 Hanyun Zhang1†
Hanyun Zhang1† Chao Zhou
Chao Zhou Changgang Sun
Changgang Sun- 1College of First Clinical Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2College of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
- 3State Key Laboratory of Quality Research in Chinese Medicine and Faculty of Chinese Medicine, Macau University of Science and Technology, Macau, China
- 4Department of Oncology, Weifang Traditional Chinese Hospital, Weifang, China
- 5College of Traditional Chinese Medicine, Weifang Medical University, Weifang, China
Acute myeloid leukemia (AML) is a highly aggressive hematologic malignancy with a 5-year survival rate of less than 30%. Continuous updating of diagnostic and therapeutic strategies has not been effective in improving the clinical benefit of AML. AML cells are prone to iron metabolism imbalance due to their unique pathological characteristics, and ferroptosis is a novel cell death mode that is dominated by three cellular biological processes: iron metabolism, oxidative stress and lipid metabolism. An in-depth exploration of the unique ferroptosis mechanism in AML can provide new insights for the diagnosis and treatment of this disease. This study summarizes recent studies on ferroptosis in AML cells and suggests that the metabolic characteristics, gene mutation patterns, and dependence on mitochondria of AML cells greatly increase their susceptibility to ferroptosis. In addition, this study suggests that AML cells can establish a variety of strategies to evade ferroptosis to maintain their survival during the process of occurrence and development, and summarizes the related drugs targeting ferroptosis pathway in AML treatment, which provides development directions for the subsequent mechanism research and clinical treatment of AML.
1 Introduction
AML is a cancer originating from the hematopoietic stem cell myeloid lineage. It is characterized by the rapid proliferation of primitive cells and their replacement of hematopoietic tissue within the bone marrow, ultimately causing disruption to the normal bone marrow microenvironment. Within this pathological context, hematopoietic progenitor cells undergo clonal expansion, resulting in a blockage of their differentiation and maturation at various stages of development. The incidence of AML has increased annually over the past few decades, and the mortality rate has remained high owing to its aggressive nature and high recurrence rate. Currently, hematopoietic stem cell transplantation (HSCT) and the traditional “3 + 7”procedure, which was developed in the 1970s, are still used to treat AML. Demethylation medicines, IDH inhibitors, FLT3 inhibitors, and BCL-2 inhibitors are just a few medications that have recently been developed as a result of an improved understanding of the pathogenic pathways causing AML, including genetic characteristics and epigenetic changes. The introduction of these medications has effectively enhanced patient survival and quality of life by bringing AML treatment into the realm of precision therapy (Dohner et al., 2021). However, there has been no breakthrough in the overall survival rate of patients with AML. Therefore, new treatment strategies for AML are urgently needed.
In 2012, Stockwell introduced the concept of “ferroptosis” and described it as a type of cell death caused by uncontrolled lipid peroxidation and secondary membrane damage (Gao et al., 2016). Mechanistically, reactive oxygen species (ROS), phospholipid peroxides (PLOOH), and other metabolites that are inevitably produced during cell growth and differentiation react with polyunsaturated fatty acids (PUFAs), which are susceptible to peroxidation, causing membrane peroxidation, loss of permeability, and an increase in toxic products, which further destroys the cellular structure (Jiang et al., 2021). There is great potential in the treatment of cancer and other diseases that are closely related to ferroptosis by inducing ferroptosis in cells (Stockwell, 2022). Currently, the induction of ferroptosis to cause cancer cell death in hematological tumors, including AML, is considered a promising new therapeutic strategy (Zhang et al., 2022a). When AML is first diagnosed, it is commonly accompanied by iron overload, manifested mainly by elevated ferritin levels due to insufficient erythropoiesis and inflammatory signaling, which is aggravated by red blood cell infusion in late chemotherapy, providing AML cells an environmental basis for ferroptosis to occur (Weber et al., 2020). High expression of SLC7A11 and GPX4 are risk factors for AML patients and may also serve as prognostic markers (Han et al., 2022). In addition, Erastin, RSL3 were found to increase the anticancer potency of anthracyclines and cytarabine and inhibit AML cell proliferation (Skouta et al., 2014; Yu et al., 2023). Altered AML cell metabolism, genetic mutations, and dependence on mitochondria make AML cells appear to be highly susceptible to ferroptosis. However, the ensuing evasion strategy established by AML cells adds to the uncertainty regarding the occurrence of ferroptosis.
This paper provides a review of the core mechanism of ferroptosis and its impact on AML. It also highlights why AML is susceptible to ferroptosis and discusses strategies to prevent its occurrence. In addition, the paper discusses the use of iron-death-related drugs in AML treatment. The future application prospects of ferroptosis in AML treatment are also explored, along with potential challenges and corresponding suggestions.
2 Ferroptosis core mechanisms
Ferroptosis is commonly thought to be one of the oldest and most prevalent types of cell death and has been observed not only in mammals but also in plants, protozoa, and fungi (Hadian and Stockwell, 2020). Ferroptosis has been defined as a highly complicated process requiring the coordinated engagement of various metabolisms and pathways, with three primary features of iron metabolism imbalance, lipid peroxide formation, and antioxidant system breakdown (Figure 1).
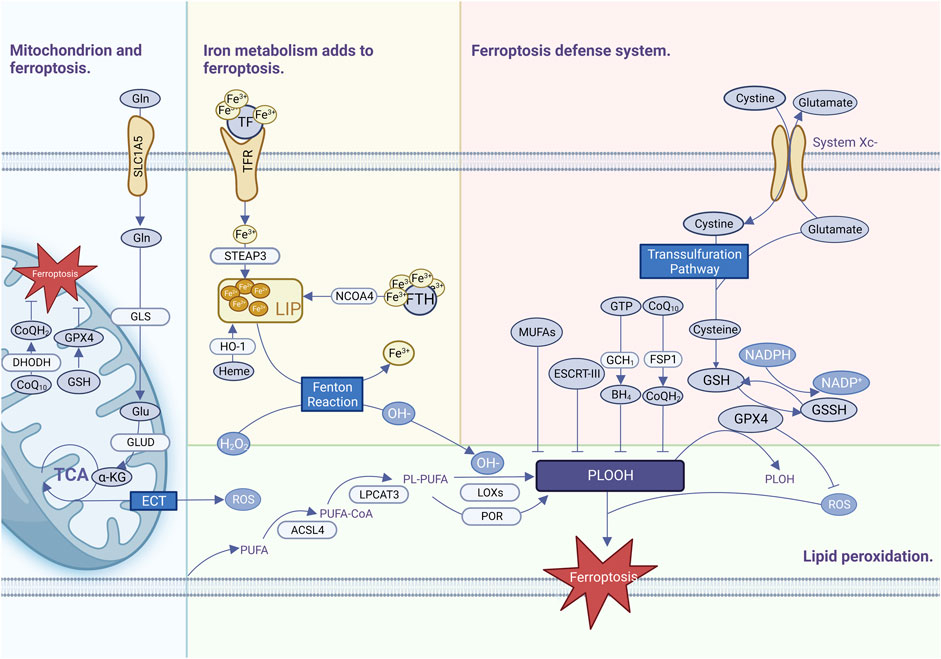
FIGURE 1. Ferroptosis occurrence and defense mechanisms.
2.1 Abnormal iron metabolism
Iron is an essential trace element involved in many physiological activities in the human body, is extremely oxidizing, generates excess ROS through the Fenton reaction, and is an important cofactor involved in the synthesis of enzymes responsible for lipid peroxidation and oxygen homeostasis (Holbein and Lehmann, 2023). Iron is usually bound to a complex such as heme (a prosthesis of numerous proteins such as hemoglobin), iron-sulfur clusters (a cofactor for many enzymes), or ferritin (a specialized iron storage protein) (Stockwell, 2022). A small amount of intracellular iron that is not bound as a complex is called the labile iron pool (LIP), which is highly reactive and oxidizable. Fe3+ in food is reduced to Fe2+ by duodenal cytochrome B, which is transferred to LIP via the divalent metal transporter (DMT1) (Donovan et al., 2000). Ferritin is recognized by nuclear receptor coactivator 4 (NCOA4), which directs ferritin into the autophagosome for degradation to release Fe2+ into the LIP (Yu et al., 2022). The LIP generates a large number of free radicals via the Fenton reaction, which damages cellular lipids and proteins due to the instability and easy redox potential of hydroxyl radicals (Dixon et al., 2012). The proteins and pathways involved in iron uptake, storage, transport, and transformation are diverse, and affect the susceptibility of cells to ferroptosis to varying extents (Stockwell, 2022). Despite a growing understanding of iron metabolism and ferroptosis, it is still impossible to quantify the amount of iron required by tumors and normal tissues, and it is difficult to measure the level of unstable iron pools.
2.2 Lipid peroxidation reaction
Uncontrolled lipid peroxidation is a typical characteristic of ferroptosis. The lipid peroxidation substrates of ferroptosis are PUFAs, which are important components of cell membrane organelles and have unstable carbon–carbon double bonds with a strong affinity for free radicals. The hydrogen atoms between the double bonds are easily oxidized by free radicals and subsequently prone to cascade-type reactions, which expand the scope of ferroptosis (Yang et al., 2016). PUFAs break down and oxidize. Subsequently, they form lipid radicals, lipid hydroperoxides, reactive aldehydes, and other toxic substances that cause further damage to cells (Xin and Schick, 2023). Acyl-CoA synthetase long-chain family member 4 (ACSL4) promotes the activation of PUFAs such as arachidonoyl (AA) and adrenal to synthesize PUFA-CoA (Kagan et al., 2017), and then lysophosphatidylcholine acyltransferase 3 (LPCAT3) binds PUFA-COA to membrane phospholipids (PLs) to catalyze the formation of phospholipids containing PUFA-PLs (Kagan et al., 2017). Eventually, PUFA-PLs are oxidized by iron and lipoxygenases (LOXs) to generate PUFA-PL-OOH, which trigger ferroptosis (Kagan et al., 2017). ROS produced by electron leakage from the mitochondrial electron transport chain (ETC.) may also contribute to lipid peroxidation during ferroptosis in some situations (Gao et al., 2019). Monounsaturated fatty acyl (MUFA) can compete with PUFAs to bind PL via ACSL3 to prevent erastin-induced ferroptosis, or exogenous MUFA can reorganize intracellular lipid metabolism so that excess lipids are stored in organelles such as triglycerides (TAG), a form of lipid that is less susceptible to oxidative reactions (Liang et al., 2022).
2.3 Antioxidant defense mechanisms
To meet high proliferative demands, cancer cells need to integrate multiple metabolic pathways including amino acid synthesis, the tricarboxylic acid cycle, oxidative phosphorylation, and iron metabolism. These active and complex metabolic pathways are usually accompanied by an imbalance of oxidative stress, which makes a robust ferroptosis defense mechanism essential for cancer cells. The XC-GPX4-GSH pathway plays a central role in ferroptosis defense, and system XC- is an amino acid transporter consisting of two key components: disulfide-linked solute carrier family 3 member 2 (SLC3A2) and solute carrier family 7 member 11 (SLC7A11) (Koppula et al., 2021). System XC exchanges extracellular cystine and intracellular glutamate in a 1:1 ratio. After entry into the cell, cystine is converted to cysteine, which is then converted to glycine with glutamate by cysteine-glutamate ligase, and then reduced to glutathione (GSH) catalyzed by glutathione synthetase (GSS) (Jiang et al., 2021). GPX4-GSH scavenges free radicals in the body and maintains intracellular free radical stability. Glutathione is the most important antioxidant in cells and a cofactor of GPX4. GPX4 is a selenoprotein that contains glutathione and selenium to maintain its activity (Liu et al., 2023a). GPX4 is the most important intracellular anti-lipid peroxidase, which uses GSH to participate in the LOOH reduction reaction to catalyze the conversion of harmful lipid peroxides to harmless lipid alcohols and prevents the occurrence of ferroptosis. GPX4 activity can be suppressed by inhibiting the XC upstream system (Zhang et al., 2021). Erastin, a ferroptosis inhibitor, acts on the XC-inhibiting cystine uptake system, leading to inhibition of intracellular GSH synthesis (Dixon et al., 2012). RSL3, a ferroptosis inhibitor, acts on GPX4 to inactivate it, resulting in a large accumulation of intracellular lipid peroxides, ultimately leading to the onset of ferroptosis (Zheng et al., 2023). In summary, the XC-GPX4-GSH system is a complex network in which various molecules affect ferroptosis indirectly or directly via the XC-GPX4-GSH system. In addition to ferroptosis, it affects apoptosis, pyroptosis, autophagy, and other cell-death mechanisms (Kang et al., 2023).
Some cancers resist ferroptosis after GPX4 inactivation, suggesting the existence of additional defense mechanisms. Recently, FSP1-coenzyme Q10 (COQ10) (Bersuker et al., 2019), DHODH-COQ10 (Mao et al., 2021), GCH1-tetrahydrobiopterin (BH4) (Vasquez-Vivar et al., 2022), and endosomal sorting complexes in retrograde transport-III (ESCRT-III) have been identified as ferroptosis defense mechanisms that are not dependent on the XC-GPX4-GSH system and function in parallel with the XC-GPX4-GSH system for parallel defense against ferroptosis (Pedrera et al., 2021).
2.4 Mitochondria and ferroptosis
The mitochondrial OXPHOS system is at the center of cellular metabolism and is critical for energy production in eukaryotic cells (Vercellino and Sazanov, 2022). Ferroptosis is promoted by metabolites during mitochondrial biosynthesis, e.g., ferroptosis is driven by the generation of ROS, ATP, and/or PUFA-PL produced during the TCA cycle. Inhibition of the mitochondrial TCA cycle or the, ETC., attenuates mitochondrial membrane potential hyperpolarization and inhibits lipid peroxide accumulation and ferroptosis (Gao et al., 2019). Accordingly, the ferroptosis defense system in mitochondria is mainly composed of GPX4-GSH, dihydroorganic acid dehydrogenase (DHODH)-COQ10 (Zheng and Conrad, 2020), while mitochondrial ferritin overexpression can also regulate iron homeostasis in mitochondria to inhibit Erastin-induced ferroptosis (Wang et al., 2016).
3 Susceptibility of AML cells to ferroptosis
3.1 Metabolic characteristics of AML cells confer susceptibility to ferroptosis
AML is distinguished by the presence of an abnormally high number of immature myeloid cells in the bone marrow that are unable to differentiate and mature properly, proliferate rapidly, or occupy space for normal cell survival. To maintain their highly proliferative and highly differentiated state, AML cells have growth characteristics of extremely high energy demand, susceptibility to oxidative stress, and accumulation of metabolites, which provide the conditions for ferroptosis to occur (Figure 2).
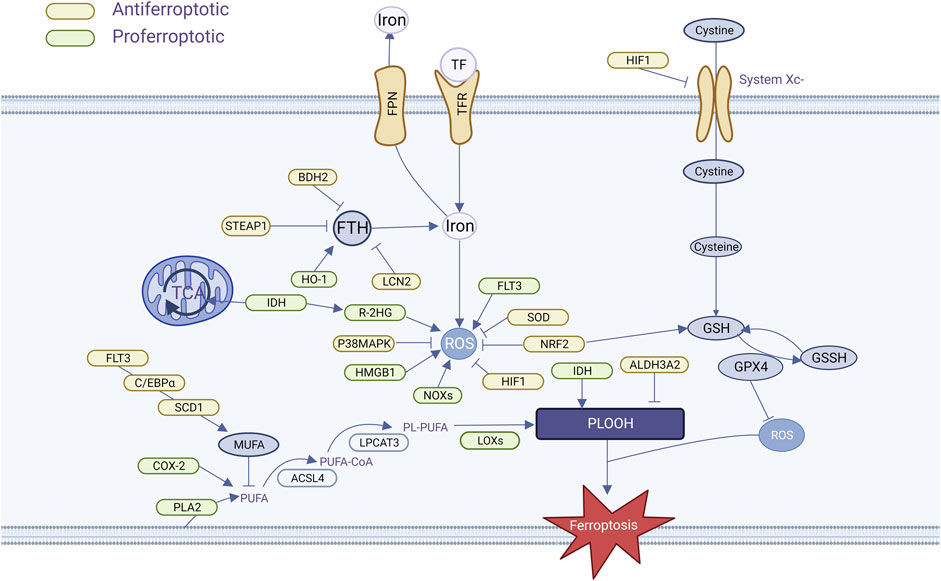
FIGURE 2. AML cells regulate ferroptosis pathways.
3.1.1 Iron overload and iron regulation imbalance
AML cells are particularly susceptible to iron overload. Patients with AML generally have high serum ferritin levels at initial diagnosis, with levels positively correlated with tumor load, which drop to normal levels after remission, and elevated levels may present with recurrence (Lebon et al., 2015). The most common cause of iron overload is multiple infusions of red blood cells during chemotherapy. One unit of red blood cells contains approximately 200 mg of iron (Wang et al., 2021), part of the iron in the transfused erythrocytes is recycled and stored in the tissues by macrophages in the liver and spleen, and the heme iron that cannot be stored is released outside the cell, increasing the amount of unstable iron pool. Measurement of iron overload by means of biomagnetism, magnetic resonance imaging, or organ biopsy has revealed the presence of iron overload in patients with MDS/AML and has led to a better understanding of the characteristics of their iron metabolism (Jensen, 2004). Heme iron overload damages bone marrow hematopoietic stem cells and affects bone marrow hematopoietic function via the ROS pathway (Lu et al., 2013; Chai et al., 2015). In turn, the decreased hematopoietic function becomes dependent on blood transfusions, creating a vicious cycle. In addition, increased heme iron increases the risk of infection-related complications, and Petzer et al. observed a strong correlation between Aspergillus growth and heme in AML patients undergoing allogeneic HSCT (Petzer et al., 2019). Usually, in order to eliminate the negative effects of iron overload, iron stingers such as Deferasirox and Deferiprone are applied clinically to reduce the iron load in AML patients. Chang et al. found that Deferasirox has a pronounced antileukemic effect, which induces apoptosis in AML cells by stinging unstable iron pools, generating large amounts of ROS, and blocking cell signaling pathways (Chang et al., 2017). We hypothesized that iron stingers such as Deferasirox may hinder the onset of AML cell ferroptosis when applied, which has been demonstrated in hepatocellular carcinoma (Jomen et al., 2022).
Transferrin (TF) and transferrin receptor (TFR) bind to form a complex that is involved in intracellular iron transport and is the main pathway for iron uptake by cancer cells, which was found to be essential for the induction of cellular ferroptosis by glutamine depletion assays (Gao et al., 2015). There is much evidence that AML cells are highly bound to TF and are highly expressed in AML cells (Yeh et al., 1984), but the specific effect on AML cells is unknown. TFR play important roles in ferroptosis. By upregulating TFR, cells with RAS mutations have increased intracellular iron content, making them more sensitive to elastin-induced ferroptosis, while knocking out TFR makes them less sensitive to erastin-induced ferroptosis (Yang and Stockwell, 2008). Two TFRs have been identified: TFR1 and TFR2. Iron enters the cell mainly via TFR1 and is transported via NCOA4 receptor-mediated endocytosis. TFR1 has been validated as a marker of ferroptosis (Feng et al., 2020). TFR1 expression is elevated in AML cells compared to normal cells, and TFR cell expression is elevated as malignant cell differentiation decreases (Liu et al., 2014; Lyons and Pappas, 2019). In addition, it has been suggested that TFR1 is associated with anemia, thrombocytopenia, and complex cytogenetics in AML (Wu et al., 2016), but not with poor AML prognosis. In bone marrow specimens from AML and MDS patients, however, the researchers found that high TFR2 mRNA expression had a relatively favorable prognosis, suggesting that TRF2 may alter iron metabolism and increase AML cell sensitivity to chemotherapy (Nakamaki et al., 2004; Di Savino et al., 2017).
The ferritin-ferromodulin (FPN) system is an important regulator of iron homeostasis, and is associated with redox control. Ferritin, which consists of FTH and FTL, inhibits iron efflux, keeps the LPI pool stable, and inhibits ferroptosis. Ferritin deficiency decreased the regulatory effects of SLC7A11 and causes ferroptosis (Fang et al., 2020). Compared with normal HSCs, FTH and FTL were overexpressed in both AML cells and LSCs, and FTH overexpression was usually accompanied by NF-κB-related gene expression, which reduced chemosensitivity (Bertoli et al., 2019). Thus, elevated ferritin levels are indicative of disease progression. FPN is the sole protein responsible for controlling intracellular iron efflux. When FPN is overexpressed, it reduces he amount of iron within cells and hinders the growth of cancer cells. Therefore, FPN could potentially serve as a vulnerability for cancer cells that heavily rely on iron. Studies have shown that FPN expression is reduced in AML compared to normal cells, up to 100,000-fold in some cell lines. The researchers concluded that low-FPN cells retain more iron and have an increased susceptibility to ROS. Follow-up experiments verified this hypothesis via ex vivo experiments that the application of iron oxide nanoparticles to AML cells with low FPN showed better anti-leukemic activity, which could explain the greater sensitivity of AML cells with low FPN to chemotherapeutic drugs. In addition, iron-regulator expression in AML is associated with an inflammatory milieu, and AML patients with low levels of FPN may have reduced FPN expression due to greater autocrine or paracrine secretion of inflammatory cytokines (such as IL-6) (Vela et al., 2018). Increased intracellular iron levels, coupled with an imbalance in iron metabolism, further increase the vulnerability of AML cells to ferroptosis.
3.1.2 Increased reactive oxygen species
As an inhomogeneous group of molecules and free radicals, reactive oxygen species (ROS) are frequently involved in various cellular processes. It is frequently found to be elevated in a variety of cancer types (Trachootham et al., 2009), and AML is no exception (Zhang et al., 2014; Hole et al., 2011). In AML, ROS are generally acquired via altered metabolic pathways such as xanthine oxidoreductase, non-coupled nitric oxide (NO) synthase (NOS), cytochrome P450 monooxygenase (CYP), cyclooxygenase (COX), and NADPH oxidase (NOX). The high frequency of ROS overproduction in cancer and leukemia strongly suggests that ROS are associated with diseases pathogenesis of these diseases is inseparable (Hole et al., 2011). In hematological malignancies, ROS plays a dual role in disease progression (Udensi and Tchounwou, 2014). In the presence of high levels of ROS, chemotherapeutic agents inhibit tumor growth and cause apoptosis by producing high levels of ROS (Khoshtabiat et al., 2016). In contrast, low ROS levels protected AML cells from apoptosis and promoted cell proliferation, survival, growth, migration, and drug resistance (Kaweme et al., 2020).
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOXS), a class of membrane-bound enzyme complexes that catalyze the production of superoxide, contributes to iron-dependent lipid peroxidation accumulation during ferroptosis (Yang et al., 2020), and in 60% of the patients, AML cells constitutively produce a large number of extracellular superoxides through the NOx family of NADPH oxidases, such as NOX1, NOX2, NOX4, and NOX5, with NOX2 being the main source of superoxide in AML (Irwin et al., 2013), with evidence showing strong expression of NOX4 in a subpopulation of patients with high ROS (Hole et al., 2013). Accordingly, reactive oxygen species produced by the NOx family hhavebeen linked to leukemic progression, DNA damage, and cellular transformation in many ways (Jayavelu et al., 2016a; Jayavelu et al., 2016b).
High mobility group box 1 (HMGB1) is a transcriptional protein sensitive to oxidative stress and is involved in multiple processes of cancer development, such as cell proliferation, angiogenesis, cell migration, and adhesion (Chen et al., 2023). Some researchers have found that HMGB1 is a central factor necessary for the occurrence of ferroptosis. Reducing ROS levels by SOD1 depletion or NAC significantly affected HMGB1 transport, whereas inhibition of HMGB1 expression prevented erythroid-induced GPX4 degradation, TFR1 expression, and subsequent ferroptosis. This suggests that HMGB1-dependent ROS is directly involved in the occurrence and maintenance of erythroid-induced ferroptosis in AML cells (Ye et al., 2019).
3.1.3 Altered lipid metabolism
AML cells exhibited enhanced cholesterol utilization and synthesis rates, decreased total cholesterol and cholesterol-to-phospholipid ratios, increased phosphatidylcholine and phosphatidylinositol, and increased unsaturated fatty acids (Vitols et al., 1990). Similar to these results, Pabst et al. found depletion of total plasma fatty acids and cholesterol in AML cells, but an increase in certain free fatty acids such as sphingolipids, choline phosphate, and a decrease in triglycerides and cholesteryl esters may be caused by enhanced fatty acid oxidation in AML cells (Pabst et al., 2017). Importantly, free fatty acids (such as arachidonic acid 20:4 n-6 and the corresponding precursors gamma-linolenic acid 18:3 n-6 and 8,11,14-eicosatrienoic acid 20:3 n-6) were found to be increased in AML cells compared to normal samples, and successively, transcript levels of soluble phospholipase A2 (PLA2) isoforms IB and X were found to be both in AML cells showed upregulation (Fiancette et al., 2009). This finding is crucial because the enzymatic activity of PLA2 catalyzes the release of AA from membrane phospholipids, which in turn generates lipoxygenase and its derived lipid mediators. LOXs drive ROS to oxidize AA and AdA-PE, and the subsequent generation of AA- and AdA-PE-OOH causes ferroptosis (Kagan et al., 2017). AA is important for the induction of ferroptosis, and AML cells with high AA expression are more malignant.
Cyclooxygenase 2 (COX-2) is a key enzyme in prostaglandin biosynthesis with dual roles as a peroxidase and dioxygenase that can promote PUFA lipid peroxidation in organelles involved in ferroptosis. The inhibition of COX-2 is an effective way to alleviate ferroptosis caused by ferritin overload. Multiple experiments have demonstrated that COX-2 production can be induced with LPS in AML cellsand stimulated HL-60 cells also express COX-2 (Vincent et al., 2008a; Puhlmann et al., 2005), which may promote ferroptosis.
3.2 Genetic mutations in AML cells confer susceptibility to ferroptosis
Somatic mutations in isocitrate dehydrogenases 1/2 (IDH1/2, respectively) are found in approximately 20% of patients with AML. Both mutants conferred novel morphological activity for the NADPH-dependent reduction of ketoglutarate (KG) to the oncogenic metabolite (R)-2-hydroxyglutarate (2HG) (Dang et al., 2009). Interestingly, one study found that IDH1 and IDH2 mutant AML cells produce the metabolite R-2HG that rapidly increases intracellular ROS, which in turn affects NF-κB protein stability and transcriptional activity (Chen et al., 2016). A study found that IDH-mutant AML cells exhibit enhanced mitochondrial oxidative metabolism and that fatty acid oxidation in IDH-mutant cells is further increased with increased TCA cycle products (Stuani et al., 2021). A recent experiment identified a new metabolic vulnerability in AML cells mutated in IDH1, which significantly disrupts lipid metabolism and is characterized by a lack of monoacylglycerides and lysophospholipids and an increase in acylcarnitine species. This lipid metabolism abnormality is more pronounced in IDH1 than in IDH2 (Thomas et al., 2023).
Fms-like tyrosine kinase 3 (FLT3) is mutated in approximately 90% of AML patients, and FLT3-ITD is a common mutation in FLT3, accounting for approximately 20% of AML patients, and is more malignant. Sabatier et al. suggested that FLT3-ITD mutant AML cells regulate fatty acid synthesis by modulating downstream CCAAT/en-hancer-binding protein α (C/EBPα)a and steroyl-CoA desaturase (SCD, mentioned below). When C/EBPα was inhibited in FLT3-ITD mutant AML cells, SCD expression was downregulated, resulting in decreased MUFA formation and increased production of PUFAs, which led to iron cell death (Sabatier et al., 2023). FLT3-ITD mutated cell lines with upregulated heme oxygenase-1 (HO-1) and nuclear factor erythroid 2-related factor 2 (Nrf2) expression produce higher levels of ROS (Kannan et al., 2022), DNA oxidation, and double-strand breaks compared to wild-type FLT3 patients (Sallmyr et al., 2008; Godfrey et al., 2012). The major source of ROS in FLT3-ITD mutant AML cells is the NOx family. Excessive ROS accumulation has signaling functions that promotes cell proliferation and migration, thus contributing to leukemic cell transformation (Godfrey et al., 2012; R et al., 2011). Targeting the Nrf2-HO-1 antioxidant pathway using the ferroptosis-related drug brusatol improved the inhibition of FLT3-ITD mutant cell proliferation (Kannan et al., 2022). IDH1/2 and FLT3-IDT mutations in AML lead to abnormalities in ROS, lipid metabolism, and increased susceptibility to ferroptosis, but it was not found whether these two mutations affect iron metabolism, which can be further explored as a direction for future research.
3.3 Mitochondria-dependent conferral of ferroptosis susceptibility in AML cells
Independent of the mutational spectrum and cytogenetic influences, mitochondrial metabolism dominates AML metabolism, is the main source of cellular ROS, and is the main site of OXPHOS (Gao et al., 2021). Many studies have shown that mitochondrial energy metabolism is significantly altered in AML cells, influencing the occurrence of ferroptosis in AML cells in terms of bioenergetics, biosynthesis, and ROS regulation.
While reducing OXPHOS could provide a survival advantage for cancer cells, AML cells do not want to do so. Compared to hematopoietic stem cells, AML cells show an increased dependence on mitochondrial metabolism and OXPHOS, and increased mitochondrial mass (Skrtic et al., 2011). Additionally, AML cells lack metabolic flexibility and struggle to meet their energy demands by inhibiting fatty acid oxidation to promote glycolysis. Consequently, AML cells depend heavily on mitochondrial OXPHOS for energy supply (Gao et al., 2021). This facilitates the onset of ferroptosis: connecting cancer cell metabolism to OXPHOS makes highly aggressive cancer cells more sensitive to ferroptosis by disrupting glycolysis (Zdralevic et al., 2018). However, OXPHOS overactivity also renders AML cells more vulnerable to resistance against cytarabine, suggesting the presence of a more robust antioxidant system that protects AML cells (Farge et al., 2017). In addition, overactivation of the mitochondrial protease CLPP can degrade respiratory chain proteins mainly conforming to the I subunit, leading to an increase in ROS in mitochondria (Ishizawa et al., 2019). Meanwhile, during the rapid growth phase of cancer cells, more iron is translocated into mitochondria to meet the exuberant energy demand of cancer cells (Duan et al., 2022). Due to the unique metabolic dependence on mitochondria, AML cells are more prone to ferroptosis.
4 AML cells escape ferroptosis
The susceptibility of AML cells to ferroptosis is a barrier to their survival and development. To avoid the risk of ferroptosis when lipid peroxidation occurs to a certain extent, AML cells construct a number of iron avoidance strategies to cope with ferroptosis, including (1) limiting the unstable iron pool, (2) limiting oxidative stress due to excessive ROS, and (3) limiting PUFA synthesis (Figure 3), which makes it difficult to induce ferroptosis in AML cells. Therefore, it is necessary to understand the ferroptosis evasion strategies in AML cells.
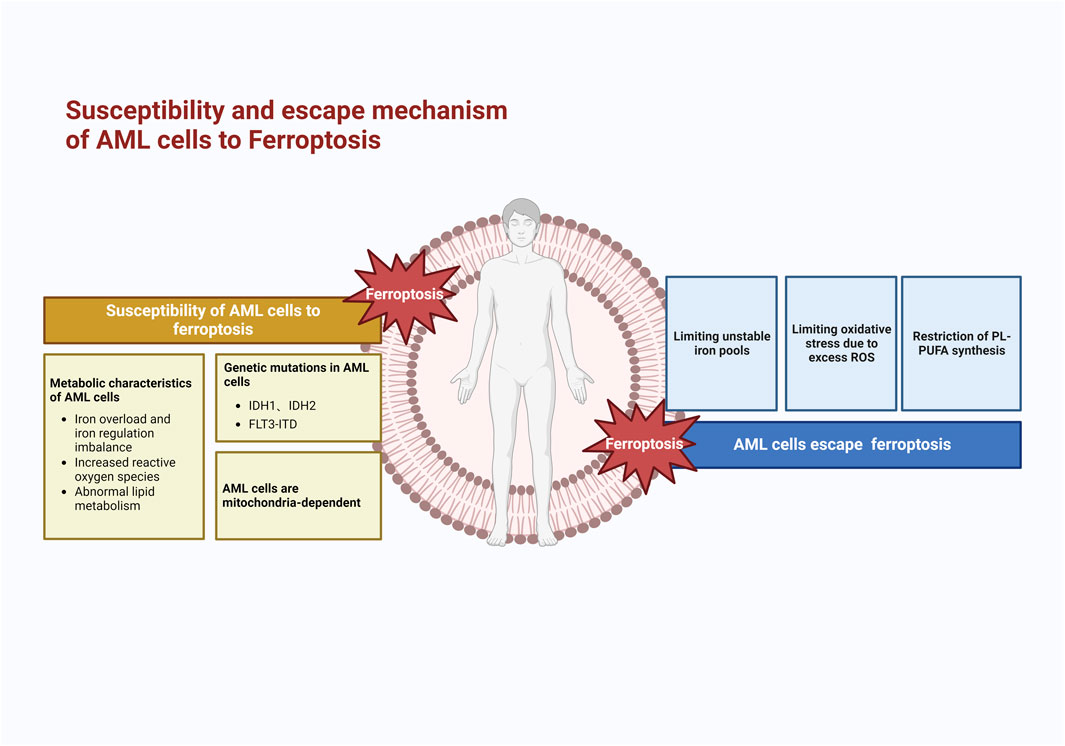
FIGURE 3. Mechanism of susceptibility to ferroptosis and escape mechanism of AML cells. AML cells are highly susceptible to ferroptosis because of their metabolic and genetic alterations; therefore, AML cells develop evasion strategies such as limiting unstable iron pools, oxidative stress, and lipid peroxidation reactions.
4.1 Limiting LIP
Elevated serum ferritin, an iron storage protein, is commonly found in most patients with new-onset AML, and this abnormality is not dependent on transfusion. It has been observed that increased ferritin levels lead to resistance to cytarabine, thus increasing the risk of HSCT recurrence and decreasing overall survival (Bertoli et al., 2019). Furthermore, patients with high ferritin expression have shown lower response rates when treated with decitabine for new-onset AML (Park et al., 2017). It is hypothesized that ferritin might contribute to methylation resistance. The presence of ferritin limits the expansion of LIP, reduces the formation of ROS, and prevents ferroptosis from occurring to a certain extent. How to break the iron homeostasis and convert the large amount of stored iron in ferritin into unstable free iron making it more likely to induce ferroptosis is a direction that can be studied in the future.
Lipid transport protein-2 (LCN2) belongs to a family of lipid transport proteins that are widely involved in the development of many types of inflammatory malignant diseases (Chi et al., 2020). This mechanism has been shown to function as an iron regulatory protein that is strongly bound to trivalent iron (Kjeldsen et al., 2000). Patients with many hematologic tumors, including AML, have expression of LCN2 (Bouchet and Bauvois, 2014; Candido et al., 2014), which is required for the BCR-ABL-induced mouse leukemia model and is involved in damaging normal hematopoietic cells and inducing tissue invasion by leukemia cells (Leng et al., 2008). Multiple experiments have found lower levels of LCN2 expression in bone marrow specimens from AML patients than in normal subjects (Candido et al., 2014; Yang et al., 2013a). Surprisingly, LCN2 expression levels gradually increased with remission in AML patients, and refractory AML patients showed low LCN2 levels (Yang et al., 2013a). High LCN2 expression in bone marrow is associated with a favorable prognosis, especially in patients with FLT3 mutations (Yang et al., 2013b). BDH2 is a novel cytosolic-type 2-hydroxybutyrate de hydrogenase involved in lipid metabolism pathways and iron transport, which also participates in the mitochondrial and tricarboxylic acid cycles (Guo et al., 2006). Devireddy et al. found that BDH2 accompanies 2,3-DHBA production during enterobactin synthesis, and contributes to iron transport and apoptosis via the LCN2-mediated pathway (Devireddy et al., 2010). In contrast to LCN2, BDH2 high expression is an independent poor prognostic factor in AML patients, with a pro-proliferative effect on AML cells and reduced sensitivity to chemotherapeutic agents (Yang et al., 2013a). It can be further hypothesized that cells with high LCN2 expression increase iron content through its iron transport function, leading to lipid peroxidation and thus ferroptosis, and high BDH2 expression prevents cytoplasmic iron overload and reduces the likelihood of ferroptosis.
STEAP3, an iron reductase within the STEAP family, mediates the conversion of trivalent to divalent iron and is an important regulator of ferroptosis. Although STEAP1 has no iron-reducing function (Ohgami et al., 2005), STEAP1 expression in Ewing tumors correlates favorably with cellular reactive oxygen species, which stimulates the production of oxidative stress-sensitive and invasive genes, which is consistent with earlier research: Thyroid epithelial cell proliferation is aided by increased STEAP1 expression (Pan et al., 2008). Another study found that STEAP1 covalently localizes to TFRC to participate in iron metabolism (Ohgami et al., 2006), suggesting that it also plays an important role in ferroptosis. In AML, researchers found that STEAP1 overexpression is associated with poor prognosis, and presumably, STEAP1 favors proliferation and survival in AML (Chen et al., 2021).
4.2 Limiting oxidative stress due to excess ROS
Collapse of the redox system is one of the triggers for the occurrence of ferroptosis. To limit reactive oxygen species, AML cells establish an antioxidant system to prevent ferroptosis from occurring.
NRF2 is a redox-dependent transcription factor involved in the pathogenesis and drug resistance mechanisms of many tumors, and affects AML cells to evade ferroptosis in two main ways. First, NRF2 overexpression can activate antioxidant function to help AML cells resist chemical damage. NRF2 is significantly expressed in high tumor load and drug-resistant AML, and is highly expressed in patients with poor prognosis (Hu et al., 2022). Correspondingly, overexpression of NRF2 also induced gene instability-dependent drug resistance in AML (Liu et al., 2021). By reducing ROS, NRF2 renders AML resistant to cytarabine and zorubicin (Karathedath et al., 2017). Further studies revealed that inactivation of the NRF2 pathway leads to reduced production of antioxidants (e.g., SOD2), which shifts the ratio of GSH/GSSG to the oxidized form, thereby increasing AML cellular chemosensitivity (Niu et al., 2021). Second, NRF2 regulates transcriptionally almost all genes related to ferroptosis, including regulation of GPX4 activity and glutathione production, which affects NADPH regeneration, glutathione-dependent thioredoxin and peroxiredoxin reduction (Anandhan et al., 2020). NRF2 also regulates genes related to iron metabolism, preventing the accumulation of free iron to avoid ferroptosis (Harada et al., 2011). Liu et al. identified a novel mechanism by which in AML Liu et al. found a new mechanism that in AML, GPX4 is an important target gene of NRF2, and activation of NRF2 can cause GPX4 to be overexpressed in AML cells, thus avoiding ferroptosis (Liu X. et al., 2023). It has been shown that the use of ferroptosis inducers generates large amounts of ROS in AML cells, which resist oxidative stress by activating NRFR/HO-1 through nuclear translocation of the NRF2 protein, and that downregulation of NRF2 expression markedly increases the killing effect of ferroptosis inhibitors (Ali et al., 2016). Therefore, NRF2 is a very important pathway target for AML cells to evade ferroptosis.
HO-1, an enzyme that catalyzes the conversion of heme to bilirubin, is a downstream target of Nrf2 (Kerins and Ooi, 2018). HO-1 is also involved in accelerating erastin-induced ferroptosis (Chang et al., 2018). The Nrf2-HO-1 pathway also has a regulatory effect on intracellular iron concentration. Nrf2 degrades HO-1 to biliverdin/bilirubin, nitric oxide, and divalent iron, and HO-1-induced ferroptosis is achieved by degradation of ferritin, which increases free iron (Tomiotto-Pellissier et al., 2018). However, HO-1 in ferroptosis appears to be a double-edged sword, depending on the metabolic state of the tumor and the tumor microenvironment. HO-1 appears to play a role in evading ferroptosis and protecting AML cells. Wei et al. demonstrated that HO-1 levels are elevated in AML patients (Wei S. et al., 2015). Numerous experiments have shown that HL60-resistant cells have higher expression of HO-1 than HL-60-sensitive cells, identifying a correlation between HO-1 overexpression and AML resistance, and subsequent experiments have hypothesized that it may be through a ROS-dependent pathway thereby allowing AML cells to escape chemotherapy-induced apoptosis (Zhe et al., 2015; Heasman et al., 2011). After the inhibition of HO-1 expression, resistant and refractory cell lines showed sensitivity to cytarabine and erythromycin. This is consistent with the results of another team’s experiments: downregulation of HO-1 by gene silencing and the classical HO-1 inhibitor ZnPPIX, which found that HO-1 silencing greatly sensitized AML cells to Zoerythromycin (Wei et al., 2014; Wei S. X. et al., 2015).
Large amounts of ROS burst in new-onset AML cells, and excess ROS or the resulting manifestation of antioxidant defense deficiency leads to cellular damage and enhanced antioxidant signaling; thus, normal cells activate stress protein kinases (SAPKs) including P38-MAPK mitogen-activated protein kinase (MAPK), which responds to reactive oxygen stress through phosphorylation and thus affects the cell cycle (Cuadrado and Nebreda, 2010; Bulavin and Fornace, 2004). It has been shown that AML cells will adapt to a high ROS state and downregulation of P38-MAPK does not affect normal cellular activity. Because excessive ROS block normal hematopoiesis (Juntilla et al., 2010; Ito et al., 2004), AML cells also gain an advantage in competition with normal cells through the ROS paracrine mode (Hole et al., 2013).
Superoxide dismutases (SODs) can catalyze the conversion of superoxide to oxygen and hydrogen peroxide, control the levels of reactive oxygen and reactive nitrogen in the body, limit the potential toxicity of these substances while controlling reactive substance signaling, and are the first line of defense against oxygen radicals (Wang et al., 2018). SODs are commonly expressed in other hematological tumors and are highly expressed in AML (Chen and Kan, 2015). In addition, other factors significantly interfere with SODs expression, such as a slight increase in SOD1 levels and a significant decrease in SOD2 in AML cells with aberrant expression of the transcription factor GATA-1 (Lu et al., 2007). This adds to the complexity of the problem, but what remains constant is that the detoxifying superoxide action of SODs is essential in AML (Huang et al., 2000), and inhibition of SODs levels can lead to cell death (Chen and Kan, 2015).
Hypoxia is a hallmark of the tumor microenvironment, and the hypoxia-inducible factor (HIF) transcript is a major regulator of hypoxia (Fuhrmann and Brune, 2022), which controls tumor glycolysis, angiogenesis, and cell proliferation and migration by affecting downstream targets (Chen et al., 2022). Hematopoietic stem cells in the bone marrow require a hypoxic environment to maintain quiescence and preserve their self-renewal potential. HIF-1 increases the transcription of SLC7A11 and HO-1 to prevent ferroptosis (Feng et al., 2021; Yuan et al., 2022), unlike HIF-2, which promotes lipid peroxidation to induce ferroptosis (Singhal et al., 2021). In AML cells, both HIF1 and HIF-2 are more highly expressed than in normal cells (Wang et al., 2011; Rouault-Pierre et al., 2013), and their silencing induces apoptosis in LSCs and disrupts AML cells repair reconstruction in xenograft mice (Wang et al., 2011). A study on MDS/AML found that HIF-1 reduced ROS in iron-overloaded MDS/AML cells, prevented ferroptosis, and prompted cells to enter the S-phase-and G2/M-phase cell cycles. In contrast, the inhibition of HIF-1 expression in an iron overload model resulted in a significant increase in ROS, causing cellular injury (Zheng et al., 2017). Thus, HIF-1 is important for regulating ROS in AML cells and circumventing the excessive accumulation of oxides.
4.3 Restriction of PL-PUFA synthesis
LOXs are non-heme iron dioxygenases (Kuhn et al., 2005) named ALOX, and are widely believed to be central players in the prolipid peroxidation process, of which ALOX5, ALOX12, and ALOX15 have been shown to trigger ferroptosis (Shah et al., 2018). In AML cells, the expression of ALOX5/12/15 transcripts is lower than that in normal freshly isolated plasma (Vincent et al., 2008b). This may be a means for AML cells to avoid ferroptosis, limiting PUFA synthesis and peroxidation. However, the mechanism that inhibits reduced ALOX production is not known.
Stearoyl-CoA desaturase 1 (SCD1), an enzyme that catalyzes the rate-limiting step of monounsaturated fatty acid synthesis in cancer cells, is an important regulator of lipid metabolism. Normally, SCD1 overexpression in cancer cells promotes monounsaturated MUFAs production and significantly reduces lipid peroxide levels (Yi et al., 2020). SCD is a ferroptosis-protective gene in AML, and CIRCZBTB46 protects AML cells from ferroptosis by upregulating SCD1 expression in a mouse xenograft model. CIRCZBTB46 is an important circRNA that is frequently expressed in AML cells and regulates cell proliferation and ferroptosis (Long et al., 2023). Recently researchers have found that the FLT3-C/EBPα-SCD axis affects fatty acid synthesis oxidation in AML patients with FLT3-ITD mutations, and that inhibition of FLT3 mutations leads to decreased expression of SCD1, resulting in an increase in PUFAs compared to MUFAs, and ultimately sensitization of AML cells to ferroptosis (Sabatier et al., 2023). Thus SCD1 plays an important role in AML cells in avoiding the occurrence of ferroptosis by limiting PUFA synthesis.
Aldehyde dehydrogenase 3a2 (ALDH3A2), an enzyme responsible for detoxifying fatty aldehydes and producing C16-18 fatty acids, is expressed in leukemic stem cells with specific metabolic vulnerabilities. Aldehydes are metabolites of oxidative phosphorylation and nucleotide synthesis in cancer that are produced by lipid peroxidation and form the basis of ferroptosis (Monteiro et al., 2022). Yusuf et al. found a unique role for ALDH3A2 in reducing the susceptibility of leukemic stem cells to ferroptosis. The researchers inhibited GPX4 activity and increased the number of ROS, following which increased ALDH3A2 depletion and ferroptosis occurred. They found that ALDH3A2 and GPX4 act in parallel: in AML, simultaneous inhibition of ALDH3A2 and GPX4 activity has a far greater cell-killing effect than inhibition of GPX4 activity alone. ALDH3A2 and GPX4 differ in that ALDH3A2 is involved in fatty acid synthesis and, via its enzymatic reactions, both replenish damaged fatty acids and limit continued fatty acid damage. Thus, compared to GPX4, ALDH3A2 inhibited ferroptosis in AML cells more strongly.
5 Ferroptosis-related drugs in AML
In recent years, drugs for the treatment of AML ferroptosis, including small molecule inhibitors, natural compounds, and nano-delivery, are being developed and utilized, and new advances are being made, and here we summarize the mechanisms and targets of drugs targeting AML ferroptosis (Table 1).
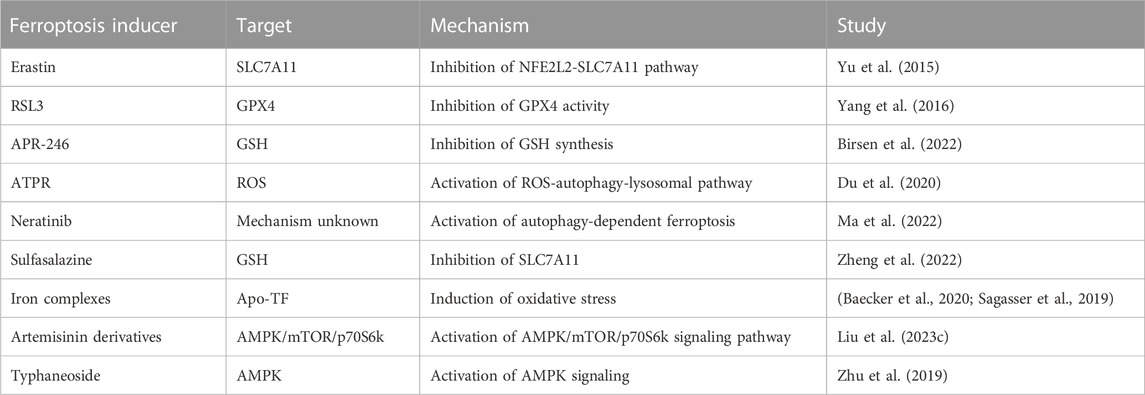
TABLE 1. Ferroptosis inducers for AML treatment.
5.1 Ferroptosis inducer
Ferroptosis inducers can be divided into two categories: those targeting systemic XC, such as Erastin and those targeting GPX4, such as RSL3. Erastin directly inhibits systemic XC-inhibited cystine uptake and reduces GSH synthesis. It has a good ferroptosis-inducing effect in many cancers (Hu et al., 2023; Xu et al., 2023). In AML cells, erastin inhibits cell growth and proliferation and has good antitumor potential (Yu et al., 2015). The ferroptosis inducer RSL3 directly inhibits GPX4 activity by covalently binding to selenocysteine (Yang et al., 2016). Studies have confirmed that the application of RSL3, an inhibitor targeting GPX4, in AML, triggers multiple cell deaths in AML cells, including ferroptosis. RSL3 can also be combined with chemotherapeutic agents such as cytarabine and adriamycin to improve the response rate to chemotherapy (Yu et al., 2015).
5.2 APR-246
A previous study found that APR-246 activates the TP53 pathway in MDS/AML cells with TP53 mutations, induces apoptotic transcription, and enhances the antitumor effects of azacitidine (Lehmann et al., 2012). The new study found that APR-246 decreased intracellular GSH content and induced lipid peroxidation to occur, while the anti-ferroptosis mechanism in AML cells enhanced the anti-leukemic activity of APR-246. In addition, this study further revealed that APR-246 action is not limited to TP53 mutated cells, which allows treatment to be applied to a wider range of patients with AML (Birsen et al., 2022). A phase II clinical trial of APR-246 combined with azacitidine in patients with TP53-mutated MDS/AML yielded good results (Mishra et al., 2022).
5.3 ATPR
ATPR is a novel all-trans retinoic acid derivative (Li et al., 2017) that shows superior anti-cancer efficacy compared to ATRA, and is expected to be a novel drug for the treatment of cancer (Du et al., 2018). Previous studies have shown that ATPR induces cell proliferation and differentiation by ROS accumulation and that ATPR also induces apoptotic autophagy in leukemia (Du et al., 2018). A recent study found that ATPR increases cellular autophagy by increasing ROS levels, which in turn regulate iron homeostasis and Nrf2, causing ferroptosis. Targeting iron homeostasis simultaneously induces ATPR-induced differentiation of AML cells and inhibits cell proliferation (Du et al., 2020).
5.4 Neratinib
Neratinib is a tyrosinase inhibitor, which has good anti-tumor effects in lung cancer and breast cancer. Applying neratinib to AML can inhibit the proliferation of HL-60 cells, induce G0/G1 cell cycle arrest, and undergo cellular autophagic ferroptosis. Researchers further explored the mechanism of action of neratinib on AML cells and found that the addition of autophagy inhibitors could attenuate the ferroptosis induced by neratinib on HL-60 cells, suggesting that neratinib inhibits the proliferation of AML through autophagy-dependent ferroptosis (Ma et al., 2022).
5.5 Sulfasalazine
Sulfasalazine is an anti-inflammatory drug that induces ferroptosis in cancer cells by inhibiting the function of SLA7A11 and preventing GSH synthesis (Zhu et al., 2022). Similarly, application of the systemic XC inhibitor sulfasalazine to AML cells decreases GSH levels and GPX4 activity (Zheng et al., 2022), inducing ferroptosis in the cells. Moreover, the synergistic action of sulfasalazine with anthracyclines and erythromycin can increase the anti-leukemic activity (Pardieu et al., 2022).
5.6 Iron complexes
Chlorido [4-carboxy-1,2-disalicylideneaminobenzene]iron (III) is an iron complex consisting of anti-leukemic activity of the same anti-leukemic effect of chlorido [N, N′-disalicylidene-1,2- phenylenediamine]iron (III) with the introduction of a 4-COOH group in the 1,2-phenylenediamine portion (Baecker et al., 2020), both of which are anti-AML cell metabolism and proliferation via ferroptosis (Sagasser et al., 2019), chlorido [4-carboxy-1,2-disalicylideneaminobenzene]iron (III) was more active than chlorido [N, N′-disalicylidene-1,2-phenylenediamine]iron (III) and also showed a good effect on perphenazine and erythromycin-resistant AML cells.
5.7 Artemisinin derivatives
Artemisinin is an antimalarial drug with more than ten derivatives including artesunate and dihydroartemisinin DHA, whose drug activity is mediated by ROS induction, has a remarkable anticancer effect, and performs excellently in AML cells (Kagan et al., 2021; Zhang et al., 2022b). DHA is an FAD-approved antimalarial drug with good anticancer efficacy against different human cancer cells. Previous studies have shown that DHA induces cancer cell death via ferroptosis in head and neck tumors and pancreatic cancer (Lin et al., 2016; Du et al., 2019). An experimental study on AML cells showed that DHA-induced autophagy in AML cells affected the AMPK/mTOR/p70S6k signaling pathway, degraded ferritin, increased the intracellular unstable iron pool, accumulated large amounts of ROS, induced lipid peroxidation, and ultimately led to ferroptosis (Du et al., 2019). A further study found that DHA induced early ferroptosis by promoting ferritin phagocytosis and iron overload and activated zinc metabolism to upregulate metallothionein and regulate GSH content, effectively promoting ferroptosis in AML cells (Grignano et al., 2023). Artesunate has a significant inhibitory effect on AML cells, and some experiments have shown that the anti-AML cellular activity of artesunate mediates cell differentiation mainly by modulating the ROS/Bim pathway to induce apoptosis and by relying on TFR cells to regulate intracellular iron homeostasis. Thus, artesunate derivatives have the potential to induce ferroptosis in AML cells (Liu et al., 2023c).
5.8 Typhaneoside
Typhaneosides extracted from Cyperus rotundus pollen have unique pharmacological activities. Zhu et al. found that TYP significantly inhibited the proliferation of AML cells and stalled the cell cycle in the G2/M phase. TYP-treated AML cells undergo autophagy, cause mitochondrial damage, and significantly increase intracellular ROS production (Zhu et al., 2019). It has been shown that autophagic signaling induces ferroptosis through ferritin degradation (Hou et al., 2016). Subsequently, Zhu et al. subsequently found that TYP enhanced lipid peroxidation, decreased GSH and GPX4 activity and induced ferroptosis in AML cells. At the same time, TYP did not affect normal cells, indicating that TYP is a low-toxicity and an efficient inducer of ferroptosis.
6 Conclusion
In recent years, the study of ferroptosis in AML has made significant progress. This new form of cell death differentiates itself from conventional cell death and holds great potential for anti-tumor applications. Metabolic alterations in AML cells, as well as genetic changes like IDH and FLT3, create a favorable environment for ferroptosis to occur. Several molecular targets closely associated with AML, including SLC7A11, FSP1, GPX4, and DPP4, have been identified through the construction of AML models (Song et al., 2021). Furthermore, certain drugs such as APR-246, ATPR, and other natural compounds have been discovered to induce ferroptosis in AML cells. As ferroptosis is closely linked to the metabolism and oxidative stress of AML cells, exploring the relationships between ferroptosis and the pathogenesis, progression, and drug resistance of AML could shed light on new avenues for treatment. Nevertheless, the established ferroptosis defense system of AML cells adds uncertainty to the occurrence of ferroptosis and the application of ferroptosis in AML treatment faces some difficulties (Figure 4).

FIGURE 4. Difficulties and recommendations in applying ferroptosis to AML treatment.
First, based on the available literature, we believe that ferroptosis in AML is a multiparty process. The induction of ferroptosis by a single pathway is likely to evoke a ferroptosis evasion mechanism in AML, leading to failure of the ferroptosis induction process. The paucity of available drugs targeting ferroptosis in AML cells also supports this. Therefore, the combined multi-pathway inhibition of ferroptosis evasion mechanisms should be sought to ensure that ferroptosis can occur successfully. Second, drugs such as Sorafenib and Brusatol have been validated as effective inducers of ferroptosis in some cancers (Tang et al., 2023; Ballout et al., 2022); however, they do not cause AML cell death by inducing ferroptosis, and it is necessary to dig deeper into the mechanism of the inability of ferroptosis to occur with these drugs, and to carry out a systematic ferroptosis study for AML. Third, there are interactions between ferroptosis and other cell death pathways, such as ferritin degradation associated with autophagy, increased iron and ROS levels (Hou et al., 2016), and the activation of excess ROS, which may cause cellular autophagy and chemotherapy resistance (Gu et al., 2020). Therefore, balancing these relationships is important. In addition to its association with cancer, ferroptosis is closely linked to pathological cell death in neurodegenerative and ischemic diseases, and the induction of ferroptosis may lead to additional toxicity and injury. At the same time, ferroptosis can also modulate inflammatory and immune responses (Zhong et al., 2022), which may pose challenges in AML treatment. Therefore, we believe that the development of nanotechnology allows iron NP inducers to precisely target cancer cells to avoid systemic adverse effects while inducing ferroptosis in AML cells (Liu K. et al., 2023). Further exploration of labeled AML-specific ferroptosis markers could accurately predict the effectiveness of ferroptosis therapy and improve the potential of ferroptosis in clinical applications.
By reviewing the alterations in metabolism and gene mutations that generate the susceptibility of AML cells to ferroptosis, this article provides insights into the therapeutic strategies for AML. It also highlights the coping strategies established by AML cells to avoid ferroptosis, as well as the potential therapeutic options against ferroptosis. Although the understanding of the pathogenesis and drug resistance mechanisms of AML from the perspective of ferroptosis remains to be improved, the application of ferroptosis to AML, including small molecule inhibitors, natural compounds, and nano-delivery of drugs may be a new direction for the treatment of AML in the future.
Author contributions
HZ: Conceptualization, Investigation, Writing–original draft. CS: Conceptualization, Investigation, Writing–original draft. QS: Supervision, Writing–review and editing. YL: Supervision, Writing–review and editing. CZ: Supervision, Writing–review and editing. CS: Writing–review and editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81973677; 82174222).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ali, D., Mohammad, D. K., Mujahed, H., Jonson-Videsater, K., Nore, B., Paul, C., et al. (2016). Anti-leukaemic effects induced by APR-246 are dependent on induction of oxidative stress and the NFE2L2/HMOX1 axis that can be targeted by PI3K and mTOR inhibitors in acute myeloid leukaemia cells. Br. J. Haematol. 174, 117–126. doi:10.1111/bjh.14036
Anandhan, A., Dodson, M., Schmidlin, C. J., Liu, P., and Zhang, D. D. (2020). Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell. Chem. Biol. 27, 436–447. doi:10.1016/j.chembiol.2020.03.011
Baecker, D., Ma, B. N., Sagasser, J., Schultz, L., Horschlager, C., Weinreich, M., et al. (2020). Amide and ester derivatives of chlorido[4-carboxy-1,2-disalicylideneaminobenzene]iron(iii) as necroptosis and ferroptosis inducers. Dalton Trans. 49, 6842–6853. doi:10.1039/d0dt00168f
Ballout, F., Lu, H., Chen, Z., Hu, T., Chen, L., Washington, M. K., et al. (2022). Targeting NRF2 sensitizes esophageal adenocarcinoma cells to cisplatin through induction of ferroptosis and apoptosis. Antioxidants (Basel) 11, 1859. doi:10.3390/antiox11101859
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi:10.1038/s41586-019-1705-2
Bertoli, S., Paubelle, E., Berard, E., Saland, E., Thomas, X., Tavitian, S., et al. (2019). Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur. J. Haematol. 102, 131–142. doi:10.1111/ejh.13183
Birsen, R., Larrue, C., Decroocq, J., Johnson, N., Guiraud, N., Gotanegre, M., et al. (2022). APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 107, 403–416. doi:10.3324/haematol.2020.259531
Bouchet, S., and Bauvois, B. (2014). Neutrophil gelatinase-associated lipocalin (NGAL), pro-matrix metalloproteinase-9 (pro-MMP-9) and their complex pro-MMP-9/NGAL in leukaemias. Cancers (Basel) 6, 796–812. doi:10.3390/cancers6020796
Bulavin, D. V., and Fornace, A. J. (2004). p38 MAP kinase's emerging role as a tumor suppressor. Adv. Cancer Res. 92, 95–118. doi:10.1016/S0065-230X(04)92005-2
Candido, S., Maestro, R., Polesel, J., Catania, A., Maira, F., Signorelli, S. S., et al. (2014). Roles of neutrophil gelatinase-associated lipocalin (NGAL) in human cancer. Oncotarget 5, 1576–1594. doi:10.18632/oncotarget.1738
Chai, X., Li, D., Cao, X., Zhang, Y., Mu, J., Lu, W., et al. (2015). ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci. Rep. 5, 10181. doi:10.1038/srep10181
Chang, Y. C., Lo, W. J., Huang, Y. T., Lin, C. L., Feng, C. C., Lin, H. T., et al. (2017). Deferasirox has strong anti-leukemia activity but may antagonize theanti-leukemia effect of doxorubicin. Leuk. Lymphoma 58, 1–12. doi:10.1080/10428194.2017.1280604
Chang, L. C., Chiang, S. K., Chen, S. E., Yu, Y. L., Chou, R. H., and Chang, W. C. (2018). Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137. doi:10.1016/j.canlet.2017.12.025
Chen, Y. L., and Kan, W. M. (2015). Down-regulation of superoxide dismutase 1 by PMA is involved in cell fate determination and mediated via protein kinase D2 in myeloid leukemia cells. Biochim. Biophys. Acta 1853, 2662–2675. doi:10.1016/j.bbamcr.2015.07.025
Chen, J. Y., Lai, Y. S., Tsai, H. J., Kuo, C. C., Yen, B. L., Yeh, S. P., et al. (2016). The oncometabolite R-2-hydroxyglutarate activates NF-κB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. 6, 32428. doi:10.1038/srep32428
Chen, W. J., Wu, H. T., Li, C. L., Lin, Y. K., Fang, Z. X., Lin, W. T., et al. (2021). Regulatory roles of six-transmembrane epithelial antigen of the prostate family members in the occurrence and development of malignant tumors. Front. Cell. Dev. Biol. 9, 752426. doi:10.3389/fcell.2021.752426
Chen, G., Wu, K., Li, H., Xia, D., and He, T. (2022). Role of hypoxia in the tumor microenvironment and targeted therapy. Front. Oncol. 12, 961637. doi:10.3389/fonc.2022.961637
Chen, R., Zou, J., Kang, R., and Tang, D. (2023). The redox protein HMGB1 in cell death and cancer. Antioxid. Redox Signal. doi:10.1089/ars.2023.0007
Chi, Y., Remsik, J., Kiseliovas, V., Derderian, C., Sener, U., Alghader, M., et al. (2020). Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 369, 276–282. doi:10.1126/science.aaz2193
Cuadrado, A., and Nebreda, A. R. (2010). Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417. doi:10.1042/BJ20100323
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. doi:10.1038/nature08617
Devireddy, L. R., Hart, D. O., Goetz, D. H., and Green, M. R. (2010). A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell. 141, 1006–1017. doi:10.1016/j.cell.2010.04.040
Di Savino, A., Gaidano, V., Palmieri, A., Crasto, F., Volpengo, A., Lorenzatti, R., et al. (2017). Clinical significance of TFR2 and EPOR expression in bone marrow cells in myelodysplastic syndromes. Br. J. Haematol. 176, 491–495. doi:10.1111/bjh.13968
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Dohner, H., Wei, A. H., and Lowenberg, B. (2021). Towards precision medicine for AML. Nat. Rev. Clin. Oncol. 18, 577–590. doi:10.1038/s41571-021-00509-w
Donovan, A., Brownlie, A., Zhou, Y., Shepard, J., Pratt, S. J., Moynihan, J., et al. (2000). Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781. doi:10.1038/35001596
Du, Y., Li, L. L., Chen, H., Wang, C., Qian, X. W., Feng, Y. B., et al. (2018). A novel all-trans retinoic acid derivative inhibits proliferation and induces apoptosis of myelodysplastic syndromes cell line SKM-1 cells via up-regulating p53. Int. Immunopharmacol. 65, 561–570. doi:10.1016/j.intimp.2018.10.041
Du, J., Wang, T., Li, Y., Zhou, Y., Wang, X., Yu, X., et al. (2019). DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 131, 356–369. doi:10.1016/j.freeradbiomed.2018.12.011
Du, Y., Bao, J., Zhang, M. J., Li, L. L., Xu, X. L., Chen, H., et al. (2020). Targeting ferroptosis contributes to ATPR-induced AML differentiation via ROS-autophagy-lysosomal pathway. Gene 755, 144889. doi:10.1016/j.gene.2020.144889
Duan, G., Li, J., Duan, Y., Zheng, C., Guo, Q., Li, F., et al. (2022). Mitochondrial iron metabolism: the crucial actors in diseases. Molecules 28, 29. doi:10.3390/molecules28010029
Fang, X., Cai, Z., Wang, H., Han, D., Cheng, Q., Zhang, P., et al. (2020). Loss of cardiac ferritin H facilitates cardiomyopathy via slc7a11-mediated ferroptosis. Circ. Res. 127, 486–501. doi:10.1161/CIRCRESAHA.120.316509
Farge, T., Saland, E., de Toni, F., Aroua, N., Hosseini, M., Perry, R., et al. (2017). Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 7, 716–735. doi:10.1158/2159-8290.CD-16-0441
Feng, H., Schorpp, K., Jin, J., Yozwiak, C. E., Hoffstrom, B. G., Decker, A. M., et al. (2020). Transferrin receptor is a specific ferroptosis marker. Cell. Rep. 30, 3411–3423. e3417. doi:10.1016/j.celrep.2020.02.049
Feng, X., Wang, S., Sun, Z., Dong, H., Yu, H., Huang, M., et al. (2021). Ferroptosis enhanced diabetic renal tubular injury via HIF-1α/HO-1 pathway in db/db mice. Front. Endocrinol. (Lausanne) 12, 626390. doi:10.3389/fendo.2021.626390
Fiancette, R., Vincent, C., Donnard, M., Bordessoule, D., Turlure, P., Trimoreau, F., et al. (2009). Genes encoding multiple forms of phospholipase A(2) are expressed in immature forms of human leukemic blasts. Leukemia 23, 1196–1199. doi:10.1038/leu.2009.36
Fuhrmann, D. C., and Brune, B. (2022). A graphical journey through iron metabolism, microRNAs, and hypoxia in ferroptosis. Redox Biol. 54, 102365. doi:10.1016/j.redox.2022.102365
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell. 59, 298–308. doi:10.1016/j.molcel.2015.06.011
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., and Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell. Res. 26, 1021–1032. doi:10.1038/cr.2016.95
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of mitochondria in ferroptosis. Mol. Cell. 73, 354–363. doi:10.1016/j.molcel.2018.10.042
Gao, J., Zhou, Q., Wu, D., and Chen, L. (2021). Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta 513, 6–12. doi:10.1016/j.cca.2020.12.005
Godfrey, R., Arora, D., Bauer, R., Stopp, S., Muller, J. P., Heinrich, T., et al. (2012). Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood 119, 4499–4511. doi:10.1182/blood-2011-02-336446
Grignano, E., Cantero-Aguilar, L., Tuerdi, Z., Chabane, T., Vazquez, R., Johnson, N., et al. (2023). Dihydroartemisinin-induced ferroptosis in acute myeloid leukemia: links to iron metabolism and metallothionein. Cell. Death Discov. 9, 97. doi:10.1038/s41420-023-01371-8
Gu, Y., Han, J., Jiang, C., and Zhang, Y. (2020). Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 59, 101036. doi:10.1016/j.arr.2020.101036
Guo, K., Lukacik, P., Papagrigoriou, E., Meier, M., Lee, W. H., Adamski, J., et al. (2006). Characterization of human DHRS6, an orphan short chain dehydrogenase/reductase enzyme: A novel, cytosolic type 2 R-beta-hydroxybutyrate dehydrogenase. J. Biol. Chem. 281, 10291–10297. doi:10.1074/jbc.M511346200
Hadian, K., and Stockwell, B. R. (2020). SnapShot: ferroptosis. Cell. 181, 1188. e1181. doi:10.1016/j.cell.2020.04.039
Han, C., Zheng, J., Li, F., Guo, W., and Cai, C. (2022). Novel prognostic signature for acute myeloid leukemia: bioinformatics analysis of combined CNV-driven and ferroptosis-related genes. Front. Genet. 13, 849437. doi:10.3389/fgene.2022.849437
Harada, N., Kanayama, M., Maruyama, A., Yoshida, A., Tazumi, K., Hosoya, T., et al. (2011). Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 508, 101–109. doi:10.1016/j.abb.2011.02.001
Heasman, S. A., Zaitseva, L., Bowles, K. M., Rushworth, S. A., and Macewan, D. J. (2011). Protection of acute myeloid leukaemia cells from apoptosis induced by front-line chemotherapeutics is mediated by haem oxygenase-1. Oncotarget 2, 658–668. doi:10.18632/oncotarget.321
Holbein, B. E., and Lehmann, C. (2023). Dysregulated iron homeostasis as common disease etiology and promising therapeutic target. Antioxidants (Basel) 12, 671. doi:10.3390/antiox12030671
Hole, P. S., Darley, R. L., and Tonks, A. (2011). Do reactive oxygen species play a role in myeloid leukemias? Blood 117, 5816–5826. doi:10.1182/blood-2011-01-326025
Hole, P. S., Zabkiewicz, J., Munje, C., Newton, Z., Pearn, L., White, P., et al. (2013). Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 122, 3322–3330. doi:10.1182/blood-2013-04-491944
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Hu, T., Pan, C., Zhang, T., Ni, M., Wang, W., Zhang, S., et al. (2022). Nrf2 overexpression increases the resistance of acute myeloid leukemia to cytarabine by inhibiting replication factor C4. Cancer Gene Ther. 29, 1773–1790. doi:10.1038/s41417-022-00501-1
Hu, Q., Dai, J., Zhang, Z., Yu, H., Zhang, J., Zhu, X., et al. (2023). ASS1-Mediated reductive carboxylation of cytosolic glutamine confers ferroptosis resistance in cancer cells. Cancer Res. 83, 1646–1665. doi:10.1158/0008-5472.CAN-22-1999
Huang, P., Feng, L., Oldham, E. A., Keating, M. J., and Plunkett, W. (2000). Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407, 390–395. doi:10.1038/35030140
Irwin, M. E., Rivera-Del Valle, N., and Chandra, J. (2013). Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal 18, 1349–1383. doi:10.1089/ars.2011.4258
Ishizawa, J., Zarabi, S. F., Davis, R. E., Halgas, O., Nii, T., Jitkova, Y., et al. (2019). Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell. 35, 721–737. doi:10.1016/j.ccell.2019.03.014
Ito, K., Hirao, A., Arai, F., Matsuoka, S., Takubo, K., Hamaguchi, I., et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002. doi:10.1038/nature02989
Jayavelu, A. K., Muller, J. P., Bauer, R., Bohmer, S. A., Lassig, J., Cerny-Reiterer, S., et al. (2016a). NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 30, 473–483. doi:10.1038/leu.2015.234
Jayavelu, A. K., Moloney, J. N., Bohmer, F. D., and Cotter, T. G. (2016b). NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 44, 1113–1122. doi:10.1016/j.exphem.2016.08.008
Jensen, P. D. (2004). Evaluation of iron overload. Br. J. Haematol. 124, 697–711. doi:10.1111/j.1365-2141.2004.04838.x
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 22, 266–282. doi:10.1038/s41580-020-00324-8
Jomen, W., Ohtake, T., Akita, T., Suto, D., Yagi, H., Osawa, Y., et al. (2022). Iron chelator deferasirox inhibits NF-κB activity in hepatoma cells and changes sorafenib-induced programmed cell deaths. Biomed. Pharmacother. 153, 113363. doi:10.1016/j.biopha.2022.113363
Juntilla, M. M., Patil, V. D., Calamito, M., Joshi, R. P., Birnbaum, M. J., and Koretzky, G. A. (2010). AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 115, 4030–4038. doi:10.1182/blood-2009-09-241000
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and arsenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi:10.1038/nchembio.2238
Kagan, A. B., Moses, B. S., Mott, B. T., Rai, G., Anders, N. M., Rudek, M. A., et al. (2021). A novel 2-carbon-linked dimeric artemisinin with potent antileukemic activity and favorable pharmacology. Front. Oncol. 11, 790037. doi:10.3389/fonc.2021.790037
Kang, N., Son, S., Min, S., Hong, H., Kim, C., An, J., et al. (2023). Stimuli-responsive ferroptosis for cancer therapy. Chem. Soc. Rev. 52, 3955–3972. doi:10.1039/d3cs00001j
Kannan, S., Irwin, M. E., Herbrich, S. M., Cheng, T., Patterson, L. L., Aitken, M. J. L., et al. (2022). Targeting the NRF2/HO-1 antioxidant pathway in FLT3-ITD-positive AML enhances therapy efficacy. Antioxidants (Basel) 11, 717. doi:10.3390/antiox11040717
Karathedath, S., Rajamani, B. M., Musheer Aalam, S. M., Abraham, A., Varatharajan, S., Krishnamurthy, P., et al. (2017). Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS One 12, e0177227. doi:10.1371/journal.pone.0177227
Kaweme, N. M., Zhou, S., Changwe, G. J., and Zhou, F. (2020). The significant role of redox system in myeloid leukemia: from pathogenesis to therapeutic applications. Biomark. Res. 8, 63. doi:10.1186/s40364-020-00242-z
Kerins, M. J., and Ooi, A. (2018). The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal 29, 1756–1773. doi:10.1089/ars.2017.7176
Khoshtabiat, L., Mahdavi, M., Dehghan, G., and Rashidi, M. R. (2016). Oxidative stress-induced apoptosis in chronic myelogenous leukemia K562 cells by an active compound from the dithio- carbamate family. Asian Pac J. Cancer Prev. 17, 4267–4273.
Kjeldsen, L., Cowland, J. B., and Borregaard, N. (2000). Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta 1482, 272–283. doi:10.1016/s0167-4838(00)00152-7
Koppula, P., Zhuang, L., and Gan, B. (2021). Cystine transporter slc7a11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 12, 599–620. doi:10.1007/s13238-020-00789-5
Kuhn, H., Saam, J., Eibach, S., Holzhutter, H. G., Ivanov, I., and Walther, M. (2005). Structural biology of mammalian lipoxygenases: enzymatic consequences of targeted alterations of the protein structure. Biochem. Biophys. Res. Commun. 338, 93–101. doi:10.1016/j.bbrc.2005.08.238
Lebon, D., Vergez, F., Bertoli, S., Harrivel, V., De Botton, S., Micol, J. B., et al. (2015). Hyperferritinemia at diagnosis predicts relapse and overall survival in younger AML patients with intermediate-risk cytogenetics. Leuk. Res. 39, 818–821. doi:10.1016/j.leukres.2015.05.001
Lehmann, S., Bykov, V. J., Ali, D., Andren, O., Cherif, H., Tidefelt, U., et al. (2012). Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 30, 3633–3639. doi:10.1200/JCO.2011.40.7783
Leng, X., Lin, H., Ding, T., Wang, Y., Wu, Y., Klumpp, S., et al. (2008). Lipocalin 2 is required for BCR-ABL-induced tumorigenesis. Oncogene 27, 6110–6119. doi:10.1038/onc.2008.209
Li, Y., Li, G., Wang, K., Xie, Y. Y., Zhou, R. P., Meng, Y., et al. (2017). Autophagy contributes to 4-Amino-2-Trifluoromethyl-Phenyl Retinate-induced differentiation in human acute promyelocytic leukemia NB4 cells. Toxicol. Appl. Pharmacol. 319, 1–11. doi:10.1016/j.taap.2017.01.016
Liang, D., Minikes, A. M., and Jiang, X. (2022). Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell. 82, 2215–2227. doi:10.1016/j.molcel.2022.03.022
Lin, R., Zhang, Z., Chen, L., Zhou, Y., Zou, P., Feng, C., et al. (2016). Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 381, 165–175. doi:10.1016/j.canlet.2016.07.033
Liu, Q., Wang, M., Hu, Y., Xing, H., Chen, X., Zhang, Y., et al. (2014). Significance of CD71 expression by flow cytometry in diagnosis of acute leukemia. Leuk. Lymphoma 55, 892–898. doi:10.3109/10428194.2013.819100
Liu, P., Ma, D., Wang, P., Pan, C., Fang, Q., and Wang, J. (2021). Nrf2 overexpression increases risk of high tumor mutation burden in acute myeloid leukemia by inhibiting MSH2. Cell. Death Dis. 12, 20. doi:10.1038/s41419-020-03331-x
Liu, Y., Wan, Y., Jiang, Y., Zhang, L., and Cheng, W. (2023a). GPX4: the hub of lipid oxidation, ferroptosis, disease and treatment. Biochim. Biophys. Acta Rev. Cancer 1878, 188890. doi:10.1016/j.bbcan.2023.188890
Liu, X., Zhong, S., Qiu, K., Chen, X., Wu, W., Zheng, J., et al. (2023b). Targeting NRF2 uncovered an intrinsic susceptibility of acute myeloid leukemia cells to ferroptosis. Exp. Hematol. Oncol. 12, 47. doi:10.1186/s40164-023-00411-4
Liu, Y., Li, H., Luo, Z., Yu, Y., Yang, J., Zhang, M., et al. (2023c). Artesunate, a new antimalarial clinical drug, exhibits potent anti-AML activity by targeting the ROS/Bim and TFRC/Fe(2+) pathways. Br. J. Pharmacol. 180, 701–720. doi:10.1111/bph.15986
Liu, K., Huang, L., Qi, S., Liu, S., Xie, W., Du, L., et al. (2023d). Ferroptosis: the entanglement between traditional drugs and nanodrugs in tumor therapy. Adv. Healthc. Mater 12, e2203085. doi:10.1002/adhm.202203085
Long, F., Lin, Z., Long, Q., Lu, Z., Zhu, K., Zhao, M., et al. (2023). CircZBTB46 protects acute myeloid leukemia cells from ferroptotic cell death by upregulating SCD. Cancers (Basel) 15, 459. doi:10.3390/cancers15020459
Lu, J., Chew, E. H., and Holmgren, A. (2007). Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U. S. A. 104, 12288–12293. doi:10.1073/pnas.0701549104
Lu, W., Zhao, M., Rajbhandary, S., Xie, F., Chai, X., Mu, J., et al. (2013). Free iron catalyzes oxidative damage to hematopoietic cells/mesenchymal stem cells in vitro and suppresses hematopoiesis in iron overload patients. Eur. J. Haematol. 91, 249–261. doi:10.1111/ejh.12159
Lyons, V. J., and Pappas, D. (2019). Affinity separation and subsequent terminal differentiation of acute myeloid leukemia cells using the human transferrin receptor (CD71) as a capture target. Analyst 144, 3369–3380. doi:10.1039/c8an02357c
Ma, H., Liu, Y., Miao, Z., Cheng, S., Zhu, Y., Wu, Y., et al. (2022). Neratinib inhibits proliferation and promotes apoptosis of acute myeloid leukemia cells by activating autophagy-dependent ferroptosis. Drug Dev. Res. 83, 1641–1653. doi:10.1002/ddr.21983
Mao, C., Liu, X., Zhang, Y., Lei, G., Yan, Y., Lee, H., et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. doi:10.1038/s41586-021-03539-7
Mishra, A., Tamari, R., DeZern, A. E., Byrne, M. T., Gooptu, M., Chen, Y. B., et al. (2022). Eprenetapopt plus azacitidine after allogeneic hematopoietic stem-cell transplantation for TP53-mutant acute myeloid leukemia and myelodysplastic syndromes. J. Clin. Oncol. 40, 3985–3993. doi:10.1200/JCO.22.00181
Monteiro, R. R. C., da Silva, S. S. O., Cavalcante, C. L., de Luna, F. M. T., Bolivar, J. M., Vieira, R. S., et al. (2022). Biosynthesis of alkanes/alkenes from fatty acids or derivatives (triacylglycerols or fatty aldehydes). Biotechnol. Adv. 61, 108045. doi:10.1016/j.biotechadv.2022.108045
Nakamaki, T., Kawabata, H., Saito, B., Matsunawa, M., Suzuki, J., Adachi, D., et al. (2004). Elevated levels of transferrin receptor 2 mRNA, not transferrin receptor 1 mRNA, are associated with increased survival in acute myeloid leukaemia. Br. J. Haematol. 125, 42–49. doi:10.1111/j.1365-2141.2004.04866.x
Niu, L. T., Wang, Y. Q., Wong, C. C. L., Gao, S. X., Mo, X. D., and Huang, X. J. (2021). Targeting IFN-gamma-inducible lysosomal thiol reductase overcomes chemoresistance in AML through regulating the ROS-mediated mitochondrial damage. Transl. Oncol. 14, 101159. doi:10.1016/j.tranon.2021.101159
Ohgami, R. S., Campagna, D. R., Greer, E. L., Antiochos, B., McDonald, A., Chen, J., et al. (2005). Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 37, 1264–1269. doi:10.1038/ng1658
Ohgami, R. S., Campagna, D. R., McDonald, A., and Fleming, M. D. (2006). The Steap proteins are metalloreductases. Blood 108, 1388–1394. doi:10.1182/blood-2006-02-003681
Pabst, T., Kortz, L., Fiedler, G. M., Ceglarek, U., Idle, J. R., and Beyoglu, D. (2017). The plasma lipidome in acute myeloid leukemia at diagnosis in relation to clinical disease features. BBA Clin. 7, 105–114. doi:10.1016/j.bbacli.2017.03.002
Pan, Y. Z., Li, Y., Guo, L. R., Zhao, Y. Y., and Zhao, X. J. (2008). Influence of expression of six transmembrane epithelial antigen of the prostate-1 on intracellular reactive oxygen species level and cell growth: an in vitro experiment. Zhonghua Yi Xue Za Zhi 88, 641–644.
Pardieu, B., Pasanisi, J., Ling, F., Dal Bello, R., Penneroux, J., Su, A., et al. (2022). Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 36, 1585–1595. doi:10.1038/s41375-022-01573-6
Park, H., Chung, H., Lee, J., Jang, J., Kim, Y., Kim, S. J., et al. (2017). Decitabine as a first-line treatment for older adults newly diagnosed with acute myeloid leukemia. Yonsei Med. J. 58, 35–42. doi:10.3349/ymj.2017.58.1.35
Pedrera, L., Espiritu, R. A., Ros, U., Weber, J., Schmitt, A., Stroh, J., et al. (2021). Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell. Death Differ. 28, 1644–1657. doi:10.1038/s41418-020-00691-x
Petzer, V., Wermke, M., Tymoszuk, P., Wolf, D., Seifert, M., Ovacin, R., et al. (2019). Enhanced labile plasma iron in hematopoietic stem cell transplanted patients promotes Aspergillus outgrowth. Blood Adv. 3, 1695–1700. doi:10.1182/bloodadvances.2019000043
Puhlmann, U., Ziemann, C., Ruedell, G., Vorwerk, H., Schaefer, D., Langebrake, C., et al. (2005). Impact of the cyclooxygenase system on doxorubicin-induced functional multidrug resistance 1 overexpression and doxorubicin sensitivity in acute myeloid leukemic HL-60 cells. J. Pharmacol. Exp. Ther. 312, 346–354. doi:10.1124/jpet.104.071571
Reddy, M. M., Fernandes, M. S., Salgia, R., Levine, R. L., Griffin, J. D., and Sattler, M. (2011). NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 25, 281–289. doi:10.1038/leu.2010.263
Rouault-Pierre, K., Lopez-Onieva, L., Foster, K., Anjos-Afonso, F., Lamrissi-Garcia, I., Serrano-Sanchez, M., et al. (2013). HIF-2α protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell. Stem Cell. 13, 549–563. doi:10.1016/j.stem.2013.08.011
Sabatier, M., Birsen, R., Lauture, L., Mouche, S., Angelino, P., Dehairs, J., et al. (2023). C/EBPα confers dependence to fatty acid anabolic pathways and vulnerability to lipid oxidative stress-induced ferroptosis in FLT3-mutant leukemia. Cancer Discov. 13, 1720–1747. doi:10.1158/2159-8290.CD-22-0411
Sagasser, J., Ma, B. N., Baecker, D., Salcher, S., Hermann, M., Lamprecht, J., et al. (2019). A new approach in cancer treatment: discovery of chlorido[N,N'-disalicylidene-1,2-phenylenediamine]iron(III) complexes as ferroptosis inducers. J. Med. Chem. 62, 8053–8061. doi:10.1021/acs.jmedchem.9b00814
Sallmyr, A., Fan, J., Datta, K., Kim, K. T., Grosu, D., Shapiro, P., et al. (2008). Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood 111, 3173–3182. doi:10.1182/blood-2007-05-092510
Shah, R., Shchepinov, M. S., and Pratt, D. A. (2018). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396. doi:10.1021/acscentsci.7b00589
Singhal, R., Mitta, S. R., Das, N. K., Kerk, S. A., Sajjakulnukit, P., Solanki, S., et al. (2021). HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest. 131, e143691. doi:10.1172/JCI143691
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi:10.1021/ja411006a
Skrtic, M., Sriskanthadevan, S., Jhas, B., Gebbia, M., Wang, X., Wang, Z., et al. (2011). Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 20, 674–688. doi:10.1016/j.ccr.2011.10.015
Song, Y., Tian, S., Zhang, P., Zhang, N., Shen, Y., and Deng, J. (2021). Construction and validation of a novel ferroptosis-related prognostic model for acute myeloid leukemia. Front. Genet. 12, 708699. doi:10.3389/fgene.2021.708699
Stockwell, B. R. (2022). Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 185, 2401–2421. doi:10.1016/j.cell.2022.06.003
Stuani, L., Sabatier, M., Saland, E., Cognet, G., Poupin, N., Bosc, C., et al. (2021). Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J. Exp. Med. 218, e20200924. doi:10.1084/jem.20200924
Tang, D., Kroemer, G., and Kang, R. (2023). Ferroptosis in hepatocellular carcinoma: from bench to bedside. Hepatology Publish Ahead of Print. doi:10.1097/HEP.0000000000000390
Thomas, D., Wu, M., Nakauchi, Y., Zheng, M., Thompson-Peach, C. A. L., Lim, K., et al. (2023). Dysregulated lipid synthesis by oncogenic IDH1 mutation is a targetable synthetic lethal vulnerability. Cancer Discov. 13, 496–515. doi:10.1158/2159-8290.CD-21-0218
Tomiotto-Pellissier, F., Alves, D. R., Miranda-Sapla, M. M., de Morais, S. M., Assolini, J. P., da Silva Bortoleti, B. T., et al. (2018). Caryocar coriaceum extracts exert leishmanicidal effect acting in promastigote forms by apoptosis-like mechanism and intracellular amastigotes by Nrf2/HO-1/ferritin dependent response and iron depletion: leishmanicidal effect of caryocar coriaceum leaf exracts. Biomed. Pharmacother. 98, 662–672. doi:10.1016/j.biopha.2017.12.083
Trachootham, D., Alexandre, J., and Huang, P. (2009). Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591. doi:10.1038/nrd2803
Udensi, U. K., and Tchounwou, P. B. (2014). Dual effect of oxidative stress on leukemia cancer induction and treatment. J. Exp. Clin. Cancer Res. 33, 106. doi:10.1186/s13046-014-0106-5
Vasquez-Vivar, J., Shi, Z., and Tan, S. (2022). Tetrahydrobiopterin in cell function and death mechanisms. Antioxid. Redox Signal 37, 171–183. doi:10.1089/ars.2021.0136
Vela, D., and Vela-Gaxha, Z. (2018). Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp. Mol. Med. 50, e436. doi:10.1038/emm.2017.273
Vercellino, I., and Sazanov, L. A. (2022). The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell. Biol. 23, 141–161. doi:10.1038/s41580-021-00415-0
Vincent, C., Donnard, M., Bordessoule, D., Turlure, P., Trimoreau, F., and Denizot, Y. (2008a). Cyclooxygenase-2 (Cox-2) and blast cells of patients with acute leukemia. Leuk. Res. 32, 671–673. doi:10.1016/j.leukres.2007.08.005
Vincent, C., Fiancette, R., Donnard, M., Bordessoule, D., Turlure, P., Trimoreau, F., et al. (2008b). 5-LOX, 12-LOX and 15-LOX in immature forms of human leukemic blasts. Leuk. Res. 32, 1756–1762. doi:10.1016/j.leukres.2008.05.005
Vitols, S., Angelin, B., Ericsson, S., Gahrton, G., Juliusson, G., Masquelier, M., et al. (1990). Uptake of low density lipoproteins by human leukemic cells in vivo: relation to plasma lipoprotein levels and possible relevance for selective chemotherapy. Proc. Natl. Acad. Sci. U. S. A. 87, 2598–2602. doi:10.1073/pnas.87.7.2598
Wang, Y., Liu, Y., Malek, S. N., Zheng, P., and Liu, Y. (2011). Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell. Stem Cell. 8, 399–411. doi:10.1016/j.stem.2011.02.006
Wang, Y. Q., Chang, S. Y., Wu, Q., Gou, Y. J., Jia, L., Cui, Y. M., et al. (2016). The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8, 308. doi:10.3389/fnagi.2016.00308
Wang, Y., Branicky, R., Noe, A., and Hekimi, S. (2018). Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J. Cell. Biol. 217, 1915–1928. doi:10.1083/jcb.201708007
Wang, X., Li, Y., Han, L., Li, J., Liu, C., and Sun, C. (2021). Role of flavonoids in the treatment of iron overload. Front. Cell. Dev. Biol. 9, 685364. doi:10.3389/fcell.2021.685364
Weber, S., Parmon, A., Kurrle, N., Schnutgen, F., and Serve, H. (2020). The clinical significance of iron overload and iron metabolism in myelodysplastic syndrome and acute myeloid leukemia. Front. Immunol. 11, 627662. doi:10.3389/fimmu.2020.627662
Wei, S., Wang, Y., Chai, Q., Fang, Q., Zhang, Y., and Wang, J. (2014). Potential crosstalk of Ca2+-ROS-dependent mechanism involved in apoptosis of Kasumi-1 cells mediated by heme oxygenase-1 small interfering RNA. Int. J. Oncol. 45, 2373–2384. doi:10.3892/ijo.2014.2661
Wei, S., Wang, Y., Chai, Q., Fang, Q., Zhang, Y., and Wang, J. (2015a). In vivo and in vitro effects of heme oxygenase-1 silencing on the survival of acute myelocytic leukemia-M2 cells. Exp. Ther. Med. 9, 931–940. doi:10.3892/etm.2015.2209
Wei, S. X., Wang, Y. T., Chai, Q. X., Fang, Q., Zhang, Y. M., and Wang, J. S. (2015b). Proapoptotic effects of heme oxygenase-1 inhibitor on Kasumi-1 cells via the ATF4/CHOP/Ire-1α pathway. Genet. Mol. Res. 14, 5994–6002. doi:10.4238/2015.June.1.17
Wu, B., Shi, N., Sun, L., and Liu, L. (2016). Clinical value of high expression level of CD71 in acute myeloid leukemia. Neoplasma 63, 809–815. doi:10.4149/neo_2016_519
Xin, S., and Schick, J. A. (2023). PUFAs dictate the balance of power in ferroptosis. Cell. Calcium 110, 102703. doi:10.1016/j.ceca.2023.102703
Xu, C., Li, S., Chen, J., Wang, H., Li, Z., Deng, Q., et al. (2023). Doxorubicin and erastin co-loaded hydroxyethyl starch-polycaprolactone nanoparticles for synergistic cancer therapy. J. Control Release 356, 256–271. doi:10.1016/j.jconrel.2023.03.001
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi:10.1016/j.chembiol.2008.02.010
Yang, W. C., Tsai, W. C., Lin, P. M., Yang, M. Y., Liu, Y. C., Chang, C. S., et al. (2013a). Human BDH2, an anti-apoptosis factor, is a novel poor prognostic factor for de novo cytogenetically normal acute myeloid leukemia. J. Biomed. Sci. 20, 58. doi:10.1186/1423-0127-20-58
Yang, W. C., Lin, P. M., Yang, M. Y., Liu, Y. C., Chang, C. S., Chou, W. C., et al. (2013b). Higher lipocalin 2 expression may represent an independent favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Leuk. Lymphoma 54, 1614–1625. doi:10.3109/10428194.2012.749402
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 113, E4966–E4975. doi:10.1073/pnas.1603244113
Yang, W. H., Huang, Z., Wu, J., Ding, C. C., Murphy, S. K., and Chi, J. T. (2020). A TAZ-ANGPTL4-NOX2 Axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol. Cancer Res. 18, 79–90. doi:10.1158/1541-7786.MCR-19-0691
Ye, F., Chai, W., Xie, M., Yang, M., Yu, Y., Cao, L., et al. (2019). HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am. J. Cancer Res. 9, 730–739.
Yeh, C. J., Taylor, C. G., and Faulk, W. P. (1984). Transferrin binding by peripheral blood mononuclear cells in human lymphomas, myelomas and leukemias. Vox Sang. 46, 217–223. doi:10.1111/j.1423-0410.1984.tb00078.x
Yi, J., Zhu, J., Wu, J., Thompson, C. B., and Jiang, X. (2020). Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. U. S. A. 117, 31189–31197. doi:10.1073/pnas.2017152117
Yu, Y., Xie, Y., Cao, L., Yang, L., Yang, M., Lotze, M. T., et al. (2015). The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2, e1054549. doi:10.1080/23723556.2015.1054549
Yu, F., Zhang, Q., Liu, H., Liu, J., Yang, S., Luo, X., et al. (2022). Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell. Discov. 8, 40. doi:10.1038/s41421-022-00390-6
Yu, Y., Meng, Y., Xu, X., Tong, T., He, C., Wang, L., et al. (2023). A ferroptosis-inducing and leukemic cell-targeting drug nanocarrier formed by redox-responsive cysteine polymer for acute myeloid leukemia therapy. ACS Nano 17, 3334–3345. doi:10.1021/acsnano.2c06313
Yuan, S., Wei, C., Liu, G., Zhang, L., Li, J., Li, L., et al. (2022). Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell. Prolif. 55, e13158. doi:10.1111/cpr.13158
Zdralevic, M., Vucetic, M., Daher, B., Marchiq, I., Parks, S. K., and Pouyssegur, J. (2018). Disrupting the 'Warburg effect' re-routes cancer cells to OXPHOS offering a vulnerability point via 'ferroptosis'-induced cell death. Adv. Biol. Regul. 68, 55–63. doi:10.1016/j.jbior.2017.12.002
Zhang, H., Fang, H., and Wang, K. (2014). Reactive oxygen species in eradicating acute myeloid leukemic stem cells. Stem Cell. Investig. 1, 13. doi:10.3978/j.issn.2306-9759.2014.04.03
Zhang, Y., Swanda, R. V., Nie, L., Liu, X., Wang, C., Lee, H., et al. (2021). mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 12, 1589. doi:10.1038/s41467-021-21841-w
Zhang, J., Liu, Y., Li, Q., Xu, A., Hu, Y., and Sun, C. (2022a). Ferroptosis in hematological malignancies and its potential network with abnormal tumor metabolism. Biomed. Pharmacother. 148, 112747. doi:10.1016/j.biopha.2022.112747
Zhang, J., Wang, Y., Yin, C., Gong, P., Zhang, Z., Zhao, L., et al. (2022b). Artesunate improves venetoclax plus cytarabine AML cell targeting by regulating the Noxa/Bim/Mcl-1/p-Chk1 axis. Cell. Death Dis. 13, 379. doi:10.1038/s41419-022-04810-z
Zhe, N., Wang, J., Chen, S., Lin, X., Chai, Q., Zhang, Y., et al. (2015). Heme oxygenase-1 plays a crucial role in chemoresistance in acute myeloid leukemia. Hematology 20, 384–391. doi:10.1179/1607845414Y.0000000212
Zheng, J., and Conrad, M. (2020). The metabolic underpinnings of ferroptosis. Cell. Metab. 32, 920–937. doi:10.1016/j.cmet.2020.10.011
Zheng, Q. Q., Zhao, Y. S., Guo, J., Zhao, S. D., Song, L. X., Fei, C. M., et al. (2017). Iron overload promotes erythroid apoptosis through regulating HIF-1a/ROS signaling pathway in patients with myelodysplastic syndrome. Leuk. Res. 58, 55–62. doi:10.1016/j.leukres.2017.04.005
Zheng, Z., Hong, X., Huang, X., Jiang, X., Jiang, H., Huang, Y., et al. (2022). Comprehensive analysis of ferroptosis-related gene signatures as a potential therapeutic target for acute myeloid leukemia: A bioinformatics analysis and experimental verification. Front. Oncol. 12, 930654. doi:10.3389/fonc.2022.930654
Zheng, C., Wang, C., Sun, D., Wang, H., Li, B., Liu, G., et al. (2023). Structure-activity relationship study of RSL3-based GPX4 degraders and its potential noncovalent optimization. Eur. J. Med. Chem. 255, 115393. doi:10.1016/j.ejmech.2023.115393
Zhong, F. M., Yao, F. Y., Liu, J., Zhang, H. B., Zhang, J., Zhang, N., et al. (2022). Ferroptosis-related molecular patterns reveal immune escape, inflammatory development and lipid metabolism characteristics of the tumor microenvironment in acute myeloid leukemia. Front. Oncol. 12, 888570. doi:10.3389/fonc.2022.888570
Zhu, H. Y., Huang, Z. X., Chen, G. Q., Sheng, F., and Zheng, Y. S. (2019). Typhaneoside prevents acute myeloid leukemia (AML) through suppressing proliferation and inducing ferroptosis associated with autophagy. Biochem. Biophys. Res. Commun. 516, 1265–1271. doi:10.1016/j.bbrc.2019.06.070
Zhu, X., Chen, Q., Xie, L., Chen, W., Jiang, Y., Song, E., et al. (2022). Iron ion and sulfasalazine-loaded polydopamine nanoparticles for Fenton reaction and glutathione peroxidase 4 inactivation for enhanced cancer ferrotherapy. Acta Biomater. 145, 210–221. doi:10.1016/j.actbio.2022.04.024
Glossary

Keywords: ferroptosis, acute myeloid leukemia, iron metabolism, ROS, lipid metabolism
Citation: Zhang H, Sun C, Sun Q, Li Y, Zhou C and Sun C (2023) Susceptibility of acute myeloid leukemia cells to ferroptosis and evasion strategies. Front. Mol. Biosci. 10:1275774. doi: 10.3389/fmolb.2023.1275774
Received: 10 August 2023; Accepted: 15 September 2023;
Published: 25 September 2023.
Edited by:
Michela Grosso, University of Naples Federico II, ItalyReviewed by:
Archita Venugopal Menon, Northeastern University, United StatesRajni Kumari, Albert Einstein College of Medicine, United States
Copyright © 2023 Zhang, Sun, Sun, Li, Zhou and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Zhou, Mjg1NzU4MDc4QDE2My5jb20=; Changgang Sun, c2NnZG9jdG9yQDEyNi5jb20=
†These authors have contributed equally to this work