 Pratap Vydyam
Pratap Vydyam Nabamita Roy
Nabamita Roy Mrinal Kanti Bhattacharyya
Mrinal Kanti Bhattacharyya
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 01 August 2023
Sec. Genome Organization and Dynamics
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1223682
This article is part of the Research Topic Microbial growth and post-exponential processes: implications for pathogenesis, survival and evolution View all articles
The human malaria parasite Plasmodium falciparum maintains the chronicity of infections through antigenic variation, a well-coordinated immune evasion mechanism. The most prominent molecular determinant of antigenic variation in this parasite includes the members of the var multigene family. Homologous recombination (HR)-mediated genomic rearrangements have been implicated to play a major role in var gene diversification. However, the key molecular factors involved in the generation of diversity at var loci are less known. Here, we tested the hypothesis that PfRad51 could carry out recombination between var genes that are not homologous but homeologous in nature. We employed the whole-genome sequencing (WGS) approach to investigate recombination events among var sequences over 100 generations and compared the rate of sequence rearrangement at the var loci in both PfRad51-proficient and -deficient parasite lines. This brief report provides evidence that the loss of the key recombinase function renders the parasite with inefficient HR and results in fewer recombination events among the var sequences, thereby impacting the diversification of the var gene repertoire.
Malaria is one of the leading life-threatening problems worldwide. This severity is attributed to chronic infections and the innate immune evasion strategy possessed by Plasmodium falciparum. One of the remarkable biological features of P. falciparum is the expression of polymorphic family proteins on the surface of infected erythrocytes (IE) called P. falciparum erythrocyte membrane protein 1 (PfEMP1), a major antigenic determinant that enables IE binding and evades the host antibody response by clonal antigenic variation (Kyes et al., 2001). Antigenic diversity generated by the polymorphic, var multigene family is the foundation for Plasmodium immune evasion (Scherf et al., 2008; Petter and Duffy, 2015). PfEMP1 is encoded by a family of hypervariable genes with long ORFs, namely, var genes. In Plasmodium, nearly 60 copies of var genes per haploid genome are clustered majorly at the sub-telomeric region, with a diverse structure, having a large exon-1 (4–10 kb) that encodes a variable extracellular domain and a small conserved exon-2 encoding the cytosolic domain of PfEMP1 which facilitates anchoring to the host erythrocyte membrane (Baruch et al., 1995; Su et al., 1995; Gardner et al., 2002). At the subdomain level, there are homology blocks within the PfEMP1 encoding region that are separated by hypervariable regions, which presumably facilitates recombination between semi-homologous genes (Bull et al., 2008). The lesser degree of sequence conservation in the hypervariable var loci, in comparison to the rest of the genome, suggests that they must undergo a high rate of mutation or recombination (Bockhorst et al., 2007). The majority of recombination events take place within or near var genes, and these are counted as recombination hotspots (Taylor et al., 2000; Mu et al., 2005; Mu et al., 2007). During asexual replication of parasites, these var genes undergo gene recombination through mitosis, resulting in diversification. An increase in diversity is achieved when ectopic (non-allelic) recombination events take place between var genes (Duffy et al., 2009). The frequency of mitotic recombination and the generation of novel var sequences were determined through whole-genome sequencing and microarray analyses (Bopp et al., 2013; Claessens et al., 2014). A high frequency of var gene rearrangements was observed in the asexual stage of parasites (Claessens et al., 2014). In the progeny generated from an experimental genetic cross between two different strains of Plasmodium, it was observed that the frequency of var recombination is much higher than the overall meiotic recombination rate for the bulk of the genome (Freitas-Junior et al., 2000). Mitotic recombination during asexual replication is thought to stem from the necessity of repairing DNA double-strand breaks that are spontaneously generated during the normal course of the development of the parasite (Kolodner et al., 2002; Moynahan and Jasin, 2010). Apparently, in P. falciparum, DSB repair is considered to rely almost exclusively on homologous recombination (HR) due to a lack of efficient classical non-homologous end joining (NHEJ) (Kirkman and Deitsch, 2012; Kirkman et al., 2014). The genome of Plasmodium is haploid in most of its life cycle (except for a short period during the mosquito stage). Hence, the unbroken sister chromatid acts as homologous templates for the HR mechanism. However, for the repair of the genes belonging to the multigene family, several additional homologous templates are available. Such a recombination event potentially gives rise to the generation of new mosaic sequences.
During DSB repair, strand exchange protein Rad51 utilises the unbroken homologous DNA template, such as the sister chromatid or homologous chromosome, to repair the broken chromosome. Rad51 can distinguish the homologous template from the non-homologous template during this process. However, the stringency of such discrimination is not complete. For example, Rad51 could carry out the strand-exchange reaction between the maternal allele and the paternal allele during meiotic recombination. Thus, Rad51 can tolerate and bypass short stretches of heterologous sequences that are present in the long stretch of homologous sequences. Such templates that contain short blocks of heterologous sequences in the homologous templates are called homeologous sequences. In the model eukaryote Saccharomyces cerevisiae, Rad51 could bypass 6–9 bp heterologous blocks and in E. coli, RecA could bypass up to 200-bp-long heterologous blocks (Iype et al., 1994).
In P. falciparum, the recombinase orthologue PfRad51 was identified, and its strand-exchange activity was characterised (Bhattacharyya and Kumar, 2003; Bhattacharyya et al., 2005). A chemical inhibitor of PfRad51 was found to block the ATPase activity, strand-exchange activity, and multimerization of the PfRad51 protein (Vydyam et al., 2019). In a genetic study, the overexpression of a dominant negative mutant of PfRad51K143R abrogated DSB repair in Plasmodium berghei, emphasising the fact that HR is the major DSB repair mechanism in this parasite. Rad51 functions as a hexameric protein, with each monomer requiring ATPase activity. The dominant negative mutant PfRad51K143R has the ability to interact within the hexameric ring along with wild-type proteins, effectively suppressing the activity of the wild-type Rad51 protein. Thus, this mutant exerts a dominant negative effect, inhibiting the function of Rad51 (Roy et al., 2014).
The following two questions remain unanswered: whether the PfRad51-mediated HR pathway actually contributes to var gene rearrangement? Whether the role of PfRad51 could be compensated by another HR protein? In this study, aiming at answering the aforementioned questions, we used a transgenic parasite strain harbouring a dominant negative allele of PfRad51. Initially, we determined the frequencies and length of heterologous blocks within var sequences. We generated dominant negative Pfrad51 (PfRad51K143R) parasite lines and monitored the rate of rearrangements over hundred generations for both PfRad51wild-type and mutant parasite lines. Whole-genome sequencing analyses revealed a significant decrease in the rate of change of nucleotide variations (NVs) and structural variations (SVs) at var genes in PfRad51 mutant parasites, suggesting a potential role in var gene diversity.
To study the nature of heterology blocks, we obtained the genomic DNA sequence from the Plasmodium genome database, PlasmoDB. We retrieved the var gene sequences and grouped them according to their upstream promoter sequence (UPS) region. Sequence alignment was carried out to get the heterologous blocks among var genes using the bioinformatics tool CLUSTALW. We analysed two types of heterologous blocks: nucleotide substitution and insertion/deletion. The length and the occurrence frequencies of the heterologous blocks were plotted using GraphPad Prism.
Synchronous early ring-stage parasites with 6%–8% parasitaemia resuspended in pre-warmed Cytomix buffer (10 mM K2HPO4 pH 7.6, 120 mM KCl, 0.15 mM CaCl2, 25 mM HEPES pH 7.6, 2 mM EGTA pH 7.6, and 5 mM MgCl2) were electroporated with 100 µg plasmid DNA (PfCENV3–PfRad51K143R) using a Bio-Rad Gene Pulsar (0.31 kV, 950 µF, and ∝ resistance). Post-transfection, once the parasitaemia reached 4%–8%, the cells were kept under blasticidine (Sigma-Aldrich) pressure (2.4 μg/mL) until the transfectants appeared. The presence of the plasmid PfCENV3–PfRad51K143R was confirmed by amplifying the PCR fragment size of 1,325 bp using primer pairs OMKB 198 (GGT GAG CTA GCT AAT AGG C) and OMKB 462 (TTA CAC TTT ATG CTT CCG GC). Similarly, the transfectants possessing the empty PfCENV3 vector were confirmed with vector-specific primers OMKB 501 (ACA ACC ATT ACC TGT CCA C) and OMKB 462 that yielded an amplicon of 1,171 bp.
The parasite culture with 6%–8% parasitaemia was harvested and treated with saponin (0.15%) (Sigma-Aldrich) at 37°C for 30 min. The parasite pellet was resuspended in the lysis buffer (10 mM Tris-HCl pH 8, 20 mM EDTA pH 8, 0.5% SDS, and 0.1 mg proteinase K) and incubated for 3 h with intermittent mixing. After incubation, equal volumes of PCIA [phenol, chloroform, and isoamyl alcohol at a ratio of 25:24:1 (v/v/v)] were added, and the aqueous layer was collected upon centrifugation at 12,000 rpm for 15 min at room temperature. The aqueous layer was treated with RNase for 30 min at 37°C, followed by PCIA treatment. The sample was subjected to precipitation by addition of sodium acetate (1/10th vol) and 100% ethanol (2.2 vol) and incubated at −80°C overnight, followed by centrifugation at 12,000 rpm at 4°C, and the DNA were resuspended in 1x TE buffer.
The previously published protocol was followed for cloning and re-cloning parasite lines using the limiting dilution method (Butterworth et al., 2011). Briefly, parasite strains of Pf3D7, PfCENV3, and PfDNRad51 were cultured routinely in vitro. All isogenic lines were subjected to re-cloning at their designated time points, including the 0th, 50th, and 100th generation. It was made sure that the culture had >90% of the singly infected erythrocytes. Parasite cultures were seeded at 0.2 to 0.5 parasites per well in a 96-well plate in 100 µL with 2% HCT Complete medium. The culture media were changed on days 4, 7, 11, 14, and 18. Sufficient number of parasites was visible after 21 days, and the culture was utilised for further experiments. It is worth mentioning that each re-cloning process, from the time of seeding to the time of harvesting parasites for DNA isolation, took approximately 10 generations time. The Nth generation is defined as the day of seeding and not the day of harvesting parasites.
A measure of 2 μg of genomic DNA samples from each time point was used for whole-genome shotgun (WGS) library preparation with a fragment length ranging from 300 to 500 bp. The generated DNA library was subjected to paired-end sequencing of 150 bp using the Illumina NextSeq (150 × 2) sequencing platform (Genotypic Technology, Bangalore, India). The WGS analysis entailed the sequencing of the 23-MB Plasmodium falciparum genome using 150-bp reads, which resulted in a genome coverage of ×20. This sequencing approach yielded an adequate quantity of high-quality reads, exceeding 2 million reads per clone for both the wild-type and mutant parasite lines. The extensive dataset provided a comprehensive perspective of the genomic landscape, facilitating a thorough examination of the genetic composition and variations. These high-quality reads were aligned with the reference genome sequence of Pf3D7 (PlasmoDB.), with the coverage of each sample at a depth of ×10 and ×20. We identified both nucleotide variations that include single-nucleotide polymorphisms (SNPs) and insertions/deletions (INDELs) between 1 and 20 bp size and structural variations (insertions, deletions, translocations, and inversions with the size of >300 bp) using SAM, BCF, and DELLY tools.
To visualise the WGS data for the easy identification of similarities and differences between genomic sequences, we prepared Circos plots for each sample using an online tool Circa (http://circos.ca). The circular ideogram layout facilitates the display of all chromosomes and the position of SNPs, INDELs, and structural variations on each chromosome. The data were represented with identified SNPs, INDELs, and translocations of six different samples from both wild-type and PfRad51 mutant parasites in six different plots. A typical Circos plot contained all 14 chromosomes of P. falciparum arranged in an ideogram track. Both SNPs and INDELs in the core genome and var genes are represented as a histogram and Scatchard plot, respectively. The translocations are represented in blue and orange lines for the core genome and var genes, respectively.
We estimated the rate of change of both nucleotide variations (SNPs and INDELs) and structural variations per generation per nucleotide in wild-type and PfRad51 mutant parasites. The rate of change was calculated by taking the total number of unique changes in nucleotide or structural variations present in that particular strain per generation per nucleotide. The genome sequences of the 0th generation of both wild-type and PfRad51 mutant parasites were taken as the reference, and any new or missing nucleotide variations or structural variations were taken as changes. The rate of change for the var gene region was calculated by considering the total size of the var genes, while the rate of change for the rest of the genome was calculated considering the genome size.
As var sequences are homeologous in nature with respect to each other, it is important to estimate the length and the frequency of such heterologous blocks at var genes. It is intitutive that shorter heterologous blocks could be bypassed during recombination events between var sequences, while the longer blocks may not be tolerated. To analyse var genes, we retrieved var gene sequences (P. falciparum 3D7 genome) from the PlasmoDB database. Sequence alignment was carried out for each UPS group of var genes using CLUSTALW (DNASTAR software), and the length of the heterologous blocks was analysed. After the alignment of the sequence, deletions/insertions or substitutions of the base pair that caused sequence divergence were identified as heterologous blocks. Every single-base substitution from each UPS group was analysed and represented in graphs by taking the length of heterologous blocks and the total number within the group (Figures 1A–E). The consensus sequences from each UPS group were also analysed to determine the lengths and frequencies of heterologous blocks across UPS groups (Figure 1F). Similarly, insertions/deletions from each UPS group and across UPS groups were also analysed (Figures 2A–F). From the data, it is evident that var genes mostly have short heterologous blocks, ranging from 1 to 6 bp. Additionally, the length of such heterologous blocks was inversely proportional to the frequency of their occurrence. There are very few base substitution blocks with lengths greater than 6 bps, and a similar pattern was also observed with insertions/deletions.
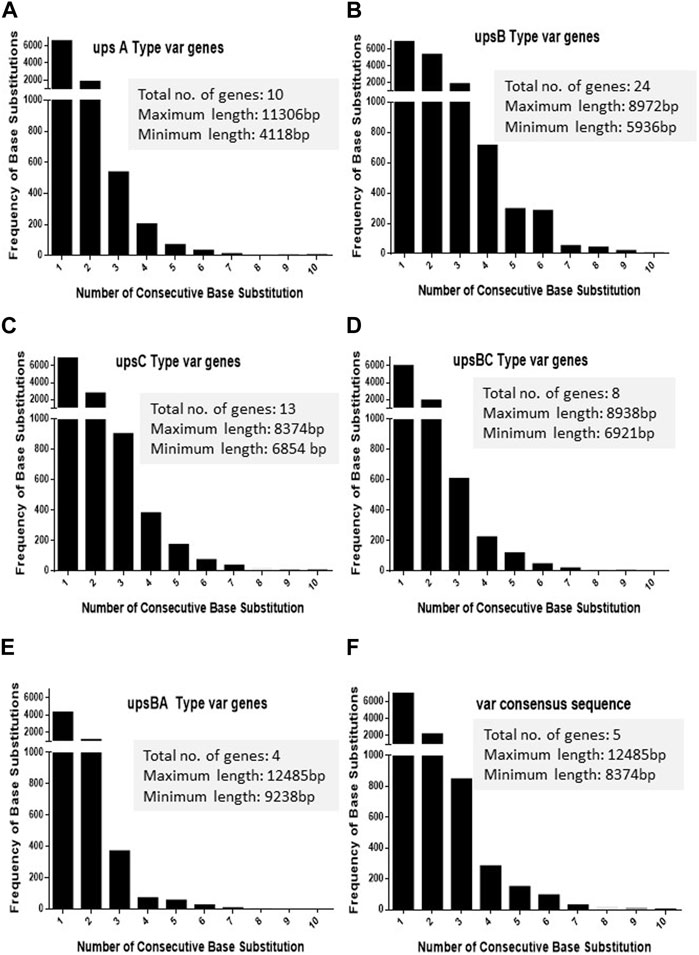
FIGURE 1. Distribution of heterologous blocks due to consecutive base substitutions in var genes. The var genes were divided into five groups according to their UPS type. (A–E) The number of consecutive base substitutions were determined and plotted against the frequency of their occurrence. The total number of genes in each group, along with the maximum and minimum lengths of the genes is mentioned in the box. (F) The consensus var sequences from each UPS group were compared against each other, and the frequencies of consecutive base substitutions were plotted.
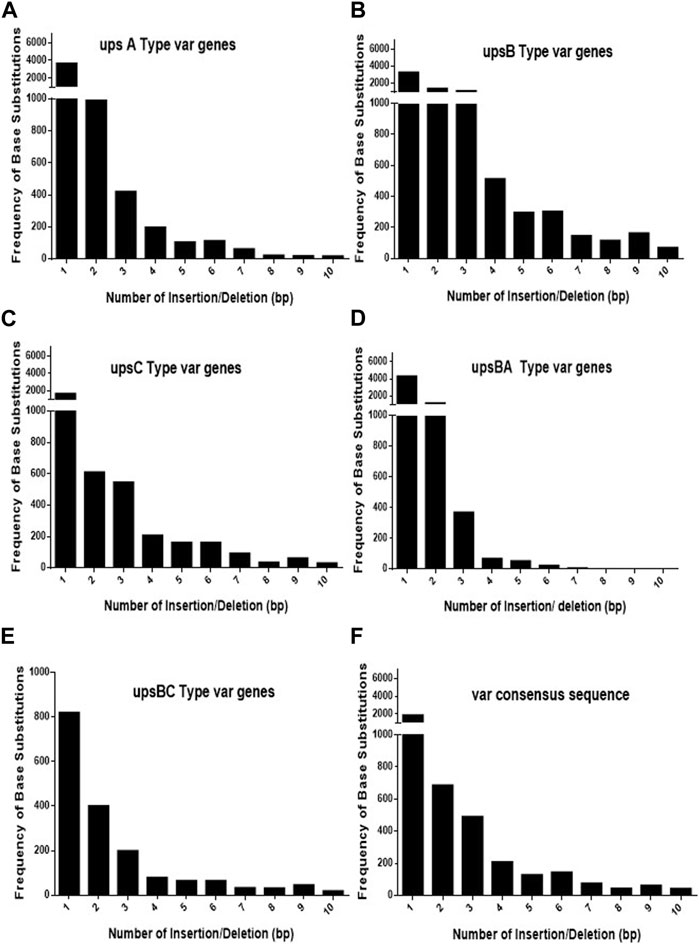
FIGURE 2. Distribution of heterologous blocks due to consecutive insertions or deletions of bases in var genes. (A–E) The var genes grouped into five UPS groups were inspected for the number of consecutive insertions/deletions of bases and plotted against the frequency of their occurrence. (F) The consensus var sequences from each UPS group were compared against each other, and the frequencies of the consecutive INDELs were plotted.
Since the lengths of heterologous blocks were found to be below the permissible limit of heterology bypass by eukaryotic Rad51, we hypothesised that PfRad51 should be able to carry out recombination between homeologous var sequences. To investigate the role of PfRad51 in var gene recombination, we have expressed a dominant negative allele of PfRad51K143R in the P. falciparum in vitro culture. This particular mutant was reported to be defective in DSB repair (Jha et al., 2021). We have chosen a Pf3D7 strain for our investigation of gene diversification studies because of its complete annotated genome (Gardner et al., 2002). We sought to identify nucleotide and structural variations at the var loci and in the core genome. To this end, we successively cultured isogenic lines of both wild-type and mutant parasites for 100 generations. At designated time intervals (0th, 50th, and 100th generation), we re-cloned both parasite lines and isolate genomic DNA from individual clones, and representative clones were subjected to Illumina platform-based WGS to obtain the mutation score as described in methodology. The total number of nucleotide variants (SNPs and micro-INDELs with the size of <20 bp) and structural variations (translocations, inversions, and duplications with >300 bp) distributed throughout the genome were identified at all the time points of both parasite lines using SAM, DELLY, and BCF tools (Morel et al., 1994; Holmes et al., 2001). A detailed inspection of NVs generated in both wild-type and DN parasites were represented as a histogram on all 14 chromosomes in Circos plots (Figure 3). Similarly, structural variations are represented as blue and orange lines in Circos maps of the respective generation for the core genome and var gene region, respectively, of both parasite lines (Figure 3).
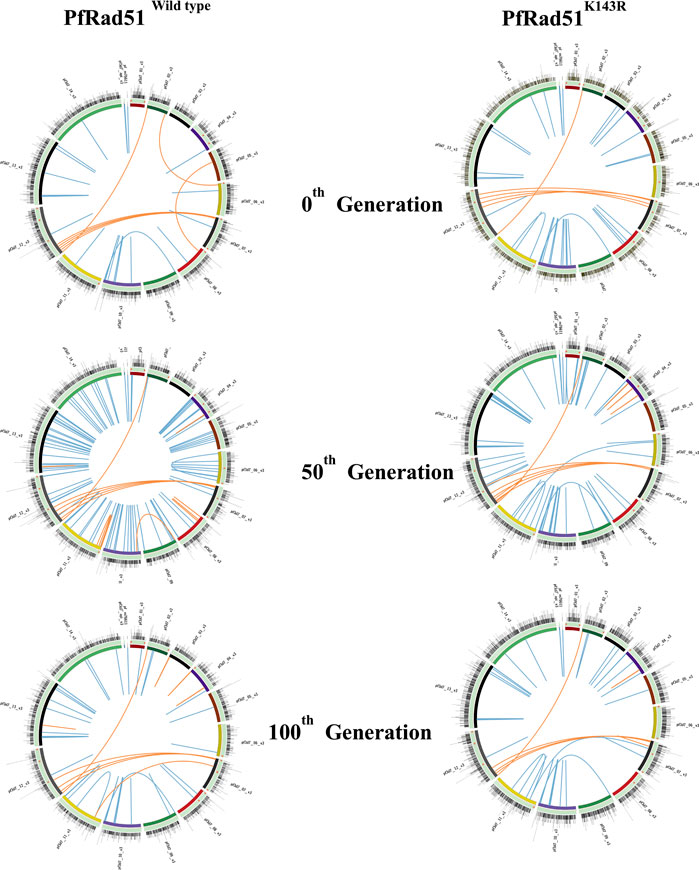
FIGURE 3. Visualisation of genomic variations on chromosomes of both wild-type and HR-deficient parasites. Each Circos plot illustrated here is the representative image of the respective generation of both wild-type (PfRad51wild-type) and HR-deficient (PfRad51K143R) parasite lines. All chromosomes in each Circos are represented as arcs in a circle, arranged in the different coloured ideogram. The outer most track with nucleotide variations (SNPs and INDELs) scattered in the genome is represented as histograms. The Scatchard plot in each track represents NVs at var genes (orange dots). Structural variations (translocations, inversions, deletions, and duplications >300 bp) are depicted at the innermost track of the core genome (blue lines) and var gene region (orange lines).
To compare the rate of rearrangement between wild-type and mutant parasite lines, we scored the unique change and calculated the rate of change in nucleotide and structural variations. We carried out detailed inspections of NVs and SVs generated in both wild-type and DN parasites. By taking the 0th generation as a reference, the appearance or disappearance of any variation was considered a single change. We scored all such novel and missing NVs or SVs from WGS of both parasite lines in subsequent generations using BCF tools. Taking the 0th generation as the reference point, we estimated the genome-wide rate of change of NVs and SVs per generation per nucleotide in both parasite lines and observed nearly a 30% reduction in the rate of change in HR-deficient parasites (Figures 4A, B).
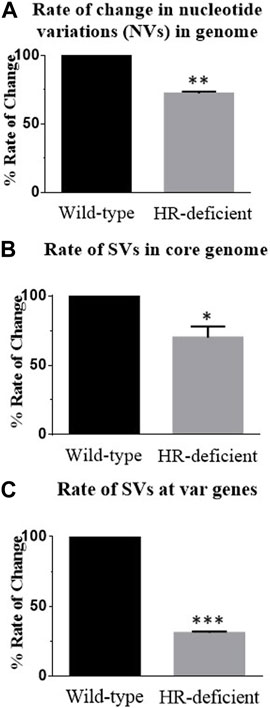
FIGURE 4. Reduced rate of change of nucleotide variations and structural variations at both the core genome and var genes in the HR-deficient parasite line. (A) Bar diagram showing the rate of change of nucleotide variations observed in the entire genome of wild-type and HR-deficient parasites. Total unique NVs present in the strain/per generations/genome size gives the rate of change in NVs. (B) Bar diagram showing the rate of change of structural variations (SVs) observed in the entire genome of both wild-type and HR-deficient parasites. Total unique SVs observed in the strain/number of generations/genome size give the rate of change in SVs. (C) The graph represents the rate of change of SVs in var genes calculated by taking the unique SVs generated only in var genes/generations/base pairs. The mean value ± SEM is plotted. Student’s t-tests were performed for determining the significance. Asterisks indicate values significantly different from the control, given as follows: ***p <0.001, **p <0.01, and *p <0.05.
To gain insights into recombination events at the var loci, we estimated the rate of change of SVs at var genes. After normalisation with the size of the core genome, the total number of unique SVs was found to be more at the var region than the core genome, and it was found that in HR-deficient parasites, nearly a 70% reduction in the rate of change at var genes was observed (Figure 4C). Together these results suggest that PfRad51-deficient parasites undergo fewer recombination events and thereby decrease the rate of change. In conclusion, the lack of a functional PfRad51 led to a significant decrease in the rate of var gene diversity in Plasmodium.
Here, we provide evidence that the homologous recombination efficiency is positively correlated with var gene recombination in P. falciparum. Our results corroborate well with a previous finding in which a link between DSB repair by the HR mechanism and a rapid diversification of the sub-telomeric var repertoire was revealed (Zhang et al., 2019). We also revealed that the functional loss of the key recombinase PfRad51 severely compromises the rate of var gene recombination. A previous study demonstrated that RecQ helicase PfBLM, another important protein of the Plasmodium HR pathway, plays a negative regulatory role in var gene recombination (Claessens et al., 2018). Thus, the interplay between Plasmodium HR proteins appears to regulate the outcome of var gene recombination.
In the absolute sense, if the PfRad51-mediated HR mechanism is the only means for generating var gene diversification, then one would expect a complete reduction in the rate of rearrangement in the PfRad51 loss-of-function mutant parasite line. Here, we observed a marked decrease in the rate, but var rearrangements were not abrogated. This could be explained by the fact that we did not conduct the experiment on a PfRad51 null mutant; instead, we overexpressed a dominant negative allele of PfRad51K143R, which does not allow the PfRad51wild-type protein to function. As PfRad51 works as a multimeric protein, the inclusion of even one molecule of the mutant protein in the multimeric complex could inactivate its function (Roy et al., 2014). However, there is always a possibility that a certain multimeric PfRad51 complex may consist of all the PfRad51wild-type monomers, and such a complex will be functionally active and capable of var rearrangements. Another possibility is that the absence of PfRad51 can be compensated by the Rad55–Rad57 complex. Indeed, in the P. falciparum genome, a putative Rad57-like protein has been identified (PF3D7_0419300). However, a Rad55-like protein seems to be absent in P. falciparum. A third possibility could be that the loss of HR activity could be compensated by the less frequent alt-NHEJ mechanism (Kirkman et al., 2014). This possibility seems very unlikely, as the loss of HR could not be compensated by any other mechanism for methyl methane sulfonate (MMS)-induced DSB repair (Roy et al., 2014). Nonetheless, our finding suggests that PfRad51-mediated HR plays a major role in var recombination.
We observed higher recombination events in the var loci compared to the rest of the genome. This is rather intuitive, as in the haploid P. falciparum genome, the homologous template for recombination comes only from sister chromatids, whereas for var recombination, there are several possible homologous templates. As depicted in the model (Figure 5), any crossover event between the identical var genes coming from sister chromatids would not produce recombinant var genes. On the other hand, a recombination between two non-identical var genes would produce recombinant var sequences, even though the two recombining var genes are coming from sister chromatids. Moreover, the recombination between two non-identical var genes belonging to different chromosomes would also produce recombinant var genes. Due to the clustering of multiple telomeres on the nuclear membrane, the juxtaposition of two var genes from different chromosomes is feasible, and hence, the recombination between them is also possible.
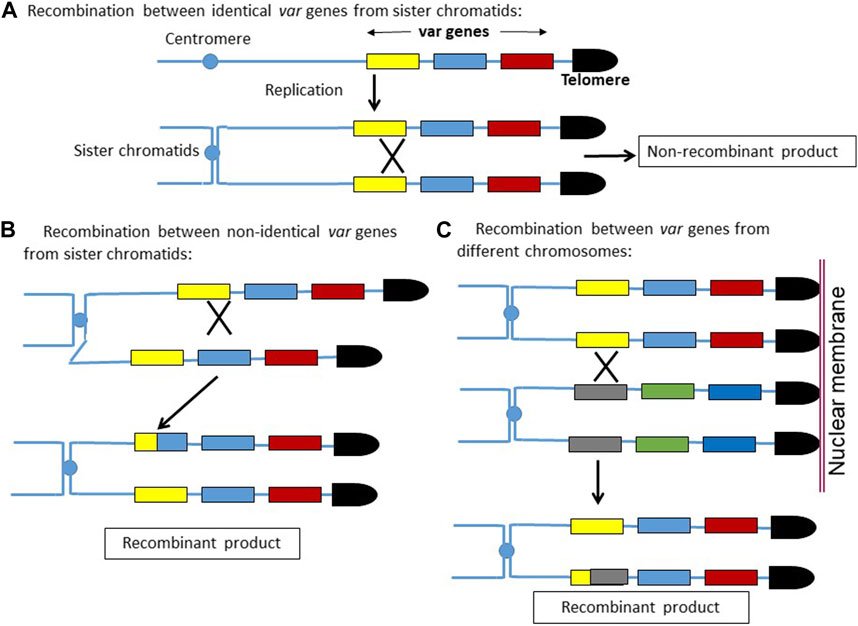
FIGURE 5. Model depicting how homologous recombinations can create new var gene sequences. (A) Recombination between two identical var sequences coming from sister chromatids would result in non-recombinant products. (B,C) Recombination between two non-identical var sequences coming from sister chromatids or from non-homologous sub-telomeric regions would result in newer var sequences.
PfEMP1, encoded by the var multigene family, is the most prominent determinant of antigenic variation in P. falciparum and, hence, involved in immune evasion. Out of the 60 or so var genes, only one is expressed at a given time and the rest are silenced. The rate of switching from one var gene expression to another is approximately 2% per generation. Thus, it is expected that after a finite number of generations, the entire var repertoire will be expressed, seriously compromising the parasite’s ability to manifest antigenic variations. This does not happen because of recombination events occurring between different var genes that give rise to newer var sequences and, thus, result in a never-ending repertoire of var sequences. Our finding that the PfRad51-mediated HR mechanism is involved in var gene recombination implies that blocking the P. falciparum HR pathway could be an excellent strategy to restrict var gene diversification in P. falciparum.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
MB conceived the idea and designed the experiments. PV and NR performed the experiments. MB, PV, and NR analysed the data. MB, PV, and NR wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from DST-SERB, Government of India (DST-SERB/2015/000999), to MB.
The authors acknowledge support from DST-FIST and UGC-SAP to the Department of Biochemistry. PV and NR were, respectively, supported by the Senior Research Fellowship from the University Grants Commission, India, and the Council for Scientific and Industrial Research, India.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Baruch, D. I., Pasloske, B. L., Singh, H. B., Bi, X., Ma, X. C., Feldman, M., et al. (1995). Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 82 (1), 77–87. doi:10.1016/0092-8674(95)90054-3
Bhattacharyya, M. K., Bhattacharyya nee Deb, S., Jayabalasingham, B., and Kumar, N. (2005). Characterization of kinetics of DNA strand-exchange and ATP hydrolysis activities of recombinant PfRad51, a Plasmodium falciparum recombinase. Mol. Biochem. Parasitol. 139 (1), 33–39. doi:10.1016/j.molbiopara.2004.09.007
Bhattacharyya, M. K., and Kumar, N. (2003). Identification and molecular characterisation of DNA damaging agent induced expression of Plasmodium falciparum recombination protein PfRad51. Int. J. Parasitol. 33 (12), 1385–1392. doi:10.1016/s0020-7519(03)00212-1
Bockhorst, J., Lu, F., Janes, J. H., Keebler, J., Gamain, B., Awadalla, P., et al. (2007). Structural polymorphism and diversifying selection on the pregnancy malaria vaccine candidate VAR2CSA. Mol. Biochem. Parasitol. 155 (2), 103–112. doi:10.1016/j.molbiopara.2007.06.007
Bopp, S. E., Manary, M. J., Bright, A. T., Johnston, G. L., Dharia, N. V., Luna, F. L., et al. (2013). Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 9 (2), e1003293. doi:10.1371/journal.pgen.1003293
Bull, P. C., Buckee, C. O., Kyes, S., Kortok, M. M., Thathy, V., Guyah, B., et al. (2008). Plasmodium falciparum antigenic variation. Mapping mosaic var gene sequences onto a network of shared, highly polymorphic sequence blocks. Mol. Microbiol. 68 (6), 1519–1534. doi:10.1111/j.1365-2958.2008.06248.x
Butterworth, A. S., Robertson, A. J., Ho, M. F., Gatton, M. L., McCarthy, J. S., and Trenholme, K. R. (2011). An improved method for undertaking limiting dilution assays for in vitro cloning of Plasmodium falciparum parasites. Malar. J. 10, 95. doi:10.1186/1475-2875-10-95
Claessens, A., Hamilton, W. L., Kekre, M., Otto, T. D., Faizullabhoy, A., Rayner, J. C., et al. (2014). Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 10 (12), e1004812. doi:10.1371/journal.pgen.1004812
Claessens, A., Harris, L. M., Stanojcic, S., Chappell, L., Stanton, A., Kuk, N., et al. (2018). RecQ helicases in the malaria parasite Plasmodium falciparum affect genome stability, gene expression patterns and DNA replication dynamics. PLoS Genet. 14 (7), e1007490. doi:10.1371/journal.pgen.1007490
Duffy, M. F., Byrne, T. J., Carret, C., Ivens, A., and Brown, G. V. (2009). Ectopic recombination of a malaria var gene during mitosis associated with an altered var switch rate. J. Mol. Biol. 389 (3), 453–469. doi:10.1016/j.jmb.2009.04.032
Freitas-Junior, L. H., Bottius, E., Pirrit, L. A., Deitsch, K. W., Scheidig, C., Guinet, F., et al. (2000). Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 407 (6807), 1018–1022. doi:10.1038/35039531
Gardner, M. J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R. W., et al. (2002). Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419 (6906), 498–511. doi:10.1038/nature01097
Holmes, V. F., Benjamin, K. R., Crisona, N. J., and Cozzarelli, N. R. (2001). Bypass of heterology during strand transfer by Saccharomyces cerevisiae Rad51 protein. Nucleic acids Res. 29 (24), 5052–5057. doi:10.1093/nar/29.24.5052
Iype, L. E., Wood, E. A., Inman, R. B., and Cox, M. M. (1994). RuvA and RuvB proteins facilitate the bypass of heterologous DNA insertions during RecA protein-mediated DNA strand exchange. J. Biol. Chem. 269 (40), 24967–24978. doi:10.1016/s0021-9258(17)31484-9
Jha, P., Gahlawat, A., Bhattacharyya, S., Dey, S., Kumar, K. A., and Bhattacharyya, M. K. (2021). Bloom helicase along with recombinase Rad51 repairs the mitochondrial genome of the malaria parasite. mSphere 6 (6), e0071821. doi:10.1128/mSphere.00718-21
Kirkman, L. A., and Deitsch, K. W. (2012). Antigenic variation and the generation of diversity in malaria parasites. Curr. Opin. Microbiol. 15 (4), 456–462. doi:10.1016/j.mib.2012.03.003
Kirkman, L. A., Lawrence, E. A., and Deitsch, K. W. (2014). Malaria parasites utilize both homologous recombination and alternative end joining pathways to maintain genome integrity. Nucleic acids Res. 42 (1), 370–379. doi:10.1093/nar/gkt881
Kolodner, R. D., Putnam, C. D., and Myung, K. (2002). Maintenance of genome stability in Saccharomyces cerevisiae. Sci. (New York, NY) 297 (5581), 552–557. doi:10.1126/science.1075277
Kyes, S., Horrocks, P., and Newbold, C. (2001). Antigenic variation at the infected red cell surface in malaria. Annu. Rev. Microbiol. 55, 673–707. doi:10.1146/annurev.micro.55.1.673
Morel, P., Stasiak, A., Ehrlich, S. D., and Cassuto, E. (1994). Effect of length and location of heterologous sequences on RecA-mediated strand exchange. J. Biol. Chem. 269 (31), 19830–19835. doi:10.1016/s0021-9258(17)32095-1
Moynahan, M. E., and Jasin, M. (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell. Biol. 11 (3), 196–207. doi:10.1038/nrm2851
Mu, J., Awadalla, P., Duan, J., McGee, K. M., Joy, D. A., McVean, G. A., et al. (2005). Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 3 (10), e335. doi:10.1371/journal.pbio.0030335
Mu, J., Awadalla, P., Duan, J., McGee, K. M., Keebler, J., Seydel, K., et al. (2007). Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat. Genet. 39 (1), 126–130. doi:10.1038/ng1924
Petter, M., and Duffy, M. F. (2015). Antigenic variation in Plasmodium falciparum. Results Probl. Cell. Differ. 57, 47–90. doi:10.1007/978-3-319-20819-0_3
Roy, N., Bhattacharyya, S., Chakrabarty, S., Laskar, S., Babu, S. M., and Bhattacharyya, M. K. (2014). Dominant negative mutant of Plasmodium Rad51 causes reduced parasite burden in host by abrogating DNA double-strand break repair. Mol. Microbiol. 94 (2), 353–366. doi:10.1111/mmi.12762
Scherf, A., Riviere, L., and Lopez-Rubio, J. J. (2008). SnapShot: Var gene expression in the malaria parasite. Cell. 134 (1), 190. doi:10.1016/j.cell.2008.06.042
Su, X. Z., Heatwole, V. M., Wertheimer, S. P., Guinet, F., Herrfeldt, J. A., Peterson, D. S., et al. (1995). The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 82 (1), 89–100. doi:10.1016/0092-8674(95)90055-1
Taylor, H. M., Kyes, S. A., and Newbold, C. I. (2000). Var gene diversity in Plasmodium falciparum is generated by frequent recombination events. Mol. Biochem. Parasitol. 110 (2), 391–397. doi:10.1016/s0166-6851(00)00286-3
Vydyam, P., Dutta, D., Sutram, N., Bhattacharyya, S., and Bhattacharyya, M. K. (2019). A small-molecule inhibitor of the DNA recombinase Rad51 from Plasmodium falciparum synergizes with the antimalarial drugs artemisinin and chloroquine. J. Biol. Chem. 294 (20), 8171–8183. doi:10.1074/jbc.RA118.005009
Keywords: malaria, Plasmodium falciparum, homeologous recombination, var gene rearrangement, antigenic variation, PfRad51
Citation: Vydyam P, Roy N and Bhattacharyya MK (2023) Uncovering the role of Rad51 in homologous recombination-mediated antigenic diversification in the human malaria parasite Plasmodium falciparum. Front. Mol. Biosci. 10:1223682. doi: 10.3389/fmolb.2023.1223682
Received: 16 May 2023; Accepted: 13 July 2023;
Published: 01 August 2023.
Edited by:
Anand Srivastava, National Institute of Animal Biotechnology (NIAB), IndiaReviewed by:
Kirk Deitsch, Cornell University, United StatesCopyright © 2023 Vydyam, Roy and Bhattacharyya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mrinal Kanti Bhattacharyya, bWtic2xAdW9oeWQuYWMuaW4=
†ORCID: Pratap Vydyam, orcid.org/0000-0003-4944-6006
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.