The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285. doi:10.1038/nsmb.1700
Tao, M., Pandey, N. K., Barnes, R., Han, S., and Langen, R. (2019). Structure of membrane-bound huntingtin exon 1 reveals membrane interaction and aggregation mechanisms. Structure 27, 1570–1580. doi:10.1016/j.str.2019.08.003
Thakur, A. K., Jayaraman, M., Mishra, R., Thakur, M., Chellgren, V. M., Byeon, I.-J. L., et al. (2009). Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389. doi:10.1038/nsmb.1570
Thompson, L. M., Aiken, C. T., Kaltenbach, L. S., Agrawal, N., Illes, K., Khoshnan, A., et al. (2009). IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell. Biol. 187, 1083–1099. doi:10.1083/jcb.200909067
Urbanek, A., Popovic, M., Morató, A., Estaña, A., Elena-Real, C. A., Mier, P., et al. (2020). Flanking regions determine the structure of the poly-glutamine in huntingtin through mechanisms common among glutamine-rich human proteins. Structure 28, 733–746. doi:10.1016/j.str.2020.04.008
van Meer, G., Voelker, D. R., and Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell. Biol. 9, 112–124. doi:10.1038/nrm2330
Veldman, M. B., Rios-Galdamez, Y., Lu, X.-H., Gu, X., Qin, W., Li, S., et al. (2015). The N17 domain mitigates nuclear toxicity in a novel zebrafish Huntington’s disease model. Mol. Neurodegener. 10, 67. doi:10.1186/s13024-015-0063-2
Vieweg, S., Mahul-Mellier, A.-L., Ruggeri, F. S., Riguet, N., DeGuire, S. M., Chiki, A., et al. (2021). The Nt17 domain and its helical conformation regulate the aggregation, cellular properties and neurotoxicity of mutant huntingtin exon 1. J. Mol. Biol. 433, 167222. doi:10.1016/j.jmb.2021.167222
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H., et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell. 12, 1393–1407. doi:10.1091/mbc.12.5.1393
Yang, H., Yang, S., Jing, L., Huang, L., Chen, L., Zhao, X., et al. (2020). Truncation of mutant huntingtin in knock-in mice demonstrates exon1 huntingtin is a key pathogenic form. Nat. Commun. 11, 2582. doi:10.1038/s41467-020-16318-1
Yaron-Barir, T. M., Joughin, B. A., Huntsman, E. M., Kerelsky, A., Cizin, D. M., Cohen, B. M., et al. (2024). The intrinsic substrate specificity of the human tyrosine kinome. Nature 629, 1174–1181. doi:10.1038/s41586-024-07407-y
Yu, Z.-X., Li, S.-H., Evans, J., Pillarisetti, A., Li, H., and Li, X.-J. (2003). Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington’s disease. J. Neurosci. 23, 2193–2202. doi:10.1523/JNEUROSCI.23-06-02193.2003
Keywords: Huntington’s disease, Huntingtin, N17 domain, aggregation, post-translational modification (PTM)
Citation: Cho H (2025) The N17 domain of huntingtin as a multifaceted player in Huntington’s disease. Front. Mol. Biosci. 11:1527313. doi: 10.3389/fmolb.2024.1527313
Received: 13 November 2024; Accepted: 19 December 2024;
Published: 07 January 2025.
Edited by:
Chiara Giacomelli, University of Pisa, ItalyReviewed by:
Partha Sarathi Sarkar, University of Texas Medical Branch at Galveston, United StatesLaura Lagartera, Spanish National Research Council (CSIC), Spain
Copyright © 2025 Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyunju Cho, aGpjaG9AaWJzLnJlLmty