 Smitha Ijee1,2Karthik Chambayil1,3†
Smitha Ijee1,2Karthik Chambayil1,3† Anurag Dutta Chaudhury4,5†Abhirup Bagchi1†
Anurag Dutta Chaudhury4,5†Abhirup Bagchi1† Kirti Modak4,5†
Kirti Modak4,5† Saswati Das2,4†Esther Sathya Bama Benjamin3,4Sonam Rani1,2Daniel Zechariah Paul4,6Aneesha Nath1Debanjan Roy4,6Dhavapriya Palani1Sweety Priyanka4Rakshini Ravichandran4Betty K. Kumary4Yazhini Sivamani4Vijayanand S.2Dinesh Babu1
Saswati Das2,4†Esther Sathya Bama Benjamin3,4Sonam Rani1,2Daniel Zechariah Paul4,6Aneesha Nath1Debanjan Roy4,6Dhavapriya Palani1Sweety Priyanka4Rakshini Ravichandran4Betty K. Kumary4Yazhini Sivamani4Vijayanand S.2Dinesh Babu1 Yukio Nakamura7
Yukio Nakamura7 Vasanth Thamodaran1,8
Vasanth Thamodaran1,8 Poonkuzhali Balasubramanian4
Poonkuzhali Balasubramanian4 Shaji R. Velayudhan1,4*
Shaji R. Velayudhan1,4*- 1Centre for Stem Cell Research (A Unit of inStem, Bengaluru), Christian Medical College Campus, Vellore, India
- 2Department of Biotechnology, Thiruvalluvar University, Vellore, India
- 3Sree Chitra Tirunal Institute of Science and Medical Technology, Thiruvananthapuram, India
- 4Department of Haematology, Christian Medical College Campus, Vellore, India
- 5Regional Centre for Biotechnology, New Delhi, India
- 6Manipal Academy of Higher Education, Manipal, India
- 7Cell Engineering Division, RIKEN BioResource Research Center, Tsukuba, Japan
- 8Tata Institute of Genetics and Society, Bengaluru, India
MicroRNAs (miRNAs) are short non-coding RNAs that play crucial roles in gene regulation, exerting post-transcriptional silencing, thereby influencing cellular function, development, and disease. Traditional loss-of-function methods for studying miRNA functions, such as miRNA inhibitors and sponges, present limitations in terms of specificity, transient effects, and off-target effects. Similarly, CRISPR/Cas9-based editing of miRNAs using single guide RNAs (sgRNAs) also has limitations in terms of design space for generating effective gRNAs. In this study, we introduce a novel approach that utilizes CRISPR/Cas9 with dual guide RNAs (dgRNAs) for the rapid and efficient generation of short deletions within miRNA genomic regions. Through the expression of dgRNAs through single-copy lentiviral integration, this approach achieves over a 90% downregulation of targeted miRNAs within a week. We conducted a comprehensive analysis of various parameters influencing efficient deletion formation. In addition, we employed doxycycline (Dox)-inducible expression of Cas9 from the AAVS1 locus, enabling homogeneous, temporal, and stage-specific editing during cellular differentiation. Compared to miRNA inhibitory methods, the dgRNA-based approach offers higher specificity, allowing for the deletion of individual miRNAs with similar seed sequences, without affecting other miRNAs. Due to the increased design space, the dgRNA-based approach provides greater flexibility in gRNA design compared to the sgRNA-based approach. We successfully applied this approach in two human cell lines, demonstrating its applicability for studying the mechanisms of human erythropoiesis and pluripotent stem cell (iPSC) biology and differentiation. Efficient deletion of miR-451 and miR-144 resulted in blockage of erythroid differentiation, and the deletion of miR-23a and miR-27a significantly affected iPSC survival. We have validated the highly efficient deletion of genomic regions by editing protein-coding genes, resulting in a significant impact on protein expression. This protocol has the potential to be extended to delete multiple miRNAs within miRNA clusters, allowing for future investigations into the cooperative effects of the cluster members on cellular functions. The protocol utilizing dgRNAs for miRNA deletion can be employed to generate efficient pooled libraries for high-throughput comprehensive analysis of miRNAs involved in different biological processes.
Introduction
It has been estimated that approximately 20,000 protein-coding genes constitute only 2% of the human genome, with the remainder being those encoding non-coding RNAs (Carninci et al., 2005). MicroRNAs (miRNAs) are a class of short non-coding RNAs (20–24 nt) that regulate gene expression at the post-transcriptional and translational levels by binding to the 3′ untranslated regions of messenger RNAs (mRNAs) (Bassett et al., 2014). They play crucial roles in cellular function, development, and disease (Gebert and MacRae, 2019). Due to the short seed sequences that determine miRNA target recognition, a single miRNA can regulate hundreds to thousands of target mRNAs, while multiple miRNAs can regulate a single mRNA (Kehl et al., 2017). It is estimated that miRNAs can regulate up to 60% of all human protein-coding genes (Friedman et al., 2009).
Despite identifying over 3,500 human miRNAs across various cell types (Kozomara and Griffiths-Jones, 2014), the biological functions of the majority of miRNAs remain unknown. Functional characterization of miRNAs can be accomplished using gain-of-function and loss-of-function approaches. Gain-of-function methods involve the introduction of miRNAs into cells either through the transfection of double-stranded miRNA mimics (Nogimori et al., 2019) or through the utilization of vectors for miRNA expression (Fan et al., 2019). Loss-of-function approaches rely on synthetic antisense oligonucleotide miRNA inhibitors, which bind to and inhibit endogenous miRNAs, preventing their interaction with the target mRNAs (Stenvang et al., 2012; Lima et al., 2018; Bajan and Hutvagner, 2020). Additionally, miRNA sponges and decoys, which contain multiple binding sites of miRNAs, competitively inhibit endogenous miRNAs by sequestering them, thereby reducing their availability to bind target mRNAs and dampening their regulatory activity (Ebert et al., 2007; Gentner et al., 2009). Nonetheless, these methodologies have certain limitations, such as transient effects, specificity concerns, toxicity, and variable efficiency (Sayed et al., 2021; Bayraktar et al., 2023). Additionally, given the presence of highly homologous seed sequences among some miRNAs, ensuring specific repression of the targeted miRNAs is crucial to avoid biased phenotypic outcomes (Stenvang et al., 2012).
The CRISPR/Cas9 genome editing tool, which employs Cas9 nuclease and guide RNAs (gRNAs) to modify the specific genomic regions, operates by inducing DNA double-strand breaks (DSBs) and relies on error-prone non-homologous end joining (NHEJ) DNA repair, leading to the efficient generation of small insertions and deletions (indels) at the targeted sites (Ran et al., 2013). Recent studies have utilized CRISPR/Cas9 with single guide RNAs (sgRNAs) targeting crucial functional sequences of miRNAs, including their seed sequences, loop regions, or biogenesis processing sites. The results demonstrated significant reductions in the expression of the targeted miRNAs (Jiang et al., 2014; Chang et al., 2016; Wallace et al., 2016; Huo et al., 2017; Zhang et al., 2018; Li et al., 2020). Importantly, unlike miRNA inhibitory methods that employ antisense oligonucleotides to deplete miRNA transcripts, CRISPR/Cas9 genomic editing leads to permanent disruption of miRNA expression. However, genome editing might inadvertently yield unpredictable and unintended indels, potentially outside critical regions for miRNA function, thus not disrupting the expression of targeted miRNAs (Bi et al., 2020; Vidigal and Ventura, 2014; Ferreira and Reis, 2023). Furthermore, indels occurring within the stem regions of miRNAs can give rise to non-naturally occurring miRNA transcripts (Bhattacharya et al., 2012). Although CRISPR/Cas9 mediated mutations at miRNA biogenesis processing sites have demonstrated the capability to reduce mature miRNAs by as much as 90% (Chang et al., 2016), a significant challenge arises in designing suitable gRNAs for all the miRNAs under study due to the limited design space available for this approach. We were unable to identify suitable gRNAs at the biogenesis sites for more than 40% of the miRNAs that we analyzed.
In this study, we report the development of a rapid and efficient protocol utilizing dual gRNAs (dgRNAs) to generate short deletions in human miRNA genomic regions. When two gRNAs bind to a genomic region, a short distance apart, they can trigger the simultaneous creation of DSBs, resulting in the generation of deletions between the binding sites of these gRNAs (Aparicio-prat et al., 2016). This method has been used to generate deletions within long non-coding RNAs (Yin et al., 2015; Aparicio-prat et al., 2016; Zhu et al., 2016; Chaudhary et al., 2017; Hao et al., 2020). Zhu et al. demonstrated efficient deletion formation (>90%) within a pool of cells that were lentivirally transduced with dgRNAs targeting one specific non-coding RNA (Zhu et al., 2016). However, this high efficiency required more than 2 weeks to achieve, and it remains uncertain whether comparable levels of efficiency could be attained for multiple non-coding RNAs. Deletion of miRNAs in mammalian cells using dgRNA expression vectors showed extremely low efficiency, necessitating single-cell sorting to isolate clones with the deletions of the targeted miRNAs for subsequent functional studies (Ho et al., 2015; Liu et al., 2016; Yan et al., 2019; Hong et al., 2020). Therefore, we assessed various parameters, including the on-target efficiency of gRNAs and the orientation and distance between the dgRNAs, which influence the efficiency of deletion formation. Through single-copy lentiviral integration of dgRNAs, we achieved a downregulation of over 90% for the targeted miRNAs within a week. The utilization of the CRISPR/Cas9 dgRNA strategy enables the specific targeting and investigation of individual miRNAs. This approach holds significant promise for gaining valuable insights into the functional roles of these molecules.
Methods
Cell culture
HUDEP2 (HUDEP-2) cells (a kind gift from Yukio Nakamura) (Kurita et al., 2013) were cultured following previously reported protocols (Kurita et al., 2013; Bagchi et al., 2021). Briefly, the cells were expanded in StemSpan SFEM-II medium (Stem Cell Technologies) supplemented with 1 µM dexamethasone, 1 μg/mL doxycycline (Dox), 50 ng/mL recombinant human stem cell factor (rh SCF), 3 units/mL recombinant human erythropoietin (rh EPO), 10 ng/mL recombinant human interleukin 3 (rh IL-3) and 1% penicillin/streptomycin. The AAVS1-Tet-On-Cas9 iPSCs generated in our laboratory (Thamodaran et al., 2022) were cultured in Matrigel (Corning) - coated plates containing mTeSR medium (STEMCELL Technologies). Upon reaching 70%–80% confluency, colonies were dissociated with Versene (Thermo Fisher Scientific) following the manufacturer’s protocol and passaged at a 1:4 ratio.
Differentiation of HUDEP-2 cells
The differentiation of HUDEP-2 cells was performed following a previously described protocol (Hawksworth et al., 2018; Bagchi et al., 2021). Initially, the cells were seeded at a density of 2 × 105 cells/mL in the differentiation medium consisting of Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 2% (v/v) fetal bovine serum (Thermo Fischer Scientific), 3% (v/v) AB Serum (MP Biomedicals), 10 μg/mL insulin (Sigma Aldrich), 3 U/mL heparin (Sigma Aldrich), 200 μg/mL human holotransferrin (Sigma Aldrich), 1 ng/mL rh IL-3 (Peprotech), 10 ng/mL rh SCF (Peprotech), 3 U/mL rh Epo (Peprotech) and 1 μg/mL Dox (Sigma-Aldrich). After 2 days, the cells were reseeded at a density of 3.5 × 105 cells/mL in a fresh medium. On day 4, the cells were further reseeded at a density of 5 × 105 cells/mL in a fresh medium without Dox. Subsequently, on day 6, a complete medium change was performed, and the cells were reseeded at a density of 1 × 106 cells/mL with an increased concentration of holotransferrin (500 μg/mL). On day 8, the cells were transferred to a differentiation medium devoid of rh SCF, rh IL-3, or Dox and maintained at a density of 1 × 106 cells/mL with complete medium changes every 2 days until day 10.
gRNA design
The stem-loop and flanking 30 nucleotide sequences of miRNAs were obtained from miRBase v21.1 (Kozomara and Griffiths-Jones, 2014) and the UCSC genome browser. To design the gRNAs, we utilized CRISPOR (Concordet and Haeussler, 2018) (http://crispor.tefor.net/), which provided on-target editing efficiency (Doench et al., 2016) and off-target effect scores. For miRNA knockout experiments using sgRNAs, we designed the gRNAs to be adjacent to the miRNA seed sequences or Drosha and Dicer biogenesis processing sites. For dgRNA knockout experiments, two gRNAs were designed to encompass the genomic sequences of either the 5p or 3p arms of the miRNAs. The gRNAs designed to be cloned in pKLV2.2 lentiviral vectors were synthesized commercially. The complimentary oligos, to be cloned into the hU6gRNA5(BbsI) cassette of the pKLV2.2 vectors were synthesized with tag sequences top strand oligo- 5′- CACCNNNNNNNNNNNNNNNNNNNN-3′ and bottom strand oligo—3′-NNNNNNNNNNNNNNNNNNNNCAAA-5′. The complimentary oligos, to be cloned into the h7SKgRNA5(BbsI) cassette were synthesized with tag sequences in the top strand oligo and bottom strand oligo as described earlier (Tzelepis et al., 2016): Top strand oligo- 5′- CTCNNNNNNNNNNNNNNNNNNNN-3′ and bottom strand oligo—3′-NNNNNNNNNNNNNNNNNNNNCAA-5′. The overhangs for cloning in LentiCRISPR V2 and pL.CRISPR.EFS.GFP lentiviral vectors were designed as described earlier (Tzelepis et al., 2016; Heckl et al., 2014).
Generation of single gRNA lentiviral vectors
Single gRNAs targeting the miRNA regions were cloned into LentiCRISPR V2 (a gift from Feng Zhang, Addgene plasmid #52961) or pKLV2.2-h7SKgRNA5(SapI)-hU6gRNA5(BbsI)-PGKpuroBFP-W (Addgene plasmid # 72666) or pKLV2.2-mU6gRNA5(SapI)-hU6gRNA5(BbsI)-PGKpuroBFP-W (Addgene no#72666) (gifts from Kosuke Yusa) (Tzelepis et al., 2016; Ihry et al., 2018). The gRNAs were cloned at the BsmBI site for LentiCRISPR V2 plasmid and SapI or BbsI sites for the pKLV2.2 plasmids as previously described (Sanjana et al., 2014; Tzelepis et al., 2016). Single gRNAs targeting the γ globin promoter region were cloned into pL.CRISPR.EFS.GFP (Addgene plasmid #57818) (gift from Benjamin Ebert) at the BsmBI site (Heckl et al., 2014).
Generation of dgRNA expression lentiviral vectors
To generate pKLV2.2 dgRNA expression lentiviral vectors, the dgRNAs were cloned in the pKLV2.2-h7SKgRNA5(SapI)-hU6gRNA5(BbsI)-PGKpuroBFP-W (Addgene plasmid # 72666; a gift from Kosuke Yusa) (Tzelepis et al., 2016). The plasmid was digested with SapI first for cloning the first gRNA and then digested with BbsI for cloning the second gRNA. The oligos for gRNA1 and gRNA2 were synthesized with specific 5′ overhangs for cloning at the SapI and BbsI sites of the plasmid (Tzelepis et al., 2016).
Preparation of lentiviruses
For the generation of lentiviruses, the gRNA and Cas9 expression plasmids were co-transfected with pMD2.G envelope plasmid (Addgene 12259) and psPAX2 packaging plasmid (Addgene 12260) (gifts from Didier Trono). The virus supernatants were collected after 48, 60 and 72 h, pooled together, and concentrated 100 times using the Lenti-X Concentrator (Takara Bio), and stored at −80°C as aliquots. Approximately 1–2 × 105 HUDEP2 cells/mL were transduced with the lentiviruses in the presence of 8 μg/mL polybrene (Sigma-Aldrich) by spinfection at 2,250 rpm for 1.5 h at room temperature.
Generation of Cas9 expressing cell lines
Cas9-HUDEP2 cells were generated by transducing HUDEP2 cells with pLentiCas9-T2A-BFP lentiviral vector (Addgene 78547, a gift from Roderic Guigo and Rory Johnson). The transduced cells that express blue fluorescence protein (BFP+ cells) were flow-sorted and further selected with 10 μg/mL blasticidin to ensure uniform Cas9 expression in the cells. Cas9-THP1, Cas9-K562, and Cas9-EM2 cell lines were generated by transducing THP1, K562, and EM2 cells, respectively, with pCLIP-Cas9-Nuclease-EFS-Blasticidin (Transomic, Cat No. V085) lentiviral vector, and the transduced cells were selected with 2 μg/mL of blasticidin from day 5 after transduction for 10 days. AAVS1-TetOn-Cas9-HUDEP2 cells were generated by transfecting HUDEP2 cells with 3 plasmids, pAAVS1-PDi-CRISPRn (Addgene ID 73500, gift from Bruce Conklin Bruce), pZT-AAVS1-R1 (Addgene ID 52638, gift from Mahendra Rao and Jizhong Zou) and pZT-AAVS1-L1 (Addgene ID 52637, gift from Mahendra Rao and Jizhong Zou) as described earlier (Luo et al., 2014; Thamodaran et al., 2022). The cells with the successful integration of the Tet-On-Cas9 cassette at the AAVS1 site were selected by puromycin selection. The puromycin-selected cells were further subjected to single-cell sorting, and the single-cell clones were propagated in the presence of puromycin to develop clonal cell lines. One of the clones exhibiting homogeneous Cas9 expression referred to as AAVS1-TetOn-Cas9-HUDEP2 cells, was selected for genome editing experiments.
Transduction of gRNA lentiviral vectors in cell lines
Concentrated viruses generated with pLentiCRISPRV2 and pKLV2.2 lentiviral vectors with cloned sgRNAs were transduced into 2 × 105 HUDEP2 and Cas9-HUDEP2 cells/mL, respectively, in the presence of 8 μg/mL polybrene by spinfection at 2,250 rpm for 1.5 h at room temperature. After 5 days, puromycin selection (for pLentiCRISPRV2) or flow sorting of BFP+ cells (for pKLV2.2) was performed to select the transduced cells. Cas9-HUDEP2 cells were transduced with LVMUsg2PG dual-gRNA lentiviral vectors and the cells that expressed green fluorescence protein (GFP+ cells) were flow-sorted and cultured for 3–4 weeks to estimate the mutations in the targeted regions. Similarly, AAVS1-Tet-On-Cas9-HUDEP2 cells were transduced with pKLV2.2 dgRNA viruses, and the BFP+ transduced cells were flow-sorted 5–6 days after transduction to determine the formation of deletions in the targeted regions. AAVS1-Tet-On-Cas9 iPSCs (Thamodaran et al., 2022) were cultured in mTeESR medium (Stem Cell Technologies) in 12 well plates. When the cells reached ∼75% confluency, they were transduced with pKLV2.2 dgRNA lentiviruses. After 5–6 days of transduction, the BFP+ transduced cells were flow-sorted and cultured in the presence and absence of Dox. DNA samples were collected on day 3 and day 6 after Dox supplementation for mutation analysis. Similarly, the Cas9-THP1, Cas9-K562, and Cas9-EM2 cell lines were transduced with pKLV2.2 dgRNA viruses in the presence of 8 μg/mL polybrene by spinfection at 2,250 rpm for 1.5 h at room temperature. After 6 days, BFP+ cells were flow sorted and further cultured to estimate mutations caused by the dgRNAs at the targeted genomic region.
CRISPR/Cas9 mutation analysis by T7EN1 assay and DECODR
Genomic DNA was extracted from the transduced cells using Gentra Puregene High Molecular Weight DNA Extraction Kit (Qiagen). DNA fragments spanning the target sites of CRISPR/cas9 were amplified using EmeraldAmp GT PCR Master Mix (Takara Bio), and PCR products were purified using a PCR product purification Kit (Machery Nagel). For the T7EN1 assay, 200–500 ng of purified PCR products in NEBuffer 2 (New England Biolabs) were denatured at 95°C for 5 min, then annealed at 95°C–85°C at 2°C/s, followed by 85°C–25°C at −0.1°C/s in a thermocycler (Y. Niu et al., 2014). Subsequently, 10 units of T7EN1 enzyme (New England Biolabs) were added to the annealed PCR products, and the reaction mix was incubated at 37°C for 15 min. The resulting products were then analyzed on a 2% agarose gel to estimate the cleavage rate. For analyzing the CRISPR edits (insertions and deletions), the PCR-amplified products from the targeted regions were subjected to Sanger sequencing. The amplified products were purified and sequenced using BrilliantDye™ v3.1 Terminator Cycle Sequencing Kit (Nimagen) on the 3,500 Genetic Analyzer (Thermo Fisher Scientific). The sequencing results were aligned to the normal sequences using SnapGene (Dotmatics) to estimate the indels or short deletions generated by the dgRNAs. The editing efficiency was quantitated using the DECODR tool (Deconvolution of Complex DNA Repair) (Bloh et al., 2021) or ICE (Inference of CRISPR Edits) (Conant et al., 2022) by comparing the sequencing chromatogram trace files of the edited and the unedited samples.
Deletion detection by capillary electrophoresis (CE)
The first round of PCR was carried out with target genomic locus-specific primers (Supplementary Table S4) that flank the gRNA binding regions. The forward primers contained a 5′-CACTCTTTCCCTACACGACGCTCTTCCGATCT-3′ tag sequence. The second round of PCRs was performed using a 5′ FAM labeled forward primer 5′-TTTCCCTACACGACGCTCTT-3′ and the same reverse primer used in the first round of PCR. The second round PCR products (∼2 µL) were denatured in deionized formaldehyde and analyzed by capillary electrophoresis using an ABI 3500 Genetic Analyzer (Thermo Fischer Scientific). The peak sizes and peak heights of the amplified products were determined using the Peak Scanner software (Thermo Fisher Scientific).
Next-generation sequencing to quantitate dgRNA-mediated mutations
Genomic DNA was extracted from transduced BFP+ sorted cells and subjected to a two-step PCR process for mutation quantitation via NGS. Both forward and reverse primers for the first and second rounds of PCR were designed to amplify the target genomic regions, incorporating Illumina adapter sequences. In the first round of PCR, primer sequences included the miRNA genomic site-binding sequence coupled with part of the NGS primer binding site (Forward: 5′-TACACGACGCTCTTCCGATCT + miRNA genomic locus-specific forward sequence; Reverse: 5′-AGACGTGTGCTCTTCCGATCT + miRNA locus-specific reverse sequence). The second round of PCR utilized primers composed of the flow cell binding region (5′-AATGATACGGCGACCACCGAGATCTACAC) + a unique 6 bp index (NNNNNN)+ and the NGS primer binding site. After secondary PCR, the PCR products with different indices were pooled and purified using a PCR product purification kit (Machery Nagel) and quantitated using Nanodrop and used for sequencing. The prepared libraries were sequenced on the Illumina HiSeq X platform, generating raw data with a read length of 2 bp × 150 bp. Sequencing data was processed to produce Fastq files, which were analyzed using Cas-Analyzer (https://www.rgenome.net/cas-analyzer) (Park et al., 2017).
Real-time PCR analysis of miRNA expression
Total RNA was extracted from 0.3 to 0.5 million cells using the Nucleozol RNA extraction reagent (Takara Bio). Subsequently, 1 μg RNA was reverse transcribed using the Mir-X™ miRNA First-Strand Synthesis Kit (Takara Bio) as per the manufacturer’s protocol. The quantitative RT-PCR reaction was set up with Go Taq qPCR Master Mix (Promega) using a miRNA-specific forward primer and a common reverse primer provided in the Mir-X™ miRNA First Strand Synthesis Kit (Takara Bio). The miRNA-specific forward primers were designed using miRprimer2 (Busk, 2014). The assay was carried out on QuantStudio 12K Flex Real-Time PCR equipment and software (Thermo Fisher Scientific). Melt/dissociation curves were analyzed before the quantification of miRNAs.
Western blot
Whole-cell lysates from HUDEP2 and AAVS1-Tet-on-Cas9-HUDEP2 cells were prepared using radioimmunoprecipitation assay (RIPA) buffer (150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8), supplemented with Halt Protease Inhibitor Cocktail (Thermo Scientific) and phenylmethanesulfonyl fluoride (PMSF) (Sigma Aldrich). Approximately 15 μg of the lysate was loaded on a 7% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by western blot using primary antibodies, anti-Cas 9 monoclonal antibody (1:5000 dilution) (Cell Signaling Technology), anti-human FANCA (Santa Cruz Biotechnology), anti-human CBR1 (Abclonal), anti-human CTSG (Cell Signaling Technology) and anti-human Actin monoclonal antibody (1: 5000 dilution) (BD Pharmingen) and secondary antibodies, anti-mouse IgG HRP (Cell Signaling Technologies) and anti-rabbit IgG HRP (Invitrogen Corporation Camarillo). The chemiluminescence detection of the protein was performed using the Westar Supernova (Cyanagen) and FluorChemE gel documentation system (Protein Simple).
Results
Evaluation of CRISPR/Cas9 sgRNA efficiency in targeting miRNA expression
Previous studies have demonstrated the efficient knockdown of miRNAs through the use of sgRNAs that target crucial functional sequences of miRNAs, such as seed sequences and biogenesis processing sites (Jiang et al., 2014; Chang et al., 2016; Wallace et al., 2016; Huo et al., 2017; Z; Zhang et al., 2018). However, the feasibility of designing efficient sgRNAs within the constrained design space (under 30 nucleotides), which includes these crucial sequences has not been thoroughly investigated for a large number of miRNAs. We analyzed sgRNAs capable of inducing indels at the biogenesis sites and seed sequences of 30 miRNAs (Supplementary Table S1).
The miRNA biogenesis process involves Drosha cleaving at the basal stem junction and Dicer executing cleavage near the loop of the miRNA hairpin structure. Considering the potential variability in the positions of the mutations induced by gRNAs, we included all gRNAs targeting a range of +2 to −2 nucleotides relative to the terminal bases of the 5p and 3p arms (Figure 1A) to encompass all potential gRNAs that could impact miRNA biogenesis. Our analysis identified sgRNAs with high off-target scores (less off-target effects) and high on-target scores (Doench et al., 2016) targeting the 5′ or 3′ Drosha or Dicer sites in 53% (16/30) of the miRNA genomic regions. For seed sequences, effective sgRNAs were found for 30% (18/60) of the miRNAs, taking into account both the 5p and 3p variants from each miRNA genomic region (Figure 1A) (Supplementary Table S1).
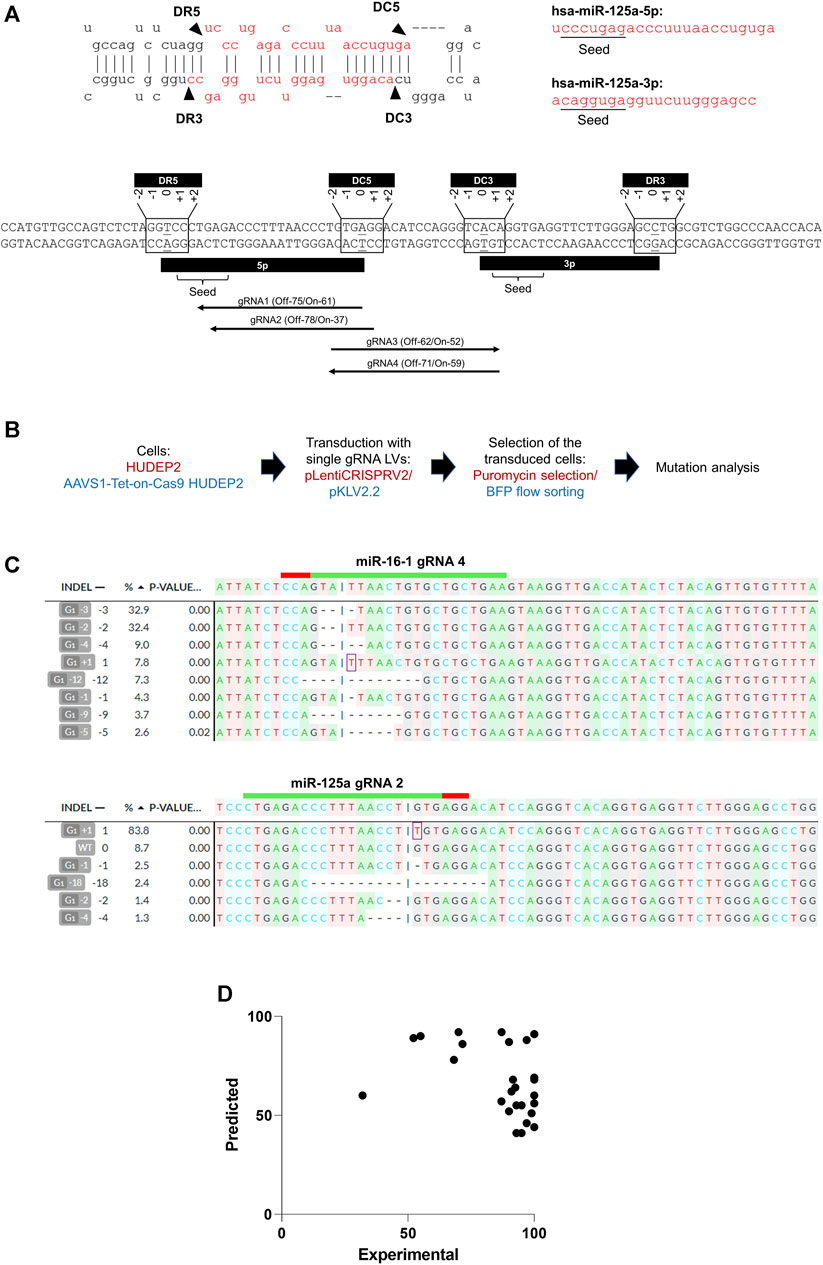
Figure 1. CRISPR/Cas9 editing of miRNA genomic regions using sgRNAs. (A) (Upper panel) Secondary structure of a representative miRNA (miR-125a), displaying its biogenesis sites and seed sequences of the 5p and 3p transcripts. DR3 and DR5 represent the 3′ and 5′ Drosha sites, respectively, and DC3 and DC5 represent the 3′ and 5′ of the Dicer sites, respectively. (Lower panel) Genomic regions of the miR-125a highlighting the seed sequences and biogenesis processing sites. The biogenesis sites encompass a 5-base region (−2 to +2) at the 5′ and 3′ ends of the coding regions of the 5p and 3p miRNA transcripts. High off-target score gRNAs with low off-target effects at the seed sequences (gRNA1 and gRNA 2) and the biogenesis sites (gRNA3 targeting DC3 and gRNA4 targeting DC5) are shown. The predicted off-target (“Off”) and on-target (“On”) scores of the gRNAs are indicated. (B) Overview of genome editing in HUDEP2 cells using lentivirally expressed sgRNAs. HUDEP2 cells were transduced with the LentiCRISPRV2 lentiviral vector expressing Cas9 and cloned sgRNAs. AAVS1-Tet-On-Cas9 HUDEP2 cells were transduced with pKLV2.2 LV harboring cloned sgRNAs. The transduced cells were selected using puromycin selection (plentiCRISPRV2) or BFP+ flow sorting (pKLV2.2) before mutation analysis. (C) Representative results from DECODR analysis of mutations created in two representative miRNA genomic regions (miR-16-1 and miR-125a) by sgRNAs. (D) Graph representing the lack of correlation between the predicted and experimental on-target efficiencies of sgRNAs.
The efficacy of gRNAs in inducing specific insertions and deletions (indels) at target sites is largely determined by their on-target editing efficiencies (Xiang et al., 2021). A number of computational tools have been developed for predicting gRNAs with optimal on-target efficiencies (Anthon et al., 2022; Konstantakos et al., 2022). We selected 1 or 2 gRNAs with high off-target scores (score >50) (i.e., low off-target effects) (Hsu et al., 2013), and varying on-target efficiency scores (range: 41–92) (Doench et al., 2016) (Supplementary Table S2) for cloning into lentiviral vectors (Figure 1B). For experiments using the pKLV2.2 lentiviral vector, which does not express Cas9, we generated AAVS1-Tet-On-Cas9 HUDEP2 cells by integrating a tetracycline/doxycycline (Dox)-inducible (Tet-On) Cas9 cassette at the AAVS1 safe harbor site, which is known for sustained and consistent transgene expression (J. R. Smith et al., 2008). A single-cell clone exhibiting robust Cas9 expression post-Dox treatment was expanded to establish a cell line for genome editing (Supplementary Figure S1). Subsequent genome editing experiments conducted with this cell line demonstrated 100% editing efficiency with multiple sgRNAs (Supplementary Table S2) and dgRNAs (Figure 3), validating the homogeneous Cas9 expression facilitating efficient genome editing.
Post-selection (puromycin or flow sorting) of the sgRNA-transduced cells, we conducted the T7EN1 cleavage assay to detect indels at the targeted regions (Supplementary Figure S2) and DECODR analysis (Bloh et al., 2021) to quantitate the mutations (Figure 1C). Although 30% of the guide RNAs (6 out of 20) had lower predicted on-target efficiency scores (<50) (Haeussler et al., 2016), they demonstrated high experimental editing efficiencies, with an average efficiency of 91.93% ± 16.033% (Supplementary Table S2). To further investigate, we transduced HUDEP2 cells with 12 sgRNAs, cloned into the pL-CRISPR.EFS.GFP gRNA and Cas9 expression vector, to target the duplicated human γ globin promoter sequences and compared the predicted and experimental on-target efficiencies (Supplementary Table S2). The collective data from 32 sgRNAs demonstrated a mean editing efficiency of 92 ± 16.03 (ranging from 32% to 100%) although their on-target efficiencies ranged from 41 to 92 (Supplementary Table S2) (Figure 1D). Our findings suggest that a significant number of miRNAs may not be amenable to editing at the biogenesis sites and seed sequences with sgRNAs with high on-target and off-target scores. Additionally, we observed a lack of correlation between the predicted and actual on-target efficiencies of lentivirally expressed gRNAs (Figure 1D), suggesting the possibility of including gRNAs with low on-target efficiencies when employing lentiviral vectors for gRNA expression.
Optimization of efficient dgRNA-mediated deletions of miRNAs in HUDEP2 cells
To simplify the generation of dgRNA expression vectors (Nath et al., 2021) and prevent intra-plasmid recombination (Vidigal and Ventura, 2015), we employed a single lentiviral plasmid featuring two distinct RNA polymerase III promoters and unique restriction enzyme sites for sequential cloning of gRNAs (Supplementary Figure S3A). We assessed the gene editing efficiencies of three gRNA expression promoters [human U6 (hU6), mouse U6 (mU6), and human 7SK (h7SK)] by cloning two gRNAs with varied on-target efficiency scores (scores of 6 and 73) under each promoter (Supplementary Figure S3B). Editing efficiency analysis in transduced HUDEP2 cells showed no significant differences among the promoters (Supplementary Figures S4A, B). Consequently, for our experiments to create short genomic deletions, we used the pKLV2.2 vector with h7SK and hU6 promoters for the expression of the dgRNAs.
We designed 33 dgRNA pairs to target 23 miRNA genomic regions. This included 10 miRNAs targeted by two dgRNA pairs and 13 miRNAs targeted by a single pair of dgRNAs (Supplementary Table S3). The selection of these miRNAs was based on their differential expression during erythroid differentiation (Supplementary Figure S5). The designed gRNA pairs had low off-target effects, while their on-target efficiency scores varied (CRISPOR Doench scores range: 31–78) (Doench et al., 2016). They were designed to flank 5p or 3p arms of the targeted miRNAs (Supplementary Figure S6) and to bind either to the same or opposite DNA strands. Their PAM offsets (the distance between the PAMs of the dgRNAs) ranged from 36 bp to 79 bp and gRNA offsets (the distance between the dgRNAs) ranged from 2 bp to 101 bp (Supplementary Table S3). Two pairs contained overlapping gRNAs (gRNAs 3 and 4 for miR-15 and gRNAs 1 and 2 for miR-223) (Supplementary Table S3) (Supplementary Figure S7A).
After AAVS1-Tet-On-Cas9 HUDEP2 cells were transduced with individual dgRNA lentiviruses, BFP+ cells were flow-sorted and then analyzed for mutations at the targeted regions. Agarose gel electrophoresis (AGE) detected deletion formation for 30 out of 33 dgRNA pairs, with complete deletions observed only in 5 pairs (Supplementary Figure S8). Capillary electrophoresis of fluorescently labeled PCR products (FL-PCR-CE) quantified deletion formation, revealing a higher percentage of deletions (mean ± SD: 87.5578 ± 24.66; range: 0%–100%) compared to AGE, with 27 pairs achieving ≥90% deletion efficiency (mean ± SD: 96.90 ± 2.705), out of which 7 pairs demonstrating 100% deletion efficiency (Figure 2A) (Figure 2B) (Supplementary Table S3). Sanger sequencing followed by DECODR analysis (Bloh et al., 2021) of the targeted regions of 25 samples showed a strong correlation with the CE results (Figure 2C) (Supplementary Table S3). We also performed next-generation sequencing of the amplified products from 10 miRNA genomic regions targeted by dgRNAs, and the data confirmed high rate of deletion formation in the targeted miRNAs (Supplementary Figure S9). The overlapping gRNA pairs predominantly resulted in indels rather than deletions (Supplementary Figure S7B). Excluding these pairs, the overall percentage of deletions was 93.206 ± 11.17 (Supplementary Table S3). Real-time PCR analysis of the 17 targeted miRNAs, which had significant expression levels in HUDEP2 cells, revealed reductions in their expression levels (mean: 94.75 ± 8.67; range: 73%–100%) (Figure 2D).
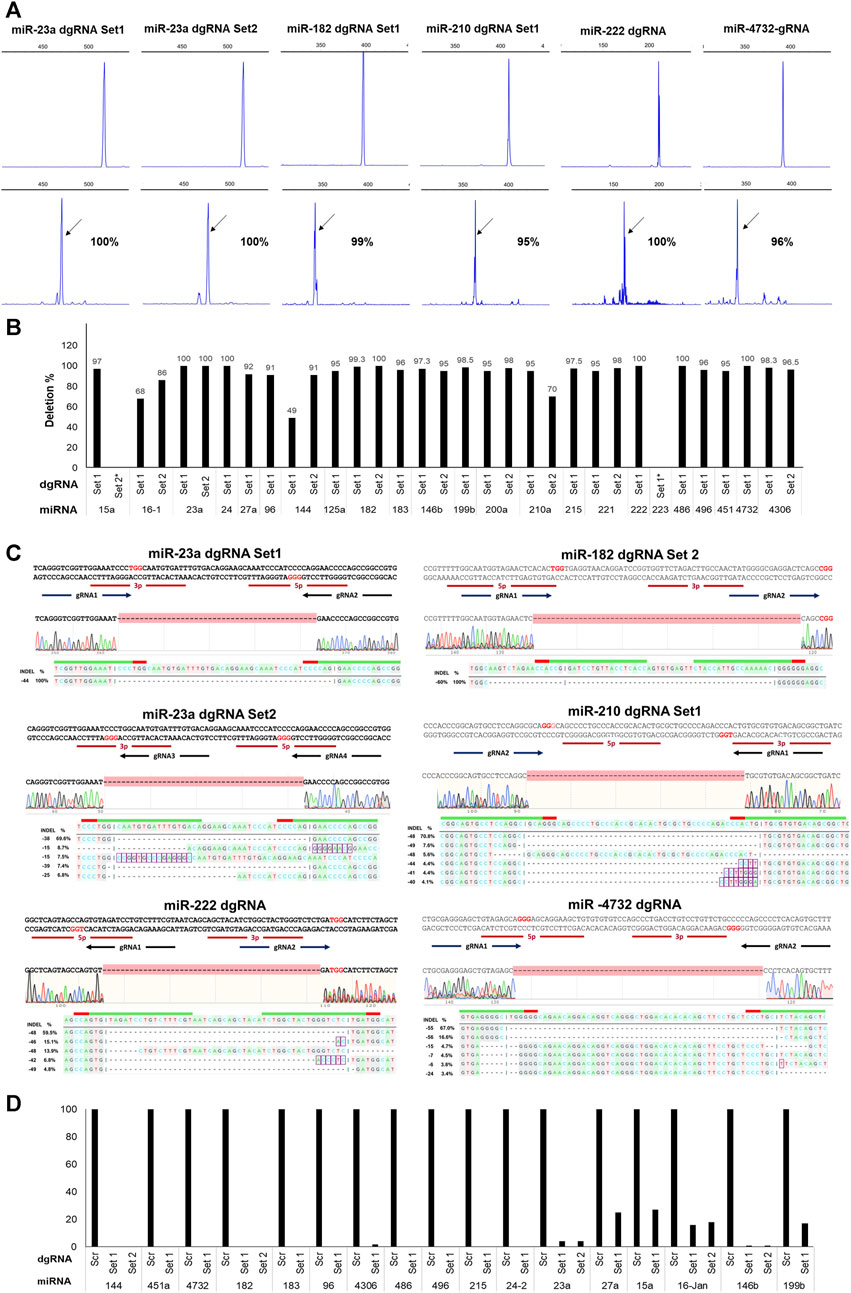
Figure 2. Efficient deletion formation in HUDEP2 cells using dgRNA expressing lentiviral vectors. (A) Representative FL-PCR-CE results of 6 miRNA genomic regions targeted by dgRNAs. The upper panel displays fragments formed from the unedited cells and the lower panel shows fragments from the respective edited cells. The arrows indicate specific fragments resulting from deletions. The corresponding percentages of deletions are also shown. (B) Graph depicting the deletion percentages detected by FL-PCR-CE in different miRNA regions (C) Sanger sequencing alignment and DECODR mutation analysis of the PCR products showing deletions in the targeted regions. (D) Real-time PCR analysis results showing a significant reduction in expression levels of targeted miRNAs in the knockout cells.
Despite low BFP+ cell percentages (ranging from 9% to 30%) in 13 dgRNA transductions, we observed high deletion formation in the flow-sorted BFP+ cells (mean ± SD: 99.08 ± 2.055; range: 93%–100%), demonstrating the efficacy of short genomic deletion formation even with low lentiviral integration of dgRNAs. Given the rapid differentiation of certain human primary cells, such as hematopoietic progenitors, in culture, quickly achieving miRNA deletions is crucial for studying the phenotypic effects of miRNA knockouts in undifferentiated cells. Therefore, we assessed the deletion formation ability of 10 dgRNA pairs that showed very high deletion percentages (mean ± SD: 97 ± 2.738; range: 93%–100%) 5 days post-transduction and observed similar high deletion percentages (mean ± SD: 99.3 ± 1.417; range: 96%–100%). Efficient deletion formation of individual miRNAs was obtained gRNA offset of up to 71 bp. Overall, our findings highlight the effectiveness of dgRNA design, the selection of lentiviral vectors for their expression, and the homogeneous expression of Cas9 for rapid and efficient generation of short genomic deletions.
Evaluating the efficacy of lentiviral dgRNA-mediated miRNA deletions in iPSCs
Induced pluripotent stem cells (iPSCs), reprogrammed from somatic cells (Takahashi et al., 2007), possess the unique capability to remain undifferentiated and can be directed to differentiate into various cell lineages, including hematopoietic progenitors (Sturgeon et al., 2014; Netsrithong et al., 2020; Dannenmann and Skokowa, 2022). Inducing miRNA deletions in undifferentiated iPSCs and during hematopoietic differentiation provides an opportunity to explore the roles of specific miRNAs in different hematopoietic cell lineages, such as erythroid cells. We aimed to assess the efficacy of lentiviral dgRNA-mediated miRNA deletions in the AAVS1-Tet-On-Cas9 iPSC line, where the Dox-inducible Cas9 cassette is integrated at the AAVS1 site (Thamodaran et al., 2022). We focused on deleting 7 miRNAs from the miR-144/451, miR-183/96/182, and miR-23a/27a/24-2 clusters. Notably, the miRNAs from the miR-144/451 and miR-183/96/182 clusters are the most upregulated miRNAs during human erythropoiesis (Nath et al., 2021). The members of the miR-144/451 cluster are involved in erythroid maturation, and those of miR-23a/27a/24-2 clusters are associated with the regulation of various aspects of hematopoietic stem cell differentiation and lineage commitment (Kurkewich et al., 2017) and lineage development (Cho et al., 2016).
The AAVS1-Tet-On-Cas9 iPSCs were transduced with pLKV2.2 lentiviruses expressing dgRNAs, and the flow-sorted BFP+ cells were cultured with Dox. Deletion formation was analyzed at 3 and 5 days after Dox supplementation by AGE (Supplementary Figure S10) and FL-PCR-CE (Figure 3). FL-PCR-CE detected higher percentages of deletions than AGE, consistent with observations in HUDEP2 cells. On day 3, deletion percentages ranged from 62% to 96% (mean ± SD: 76.29 ± 11.30) as determined by CE, and increased to 81%–100% (mean ± SD: 92.57 ± 5.78) (Figure 3) by day 5. Notably, significant cell death (>90% cells) was observed in cultures with dgRNAs targeting miR27a and miR23a, suggesting the critical role of these miRNAs in the survival of pluripotent stem cells. These results emphasize the efficacy of lentiviral dgRNA-mediated miRNA deletion in iPSCs, showing great potential for investigating the specific roles of miRNAs in iPSC maintenance and differentiation.
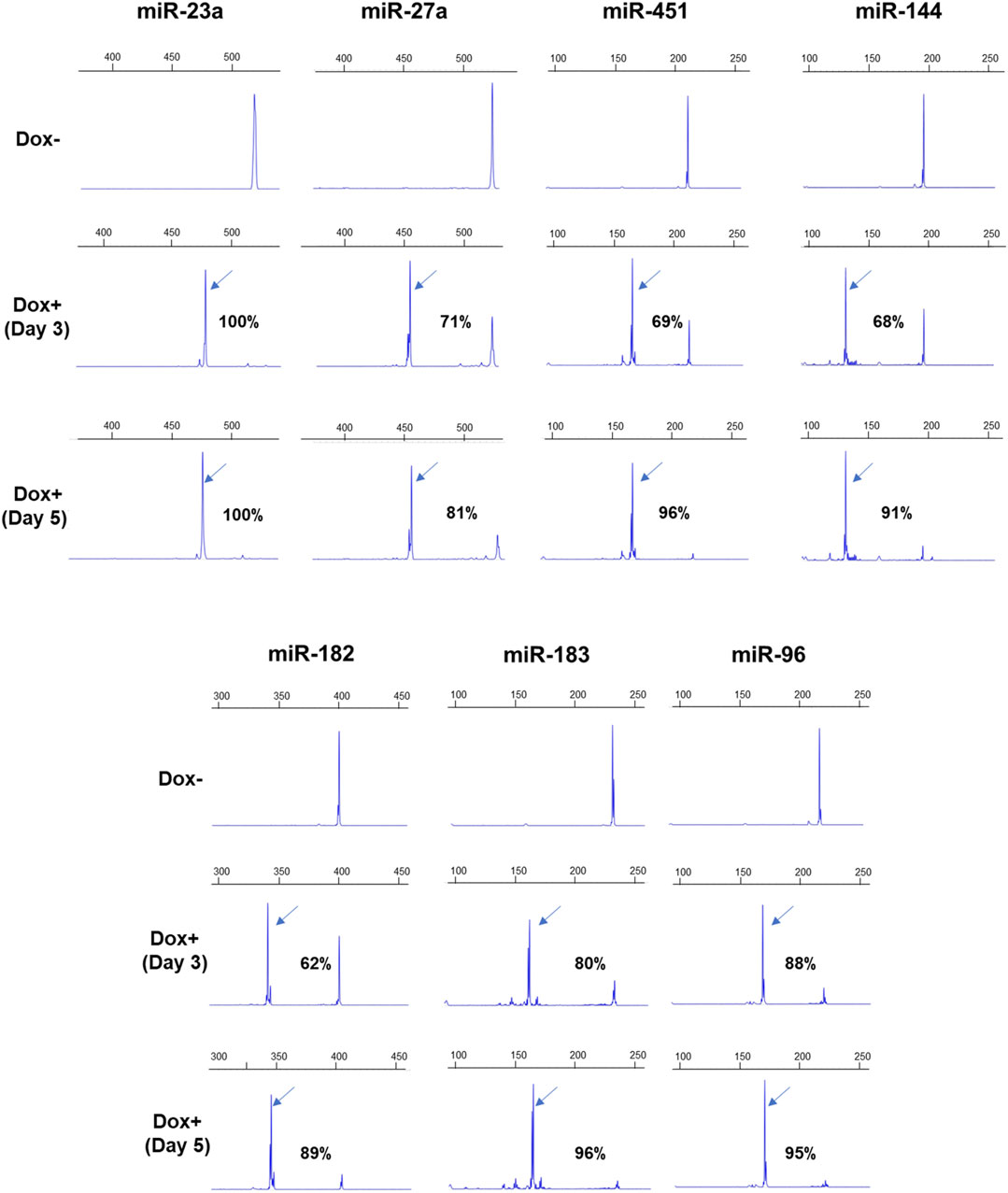
Figure 3. FL-PCR-CE results of 7 miRNA genomic regions targeted in iPSCs by dgRNAs. The upper panel displays the electropherograms of PCR fragments formed from the targeted regions in the unedited iPSCs, cultured without Dox (-Dox). The middle and lower panels show electropherograms of fragments derived from edited regions after 3 (Day 3) and 5 (Day 5) days of Dox supplementation, respectively. The arrows indicate specific fragments resulting from deletions. The corresponding percentages of deletions are also shown.
Analyzing the impact of miRNA deletions on erythroid differentiation in HUDEP2 cells
To assess the effectiveness of our miRNA deletion strategy in elucidating the role of miRNAs in erythroid differentiation, we utilized HUDEP2 cells, known for their differentiation capacity into late-stage erythroid cells. Among the tested miRNAs for deletion formation, miR-144 and miR-451 are known to significantly influence erythroid differentiation (Rasmussen et al., 2010; Kim et al., 2015). AAVS1-Tet-On-Cas9 HUDEP2 cells transduced with dgRNAs targeting miR-144, miR-182, miR-183, miR-451, miR-4732, miR-96, miR-4306, miR-215, miR-486, miR-496, miR-27a, miR-23a, and miR-24-2 were used for the experiment (Figure 4A). We examined CD71 and CD235a expression, two erythroid surface markers, to understand miRNA knockout impacts on erythroid differentiation kinetics. Undifferentiated HUDEP2 cells co-express CD71 and CD235a surface markers, but during differentiation CD71 expression undergoes a decline, whereas CD235a expression remains stable throughout differentiation. Defects in erythroid differentiation can be identified by observing a slower or no decrease in CD71 expression, indicating potential disruptions in the maturation of erythroid cells.
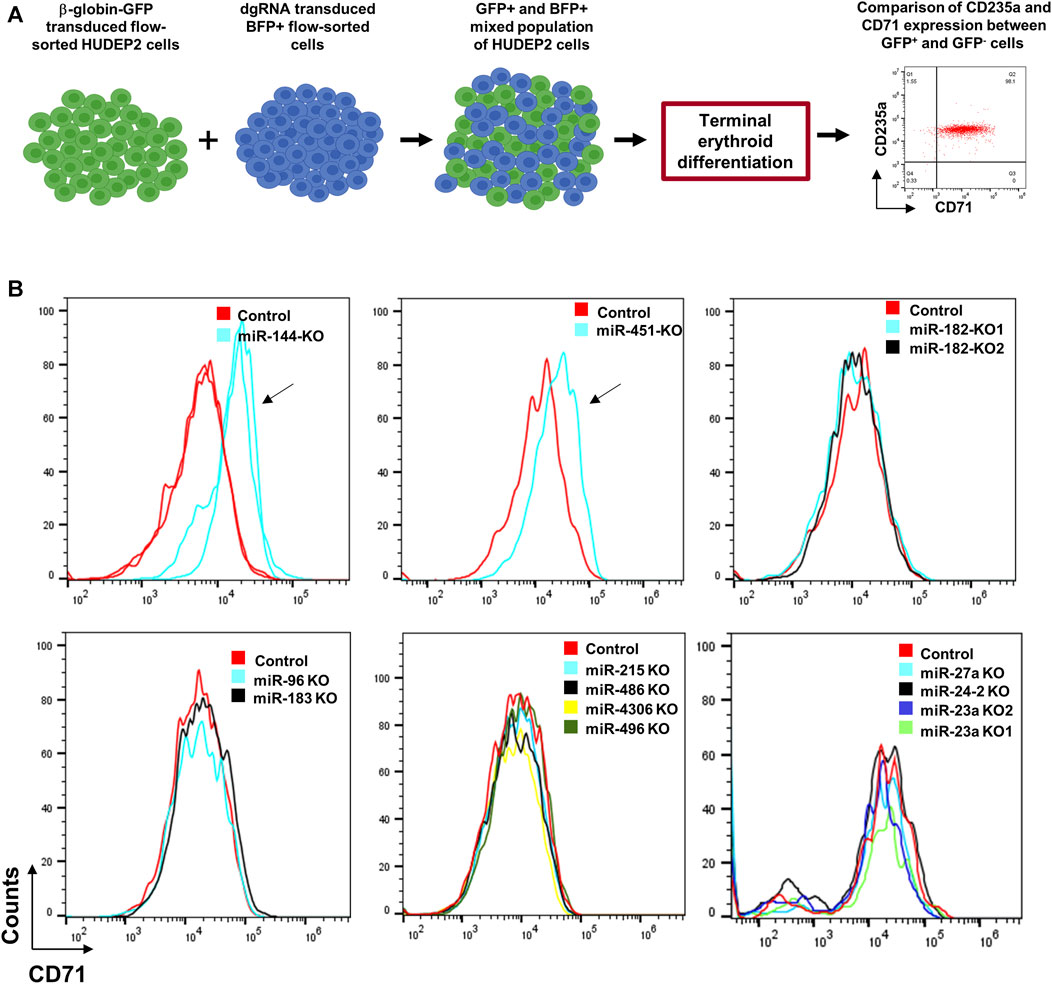
Figure 4. Differentiation of miRNA-knocked out HUDEP2 cells. (A) Experimental setup to study the effect of miRNA deletions on differentiation of HUDEP2 cells. The dgRNA transduced BFP+ HUDEP2 cells were mixed with HUDEP2 cells transduced with β-globin promoter-GFP lentiviral vector. The mixed population was then subjected to terminal differentiation and CD71 and CD235a expressions were compared in the GFP- and GFP+ cells. (B) Flow cytometry data showing CD71 expression in the terminally differentiated unedited and edited cells. The miRNA knockouts included miR-182, miR-183, miR-96, miR-144, miR-451a, miR-4732, miR-4306, miR-215, miR-486, miR-496, miR-23a, miR-24-2 and miR-27a. The arrows are used to indicate the difference in differentiation kinetics in miR-144 and miR-451 knockout HUDEP2 cells compared to the control cells. KO1 and KO2 refer to knockouts using dgRNA set 1 and set 2, respectively.
Due to the gradual decline in BFP expression during differentiation, likely resulting from transgene silencing by the PGK (phosphoglycerate kinase 1) promoter, analyzing differentiation kinetics between BFP+ and BFP- cells in a mixed population proved challenging. To address this issue, we combined the dgRNA-transduced cells with HUDEP2 cells transduced with a lentiviral vector, which expresses GFP under the β-globin promoter (Figure 4A). Notably, the β-globin promoter is renowned for its strong transcriptional activity during erythroid differentiation (Bagchi et al., 2022). Upon subjecting the mixed cell population to terminal differentiation, we analyzed the expression of CD71 and CD235a in both GFP+ (β-globin promoter-transduced) and GFP- (dgRNA-transduced) cells. Knockout of miR-144 and miR-451 led to arrested differentiation (Figure 4B), whereas other miRNA knockouts did not significantly alter differentiation kinetics compared to control cells, despite their upregulation during erythroid differentiation (Figure 4B).
Efficacy of lentiviral dgRNAs in inducing deletions across diverse genomic regions in multiple cell types
Following the successful induction of deletions in the miRNA genomic regions, we assessed the capacity of lentivirally transduced dgRNAs to create deletions at other genomic regions, including both protein-coding genes and intergenic regions (Figures 5A, B). We first developed THP1, K562, and EM2 cell lines with stable Cas9 expression through lentiviral transduction. We targeted LEF1, CTSG, and CBR1 genes in THP1-Cas9 cells, miR-30b and CSTG in EM2-Cas9 cells, and miR-30b in K562-Cas9 cells (Figure 5A). Additionally, we targeted the protein-coding genes FANCA, FANCB, FANCC, and FANCD1 in AAVS1-Tet-On-Cas9 iPSCs (Figure 5B). We also targeted the DNase I hypersensitive sites HS1, HS2, HS3, and HS4, which constitute transcriptional enhancer elements in the β-globin cluster in AAVS1-Tet-On-HUDEP2 cells. The dgRNA-transduced cells exhibited 76%–100% deletion formation (mean + SD = 95.4 + 7.5) in the target regions within a week across all cell types (Figure 5A). Immunoblot analysis of four targeted proteins revealed a notable decrease in expression, with three exhibiting a complete absence of expression (Figure 5C). In summary, our research effectively showcased the efficiency of lentivirally transduced dgRNAs in producing deletions, extending beyond miRNA genomic regions to include both protein-coding genes and intergenic regions. These findings underscore the potential of our lentiviral approach for efficient genome editing across diverse genomic and cellular contexts.
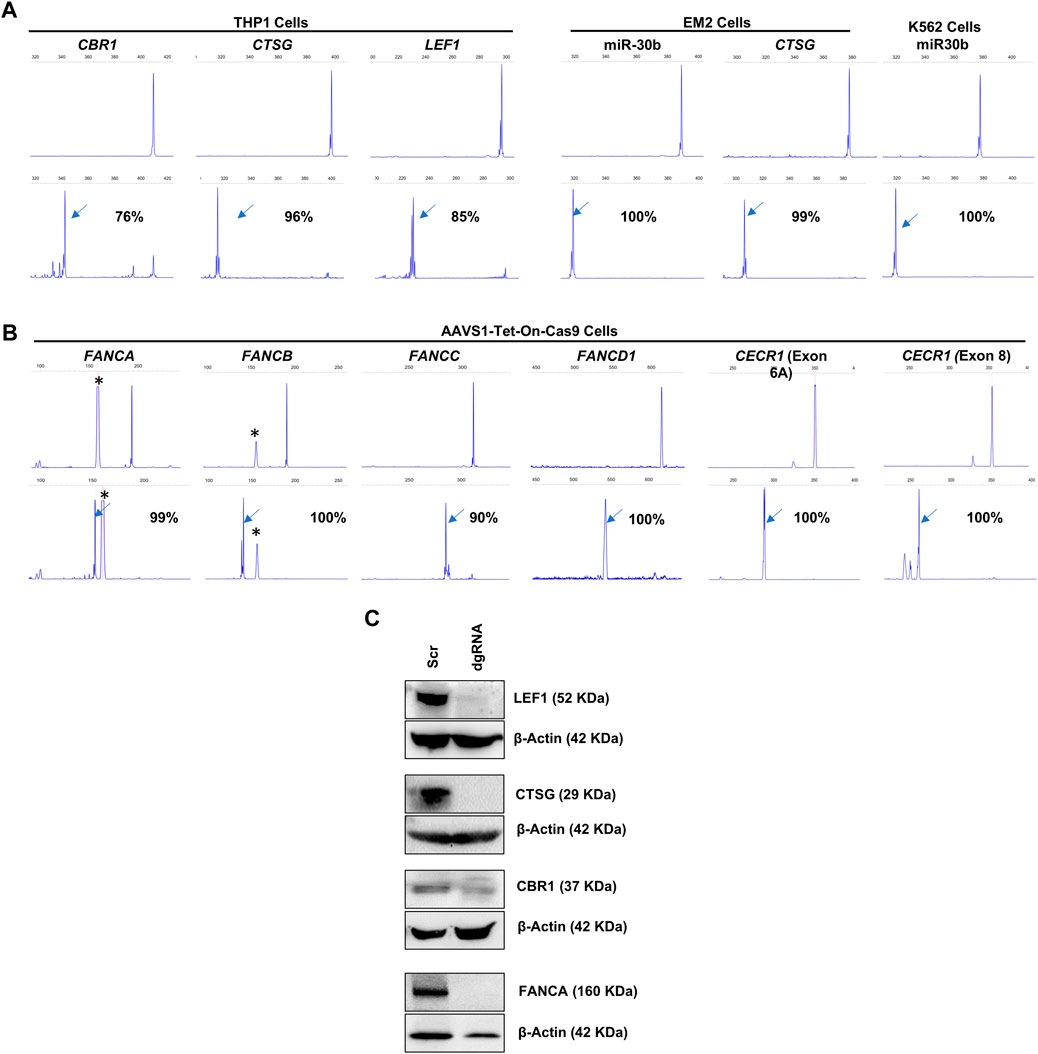
Figure 5. FL-PCR-CE analysis of dgRNA mediated deletion formation in protein-coding genes and intergenic regions. (A) Targeting CBR1 (exon 1), CTSG (exon 1) and LEF1 (exon 4) in THP1-Cas9 cells and CTSG (exon 1) in EM2-Cas9 cells, using dgRNAs Targeting miR-30b in EM2-Cas9 and K562-Cas9 cells are also shown. (B) Targeting FANCA (exon 2), FANCB (exon 1), FANCC (exon 5), FANCD1 (exon 10) and CECR1 (Exon 6A and Exon 8) with dgRNAs in AAVS1-Tet-On-Cas9 iPSCs. The upper panels of (A,B) display fragments formed from the unedited cells and the lower panel shows fragments from the edited cells. The arrows indicate specific fragments resulting from deletions. The corresponding percentages of deletions are shown. Asterisks (*) indicate non-specific peaks present in the PCR products from both edited and unedited cells. (C) Immunoblot analysis to assess protein expression from the dgRNA-targeted genes.
Discussion
Understanding the functional roles of miRNAs is crucial for unraveling the complexities of gene regulation that govern various cellular processes. The extensive impact of miRNAs has been implicated in their dysregulation associated with various diseases, including cancer, cardiovascular disorders, and neurological conditions (Calin and Croce, 2006). For miRNA loss-of-function studies, selectively suppressing specific miRNAs, while minimizing the impact on similar off-target miRNAs, is critical. In this study, we introduce a novel approach utilizing CRISPR/Cas9 and dgRNAs for the efficient generation of miRNA deletions within a short timeframe (approximately 5 days). This method achieves highly efficient knockdown (>90%) of targeted miRNAs with single-copy lentiviral integration of dgRNAs in human cell lines.
Traditional miRNA inhibitory approaches, such as antisense oligonucleotides (Hutvágner et al., 2004; Krützfeldt et al., 2006) and sponges/decoys (Ebert et al., 2007; Ebert and Sharp, 2010), effectively induce miRNA loss of function but lack specificity when targeting miRNAs with similar seed sequences. Conversely, sgRNA-based CRISPR/Cas9 systems offer greater specificity but are limited by a design space of about 25 bases, encompassing the miRNA biogenesis sites and seed sequences. Our CRISPR/Cas9 method with dgRNAs shows exceptional efficiency in miRNA knockout and allows more flexible gRNA design, enabling targeting within the entire miRNA region, beyond just the biogenesis processing sites or seed sequences. Targeting either the 3p or 5p genomic region of the miRNAs, which form their double-stranded stem structure, impacts the expression of the other region, thus broadening the design scope for specific miRNA modulation. The efficacy of this method is partly due to the robustness of the lentiviral dual-gRNA approach and the use of cell lines with integrated Cas9, which ensures consistent expression of the genes with minimal transgene silencing (J. R. Smith et al., 2008). The use of Dox-inducible Cas9 expression allows for regulated and stage-specific miRNA knockout in various cell types, including undifferentiated stem cells, progenitors, and differentiated cells. This versatility provides valuable insights into miRNA roles across different cell types. While previous studies have explored dgRNAs for miRNA knockouts, they often show low efficiency, necessitating single-cell sorting to create cell lines with miRNA knockouts (Ho et al., 2015; Liu et al., 2016; Yan et al., 2019; Hong et al., 2020). Our study found that following the establishment of Cas9-expressing cell lines (approximately 1 month), the transduction of dgRNAs and generation of deletions could be achieved in under a week.
MicroRNAs (miRNAs) are pivotal in regulating transcription signals, growth factors, and epigenetic cell control (Zahnow and Baylin, 2010; Gruber and Zavolan, 2013; Pagano et al., 2013). This study employs a dgRNA-mediated approach for miRNA deletion in two human cell lines: HUDEP2, used in erythropoiesis research, and induced pluripotent stem cells (iPSCs), valuable for studying cellular differentiation and disease mechanisms. Our findings in HUDEP2 cells show that the knockout of miR-144 and miR-451a significantly alters erythroid differentiation. By efficiently knocking out the target miRNAs, we discovered that the loss of miR-27a and miR-23a expression led to pronounced cell death, indicating the importance of these miRNAs for the survival of pluripotent stem cells. Dox-inducible miRNA knockout in hematopoietic stem and progenitor cells (HSPCs) and erythroid cells will enhance our understanding of the function of these miRNAs in various cellular contexts.
In our systematic assessment of the dgRNA system, we evaluated parameters including on-target gRNA efficiencies and the orientation and distance between gRNAs, to discern their effects on deletion efficiency. We observed over 80% efficiency in deletion formation with gRNA offsets ranging from 2 to 101 bases. Interestingly, the orientation and binding position of the gRNAs, whether on the same or opposite strands, did not significantly affect the efficiency of deletion formation. While AGE was insufficient in detecting substantial deletion formation in our targets, FL-PCR-CE, analyzing single-stranded PCR products, successfully identified a high rate of deletions in most targeted miRNAs. This discrepancy is likely due to the formation of heteroduplexes between PCR products with different sequence alterations at the deletion breakpoints. Sanger sequencing of the targeted regions corroborated the presence of various deletion breakpoints, supporting the heteroduplex formation hypothesis. FL-PCR-CE proved to be a highly sensitive method for detecting short deletions caused by dgRNAs, presenting benefits of speed, cost-efficiency, and ease of use, compared to next-generation sequencing (NGS) methods. Among the 31 dgRNAs that showed deletion formation in HUDEP2 cells, 3 (10%) exhibited deletion percentages below 80% (Supplementary Table S3). For these cells, single-cell sorting might be necessary to isolate populations with higher deletion percentages. The high efficiency of genomic deletions achieved by dgRNAs was further substantiated through protein-coding sequence editing and subsequent protein expression analysis. We also enhanced the dgRNA lentiviral vectors by creating several variants. These modifications included substituting BFP with green (GFP) and red fluorescence proteins (RFP) for compatibility with different fluorescence markers or antibodies when multi-fluorescence experiments are performed (Supplementary Figure S11A), and replacing the PGK promoter with the EF1α and MND promoters to enhance fluorescence protein expression in stem cells (Supplementary Figure S11B). Additionally, we substituted short stuffer sequences with longer ones to ensure complete digestion of the backbone before gRNA cloning (Supplementary Figure S11C).
Future research will benefit from experiments aimed at determining the maximum effective distance between guide RNAs (gRNAs) for efficient miRNA deletion. This is crucial for developing protocols to simultaneously delete multiple miRNAs within a cluster. Targeting individual miRNAs in a cluster might result in limited phenotypic changes, but the collective downregulation of all members in the cluster can have significant effects, given their cooperative amplification of regulatory impacts (Dylla and Jedlicka, 2013). The dgRNA strategy proposed here can facilitate the creation of CRISPR/gRNA libraries. These libraries could be instrumental for high-throughput screening of all known miRNAs, thereby serving as a potent tool in identifying miRNAs associated with specific biological processes and elucidating their complex roles in disease pathogenesis, cellular differentiation, and other key biological events. However, before conducting such high-throughput assays, it is essential to evaluate the off-target effects of designed gRNAs. Employing advanced algorithms to select gRNAs with minimal off-target consequences (Concordet and Haeussler, 2018; Dhanjal et al., 2020; Trivedi et al., 2020; Dhanjal et al., 2020; Niu et al., 2021; Z.-R; Zhang and Jiang, 2022) is essential for maintaining the specificity of the CRISPR/Cas9 system. Additionally, the application of Cas9 variants known for reduced off-target activity, such as enhanced S. pyogenes Cas9 (eSpCas9) (Slaymaker et al., 2016) and high-fidelity SpCas9-HF1 (P. H. Smith et al., 2016), could significantly enhance the precision of CRISPR/Cas9-mediated miRNA editing. Exploring other Cas variants, like Cas12a (Ma et al., 2022) and CasX (Tsuchida et al., 2022), may offer further advancements in the disruption of miRNA genomic regions.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: The small RNA sequencing discussed in this manuscript has been deposited in NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO Series accession number GSE185685.
Author contributions
SI: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing, Visualization. KC: Data curation, Formal Analysis, Methodology, Writing–review and editing. AC: Methodology, Writing–review and editing, Data curation, Formal Analysis. AB: Formal Analysis, Methodology, Visualization, Writing–review and editing. KM: Formal Analysis, Methodology, Writing–review and editing, Data curation. SD: Data curation, Methodology, Writing–review and editing, Formal Analysis. EB: Data curation, Methodology, Writing–review and editing, Formal Analysis. SR: Formal Analysis, Methodology, Writing–review and editing. DaP: Methodology, Writing–review and editing. AN: Methodology, Writing–review and editing. DhP: Methodology, Writing–review and editing. SP: Methodology, Writing–review and editing. RR: Methodology, Writing–review and editing. BK: Methodology, Writing–review and editing. YS: Methodology, Writing–review and editing. VS: Methodology, Writing–review and editing. DB: Data curation, Methodology, Writing–review and editing, Formal Analysis. YN: Resources, Writing–review and editing. VT: Data curation, Methodology, Writing–review and editing, Formal Analysis, Investigation. PB: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing–review and editing. SV: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. KC was supported by the research fellowship from the Department of Biotechnology, Government of India. AN was supported by the research fellowship from the University Grants Commission, Government of India. The research funding for this study was funded by the Department of Biotechnology, Government of India (Grant No. BT/PR17316/MED/31/326/2015) to SV. SV is additionally funded by the Department of Biotechnology (DBT)—Wellcome Trust Alliance Senior Research Fellowship (Grant No. IA/S/17/1/503118).
Acknowledgments
The authors thank Sandhya Rani for helping us with the flow cytometry analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1295507/full#supplementary-material
References
Anthon, C., Corsi, G. I., and Gorodkin, J. (2022). CRISPRon/off: CRISPR/Cas9 on- and off-target gRNA design. Bioinformatics 38 (24), 5437–5439. doi:10.1093/bioinformatics/btac697
Aparicio-prat, E., Arnan, C., Sala, I., Bosch, N., Guigó, R., and Johnson, R. (2016). DECKO: single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genomics 16. doi:10.1186/s12864-015-2086-z
Bagchi, A., Devaraju, N., Chambayil, K., Rajendiran, V., Venkatesan, V., Sayed, N., et al. (2022). Erythroid lineage-specific lentiviral RNAi vectors suitable for molecular functional studies and therapeutic applications. Sci. Rep. 12 (1). doi:10.1038/s41598-022-13783-0
Bagchi, A., Nath, A., Thamodaran, V., Ijee, S., Palani, D., Rajendiran, V., et al. (2021). Direct generation of immortalized erythroid progenitor cell lines from peripheral blood mononuclear cells. Cells 10 (3). doi:10.3390/cells10030523
Bajan, S., and Hutvagner, G. (2020). RNA-based therapeutics: from antisense oligonucleotides to miRNAs. Cells 9 (1). doi:10.3390/cells9010137
Bassett, A. R., Azzam, G., Wheatley, L., Tibbit, C., Rajakumar, T., McGowan, S., et al. (2014). Understanding functional miRNA-target interactions in vivo by site-specific genome engineering. Nat. Commun. 5, 1–11. doi:10.1038/ncomms5640
Bayraktar, E., Bayraktar, R., Oztatlici, H., Lopez-Berestein, G., Amero, P., and Rodriguez-Aguayo, C. (2023). Targeting miRNAs and other non-coding RNAs as a therapeutic approach: an update. Non-Coding RNA 9 (2), 27. doi:10.3390/ncrna9020027
Bhattacharya, A., Ziebarth, J. D., and Cui, Y. (2012). Systematic analysis of microRNA targeting impacted by small insertions and deletions in human genome. PLoS ONE 7 (9), e46176. doi:10.1371/journal.pone.0046176
Bi, H., Fei, Q., Li, R., Liu, B., Xia, R., Char, S. N., et al. (2020). Disruption of miRNA sequences by TALENs and CRISPR/Cas9 induces varied lengths of miRNA production. Plant Biotechnol. J. 18 (7), 1526–1536. doi:10.1111/pbi.13315
Bloh, K., Kanchana, R., Bialk, P., Banas, K., Zhang, Z., Yoo, B. C., et al. (2021). Deconvolution of complex DNA repair (DECODR): establishing a novel deconvolution algorithm for comprehensive analysis of CRISPR-edited sanger sequencing data. CRISPR J. 4 (1), 120–131. doi:10.1089/crispr.2020.0022
Busk, P. K. (2014). A tool for design of primers for microRNA-specific quantitative RT-qPCR. BMC Bioinforma. 15 (1), 29–9. doi:10.1186/1471-2105-15-29
Calin, G. A., and Croce, C. M. (2006). MicroRNA signatures in human cancers. Nat. Rev. Cancer 6 (11), 857–866. doi:10.1038/nrc1997
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., et al. (2005). The transcriptional landscape of the mammalian genome. Science 309 (5740), 1559–1563. doi:10.1126/SCIENCE.1112014
Chang, H., Yi, B., Ma, R., Zhang, X., Zhao, H., and Xi, Y. (2016). CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 6, 22312–12. doi:10.1038/srep22312
Chaudhary, R., Gryder, B., Woods, W. S., Subramanian, M., Jones, M. F., Li, X. L., et al. (2017). Prosurvival long noncoding RNA PINCR regulates a subset of p53 targets in human colorectal cancer cells by binding to Matrin 3. eLife 6, e23244. doi:10.7554/ELIFE.23244
Cho, S., Wu, C. J., Yasuda, T., Cruz, L. O., Khan, A. A., Lin, L. L., et al. (2016). miR-23∼27∼24 clusters control effector T cell differentiation and function. J. Exp. Med. 213 (2), 235–249. doi:10.1084/JEM.20150990
Conant, D., Hsiau, T., Rossi, N., Oki, J., Maures, T., Waite, K., et al. (2022). Inference of CRISPR edits from sanger trace data. CRISPR J. 5 (1), 123–130. doi:10.1089/crispr.2021.0113
Concordet, J. P., and Haeussler, M. (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46 (W1), W242–W245. doi:10.1093/nar/gky354
Dannenmann, B., and Skokowa, J. (2022). Generation, expansion, and drug treatment of hematopoietic progenitor cells derived from human iPSCs. Star. Protoc. 3 (2), 101400. doi:10.1016/j.xpro.2022.101400
Dhanjal, J. K., Dammalapati, S., Pal, S., and Sundar, D. (2020). Evaluation of off-targets predicted by sgRNA design tools. Genomics 112 (5), 3609–3614. doi:10.1016/j.ygeno.2020.04.024
Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. doi:10.1038/nbt.3437
Dylla, L., and Jedlicka, P. (2013). Growth-promoting role of the miR-106a∼363 cluster in Ewing sarcoma. PLoS ONE 8 (4), e63032. doi:10.1371/journal.pone.0063032
Ebert, M. S., Neilson, J. R., and Sharp, P. A. (2007). MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4 (9), 721–726. doi:10.1038/nmeth1079
Ebert, M. S., and Sharp, P. A. (2010). Emerging roles for natural microRNA sponges. Curr. Biol. 20 (19), R858–R861. doi:10.1016/j.cub.2010.08.052
Fan, J., Feng, Y., Zhang, R., Zhang, W., Shu, Y., Zeng, Z., et al. (2019). A simplified system for the effective expression and delivery of functional mature microRNAs in mammalian cells. Cancer Gene Ther. 27 (6), 424–437. doi:10.1038/s41417-019-0113-y
Friedman, R. C., Farh, K. K., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. doi:10.1101/gr.082701.108
Gebert, L. F. R., and MacRae, I. J. (2019). Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 20 (1), 21–37. doi:10.1038/s41580-018-0045-7
Gentner, B., Schira, G., Giustacchini, A., Amendola, M., Brown, B. D., Ponzoni, M., et al. (2009). Stable knockdown of microRNA in vivo by lentiviral vectors. Nat. Methods 6 (1), 63–66. doi:10.1038/nmeth.1277
Gruber, A. J., and Zavolan, M. (2013). Modulation of epigenetic regulators and cell fate decisions by miRNAs. Epigenomics 5 (6), 671–683. doi:10.2217/epi.13.65
Hao, Q., Zong, X., Sun, Q., Lin, Y. C., Song, Y. J., Hashemikhabir, S., et al. (2020). The S-phase-induced lncRNA SUNO1 promotes cell proliferation by controlling YAP1/Hippo signaling pathway. eLife 9, e55102–e55133. doi:10.7554/eLife.55102
Haeussler, M., Schönig, K., Eckert, H., Eschstruth, A., Mianné, J., Renaud, J. B., et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17 (1), 148. doi:10.1186/s13059-016-1012-2
Heckl, D., Kowalczyk, M. S., Yudovich, D., Belizaire, R., Puram, R. V., McConkey, M. E., et al. (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 32 (9), 941–946. doi:10.1038/nbt.2951
Ho, T. T., Zhou, N., Huang, J., Koirala, P., Xu, M., Fung, R., et al. (2015). Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 43 (3), e17. doi:10.1093/nar/gku1198
Hong, H., Yao, S., Zhang, Y., Ye, Y., Li, C., Hu, L., et al. (2020). In vivo miRNA knockout screening identifies miR-190b as a novel tumor suppressor. PLoS Genet. 16 (11), e1009168. doi:10.1371/journal.pgen.1009168
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31 (9), 827–832. doi:10.1038/nbt.2647
Huo, W., Zhao, Gg., Yin, J., Ouyang, X., Wang, Y., Yang, C., et al. (2017). Lentiviral CRISPR/Cas9 vector mediated miR-21 gene editing inhibits the epithelial to mesenchymal transition in ovarian cancer cells. J. Cancer 8 (1), 57–64. doi:10.7150/jca.16723
Hutvágner, G., Simard, M. J., Mello, C. C., and Zamore, P. D. (2004). Sequence-specific inhibition of small RNA function. PLoS Biol. 2 (4), E98–E475. doi:10.1371/journal.pbio.0020098
Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., et al. (2018). p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 24 (7), 939–946. doi:10.1038/s41591-018-0050-6
Jiang, Q., Meng, X., Meng, L., Chang, N., Xiong, J., Cao, H., et al. (2014). Small indels induced by CRISPR/Cas9 in the 5′ region of microRNA lead to its depletion and Drosha processing retardance. RNA Biol. 11 (10), 1243–1249. doi:10.1080/15476286.2014.996067
Kehl, T., Backes, C., Kern, F., Fehlmann, T., Ludwig, N., Meese, E., et al. (2017). About miRNAs, miRNA seeds, target genes and target pathways. Oncotarget 8 (63), 107167–107175. doi:10.18632/oncotarget.22363
Kim, M., Tan, Y. S., Cheng, W. C., Kingsbury, T. J., Heimfeld, S., and Civin, C. I. (2015). MIR144 and MIR451 regulate human erythropoiesis via RAB14. Br. J. Haematol. 168 (4), 583–597. doi:10.1111/bjh.13164
Konstantakos, V., Nentidis, A., Krithara, A., and Paliouras, G. (2022). CRISPR-Cas9 gRNA efficiency prediction: an overview of predictive tools and the role of deep learning. Nucleic Acids Res. 50 (7), 3616–3637. doi:10.1093/nar/gkac192
Kozomara, A., and Griffiths-Jones, S. (2014). MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42 (D1), 68–73. doi:10.1093/nar/gkt1181
Krützfeldt, J., Poy, M. N., and Stoffel, M. (2006). Strategies to determine the biological function of micrornas. Nat. Genet. 38 (6S), S14–S19. doi:10.1038/ng1799
Kurita, R., Suda, N., Sudo, K., Miharada, K., Hiroyama, T., Miyoshi, H., et al. (2013). Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 8 (3), e59890. doi:10.1371/journal.pone.0059890
Kurkewich, J. L., Hansen, J., Klopfenstein, N., Zhang, H., Wood, C., Boucher, A., et al. (2017). The miR-23a27a24-2 microRNA cluster buffers transcription and signaling pathways during hematopoiesis. PLoS Genet. 13 (7), 1–28. doi:10.1371/journal.pgen.1006887
Li, J., Wang, L., Hua, X., Tang, H., Chen, R., Yang, T., et al. (2020). CRISPR/Cas9-Mediated miR-29b editing as a treatment of different types of muscle atrophy in mice. Mol. Ther. 28 (5), 1359–1372. doi:10.1016/j.ymthe.2020.03.005
Lima, J. F., Cerqueira, L., Figueiredo, C., Oliveira, C., and Azevedo, N. F. (2018). Anti-miRNA oligonucleotides: a comprehensive guide for design. RNA Biol. 15 (3), 338–352. doi:10.1080/15476286.2018.1445959
Liu, Z., Hui, Y., Shi, L., Chen, Z., Xu, X., Chi, L., et al. (2016). Efficient CRISPR/Cas9-Mediated versatile, predictable, and donor-free gene knockout in human pluripotent stem cells. Stem Cell Rep. 7 (3), 496–507. doi:10.1016/j.stemcr.2016.07.021
Luo, Y., Liu, C., Cerbini, T., San, H., Lin, Y., Chen, G., et al. (2014). Stable enhanced green fluorescent protein expression after differentiation and transplantation of reporter human induced pluripotent stem cells generated by AAVS1 transcription activator-like effector nucleases. Stem Cells Transl. Med. 3 (7), 821–835. doi:10.5966/sctm.2013-0212
Ma, E., Chen, K., Shi, H., Stahl, E. C., Adler, B., Trinidad, M., et al. (2022). Improved genome editing by an engineered CRISPR-Cas12a. Nucleic Acids Res. 50 (22), 12689–12701. doi:10.1093/nar/gkac1192
Nath, A., Rayabaram, J., Ijee, S., Bagchi, A., Dutta Chaudhury, A., Roy, D., et al. (2021). Comprehensive analysis of microRNAs in human adult erythropoiesis. Cells 10 (11), 3018–3021. doi:10.3390/cells10113018
Netsrithong, R., Suwanpitak, S., Boonkaew, B., Trakarnsanga, K., Chang, L. J., Tipgomut, C., et al. (2020). Multilineage differentiation potential of hematoendothelial progenitors derived from human induced pluripotent stem cells. Stem Cell Res. Ther. 11 (1), 1–15. doi:10.1186/s13287-020-01997-w
Niu, R., Peng, J., Zhang, Z., and Shang, X. (2021). R-CRISPR: a deep learning network to predict off-target activities with mismatch, insertion and deletion in CRISPR-Cas9 system. Genes 12 (12), 1878. doi:10.3390/genes12121878
Niu, Y., Shen, B., Cui, Y., Chen, Y., Wang, J., Wang, L., et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156 (4), 836–843. doi:10.1016/j.cell.2014.01.027
Nogimori, T., Furutachi, K., Ogami, K., Hosoda, N., and Hoshino, S. ichi. (2019). A novel method for stabilizing microRNA mimics. Biochem. Biophys. Res. Commun. 511 (2), 422–426. doi:10.1016/j.bbrc.2019.02.075
Pagano, F., De Marinis, E., Grignani, F., and Nervi, C. (2013). Epigenetic role of miRNAs in normal and leukemic hematopoiesis. Epigenomics 5 (5), 539–552. doi:10.2217/epi.13.55
Park, J., Lim, K., Kim, J.-S., and Bae, S. (2017). Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics 33, 286–288. doi:10.1093/bioinformatics/btw561
Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., et al. (2013). Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 154 (6), 1380–1389. doi:10.1016/j.cell.2013.08.021
Rasmussen, K. D., Simmini, S., Abreu-Goodger, C., Bartonicek, N., Di Giacomo, M., Bilbao-Cortes, D., et al. (2010). The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 207 (7), 1351–1358. doi:10.1084/jem.20100458
Sanjana, N. E., Shalem, O., and Zhang, F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 11 (8), 783–784. doi:10.1038/nmeth.3047
Sayed, S. R. E., Cristante, J., Guyon, L., Denis, J., Chabre, O., and Cherradi, N. (2021). Microrna therapeutics in cancer: current advances and challenges. Cancers 13 (11), 1–28. doi:10.3390/cancers13112680
Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X., and Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science 351 (6268), 84–88. doi:10.1126/science.aad5227
Smith, J. R., Maguire, S., Davis, L. A., Alexander, M., Yang, F., Chandran, S., et al. (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 26 (2), 496–504. doi:10.1634/STEMCELLS.2007-0039
Smith, P. H., Bessette, A. J., Weinberger, A. H., Sheffer, C. E., and Mckee, S. A. (2016). Sex/gender differences in smoking cessation: a review. Physiol. Behav. 92 (3), 135–140. doi:10.1016/j.ypmed.2016.07.013
Stenvang, J., Petri, A., Lindow, M., Obad, S., and Kauppinen, S. (2012). Inhibition of microRNA function by antimiR oligonucleotides. Silence 3 (1), 1. doi:10.1186/1758-907X-3-1
Sturgeon, C. M., Ditadi, A., Awong, G., Kennedy, M., and Keller, G. (2014). Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 32 (6), 554–561. doi:10.1038/nbt.2915
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 (5), 861–872. doi:10.1016/j.cell.2007.11.019
Thamodaran, V., Rani, S., and Velayudhan, S. R. (2022). Gene editing in human induced pluripotent stem cells using doxycycline-inducible CRISPR-cas9 system. Methods Mol. Biol. 2454, 755–773. doi:10.1007/7651_2021_348
Trivedi, T. B., Boger, R., Kamath, G., Evangelopoulos, G., Cate, J., Doudna, J., et al. (2020). Crispr2vec: machine learning model predicts off-target cuts of CRISPR systems. BioRxiv, 1–19. doi:10.1101/2020.10.28.359885
Tsuchida, C. A., Zhang, S., Doost, M. S., Zhao, Y., Wang, J., O’Brien, E., et al. (2022). Chimeric CRISPR-CasX enzymes and guide RNAs for improved genome editing activity. Mol. Cell 82 (6), 1199–1209.e6. doi:10.1016/j.molcel.2022.02.002
Tzelepis, K., Koike-Yusa, H., De Braekeleer, E., Li, Y., Metzakopian, E., Dovey, O. M., et al. (2016). A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 17 (4), 1193–1205. doi:10.1016/j.celrep.2016.09.079
Vidigal, J. A., and Ventura, A. (2014). The biological functions of miRNAs: lessons from in vivo studies an elusive role for microRNAs. Trends Cell Biol. Trends Cell Biol. 25 (3), 137–147. doi:10.1016/j.tcb.2014.11.004
Vidigal, J. A., and Ventura, A. (2015). Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun. 6 (1), 8083–8087. doi:10.1038/ncomms9083
Wallace, J., Hu, R., Mosbruger, T. L., Dahlem, T. J., Stephens, W. Z., Rao, D. S., et al. (2016). Genome-wide CRISPR-cas9 screen identifies MicroRNAs that regulate myeloid leukemia cell growth. PLoS ONE 11 (4), 0153689. 11. doi:10.1371/journal.pone.0153689
Xiang, X., Corsi, G. I., Anthon, C., Qu, K., Pan, X., Liang, X., et al. (2021). Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 12 (1), 3238–3239. doi:10.1038/s41467-021-23576-0
Yan, Y., Qin, D., Hu, B., Zhang, C., Liu, S., Wu, D., et al. (2019). Deletion of miR-126a promotes hepatic aging and inflammation in a mouse model of cholestasis. Mol. Ther. Nucleic Acids 16, 494–504. doi:10.1016/j.omtn.2019.04.002
Yin, Y., Yan, P., Lu, J., Song, G., Zhu, Y., Li, Z., et al. (2015). Opposing roles for the lncRNA haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell 16 (5), 504–516. doi:10.1016/j.stem.2015.03.007
Zahnow, C. A., and Baylin, S. B. (2010). Epigenetic networks and miRNAs in stem cells and cancer. Mol. Cell 39 (5), 661–663. doi:10.1016/j.molcel.2010.08.036
Zhang, Z., Ursin, R., Mahapatra, S., and Ian Gallicano, G. (2018). CRISPR/CAS9 ablation of individual miRNAs from a miRNA family reveals their individual efficacies for regulating cardiac differentiation. Mech. Dev. 150, 10–20. doi:10.1016/j.mod.2018.02.002
Zhang, Z.-R., and Jiang, Z.-R. (2022). Effective use of sequence information to predict CRISPR-Cas9 off-target. Comput. Struct. Biotechnol. J. 20, 650–661. doi:10.1016/j.csbj.2022.01.006
Keywords: miRNA, CRISPR-Cas9, dual-gRNA, iPSCs, erythroid
Citation: Ijee S, Chambayil K, Chaudhury AD, Bagchi A, Modak K, Das S, Benjamin ESB, Rani S, Paul DZ, Nath A, Roy D, Palani D, Priyanka S, Ravichandran R, Kumary BK, Sivamani Y, S. V, Babu D, Nakamura Y, Thamodaran V, Balasubramanian P and Velayudhan SR (2024) Efficient deletion of microRNAs using CRISPR/Cas9 with dual guide RNAs. Front. Mol. Biosci. 10:1295507. doi: 10.3389/fmolb.2023.1295507
Received: 16 September 2023; Accepted: 27 December 2023;
Published: 02 April 2024.
Edited by:
Sumana Venkat, University of Texas Southwestern Medical Center, United StatesReviewed by:
Debojyoti Chakraborty, Council of Scientific and Industrial Research (CSIR), IndiaGiriraj Ratan Chandak, Centre for Cellular and Molecular Biology (CCMB), India
Copyright © 2024 Ijee, Chambayil, Chaudhury, Bagchi, Modak, Das, Benjamin, Rani, Paul, Nath, Roy, Palani, Priyanka, Ravichandran, Kumary, Sivamani, S., Babu, Nakamura, Thamodaran, Balasubramanian and Velayudhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaji R. Velayudhan, cnZzaGFqaUBjbWN2ZWxsb3JlLmFjLmlu
†These authors have contributed equally to this work