 Kejia Yuan†
Kejia Yuan† Lei Peng
Lei Peng Qi Yi
Qi Yi- Key Laboratory of Model Animals and Stem Cell Biology, Hunan Normal University School of Medicine, Changsha, Hunan, China
The transcriptional regulator ATRX, a genetic factor, is associated with a range of disabilities, including intellectual, hematopoietic, skeletal, facial, and urogenital disabilities. ATRX mutations substantially contribute to the pathogenesis of ATRX syndrome and are frequently detected in gliomas and many other cancers. These mutations disrupt the organization, subcellular localization, and transcriptional activity of ATRX, leading to chromosomal instability and affecting interactions with key regulatory proteins such as DAXX, EZH2, and TERRA. ATRX also functions as a transcriptional regulator involved in the pathogenesis of neuronal disorders and various diseases. In conclusion, ATRX is a central protein whose abnormalities lead to multiple diseases.
1 Introduction
The ATRX gene was first identified as a pathogenic gene for a rare hereditary disease that causes intellectual disability and α-thalassemia more than two decades ago (Weatherall et al., 1981; Gibbons et al., 2008; Watson et al., 2015; León and Harley, 2021). ATRX belongs to the switch/sucrose nonfermentable (SWI-SNF) protein clan and functions as a chromatin remodeler, many of which can slide, modify, or remove histones in vitro (Stayton et al., 1994; Picketts et al., 1996; Argentaro et al., 2007; Clynes et al., 2013). Recent research indicates that ATRX is involved in various biological activities, such as nucleosome remodeling (Gibbons et al., 1995; Xue et al., 2003; Bérubé et al., 2008), transcriptional regulation (Li et al., 2016; Chu et al., 2017; Schenkel et al., 2017; Truch et al., 2022), recombination (Bérubé et al., 2002; Clynes et al., 2015; Gramatzki et al., 2016; Teng et al., 2021; Liu and Malkova, 2022), DNA repair (Nasmyth and Haering, 2009; Clynes et al., 2015; Han et al., 2020), and tumorigenesis (Picketts et al., 1996; Danussi et al., 2018; Ohba et al., 2020; Gulve et al., 2022; Hackeng et al., 2022).
ATRX syndrome includes five main typical clinical manifestations: facial dysmorphism, hypotonia, skeletal, genitourinary system and hematopoietic abnormalities. Mental retardation, which is a consistent clinical characteristic in all ATRX syndrome patients, is the most basic clinical manifestation of the ATRX syndrome diagnosis (León and Harley, 2021). ATRX syndrome is usually asymptomatic in women due to significant skewing of X chromosome offset inactivation, while the symptoms are more obvious in men due to the absence of alleles (Wada et al., 2005). ATRX syndrome is primarily caused by ATRX mutations and characterized by ATRX deletion or missense mutations (Masliah-Planchon et al., 2018; Fan et al., 2019). This syndrome arises from several pathways, including neuronal differentiation, telomere stability, DNA damage response (DDR) pathways, and altered expression and structure of ATRX-interacting proteins (Hong et al., 2009; De La Fuente et al., 2011; Lacoste et al., 2014; Li et al., 2016; Garbarino et al., 2021). In addition, studies have shown that patients with ATRX syndrome have a genetic predisposition to osteosarcoma, which offers a novel avenue for diagnosing ATRX syndrome as a nonclassical phenotype of ATRX syndrome (Ji et al., 2017).
ATRX mutations are identified in a range of malignant tumors, such as neuroblastoma, pancreatic neuroendocrine tumors, and osteosarcoma. Up to 25% of gliomas exhibit ATRX mutations, with specific percentages including 67% of WHO grade II astrocytomas, 73% of WHO grade III astrocytomas, and 57% of secondary glioblastomas (Olar and Sulman, 2015).
In approximately 10%–15% of tumors, telomere length is maintained via alternative lengthening of telomeres (ALT) rather than telomerase activation (Shay and Wright, 2019). The ALT pathway is activated by telomere replication stress and DNA damage and relies on break-induced replication (BIR) for complete DNA replication. This process promotes the formation of ALT-associated PML (APB) bodies, which in turn trigger BIR to elongate telomeres (Dilley et al., 2016). In the past decade, high-throughput exon and whole-genome sequencing has shown that mutations in the histone protein ATRX and its partner DAXX are associated with an increased incidence of ALT activation in various cancer types (Heaphy et al., 2011; Schwartzentruber et al., 2012; Ceccarelli et al., 2016). ATRX or DAXX mutations in human tumors are mutually exclusive to TERT promoter mutations (Killela et al., 2013; Brat et al., 2015; Eckel-Passow et al., 2015; Barthel et al., 2017). A survey of 22 immortalized human ALT cell lines indicated that approximately 90% of the cells exhibited abnormal expression of the ATRX and/or DAXX proteins (Lovejoy et al., 2012), confirming the association between the ATRX/DAXX status and ALT activation.
Due to the crucial role of telomere length maintenance in cell perpetuation and tumorigenesis, the strong correlation between ATRX deficiency and ALT activation suggests that ATRX might suppress tumors by directly inhibiting ALT (Pickett and Reddel, 2015; Maciejowski and de Lange, 2017). Restoring ATRX expression in ATRX-negative ALT cell lines significantly reduced ALT-associated phenotypes (Clynes et al., 2015; Napier et al., 2015). ATRX is proposed to suppress ALT through multiple nuclear functions, including telomere H3.3 loading (Wong et al., 2010), telomere condensation resolution (Ramamoorthy and Smith, 2015), protection of DNA replication from stress or G-quadruplex formation (Law et al., 2010; Leung et al., 2013; Clynes et al., 2015) and telomere R-loop repression (Nguyen et al., 2017).
ATRX mutations are also associated with IDH (isocitrate dehydrogenase)-mutant tumors, which have a high incidence of ATRX mutations (Kannan et al., 2012). ATRX loss, attributed to mutations, deletions, or gene fusions, is linked to other distinct molecular alterations, such as the ALT phenotype (Nandakumar et al., 2017).
This paper highlights the various abnormalities associated with the ATRX protein, from its structure, interactive proteins, and abnormal expression to ATRX as a transcription factor affecting protein expression. In addition, the role of ATRX in tumorigenesis and its mechanism is described to comprehend its role in normal biology and tumors and explore potential research directions in pathogenesis of ATRX syndrome and cancer. Gaining insight into ATRX’s role in cancer will help develop more effective and targeted anti-cancer therapies.
2 Main text
In order to explore the role of ATRX in ATRX syndrome and tumors, we introduced the structural mutations of ATRX, abnormal interacting proteins and transcriptional regulation, the effect of deletion on disease, the effect of mutations on ALT formation, tumor immunity, post-translational modification. We also summarized the effects of various ATRX mutation types on the development of disease, and investigate future directions for ATRX as a potential therapeutic target or prognostic biomarker.
2.1 Structural mutations of ATRX
ATRX is composed of two separate regions: a cysteine-rich ATRX-DNMT3-DNMT3L (ADD) motif domain at the N-terminus and an ATP-dependent helicase domain at the C-terminus (Olar and Sulman, 2015). The ADD domain features two distinctive fingers, a GATA-like finger at the N-terminal end and a PHD finger at the C-terminal end, which are involved in chromatin localization (Gibbons et al., 2008; Iwase et al., 2011). Previous research has shown that ATRX is attracted to pericentromeric heterochromatin (PCH) via its binding to histone H3 trimethylated on lysine 9 (H3K9me3) through the ADD domain but is inhibited by histone H3 trimethylated on lysine 4 (H3K4me3) (Iwase et al., 2011). The majority of missense mutations leading to ATRX syndrome occur within the ADD domains in ATRX patients (Gibbons et al., 2008; Iwase et al., 2011). Mutations in the ADD domain caused a marked reduction in binding to H3K9me3, consequently disrupting the subnuclear localization of ATRX. These aberrations are closely correlated with severe impairments in neuronal differentiation, psychomotor function, and urogenital development (Badens et al., 2006; Gibbons et al., 2008; Dhayalan et al., 2011; Palaniappan and Ramalingam, 2017; Bieluszewska et al., 2022).
The C-terminus of ATRX harbors a septet of helicase domains responsible for ATPase and helicase functions. The C-terminal helicase domain has been implicated in both ATRX syndrome and cancer (Gibbons et al., 2008; Clynes et al., 2013; Bieluszewska et al., 2022). Importantly, ATRX syndrome is attributed to recurrent substitutions in a highly conserved residue, residue 2,085, within the helicase domain of ATRX (Lacoste et al., 2014). Additionally, the K1584R mutation induces mouse embryonic stem cells (mESCs) to differentiate into neural progenitor cells (NPCs) while disrupting glucose metabolic pathways, resulting in distinct neurodevelopmental defects (Bieluszewska et al., 2022). The K1584R mutant is unable to hydrolyze ATP, thus increasing binding to nucleic acids, which results in increased enrichment of ATRX at target sites but decreased localization to pericentromeres (Mitson et al., 2011; Sarma et al., 2014; Bieluszewska et al., 2022) (Figure 1). Mutations associated with ATRX syndrome primarily manifest as missense mutations in the regions described above. However, the precise mechanisms underlying ATRX syndrome resulting from mutations in distinct regions are inadequately explored. Further investigation is warranted to elucidate how these mutations impact the subnuclear localization of ATRX, the pathological consequences of aberrant ATRX localization, and the differential effects of mutations across various regions on the development of intellectual impairment.
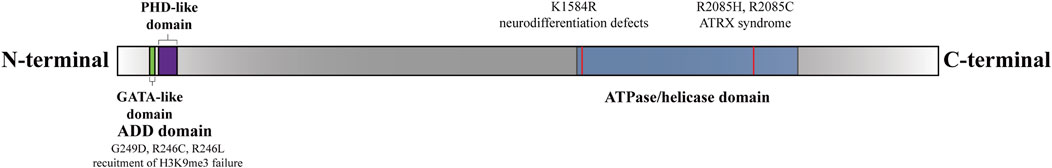
Figure 1. Functional domains and sites of the ATRX (Gibbons et al., 2008). There are two functional domains of ATRX: The N-terminal ADD-domain and the C-terminal ATPase/helicase-domain. The ADD domain bears the GATA-like finger followed by a PHD finger. ADD-domain is required for targeting pericentromeric heterochromatin (PCH) by binding to H3K9me3. The mutations G249D, R246C, and R246L hinder ATRX’s capability to bind to H3K9me3 (Dhayalan et al., 2011). The K1584R mutant induces specific neurodevelopmental abnormalities (Bieluszewska et al., 2022). The R2085H and R2085C mutants are potential genetic triggers of ATRX syndrome (Lacoste et al., 2014).
In the cBioPortal datasets, 884 ATRX mutations have been identified in various cancers. Among these mutations, 267 (30.2%) are associated with glioblastoma multiforme (GBM), oligoastrocytoma, oligodendroglioma, low-grade glioma (NOS), and astrocytoma. In the TCGA glioma datasets, approximately 21% of the samples exhibited ATRX mutations, with truncating mutations and deletions most commonly occurring. The presence of ATRX mutations is linked to various pivotal molecular occurrences (Xie et al., 2019). However, the precise roles of missense, truncating, inframe, splice, and fusion variants in these tumors have not been well investigated. These changes, particularly truncating mutations, occur throughout multiple protein domains and result in decreased protein expression. ATRX mutation and aberrant p53 expression are frequently present in IDH-mutant astrocytomas, which exhibit chromosomal instability and the ALT phenotype, eventually progressing to glioma (Heaphy et al., 2011; Marinoni et al., 2014; Ohba et al., 2020) (Figure 2).
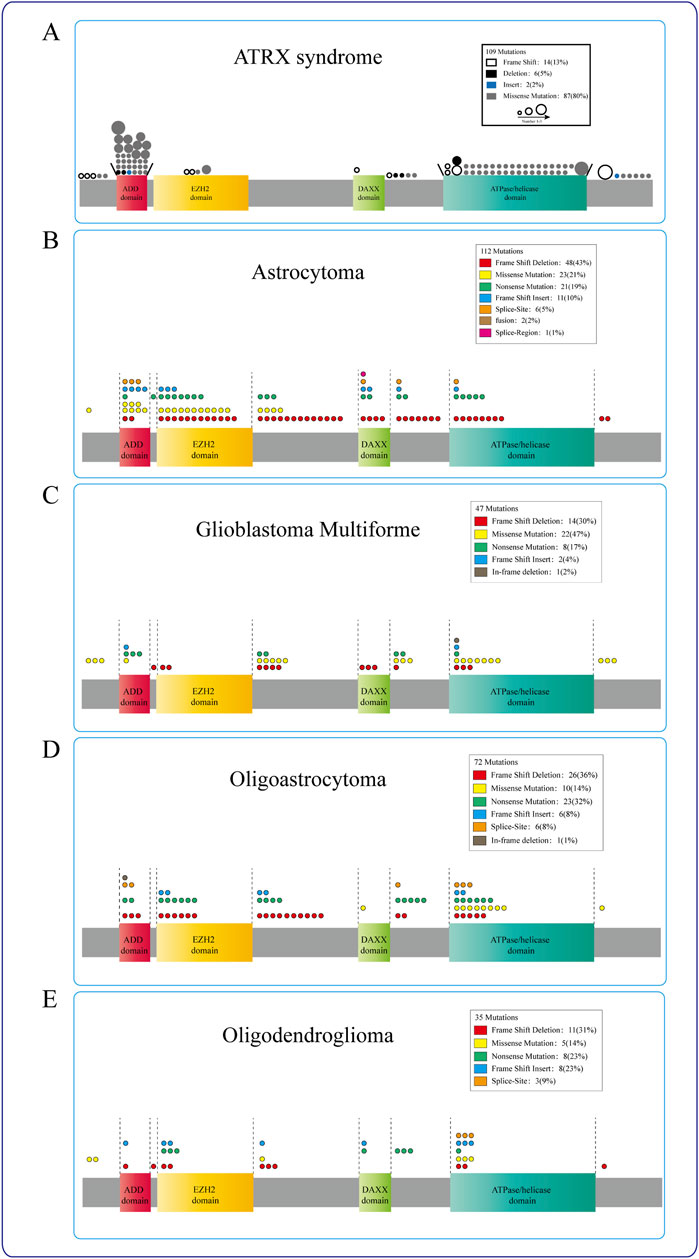
Figure 2. ATRX mutations in diseases. (A) ATRX patient mutations (circle) are predominantly found in either the ADD or the C-terminal ATP-dependent helicase domain, different circles indicate different ways of mutation, and the circle size represents the mutation frequency at the same locus. (B–E) The mutation sites of ATRX in Astrocytoma, Glioblastoma multiforme, Oligoastrocytoma, and Oligodendroglioma were mostly concentrated in the ATRX functional domain. Different colors represent different mutation modes, and the number of circles indicates the frequency with which this mutation mode occurs in this region.
2.2 Interacting proteins
Numerous proteins interact with ATRX in cells, collectively participating in various cellular processes. These proteins mediate H3.3 deposition, DNA methylation, the regulation of heterochromatin epigenetics and DNA transcription. These proteins facilitate the localization of ATRX to PCH, aiding chromosome cohesion during mitosis, and contribute to DNA double-strand break repair, DNA replication, and telomere maintenance. Mutations in these interacting proteins can disrupt the function of ATRX, causing unusual transcription and the onset of the ALT phenotype. This phenotype is strongly linked to the manifestation of ATRX syndrome and a range of tumors (Table 1).
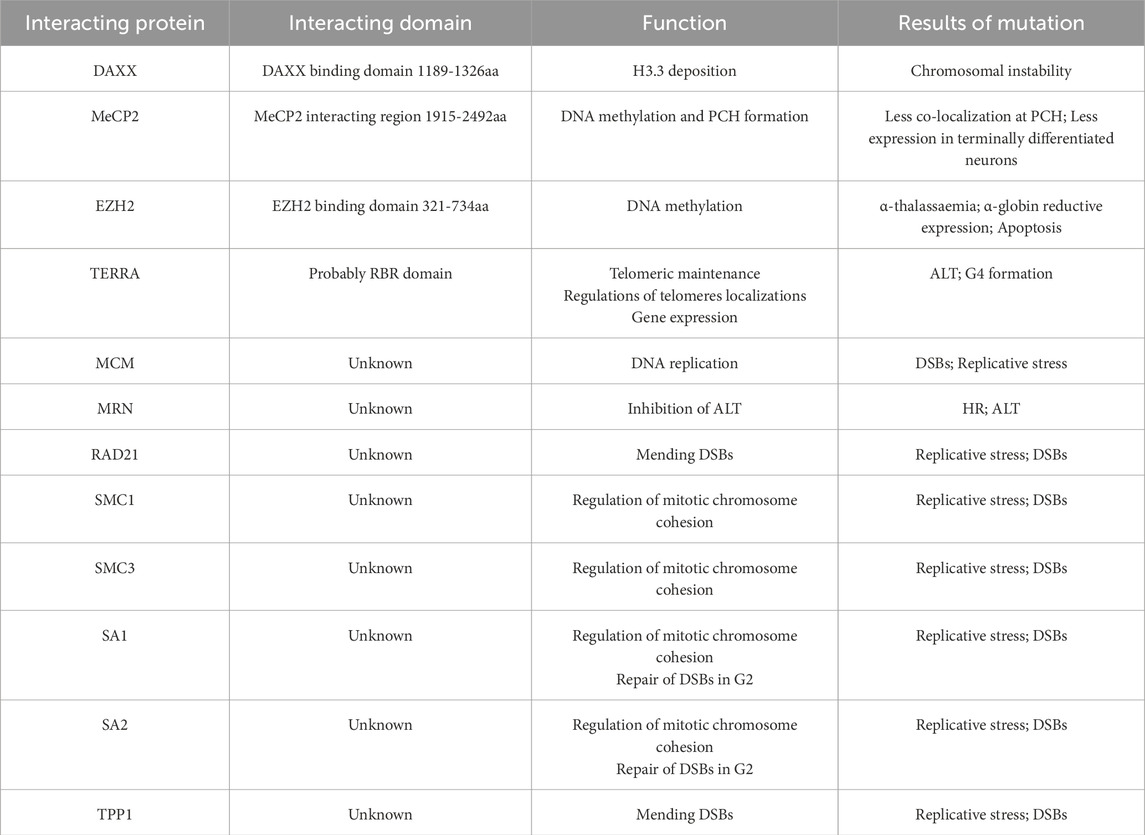
Table 1. ATRX interacting proteins, relative binding domains, function, and results of mutation.
2.2.1 DAXX
Initially, identified as a mediator of Fas-induced apoptosis, death domain-associated protein (DAXX) was suggested to directly interact with Fas. ATRX interacts with DAXX, acting as a histone chaperone to form a chromatin remodeling complex. This complex facilitates the inclusion of the replication-independent histone variant H3.3 in transcriptionally repressed regions such as PCH, telomeres, and other repetitive elements, which are crucial for maintaining chromatin stability (Goldberg et al., 2010; Bogolyubova and Bogolyubov, 2021). These regions are often enriched with histone marks such as H3K9me3, H4K20me3, and DNA methylation (Yang et al., 1997; Xue et al., 2003; Tang et al., 2004; Goldberg et al., 2010; Law et al., 2010; Lewis et al., 2010; Voon et al., 2015). Unlike histone H3, histone H3.3 is not involved in DNA synthesis in the S phase (Goldberg et al., 2010). H3.3 serves as a “replacement” histone in genomic regions lacking histones during phases outside of S phase, restoring chromatin status (Wu et al., 1982; Henikoff et al., 2004).
DAXX specifically binds to H3.3, acting as a chaperone, while ATRX directs the DAXX-mediated incorporation of H3.3 into H3K9me3-enriched chromatin regions via its ADD domain (Iwase et al., 2011). The histone variant H3.3 is present in both transcribed genes and constitutive heterochromatin, but its deposition in these regions involves distinct mechanisms (Goldberg et al., 2010; Wong et al., 2010). When ATRX/DAXX is lacking, histone cell cycle regulator (HIRA) can substitute for the deposition of H3.3 in telomeric heterochromatin (Ahmad and Henikoff, 2002; Tagami et al., 2004). Unlike ATRX/DAXX, HIRA specifically deposits H3.3 in transcriptionally active genome regions, like gene bodies, promoters, and enhancers (Tagami et al., 2004; Wirbelauer et al., 2005; Goldberg et al., 2010).
When ATRX is lost, heterochromatin regions fail to deposit H3.3, causing DNA damage, telomeric end fusions, and genomic instability, ultimately affecting the transcription of tandem repeats and telomere functions (Haase et al., 2018). Additionally, ATRX colocalizes with SETDB1, an H3K9 histone methyltransferase. The ATRX and DAXX complex maintains H3K9me3 at transcriptional sites, but when one component of the complex or H3.3 is depleted, H3K9me3 is reduced among repetitive regions.
ATRX and DAXX act as tumor suppressor proteins and are typically mutated in cells exhibiting ALT-like characteristic gliomas (Lovejoy et al., 2012; Gulve et al., 2022; Clatterbuck Soper and Meltzer, 2023). These mutations lead to the downregulation of the p53 pathway and a diminished response to DNA damage (Gulve et al., 2022).
2.2.2 PML-NBs
The ATRX and DAXX complex is enriched in promyelocytic leukemia nuclear bodies (PML-NBs) (Ishov et al., 1999; Xue et al., 2003). PML-NBs harbor a multitude of proteins, consisting of the ATRX, DAXX and H3.3 complex, as well as SUMO1/2/3 (Ishov et al., 1999; Delbarre et al., 2013). PML, the primary component of PML-NBs, aids H3.3 chromatin assembly during DNA replication and inhibits DAXX-mediated assembly by recruitment to H3.3 in heterochromatin in the S phase. PML is also a crucial site for mutation and dysregulation that affects the epigenetic inheritance of heterochromatin and induces a variety of diseases (Shastrula et al., 2019).
Two methods have been proposed to connect ATRX/DAXX/H3.3 deposition with PML-NBs. In the traditional method, DAXX is drawn to PML bodies by its SUMO-interacting motif (SIM) domains (Lin et al., 2006; Santiago et al., 2009), while the localization of ATRX to PML bodies hinges on its interaction with DAXX (Tang et al., 2004). PML bodies store soluble H3.3-H4 heterodimers, after which ATRX and DAXX are deposited (Delbarre et al., 2013). Recent research has indicated that PML-associated domains (PADs) are extensive heterochromatic regions where ATRX/DAXX/H3.3 complexes are deposited by PML-NBs (Delbarre et al., 2017). Without PML, the ATRX and DAXX complex lacks H3.3 deposition in these regions (Delbarre et al., 2017). At the same time, the epigenetic signature of the pad is altered, with increased H3K27me3 and loss of H3K9me3 (Delbarre et al., 2017). Both H3K9me3 and H3K27me3 are involved in maintaining genomic stability, and this genetic shift serves as a compensatory mechanism when ATRX cannot target PADs (Delbarre et al., 2017). However, increasing H3K27me3 to maintain heterochromatin in PADs might depend on the ability of ATRX to recruit polycomb repressive complex 2 (PCR2) complexes to heterochromatin (Sarma et al., 2014; Delbarre et al., 2017) (Figure 3A).
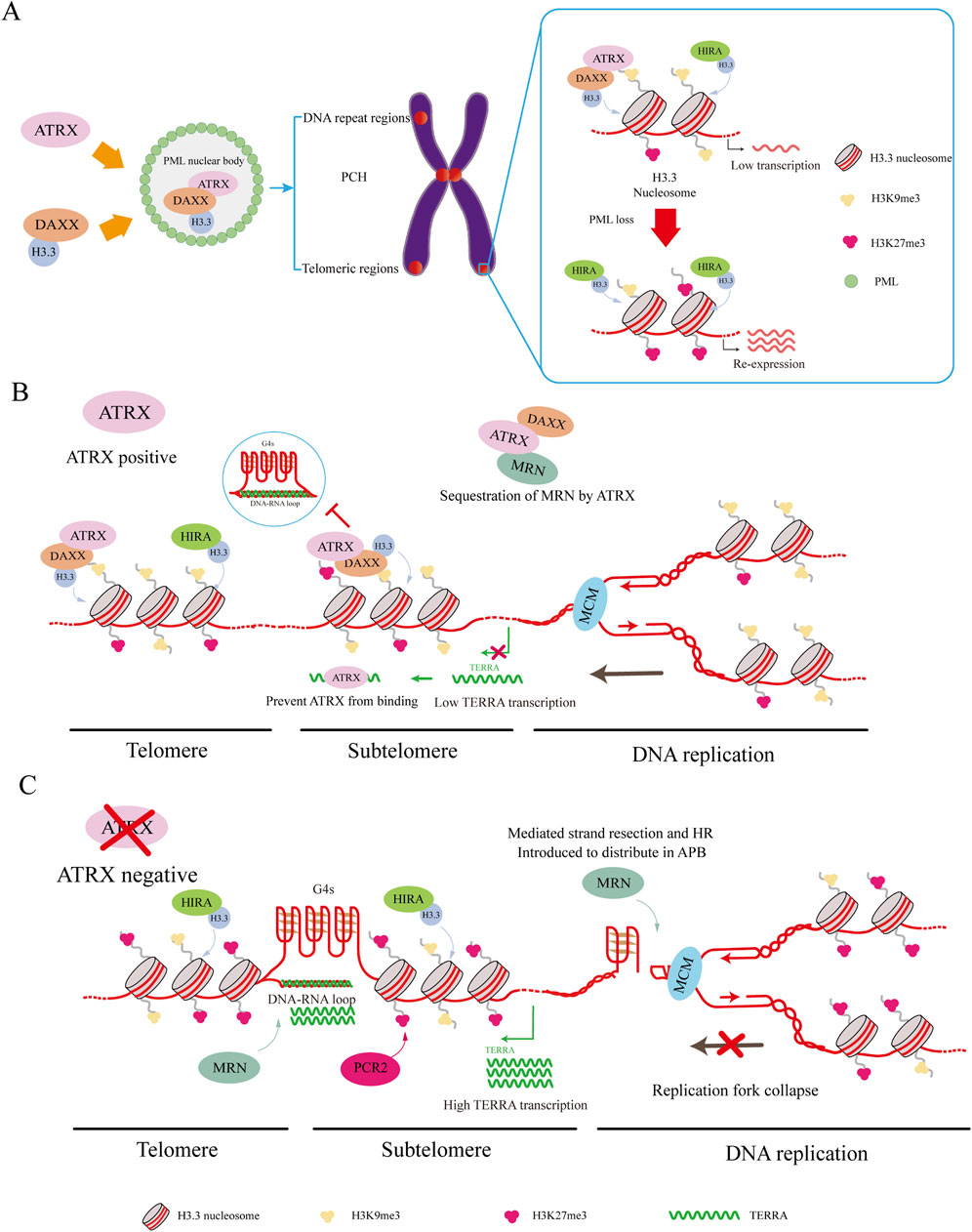
Figure 3. ATRX interacting proteins. (A) DAXX binds to H3.3 and carries it to PML-NBs for complex assembly. This complex targets H3K9me3, leading to the deposition of H3.3 in regions such as PCH, telomeric regions, and other DNA repeat regions, resulting in silent gene expression. HIRA can also deposit H3.3 in these regions. Loss of PML-NBs causes loss of H3K9me3 and increased H3K27me3 misdeposition, which leads to re-expression of silenced genes. (B) ATRX maintains telomeric and subtelomeric chromatin integrity while aiding DNA replication. ATRX facilitates the relocation of the MRN complex from telomeric DNA. Additionally, it resolves G4 DNA structures encountered by the replicative MCM helicase complex and deposits H3.3 to promote MCM progression. Meanwhile, ATRX binds to TERRA to suppress its expression and down-regulate the co-localized genes. (C) In ATRX deficiency, elevated TERRA expression leads to the formation of DNA: RNA hybrids called R-loops. These R-loops hinder telomere replication. Meanwhile, G4 formation causes replication fork collapse, redirecting MRN to the APB site. These promote the occurrence of ALT and recombination-mediated processes at the telomere.
This discovery underscores the unique connection between ATRX/DAXX and PML, elucidating why the loss of PML leads to alterations in the epigenetic marks of PADs. Unlike ATRX, HIRA maintains its capacity for H3.3 deposition into PADs without elevating H3K27me3, supporting a mechanism to fill gaps and maintain chromatin stability (Ray-Gallet et al., 2011; Delbarre et al., 2017). The shift of H3K9me3 to H3K27me3 can preserve heterochromatin, but changes in some regions can lead to the misdeposition of H3K27me3, causing impoverishment of H3K27me3 in some regions. As a result, some tumors exhibit re-expression of silenced genes when ATRX is depleted (Sarma et al., 2014; Danussi et al., 2018; Oppel et al., 2019; Han et al., 2020) (Figure 3A).
2.2.3 MeCP2
Through epigenetic imprinting and chromatin condensation, methyl-CpG binding protein 2 (MeCP2) modulates gene expression and functions as a transcriptional regulator (Haase et al., 2022). Functioning as a transcriptional silencing factor, MeCP2 promotes the attraction of histone deacetylases and comodulators by binding specifically to methylated DNA (Marano et al., 2019). In mature neurons, MeCP2 and a major satellite forwards transcript target ATRX to the PCH and regulate its expression (Marano et al., 2019). Pathological alterations leading to intellectual disability and Rett syndrome (RTT) may arise from point mutations in the MeCP2 gene, which disrupt the MeCP2-ATRX interaction (Nan et al., 2007; Gibbons et al., 2008). Similar results have been predicted for mice (Nan et al., 2007; Kernohan et al., 2010; Kernohan et al., 2014; Haase et al., 2022). Kristin’s study found that ATRX collaborates with MeCP2 to repress a specific group of imprinted genes in the postnatal mouse brain. Mutated MeCP2 interferes with the brain’s repression of imprinted genes, increasing the cognitive deficits associated with RTT and ATRX syndromes (Nan et al., 2007; Kernohan et al., 2010).
2.2.4 EZH2
EZH2 plays a pivotal role in catalyzing H3K27me3 modification, which inhibits target gene transcription by causing chromatin condensation. ATRX interacts with EZH2 through its SET domain to sequester EZH2 to limit the repressive function of EZH2 (Cardoso et al., 1998; Zeng et al., 2022). Prior research has demonstrated a correlation between EZH2 and VAV1, a proto-oncogene, which forms the VAV1-EZH2-ATRX complex. This complex plays a crucial role in hematopoiesis and has been associated with α-thalassemia. The helicase activity of ATRX opposes EZH2 repression, thus promoting α-globin gene expression. Therefore, ATRX deficiency suppresses α-globin gene expression, contributing to disease development (Ye and Worman, 1996; Cardoso et al., 1998; Bieluszewska et al., 2022). Furthermore, inhibiting EZH2 in neuroblastoma (NB) patients upregulates genes related to neuronal maturation, inducing apoptosis in ATRX in-frame fusion (IFF) NB cells (Qadeer et al., 2019). Temozolomide (TMZ) serves as the primary chemotherapeutic agent for glioma treatment. The chemoresistance observed in gliomas is related to ATRX. Compared to parental cells, TMZ-resistant glioma cells expressed higher levels of ATRX and were enriched in EZH2 (Han et al., 2020). This promotes DNA damage repair mechanisms and facilitates the proliferation of glioma cells (Han et al., 2020).
2.2.5 TERRA
ATRX acts as a chromatin remodeling element, regulating gene expression in regions of DNA called G-quadruplexes (G4s), which are found in G-rich repetitive sequences of DNA, including telomeric repeats (Law et al., 2010; Yeung et al., 2013). These G4 structures may lead to DNA damage, acting as substrates for homologous recombination (HR) and inducing a process called ALT during DNA replication (Gramatzki et al., 2016; Teng et al., 2021). ATRX is supposed to prevent this by resolving G4 motifs preceding the replication fork; however, it lacks the ability to resolve these structures on its own (Law et al., 2010; Watson et al., 2013; Clynes et al., 2014; Clynes et al., 2015; Wang et al., 2019). Another molecule that maintains telomeres is the long noncoding RNA telomeric repeat-containing RNA (TERRA), which is composed of the repetitive RNA sequence UUAGGG (Azzalin et al., 2007; Schoeftner and Blasco, 2008). Its synthesis occurs from subtelomeric regions towards telomeric ends (Azzalin et al., 2007; Nergadze et al., 2009). As TERRA invades double-stranded DNA, it forms an RNA‒DNA hybrid (R-loop) at its target site, which aids in the generation of DNA G4s (Tsai et al., 2022).
TERRA and ATRX share hundreds of genes, and their competition for binding at the telomeric region suppresses ATRX localization and maintains telomeric stability (Chu et al., 2017). TERRA can also regulate gene expression in two ways: cis and trans. In cis, ATRX, which is enriched in subtelomeric genes, contributes to H3K9me3-mediated chromatin condensation and downregulates gene expression after TERRA deletion. In trans, TERRA promotes the formation of G4 structures at transcription start sites (TSSs) and prevents ATRX from binding, which leads to the upregulation of gene expression. When TERRA is deleted, the level of ATRX near the TSS increases, resulting in G4 DNA unwinding and gene repression (Tsai et al., 2022).
TERRA strongly binds to DNA via H3K27me3, which is modified by EZH2, while ATRX is associated with H3K9me3 (Sarma et al., 2014; Clynes et al., 2015). When TERRA is deleted, ATRX and H3K9me3 occupy subtelomere regions more frequently, which leads to the downregulation of certain genes that are targeted by both TERRA and ATRX, including Edr1, Mid1, Asmt, Fyco1, and Lphn2 (Chu et al., 2017). Knocking down the ATRX protein by siRNA largely leads to the upregulation of the genes mentioned above (Chu et al., 2017).
According to previous studies, R-loops and G4s can inhibit DNA replication, increase telomere replication stress, and initiate the ALT pathway. However, studies have also demonstrated that ATRX can repress the formation of R-loops both in vivo and in vitro (Tsai et al., 2022; Yan et al., 2022). By inhibiting R-loop formation, ATRX prevents G-rich single-stranded DNA extrusion, preventing G4 formation (Tsai et al., 2022) (Figures 3B, C).
2.2.6 Cohesion
Canonical cohesion is a ring-shaped complex consisting of structural maintenance of chromosomes 1 and 3 (SMC1, SMC3), the Rad21 cohesion complex (RAD21), and either stromal antigen 1 or 2 (SA1 or SA2) (Liu et al., 2023). This complex is crucial for accurate sister chromatid segregation during mitosis and double-strand break (DSB) repair in G2 (Nasmyth and Haering, 2005; Hirano, 2006; Onn et al., 2008; Peters et al., 2008; Nasmyth and Haering, 2009). Among these proteins, ATRX and SA1 can repair DSBs in a similar way by collaborating with the ACD shelterin complex subunit and telomerase recruitment factor (TPP1) to promote the cohesion of telomeres and inhibit nonallelic telomere associations. In addition, SA1 and SA2 cooperate with ATRX-DAXX, which facilitates telomeric DSB repair (Lovejoy et al., 2020). Therefore, in many telomerase-deficient cancers, ATRX inhibits the ALT pathway by promoting cohesion functions in both sister chromatid segregation and telomeric repair of DSBs during G2. However, the detailed mechanisms underlying the interactions between these components warrant further investigation.
2.2.7 MRN
The MRE11-RAD50-NBS1 (MRN) complex is crucial for maintaining the stability of the genome and supporting DNA replication. This complex serves as a DNA damage sensor and helps restart replication forks and repair DNA damage (Clynes et al., 2015). Moreover, the ATRX complex aids in the removal of DNA ends from 5′ to 3′, generating a 3′overhang crucial for subsequent strand invasion (Bérubé et al., 2005; Clynes et al., 2015). ATRX presence reorganizes the MRN complex, relocating it from telomeric DNA and PML nuclear bodies (Clynes et al., 2015). If ATRX is not expressed, the MRN complex is reassigned to the APB site, where it may promote HR (Bérubé et al., 2002; Clynes et al., 2015) (Figures 3B, C).
2.2.8 MCM
The minichromosome maintenance complex (MCM) is a DNA helicase that is essential to initiate DNA replication and elongation (Wu et al., 2018). Studies indicate a connection between ATRX and MCM, and ATRX can utilize its helicase activity to resolve G4 DNA to promote MCM progression in the presence of G4 DNA. At the same time, DAXX mediates variant H3.3 deposition to contain the G4 region in closed heterochromatin, reducing chromatin accessibility and preventing chromosome instability (Teng et al., 2021). Cells are protected from replication stress caused by G4 structures by these activities occurring in the upper reaches of heterochromatin formation mediated by ESET (Figures 3B, C).
2.3 ATRX acts as a transcriptional factor
α-Thalassaemia is a phenotype of ATRX syndrome characterized by decreased expression of α-globin (Truch et al., 2022). ATRX acts as a transcription factor pivotal in regulating α-globin expression. Therefore, mutations in ATRX are significant contributors to alpha thalassemia myelodysplastic syndrome (ATMDS), which is a severe form of α-thalassemia (Gibbons et al., 2003; Steensma et al., 2005; Nguyen et al., 2023).
The variable number of tandem repeats (VNTR) sequence, with the potential G-quadruplex-forming sequence CGC(GGGGCGGGG) n, contributes to the downregulation of α-globin expression (Li et al., 2016). ATRX and DAXX bind collaboratively to the VNTR sequence in the α-globin gene cluster promoter, resolving G4 and promoting the transcription of α-globin (Li et al., 2016). Additionally, using its ADD domain, ATRX identifies H3K9me3-containing nucleosomes (H3.1 and H3.2) and swaps macroH2A for H3.3 (via DAXX) at the α-globin locus. This process promotes a switch towards open chromatin and transcription of α-globin (Ratnakumar and Bernstein, 2013). For patients with ATRX syndrome, mutations or the absence of ATRX can alter VNTR sequence lengths, increasing the presence of variant macroH2A at the α-globin locus and leading to transcriptional repression of the hemoglobin α (HBA) gene at the same locus (Pan and Sawalha, 2009; Ratnakumar et al., 2012; Truch et al., 2022; Aguilera and López-Contreras, 2023).
Moreover, ATRX-mediated regulation of transcription with TERRA at telomeres has been described above, but the specific protein regulating transcription by ATRX is closely related to tumorigenesis and deserves further study. Overall, ATRX, which functions as a transcription factor, regulates the transcription of downstream proteins by deregulating aberrant DNA structures that affect transcription, changing histone deposition, and competing sites for transcription. More modes of regulation remain to be investigated.
2.4 Aberrant expression of ATRX
Brain and neuronal development rely on ATRX, and its aberrant expression contributes to intellectual disability (Bérubé et al., 2005; Ritchie et al., 2008; Medina et al., 2009). Several developmental defects and strong apoptosis were observed in Drosophila neurons when ATRX was inhibited, resulting in caspase-dependent apoptosis via the JNK and dFOXO pathways (Hong et al., 2009). ATRX syndrome is associated with the overexpression of ATRX in the nucleus. Excessive levels of ATRX have been shown to induce growth impairment, neural tube malformations, and heightened embryonic death in transgenic mice (Bérubé et al., 2002). Furthermore, the absence of the ATRX protein can exacerbate neuronal loss, culminating in severe intellectual disability in humans (Bérubé et al., 2005). ATRX deficiency can trigger p53-dependent neuronal apoptosis. Despite the use of ATRX-p53 double knockout mice to rescue embryonic cortex cell death and basal ganglia, this model cannot address all aspects of ATRX deficiency in the developing forebrain (Seah et al., 2008). Postnatal forebrain excitatory neurons lacking ATRX (ATRX-cKO) are less able to transmit synaptic signals and lack plasticity in the hippocampal formation (Gugustea et al., 2020).
ATRX/DAXX/H3.3 are crucial for preserving telomere structural integrity (Ahmad and Henikoff, 2002; Heaphy et al., 2011). Loss of ATRX impairs nucleosome histone H3.3 loading to human telomeres, reducing nucleosome assembly during telomere replication and causing a progressive increase in the length of unbound DNA segments between nucleosomes (Clynes et al., 2014; Koschmann et al., 2016; Li et al., 2019). When these DNA segments reach a certain length, they break or form secondary structures such as G4, impeding telomere replication and provoking a DNA damage response. Retaining telomeric DNA damage can hamper cell growth, accelerate aging, and impede the proliferation of cells (Li et al., 2019). ATRX-deficient tumors always present with ALT (Lovejoy et al., 2012; Amorim et al., 2016; Koschmann et al., 2016). However, the absence of ATRX leads to the concurrent stabilization of G4 structures, impeding these repair pathways, thereby exacerbating DNA damage and precipitating cell death (Wang et al., 2019).
2.5 ATRX mutations in cancer
ATRX is known to function as a tumor suppressor, with mutations identified in many cancers (Pang et al., 2023). This summary highlights the impact of ATRX mutations on gliomas, neuroblastomas, pancreatic neuroendocrine tumors, and osteosarcomas. ATRX may be a potentially effective target for tumour therapy (Figure 4).
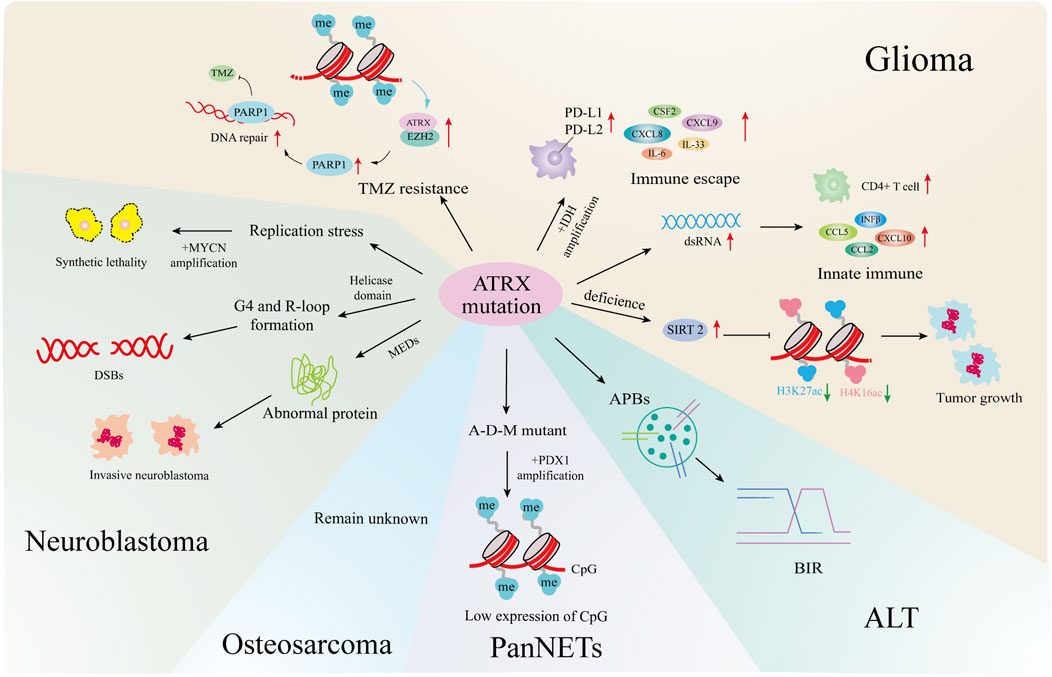
Figure 4. ATRX mutations and cancers. In gliomas, ATRX mutations is involved in the development of ALT, the formation of TMZ resistant strains, the changes of the immune microenvironment, and affects tumor growth and senescence through epigenetics. In neuroblastoma, ATRX processes cause synthetic lethality by MYCN amplification, helicase domain mutations cause DNA double-strand breaks, and multiple exon deletion leads to dysregulated protein expression and increased tumor aggressiveness. In PanNETs, ATRX promotes tumor progression by affecting downstream gene expression through combined mutations with IDH, MEN1. In osteosarcoma, ATRX mutations cause the formation of an ALT. All four tumors are closely related to the formation of ALT.
2.5.1 Glioma
In astroglioma (Koschmann et al., 2016), IDH1R132H combined with ATRX knockout induces APBs, the primary sites of de novo telomere synthesis in ALT-positive cells (Udugama et al., 2021). ATRX mutations occur in 50% of ALT-positive pediatric high-grade glioma (pHGG) patients (Stundon et al., 2023). Other pathways may drive ALT formation in various glioma types, but the mechanism linking ATRX loss to ALT remains to be further explored (Becher, 2023).
The combination of IDH1R132H/p53mut and ATRX loss induces immune escape by upregulating immune checkpoint proteins such as PD-L1, PD-L2, and the BET protein BRD3/4 in astrogliomas, as well as cytokines and chemokines such as IL-33, CXCL8, CSF2, IL-6, and CXCL9 (Hu et al., 2022).
Grade WHO II/III IDH-mutant gliomas, including oligodendrogliomas (IDH-O) with 1p/19q codeletion and astrocytomas (IDH-A) with intact 1p/19q, show differences in chromatin accessibility and gene expression that correlate with the extent of ATRX loss. IDH-A exhibits a greater incidence of ATRX loss, influencing the regulation of various chemokines, cytokines, and upstream transcription factors to sustain distinct tumor microenvironments (Babikir et al., 2021).
In most gliomas, ATRX deletion in combination with IDH mutation leads to immunosuppression, whereas IDH-mutant, ATRX-mutant astrocytomas exhibit greater immune cell infiltration than IDH-mutant, ATRX wild-type oligodendrogliomas (Venteicher et al., 2017). Studies indicate that ATRX deficiency alters the tumor microenvironment by inducing innate immune responses to dsRNA, upregulating chemokines such as CCL2, CCL5, CXCL10 and IFNβ in innate immune pathways, and enhancing immune CD4+Tcell infiltration (Hariharan et al., 2024). The contrasting immunological effects of ATRX deletion are attributed to the sequence and timing of ATRX mutations relative to IDH mutations, influencing distinct epigenetic alterations in chromatin.
In primary gliomas, DNA methylation downregulates ATRX, inhibiting DNA damage repair through the ATRX/EZH2 complex-PARP1 axis and increasing sensitivity to TMZ. Conversely, in TMZ-resistant glioma cells, DNA demethylation enhances ATRX expression, promoting DNA damage repair to counteract drug effects like TMZ (Han et al., 2020).
SIRT2, which was identified through multiomics synthesis as a potential driver gene in ATRX-null gliomas, is overexpressed. Inhibitors targeting SIRT2 induce epigenome reprogramming in ATRX-deficient glioma models by modifying H3K27ac and H4K16ac marks on chromatin and causing genome-wide changes via enhancer-associated modifications, promoting senescence in the ATRX-deficient glioma model system (Malgulwar et al., 2024).
2.5.2 Neuroblastoma
According to genome aberrations, high-risk neuroblastomas can be divided into four categories: MYCN amplification (37%), TERT rearrangement (23%), lack of ATRX (11%), and not mutant (29%) (Brodeur, 2003; Cheung and Dyer, 2013; Zeineldin et al., 2020). Previous genomic analysis of stage 4 pediatric neuroblastoma samples identified ATRX mutations occurred among patients who were typically older than 5 years of age, had an indolent disease course, and poor overall survival (OS) (Cheung et al., 2012; Cheung and Dyer, 2013; Mossé et al., 2014).
However, MYCN expression and ATRX mutations are incompatible. Elevated MYCN levels promote metabolic reprogramming, mitochondrial dysfunction, reactive-oxygen species generation, and DNA-replicative stress. The combination of replicative stress caused by defects in the ATRX–histone chaperone complex, and that induced by MYCN-mediated metabolic reprogramming, leads to synthetic lethality (Zeineldin et al., 2020). Therefore, ATRX and MYCN represent an unusual example, where inactivation of a tumor-suppressor gene and activation of an oncogene are incompatible. This synthetic lethality may eventually be exploited to improve outcomes for patients with high-risk neuroblastoma.
ATRX aberrations were classified as nonsense mutations, missense mutations, or multiexon deletions (MEDs), with 68% identified as MEDs. Among these MEDs, 75% were predicted to be in-frame. The predominant deletion patterns involved the absence of exons 8-9 or exons 11-12 (Zeineldin et al., 2020).
Nonsense mutations in ATRX are scattered across the gene and can result in the absence of ATRX protein production; however, the impact of these mutations on neuroblastoma remains insufficiently studied (Zeineldin et al., 2020). Among these patients, those with ATRX missense mutations may have slightly better prognoses than those with the other two types of mutations (Zeineldin et al., 2020). It is unclear whether the missense mutation leads to reduced or missing expression of the neuroblastoma ATRX protein. Interestingly, in patients with ATRX syndrome, which is characterized by mild α-thalassemia symptoms and moderate intellectual disability, most missense mutations cause protein instability and increased susceptibility to degradation, resulting in reduced protein levels (Ratnakumar et al., 2012; Bieluszewska et al., 2022). ATRX mutations predominantly occur in the helicase domain, which is crucial for ATRX DNA translocation activity and can disrupt processes such as histone variant H3.3 incorporation and G-quadruplex or R-loop resolution (Zeineldin et al., 2020). In addition to point mutations and indels at the ATRX locus, large deletions at the N-terminus of ATRX can produce in-frame fusion proteins (IFFs). These IFFs lack critical chromatin interaction domains and contribute to aggressive neuroblastoma by altering chromatin structure and causing transcriptional dysregulation.
Some common chromosome aberrations in neuroblastoma, including recurrent distortions such as gain of 17q and loss of 11q, 2p, and 1p, are associated with poor prognosis (Brady et al., 2020). Loss of 11q occurs more frequently only in tumors lacking ATRX than in those with the other two mutation subtypes (Zeineldin et al., 2020). The region lost on 11q contains numerous genes involved in cell apoptosis and cell cycle progression. ATRX mutations are known to lead to increased replication-associated DNA damage. Therefore, the acquisition of 11q deletions may confer an evolutionary advantage when ATRX is abnormal, potentially reducing the level of DNA damage-induced apoptosis.
Almost all neuroblastoma patients with ATRX distortion suffer from ALT (Zeineldin et al., 2020; van Gerven et al., 2022). ATRX mutations contribute to tumorigenesis in two ways. First, they cause defects in telomeric H3.3 deposition, leading to telomere maintenance via ALT (Li et al., 2019). Second, ATRX mutations induce abnormal H3.3 deposition at gene promoters and enhancers involved in neuronal differentiation (including retinoic acid-responsive genes), resulting in structural defects at G4 sites. This attenuation of gene expression allows ATRX-mutated neuroblastoma cells to proliferate continuously (Zeineldin et al., 2020). Although DAXX mutations can also promote ALT, they do not affect the deposition of H3.3 at G4 structures.
2.5.3 PanNETs
A total of 43% of PanNET patients had DAXX (25%) or ATRX (18%) mutations, and some exhibited ATRX loss (10%) (Heaphy and Singhi, 2023). The establishment of ATRX-KO mutants in pancreatic β cells via a genetically engineered mouse model did not directly cause PanNETs (Chan et al., 2018). However, simultaneous deletion of ATRX, MEN1, and PTEN resulted in the development of high-grade PanNETs in mice (Fuentes et al., 2024). The role of ATRX mutations in PanNETs is not well understood, but it is widely acknowledged that the simultaneous loss of multiple tumor suppressors is pivotal for PanNET development.
Additionally, DAXX/ATRX and ALT serve as prognostic biomarkers for PanNETs, with DAXX/ATRX mutations associated with a better prognosis. ALT-positive NF-PanNETs are significantly correlated with larger tumor size, increased perineural invasion, lymphovascular invasion, advanced pathological T stage and grade, regional lymph node metastases and postoperative distant metastasis/recurrence (p < 0.001) (Heaphy and Singhi, 2023).
ATRX mutations in PanNETs also influence gene expression through epigenetic mechanisms. Specifically, ATRX, DAXX, and MEN1 mutants (A-D-M mutant PanNETs) exhibit reduced histone methylation levels compared to wild-type PanNETs (Chan et al., 2018). As a critical transcription factor in pancreatic development and β-cell maturation, PDX1 exhibits hypermethylation and low expression of CpG sites in A-D-M mutant PanNETs, promoting a pancreatic “α-cell-like” phenotype thought to be the tumor’s origin (Chan et al., 2018).
2.5.4 Osteosarcoma
Osteosarcoma is a rare and highly metastatic bone cancer originating from mesenchymal cells that predominantly affects children and adolescents (Lindsey et al., 2017). It is characterized by chromosomal instability and genetic alterations leading to aneuploidy and increased tumor aggressiveness (Leibowitz et al., 2015). In exome sequencing studies involving 1,244 osteosarcoma patients, ATRX was identified as a susceptibility gene, and patients with ATRX syndrome exhibited more aggressive forms of osteosarcoma (Mirabello et al., 2020).
Loss of ATRX in overall survival (OS) tumors has been reported to promote migration and invasion by upregulating NF-κB signaling and enhancing integrin binding (Bartholf DeWitt et al., 2022). In osteosarcoma, the ALT-positive phenotype, which is typically associated with ATRX mutation or loss of expression, is also observed in high-grade pediatric osteosarcoma, where telomere maintenance occurs despite wild-type ATRX expression. This is achieved through TOP3A amplification or overexpression, enabling ALT activation, overcoming replicative senescence, and promoting tumor progression (de Nonneville et al., 2022). The specific impact of ATRX mutation on osteosarcoma remains unstudied, but understanding this effect and the factors influencing ALT formation will be crucial for diagnosing and treating osteosarcoma.
3 Discussion
Studies have unequivocally established ATRX as a chromatin remodeler and a critical participant in various vital cellular processes.
ATRX syndrome manifests as multiple bodily system abnormalities (Badens et al., 2006; Gibbons et al., 2008; Dhayalan et al., 2011; Palaniappan and Ramalingam, 2017; Bieluszewska et al., 2022), which are related to various factors, such as ATRX structural alterations, interacting proteins, and the regulation of gene expression. ATRX syndrome primarily arises from ATRX mutations, which extensively disrupt neuronal cell development (Bérubé et al., 2005; Ritchie et al., 2008; Medina et al., 2009) and dysregulate α-globin transcription, mirroring the clinical manifestations observed in individuals with α-thalassemia (Ratnakumar et al., 2012; Li et al., 2016). The intellectual retardation and gonadal abnormalities displayed by the ATRX syndrome model constructed by knockout in different cells/tissues indicate that ATRX is widely involved in the development of the brain and gonads (León and Harley, 2021). Studies have shown that mutations in ATRX affect DNA repair by affecting cell cycle progression by affecting the abnormality of neuronal stem cells and gonad cell development (Bagheri-Fam et al., 2011; Han et al., 2020). Therefore, more research on the function of ATRX in chromosome segregation and the cell cycle is warranted. ATRX syndrome is closely related to mutations in ATRX, and its detection and risk of in carriers is particularly important because of its significant impact on child intelligence (León and Harley, 2021). However, the specific mechanism remains undefined, and the etiology of other systemic abnormalities remains under investigation.
Currently, studies have demonstrated that ATRX plays a significant role in tumorigenesis (Picketts et al., 1996; Danussi et al., 2018; Ohba et al., 2020; Gulve et al., 2022; Hackeng et al., 2022). Among the myriad factors implicated in tumor initiation and progression, abnormalities in ALT are notable features associated with ATRX dysregulation. Moreover, ATRX is involved in the development of cancers involving ALT, such as neuroblastoma, pancreatic neuroendocrine tumors and osteosarcoma (Olar and Sulman, 2015; Li et al., 2019). Nevertheless, the underlying causes of ALT remain to be elucidated through further studies.
G4s are predominantly observed in heterochromatic repeat regions, telomeres, pericentric repeat regions, rDNA repeats and endogenous retroviral elements (ERVs) (Law et al., 2010; Sarma et al., 2014; He et al., 2015; Sadic et al., 2015; Voon et al., 2015; Danussi et al., 2018; Udugama et al., 2018), which have adverse effects on gene expression, the progression of replication and the stability of DNA duplexes (Law et al., 2010; Clynes et al., 2015; Teng et al., 2021). ATRX prevents G4 structure formation by maintaining heterochromatin in G4-rich regions through DAXX-mediated incorporation of variant H3.3. The condensation of heterochromatin is crucial for protecting cells from the harmful effects of G4-induced stress (Clynes et al., 2014; Teng et al., 2021). In addition, ATRX modulates gene expression not only directly but also in collaboration with other proteins (Medina et al., 2009; Law et al., 2010; Clynes et al., 2014; Chu et al., 2017; Yan et al., 2022). This action prevents further double-strand breaks and ALT formation, thereby reducing cancer occurrence (Law et al., 2010; Leung et al., 2013; Clynes et al., 2015; Ramamoorthy and Smith, 2015; Nguyen et al., 2017). However, the specific etiological factors underlying ALT levels are unknown and require further investigation.
The MRN and MCM proteins discussed in this paper represent only a fraction of the many proteins implicated in the mechanism of ALT. However, the specific mechanism by which these proteins interact with ALT remains to be further studied. It has also been reported that the overexpression of Sp100, another PML nucleosome component, can inhibit ALT by separating MRN components from APB (Jiang et al., 2005). Hence, ATRX expression could similarly influence Sp100 overexpression, limiting ALT by relocating the MRN complex from the telomeric recombination site. Notably, the MRN complex favors G4 structures as its substrate (Ghosal and Muniyappa, 2005), potentially directly cleaving persistent G4 structures in ALT tumors after DNA replication or transcription, leading to DSB formation and HDR (Seah et al., 2008). The mechanism involved in the interaction between ATRX and MCM through H3.3 remains unclear.
Studies have shown that ALT can develop in the absence of ATRX mutations, rendering ATRX-targeted therapies ineffective for some ALT-positive gliomas, necessitating alternative treatments (Stundon et al., 2023). Additional ALT mechanisms may exist but have yet been investigated.
In glioma, ATRX/IDH/p53 combined mutations and individual mutations exhibit opposite effects on the immune microenvironment, which encourages tumor proliferation and invasion (Hu et al., 2022; Hariharan et al., 2024). Our hypothesis is that these mutations cooperate to affect chemokine and signal transducer expression and, consequently, the tumor microenvironment. As a therapeutic drug for glioma, TMZ inhibits tumor cell proliferation mainly by inhibiting DNA damage repair in ATRX mutation-positive glioma (Han et al., 2020). The resistant strain maintains DNA repair by altering ATRX transcription levels through demethylation of its transcription factors (Han et al., 2020). Studies suggest that TMZ + γ-radiotherapy enhances the immunosuppressive effects in the absence of ATRX, inducing an immunosuppressive microenvironment (Hu et al., 2022). This proposes modifying the demethylation levels of glioma chemoradiotherapy or combining it with immunomodulatory medications to improve efficacy.
As a transcription factor, ATRX plays a broad role in epigenetic development (Haase et al., 2018). It controls gene transcription via histone modifications, promoting tumor initiation and progression, while its own post-translational modifications (PTMs) are undefined (Delbarre et al., 2017; Malgulwar et al., 2024). Studies report that phosphorylation at multiple sites of ATRX is strongly related its role in the cell cycle (Bérubé et al., 2000). The impact of the modification of ATRX itself on its function remains to be thoroughly investigated.
Through their effects on the immune microenvironment and cytokine production, IDH and ATRX mutations in glioblastoma and astrocytoma promote immune suppression, and may be a potential therapeutic target for prolonging patient survival (Hu et al., 2022; Hariharan et al., 2024; Squalli Houssaini et al., 2024). In addition, in pancreatic neuroendocrine tumors, ATRX/DAXX mutations are considered prognostic biomarkers in patients with poor disease-free survival (DFS), but opposite results in advanced/metastatic patients (Wang et al., 2021), indicating that ATRX mutations at different stages may have different effects and have implications for prognostic judgment. With the development of medicine, how ATRX mediates ALT, affects immune functions, epigenetic and its role in tumor development will be studied in the near future. ATRX opens avenues for discovering novel precision medicine-based therapeutic approaches.
Author contributions
KY: Conceptualization, Data curation, Visualization, Writing–original draft, Writing–review and editing. YT: Data curation, Visualization, Writing–original draft, Writing–review and editing. ZD: Writing–review and editing. LP: Writing–review and editing. JZ: Writing–review and editing. HW: Supervision, Writing–review and editing. QY: Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by National Natural Science Foundation of China, grant number 32000499; Natural Science Foundation of Hunan Provincial of China, grant number 2021JJ40346; Hunan Students’ Platform for Innovation and Entrepreneurship Training Program, grant number S202310542208.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguilera, P., and López-Contreras, A. J. (2023). ATRX, a guardian of chromatin. Trends Genet. 39 (6), 505–519. doi:10.1016/j.tig.2023.02.009
Ahmad, K., and Henikoff, S. (2002). The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9 (6), 1191–1200. doi:10.1016/s1097-2765(02)00542-7
Amorim, J. P., Santos, G., Vinagre, J., and Soares, P. (2016). The role of ATRX in the alternative lengthening of telomeres (ALT) phenotype. Genes (Basel) 7 (9), 66. doi:10.3390/genes7090066
Argentaro, A., Yang, J. C., Chapman, L., Kowalczyk, M. S., Gibbons, R. J., Higgs, D. R., et al. (2007). Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc. Natl. Acad. Sci. U. S. A. 104 (29), 11939–11944. doi:10.1073/pnas.0704057104
Azzalin, C. M., Reichenbach, P., Khoriauli, L., Giulotto, E., and Lingner, J. (2007). Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318 (5851), 798–801. doi:10.1126/science.1147182
Babikir, H., Wang, L., Shamardani, K., Catalan, F., Sudhir, S., Aghi, M. K., et al. (2021). ATRX regulates glial identity and the tumor microenvironment in IDH-mutant glioma. Genome Biol. 22 (1), 311. doi:10.1186/s13059-021-02535-4
Badens, C., Lacoste, C., Philip, N., Martini, N., Courrier, S., Giuliano, F., et al. (2006). Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin. Genet. 70 (1), 57–62. doi:10.1111/j.1399-0004.2006.00641.x
Bagheri-Fam, S., Argentaro, A., Svingen, T., Combes, A. N., Sinclair, A. H., Koopman, P., et al. (2011). Defective survival of proliferating Sertoli cells and androgen receptor function in a mouse model of the ATR-X syndrome. Hum. Mol. Genet. 20 (11), 2213–2224. doi:10.1093/hmg/ddr109
Barthel, F. P., Wei, W., Tang, M., Martinez-Ledesma, E., Hu, X., Amin, S. B., et al. (2017). Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 49 (3), 349–357. doi:10.1038/ng.3781
Bartholf DeWitt, S., Hoskinson Plumlee, S., Brighton, H. E., Sivaraj, D., Martz, E. J., Zand, M., et al. (2022). Loss of ATRX promotes aggressive features of osteosarcoma with increased NF-κB signaling and integrin binding. JCI Insight 7 (17), e151583. doi:10.1172/jci.insight.151583
Becher, O. J. (2023). A new path to alternative lengthening of telomeres?. Neuro Oncol. 25 (7), 1343–1344. doi:10.1093/neuonc/noad054
Bérubé, N. G., Healy, J., Medina, C. F., Wu, S., Hodgson, T., Jagla, M., et al. (2008). Patient mutations alter ATRX targeting to PML nuclear bodies. Eur. J. Hum. Genet. 16 (2), 192–201. doi:10.1038/sj.ejhg.5201943
Bérubé, N. G., Jagla, M., Smeenk, C., De Repentigny, Y., Kothary, R., and Picketts, D. J. (2002). Neurodevelopmental defects resulting from ATRX overexpression in transgenic mice. Hum. Mol. Genet. 11 (3), 253–261. doi:10.1093/hmg/11.3.253
Bérubé, N. G., Mangelsdorf, M., Jagla, M., Vanderluit, J., Garrick, D., Gibbons, R. J., et al. (2005). The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J. Clin. Invest 115 (2), 258–267. doi:10.1172/jci22329
Bérubé, N. G., Smeenk, C. A., and Picketts, D. J. (2000). Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum. Mol. Genet. 9 (4), 539–547. doi:10.1093/hmg/9.4.539
Bieluszewska, A., Wulfridge, P., Doherty, J., Ren, W., and Sarma, K. (2022). ATRX histone binding and helicase activities have distinct roles in neuronal differentiation. Nucleic Acids Res. 50 (16), 9162–9174. doi:10.1093/nar/gkac683
Bogolyubova, I., and Bogolyubov, D. (2021). DAXX is a crucial factor for proper development of mammalian oocytes and early embryos. Int. J. Mol. Sci. 22 (3), 1313. doi:10.3390/ijms22031313
Brady, S. W., Liu, Y., Ma, X., Gout, A. M., Hagiwara, K., Zhou, X., et al. (2020). Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 11 (1), 5183. doi:10.1038/s41467-020-18987-4
Brat, D. J., Verhaak, R. G., Aldape, K. D., Yung, W. K., Salama, S. R., Cooper, L. A., et al. (2015). Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372 (26), 2481–2498. doi:10.1056/NEJMoa1402121
Brodeur, G. M. (2003). Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer 3 (3), 203–216. doi:10.1038/nrc1014
Cardoso, C., Timsit, S., Villard, L., Khrestchatisky, M., Fontès, M., and Colleaux, L. (1998). Specific interaction between the XNP/ATR-X gene product and the SET domain of the human EZH2 protein. Hum. Mol. Genet. 7 (4), 679–684. doi:10.1093/hmg/7.4.679
Ceccarelli, M., Barthel, F. P., Malta, T. M., Sabedot, T. S., Salama, S. R., Murray, B. A., et al. (2016). Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164 (3), 550–563. doi:10.1016/j.cell.2015.12.028
Chan, C. S., Laddha, S. V., Lewis, P. W., Koletsky, M. S., Robzyk, K., Da Silva, E., et al. (2018). ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 9 (1), 4158. doi:10.1038/s41467-018-06498-2
Cheung, N. K., and Dyer, M. A. (2013). Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13 (6), 397–411. doi:10.1038/nrc3526
Cheung, N. K., Zhang, J., Lu, C., Parker, M., Bahrami, A., Tickoo, S. K., et al. (2012). Association of age at diagnosis and genetic mutations in patients with neuroblastoma. Jama 307 (10), 1062–1071. doi:10.1001/jama.2012.228
Chu, H. P., Cifuentes-Rojas, C., Kesner, B., Aeby, E., Lee, H. G., Wei, C., et al. (2017). TERRA RNA antagonizes ATRX and protects telomeres. Cell 170 (1), 86–101. doi:10.1016/j.cell.2017.06.017
Clatterbuck Soper, S. F., and Meltzer, P. S. (2023). ATRX/DAXX: guarding the genome against the hazards of ALT. Genes (Basel) 14 (4), 790. doi:10.3390/genes14040790
Clynes, D., Higgs, D. R., and Gibbons, R. J. (2013). The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem. Sci. 38 (9), 461–466. doi:10.1016/j.tibs.2013.06.011
Clynes, D., Jelinska, C., Xella, B., Ayyub, H., Scott, C., Mitson, M., et al. (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 6, 7538. doi:10.1038/ncomms8538
Clynes, D., Jelinska, C., Xella, B., Ayyub, H., Taylor, S., Mitson, M., et al. (2014). ATRX dysfunction induces replication defects in primary mouse cells. PLoS One 9 (3), e92915. doi:10.1371/journal.pone.0092915
Danussi, C., Bose, P., Parthasarathy, P. T., Silberman, P. C., Van Arnam, J. S., Vitucci, M., et al. (2018). Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat. Commun. 9 (1), 1057. doi:10.1038/s41467-018-03476-6
De La Fuente, R., Baumann, C., and Viveiros, M. M. (2011). Role of ATRX in chromatin structure and function: implications for chromosome instability and human disease. Reproduction 142 (2), 221–234. doi:10.1530/rep-10-0380
Delbarre, E., Ivanauskiene, K., Küntziger, T., and Collas, P. (2013). DAXX-dependent supply of soluble (H3.3-H4) dimers to PML bodies pending deposition into chromatin. Genome Res. 23 (3), 440–451. doi:10.1101/gr.142703.112
Delbarre, E., Ivanauskiene, K., Spirkoski, J., Shah, A., Vekterud, K., Moskaug, J., et al. (2017). PML protein organizes heterochromatin domains where it regulates histone H3.3 deposition by ATRX/DAXX. Genome Res. 27 (6), 913–921. doi:10.1101/gr.215830.116
de Nonneville, A., Salas, S., Bertucci, F., Sobinoff, A. P., Adélaïde, J., Guille, A., et al. (2022). TOP3A amplification and ATRX inactivation are mutually exclusive events in pediatric osteosarcomas using ALT. EMBO Mol. Med. 14 (10), e15859. doi:10.15252/emmm.202215859
Dhayalan, A., Tamas, R., Bock, I., Tattermusch, A., Dimitrova, E., Kudithipudi, S., et al. (2011). The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum. Mol. Genet. 20 (11), 2195–2203. doi:10.1093/hmg/ddr107
Dilley, R. L., Verma, P., Cho, N. W., Winters, H. D., Wondisford, A. R., and Greenberg, R. A. (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539 (7627), 54–58. doi:10.1038/nature20099
Eckel-Passow, J. E., Lachance, D. H., Molinaro, A. M., Walsh, K. M., Decker, P. A., Sicotte, H., et al. (2015). Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 372 (26), 2499–2508. doi:10.1056/NEJMoa1407279
Fan, H. C., Chen, C. M., Chi, C. S., Tsai, J. D., Chiang, K. L., Chang, Y. K., et al. (2019). Targeting telomerase and ATRX/DAXX inducing tumor senescence and apoptosis in the malignant glioma. Int. J. Mol. Sci. 20 (1), 200. doi:10.3390/ijms20010200
Fuentes, M. E., Lu, X., Flores, N. M., Hausmann, S., and Mazur, P. K. (2024). Combined deletion of MEN1, ATRX and PTEN triggers development of high-grade pancreatic neuroendocrine tumors in mice. Sci. Rep. 14 (1), 8510. doi:10.1038/s41598-024-58874-2
Garbarino, J., Eckroate, J., Sundaram, R. K., Jensen, R. B., and Bindra, R. S. (2021). Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl. Oncol. 14 (9), 101147. doi:10.1016/j.tranon.2021.101147
Ghosal, G., and Muniyappa, K. (2005). Saccharomyces cerevisiae Mre11 is a high-affinity G4 DNA-binding protein and a G-rich DNA-specific endonuclease: implications for replication of telomeric DNA. Nucleic Acids Res. 33 (15), 4692–4703. doi:10.1093/nar/gki777
Gibbons, R. J., Pellagatti, A., Garrick, D., Wood, W. G., Malik, N., Ayyub, H., et al. (2003). Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS). Nat. Genet. 34 (4), 446–449. doi:10.1038/ng1213
Gibbons, R. J., Picketts, D. J., Villard, L., and Higgs, D. R. (1995). Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 80 (6), 837–845. doi:10.1016/0092-8674(95)90287-2
Gibbons, R. J., Wada, T., Fisher, C. A., Malik, N., Mitson, M. J., Steensma, D. P., et al. (2008). Mutations in the chromatin-associated protein ATRX. Hum. Mutat. 29 (6), 796–802. doi:10.1002/humu.20734
Goldberg, A. D., Banaszynski, L. A., Noh, K. M., Lewis, P. W., Elsaesser, S. J., Stadler, S., et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140 (5), 678–691. doi:10.1016/j.cell.2010.01.003
Gramatzki, D., Frei, K., Cathomas, G., Moch, H., Weller, M., and Mertz, K. D. (2016). Interleukin-33 in human gliomas: expression and prognostic significance. Oncol. Lett. 12 (1), 445–452. doi:10.3892/ol.2016.4626
Gugustea, R., Tamming, R. J., Martin-Kenny, N., Bérubé, N. G., and Leung, L. S. (2020). Inactivation of ATRX in forebrain excitatory neurons affects hippocampal synaptic plasticity. Hippocampus 30 (6), 565–581. doi:10.1002/hipo.23174
Gulve, N., Su, C., Deng, Z., Soldan, S. S., Vladimirova, O., Wickramasinghe, J., et al. (2022). DAXX-ATRX regulation of p53 chromatin binding and DNA damage response. Nat. Commun. 13 (1), 5033. doi:10.1038/s41467-022-32680-8
Haase, F., Singh, R., Gloss, B., Tam, P., and Gold, W. (2022). Meta-analysis identifies BDNF and novel common genes differently altered in cross-species models of Rett syndrome. Int. J. Mol. Sci. 23 (19), 11125. doi:10.3390/ijms231911125
Haase, S., Garcia-Fabiani, M. B., Carney, S., Altshuler, D., Núñez, F. J., Méndez, F. M., et al. (2018). Mutant ATRX: uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 22 (7), 599–613. doi:10.1080/14728222.2018.1487953
Hackeng, W. M., Brosens, L. A. A., Kim, J. Y., O'Sullivan, R., Sung, Y. N., Liu, T. C., et al. (2022). Non-functional pancreatic neuroendocrine tumours: ATRX/DAXX and alternative lengthening of telomeres (ALT) are prognostically independent from ARX/PDX1 expression and tumour size. Gut 71 (5), 961–973. doi:10.1136/gutjnl-2020-322595
Han, B., Meng, X., Wu, P., Li, Z., Li, S., Zhang, Y., et al. (2020). ATRX/EZH2 complex epigenetically regulates FADD/PARP1 axis, contributing to TMZ resistance in glioma. Theranostics 10 (7), 3351–3365. doi:10.7150/thno.41219
Hariharan, S., Whitfield, B. T., Pirozzi, C. J., Waitkus, M. S., Brown, M. C., Bowie, M. L., et al. (2024). Interplay between ATRX and IDH1 mutations governs innate immune responses in diffuse gliomas. Nat. Commun. 15 (1), 730. doi:10.1038/s41467-024-44932-w
He, Q., Kim, H., Huang, R., Lu, W., Tang, M., Shi, F., et al. (2015). The daxx/atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell 17 (3), 273–286. doi:10.1016/j.stem.2015.07.022
Heaphy, C. M., de Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., Shi, C., et al. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333 (6041), 425. doi:10.1126/science.1207313
Heaphy, C. M., and Singhi, A. D. (2023). Reprint of: the diagnostic and prognostic utility of incorporating DAXX, ATRX, and alternative lengthening of telomeres (ALT) to the evaluation of pancreatic neuroendocrine tumors (PanNETs). Hum. Pathol. 132, 1–11. doi:10.1016/j.humpath.2023.01.004
Henikoff, S., Furuyama, T., and Ahmad, K. (2004). Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 20 (7), 320–326. doi:10.1016/j.tig.2004.05.004
Hirano, T. (2006). At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7 (5), 311–322. doi:10.1038/nrm1909
Hong, Y. K., Lee, N. G., Lee, M. J., Park, M. S., Choi, G., Suh, Y. S., et al. (2009). dXNP/DATRX increases apoptosis via the JNK and dFOXO pathway in Drosophila neurons. Biochem. Biophys. Res. Commun. 384 (2), 160–166. doi:10.1016/j.bbrc.2009.04.112
Hu, C., Wang, K., Damon, C., Fu, Y., Ma, T., Kratz, L., et al. (2022). ATRX loss promotes immunosuppressive mechanisms in IDH1 mutant glioma. Neuro Oncol. 24 (6), 888–900. doi:10.1093/neuonc/noab292
Ishov, A. M., Sotnikov, A. G., Negorev, D., Vladimirova, O. V., Neff, N., Kamitani, T., et al. (1999). PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147 (2), 221–234. doi:10.1083/jcb.147.2.221
Iwase, S., Xiang, B., Ghosh, S., Ren, T., Lewis, P. W., Cochrane, J. C., et al. (2011). ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 18 (7), 769–776. doi:10.1038/nsmb.2062
Ji, J., Quindipan, C., Parham, D., Shen, L., Ruble, D., Bootwalla, M., et al. (2017). Inherited germline ATRX mutation in two brothers with ATR-X syndrome and osteosarcoma. Am. J. Med. Genet. A 173 (5), 1390–1395. doi:10.1002/ajmg.a.38184
Jiang, W. Q., Zhong, Z. H., Henson, J. D., Neumann, A. A., Chang, A. C., and Reddel, R. R. (2005). Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol. Cell Biol. 25 (7), 2708–2721. doi:10.1128/mcb.25.7.2708-2721.2005
Kannan, K., Inagaki, A., Silber, J., Gorovets, D., Zhang, J., Kastenhuber, E. R., et al. (2012). Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3 (10), 1194–1203. doi:10.18632/oncotarget.689
Kernohan, K. D., Jiang, Y., Tremblay, D. C., Bonvissuto, A. C., Eubanks, J. H., Mann, M. R., et al. (2010). ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 18 (2), 191–202. doi:10.1016/j.devcel.2009.12.017
Kernohan, K. D., Vernimmen, D., Gloor, G. B., and Bérubé, N. G. (2014). Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 42 (13), 8356–8368. doi:10.1093/nar/gku564
Killela, P. J., Reitman, Z. J., Jiao, Y., Bettegowda, C., Agrawal, N., Diaz, L. A., et al. (2013). TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. U. S. A. 110 (15), 6021–6026. doi:10.1073/pnas.1303607110
Koschmann, C., Calinescu, A. A., Nunez, F. J., Mackay, A., Fazal-Salom, J., Thomas, D., et al. (2016). ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 8 (328), 328ra28. doi:10.1126/scitranslmed.aac8228
Lacoste, C., Leheup, B., Agouti, I., Mowat, D., Giuliano, F., and Badens, C. (2014). Mutations of codon 2085 in the helicase domain of ATRX are recurrent and cause ATRX syndrome. Clin. Genet. 86 (5), 502–503. doi:10.1111/cge.12319
Law, M. J., Lower, K. M., Voon, H. P., Hughes, J. R., Garrick, D., Viprakasit, V., et al. (2010). ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143 (3), 367–378. doi:10.1016/j.cell.2010.09.023
Leibowitz, M. L., Zhang, C. Z., and Pellman, D. (2015). Chromothripsis: a new mechanism for rapid karyotype evolution. Annu. Rev. Genet. 49, 183–211. doi:10.1146/annurev-genet-120213-092228
León, N. Y., and Harley, V. R. (2021). ATR-X syndrome: genetics, clinical spectrum, and management. Hum. Genet. 140 (12), 1625–1634. doi:10.1007/s00439-021-02361-5
Leung, J. W., Ghosal, G., Wang, W., Shen, X., Wang, J., Li, L., et al. (2013). Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J. Biol. Chem. 288 (9), 6342–6350. doi:10.1074/jbc.M112.411603
Lewis, P. W., Elsaesser, S. J., Noh, K. M., Stadler, S. C., and Allis, C. D. (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U. S. A. 107 (32), 14075–14080. doi:10.1073/pnas.1008850107
Li, F., Deng, Z., Zhang, L., Wu, C., Jin, Y., Hwang, I., et al. (2019). ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. Embo J. 38 (19), e96659. doi:10.15252/embj.201796659
Li, Y., Syed, J., Suzuki, Y., Asamitsu, S., Shioda, N., Wada, T., et al. (2016). Effect of ATRX and G-quadruplex formation by the VNTR sequence on α-globin gene expression. Chembiochem 17 (10), 928–935. doi:10.1002/cbic.201500655
Lin, D. Y., Huang, Y. S., Jeng, J. C., Kuo, H. Y., Chang, C. C., Chao, T. T., et al. (2006). Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 24 (3), 341–354. doi:10.1016/j.molcel.2006.10.019
Lindsey, B. A., Markel, J. E., and Kleinerman, E. S. (2017). Osteosarcoma overview. Rheumatol. Ther. 4 (1), 25–43. doi:10.1007/s40744-016-0050-2
Liu, H., Cheng, J., Zhuang, X., Qi, B., Li, F., and Zhang, B. (2023). Genomic instability and eye diseases. Adv. Ophthalmol. Pract. Res. 3 (3), 103–111. doi:10.1016/j.aopr.2023.03.002
Liu, L., and Malkova, A. (2022). Break-induced replication: unraveling each step. Trends Genet. 38 (7), 752–765. doi:10.1016/j.tig.2022.03.011
Lovejoy, C. A., Li, W., Reisenweber, S., Thongthip, S., Bruno, J., de Lange, T., et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 8 (7), e1002772. doi:10.1371/journal.pgen.1002772
Lovejoy, C. A., Takai, K., Huh, M. S., Picketts, D. J., and de Lange, T. (2020). ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 18 (1), e3000594. doi:10.1371/journal.pbio.3000594
Maciejowski, J., and de Lange, T. (2017). Telomeres in cancer: tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 18 (3), 175–186. doi:10.1038/nrm.2016.171
Malgulwar, P. B., Danussi, C., Dharmaiah, S., Johnson, W., Singh, A., Rai, K., et al. (2024). Sirtuin 2 inhibition modulates chromatin landscapes genome-wide to induce senescence in ATRX-deficient malignant glioma. Neuro Oncol. 26 (1), 55–67. doi:10.1093/neuonc/noad155
Marano, D., Fioriniello, S., Fiorillo, F., Gibbons, R. J., D'Esposito, M., and Della Ragione, F. (2019). ATRX contributes to MeCP2-mediated pericentric heterochromatin organization during neural differentiation. Int. J. Mol. Sci. 20 (21), 5371. doi:10.3390/ijms20215371
Marinoni, I., Kurrer, A. S., Vassella, E., Dettmer, M., Rudolph, T., Banz, V., et al. (2014). Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 146 (2), 453–460. doi:10.1053/j.gastro.2013.10.020
Masliah-Planchon, J., Lévy, D., Héron, D., Giuliano, F., Badens, C., Fréneaux, P., et al. (2018). Does ATRX germline variation predispose to osteosarcoma? Three additional cases of osteosarcoma in two ATR-X syndrome patients. Eur. J. Hum. Genet. 26 (8), 1217–1221. doi:10.1038/s41431-018-0147-x
Medina, C. F., Mazerolle, C., Wang, Y., Bérubé, N. G., Coupland, S., Gibbons, R. J., et al. (2009). Altered visual function and interneuron survival in Atrx knockout mice: inference for the human syndrome. Hum. Mol. Genet. 18 (5), 966–977. doi:10.1093/hmg/ddn424
Mirabello, L., Zhu, B., Koster, R., Karlins, E., Dean, M., Yeager, M., et al. (2020). Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol. 6 (5), 724–734. doi:10.1001/jamaoncol.2020.0197
Mitson, M., Kelley, L. A., Sternberg, M. J., Higgs, D. R., and Gibbons, R. J. (2011). Functional significance of mutations in the Snf2 domain of ATRX. Hum. Mol. Genet. 20 (13), 2603–2610. doi:10.1093/hmg/ddr163
Mossé, Y. P., Deyell, R. J., Berthold, F., Nagakawara, A., Ambros, P. F., Monclair, T., et al. (2014). Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr. Blood Cancer 61 (4), 627–635. doi:10.1002/pbc.24777
Nan, X., Hou, J., Maclean, A., Nasir, J., Lafuente, M. J., Shu, X., et al. (2007). Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. U. S. A. 104 (8), 2709–2714. doi:10.1073/pnas.0608056104
Nandakumar, P., Mansouri, A., and Das, S. (2017). The role of ATRX in glioma biology. Front. Oncol. 7, 236. doi:10.3389/fonc.2017.00236
Napier, C. E., Huschtscha, L. I., Harvey, A., Bower, K., Noble, J. R., Hendrickson, E. A., et al. (2015). ATRX represses alternative lengthening of telomeres. Oncotarget 6 (18), 16543–16558. doi:10.18632/oncotarget.3846
Nasmyth, K., and Haering, C. H. (2005). The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74, 595–648. doi:10.1146/annurev.biochem.74.082803.133219
Nasmyth, K., and Haering, C. H. (2009). Cohesin: its roles and mechanisms. Annu. Rev. Genet. 43, 525–558. doi:10.1146/annurev-genet-102108-134233
Nergadze, S. G., Farnung, B. O., Wischnewski, H., Khoriauli, L., Vitelli, V., Chawla, R., et al. (2009). CpG-island promoters drive transcription of human telomeres. Rna 15 (12), 2186–2194. doi:10.1261/rna.1748309
Nguyen, D. T., Voon, H. P. J., Xella, B., Scott, C., Clynes, D., Babbs, C., et al. (2017). The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 18 (6), 914–928. doi:10.15252/embr.201643078
Nguyen, P. C., Tiong, I. S., Westerman, D. A., and Blombery, P. (2023). A novel ATRX variant with splicing consequences in myelodysplastic syndrome with acquired alpha thalassaemia. Br. J. Haematol. 200 (1), e13–e16. doi:10.1111/bjh.18525
Ohba, S., Kuwahara, K., Yamada, S., Abe, M., and Hirose, Y. (2020). Correlation between IDH, ATRX, and TERT promoter mutations in glioma. Brain Tumor Pathol. 37 (2), 33–40. doi:10.1007/s10014-020-00360-4
Olar, A., and Sulman, E. P. (2015). Molecular markers in low-grade glioma-toward tumor reclassification. Semin. Radiat. Oncol. 25 (3), 155–163. doi:10.1016/j.semradonc.2015.02.006
Onn, I., Heidinger-Pauli, J. M., Guacci, V., Unal, E., and Koshland, D. E. (2008). Sister chromatid cohesion: a simple concept with a complex reality. Annu. Rev. Cell Dev. Biol. 24, 105–129. doi:10.1146/annurev.cellbio.24.110707.175350
Oppel, F., Tao, T., Shi, H., Ross, K. N., Zimmerman, M. W., He, S., et al. (2019). Loss of atrx cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 15 (4), e1008039. doi:10.1371/journal.pgen.1008039
Palaniappan, C., and Ramalingam, R. (2017). Deciphering the molecular effects of mutations on ATRX cause ATRX syndrome: a molecular dynamics study. J. Cell Biochem. 118 (10), 3318–3327. doi:10.1002/jcb.25984
Pan, Y., and Sawalha, A. H. (2009). Epigenetic regulation and the pathogenesis of systemic lupus erythematosus. Transl. Res. 153 (1), 4–10. doi:10.1016/j.trsl.2008.10.007
Pang, Y., Chen, X., Ji, T., Cheng, M., Wang, R., Zhang, C., et al. (2023). The chromatin remodeler ATRX: role and mechanism in biology and cancer. Cancers (Basel) 15 (8), 2228. doi:10.3390/cancers15082228
Peters, J. M., Tedeschi, A., and Schmitz, J. (2008). The cohesin complex and its roles in chromosome biology. Genes Dev. 22 (22), 3089–3114. doi:10.1101/gad.1724308
Pickett, H. A., and Reddel, R. R. (2015). Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 22 (11), 875–880. doi:10.1038/nsmb.3106
Picketts, D. J., Higgs, D. R., Bachoo, S., Blake, D. J., Quarrell, O. W., and Gibbons, R. J. (1996). ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum. Mol. Genet. 5 (12), 1899–1907. doi:10.1093/hmg/5.12.1899
Qadeer, Z. A., Valle-Garcia, D., Hasson, D., Sun, Z., Cook, A., Nguyen, C., et al. (2019). ATRX in-frame fusion neuroblastoma is sensitive to EZH2 inhibition via modulation of neuronal gene signatures. Cancer Cell 36 (5), 512–527. doi:10.1016/j.ccell.2019.09.002
Ramamoorthy, M., and Smith, S. (2015). Loss of ATRX suppresses resolution of telomere cohesion to control recombination in ALT cancer cells. Cancer Cell 28 (3), 357–369. doi:10.1016/j.ccell.2015.08.003
Ratnakumar, K., and Bernstein, E. (2013). ATRX: the case of a peculiar chromatin remodeler. Epigenetics 8 (1), 3–9. doi:10.4161/epi.23271
Ratnakumar, K., Duarte, L. F., LeRoy, G., Hasson, D., Smeets, D., Vardabasso, C., et al. (2012). ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Genes Dev. 26 (5), 433–438. doi:10.1101/gad.179416.111
Ray-Gallet, D., Woolfe, A., Vassias, I., Pellentz, C., Lacoste, N., Puri, A., et al. (2011). Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 44 (6), 928–941. doi:10.1016/j.molcel.2011.12.006
Ritchie, K., Seah, C., Moulin, J., Isaac, C., Dick, F., and Bérubé, N. G. (2008). Loss of ATRX leads to chromosome cohesion and congression defects. J. Cell Biol. 180 (2), 315–324. doi:10.1083/jcb.200706083
Sadic, D., Schmidt, K., Groh, S., Kondofersky, I., Ellwart, J., Fuchs, C., et al. (2015). Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 16 (7), 836–850. doi:10.15252/embr.201439937
Santiago, A., Godsey, A. C., Hossain, J., Zhao, L. Y., and Liao, D. (2009). Identification of two independent SUMO-interacting motifs in Daxx: evolutionary conservation from Drosophila to humans and their biochemical functions. Cell Cycle 8 (1), 76–87. doi:10.4161/cc.8.1.7493
Sarma, K., Cifuentes-Rojas, C., Ergun, A., Del Rosario, A., Jeon, Y., White, F., et al. (2014). ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 159 (4), 869–883. doi:10.1016/j.cell.2014.10.019
Schenkel, L. C., Kernohan, K. D., McBride, A., Reina, D., Hodge, A., Ainsworth, P. J., et al. (2017). Identification of epigenetic signature associated with alpha thalassemia/mental retardation X-linked syndrome. Epigenetics Chromatin 10, 10. doi:10.1186/s13072-017-0118-4
Schoeftner, S., and Blasco, M. A. (2008). Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 10 (2), 228–236. doi:10.1038/ncb1685
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482 (7384), 226–231. doi:10.1038/nature10833
Seah, C., Levy, M. A., Jiang, Y., Mokhtarzada, S., Higgs, D. R., Gibbons, R. J., et al. (2008). Neuronal death resulting from targeted disruption of the Snf2 protein ATRX is mediated by p53. J. Neurosci. 28 (47), 12570–12580. doi:10.1523/jneurosci.4048-08.2008
Shastrula, P. K., Sierra, I., Deng, Z., Keeney, F., Hayden, J. E., Lieberman, P. M., et al. (2019). PML is recruited to heterochromatin during S phase and represses DAXX-mediated histone H3.3 chromatin assembly. J. Cell Sci. 132 (6), jcs220970. doi:10.1242/jcs.220970
Shay, J. W., and Wright, W. E. (2019). Telomeres and telomerase: three decades of progress. Nat. Rev. Genet. 20 (5), 299–309. doi:10.1038/s41576-019-0099-1
Squalli Houssaini, A., Lamrabet, S., Senhaji, N., Sekal, M., Nshizirungu, J. P., Mahfoudi, H., et al. (2024). Prognostic value of ATRX and p53 status in high-grade glioma patients in Morocco. Cureus 16 (3), e56361. doi:10.7759/cureus.56361
Stayton, C. L., Dabovic, B., Gulisano, M., Gecz, J., Broccoli, V., Giovanazzi, S., et al. (1994). Cloning and characterization of a new human Xq13 gene, encoding a putative helicase. Hum. Mol. Genet. 3 (11), 1957–1964. doi:10.1093/hmg/3.11.1957
Steensma, D. P., Gibbons, R. J., and Higgs, D. R. (2005). Acquired alpha-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies. Blood 105 (2), 443–452. doi:10.1182/blood-2004-07-2792
Stundon, J. L., Ijaz, H., Gaonkar, K. S., Kaufman, R. S., Jin, R., Karras, A., et al. (2023). Alternative lengthening of telomeres (ALT) in pediatric high-grade gliomas can occur without ATRX mutation and is enriched in patients with pathogenic germline mismatch repair (MMR) variants. Neuro Oncol. 25 (7), 1331–1342. doi:10.1093/neuonc/noac278
Tagami, H., Ray-Gallet, D., Almouzni, G., and Nakatani, Y. (2004). Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116 (1), 51–61. doi:10.1016/s0092-8674(03)01064-x
Tang, J., Wu, S., Liu, H., Stratt, R., Barak, O. G., Shiekhattar, R., et al. (2004). A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 279 (19), 20369–20377. doi:10.1074/jbc.M401321200
Teng, Y. C., Sundaresan, A., O’Hara, R., Gant, V. U., Li, M., Martire, S., et al. (2021). ATRX promotes heterochromatin formation to protect cells from G-quadruplex DNA-mediated stress. Nat. Commun. 12 (1), 3887. doi:10.1038/s41467-021-24206-5
Truch, J., Downes, D. J., Scott, C., Gür, E. R., Telenius, J. M., Repapi, E., et al. (2022). The chromatin remodeller ATRX facilitates diverse nuclear processes, in a stochastic manner, in both heterochromatin and euchromatin. Nat. Commun. 13 (1), 3485. doi:10.1038/s41467-022-31194-7
Tsai, R. X., Fang, K. C., Yang, P. C., Hsieh, Y. H., Chiang, I. T., Chen, Y., et al. (2022). TERRA regulates DNA G-quadruplex formation and ATRX recruitment to chromatin. Nucleic Acids Res. 50 (21), 12217–12234. doi:10.1093/nar/gkac1114
Udugama, M., Hii, L., Garvie, A., Cervini, M., Vinod, B., Chan, F. L., et al. (2021). Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun. 12 (1), 2584. doi:10.1038/s41467-021-22543-z
Udugama, M., Sanij, E., Voon, H. P. J., Son, J., Hii, L., Henson, J. D., et al. (2018). Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl. Acad. Sci. U. S. A. 115 (18), 4737–4742. doi:10.1073/pnas.1720391115
van Gerven, M. R., Bozsaky, E., Matser, Y. A. H., Vosseberg, J., Taschner-Mandl, S., Koster, J., et al. (2022). Mutational spectrum of ATRX aberrations in neuroblastoma and associated patient and tumor characteristics. Cancer Sci. 113 (6), 2167–2178. doi:10.1111/cas.15363
Venteicher, A. S., Tirosh, I., Hebert, C., Yizhak, K., Neftel, C., Filbin, M. G., et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355 (6332), eaai8478. doi:10.1126/science.aai8478
Voon, H. P., Hughes, J. R., Rode, C., De La Rosa-Velázquez, I. A., Jenuwein, T., Feil, R., et al. (2015). ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 11 (3), 405–418. doi:10.1016/j.celrep.2015.03.036
Wada, T., Sugie, H., Fukushima, Y., and Saitoh, S. (2005). Non-skewed X-inactivation may cause mental retardation in a female carrier of X-linked alpha-thalassemia/mental retardation syndrome (ATR-X): X-inactivation study of nine female carriers of ATR-X. Am. J. Med. Genet. A 138 (1), 18–20. doi:10.1002/ajmg.a.30901
Wang, F., Xu, X., Ye, Z., Qin, Y., Yu, X., and Ji, S. (2021). Prognostic significance of altered ATRX/DAXX gene in pancreatic neuroendocrine tumors: a meta-analysis. Front. Endocrinol. (Lausanne) 12, 691557. doi:10.3389/fendo.2021.691557
Wang, Y., Yang, J., Wild, A. T., Wu, W. H., Shah, R., Danussi, C., et al. (2019). G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 10 (1), 943. doi:10.1038/s41467-019-08905-8
Watson, L. A., Goldberg, H., and Bérubé, N. G. (2015). Emerging roles of ATRX in cancer. Epigenomics 7 (8), 1365–1378. doi:10.2217/epi.15.82
Watson, L. A., Solomon, L. A., Li, J. R., Jiang, Y., Edwards, M., Shin-ya, K., et al. (2013). Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J. Clin. Invest 123 (5), 2049–2063. doi:10.1172/jci65634
Weatherall, D. J., Higgs, D. R., Bunch, C., Old, J. M., Hunt, D. M., Pressley, L., et al. (1981). Hemoglobin H disease and mental retardation: a new syndrome or a remarkable coincidence?. N. Engl. J. Med. 305 (11), 607–612. doi:10.1056/nejm198109103051103
Wirbelauer, C., Bell, O., and Schübeler, D. (2005). Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 19 (15), 1761–1766. doi:10.1101/gad.347705
Wong, L. H., McGhie, J. D., Sim, M., Anderson, M. A., Ahn, S., Hannan, R. D., et al. (2010). ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 20 (3), 351–360. doi:10.1101/gr.101477.109
Wu, R. S., Tsai, S., and Bonner, W. M. (1982). Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 31 (2 Pt 1), 367–374. doi:10.1016/0092-8674(82)90130-1
Wu, W., Rokutanda, N., Takeuchi, J., Lai, Y., Maruyama, R., Togashi, Y., et al. (2018). HERC2 facilitates BLM and WRN helicase complex interaction with RPA to suppress G-quadruplex DNA. Cancer Res. 78 (22), 6371–6385. doi:10.1158/0008-5472.Can-18-1877
Xie, Y., Tan, Y., Yang, C., Zhang, X., Xu, C., Qiao, X., et al. (2019). Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis. Cancer Biol. Med. 16 (4), 784–796. doi:10.20892/j.issn.2095-3941.2019.0143
Xue, Y., Gibbons, R., Yan, Z., Yang, D., McDowell, T. L., Sechi, S., et al. (2003). The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. U. S. A. 100 (19), 10635–10640. doi:10.1073/pnas.1937626100
Yan, Q., Wulfridge, P., Doherty, J., Fernandez-Luna, J. L., Real, P. J., Tang, H. Y., et al. (2022). Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat. Commun. 13 (1), 53. doi:10.1038/s41467-021-27722-6
Yang, X., Khosravi-Far, R., Chang, H. Y., and Baltimore, D. (1997). Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89 (7), 1067–1076. doi:10.1016/s0092-8674(00)80294-9
Ye, Q., and Worman, H. J. (1996). Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J. Biol. Chem. 271 (25), 14653–14656. doi:10.1074/jbc.271.25.14653
Yeung, Y. T., McDonald, K. L., Grewal, T., and Munoz, L. (2013). Interleukins in glioblastoma pathophysiology: implications for therapy. Br. J. Pharmacol. 168 (3), 591–606. doi:10.1111/bph.12008
Zeineldin, M., Federico, S., Chen, X., Fan, Y., Xu, B., Stewart, E., et al. (2020). MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun. 11 (1), 913. doi:10.1038/s41467-020-14682-6
Keywords: ATRX, ATRX syndrome, replication stress, ALT, transcriptional regulation, tumorigenesis
Citation: Yuan K, Tang Y, Ding Z, Peng L, Zeng J, Wu H and Yi Q (2024) Mutant ATRX: pathogenesis of ATRX syndrome and cancer. Front. Mol. Biosci. 11:1434398. doi: 10.3389/fmolb.2024.1434398
Received: 21 May 2024; Accepted: 04 October 2024;
Published: 16 October 2024.
Edited by:
Xiaojun Ren, University of North Carolina at Charlotte, United StatesReviewed by:
Robert Dilley, Massachusetts General Hospital and Harvard Medical School, United StatesIrina Nakashidze, Shota Rustaveli State University, Georgia
Copyright © 2024 Yuan, Tang, Ding, Peng, Zeng, Wu and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaying Wu, aHVheWluZ3d1NkBodW5udS5lZHUuY24=; Qi Yi, eWlxaUBodW5udS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship