
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 10 June 2024
Sec. Biological Modeling and Simulation
Volume 11 - 2024 | https://doi.org/10.3389/fmolb.2024.1414572
 Ali Raza Siddiqui1
Ali Raza Siddiqui1 Mamona Mushtaq2Madiha Sardar1Lubna Atta2Mohammad Nur-e-Alam3Aftab Ahmad4
Mamona Mushtaq2Madiha Sardar1Lubna Atta2Mohammad Nur-e-Alam3Aftab Ahmad4 Zaheer Ul-Haq2*
Zaheer Ul-Haq2*Introduction: The Macrophage Migration Inhibitory Factor (MIF), a key pro-inflammatory mediator, is responsible for modulating immune responses. An array of inflammatory and autoimmune diseases has been linked to the dysregulated activity of MIF. The significance in physiological as well as pathophysiological phenomena underscores the potential of MIF as an attractive target with pharmacological relevance. Extensive research in past has uncovered a number of inhibitors, while the ISO-1, or (S, R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester being recognized as a benchmark standard so far. Recent work by Yang and coworkers identified five promising dietary flavonoids, with superior activity compared to the standard ISO-1. Nevertheless, the exact atomic-level inhibitory mechanism is still elusive.
Methods: To improve the dynamic research, and rigorously characterize, and compare molecular signatures of MIF complexes with ISO-1 and flavonoids, principal component analysis (PCA) was linked with molecular dynamics (MD) simulations and binding free energy calculations.
Results: The results suggest that by blocking the tautomerase site these small molecule inhibitors could modify the MIF activity by disrupting the intrinsic dynamics in particular functional areas. The stability matrices revealed the average deviation values ranging from 0.27–0.32 nm while the residue level fluctuations indicated that binding of the selected flavonoids confer enhanced stability relative to the ISO-1. Furthermore, the gyration values extracted from the simulated trajectories were found in the range of 1.80–1.83 nm.
Discussion: Although all the tested flavonoids demonstrated remarkable results, the one obtained for the potent inhibitors, particularly Morin and Amentoflavone exhibited a good correlation with biological activity. The PCA results featured relatively less variance and constricted conformational landscape than others. The stable ensembles and reduced variation in turns might be the possible reasons for their outstanding performance documented previously. The results from the present exploration provide a comprehensive understanding of the molecular complexes formed by flavonoids and MIF, shedding light on their potential roles and impacts. Future studies on MIF inhibitors may benefit from the knowledge gathered from this investigation.
Discovered almost 4 decades ago while studying the Type IV hypersensitivity reaction, macrophage migration inhibitory factor (MIF) stood out among the first recognized cytokines. MIF serves various roles and acts as a signaling molecule, that is, a cytokine, chemokine, hormone, molecular chaperone, or enzyme (Zhang et al., 2022). Being an essential effector molecule, MIF initiates and coordinates the humoral and cellular responses, playing a central role in immunomodulation. MIF is regarded as a potential biomarker for several diseases presenting with inflammatory elements because it regulates both adaptive and innate responses. The multifaced roles of the MIF and its involvement in the genesis of pathologies like sepsis, cancer, and autoimmune diseases are highlighted in the literature, rendering MIF an attractive target for drug development (Kang and Bucala, 2019). The biological effects elicited by MIF are mediated by both intracellular and extracellular mechanisms. While the intracellular pathway, which is receptor independent, works by physical interaction with an array of resident proteins and enzymes, the extracellular signaling involves binding of MIF to receptor (CD74)/co-receptor complexes (CD44) (Neo and Mitchell, 2020). The attachment of MIF to the receptor activates multiple signaling cascades mediated by two mechanisms involving MAP kinase (p38, JNK, and ERK1/2) and phosphatidil-inositol-3 kinase (PI3K) dependent pathways. The process eventually results in the migration of various transcription factors, like AKT, NF-κB, and STAT3, inside the nucleus, thereby instigating the activation of genes associated with inflammation (Vázquez et al., 2023).
MIF is highly conserved among vertebrates (but is also found in arthropods, nematodes, and protozoans). MIF weighs 12.5 kDa, comprises 114 residues, and has a toroidal shape (Bilsborrow et al., 2019; Penticuff et al., 2019). In its functional form, MIF manifests as a homotrimeric configuration. Each monomer consists of two antiparallel α-helices positioned closely alongside a four-stranded β sheet. The assembly gains additional stability via the interaction of β-strands and sheets of nearby subunits forming the interface region. In general, the structure features a central channel accessible to solvent and a barrel-shaped motif, while the catalytic region is located at the interface of two adjacent monomers (Zhang et al., 2022; Zhang et al., 2024). In addition to involvement in immune responses, MIF functions as thiol-protein oxidoreductase, which depends on a four-residue motif, Cys–Ala–Leu–Cys(CXXC), occupying 56–59th position. The Cys-59 is essential for the underlying activity (Trivedi-Parmar et al., 2018; Qian et al., 2019; Mahalingam et al., 2020). Moreover, the protein under discussion also executes the phenylpyruvate tautomerase activity, catalyzing the tautomerization of phenylpyruvate tautomerase and D-dopachrome, dependent on the distinctive proline residue (Pro1) from the N-terminal of the monomer. Thus, Pro1 is critical for the interaction of MIF with other receptors on the cell surface (Yang et al., 2021; Chen et al., 2023). However, uncertainties exist concerning the correlation between enzymatic activity and physiological role. Thus, inhibitors that modify the surface characteristics of MIF remain a promising area for further exploration. In this regard, targeted efforts to inhibit the tautomerase activity of the enzyme by means of small molecules are a promising strategy, supported by experimental findings from both laboratory-based assays and animal models (Trivedi-Parmar and Jorgensen, 2018; O’Reilly et al., 2016; Sinitski et al., 2019).
From the initial identification of the reference inhibitor ISO-1, several small molecules have been suggested as potential inhibitors of MIF from different scaffolds, that is, pyrazole derivatives, biaryltriazoles, and benzoxazolones, working either as non-covalent or allosteric inhibitors (Lubetsky et al., 2002; Cournia et al., 2009; Jorgensen et al., 2010; Xu et al., 2014; Trivedi-Parmar et al., 2018). A growing body of studies has documented the potential of natural products such as curcumin, ellagic acid, epicatechins, and flavonoids (Dickerhof et al., 2014; Young et al., 2014; Sarkar et al., 2015; Sun et al., 2022). Previous studies, including the work of Garai and coworkers as an example, revealed the efficiency of the flavonoids luteolin and quercetin in interfering with the tautomerase activity of MIF (Garai and Adlercreutz, 2004). Inspired by the anticancer and anti-inflammatory features of flavonoids (Dai et al., 2012; Tuli et al., 2023), Yang and coworkers investigated a set of 24 flavonoids to assess their potential in counteracting both the tautomerase and the biological effects of MIF in the RAW264.7 cell line. They identified five promising candidates (Figure 1) with superior activity and low inhibition constants (IC50) relative to ISO-1. However, the precise mechanism for the observed inhibition for these flavonoids, particularly for the standout candidates, that is, morin and amentoflavone (IC50 = 11.01 ± 0.45 μM and 13.32 ± 0.64 μM, respectively), remains to be determined. The evolution and emergence of advanced in silico approaches have enabled the streamlining of the drug discovery pipeline alongside unraveling mechanistic insights. The precision and efficiency of in silico tools such as molecular dynamic simulation provide a wealth of benefits in understanding the actions of molecules at the atomic scale (Yuan et al., 2022). Much literature highlights the potential of computational explorations, particularly MD simulations, in revealing the mechanism of inhibition. This is exemplified by the previous work of Qureshi et al. (2022), along with many others. In a recent work, Baidya et al. (2023) used computational tools to provide a mechanistic insight into the inhibitory mechanism of proton pump inhibitors targeting choline acetyltransferase. A computational framework has been devised in the present work to uncover both the binding modes and time-dependent stability patterns for the mentioned dietary flavonoids. Following the exploration of binding modes in static and dynamic phases, the ligand-induced structural alterations triggered within the MIF were monitored at local and global levels. The second phase entails the computation of associated variance through PCA aimed at exploring the conformational landscape.
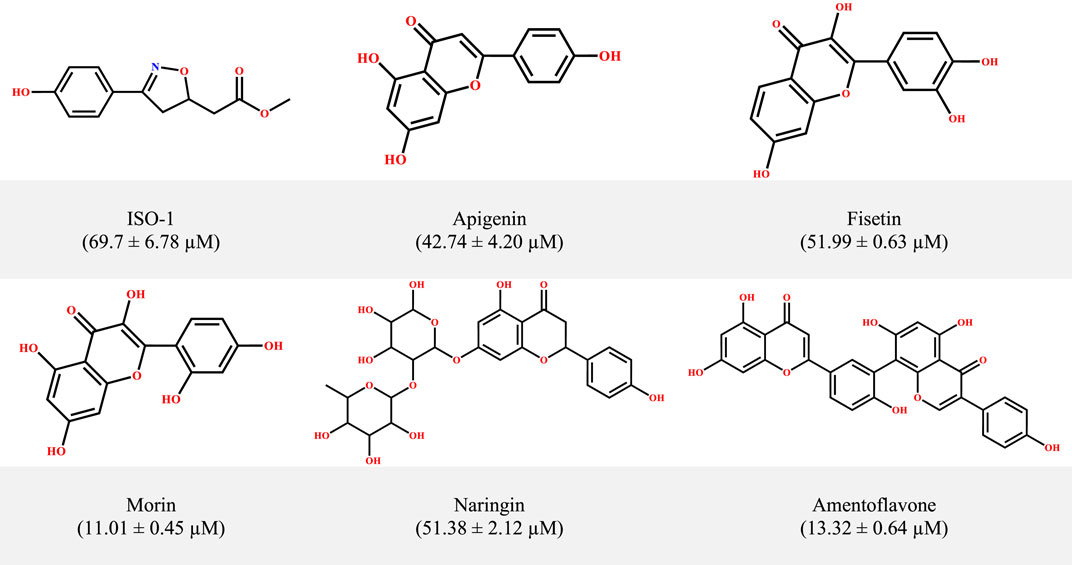
Figure 1. The structural information of MIF inhibitors, a subset within the small-molecule class of flavonoids, with corresponding inhibition concentrations (IC50) provided in parentheses.
The finding from the current work provides a comprehensive molecular-level insight into the MIF-flavonoids and ISO-1 complexes, thus shedding light on the possible reasons for the observed differences in functionalities among the tested candidates. These insights are proposed to provide valuable information that might assist in the development of MIF inhibitors.
The present work aims to identify the binding modes of the experimentally validated dietary flavonoids: apigenin (Ap1), fisetin (F9), morin (M12), naringin (N16), and amentoflavone (Am22), in the tautomerase site of MIF. It will be followed by the structural characterization, thereby elucidating the mechanism of inhibition via a detailed biophysical analysis.
The atomic coordinates of MIF in complex with ISO-1, corresponding to the PDB ID: 1LJT (r: 0.237), were retrieved from the repository of the Protein Data Bank (Lubetsky et al., 2002). The unwanted heteroatoms and the chains other than A and B were deleted. The initial preparatory steps involved the correction and protonation at the default settings in Molecular Operating Environment (MOE) version 2019 (MOE, 2019) (Chemical Computing Group Inc., 2019). Subsequently, the protein was subjected to energy minimization and assignment of partial charges using the Amber10EHT force field. The selected ligands were constructed in ChemDraw version 22.0.0, followed by preparation using MOE. In brief, lone pairs were adjusted, and hydrogens were added where required via the “Protonate 3D” module. The minimization in terms of energy and the assignment of partial charges were in accordance with the MMFF94X force field. The dielectric constant was set to 1, while the RMS gradient threshold was set to 0.1 kcal/mol/Å. The optimized compounds were then subjected to docking studies.
The active or tautomeric site is reported to be positioned at the interface of two monomers. Initially, the the docking software and protocol were benchmarked via a redocking strategy. The resultant deviation in turns indicate the fitness of the docking platform, determining the appropriate use for further work. Based on the benchmarking, the docking was executed with MOE. The “Alpha Triangle” placement method rendered the placement of the ligand poses in the targeted cavity. The resultant poses were scored with the GBVI/WSA dG scoring function (SF), following refinement with the London dG SF. The induced fit docking approach was used, thereby allowing some mobility (Khan et al., 2021; Sardar et al., 2022). Of the retained poses, the conformation featuring the lowest score was selected for interaction pattern analysis via the Protein-Ligand Interaction Profiler (PLIP) (Salentin et al., 2015). The intermolecular interaction patterns alongside the binding modes were modeled and pictured using UCSF Chimera (Pettersen et al., 2004).
In order to examine the kinetic and structural interactions present in the protein–ligand complexes, a 100 ns molecular dynamics (MD) simulation for six different systems was performed using the Gromacs 2021 software package (Abraham et al., 2015). The ACPYPE (Ante-Chamber PYthon Parser interface) server was used to create the ligand topologies described by Sousa da Silva and Vranken (2012). Meanwhile, the Amber ff99SB-ILDN force field was utilized for the target protein parameterization. The ligands and receptors were then combined to form a single, cohesive system, and each resulting complex was enclosed in a box with a cubic shape. Careful positioning was done to ensure that any solute atoms were at least 10Å away from the margins of the periodic box. The TIP3P (transferable intermolecular potential 3P) water model was used to solve the systems (Mark and Nilsson, 2001). Counter ions (Na+ and Cl−) were added to maintain system neutrality. The initial phase consisted of a 50,000-step energy reduction procedure designed to remove steric collisions. The particle-mesh Ewald (PME) approach was utilized to handle long-range electrostatic interactions, whereas van der Waals forces and short-range electrostatic interactions were terminated at 1.0 nm. Following successful convergence, all systems were subjected to pressure equilibration at 1 bar via the NPT ensemble (isothermal and isobaric) and temperature equilibration at 300 K under the NVT condition (constant temperature and volume), using periodic boundary conditions, a Parrinello–Rahman barostat, and a Berendsen thermostat. Following system equilibration, a 100 ns production run was applied. The real-time dynamics and stability of every complicated system were observed using the leap-frog integrator, which applied a time step of 2 fs. The radius of gyration (RoG), root mean square deviation (RMSD), and root mean square fluctuation (RMSF) were the stability indices that were used to track the stability pattern, followed by principal component analysis (PCA). Visual molecular dynamics (VMD) (Humphrey et al., 1996) was used for visual assessment, and the Xmgrace tool (Turner, 2005) was used to plot the graphs.
PCA is a potent statistical technique for extracting important information from large datasets. In this investigation, we want to use PCA to find and examine possible differences in a complex’s energetic and structural characteristics. The focus is on using the MDAnalysis tool (Michaud-Agrawal et al., 2011; Gowers et al., 2016) to analyze simulated data. This study focuses on five flavonoids and the ISO-1 standard, exploring their principal components. The goal is to identify subtle variations in these molecules’ dynamic behavior that may emerge during the MD simulations. Using PCA, we aimed to isolate and emphasize the key elements influencing the observed alterations in the complex’s properties. This strategy provides a means of identifying patterns and trends in the complex interactions between molecule energy and structures.
When combined with techniques for calculating free energy, molecular dynamics can yield informative energetic determinants of binding interactions. The binding free energy, or ΔG bind, is used to quantify the strength of the binding. As a result, the combination of molecular mechanics and generalized Born surface or Poisson–Boltzmann (MMGB/PBSA) has become a popular, dependable, and effective method for doing free energy simulations, especially when dealing with protein–ligand binding (Genheden and Ryde, 2015). Molecular mechanics/generalized Born surface area (MM/GBSA) was adopted in this work to evaluate the binding free energies of the selected flavonoids and the ISO-1 standard. For every simulated system, 1,000 frames were taken from the final 10 ns of the molecular dynamics trajectory. Furthermore, separate computations of different energy components were performed, such as electrostatic energy, van der Waals interactions, and polar and nonpolar solvation energies. Together, these elements aid in the thorough calculation of the total binding free energy. The calculations performed were as follows.
where
Serving an array of biological functions, MIF acts as a signaling molecule and influences both arms of the immune responses. The pleiotropic nature thus renders MIF a prospective biomarker and a promising pharmacological target for several etiologies demonstrating inflammatory elements (Kang and Bucala, 2019; Zhang et al., 2022). Yang et al. (2021) evaluated a series of 24 flavonoids to assess their inhibitory potential toward MIF. Five flavonoids with low IC50 values and, hence, superior potency to the known standard (ISO-1) were identified. However, the precise reason for the observed inhibition pattern remains elusive. The development of advanced computational tools (especially advanced molecular docking and MD simulation methods) allows for the accurate investigation of complex inhibitory mechanisms, transforming the field of drug discovery and enhancing our comprehension of biomolecular interactions (Rossetti and Mandelli, 2022; Jin and Wei, 2024). In an attempt to provide a detailed insight, a computational exploration has been made to envisage the binding conformations both in static and dynamic modes. The comprehensive findings outline the time-dependent conformational alterations, stability patterns, and dynamic interactions triggered by the binding of the ligand with the protein of interest. These understandings can be an important guide for future efforts to build MIF inhibitors.
The exploration of binding modes driven by molecular docking studies was initiated following the benchmarking of both the protocol and the docking station. As presented in Supplementary Figure S1, promising efficiency was demonstrated by London dG, GBVI/WSA dG, and Alpha Triangle as a preferred method for the initial placement of the resultant conformations and primary and secondary scoring functions, respectively. Identified originally, ISO-1 is known for its high specificity and hence used as the reference inhibitor for MIF. The binding cavity native to ISO-1 lies close to the CD74 receptor binding site and is surrounded by Y36, K66, and N109. The docking results, in correlation with biological activity, highlighted strong affinities of tested flavonoids toward MIF, with Am22 and F9 standing out among others. Docking scores of −15.28 kcal/mol and −15.39 kcal/mol were yielded for M12 and Am22, featuring the lowest IC50 values of 11.01 ± 0.45 μM and 13.32 ± 0.64 μM. The N16, Ap1, and F9, on the other hand, were presented with docking scores of −12.26 kcal/mol, −13.22 kcal/mol, and −15.91 kcal/mol, respectively, compared to the −11.95 kcal/mol recorded for ISO-1. The interaction patterns observed for the protein–ligand complexes are presented in Figure 2. Supplementary Table S1 suggested the contribution of both the hydrophobic and electrostatic contacts in providing overall stability to the complexes.
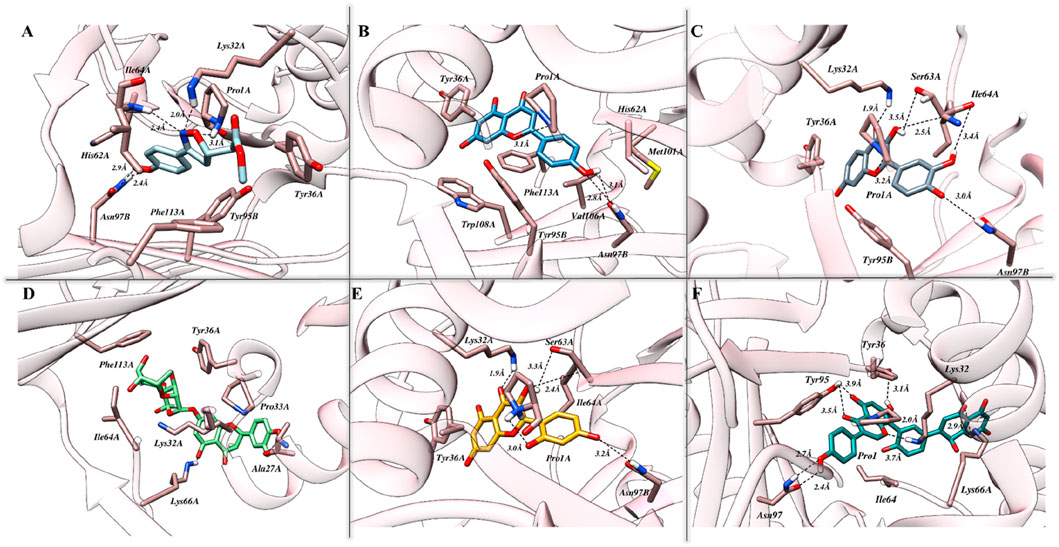
Figure 2. The intermolecular interaction patterns of MIF in complex with (A) ISO-1, (B) Ap1, (C) F9, (D) N16, (E) M12, and (F) Am22 flavonoids are shown in a three-dimensional depiction. Dotted black lines are used to depict hydrogen bonds, while sticks are used to symbolize the involved residues.
MD simulations represent a conventional approach to producing physically relevant ensembles of conformations. The generation of conformational ensembles relies on employing complementary force fields, describing the forces acting on atomic components. Herein, an all-atom dynamic simulation was conducted involving the ligand-bound complexes of MIF. In the following portions, an in-depth analysis of the stability patterns and the associated variations in the resulting conformations of the simulated systems is presented. The following sections will detail the perturbations in stability patterns and conformational landscape following the binding of the dietary flavonoids.
The multitude of conformations resulting from the simulation studies necessitates the clustering of conformations using a common metric in order to monitor the dynamical changes experienced by the simulated system with respect to time (Sargsyan et al., 2017). The RMSD, a pairwise similarity metric, was employed to compute the differences in backbone coordinates. As illustrated in Figure 3, the computation revealed that a well-converged and equilibrated state was observed following an initial phase of stabilization. The initial ∼40 ns presented substantial backbone deviations ranging from 0.25 nm to 0.32 nm, reaching a maximum height of 0.43 nm observed in the case of ISO-1-bound MIF. Secondary to this was the N16-bound system, which presented a maximum deviation peak of 0.43 nm. The ISO-1 induced significant deviations both during the initial and following the attainment of the stabilization compared to the selected flavonoids. However, an overlapping trend was noted during the last 10–20 ns among the systems except for the ISO-1- and Am22-bound MIF, which featured altered patterns (Supplementary Figure S2). Average pairwise backbone deviations of 0.28 ± 0.02 nm, 0.30 ± 0.02 nm, 0.31 ± 0.03 nm, 0.32 ± 0.03 nm, 0.34 ± 0.03 nm, and 0.35 ± 0.04 nm were computed for the converged ensemble. The calculation from the entirety of the trajectories resulted in the values of 0.27 ± 0.04 nm, 0.29 ± 0.04 nm, 0.31 ± 0.03 nm, 0.29 ± 0.05 nm, 0.32 ± 0.04 nm, and 0.34 ± 0.05 nm for F9, AP1, N16, M12, Am22, and ISO-1, respectively. The increased deviations recorded for the flavonoids N16 and Am22 are likely due to the structural complexity and associated size, hindering the proper fitting within the binding cavity. The distribution patterns for the RMSDs highlighted enhanced stability to the receptor compared to the benchmark standard.
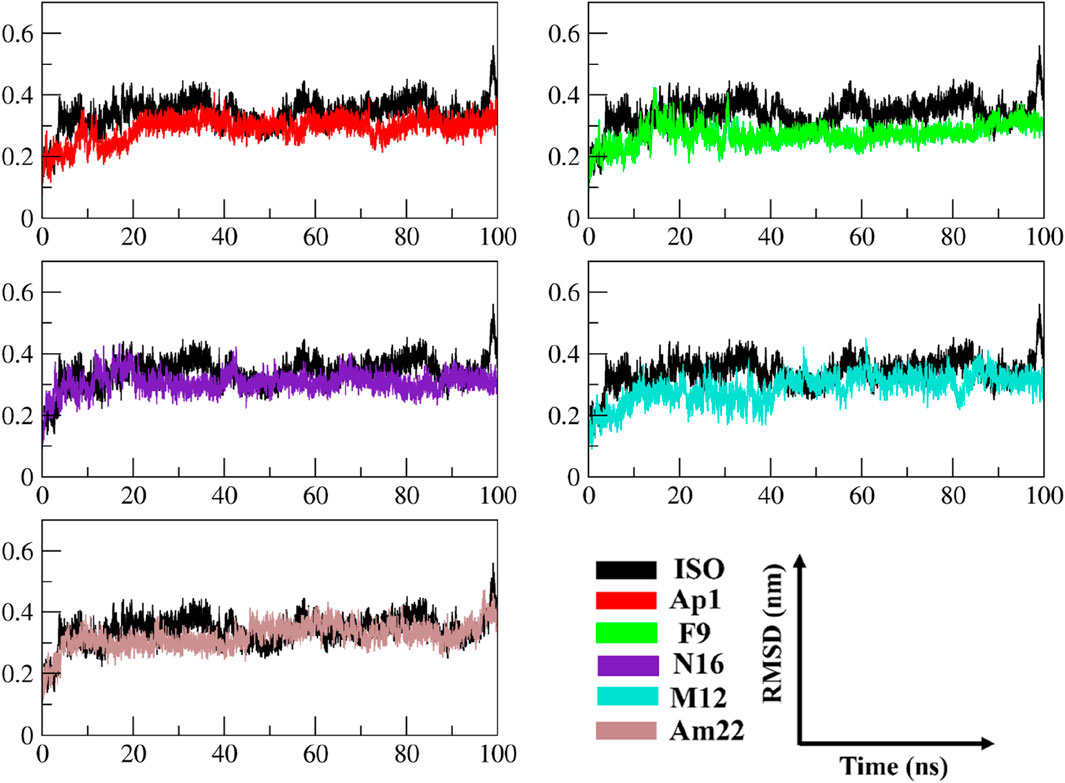
Figure 3. The time evolution of the RMSD between MIF’s bound states with dietary flavonoids and ISO-1 shed light on the protein’s structural stability and conformational changes.
The RMSF data provide significant insights into the dynamics of constituent amino acids harbored by the macromolecular system. The perturbations triggered by the binding event consequently shed light on the binding behavior of the ligand (Zhang et al., 2020). The thorough evaluation of the residue-wise fluctuations via calculating the RMSF undergone by the backbone atoms of the protein unveiled a uniform degree of stability post-ligand binding. The outcome thus underscores the capability of these ligands to impart enhanced stability by restricting the mobility of the residues in most of the cases relative to the standard, as depicted in Figure 4. An overview of the fluctuation values recorded is presented in a box plot format to provide an insight into the range of the fluctuations experienced by the different systems of MIF (Supplementary Figure S3). The maximum and the minimum height of the fluctuations were observed at 0.837 nm and 0.055 nm and originated from the MIF when complexed with Ap1 and F9, respectively. Upon computation, the mean fluctuations were 0.14 ± 0.10 nm, 0.13 ± 0.08 nm, 0.13 ± 0.09 nm, 0.13 ± 0.06 nm, and 0.15 ± 0.12 nm for Ap1, N16, M12, Am22, and F9 compared to the value of 0.16 ± 0.10 nm for the ISO-1-MIF complex. Next, the focus was directed toward the particular residues lining the binding cavity. The binding pocket residues and fluctuations from both chains are presented in Supplementary Figure S4. As expected, the residues from MIF, when bound to dietary flavonoids, featured less fluctuation than ISO-1, highlighting their contribution in rendering the system stable. Limiting the protein’s mobility, hence, could be a likely mechanism for the underlying antagonistic effect of these flavonoids. This structural alteration, in consequence, impacts the normal function of the protein.
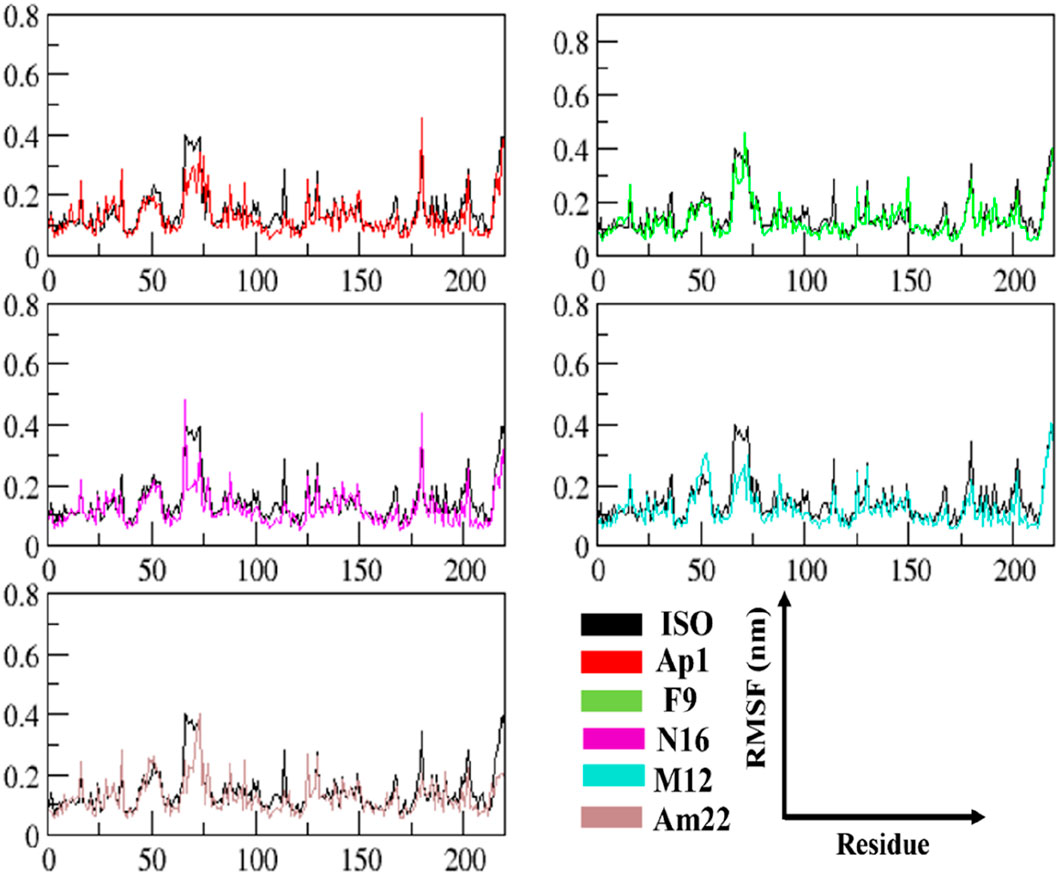
Figure 4. The analysis of time-dependent RMSF demonstrates conformational alterations and highlights MIF’s structural stability.
The radius of gyration (RoG) was used to monitor stability in ligand-bound complexes of MIF, shedding light on the time-dependent folding behavior in the presence of solvent. The stabilization of gyration curves indicates that the protein is correctly folded, while fluctuations are associated with structural changes (Li D. D. et al., 2020). Thus, lower RoG values represent a compact and well-folded conformation, while the higher values are linked to loss of compaction and an unfolded state. As presented in Figure 5, the RoG curves level off after 44 ns of the run in general, with notable fluctuations in gyration values for the ISO-MIF complex. A closer view into the RoG trend during the converged phase (Supplementary Figure S5) featured an overlapping pattern for N16 and M12-MIF complexes with recorded mean values of 1.83 ± 0.01 nm. Similar to this were the observations made for Ap1 and Am22, both with an average value of 1.82 ± 0.01 nm. In line with the pattern observed for RMSD plots, notable fluctuations in the curve were seen for the N16-bound MIF compared to the rest of the systems under investigation. In contrast, the protein bound to the F9 attained a more folded state among all, followed by Am22. The mean values extracted from the curves, when considering the whole trajectories, were 1.80 ± 0.01 nm, 1.82 ± 0.01 nm, 1.83 ± 0.01 nm, 1.83 ± 0.01 nm, and 1.83 ± 0.01 nm for F9, Am22, AP1, N16, and M12, respectively.
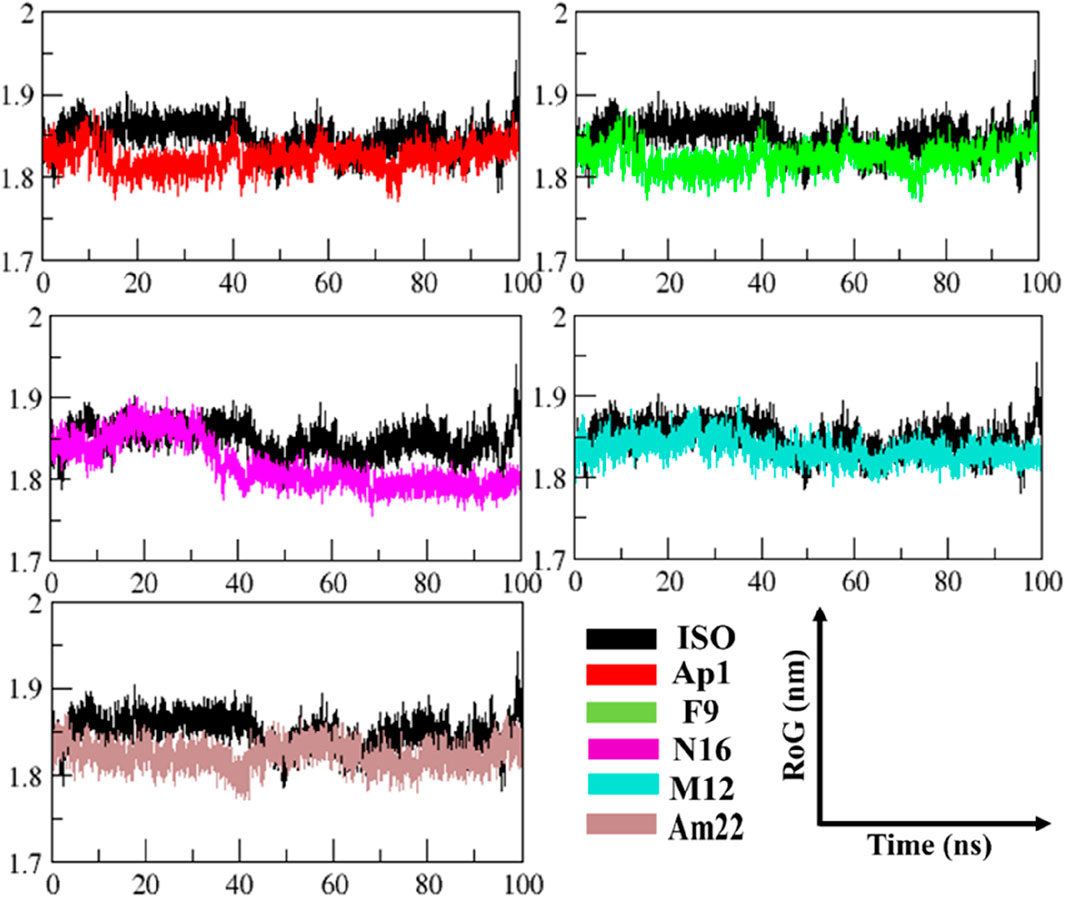
Figure 5. The pattern of gyration is shown in the MIF-simulated complexes.
Conversely, the value of 1.84 ± 0.02 nm was extracted for MIF bound to ISO-1. Note that all the test systems showcased a well-folded state, highlighting the structural integrity relative to the standard ligand used herein and signifying optimum convergence.
The binding of ligands to the cognate pocket is shaped by the intricate network of intermolecular interactions during the dynamics governing the conformational behavior and the stability of the complexes. Knowledge about these drivers provides a comprehensive insight into the structural characterization, functional relationship, and subsequent applications as therapeutics. In the succeeding section, the intermolecular interaction profile of the ligands inside the designated pocket is covered. The translocation in the initial placement observed for the hits following the simulation run can clearly be seen in Supplementary Figure S6, highlighting the ligand’s efficiency towards optimal accommodation in the binding cavity.
Electrostatic interactions, especially hydrogen bonds, are significant regarding structure maintenance and overall stability. Further, alterations in the H-bond network have been linked to pathological conditions such as Parkinson’s (Li X. et al., 2020). In this context, the formation of intermolecular H-bonds and the associated occupancies were monitored over time for the studied systems (Figure 6). The system with standard inhibitor ISO-1 presented with the formation of six hydrogen bonds; however, only three were observed with significant occupancy. N16 and Am22 formed five bonds, and three and two of these bonds had good occupancy, respectively. The MIF bound to Ap1 formed four H-bonds, with two being consistent over time. The F9, on the other hand, formed four H-bonds, of which three persisted in the majority of the frames. In contrast, M12 formed six H-bonds, though only two remained consistent over time.
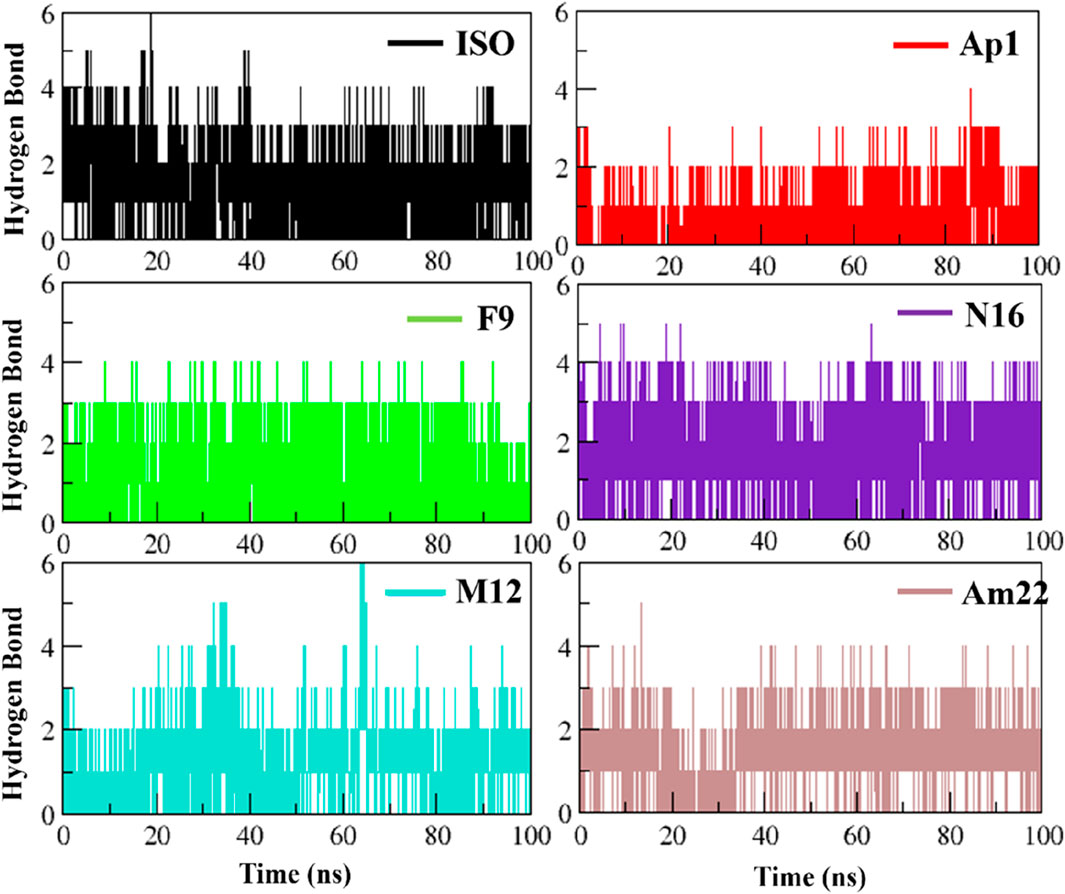
Figure 6. Plotting the evolution of hydrogen bonding over time for the selected dietary flavonoids.
The interaction pose of ISO-1 and MIF, generated from the terminal frame revealed the formation of two electrostatic contacts, with protein residues acting as H-bond donors. Side chain amines of Pro1A and Asn97B donated their hydrogen atoms to the oxazole’s oxygen and the hydroxyl’s phenol oxygen and created H-bonds with lengths of 2.40 Å and 2.11 Å, respectively (Figure 7A). The test compound AP1 displayed five H-bonds where oxygen from the chromone moiety accepted a hydrogen from the pyrrolidine’s nitrogen of the Pro1A positioned 2.2 Å from the acceptor. Another contact of length 2.7 Å was established among the phenol’s oxygen, forming an H-bond of 2.7 Å with the amine nitrogen of Met2A. The carbonyl moiety of His62A, on the other hand, acted as an H-bond acceptor from the phenol, leading to the formation of a bond of length 3.5 Å. Further, TRP95 and Asn97B donated hydrogens to the resorcinol and pyrone ring of the ligand at 3.6 Å and 3.2 Å, respectively (Figure 7B). The stability of the F9 in the binding cavity was governed by three H-bonds. The Pro1A formed two H-bonds, acting as a donor and an acceptor for the reactive ligand moieties positioned 3.6 Å and 2.1 Å from the residue. Another H-bond of 3.7 Å was formed between the Ser63A’s amine and the resorcinol’s hydroxyl group (Figure 7C). The MIF bound to N16 retained only a single H-bond after the simulation run, where the carboxylic acid from the Ala114A’s backbone mediated interaction with the phenol 1.6 Å distant from the acceptor atom (Figure 7D).
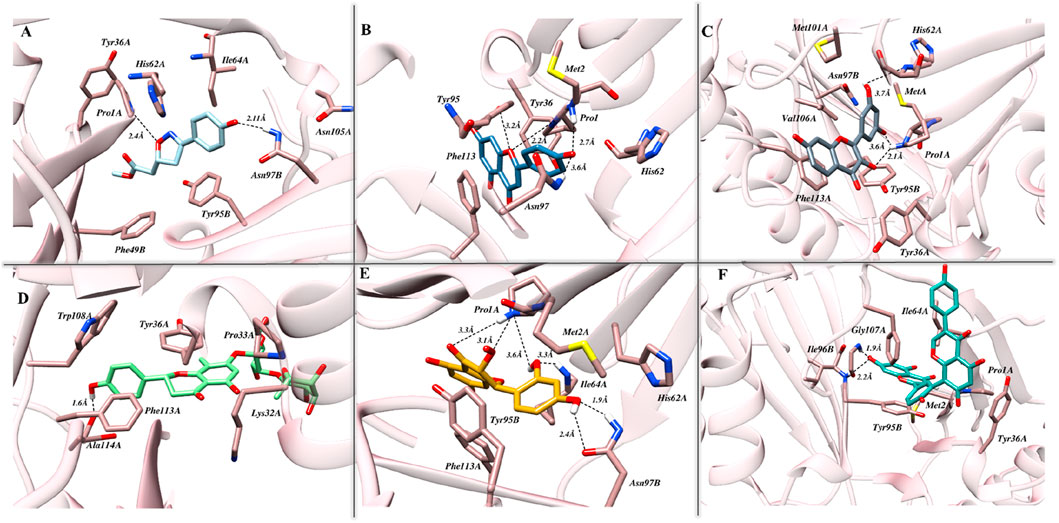
Figure 7. The pattern of intermolecular interactions between MIF in complex with (A) ISO-1, (B) Ap1, (C) F9, (D) N16, (E) M12, and (F) Am22 after the simulation run. Stick representations of the relevant residues are used, and dotted black lines show hydrogen bonding.
The M12 featured the formation of six H-bonds, two involving amide groups (-CONH2) of Asn97B with bond lengths of 1.9 Å and 2.4 Å. The Pro1 from chain A interacted with the chromone moiety and established two H-bonds with lengths of 3.3 Å and 3.1 Å, and also with the resorcinol group featuring contact with a bond length of 2.2 Å. Additional stability was provided by the 3.3 Å contact between Ile64 from chain A and the ligand’s resorcinol (Figure 7E). Am22, on the other hand, acted as an H-bond acceptor for the Gly107A and Ile64B and mediated the formation of electrostatic contacts featuring bond distances of 1.9 Å and 2.2 Å, respectively (Figure 7F). It is important to highlight here that many electrostatic contacts were observed in docking studies, though not all remained persistent in the dynamic state. The alignment of binding poses before and after the simulation indicates that almost all the tested ligands experience translocation from the initial position. Because ligands are allowed to move freely in the presence of a solvent, the observed shift is not a strange phenomenon. This obvious shift in coordinates is proposed as a possible reason for the loss of the interaction observed under static mode. However, the ligands with a promising affinity towards the cognate protein do not leave the binding pocket, and the observed fluctuations underscore the mobility of the ligands during the stabilization phase when they try to fit in an optimal manner. ISO-1 and N16 experienced significant dislocation from the original coordinates, which justifies the loss of the initial contacts.
Overall, the interaction pattern observed for the dietary flavonoids highlighted their potential to efficiently engage the binding pocket residues via numerous hydrophobic contacts. Furthermore, some ligands also demonstrated π-π stacking, providing additional strength to the complexes. The details of the interactions, including the residues involved, are presented in Supplementary Table S2.
A statistical technique called PCA is used to distill data from large datasets. It is especially helpful for lowering the dimensionality of large datasets. PCA uses a mixture of orthogonal vectors to represent each frame’s coordinates, which simplifies trajectory data in MD simulations. The trajectory’s primary uncorrelated motion is captured by the first principal component, which has the biggest variance. Subsequent components account for increasingly lower variances (Pedregosa et al., 2011; Sittel et al., 2014). The analysis of the principal components essentially provides a distilled view of the critical dynamics, presenting frame-by-frame conformational fluctuations. Through the visualization of the PCA output, we can investigate relationships and trends between the structural characteristics that impact the complex’s stability, thereby revealing critical structural elements that contribute to the observed activity. Recently, our group applied PCA to explore complex conformational studies (Mushtaq et al., 2023). The outcomes of our analysis show that M12 and Am22 have relatively fewer conformational variations than the other entities. This is illustrated in the first principal component (PC1) to the second principal component (PC2) scatter plot (Figure 8). In particular, M12 and Am22 have a cumulative variance for the PC1 of 40% and 35%, respectively (Supplementary Figure S7). The plot indicates that the complex does not diffuse widely across conformational areas, which is noteworthy. On the other hand, the standard ISO-1 exhibited a distinct pattern. The scatter plot of principal components shows how ISO-1’s points are dispersed over the conformational landscape. Three different conformations were seen throughout the simulation. For ISO-1, the PC1 cumulative variance accounts for 45% of the variation. The dispersed arrangement of dots suggests a varied investigation of conformational regions, exposing different structural dynamics during the simulation. M12 and Am22 are less scattered along the PC1 axis, which depicts reduced structural dynamics in a conformational landscape.
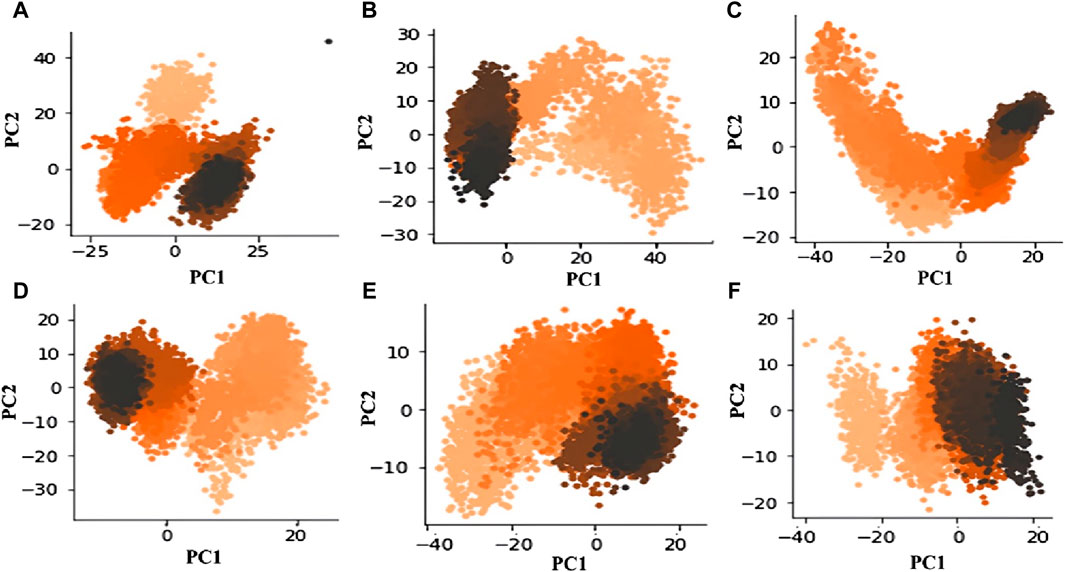
Figure 8. The conformational space is revealed by the scatter figure showing PC1 vs. PC2 for MIF bound to (A) ISO-1, (B) Ap1, (C) F9, (D) N16, (E) M12, and (F) Am22.
Accurate free energy calculations are of central significance in molecular biology because free energies guide an array of cellular processes, from the signal transduction cascades to the biomolecular degradation. In most cases, these events involve ligand binding, thereby providing key insights into molecular recognition. In this relation, the estimation of free energies (ΔGbind) remains one of the key aspects while designing a ligand for a biological target in computer-assisted drug design (CADD). Herein, the ΔGbind for the targeted flavonoids was calculated via a molecular mechanics/GBSA approach, offering a reliable and cost-effective alternative for such evaluations. The free energy calculations combined with the system’s energetics provided valuable insight into the determinants driving the complex formation. The extracted results for the ΔGbind alongside the different energy components are listed in Table 1. As evident from the observed results, a more negative value of −21.63 kcal/mol was obtained for Am22, followed by F9 featuring a ΔGbind of −13.09 kcal/mol, thus emphasizing a strong affinity towards the target protein. N16 displayed somewhat comparable results to the ISO-1, in parallel with the previous findings that highlighted its reduced stability compared to others. Further insight into the data indicated that the strength of the complex was mainly provided by the electrostatic contacts, which is also evident from the previous discussion highlighting the formation of multiple hydrogen bonds. The van der Waals interactions also served as critical determinants influencing ligand binding, which is attributed to the aromatic scaffolds present in the structures. In contrast, the positive values for ΔGsolv indicated their counteractivity in the formation of a tighter complex.
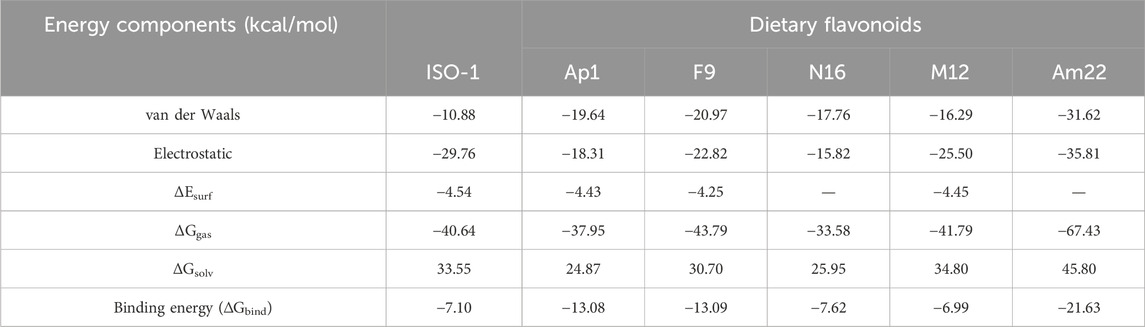
Table 1. The free energies (kcal/mol) of binding were calculated for the standard ISO-1 and selected flavonoids alongside the determinants driving the binding.
The pleiotropic nature of MIF and its potential role in the pathogenesis of several diseases, such as sepsis, cancer, and autoimmune-related ailments, is evident from the extensive literature. This emphasizes the attractiveness of MIF as a therapeutic target with pharmacological significance. Inspired by the anti-inflammatory and anticancer properties displayed by flavonoids, several studies investigated their ability to prevent MIF’s tautomerase activity. In this connection, potential dietary flavonoids with superior activity and low inhibition constants (IC50) have been reported by Yang et al. (2021). The precise mechanism for the observed impairment is still elusive. In this current work, a computational framework was utilized to explore the static and dynamic aspects of the binding mechanism of these dietary flavonoids. The target protein’s stability patterns, dynamic interactions, and time-dependent structural alterations were investigated in detail. The flavonoids featured good binding scores and interacted with Pro1A, Lys32A, Ile64A, Asn97B, and Asp95B residues from the tautomerase site. In general, these compounds exhibited stable binding during the molecular dynamic investigations. Nevertheless, N16 dislocated significantly from the tautomerase site. Although all the tested flavonoids demonstrated remarkable results, the results obtained for the potent inhibitors, particularly morin and amentoflavone, exhibited a good correlation with biological activity. The conformational landscape for these flavonoids revealed fewer structural variations. The obtained results are inconsistent with results derived from the other stability matrices utilized herein. The outcomes shed light on the functions and possible impacts of these flavonoids and MIF by offering a thorough mechanistic insight. These understandings could help direct future efforts focused on creating MIF inhibitors.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
AS: writing–original draft, methodology, investigation, formal analysis. MM: writing–original draft, methodology, formal analysis. MS: writing–original draft, validation, formal analysis. LA: writing–review and editing, validation, formal analysis. MN: writing–review and editing, validation. AA: writing–review and editing, validation. ZU-H: writing–review and editing, supervision, resources, conceptualization.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R994), King Saud University, Riyadh, Saudi Arabia. Financial support for computational resources from the Higher Education Commission (HEC) Project No. 20-17602/NRPU/R&D/HEC/2021 is also highly acknowledged.
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R994), King Saud University, Riyadh, Saudi Arabia. Financial support for computational resources from the Higher Education Commission (HEC) Project No. 20-17602/NRPU/R&D/HEC/2021 is also highly acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2024.1414572/full#supplementary-material
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. doi:10.1016/j.softx.2015.06.001
Baidya, A. T., Das, B., Devi, B., Langstrom, B., Agren, H., Darreh-Shori, T., et al. (2023). Mechanistic insight into the inhibition of choline acetyltransferase by proton pump inhibitors. ACS Chem. Neurosci. 14 (4), 749–765. doi:10.1021/acschemneuro.2c00738
Bilsborrow, J. B., Doherty, E., Tilstam, P. V., and Bucala, R. (2019). Macrophage migration inhibitory factor (MIF) as a therapeutic target for rheumatoid arthritis and systemic lupus erythematosus. Expert Opin. Ther. targets 23 (9), 733–744. doi:10.1080/14728222.2019.1656718
Chen, D., Osipyan, A., Adriana, J., Kader, M., Gureev, M., Knol, C. W., et al. (2023). Allosteric inhibitors of macrophage migration inhibitory factor (MIF) Interfere with Apoptosis-Inducing factor (AIF) Co-Localization to prevent Parthanatos. J. Med. Chem. 66, 8767–8781. doi:10.1021/acs.jmedchem.3c00397
Cournia, Z., Leng, L., Gandavadi, S., Du, X., Bucala, R., and Jorgensen, W. L. (2009). Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J. Med. Chem. 52 (2), 416–424. doi:10.1021/jm801100v
Dai, S. X., Zou, Y., Feng, Y. L., Liu, H. B., and Zheng, X. B. (2012). Baicalin down-regulates the expression of macrophage migration inhibitory factor (MIF) effectively for rats with ulcerative colitis. Phytotherapy Res. 26 (4), 498–504. doi:10.1002/ptr.3581
Dickerhof, N., Magon, N. J., Tyndall, J. D., Kettle, A. J., and Hampton, M. B. (2014). Potent inhibition of macrophage migration inhibitory factor (MIF) by myeloperoxidase-dependent oxidation of epicatechins. Biochem. J. 462 (2), 303–314. doi:10.1042/BJ20140612
Garai, J., and Adlercreutz, H. (2004). Estrogen-inducible uterine flavonoid binding sites: is it time to reconsider? J. Steroid Biochem. Mol. Biol. 88 (4-5), 377–381. doi:10.1016/j.jsbmb.2004.01.001
Genheden, S., and Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10 (5), 449–461. doi:10.1517/17460441.2015.1032936
Gowers, R. J., Linke, M., Barnoud, J., Reddy, T. J., Melo, M. N., Seyler, S. L., et al. (2016). “MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations,” in Proceedings of the 15th python in science conference (Austin, TX: SciPy), 98, 105. doi:10.25080/majora-629e541a-00e
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14 (1), 33–38. doi:10.1016/0263-7855(96)00018-5
Jin, Z., and Wei, Z. (2024). Molecular simulation for food protein–ligand interactions: a comprehensive review on principles, current applications, and emerging trends. Compr. Rev. Food Sci. Food Saf. 23 (1), 132800–e13329. doi:10.1111/1541-4337.13280
Jorgensen, W. L., Gandavadi, S., Du, X., Hare, A. A., Trofimov, A., Leng, L., et al. (2010). Receptor agonists of macrophage migration inhibitory factor. Bioorg. Med. Chem. Lett. 20 (23), 7033–7036. doi:10.1016/j.bmcl.2010.09.118
Kang, I., and Bucala, R. (2019). The immunobiology of MIF: function, genetics and prospects for precision medicine. Nat. Rev. Rheumatol. 15 (7), 427–437. doi:10.1038/s41584-019-0238-2
Khan, S. A., Zia, K., Ashraf, S., Uddin, R., and Ul-Haq, Z. (2021). Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 39 (7), 2607–2616. doi:10.1080/07391102.2020.1751298
Li, D. D., Wu, T. T., Yu, P., Wang, Z. Z., Xiao, W., Jiang, Y., et al. (2020a). Molecular dynamics analysis of binding sites of epidermal growth factor receptor kinase inhibitors. ACS Omega 5 (26), 16307–16314. doi:10.1021/acsomega.0c02183
Li, X., Ye, M., Wang, Y., Qiu, M., Fu, T., Zhang, J., et al. (2020b). How Parkinson's disease-related mutations disrupt the dimerization of WD40 domain in LRRK2: a comparative molecular dynamics simulation study. Phys. Chem. Chem. Phys. 22 (36), 20421–20433. doi:10.1039/d0cp03171b
Lubetsky, J. B., Dios, A., Han, J., Aljabari, B., Ruzsicska, B., Mitchell, R., et al. (2002). The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J. Biol. Chem. 277 (28), 24976–24982. doi:10.1074/jbc.M203220200
Mahalingam, D., Patel, M. R., Sachdev, J. C., Hart, L. L., Halama, N., Ramanathan, R. K., et al. (2020). Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. Br. J. Clin. Pharmacol. 86 (9), 1836–1848. doi:10.1111/bcp.14289
Mark, P., and Nilsson, L. (2001). Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105 (43), 9954–9960. doi:10.1021/jp003020w
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B., and Beckstein, O. (2011). MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32 (10), 2319–2327. doi:10.1002/jcc.21787
Molecular Operating Environment (MOE) (2019). Chemical computing group ULC, 1010 Sherbrooke St. West, suite #910. QC, Canada: Montreal. H3A 2R7.
Mushtaq, M., Naz, S., Ashraf, S., Doerksen, R. J., Nur-e-Alam, M., and Ul-Haq, Z. (2023). Exploring the viral protease inhibitor space driven by consensus scoring-based virtual screening. Silico Pharmacol. 12 (1), 2. doi:10.1007/s40203-023-00174-0
Noe, J. T., and Mitchell, R. A. (2020). MIF-dependent control of tumor immunity. Front. Immunol. 11, 609948. doi:10.3389/fimmu.2020.609948
O'Reilly, C., Doroudian, M., Mawhinney, L., and Donnelly, S. C. (2016). Targeting MIF in cancer: therapeutic strategies, current developments, and future opportunities. Med. Res. Rev. 36 (3), 440–460. doi:10.1002/med.21385
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830. doi:10.48550/arXiv.1201.0490
Penticuff, J. C., Woolbright, B. L., Sielecki, T. M., Weir, S. J., and Taylor III, J. A. (2019). MIF family proteins in genitourinary cancer: tumorigenic roles and therapeutic potential. Nat. Rev. Urol. 16 (5), 318–328. doi:10.1038/s41585-019-0171-9
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25 (13), 1605–1612. doi:10.1002/jcc.20084
Qian, Y., Cabeza de Vaca, I., Vilseck, J. Z., Cole, D. J., Tirado-Rives, J., and Jorgensen, W. L. (2019). Absolute free energy of binding calculations for macrophage migration inhibitory factor in complex with a druglike inhibitor. J. Phys. Chem. B 123 (41), 8675–8685. doi:10.1021/acs.jpcb.9b07588
Qureshi, U., Mir, S., Naz, S., Nur-e-Alam, M., Ahmed, S., and Ul-Haq, Z. (2022). Mechanistic insights into the inhibitory activity of FDA approved ivermectin against SARS-CoV-2: old drug with new implications. J. Biomol. Struct. Dyn. 40 (17), 8100–8111. doi:10.1080/07391102.2021.1906750
Rossetti, G., and Mandelli, D. (2024). How exascale computing can shape drug design: a perspective from multiscale QM/MM molecular dynamics simulations and machine learning-aided enhanced sampling algorithms. Curr. Opin. Struct. Biol. 86, 102814. doi:10.1016/j.sbi.2024.102814
Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., and Schroeder, M. (2015). PLIP: fully automated protein–ligand interaction profiler. Nucleic acids Res. 43 (W1), W443–W447. doi:10.1093/nar/gkv315
Sardar, M., Zia, K., Ashraf, S., Malik, H. N., Jabeen, A., Khan, K. M., et al. (2022). Interface inhibitory action on Interleukin-1β using selected anti-inflammatory compounds to mitigate the depression: a computational investigation. Comput. Biol. Chem. 101, 107774. doi:10.1016/j.compbiolchem.2022.107774
Sargsyan, K., Grauffel, C., and Lim, C. (2017). How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 13 (4), 1518–1524. doi:10.1021/acs.jctc.7b00028
Sarkar, S., Siddiqui, A. A., Mazumder, S., De, R., Saha, S. J., Banerjee, C., et al. (2015). Ellagic acid, a dietary polyphenol, inhibits tautomerase activity of human macrophage migration inhibitory factor and its pro-inflammatory responses in human peripheral blood mononuclear cells. J. Agric. food Chem. 63 (20), 4988–4998. doi:10.1021/acs.jafc.5b00921
Sinitski, D., Kontos, C., Krammer, C., Asare, Y., Kapurniotu, A., and Bernhagen, J. (2019). Macrophage migration inhibitory factor (MIF)-based therapeutic concepts in atherosclerosis and inflammation. Thrombosis Haemostasis 119 (04), 553–566. doi:10.1055/s-0039-1677803
Sittel, F., Jain, A., and Stock, G. (2014). Principal component analysis of molecular dynamics: on the use of Cartesian vs. internal coordinates. J. Chem. Phys. 141 (1), 014111–014601. doi:10.1063/1.4885338
Sousa da Silva, A. W., and Vranken, W. F. (2012). ACPYPE-Antechamber python parser interface. BMC Res. Notes 5 (1), 367–368. doi:10.1186/1756-0500-5-367
Sun, Q., Liu, Q., Zhou, X., Wang, X., Li, H., Zhang, W., et al. (2022). Flavonoids regulate tumor-associated macrophages - from structure-activity relationship to clinical potential (Review). Pharmacol. Res. 184, 106419. doi:10.1016/j.phrs.2022.106419
Trivedi-Parmar, V., and Jorgensen, W. L. (2018). Advances and insights for small molecule inhibition of macrophage migration inhibitory factor. J. Med. Chem. 61 (18), 8104–8119. doi:10.1021/acs.jmedchem.8b00589
Trivedi-Parmar, V., Robertson, M. J., Cisneros, J. A., Krimmer, S. G., and Jorgensen, W. L. (2018). Optimization of pyrazoles as phenol surrogates to yield potent inhibitors of macrophage migration inhibitory factor. ChemMedChem 13 (11), 1092–1097. doi:10.1002/cmdc.201800158
Tuli, H. S., Garg, V. K., Bhushan, S., Uttam, V., Sharma, U., Jain, A., et al. (2023). Natural flavonoids exhibit potent anticancer activity by targeting microRNAs in cancer: a signature step hinting towards clinical perfection. Transl. Oncol. 27, 101596. doi:10.1016/j.tranon.2022.101596
Turner, P. J. (2005). Xmgrace. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR, 2. Version 5.1. 19.
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A., and Moreno, E. (2021). gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 17 (10), 6281–6291. doi:10.1021/acs.jctc.1c00645
Vázquez, A., González, M. I., and Reyes, J. L. (2023). Targeting macrophage migration inhibitory factor (MIF): a promising therapy for inflammatory ocular diseases. J. Ophthalmic Inflamm. Infect. 13 (1), 37. doi:10.1186/s12348-023-00361-2
Xu, L., Zhang, Y., Zheng, L., Qiao, C., Li, Y., Li, D., et al. (2014). Discovery of novel inhibitors targeting the macrophage migration inhibitory factor via structure-based virtual screening and bioassays. J. Med. Chem. 57 (9), 3737–3745. doi:10.1021/jm401908w
Yang, L., Guo, D., and Fan, C. (2021). Identification and structure–activity relationships of dietary flavonoids as human macrophage migration inhibitory factor (MIF) inhibitors. J. Agric. Food Chem. 69 (35), 10138–10150. doi:10.1021/acs.jafc.1c03367
Young, N. A., Bruss, M. S., Gardner, M., Willis, W. L., Mo, X., Valiente, G. R., et al. (2014). Oral administration of nano-emulsion curcumin in mice suppresses inflammatory-induced NFκB signaling and macrophage migration. PloS one 9 (11), e111559. doi:10.1371/journal.pone.0111559
Yuan, Y., Deng, J., and Cui, Q. (2022). Molecular dynamics simulations establish the molecular basis for the broad allostery hotspot distributions in the tetracycline repressor. J. Am. Chem. Soc. 144 (24), 10870–10887. doi:10.1021/jacs.2c03275
Zhang, H., Zhang, X., Li, H., Wang, B., Chen, P., and Meng, J. (2024). The roles of macrophage migration inhibitory factor in retinal diseases. Neural Regen. Res. 19 (2), 309–315. doi:10.4103/1673-5374.379020
Zhang, Y., Xu, L., Zhang, Y., Pan, J., Wang, P. Q., Tian, S., et al. (2022). Discovery of novel MIF inhibitors that attenuate microglial inflammatory activation by structures-based virtual screening and in vitro bioassays. Acta Pharmacol. Sin. 43 (6), 1508–1520. doi:10.1038/s41401-021-00753-x
Keywords: macrophage migration inhibitory factor, dietary flavonoids, molecular dynamic simulation, principal component analysis, ISO-1
Citation: Siddiqui AR, Mushtaq M, Sardar M, Atta L, Nur-e-Alam M, Ahmad A and Ul-Haq Z (2024) Mechanistic insight into the mode of inhibition of dietary flavonoids; targeting macrophage migration inhibitory factor. Front. Mol. Biosci. 11:1414572. doi: 10.3389/fmolb.2024.1414572
Received: 09 April 2024; Accepted: 08 May 2024;
Published: 10 June 2024.
Edited by:
Vikram Dalal, Washington University in St. Louis, United StatesReviewed by:
Debanjan Kundu, Georgia Institute of Technology, United StatesCopyright © 2024 Siddiqui, Mushtaq, Sardar, Atta, Nur-e-Alam, Ahmad and Ul-Haq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zaheer Ul-Haq, emFoZWVyLnFhc21pQGljY3MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.