 Fatemeh Alipour
Fatemeh Alipour Connor Holmes2†
Connor Holmes2† Yang Young Lu
Yang Young Lu Kathleen A. Hill
Kathleen A. Hill
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 11 January 2024
Sec. Structural Biology
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1305506
This article is part of the Research TopicNew Approaches Toward Understanding Challenges in Molecular Virology: Computational Techniques and Machine LearningView all 5 articles
Astroviruses are a family of genetically diverse viruses associated with disease in humans and birds with significant health effects and economic burdens. Astrovirus taxonomic classification includes two genera, Avastrovirus and Mamastrovirus. However, with next-generation sequencing, broader interspecies transmission has been observed necessitating a reexamination of the current host-based taxonomic classification approach. In this study, a novel taxonomic classification method is presented for emergent and as yet unclassified astroviruses, based on whole genome sequence k-mer composition in addition to host information. An optional component responsible for identifying recombinant sequences was added to the method’s pipeline, to counteract the impact of genetic recombination on viral classification. The proposed three-pronged classification method consists of a supervised machine learning method, an unsupervised machine learning method, and the consideration of host species. Using this three-pronged approach, we propose genus labels for 191 as yet unclassified astrovirus genomes. Genus labels are also suggested for an additional eight as yet unclassified astrovirus genomes for which incompatibility was observed with the host species, suggesting cross-species infection. Lastly, our machine learning-based approach augmented by a principal component analysis (PCA) analysis provides evidence supporting the hypothesis of the existence of human astrovirus (HAstV) subgenus of the genus Mamastrovirus, and a goose astrovirus (GoAstV) subgenus of the genus Avastrovirus. Overall, this multipronged machine learning approach provides a fast, reliable, and scalable prediction method of taxonomic labels, able to keep pace with emerging viruses and the exponential increase in the output of modern genome sequencing technologies.
Astroviruses are a genetically diverse virus family notably responsible for the second most common cause of nosocomial diarrhea following rotaviruses (Meyer et al., 2015), as well as substantial economic losses in the poultry industry (Karlsson et al., 2015; Li et al., 2023). Also, astrovirus infection has been associated with encephalitis and meningitis in immunocompromised patients (Vu et al., 2017) and astrovirus infection has been shown to be present in the brains of some mammals (Chae et al., 2023). According to several studies (Resque et al., 2007; Meyer et al., 2015; Keita et al., 2023), the prevalence of astroviruses among human populations ranges from 2% to 9% at any given time. In developing countries, this percentage can be significantly higher, affecting up to 30% of the population (De Benedictis et al., 2011). Infants between 3 and 8 months of age (Herrmann et al., 1991; Shastri et al., 1998; Dennehy et al., 2001; Jeong et al., 2012; Chhabra et al., 2013), and the elderly (Babkin et al., 2012) along with immunocompromised patients are primarily infected (Grohmann et al., 1993; Palombo and Bishop, 1996; Liste et al., 2000). Outbreaks have been reported for immunocompetent adults (Midthun et al., 1993; Oishi et al., 1994; Jarchow-Macdonald et al., 2015). Astrovirus transmission occurs exclusively through the fecal-oral route (Lefkowitz et al., 2017), with notable interspecies transmission (De Benedictis et al., 2011). Astrovirus genetic diversity is linked to the proposed replication via the class III PI3K pathway during autophagy (Bub et al., 2023) and genetic recombination associated with cross-species transmission through abiotic vectors such as drinking water, sewage, and other contaminated systems (Abad et al., 1997; Le Cann et al., 2004). The increasing interspecies transmission enhances the risk of extraintestinal infections in humans as reported in animal populations (Qureshi et al., 2023). Genetic recombination elevates genetic diversity in the context of a concurrent multiplicity of infections for viruses of different genera given cross-species transmissions.
The International Committee on the Taxonomy of Viruses (ICTV) structures the family Astroviridae into two genera, Avastrovirus and Mamastrovirus (Lefkowitz et al., 2017), and determines taxonomic classification by defining species intragroups as strains with a minimum amino acid identity of 75% in the open reading frame 2 (ORF2) region. The two genera include many host-associated astroviruses and the number of known animal hosts has reached over 160, spanning 13 classes of organisms. Next-generation sequencing continues to achieve rapid detection of new astroviruses and the identification of new host species (Vu et al., 2017; Zheng et al., 2017), with a steadily increasing number of astrovirus genomes on the National Center for Biotechnology Information (NCBI) awaiting classification at the genus level.
Urgency and need to refine the taxonomy of family Astroviridae is accelerated by reports of astrovirus recombination during concomitant infections (Pantin-Jackwood et al., 2012; Lefkowitz et al., 2017), including some involving HAstVs (Vu et al., 2017). Also, avian and mammalian astrovirus species have been found in nonhuman primates (Cortez et al., 2017), and inter-species crossover between humans and various animals, such as felines, cats, pigs, California sea lions, dogs, sheep, and turkeys have been identified (Jiang et al., 1993; Meliopoulos et al., 2014; Karlsson et al., 2015; Cortez et al., 2017). These observations complicate a taxonomic classification based solely on host species and nucleotide sequence identity. Moreover, with the emergence of interspecies transmission, confusion has arisen in classifying astroviruses based on their origins. Due to classification at the species level based on sequence identity, some inter-cluster species of different genera, namely, HAstVs, show more relatedness than those of the same intra-cluster genus (Jiang et al., 1993). This has led to a call for standardized methods of classification for family Astroviridae (Cortez et al., 2017).
Recent years have seen a rapid growth in the volume of accessible genomic data, due to notable advancements in next-generation sequencing (NGS) technologies and a reduction in sequencing costs (Schwende and Pham, 2014). Consequently, there is an increasing demand for computationally efficient and scalable methods to handle large genomic datasets (Shendure et al., 2004; Katz et al., 2022). Earlier attempts to tackle genomic classification/clustering problems can be categorized into two approaches: “alignment-based” and “alignment-free” methods. The high computational cost and the reliance on sequence homology of alignment-based techniques make alignment-free methods a more suitable choice for addressing the virus classification problem. Consequently, a multitude of alignment-free classification (Solis-Reyes et al., 2018; Fabijańska and Grabowski, 2019; Randhawa et al., 2019; Jiang et al., 2023) and clustering methods (Girgis, 2022; Millán Arias et al., 2022; Millan Arias et al., 2023) suitable for viral genomic sequence datasets have emerged, and initial studies demonstrated their effectiveness and scalability compared to traditional alignment-based methods (Thompson et al., 1994; Edgar, 2004).
This paper presents a novel machine-learning classification method hereafter called the Three-Pronged Classification Method (3PCM) to classify astrovirus sequences that are as yet unclassified. The method utilizes the primary sequence composition of the entire genome in the form of k-mer frequency vectors, where k is set to 6. In this paper, k = 6 was empirically found to achieve the best balance between accuracy and computational complexity. An initial optional component was incorporated into 3PCM’s pipeline to detect potential recombinant sequences and exclude them from the analysis. This step aims to prevent any noise caused by inter-species crossover, which could otherwise confound machine-learning models. 3PCM consists of three main components: Prong 1 (a supervised classification method utilizing Quadratic SVM), Prong 2 (an unsupervised clustering technique based on K-means++); and Prong 3 (the identification of host labels at the class level from relevant literature for the as yet unclassified viral sequences). In this paper, taxonomic classification was suggested when all three prongs of 3PCM agreed on a taxonomic label. When Prong 1 and Prong 2 concurred on a classification that differed from Prong 3, a taxonomic classification was suggested, subject to independent confirmation.
The design of 3PCM utilizes genome composition information from astrovirus sequences with known taxonomic labels to classify/cluster astrovirus sequences with mammalian or avian hosts that are as yet unclassified. Although the default output of 3PCM is based on the consensus prediction of the three prongs, the individual prongs can be used independently in cases where one or two prongs are not applicable or do not agree. For instance, Prong 1 is not suitable for classifying as yet unclassified astrovirus sequences with non-mammalian non-avian hosts, due to the absence of ground truth labels which are necessary for training a supervised model. In such a situation, Prong 2 can be used in conjunction with Prong 3 to investigate the classification of the sequences. In this and other scenarios, other analyses such as genome composition analysis can be employed to validate the results.
The main contributions of this paper are:
• Proposing genus labels (Mamastrovirus or Avastrovirus) for 191 as yet unclassified astrovirus genome sequences for which the results of Prongs 1, 2, and 3 all agree.
• Suggesting genus labels (Mamastrovirus or Avastrovirus) for 8 additional as yet unclassified astrovirus genome sequences, for which incompatibility was observed between the taxonomic label proposed by Prong 1 and Prong 2, and the host label provided by Prong 3. This may be due to cross-species transmission, and further investigation is needed to resolve the contrasting labels associated with these sequences.
• Providing evidence supporting the hypothesis of the existence of a human astrovirus subgenus of the genus Mamastrovirus and a goose astrovirus subgenus of the genus Avastrovirus, through the application of the proposed machine learning-based approach, enhanced by a principal component analysis (PCA) of the sequence composition.
Overall, this multipronged machine learning approach provides a fast, reliable, and scalable prediction method of taxonomic labels, able to keep pace with emerging viruses and the exponential increase in the output of modern genome sequencing technologies.
The first part of this section, Materials, provides an overview of the dataset used in this study. The second subsection, Methods, describes the technical and implementation details of three prongs of the proposed classification method. Moreover, the evaluation metrics used to evaluate the proposed methodology will be discussed throughout the Methods section.
The dataset used in this study consists of RNA sequences from the viral family, Astroviridae downloaded from the NCBI database. In the RNA sequence, Ns replaced all sequence characters other than adenine (A), cytosine (C), guanine (G), and uracil (U). The N in an RNA sequence means that any of the four bases could occupy the position in question. All sequences were uploaded to a folder in Genbank. These were then exported as a single multifasta file for further testing.
A total of 1,039 sequences from the family Astroviridae were downloaded from the NCBI database on 27th July 2022. The sequences included in this study were between 5 and 10 kbps in length. The host for each virus sequence was identified from the literature where a publication was available. In the absence of published records, the organism listed in the NCBI database submission was considered the host. We excluded 47 out of 1,039 sequences from our analysis due to the lack of information regarding the host of the virus given collection from sewage, rivers, and streams. Patent sequences were also excluded. Following the removal of these sequences, 992 sequences were used in this study as the primary dataset (Dataset 1, described in Table 1). Among the 992 sequences in this dataset, 308 are as yet unclassified at the genus level. The individual host species were ascribed to their respective class and genus. The final dataset contains Astrovirus genomes found in 13 unique host classes and 96 unique host genera.
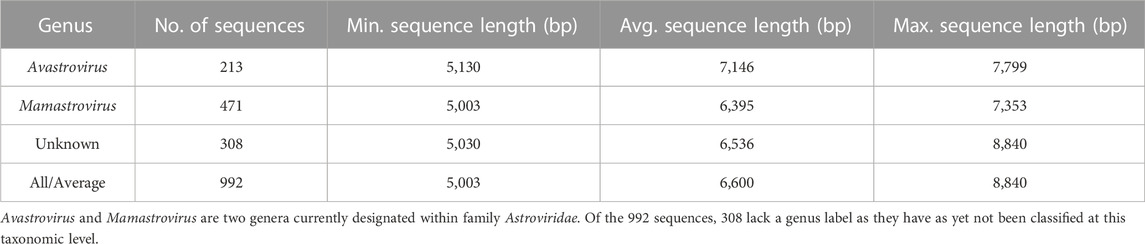
TABLE 1. Description of Dataset 1 containing 992 viral genomes belonging to the family Astroviridae.
In addition to Dataset 1, two other subsets of this dataset are used throughout the paper, as described below. Dataset 2 (described in Table 2) comprises the 684 genomes in Dataset 1 that belong to either Avastrovirus or Mamastrovirus genus. Dataset 2 was used both as the main training dataset and as the dataset employed for determining different parameters of the proposed three-pronged classification method.
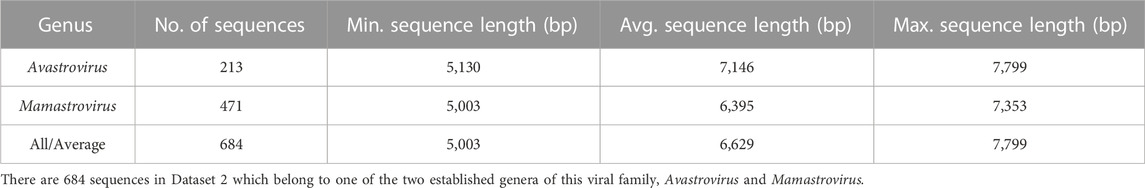
TABLE 2. Description of Dataset 2, a subset of Dataset 1 consisting of sequences with available ground truth.
With a goal to predict genus level labels for the 308 as yet unclassified sequences, we investigated the current information about these sequences, that is, their hosts. Please see Supplementary Material S1 (Analysis of Astroviruses of Unknown Genus Label), for the distribution of hosts for all the 308 Astrovirus genomes with unknown genus level labels. For purposes described in the Results section, Dataset 3 was created, consisting of 187 astrovirus genomes in Dataset 1 with unknown genus level labels and mammalian hosts, and 42 astrovirus genomes in Dataset 1 with unknown genus level labels and avian hosts (see Table 3).
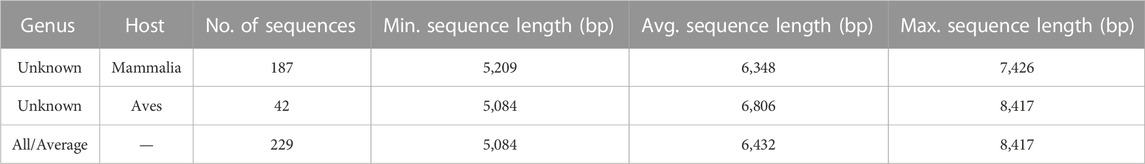
TABLE 3. Description of Dataset 3, consisting of 187 astrovirus genomes in Dataset 1 that are as yet unclassified (unknown genus) and have a mammalian host, and 42 astrovirus genomes in Dataset 1 that are as yet unclassified (unknown genus) and have an avian host.
We herein propose a three-pronged classification method (3PCM) for the taxonomic classification of emergent but as yet unclassified astrovirus sequences.
An optional initial component of 3PCM aims to eliminate recombinant sequences from the training and testing datasets, for scenarios where their presence may confound the machine learning process. The main methodological pipeline consists of three prongs, as illustrated in Figure 1:
1. Prong 1 (supervised learning): training a classification model using the whole genome sequence for astroviruses with known taxonomic classification in the training phase and leveraging the trained predictive model to predict the labels of as yet unclassified astroviruses in the testing phase.
2. Prong 2 (unsupervised learning): training a clustering model using the whole genomes of astroviruses in the training phase and using the trained predictive model to predict taxonomic labels of as yet unclassified astroviruses in the testing phase. Taxonomic labels are not used in the training phase, therefore, this model is less vulnerable to inaccuracies of current taxonomic labels and classifications.
3. Prong 3 (identifying host label): identifying the class label (Mammalia, Aves, etc.) of the host from which the as yet unclassified viral sample was obtained.
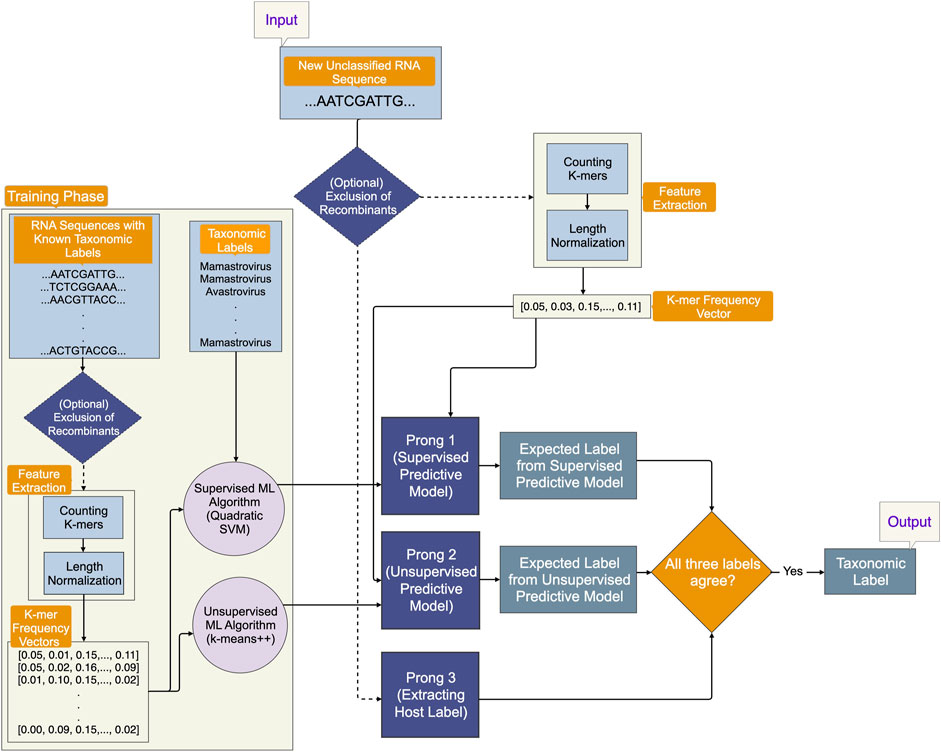
FIGURE 1. An overview of the proposed Three-Pronged Classification Method (3PCM). The input is an as yet unclassified genomic sequence. As an initial elective step, a component within the pipeline gives the option to eliminate recombinant sequences from the dataset. Prong 1 employs a supervised predictive model trained on genomic sequences with known taxonomic labels. Prong 2 uses an unsupervised predictive model trained on the same genomic sequences, but it does not use their taxonomic labels for training. Prong 3 uses the host label of the input genomic sequence. All three prongs of 3PCM must agree in their prediction, in order to produce a suggested taxonomic label.
In the event that the predictions of all three prongs agree, taxonomic labels are proposed for as yet unclassified sequences. If there is agreement only between Prong 1 and Prong 2, taxonomic labels are proposed with recommendations for further investigation.
3PCM can be used both with comprehensive datasets and with versions of those datasets where known or presumed recombinant sequences have been removed. This option was added because astroviruses exhibit genetic diversity in part through recombination, which is further complicated by concurrent infections with multiple astrovirus taxa and recombination with up to three genomes at once (Wei et al., 2023). The nature of this recombination significantly enhances the genetic diversity of astroviruses and hinders the reconstruction of astrovirus evolutionary history. To evaluate the impact of recombinant genomes on machine learning-based classification methods, the aligned sequences of this viral family were uploaded to the Recombination Detection Program RDP4 (Martin et al., 2015). RDP4 employed various tools such as RDP, GENECONV, BOOTSCAN, MaxChi, SiScan, CHIMEARA, and TOPAL to assess all possible sequence triplets and determine the recombinant sequence, major parent, and minor parent involved. In this study, the only recombination events that were considered were those where the parents were identified by two or more of the seven aforementioned tools, with a level of significance p < 0.05 (statistically significant). For further information please refer to Supplementary Material S2 (Identification and Analysis of Candidate Recombinant Astrovirus Genome Sequences).
The first prong makes use of existing and established knowledge regarding astroviruses to train a supervised machine learning model based on the complete genomes of astroviruses with known taxonomic labels (Dataset 2) in the training phase. We then used the trained model to predict the labels of as yet unclassified astroviruses (Dataset 3) during the testing phase.
The features used in the supervised learning methods in this study were the k-mer frequency vector of each astrovirus genome. The k-mers containing an N (not specifically one of the four bases in RNA) were not included. The performance of Prong 1 for values of k in the range [1, 9] in terms of classification accuracy and running time can be found in Supplementary Material S3 (Performance Results of 3PCM Using Different Classification/Clustering Algorithms). The value k = 6 was empirically found to achieve the best balance between accuracy and computational complexity for the datasets and computational experiments in this paper. To avoid potential effects of sequence length variation (astrovirus genome lengths range between 5,003 and 8,840 bp), the feature vectors were normalized to the interval [0, 1] by dividing each vector by the length of the originating RNA sequence.
By utilizing a supervised model trained on the astrovirus sequences with known taxonomic labels, we were able to take advantage of established knowledge about this virus family. The supervised classification algorithms tested in this study are 10-Nearest Neighbours (Altman, 1992), Nearest Centroid Mean (Tibshirani et al., 2002), Nearest Centroid Median (Tibshirani et al., 2002), Logistic Regression (McCullagh and Nelder, 1989), Linear Support Vector Machines (SVM) (Cristianini and Shawe-Taylor, 2000), SVM with quadratic polynomial kernel (Quadratic SVM) (Cristianini and Shawe-Taylor, 2000), SVM with cubic polynomial kernel (Cubic SVM) (Cristianini and Shawe-Taylor, 2000), SVM with stochastic gradient descent learning and linear kernel function (SGD) (Cristianini and Shawe-Taylor, 2000), Decision Tree (Breiman et al., 1984), Random Forest (Breiman, 2001), AdaBoost (Freund and Schapire, 1997), Gaussian Naive Bayes (Chan et al., 1982), Linear Discriminant Analysis (LDA) (Hastie et al., 2009), Quadratic Discriminant Analysis (QDA) (Hastie et al., 2009), and Multilayer Perceptron (MLP) (Hinton, 1990; Kingma and Ba, 2015). Python library scikit-learn’s implementations of the fifteen aforementioned classifiers (Pedregosa et al., 2011) were used. Supplementary Material S3 (Performance Results of 3PCM Using Different Classification/Clustering Algorithms) list the experimental results of these fifteen candidate algorithms used in Prong 1 of 3PCM.
Only astrovirus RNA sequences in Dataset 2 with existing labels (described in Table 2) were used for training and testing for our initial experiment in order to select the most effective supervised classification algorithm among sixteen candidates as well as to demonstrate the effectiveness of Prong 1. As the testing dataset consisted only of sequences with known taxonomic labels, we could determine classification accuracy by comparing the predicted labels with the true labels. In order to assess the accuracy of the classifiers, we used Stratified 10-Fold Cross-Validation (Refaeilzadeh et al., 2009; Pedregosa et al., 2011). Based on the results presented in Supplementary Material S3 (Performance Results of 3PCM Using Different Classification/Clustering Algorithms), most of the model’s predictions match the true label (fourteen of sixteen algorithms achieved accuracy greater than 90%). Quadratic SVM and Cubic SVM were the most accurate algorithms for classifying astrovirus whole genomes by achieving an accuracy of 99.56%. Consequently, Quadratic SVM was selected as the classification algorithm in Prong 1 for the remainder of this paper.
To further assess the performance of 3PCM’s Prong 1, we conducted a comparative analysis by benchmarking our outcomes against two leading alignment-free machine-learning genome classification methods suitable for viral classification: Machine Learning with Digital Signal Processing (ML-DSP) (Randhawa et al., 2019; Randhawa et al., 2020) and the Viral Genome Deep Classifier (VGDC) (Fabijańska and Grabowski, 2019). The performance comparison between these two methods and the proposed Prong 1 with Quadratic SVM is detailed in Table 4, based on experiments using Dataset 2 with 10-fold cross-validation. As seen in Table 4, Prong 1 achieves superior classification accuracy compared to both ML-DSP and VGDC by margins of 0.56% and 3.68%, respectively.

TABLE 4. Classification accuracy of 3PCM’s Prong 1 against two state-of-the-art alignment-free machine-learning viral genome classification methods (ML-DSP, VGDC) using 10-fold cross-validation technique.
Prong 2 of the proposed classification method is unsupervised clustering, which is agnostic to and independent of taxonomic labels and annotations. Taking into account the possibility that the current classification of viruses based solely on their host may be flawed or incomplete due to limited information, knowledge, or characterization, it was necessary to use an alternate approach that does not rely on current labels. The use of unsupervised clustering alongside Prong 1 (supervised learning) allowed for the flexible use of as yet unclassified and unannotated astrovirus genomes in the training phase. Approximately one-third of astrovirus sequences are as yet unclassified (308 out of 992) and cannot be used in supervised models as they lack “ground truth” taxonomic labels. The potential inclusion of these sequences in the clustering model allows for the examination of the hypothesis that astrovirus consists of more than two genera (Mamastrovirus and Avastrovirus), which was not possible in Prong 1.
In Prong 2, the same feature vectors as in Prong 1 (k-mer counts) were used. The performance of Prong 2 for different values of k in the range [1, 9] in terms of classification accuracy and time can be found in Supplementary Material S3 (Performance Results of 3PCM Using Different Classification/Clustering Algorithms). The value k = 6 was empirically found to achieve the best balance between accuracy and computational complexity for the datasets and computational experiments in this paper. Furthermore, to find the most suitable clustering algorithm, we calculated and normalized the feature vectors and then tested three different clustering algorithms, K-means++ (Arthur and Vassilvitskii, 2007), Gaussian Mixture Model (GMM) (Dempster et al., 1977), and Hierarchical Clustering (Bridges Jr, 1966). We used Python library scikit-learn’s implementations of the three candidate clustering algorithms (Pedregosa et al., 2011). These algorithms were chosen due to their effectiveness in RNA classification (Kraskov et al., 2005; Akhtar et al., 2007; Aleb and Labidi, 2015; Hoang et al., 2015; Bustamam et al., 2017; James et al., 2018; Mendizabal-Ruiz et al., 2018).
These three clustering algorithms were compared by calculating the silhouette coefficient (Rousseeuw, 1987) as an internal evaluation metric ranging from −1 to 1, with higher values indicative of better clustering performance. In addition, we calculated external evaluation metrics such as Normalized Mutual Information (NMI) (Strehl and Ghosh, 2002), Adjusted Rand Index (ARI) (Rand, 1971), and classification accuracy to further compare the five clustering algorithms. NMI values range from 0 to 1, with 1 indicating perfect agreement and 0 indicating no agreement between these two clusterings. ARI values range from −1 to 1, where a value of 1 indicates perfect agreement between predicted and true labels, a value of 0 indicates no agreement and negative values indicate disagreement. We calculated classification accuracy of the clustering algorithms by using the Hungarian algorithm (Kuhn, 1955) in a post hoc step. This algorithm identifies the optimal mapping between the numerical cluster labels obtained by the clustering algorithms and the true taxonomic cluster labels. In external evaluation metrics, the results of clustering are compared with some known ground truth or with a reference set of labels. Consequently, we focused on sequences that had already been established as belonging to the Mamastrovirus and Avastrovirus genera of the Astroviridae family (Dataset 2 described in Table 2) and used this information for calculating external evaluation metrics. Please refer to Supplementary Material S3 (Performance Results for 3PCM Testing of Multiple Accuracy Classifiers) to see implementation details and the performance results of the clustering of the three clustering algorithm candidates measured in terms of the aforementioned internal and external evaluation metrics.
Among the three clustering algorithms, K-means++ performs the best in terms of all four evaluation metrics. K-means++ succeeded to achieve a classification accuracy of 88.16% NMI of 0.45, ARI of 0.58, and silhouette coefficient of 0.08. Consequently, K-means++ was selected as the clustering algorithm in Prong 2 for the remainder of this study. Figure 2 displays the confusion matrix obtained from clustering Dataset 2 using the K-means++ algorithm in Prong 2 of 3PCM. According to the figure, 448 out of 471 Mamastroviruses and 161 out of 213 Avastroviruses were clustered correctly. Major misclustering occurred for 52 Avastroviruses that were grouped with the majority of Mamastroviruses. It is possible that this is the result of an over-representation of Mamastrovirus over Avastrovirus in the dataset. Another possible explanation for this misclustering is the possibility of the existence of additional genera or subgenera inside the family Astroviridae which will be investigated in the Results section.
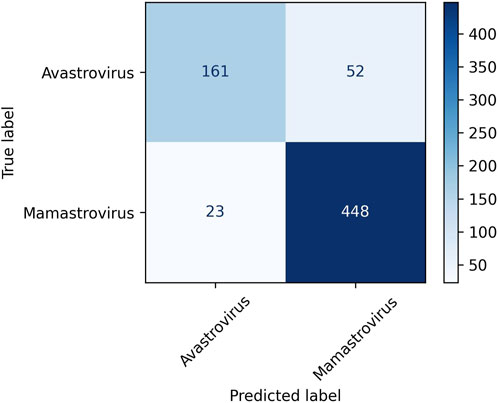
FIGURE 2. Confusion matrix for the clustering of Dataset 2, the astrovirus whole genomes with available taxonomic labels (dataset described in Table 2) into the Avastrovirus and Mamastrovirus genera using K-means++ algorithm in Prong 2 of 3PCM.
To further evaluate the performance of 3PCM’s Prong 2, we conducted a comparative analysis by contrasting our results with three state-of-the-art alignment-free machine-learning genome clustering methods suitable for viral sequences: Deep Learning for Unsupervised Classification of DNA Sequences (DeLUCS) (Millán Arias et al., 2022), its enhanced and interactive version (iDeLUCS) (Millan Arias et al., 2023), and MeShClust v3.0 (Girgis, 2022). DeLUCS and iDeLUCS rely on deep learning to uncover patterns (genomic signatures) within raw, unlabeled primary RNA/DNA sequence data, while MeShClust employs a mean-shift algorithm on pairwise alignment-free identity scores. The performance comparison of these three clustering methods and the proposed Prong 2 with K-means++ is presented in Table 5, based on experiments utilizing Dataset 2 for both the testing and the training phases.
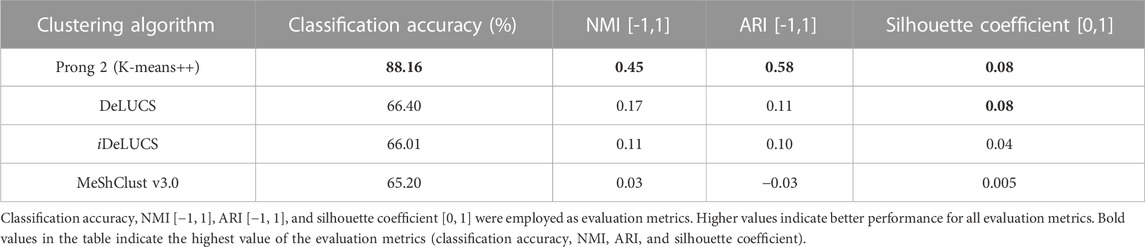
TABLE 5. Performance of 3PCM’s Prong 2 against three state-of-the-art alignment-free machine-learning viral genome clustering methods (DeLUCS, iDeLUCS, and MeShClust v3.0) for clustering DNA sequences of the family Astroviridae, with available taxonomic labels at the genera level (Dataset 2).
The computational experiments involving DeLUCS and iDeLUCS showed that increasing the mutation rate to pts = 10–3 and ptv = 0.5 × 10−3 (rather than the default values pts = 10–4 and ptv = 0.5 × 10−4) and using 9 mimic sequences (rather than the default value of 3) increased the accuracy of astrovirus genome clustering. Due to the variability in the results of DeLUCS, the results reported in Table 5 are the average values over 10 different runs. In contrast to the other methods, MeShClust does not allow for the pre-setting of the number of clusters due to its density-based nature. As a result, multiple values were examined for the identity score threshold within the range of [0, 1], and the value of 0.4005 was selected as it was the one resulting in two clusters. Overall, as shown in Table 5, Prong 2 outperforms DeLUCS, iDeLUCS, and MeShClust in terms of both internal and external evaluation metrics, with a classification accuracy that is 21.76%–22.96% higher than the other three methods.
The proposed methodology was further tested by collecting 1,450 genomes of the closest viral family to astrovirus, namely, potyvirus (Potyviridae). Using potyvirus genome sequences, a dataset consisting of all available astrovirus and potyvirus genomes was created and classification/clustering of these two viral families was tested using 3PCM. The exclusion of recombinants from this dataset was not performed due to the rarity of interspecific recombination within potyvirus. Additionally, no recombination is anticipated between potyvirus and astrovirus (Gibbs et al., 2020). Prong 1 (supervised) and Prong 2 (unsupervised) achieved accuracies of 99.8% and 93.47%, respectively. The accuracies achieved provide compelling evidence of the effectiveness of 3PCM in the classification/clustering of viral genomes at different taxonomic levels. Details of these computational experiments can be found in Supplementary Material S4 (Astrovirus Near-Neighbour Analysis: Potyvirus).
Lastly, 3PCM was used at a lower taxonomic level, for the classification/clustering of Avastrovirus and Mamastrovirus genera into different subgroups based on their host species. This test was augmented by a principal component analysis (PCA) of the k-mer composition of the astrovirus genomes, for k = 6.
The laptop used for data collection and recombinants analysis was a Lenovo L-series ThinkPad with an intel core i5 processor and 32 GB ram. Datasets consisting of Astroviridae and Patatavirales in this study were retrieved from the National Center for Biotechnology Information (NBCI). Sequences were downloaded using the application Geneious Prime 2022.1 https://www.geneious.com/ via the NCBI nucleotide database.
We empirically selected the hyperparameters of different classification/clustering algorithms that yielded the best performance during the training procedure. Both Prong 1 and Prong 2 of 3PCM are implemented in Python 3.10 and the source code, as well as all the datasets used in this paper, are publicly available in the GitHub repository https://github.com/fatemehalipour/3PCM. All of the tests were performed on Google Colab Pro environment [2 x Intel(R) Xeon(R) CPU @ 2.20 GHz, 32 GB RAM] with NVIDIA A100 GPU.
In this section, we showcase the outcomes achieved through the pipeline explained in the preceding section, Materials and Methods. First, the results of applying recombinant elimination to astrovirus sequences will be discussed. Following that, we will present the results of the novel classification method applied to as yet unclassified astroviruses. Lastly, the existence of subgenera within Mamastrovirus and Avastrovirus genera, as suggested by our observations, will be explored.
The optional component to eliminate recombinants was employed to examine Dataset 1, the primary dataset used in this study. As a result, 54 sequences (5.4% of the dataset) involved in interspecific recombination, associated with 34 recombination events, were identified. Notably, out of these 54 recombinations, 7 were intergeneric. Although the taxonomic classification task performed in this study is at the genus level, the presence of a negligible number (7) of intergeneric recombinations (which would yield noticeable variations in evaluation metrics) led to the decision to eliminate all 54 recombinants. For more detailed information, please refer to Supplementary Material S2 (Identification and Analysis of Candidate Recombinant Astrovirus Genome Sequences).
Using 3PCM, we attempted to predict taxonomic classification for as yet unclassified astrovirus sequences. The predictive models of 3PCM’s Prong 1 (supervised) and Prong 2 (unsupervised) were trained on sequences with known taxonomic labels (Mamastrovirus or Avastrovirus), and later used to predict the genus of as yet unclassified astrovirus sequences. In Prong 3, the class level host labels of the input as yet unclassified astrovirus sequences were considered. In cases where all three labels agree, those labels were proposed as genus labels for the respective as yet unclassified astrovirus genomes. When the taxonomic labels predicted by Prong 1 and Prong 2 agree, but they differ from the host label found by Prong 3, they are considered tentative and subject to further investigation.
For this analysis, only sequences with mammalian or avian hosts were investigated, since the two supervised and unsupervised predictive models were trained only on Mamastrovirus and Avastrovirus genomes. The sequences obtained from 11 other hosts were discarded, resulting in Dataset 3 comprising 229 astroviruses with mammalian or avian hosts, as the testing dataset (Table 3). Details of the distribution of hosts for the 308 as yet unclassified astrovirus RNA sequences can be found in Supplementary Material S1 (Analysis of Astroviruses of Unknown Genus Label).
Figure 3 displays the confusion matrices resulting from the use of Prong 1 (supervised) and Prong 2 (unsupervised) for the classification/clustering datasets including (top), respectively excluding (bottom) recombinant sequences. Note that, out of 54 identified recombinant sequences, 41 were present in the training set (Dataset 2), while the remaining 13 belong to the testing set (Dataset 3). Table 6 summarizes the results of the evaluation metrics for both Prong 1 (classification accuracy) and Prong 2 (clustering accuracy, NMI, ARI, Silhouette Coefficient). As seen from Figure 3 and Table 6, the accuracy of both Prong 1 and Prong 2 increased slightly when the recombinant sequences were removed from the training and test sets (by 1.59% in the case of supervised learning, and by 1.48% in the case of unsupervised clustering). This observation indicates the potential negative impact that recombination events can have on machine learning-based classification/clustering approaches. While the impact may not be highly significant in this analysis, it is crucial to recognize that this may not hold true in all cases. The effect of this elimination process will correlate with the extent to which recombination contributes to noise in the classification. This, in turn, varies based on virus biology, frequency within the dataset, and the taxonomic level of classification.
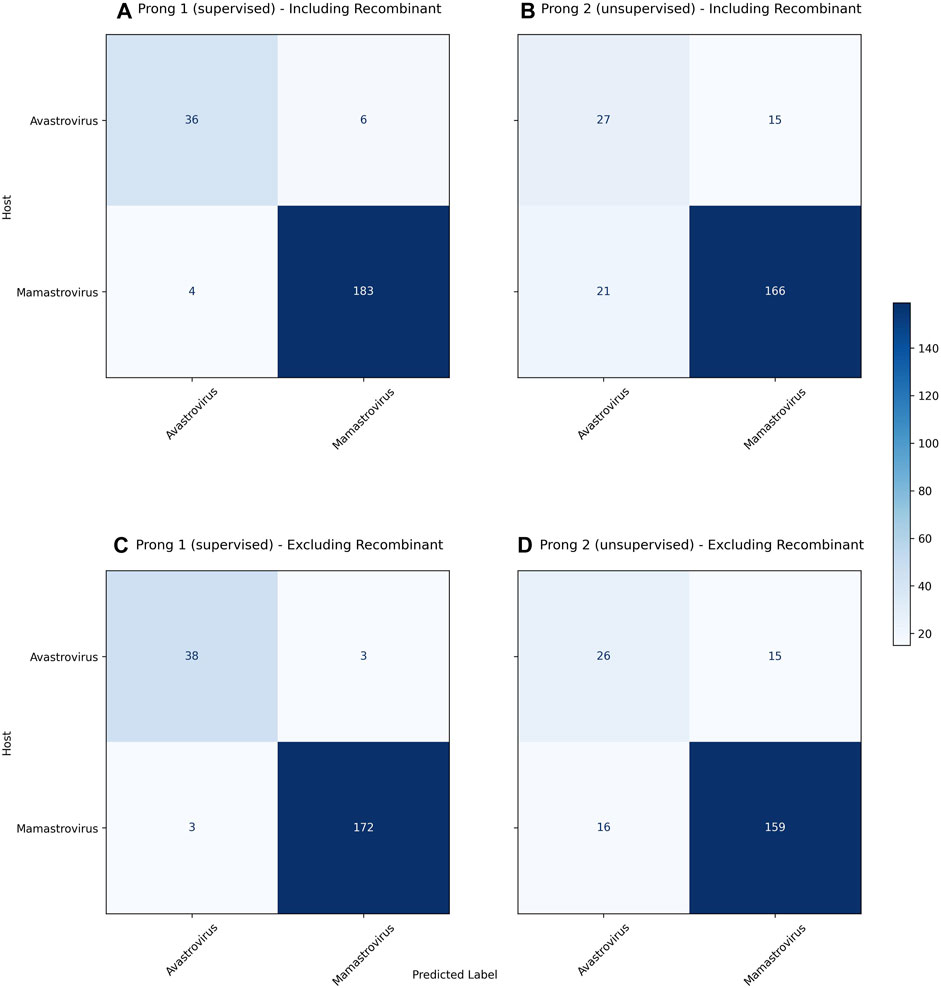
FIGURE 3. (A): Confusion matrix for the classification of as yet unclassified astroviruses including recombinants using Prong 1 (supervised). (B): Confusion matrix for clustering as yet unclassified astroviruses including recombinants using Prong 2 (unsupervised). (C): Confusion matrix for the classification of as yet unclassified astroviruses excluding recombinants using Prong 1 (supervised). (D): Confusion matrix for clustering as yet unclassified astroviruses excluding recombinants using Prong 2 (unsupervised). Prong 3, the host labels, was considered the ground truth for both confusion matrices.
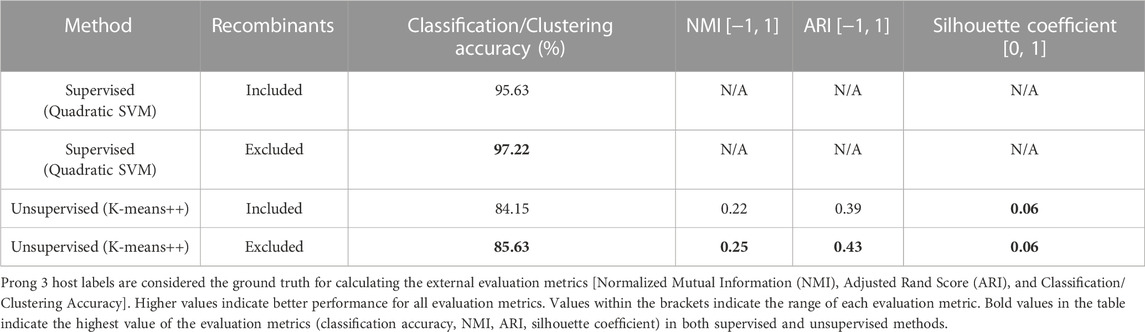
TABLE 6. Evaluation metrics of Prong 1 and Prong 2 applied to the classification/clustering unclassified astroviruses into Mamastroviruses and Avastroviruses when using datasets that include, respectively exclude recombinant sequences.
As seen in Table 7, when both the training and test datasets include recombinant sequences, all three prongs agree on 83.41% (191 out of 229) of the sequences in the testing dataset. For an additional 3% (8 out of 229) of the sequences, the Prong 1 and Prong 2 predictions agree, but differ from the Prong 3 prediction. When recombinant sequences are excluded from both the training and testing datasets, all three prongs agree on predictions for 85.65% (185 out of 216) of the sequences in the testing dataset. Similarly, for an additional 3% (6 out of 216) of the sequences, Prong 1 and Prong 2 agree in their predictions, while Prong 3 disagrees. The NCBI accession IDs of the as yet unclassified 191 + 8 astrovirus sequences when including recombinants and the as yet unclassified 185 + 6 astrovirus sequences when excluding recombinants, together with the taxonomic labels (at the genus level) predicted by 3PCM can be found in Supplementary Material S5 (Proposed Classification for as yet Unclassified Astroviruses). The group of 185 + 6 sequences, where 3PCM led to a genus level classification when excluding recombinants, is a subset of 191 + 8 sequences for which a classification was proposed using 3PCM when including recombinants, with the exception of one sequence with accession ID MT138006, for which the classification prediction was only generated when recombinants were excluded from the analysis.
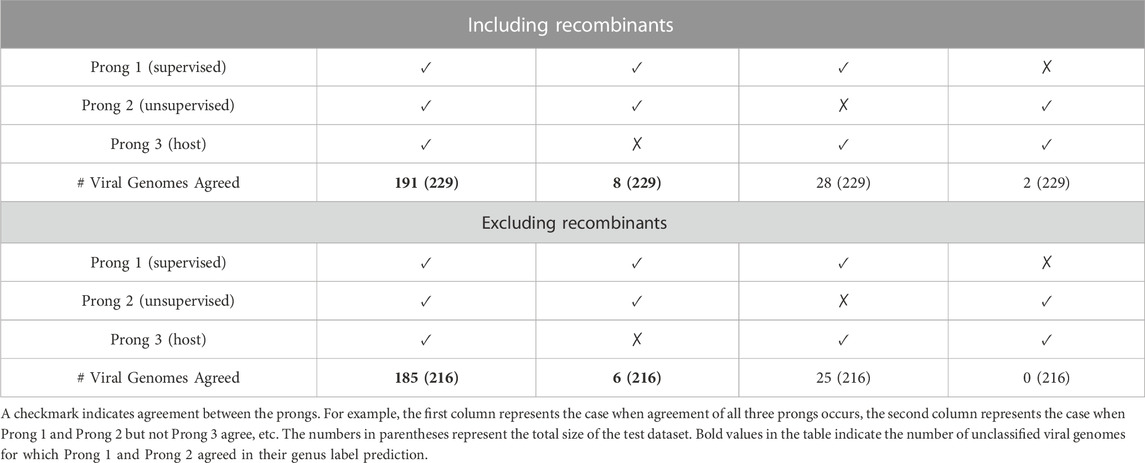
TABLE 7. Number of as yet unclassified viral genomes for which two or all three, of the prongs, agree in their genus label prediction.
Although both sets of results (with and without recombinants) are meaningful, we selected as the primary result of this paper the 191 + 8 genus label predictions obtained from the analysis that includes recombinants. This decision was mainly influenced by the fact that the majority of the recombination events observed are intrageneric, and are thus not significant at genus level classification/clustering.
For the 191 sequences for which all three prongs agree, 26 are predicted to belong to genus Avastrovirus, and the remainder 165 are predicted to belong to genus Mamastrovirus. For these 191 sequences, the proposed genus labels are more certain than for the 8 sequences for which Prong 3’s host label disagreed with Prong 1 and Prong 2’s predictions. For the latter, three out of eight sequences were classified as Avastrovirus by Prong 1 and Prong 2 despite being obtained from mammals. These three sequences are JN420353 [a California sea lion astrovirus (Li et al., 2011)], MH933754 [a human astrovirus (Yinda et al., 2019)], and NC_035758 [a human astrovirus (Orf et al., 2023)]. Regarding the remaining five sequences, obtained from an avian host, the first two prongs predicted that they belonged to the Mamastrovirus genus. The five sequences are: KP663426 (Pankovics et al., 2015), MT138010 (Shan et al., 2022), NC_027426 (Orf et al., 2023), ON304005 (French et al., 2022), MK096773 (Fernández-Correa et al., 2019). Further investigation is needed in order to determine the origin, and the spectrum of natural host species, of these eight sequences.
The genomes of as yet unclassified astroviruses with hosts other than mammals and avians were also examined, to determine whether they can be classified as belonging to the genera, Mamastrovirus or Avastrovirus or to detect whether this family of viruses has more than two genera. Prong 1 (supervised) was not applicable to this problem, due to the absence of ground truth labels in the training set. The clustering results obtained by using Prong 2 (unsupervised) showed no clear separation among the unclassified astroviruses with non-mammalian/non-avian hosts, nor was there any clear separation found between these genomes and Avastroviruses or Mamastroviruses. This could potentially be due to a lack of availability of sufficiently many genomes with non-mammalian and non-avian hosts, which can negatively affect the efficacy of machine learning methods. Details of these computational experiments can be found in Supplementary Material S1 (Analysis of Astroviruses of Unknown Genus Label).
Given the mounting evidence for different serotypes, clades, and genotypes associated with unique cross-species transmissions, different rates of evolution and intraspecific recombination for human astroviruses (HAstV) (Bosch et al., 2014; Donato and Vijaykrishna, 2017; Hargest et al., 2021; Perez et al., 2023) and goose astroviruses (GoAstV) (Fei et al., 2022; Zhu and Sun, 2022), we further investigated the sequences belonging to these two subgroups. Figure 4 displays a visualization of the 6-mer counts of the genomes in Dataset 2 (Mamastrovirus and Avastrovirus genomes with established labels), together with the 191 genomes with genus labels predicted by 3PCM with the use of principal component analysis (PCA). For visualization purposes, the first three principal components of the 6-mer counts for each genome are utilized, preserving ∼21% of the explained variance.
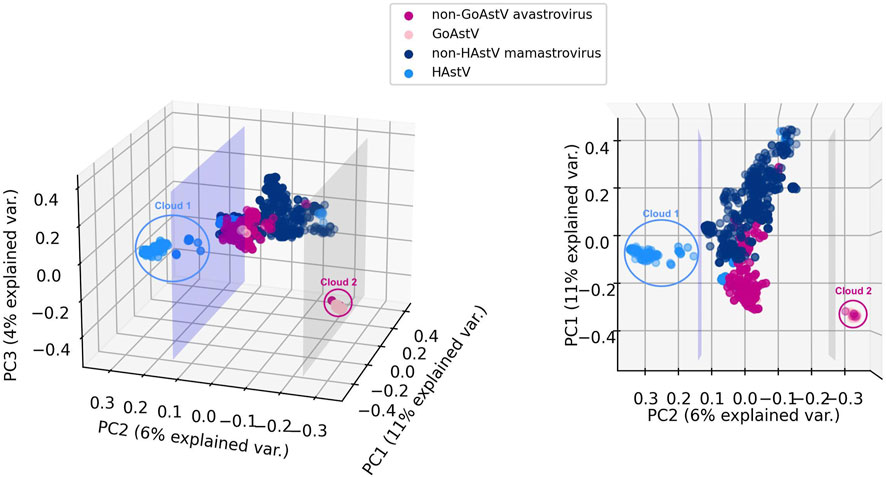
FIGURE 4. Two views of 3D PCA data visualizations of Mamastrovirus and Avastrovirus sequences k-mer frequencies: astrovirus sequences in Dataset 2 (known genus labels), together with the 191 astrovirus genomes with genus labels predicted by 3PCM. For comparison purposes, HAstV and GoAstV are highlighted with different colors compared to the rest of Mamastroviruses (non-HAstV Mamastroviruses) respectively the rest of the Avastroviruses (non-GoAstV Avastroviruses). The lavender plane illustrates the separation between two possible subgenera of Mamastrovirus. The grey plane illustrates the separation between two possible subgenera of Avastrovirus. This visualization is based on the first three principal components of 6-mer counts for the entire genome. In this figure, Clouds 1 and 2 represent possible subgenera of HAstV (Cloud 1) within genus Mamastrovirus, and GoAstV (Cloud 2) within genus Avastrovirus.
The first step in this analysis was to extract information about the host species. The available 636 Mamastrovirus sequences with a host label at the species level were obtained from 69 different species. Of these, 362 sequences were obtained from the four most representative hosts, 105 from Sus scrofa, 44 from Sus domesticus, 153 from Homo sapiens, and 60 from Bos taurus.
As seen in Figure 4, a separating plane exists that separates Cloud 1 (111 Mamastrovirus genomes) from the rest of the Mamastrovirus genomes. A closer examination reveals that, while not all 163 HAstV (human host) sequences are in Cloud 1, all 111 sequences in Cloud 1 are HAstV sequences. This suggests that a HAstV subgenus exists within the genus Mamastrovirus. A comparison between Cloud 1 and the human Mamastroviruses sequences analyzed in Perez et al. (2023) reveals that all the 91 MAstV-Sp7G3 human astrovirus sequences included in our analysis (not collected from sewage, not collected from an unknown host, etc.) are located in Cloud 1. Moreover, all 13 MAstV-Sp6G2 sequences and all 18 MAstV-Sp6G7 sequences analyzed in Perez et al. (2023) are separated from Cloud 1 and located in the main cloud (the sequences located between the two separating planes). This suggests a correspondence between the Cloud 1 sequences and MAstV-Sp7G3 sequences. The accession IDs of sequences in Cloud 1 can be found in Supplementary Material S1 (Analysis of Astroviruses of Unknown Genus Label).
To further examine this hypothesis, 3PCM was applied to the genome sequences of the Mamastrovirus genus (the aforementioned 636 Mamastrovirus genome sequences), with the labels being HAstV and Non-HAstV Mamastrovirus depending on the sequences’ host species. The accuracies of applying Prong 1 (supervised) and Prong 2 (unsupervised) to this dataset, computed using Prong 3 (host labels) as the ground truth, are shown in Table 8. The high classification accuracy of Prong 1 (99.36%) and unsupervised clustering accuracy of Prong 2 (80.88%), provide additional evidence supporting the hypothesis of the existence of a HAstV subgenus of the genus Mamastrovirus. Using the available data, no separation is evident for the other hosts of Mamastroviruses.
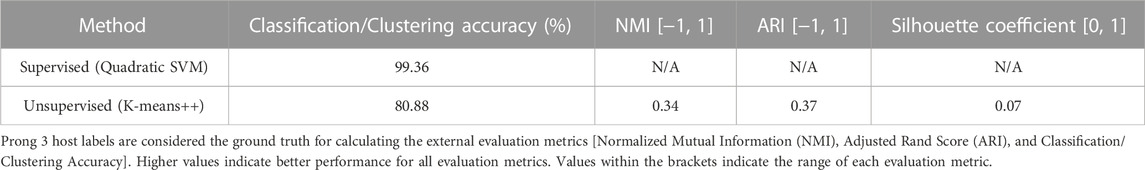
TABLE 8. Evaluation metrics of Prong 1 and Prong 2 applied to the classification/clustering of Mamastrovirus sequences into HAstV and Non-HAstV Mamastrovirus.
When investigating the host species of genus Avastrovirus, we analyzed the 239 Avastroviruses with available host species labels, obtained from 28 different species. Of these, 135 sequences were obtained from the two most representative hosts, 64 from Goose and 71 from Chicken (Gallus gallus).
As seen in Figure 4, a separating plane exists that separates Cloud 2 (66 Avastrovirus genomes) from the rest of the Avastrovirus genomes. Further investigation of Cloud 2 revealed that 59 of its sequences have a Goose host (GoAstV sequences), amounting to 92% (59 out of 64) of the available GoAstV sequences. This suggests that a GoAstV subgenus may exist within the genus Avastrovirus. A comparison between the sequences in Cloud 2 and two established genotypes of GoAstV (see Zhu and Sun, 2022) reveals that all 51 GoAstV-2 (G2) goose astrovirus sequences analyzed in Zhu and Sun (2022) are located in Cloud 2, and all 5 GoAstV-1 (G1) sequences analyzed in Zhu and Sun (2022) are separated from Cloud 2 and located in the main cloud (the sequences located between the two separating planes). This suggests that the observed separation of the Cloud 2 GoAstV sequences from the rest corresponds to the aforementioned two genotypes of the GoAstV virus analyzed in Zhu and Sun (2022). The accession IDs of sequences in Cloud 2 can be found in Supplementary Material S1 (Analysis of Astroviruses of Unknown Genus Label).
To further examine this hypothesis, 3PCM was applied to the genome sequences in this dataset, with the labels being GoAstV and Non-GoAstV Avastrovirus depending on the sequence’s host species. The classification/clustering accuracies of applying Prong 1 (supervised) and Prong 2 (unsupervised) to this dataset, computed using Prong 3 (host labels) as the ground truth, are shown in Table 9. The high classification accuracy of Prong 1 (94.96%), and unsupervised clustering accuracy of Prong 2 (94.98%), provide additional evidence supporting the hypothesis of the existence of a GoAstV subgenus of the genus Avastrovirus. According to the available data, no separation is apparent for the other hosts of Avastroviruses.
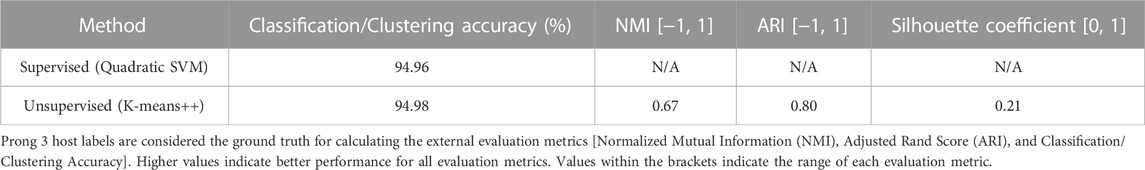
TABLE 9. Evaluation metrics of Prong 1 and Prong 2 applied to the classification/clustering of Avastrovirus sequences into GoAstV and Non-GoAstV Avastrovirus.
We introduce the Three-Pronged Classification Method (3PCM), a novel approach that integrates both supervised and unsupervised machine learning paradigms, along with information about the originating species, to classify emerging astroviruses. The main objective of this study was to suggest a classification system for 229 as yet unclassified astrovirus sequences acquired from avian and mammalian hosts. This approach was taken due to the limited number of available sequences and the lack of definitive information on other hosts. Out of the 229 as yet unclassified sequences, the three-pronged classification yielded consistent predictions for 191 of them, indicating a very high level of reliability for the proposed classification. Furthermore, among the as yet unclassified sequences, For 8 additional sequences, the computational predictions of Prong 2 and Prong 2 coincided, but were different from the host information obtained by Prong 3. In light of numerous supporting evidence regarding the possibility of cross-species infection Pankovics et al. (2015), the classification proposed by Prong 1 and Prong 2 takes precedence over Prong 3. With the investigation in literature, we were able to validate the taxonomic classification labels proposed by Prong 1 and Prong 2, confirming the existence of cross-species infection in both Mamastroviruses and Avastroviruses in these sequences.
3PCM’s versatility lies in its ability to employ each of the three prongs independently or in combination, providing a highly adaptable classification method suitable for various taxonomy tasks. The hypothesis of the existence of additional Avastrovirus and Mamastrovirus genera associated with astroviruses from Reptiles, Amphibians, and Actinopterigii hosts was explored using Prong 2 and Prong 3, which are not reliant on sequence labels. The results showed no clear separation between these three groups and Mamastrovirus and Avastroviruses, but the reason for this could be the limited availability of sequences. For a more comprehensive and accurate analysis, the identification of more Astrovirus sequences from hosts beyond avians and mammalians is necessary.
Elimination of recombinant sequences is an optional step in the 3PCM pipeline. We identified 54 instances of interspecies recombination in Dataset 1, seven of which were intergeneric. The classification/clustering accuracy of Prongs 1 and 2 increased by 1%–2% after the 54 recombinant candidates were removed. Since this study was concerned with genus level classification, of all recombination events only intergeneric recombinations could have an influence on the classification accuracy. As the number of intergeneric recombination events detected in this dataset was low (7 out of 992), the impact of the removal of recombinant sequences on classification accuracies was expected to be negligible. Nonetheless, the option to include this step in the pipeline is essential, as its impact on classification accuracy may vary, depending on the level of taxonomic classification and the frequency and nature of genetic recombination in the virus genomes being classified. The expert user can decide whether or not to include this option, based on the specific virus biology (including the propensity for recombination and whether it is intra or intergeneric), the frequency of such recombinations in the dataset, and the level of classification (e.g., genus or species).
Since the recognition of Astroviridae as a family in 1993, this group of viruses infected over 140 hosts across the globe and is the second largest cause of gastroenteritis in humans. Due to the rapid expansion of infected hosts, frequent inter-species transmission, and genetic recombination, traditional classification based solely on a host may be insufficient. This paper presented 3PCM, a novel machine-learning classification method utilizing both virus-host and whole-genome composition. To enhance the effectiveness of 3PCM, an optional component was added to the pipeline that is responsible for eliminating recombinant sequences. Following the classification of the as yet unclassified astroviruses using Prong 1 (supervised classification) and Prong 2 (unsupervised clustering), the NCBI host labels were used as possible ground truth to classify/cluster astrovirus whole genome sequences, with an accuracy of 95.63%, and 84.15%, respectively. From this classification method, we propose 26 avian-host-derived sequences and 165 mammalian host-derived sequences be added to Avastrovirus and Mamastrovirus genera, respectively. A taxonomic classification was also proposed for eight additional as yet unclassified astrovirus sequences, which are not aligned with the host species of the sequences and may be capable of transmitting across species. As anticipated, the need for a rapid and multipronged approach for astrovirus classification continues to grow (the number of unclassified genome sequences grew from 308 in July 2022 to 429 in September 2023). The 3PCM pipeline is available for ongoing classification of newly added sequences and its power increases with the informative increase in the modeling.
Furthermore, 3PCM was used to investigate the hypothesis of the existence of subgenera GoAstV and HAstV within Avastrovirus and Mamastrovirus respectively. Using 3PCM for classification/clustering of the genus Mamastrovirus into HAstV and Non-HAstV Mamastrovirus, accuracies of 99.36%, and 80.88% for Prong 1 and Prong 2 were achieved. Furthermore, the accuracy of 94.96% and 94.98% were achieved when Prong 1 and Prong 2 were used for classification/clustering of the genus Avastrovirus into GoAstV and Non-GoAstV, respectively. The results of these two experiments were further verified by an investigation of the difference in the genome composition of the subgroups. As a result, we propose that each of these subgroups is a distinct sub-genus.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author. The datasets collected and analyzed for this study can be found in the GitHub repository https://github.com/fatemehalipour/3PCM.
FA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. CH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. YL: Methodology, Supervision, Writing–review and editing. KH: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing–review and editing. LK: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing–review and editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Natural Science and Engineering Research Council of Canada Grants R3511A12 to KH and RGPIN-2023-03663 to LK. This research was enabled in part by support provided by Compute Canada RPP (Research Platforms Portals), https://www.computecanada.ca/, Grant 616 to KH and LK. The funders had no role in the preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1305506/full#supplementary-material
SUPPLEMENTARY DATA SHEET S1 | Supplementary Material S1: Analysis of Astroviruses of Unknown Genus Label.
SUPPLEMENTARY DATA SHEET S2 | Supplementary Material S2: Identification and Analysis of Candidate Recombinant Astrovirus Genome Sequences.
SUPPLEMENTARY DATA SHEET S3 | Supplementary Material S3: Performance Results of 3PCM Using Different Classification/Clustering Algorithms.
SUPPLEMENTARY DATA SHEET S4 | Supplementary Material S4: Astrovirus Near-Neighbour Analysis (Potyvirus).
SUPPLEMENTARY DATA SHEET S5 | Supplementary Material S5: Proposed Classification for as yet Unclassified Astroviruses.
Abad, F. X., Pintó, R. M., Villena, C., Gajardo, R., and Bosch, A. (1997). Astrovirus survival in drinking water. Appl. Environ. Microbiol. 63, 3119–3122. doi:10.1128/aem.63.8.3119-3122.1997
Akhtar, M., Ambikairajah, E., and Epps, J. (2007). “GMM-based classification of genomic sequences,” in 2007 15th International Conference on Digital Signal Processing, China, 1-4 July 2007 (IEEE), 103–106. doi:10.1109/ICDSP.2007.4288529
Aleb, N., and Labidi, N. (2015). An improved k-means algorithm for DNA sequence clustering. In 26th International Workshop on Database and Expert Systems Applications (DEXA) 1-4 Sept. 2015, USA, (IEEE, 39–42. doi:10.1109/DEXA.2015.27
Altman, N. S. (1992). An introduction to kernel and nearest-neighbor nonparametric regression. Am. Statistician 46, 175–185. doi:10.2307/2685209
Arthur, D., and Vassilvitskii, S. (2007). “k-means++: the advantages of careful seeding,” in In Eighteenth Annual ACM-SIAM Symposium on Discrete Algorithms, New York, January 7–9, 2007 (ACM), 1027–1035. doi:10.1145/1283383.1283494
Babkin, I. V., Tikunov, A. Y., Zhirakovskaia, E. V., Netesov, S. V., and Tikunova, N. V. (2012). High evolutionary rate of human astrovirus. Infect. Genet. Evol. 12, 435–442. doi:10.1016/j.meegid.2012.01.019
Bosch, A., Pintó, R. M., and Guix, S. (2014). Human astroviruses. Clin. Microbiol. Rev. 27, 1048–1074. doi:10.1128/CMR.00013-14
Breiman, L., Friedman, J., and Stone, C. J. (1984). Classification and regression trees. Abingdon, UK: Chapman & Hall.
Bridges, C. C. (1966). Hierarchical cluster analysis. Psychol. Rep. 18, 851–854. doi:10.2466/pr0.1966.18.3.851
Bub, T., Hargest, V., Tan, S., Smith, M., Vazquez-Pagan, A., Flerlage, T., et al. (2023). Astrovirus replication is dependent on induction of double-membrane vesicles through a pi3k-dependent, lc3-independent pathway. J. Virology 97, 010255. doi:10.1128/jvi.01025-23
Bustamam, A., Tasman, H., Yuniarti, N., Frisca, F., and Mursidah, I. (2017). Application of k-means clustering algorithm in grouping the DNA sequences of hepatitis B virus (HBV). In AIP conference proceedings China, (AIP Publishing. doi:10.1063/1.4991238
Chae, S.-B., Jeong, C.-G., Park, J.-S., Na, E.-J., and Oem, J.-K. (2023). Detection and genetic characterization of astroviruses in brain tissues of wild raccoon dogs. Viruses 15, 1488. doi:10.3390/v15071488
Chan, T. F., Golub, G. H., and LeVeque, R. J. (1982). “Updating formulae and a pairwise algorithm for computing sample variances,” in COMPSTAT 1982 5th symposium held at toulouse 1982 (Germany: Springer), 30–41. doi:10.1007/978-3-642-51461-6_3
Chhabra, P., Payne, D. C., Szilagyi, P. G., Edwards, K. M., Staat, M. A., Shirley, S. H., et al. (2013). Etiology of viral gastroenteritis in children <5 years of age in the United States, 2008–2009. J. Infect. Dis. 208, 790–800. doi:10.1093/infdis/jit254
Cortez, V., Meliopoulos, V. A., Karlsson, E. A., Hargest, V., Johnson, C., and Schultz-Cherry, S. (2017). Astrovirus biology and pathogenesis. Annu. Rev. Virology 4, 327–348. doi:10.1146/annurev-virology-101416-041742
Cristianini, N., and Shawe-Taylor, J. (2000). An introduction to support vector machines and other kernel-based learning methods. UK: Cambridge University Press.
De Benedictis, P., Schultz-Cherry, S., Burnham, A., and Cattoli, G. (2011). Astrovirus infections in humans and animals – Molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 11, 1529–1544. doi:10.1016/j.meegid.2011.07.024
Dempster, A. P., Laird, N. M., and Rubin, D. B. (1977). Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B Methodol. 39, 1–22. doi:10.1111/j.2517-6161.1977.tb01600.x
Dennehy, P., Nelson, S., Spangenberger, S., Noel, J., Monroe, S., and Glass, R. (2001). A prospective case-control study of the role of astrovirus in acute diarrhea among hospitalized young children. J. Infect. Dis. 184, 10–15. doi:10.1086/321007
Donato, C., and Vijaykrishna, D. (2017). The broad host range and genetic diversity of mammalian and avian astroviruses. Viruses 9, 102. doi:10.3390/v9050102
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi:10.1093/nar/gkh340
Fabijańska, A., and Grabowski, S. (2019). Viral genome deep classifier. IEEE Access 7, 81297–81307. doi:10.1109/ACCESS.2019.2923687
Fei, Z., Jiao, A., Xu, M., Wu, J., Wang, Y., Yu, J., et al. (2022). Genetic diversity and evolution of goose astrovirus in the east of China. Transbound. Emerg. Dis. 69, e2059. e2072. doi:10.1111/tbed.14542
Fernández-Correa, I., Truchado, D. A., Gomez-Lucia, E., Doménech, A., Pérez-Tris, J., Schmidt-Chanasit, J., et al. (2019). A novel group of avian astroviruses from Neotropical passerine birds broaden the diversity and host range of Astroviridae. Sci. Rep. 9, 9513. doi:10.1038/s41598-019-45889-3
French, R. K., Filion, A., Niebuhr, C. N., and Holmes, E. C. (2022). Metatranscriptomic comparison of viromes in endemic and introduced passerines in New Zealand. Viruses 14, 1364. doi:10.3390/v14071364
Freund, Y., and Schapire, R. E. (1997). A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 55, 119–139. doi:10.1006/jcss.1997.1504
Gibbs, A. J., Hajizadeh, M., Ohshima, K., and Jones, R. A. (2020). The potyviruses: an evolutionary synthesis is emerging. Viruses 12, 132. doi:10.3390/v12020132
Girgis, H. Z. (2022). MeShClust v3. 0: high-quality clustering of DNA sequences using the mean shift algorithm and alignment-free identity scores. BMC Genomics 23, 423. doi:10.1186/s12864-022-08619-0
Grohmann, G. S., Glass, R. I., Pereira, H. G., Monroe, S. S., Hightower, A. W., Weber, R., et al. (1993). Enteric viruses and diarrhea in HIV-infected patients. Enteric opportunistic infections working group. N. Engl. J. Med. 329, 14–20. doi:10.1056/NEJM199307013290103
Hargest, V., Davis, A. E., Tan, S., Cortez, V., and Schultz-Cherry, S. (2021). Human astroviruses: a tale of two strains. Viruses 13, 376. doi:10.3390/v13030376
Hastie, T., Tibshirani, R., Friedman, J. H., and Friedman, J. H. (2009). The elements of statistical learning: data mining, inference, and prediction, 2. New York: Springer.
Herrmann, J. E., Taylor, D. N., Echeverri, P., and Blacklow, N. R. (1991). Astroviruses as a cause of gastroenteritis in children. N. Engl. J. Med. 324, 1757–1760. doi:10.1056/NEJM199106203242501
Hinton, G. E. (1990). “Connectionist learning procedures,” in Machine learning (USA: Elsevier), 555–610. doi:10.1016/0004-3702(89)90049-0
Hoang, T., Yin, C., Zheng, H., Yu, C., Lucy He, R., and Yau, S. S.-T. (2015). A new method to cluster DNA sequences using Fourier power spectrum. J. Theor. Biol. 372, 135–145. doi:10.1016/j.jtbi.2015.02.026
James, B., Luczak, B., and Girgis, H. (2018). MeShClust: an intelligent tool for clustering DNA sequences. Nucleic Acids Res. 46, e83. doi:10.1093/nar/gky315
Jarchow-Macdonald, A. A., Halley, S., Chandler, D., Gunson, R., Shepherd, S. J., and Parcell, B. J. (2015). First report of an astrovirus type 5 gastroenteritis outbreak in a residential elderly care home identified by sequencing. J. Clin. Virology 73, 115–119. doi:10.1016/j.jcv.2015.11.006
Jeong, H. S., Jeong, A., and Cheon, D.-S. (2012). Epidemiology of astrovirus infection in children. Korean J. Pediatr. 55, 77–82. doi:10.3345/kjp.2012.55.3.77
Jiang, B., Monroe, S. S., Koonin, E. V., Stine, S. E., and Glass, R. I. (1993). RNA sequence of astrovirus: distinctive genomic organization and a putative retrovirus-like ribosomal frameshifting signal that directs the viral replicase synthesis. Proc. Natl. Acad. Sci. U. S. A. 90, 10539–10543. doi:10.1073/pnas.90.22.10539
Jiang, J.-Z., Yuan, W.-G., Shang, J., Shi, Y.-H., Yang, L.-L., Liu, M., et al. (2023). Virus classification for viral genomic fragments using PhaGCN2. Briefings Bioinforma. 24, bbac505. bbac505. doi:10.1093/bib/bbac505
Karlsson, E. A., Small, C. T., Freiden, P., Feeroz, M., Matsen, F. A., San, S., et al. (2015). Non-human Primates harbor diverse mammalian and avian astroviruses including those associated with human infections. PLoS Pathog. 11, e1005225. doi:10.1371/journal.ppat.1005225
Katz, K., Shutov, O., Lapoint, R., Kimelman, M., Brister, J. R., and O’Sullivan, C. (2022). The sequence read archive: a decade more of explosive growth. Nucleic Acids Res. 50, D387–D390. doi:10.1093/nar/gkab1053
Keita, A. M., Doh, S., Sow, S. O., Powell, H., Omore, R., Jahangir Hossain, M., et al. (2023). Prevalence, clinical severity, and seasonality of adenovirus 40/41, astrovirus, sapovirus, and rotavirus among young children with moderate-to-severe diarrhea: results from the vaccine impact on diarrhea in Africa (VIDA) study. Clin. Infect. Dis. 76, S123–S131. doi:10.1093/cid/ciad060
Kingma, D., and Ba, J. (2015). “Adam: a method for stochastic optimization,” in International conference on learning representations (ICLR) (San Diega, CA, USA: IEEE).
Kraskov, A., Stögbauer, H., Andrzejak, R. G., and Grassberger, P. (2005). Hierarchical clustering using mutual information. Europhys. Lett. 70, 278–284. doi:10.1209/epl/i2004-10483-y
Kuhn, H. W. (1955). The Hungarian method for the assignment problem. Nav. Res. Logist. Q. 2, 83–97. doi:10.1002/nav.3800020109
Le Cann, P., Ranarijaona, S., Monpoeho, S., Le Guyader, F., and Ferré, V. (2004). Quantification of human astroviruses in sewage using real-time RT-PCR. Res. Microbiol. 155, 11–15. doi:10.1016/j.resmic.2003.09.013
Lefkowitz, E. J., Dempsey, D. M., Hendrickson, R. C., Orton, R. J., Siddell, S. G., and Smith, D. B. (2017). Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 46, D708–D717. doi:10.1093/nar/gkx932
Li, H., Wan, C., Wang, Z., Tan, J., Tan, M., Zeng, Y., et al. (2023). Rapid diagnosis of duck Tembusu virus and goose astrovirus with TaqMan-based duplex real-time PCR. Front. Microbiol. 14, 1146241. doi:10.3389/fmicb.2023.1146241
Li, L., Shan, T., Wang, C., Côté, C., Kolman, J., Onions, D., et al. (2011). The fecal viral flora of California sea lions. J. Virology 85, 9909–9917. doi:10.1128/JVI.05026-11
Liste, M. B., Natera, I., Suarez, J. A., Pujol, F. H., Liprandi, F., and Ludert, J. E. (2000). Enteric virus infections and diarrhea in healthy and human immunodeficiency virus-infected children. J. Clin. Microbiol. 38, 2873–2877. doi:10.1128/JCM.38.8.2873-2877.2000
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003. doi:10.1093/ve/vev003
Meliopoulos, V. A., Kayali, G., Burnham, A., Oshansky, C. M., Thomas, P. G., Gray, G. C., et al. (2014). Detection of antibodies against Turkey astrovirus in humans. PLoS ONE 9, e96934. doi:10.1371/journal.pone.0096934
Mendizabal-Ruiz, G., Román-Godínez, I., Torres-Ramos, S., Salido-Ruiz, R. A., Vélez-Pérez, H., and Morales, J. A. (2018). Genomic signal processing for DNA sequence clustering. PeerJ 6, e4264. doi:10.7717/peerj.4264
Meyer, C. T., Bauer, I. K., Antonio, M., Adeyemi, M., Saha, D., Oundo, J. O., et al. (2015). Prevalence of classic, MLB-clade and VA-clade astroviruses in Kenya and the Gambia. Virology J. 12, 78. doi:10.1186/s12985-015-0299-z
Midthun, K., Greenberg, H. B., Kurtz, J. B., Gary, G. W., Lin, F. Y., and Kapikian, A. Z. (1993). Characterization and seroepidemiology of a type 5 astrovirus associated with an outbreak of gastroenteritis in Marin County, California. J. Clin. Microbiol. 31, 955–962. doi:10.1128/jcm.31.4.955-962.1993
Millán Arias, P., Alipour, F., Hill, K. A., and Kari, L. (2022). DeLUCS: deep learning for unsupervised clustering of DNA sequences. PLoS ONE 17, e0261531. doi:10.1371/journal.pone.0261531
Millan Arias, P., Hill, K. A., and Kari, L. (2023). iDeLUCS: a deep learning interactive tool for alignment-free clustering of DNA sequences. Bioinformatics 39, btad508. doi:10.1093/bioinformatics/btad508
Oishi, I., Yamazaki, K., Kimoto, T., Minekawa, Y., Utagawa, E., Yamazaki, S., et al. (1994). A large outbreak of acute gastroenteritis associated with astrovirus among students and teachers in Osaka, Japan. J. Infect. Dis. 170, 439–443. doi:10.1093/infdis/170.2.439
Orf, G. S., Olivo, A., Harris, B., Weiss, S. L., Achari, A., Yu, G., et al. (2023). Metagenomic detection of divergent insect-and bat-associated viruses in plasma from two African individuals enrolled in blood-borne surveillance. Viruses 15, 1022. doi:10.3390/v15041022
Palombo, E. A., and Bishop, R. F. (1996). Annual incidence, serotype distribution, and genetic diversity of human astrovirus isolates from hospitalized children in Melbourne, Australia. J. Clin. Microbiol. 34, 1750–1753. doi:10.1128/jcm.34.7.1750-1753.1996
Pankovics, P., Boros, Á., Kiss, T., Delwart, E., and Reuter, G. (2015). Detection of a mammalian-like astrovirus in bird, European roller (Coracias garrulus). Infect. Genet. Evol. 34, 114–121. doi:10.1016/j.meegid.2015.06.020
Pantin-Jackwood, M., Todd, D., and Koci, M. D. (2012). “Avian astroviruses,” in Astrovirus research. Editor S. Schultz-Cherry (New York, NY: Springer New York), 151–180. doi:10.1007/978-1-4614-4735-1_9
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830.
Perez, L. J., Forberg, K., Cloherty, G. A., and Berg, M. G. (2023). Temporal and coevolutionary analyses reveal the events driving the emergence and circulation of human mamastroviruses. Emerg. Microbes Infect. 12, 2217942. doi:10.1080/22221751.2023.2217942
Qureshi, M. I., Worthington, B. M., Liu, Y., Cheung, W. Y.-M., Su, S., Zheng, Z., et al. (2023). Discovery of novel Mamastroviruses in Bactrian camels and dromedaries reveals complex recombination history. Virus Evol. 9, veac125. doi:10.1093/ve/veac125
Rand, W. M. (1971). Objective criteria for the evaluation of clustering methods. J. Am. Stat. Assoc. 66, 846–850. doi:10.1080/01621459.1971.10482356
Randhawa, G. S., Hill, K. A., and Kari, L. (2019). ML-DSP: machine learning with digital signal processing for ultrafast, accurate, and scalable genome classification at all taxonomic levels. BMC Genomics 20, 267–321. doi:10.1186/s12864-019-5571-y
Randhawa, G. S., Hill, K. A., and Kari, L. (2020). MLDSP-GUI: an alignment-free standalone tool with an interactive graphical user interface for DNA sequence comparison and analysis. Bioinformatics 36, 2258–2259. doi:10.1093/bioinformatics/btz918
Refaeilzadeh, P., Tang, L., and Liu, H. (2009). Cross-validation. Encycl. Database Dystems 5, 532–538. doi:10.1007/978-0-387-39940-9_565
Resque, H. R., Munford, V., Castilho, J. G., Schmich, H., Caruzo, T. A. R., and Rácz, M. L. (2007). Molecular characterization of astrovirus in stool samples from children in São Paulo, Brazil. Memórias do Inst. Oswaldo Cruz 102, 969–974. doi:10.1590/S0074-02762007000800012
Rousseeuw, P. J. (1987). Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65. doi:10.1016/0377-0427(87)90125-7
Schwende, I., and Pham, T. D. (2014). Pattern recognition and probabilistic measures in alignment-free sequence analysis. Briefings Bioinforma. 15, 354–368. doi:10.1093/bib/bbt070
Shan, T., Yang, S., Wang, H., Wang, H., Zhang, J., Gong, G., et al. (2022). Virome in the cloaca of wild and breeding birds revealed a diversity of significant viruses. Microbiome 10, 60–21. doi:10.1186/s40168-022-01246-7
Shastri, S., Doane, A. M., Gonzales, J., Upadhyayula, U., and Bass, D. M. (1998). Prevalence of astroviruses in a children’s hospital. J. Clin. Microbiol. 36, 2571–2574. doi:10.1128/JCM.36.9.2571-2574.1998
Shendure, J., Mitra, R. D., Varma, C., and Church, G. M. (2004). Advanced sequencing technologies: methods and goals. Nat. Rev. Genet. 5, 335–344. doi:10.1038/nrg1325
Solis-Reyes, S., Avino, M., Poon, A., and Kari, L. (2018). An open-source k-mer based machine learning tool for fast and accurate subtyping of HIV-1 genomes. PLoS ONE 13, e0206409. doi:10.1371/journal.pone.0206409
Strehl, A., and Ghosh, J. (2002). Cluster ensembles – a knowledge reuse framework for combining multiple partitions. J. Mach. Learn. Res. 3, 583–617. doi:10.1162/153244303321897735
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi:10.1093/nar/22.22.4673
Tibshirani, R., Hastie, T., Narasimhan, B., and Chu, G. (2002). Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. 99, 6567–6572. doi:10.1073/pnas.082099299
Vu, D.-L., Bosch, A., Pintó, R., and Guix, S. (2017). Epidemiology of classic and novel human astrovirus: gastroenteritis and beyond. Viruses 9, 33. doi:10.3390/v9020033
Wei, H., Kumthip, K., Khamrin, P., Yodmeeklin, A., Jampanil, N., Phengma, P., et al. (2023). Triple intergenotype recombination of human astrovirus 5, human astrovirus 8, and human astrovirus 1 in the open reading frame 1a, open reading frame 1b, and open reading frame 2 regions of the human astrovirus genome. Microbiol. Spectr. 11, e0488822. doi:10.1128/spectrum.04888-22
Yinda, C. K., Vanhulle, E., Conceição-Neto, N., Beller, L., Deboutte, W., Shi, C., et al. (2019). Gut virome analysis of Cameroonians reveals high diversity of enteric viruses, including potential interspecies transmitted viruses. MSphere 4, 005855–e618. doi:10.1128/mSphere.00585-18
Zheng, Y., Gao, S., Padmanabhan, C., Li, R., Galvez, M., Gutierrez, D., et al. (2017). VirusDetect: an automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 500, 130–138. doi:10.1016/j.virol.2016.10.017
Keywords: machine learning, viral classification and clustering, family Astroviridae, Avastrovirus, Mamastrovirus, alignment-free classification, genomic signature, k-mer frequency
Citation: Alipour F, Holmes C, Lu YY, Hill KA and Kari L (2024) Leveraging machine learning for taxonomic classification of emerging astroviruses. Front. Mol. Biosci. 10:1305506. doi: 10.3389/fmolb.2023.1305506
Received: 01 October 2023; Accepted: 12 December 2023;
Published: 11 January 2024.
Edited by:
Jayaraman Valadi, Flame University, IndiaReviewed by:
Indira Ghosh, Jawaharlal Nehru University, IndiaCopyright © 2024 Alipour, Holmes, Lu, Hill and Kari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fatemeh Alipour, ZmFsaXBvdXJAdXdhdGVybG9vLmNh
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.