 Lamei Yuan
Lamei Yuan Xuehui Yu
Xuehui Yu Heng Xiao
Heng Xiao Sheng Deng
Sheng Deng Hong Xia
Hong Xia Hongbo Xu
Hongbo Xu Yan Yang
Yan Yang Hao Deng
Hao Deng- 1Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Center for Experimental Medicine, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Disease Genome Research Center, Central South University, Changsha, China
- 4Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
- 5Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, China
- 6Department of Emergency, The Third Xiangya Hospital, Central South University, Changsha, China
Most internal organs in humans and other vertebrates exhibit striking left-right asymmetry in position and structure. Variation of normal organ positioning results in left-right asymmetry disorders and presents as internal organ reversal or randomization. Up to date, at least 82 genes have been identified as the causative genetic factors of left-right asymmetry disorders. This study sought to discover potential pathogenic variants responsible for left-right asymmetry disorder present in a Han-Chinese family using whole exome sequencing combined with Sanger sequencing. Novel compound heterozygous variants, c.5690A>G (p.Asn1897Ser) and c.7759G>A (p.Val2587Met), in the dynein axonemal heavy chain 1 gene (DNAH1), were found in the proband and absent in unaffected family members. Conservation analysis has shown that the variants affect evolutionarily conserved residues, which may impact the tertiary structure of the DNAH1 protein. The novel compound heterozygous variants may potentially bear responsibility for left-right asymmetry disorder, which results from a perturbation of left-right axis coordination at the earliest embryonic development stages. This study broadens the variant spectrum of left-right asymmetry disorders and may be helpful for genetic counseling and healthcare management for the diagnosed individual, and promotes a greater understanding of the pathophysiology.
Introduction
Most human and other vertebrate internal organs asymmetrically orient along a left-right (L-R) axis and exhibit an elaborate L-R asymmetric pattern (McGrath et al., 2003; Blum et al., 2014). Genetic alterations of L-R signaling pathways may lead to L-R asymmetry disorders, which may be inherited in autosomal recessive, autosomal dominant, or X-linked modes (Deng et al., 2015; Perles et al., 2015; Grimes et al., 2016). Environmental modifiers and developmental randomness are also likely to play roles in L-R asymmetry disorders (Deng et al., 2015). Three broad types of internal organ positioning along the L-R axis are recognized (Levin, 2004; Best et al., 2019). Situs solitus is a condition in which all internal organs are positioned in a normal visceroatrial arrangement (Offen et al., 2016). In situs inversus (SI) and heterotaxy (HTX), there are mirror-image reversals and randomizations of visceroatrial arrangements, respectively (Offen et al., 2016; Geddes et al., 2020). SI and HTX are genetically heterogeneous disorders with reduced penetrance (Deng et al., 2015). SI with an incidence of 1 in every 8,500 live births is usually not related to congenital cardiac defects (Basu and Brueckner, 2008). Complete reversal of internal organs usually doesn’t result in discernible physiological risk, as the organs maintain their normal structures and relative positions (Bisgrove et al., 2003; Peeters and Devriendt, 2006). HTX with an incidence of 1 in 10,000 live births is related to at least 3% of all congenital cardiac disease cases (Basu and Brueckner, 2008). Other congenital anomalies in HTX usually manifest as pulmonary isomerism, intestinal malrotation, asplenia, or polysplenia (Wang et al., 2022; Wells et al., 2022).
Previously reported genetic defects implicated in L-R asymmetry disorders include complex chromosomal rearrangements, translocations, insertions/duplications, deletions, and inversions (Kosaki and Casey, 1998; Olbrich et al., 2002; Sutherland and Ware, 2009). Since the Zic family member 3 gene (ZIC3) variants in X-linked HTX were identified, at least 82 genes have been considered to be responsible for human L-R asymmetry disorders (Gebbia et al., 1997; Yu et al., 2022).
This study sought to identify the genetic factors responsible for the L-R asymmetry disorder present in a Han-Chinese family using whole exome sequencing (WES) combined with Sanger sequencing. Compound heterozygous variants in the dynein axonemal heavy chain 1 gene (DNAH1, OMIM 603332, NM_015512.5), including missense variants c.5690A>G (p.Asn1897Ser) and c.7759G>A (p.Val2587Met), may be the possible genetic etiology of the L-R asymmetry disorder. Basic Local Alignment Search Tool comparison of protein sequences revealed that p.Asn1897 and p.Val2587 were highly conserved in DNAH1 protein. Structural modeling also showed that residues are crucial to the structure or function of DNAH1 protein. These findings expand the variant spectrum of DNAH1, which may be beneficial for clinical and genetic diagnosis.
Methods
Pedigree and subjects
Nine members of a three-generation Han-Chinese pedigree containing the L-R asymmetry disorder sufferer were enrolled at the Third Xiangya Hospital, Central South University, P.R. China (Figure 1A). Peripheral blood samples and available clinical data were collected from the proband (II:2) and available unaffected pedigree members (I:2, II:1, II:4, III:1, III:2, and III:3). Written informed consents were given by all participants. This study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Third Xiangya Hospital, Central South University, Changsha, Hunan, China.
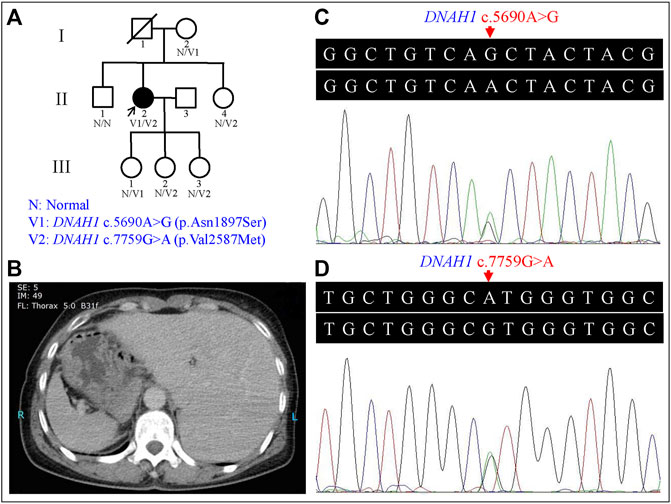
FIGURE 1. Genetic analysis of the left-right (L-R) asymmetry disorder pedigree and the representative computed tomography (CT) image of the proband. (A) Pedigree analysis of the L-R asymmetry disorder family. Squares and circles indicate males and females, respectively; open symbols indicate unaffected family members; the arrow indicates the proband; the symbol with a slash indicates a deceased member. (B) CT image of the proband showed the stomach and spleen were right-sided, and the liver was left-sided. (C) Heterozygous DNAH1 c.5690A>G (p.Asn1897Ser) variant in the proband (II:2). (D) Heterozygous DNAH1 c.7759G>A (p.Val2587Met) variant in the proband (II:2).
Sample preparation and whole exome sequencing
Genomic DNA (gDNA) was isolated from peripheral blood samples according to the previously described standard method (Xiang et al., 2018; Xiao et al., 2019). WES was carried out to detect the proband’s genetic variants by BGI-Shenzhen, China (Xiao et al., 2018). Qualified gDNA was randomly broken into fragments sized from 150 bp to 250 bp. Both ends of the end-repaired DNA fragments were ligated to adapters for amplification, purification, and hybridization. Captured products were processed to form DNA nanoballs by circularization and amplification. Agilent SureSelect Human All Exon V6, which covers about 99% of the human exonic regions, was used in the exome capture. The qualified circular DNA library was loaded on a BGISEQ-500 sequencing platform to perform high-throughput sequencing (Fan et al., 2019).
Variant analysis
Raw data (stored in FASTQ format) derived from the BGISEQ-500 sequencing platform were processed to obtain clean data. According to the strict data filtering criteria and data analysis quality control setup, the clean data were filtered from “raw data” to get access to high-quality sequencing data. Reads were cleaned during the “data cleanup” process using the following criteria: i) adapter sequence, ii) unknown base ratios more than 10%, and iii) inferior-quality base (base quality ≤5) ratios no less than 50% (Xiong et al., 2021). These clean data were mapped to the human reference genome sequence (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA, v0.7.15) software. Picard tools (v2.5.0, https://broadinstitute.github.io/picard/) were used to mark duplicated reads. Reads around insertions/deletions (indels) were realigned, and a base quality score recalibration step to improve base quality scores was conducted by Genome Analysis Toolkit (GATK, v3.3.0, https://gatk.broadinstitute.org/hc/en-us). HaplotypeCaller of GATK was applied to call a raw variant set including single nucleotide polymorphisms (SNPs) and indels. Hard-filtering methods with proper parameters were used to filter SNPs and indels (Xia et al., 2018; Xia et al., 2019). The resulting high-confident SNPs and indels were further annotated by a SnpEff tool (https://pcingola.github.io/SnpEff/). In order to find candidate variants, high-confident SNPs and indels were filtered against the 1000 Genomes Project (1000G), the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) 6500, the Exome Aggregation Consortium (ExAC), and Genome Aggregation Database (gnomAD), as well as the Single Nucleotide Polymorphism database (version 154, dbSNP154). The remaining variants with minor allele frequency (MAF) lower than 0.5% were further filtered against the BGI in-house exome databases (containing 1,943 Chinese controls without L-R asymmetry disorders).
Sanger sequencing verified the candidate variants revealed by WES in the proband and screened them in the available family members. The used primer sequences designed by Primer3 software (https://primer3.ut.ee/) for the candidate variants were 5′-TGCCCCTTGGCATAGAATAC-3′ and 5′-CATGGGTGAGGAGGTCAAAC-3′, and 5′-GAAGCTGGTCCTCTTCATGG-3′ and 5′-AAGCATGGGTCAGTCAAACC-3′, respectively. The detected variants were further classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpreting gene variants (Richards et al., 2015). Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used to carry out a conservative analysis by aligning nine homologous DNAH1 protein sequences retrieved from the National Center for Biotechnology Information Protein database (https://www.ncbi.nlm.nih.gov/protein/). The tertiary structures of wild-type and variant-type were conducted with the online SWISS-MODEL tool (https://swissmodel.expasy.org/) and further visualized structures were constructed via PyMOL software (version 2.3, Schrödinger, LLC, Portland, United States) (Xiang et al., 2019).
Results
Clinical data
The proband (II:2) is a 54-year-old female without respiratory symptoms or fertility problems. L-R asymmetry disorder was diagnosed after a routine preoperative assessment for surgical management of a multinodular goiter. From the ultrasonographic examination and computed tomography (CT) results, she was diagnosed as suffering from SI, including dextrocardia, left-sided liver, and stomach and spleen on the right side of the proband’s body (Figure 1B). Transthoracic echocardiography revealed normal characteristic morphological features and normal function of the heart, as well as normal valve morphology and function.
Genetic analysis
Proband gDNA exome sequencing produced a total of 226.50 million clean reads. After duplicate reads removal, 201.92 million effective reads were generated. Of these, 99.94% were mapped to the human reference genome. The average sequencing depth across the target region was 249.91×, and 99.41% of the target region was covered at 10×. In total, 103,286 SNPs and 18,053 indels were detected. Commonly known variants with MAF ≥0.5% recorded in the 1000G, the NHLBI ESP6500, and the dbSNP154 databases were removed. The remaining variants were further filtered against the BGI in-house exome databases. By screening all known disease-causing genes responsible for L-R asymmetry disorders, only two compound heterozygous DNAH1 gene missense variants, c.5690A>G (p.Asn1897Ser) in the exon 36 and c.7759G>A (p.Val2587Met) in the exon 49, were classified as potential disease-causing variants for the proband. Other potential disease-causing variants in at least 82 known genes associated with L-R asymmetry disorder phenotypes were ruled out in the proband, though large variants like complex rearrangement and gross deletion/duplication in these genes cannot be completely excluded. The c.5690A>G and c.7759G>A variants are documented in the dbSNP154 and have low frequencies in the global population (Table 1), indicating the compound heterozygous variants are potentially disorder-related variants.
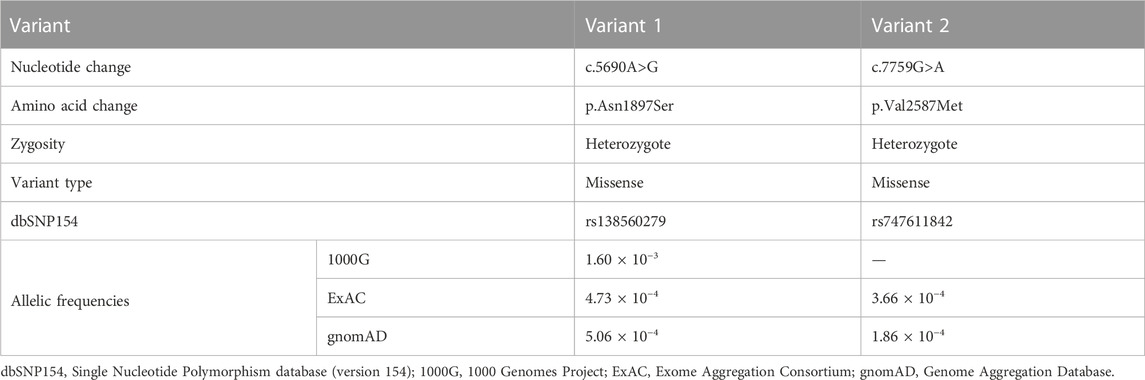
TABLE 1. Identification of the dynein axonemal heavy chain 1 gene variants in the patient.
Sanger sequencing confirmed the DNAH1 variants c.5690A>G and c.7759G>A in the proband (Figures 1C, D). In the pedigree, unaffected family members (I:2 and III:1) had the heterozygous c.5690A>G variant, and unaffected family members (II:4, III:2, and III:3) had the heterozygous c.7759G>A variant. These results indicated that the compound heterozygous variants c.5690A>G and c.7759G>A co-segregated with L-R asymmetry disorder in the pedigree. The c.7759G>A variant was absent from 1000G and the BGI in-house exome databases. Although the c.5690A>G (rs138560279) variant was recorded in the public database, the frequencies were low, with a MAF for “G” ranging from 0.0005 (ExAC) to 0.0016 (1000G). The c.5690A>G variant was also absent from the BGI in-house exome databases. The sequence variants, c.5690A>G and c.7759G>A, were classified as “likely pathogenic” following the ACMG standards and guidelines. Clustal Omega showed that the two residues p.Asn1897 and p.Val2587 in the DNAH1 protein were fully conserved among nine vertebrates (Figure 2), indicating that the two variants are probably pathogenic. A structural model showed the conformational alterations of asparagine (Asn-1897) into serine (Ser-1897) and valine (Val-2587) into methionine (Met-2587), further supporting the possible pathogenicity of the variants (Figure 3).
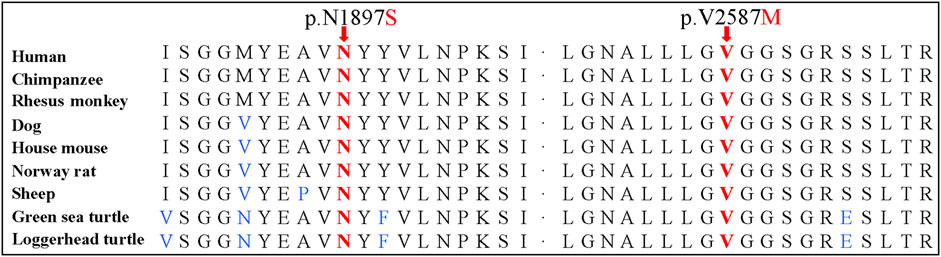
FIGURE 2. Conservation analyses of the dynein axonemal heavy chain 1 p.Asn1897 and p.Val2587 amino acid residues.
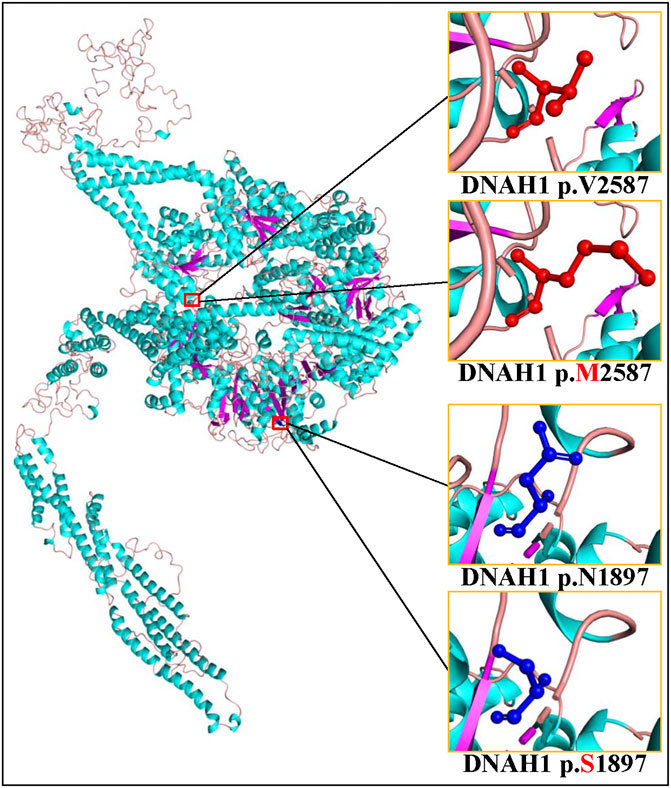
FIGURE 3. Cartoon model of the dynein axonemal heavy chain 1 (DNAH1) protein structure visualized by PyMOL based on the SWISS-MODEL. The asparagine (N) and mutated serine (S) at position 1897 and the valine (V) and mutated methionine (M) at position 2587 are indicated with ball-and-stick models.
Discussion
Human L-R asymmetry disorders may arise as a manifestation of a wide disease spectrum, such as primary ciliary dyskinesia (PCD), polycystic kidney disease 2 (PKD2), nephronophthisis, and Bardet-Biedl syndrome (Kosaki and Casey, 1998; Bataille et al., 2011; Marion et al., 2012). SI may be an incidental discovery in asymptomatic individuals during health examinations since complete mirror-image reversal of all asymmetrical structures may pose no detriment (Casey, 1998; Bisgrove et al., 2003). L-R asymmetry establishment in vertebrates generally has four phases: L-R symmetry breaking at early embryonic stages, transfer of asymmetric signals, asymmetric expression of left determinants in the left lateral plate mesoderm, and visceral organ L-R asymmetric morphogenesis (Gebbia et al., 1997; Okada et al., 2005; Shiraishi and Ichikawa, 2012). The genes encoding dynein axonemal heavy chains (DNAHs), such as DNAH5 (OMIM 603335), DNAH6 (OMIM 603336), DNAH9 (OMIM 603330), DNAH11 (OMIM 603339), and DNAH17 (OMIM 610063), are involved in L-R symmetry disorder development. Genetic variants in the above human DNAH genes have been reported to be associated with cilia and flagella dysfunction (Fliegauf et al., 2005; Hornef et al., 2006; Li et al., 2016; Xiong et al., 2021; Yu et al., 2022).
The DNAH1 gene, located on 3p21.1, comprises 78 exons and encodes an inner dynein arm heavy chain with 4,265 amino-acid residues. The DNAH1 protein functions as an ATP-dependent motor protein that generates force towards the minus end of microtubules, which is required for the bending of cilia and sperm flagellum motility (McGrath and Brueckner, 2003; Wambergue et al., 2016). Its C-terminal ∼3,500 amino-acid residues constitute the conserved dynein motor domain, which conducts the ATP-hydrolysis process in a ring of AAA+ (extended ATPases associated with various cellular activities) domains (Wendler et al., 2012; Schmidt and Carter, 2016). Dyneins play roles in mitosis, cellular transport, ciliary and flagellar motility, and L-R asymmetry determination (Supp et al., 1997; Supp et al., 1999; Basu and Brueckner, 2008; Schmidt et al., 2015). DNAH1 protein deficiencies may result in ciliary structure and function defects and impair sperm axoneme biogenesis, proposed to result in PCD and male infertility, respectively (McGrath and Brueckner, 2003; Imtiaz et al., 2015; Yang et al., 2018). Previously identified DNAH1 gene variants include at least 29 missense variants, 11 nonsense variants, 11 splicing variants, 6 frameshift variants, a deletion variant, and an extension variant (Table 2). Except for the DNAH1 gene homozygous missense variant c.3460A>C (p.Lys1154Gln) that was reported to be responsible for PCD in combination with SI (Imtiaz et al., 2015), the other known variants were involved in male infertility (Amiri-Yekta et al., 2016; Sha et al., 2017; Wang et al., 2017). Strikingly, the proband in our study denied the history of in vitro fertilization, and seems to have three biological children who inherited the possible disease-causing variants c.5690A>G (III:1) and c.7759G>A (III:2 and III:3), consistent with the reports that variants in DNAH genes may also not cause infertility (Xiong et al., 2021; Feng et al., 2022; Yu et al., 2022).
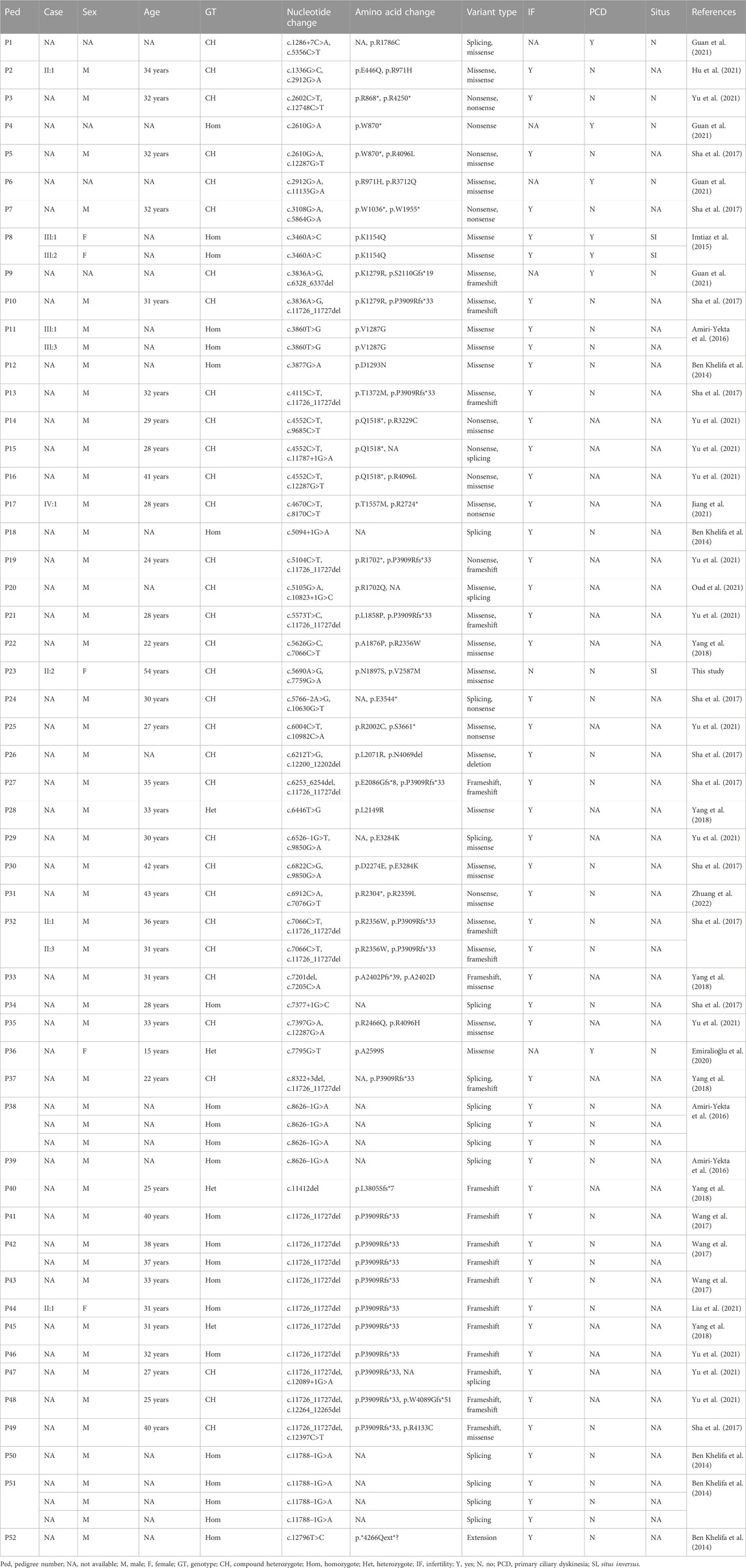
TABLE 2. Clinical data of the dynein axonemal heavy chain 1 gene variant carriers in different families.
In this study, two DNAH1 gene variants were identified in a Han-Chinese family including L-R asymmetry disorder sufferer. The DNAH1 variants c.5690A>G and c.7759G>A are located at AAA2 and a highly conserved nucleotide-binding motif (P-loop) in AAA4 (UniProt ID Q9P2D7), respectively (Mocz and Gibbons, 2001; Kon et al., 2004). These two DNAH1 variants may interfere with the rigid block formed by the whole AAA2-AAA4 region which may produce detrimental effects on the inner dynein arm heavy chains involved in generating oscillating beating in cilia (Shingyoji et al., 1998; Mocz and Gibbons, 2001; Schmidt et al., 2015). Perturbation may occur at the earliest stages in the signaling pathways that coordinate the L-R asymmetry and result in deficient embryonic nodal flow, impaired asymmetric transport of L-R signals and gene expression, and the final complete inversion of the L-R axis (Supp et al., 1997; Bisgrove et al., 2003; Peeters and Devriendt, 2006). PCD is a genetically and clinically heterogeneous disease with a diverse phenotype spectrum including chronic respiratory tract infections, L-R asymmetry disorders, and infertility (Lobo et al., 2015; Horani and Ferkol, 2018). Approximately half of PCD patients had SI or HTX (Basu and Brueckner, 2008; Deng et al., 2015). The proband in this study did not have related respiratory symptoms or fertility problems and cannot be diagnosed as typical PCD. The lack of respiratory symptoms may be due to the underlying compensation role of other dyneins that are phylogenetically close to DNAH1, such as DNAH3 (OMIM 603334), DNAH7 (OMIM 610061), and DNAH12 (OMIM 603340) (Ben Khelifa et al., 2014). Similarly, biallelic DNAH17 carriers and a few DNAH9 patients were reported to only exhibit SI or sperm flagellum defects, but without other cilia-related symptoms (Fliegauf et al., 2005; Yu et al., 2022). Our patient only has L-R asymmetry disorder, without other PCD-associated disorders, which may also be counted as a variant form of PCD suffering a mild consequence of cilia dysfunction. A limitation of this study is the lack of nasal epithelial brush biopsy samples for cilia beat and ultrastructure analysis.
Cilia are central to the initial breaking of L-R symmetry (Basu and Brueckner, 2008; Zhu et al., 2020; Little and Norris, 2021). During the development of vertebrate L-R asymmetry, motile embryonic cilia produce leftward extracellular fluid flow to initiate the event that converts early embryonic bilateral symmetry to a left-sided heart and asymmetric arrangement of visceral organs (Brody, 2004; Fliegauf et al., 2005). Structural and functional ciliary defects are related to hydrocephalus, Kartagener’s syndrome, infertility, PKD2, and randomization of the L-R axis (Ibañez—Tallon et al., 2002; Brody, 2004; Fliegauf et al., 2005). More than 100 genes may be involved in L-R asymmetry defects in model organisms (Catana and Apostu, 2017). Dnah5 mutations in mouse models result in the randomization of visceral organs’ laterality (Ibañez—Tallon et al., 2002; Olbrich et al., 2002). In Dnah1 mutant mice, abnormal sperm behavior, fertilization failure, and reduced ciliary beat frequency were observed, similar to phenotypes of patients suffering from infertility and PCD (Neesen et al., 2001; Hu et al., 2019). The identification of the DNAH1 gene variants in PCD patients with SI and the limited reports of animal models implies that more cases and animal models are warranted to fully reveal the effect of the DNAH1 gene variants on L-R asymmetry (Neesen et al., 2001; Imtiaz et al., 2015).
Early L-R asymmetry disorder diagnosis may be beneficial to patients when they need external chest compression or emergency surgery for heart attack or abdominal trauma. Plain chest radiographs, echocardiography, abdominal sonography, CT, and magnetic resonance imaging are effective means of discovering and diagnosing L-R asymmetry disorders (Winer-Muram, 1995).
In summary, the novel compound heterozygous DNAH1 gene c.5690A>G (p.Asn1897Ser) and c.7759G>A (p.Val2587Met) variants were identified in a Han-Chinese pedigree containing L-R asymmetry disorder sufferer. We present, for the first time, evidence that DNAH1 variants do not necessarily lead to female infertility. This conclusion is based on our analysis of the female proband and her female offsprings, and further discovery of more such cases, especially homozygous variants cases, may help to understand the genotype-phenotype association of DNAH1. The discovery provides new evidence of the potential association between the DNAH1 gene and L-R asymmetry disorders and extends the phenotypic spectrum of DNAH1-associated diseases. It supports the notion that laterality disorders may result from disturbances at the primary cilia level (Peeters and Devriendt, 2006). This work may promote a better understanding of the genetic causes underlying L-R asymmetry disorders and assist in genetic counseling and management of diagnosed individuals.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://db.cngb.org/, CNP0003867
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Third Xiangya Hospital, Central South University, Changsha, Hunan, China. The patients/participants provided their written informed consent to participate in this study.
Author contributions
LY, XY, and HD conceived and designed this study. XY, HXiao, SD, HXia, HXu, and YY collected the patient samples and clinical data. LY, XY, and HXiao performed the experiments. LY, XY, HXiao, and HD analyzed the data. LY, XY, and HD wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81670216, 81873686, and 81800219), Natural Science Foundation of Hunan Province (Grant Nos. 2020JJ4830, 2020JJ3057, and 2022JJ30922), Hunan Provincial Innovation Foundation for Postgraduate (Grant No. CX20210375), Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University (Grant No. YX202109), and Distinguished Professor of the Lotus Scholars Award Program of Hunan Province, China.
Acknowledgments
We appreciate our patients, their families, and referral physicians for their cooperation and contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amiri-Yekta, A., Coutton, C., Kherraf, Z. E., Karaouzène, T., Le Tanno, P., Sanati, M. H., et al. (2016). Whole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutations. Hum. Reprod. 31, 2872–2880. doi:10.1093/humrep/dew262
Basu, B., and Brueckner, M. (2008). Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr. Top. Dev. Biol. 85, 151–174. doi:10.1016/S0070-2153(08)00806-5
Bataille, S., Demoulin, N., Devuyst, O., Audrézet, M. P., Dahan, K., Godin, M., et al. (2011). Association of PKD2 (polycystic 2) mutations with left-right laterality defects. Am. J. Kidney Dis. 58, 456–460. doi:10.1053/j.ajkd.2011.05.015
Ben Khelifa, M., Coutton, C., Zouari, R., Karaouzène, T., Rendu, J., Bidart, M., et al. (2014). Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am. J. Hum. Genet. 94, 95–104. doi:10.1016/j.ajhg.2013.11.017
Best, S., Shoemark, A., Rubbo, B., Patel, M. P., Fassad, M. R., Dixon, M., et al. (2019). Risk factors for situs defects and congenital heart disease in primary ciliary dyskinesia. Thorax 74, 203–205. doi:10.1136/thoraxjnl-2018-212104
Bisgrove, B. W., Morelli, S. H., and Yost, H. J. (2003). Genetics of human laterality disorders: Insights from vertebrate model systems. Annu. Rev. Genomics Hum. Genet. 4, 1–32. doi:10.1146/annurev.genom.4.070802.110428
Blum, M., Feistel, K., Thumberger, T., and Schweickert, A. (2014). The evolution and conservation of left-right patterning mechanisms. Development 141, 1603–1613. doi:10.1242/dev.100560
Brody, S. L. (2004). Genetic regulation of cilia assembly and the relationship to human disease. Am. J. Respir. Cell Mol. Biol. 30, 435–437. doi:10.1165/rcmb.2004-0004PS
Casey, B. (1998). Two rights make a wrong: Human left-right malformations. Hum. Mol. Genet. 7, 1565–1571. doi:10.1093/hmg/7.10.1565
Catana, A., and Apostu, A. P. (2017). The determination factors of left-right asymmetry disorders-a short review. Clujul Med. 90, 139–146. doi:10.15386/cjmed-701
Deng, H., Xia, H., and Deng, S. (2015). Genetic basis of human left-right asymmetry disorders. Expert Rev. Mol. Med. 16, e19. doi:10.1017/erm.2014.22
Emiralioğlu, N., Taşkıran, E. Z., Koşukcu, C., Bilgiç, E., Atilla, P., Kaya, B., et al. (2020). Genotype and phenotype evaluation of patients with primary ciliary dyskinesia: First results from Turkey. Pediatr. Pulmonol. 55, 383–393. doi:10.1002/ppul.24583
Fan, K., Zhu, H., Xu, H., Mao, P., Yuan, L., and Deng, H. (2019). The identification of a transthyretin variant p.D38G in a Chinese family with early-onset leptomeningeal amyloidosis. J. Neurol. 266, 232–241. doi:10.1007/s00415-018-9125-z
Feng, J., Li, J., Du, Y., Shi, T., Sharma, L., and Jie, Z. (2022). Case report: Rare dynein axonemal heavy chain 9 mutations in a Han-Chinese patient with Kartagener syndrome. Front. Med. 9, 893968. doi:10.3389/fmed.2022.893968
Fliegauf, M., Olbrich, H., Horvath, J., Wildhaber, J. H., Zariwala, M. A., Kennedy, M., et al. (2005). Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 171, 1343–1349. doi:10.1164/rccm.200411-1583OC
Gebbia, M., Ferrero, G. B., Pilia, G., Bassi, M. T., Aylsworth, A., Penman-Splitt, M., et al. (1997). X-linked situs abnormalities result from mutations in ZIC3. Nat. Genet. 17, 305–308. doi:10.1038/ng1197-305
Geddes, G. C., Samudrala, S. S., and Earing, M. G. (2020). Neonatal assessment of infants with heterotaxy. Clin. Perinatol. 47, 171–182. doi:10.1016/j.clp.2019.10.011
Grimes, D. T., Keynton, J. L., Buenavista, M. T., Jin, X., Patel, S. H., Kyosuke, S., et al. (2016). Genetic analysis reveals a hierarchy of interactions between polycystic-encoding genes and genes controlling cilia function during left-right determination. PLoS Genet. 12, e1006070. doi:10.1371/journal.pgen.1006070
Guan, Y., Yang, H., Yao, X., Xu, H., Liu, H., Tang, X., et al. (2021). Clinical and genetic spectrum of children with primary ciliary dyskinesia in China. Chest 159, 1768–1781. doi:10.1016/j.chest.2021.02.006
Horani, A., and Ferkol, T. W. (2018). Advances in the genetics of primary ciliary dyskinesia: Clinical implications. Chest 154, 645–652. doi:10.1016/j.chest.2018.05.007
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T., et al. (2006). DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 174, 120–126. doi:10.1164/rccm.200601-084OC
Hu, H. Y., Wei, T. Y., Feng, Z. K., Li, S. J., Zhao, R., Yi, X. L., et al. (2021). Novel biallelic DNAH1 variations cause multiple morphological abnormalities of the sperm flagella. DNA Cell Biol. 40, 833–840. doi:10.1089/dna.2021.0097
Hu, J., Lessard, C., Longstaff, C., O'Brien, M., Palmer, K., Reinholdt, L., et al. (2019). ENU-induced mutant allele of Dnah1, ferf1, causes abnormal sperm behavior and fertilization failure in mice. Mol. Reprod. Dev. 86, 416–425. doi:10.1002/mrd.23120
Ibañez -Tallon, I., Gorokhova, S., and Heintz, N. (2002). Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum. Mol. Genet. 11, 715–721. doi:10.1093/hmg/11.6.715
Imtiaz, F., Allam, R., Ramzan, K., and Al-Sayed, M. (2015). Variation in DNAH1 may contribute to primary ciliary dyskinesia. BMC Med. Genet. 16, 14. doi:10.1186/s12881-015-0162-5
Jiang, C., Zhang, X., Zhang, H., Guo, J., Zhang, C., Li, J., et al. (2021). Novel bi-allelic mutations in DNAH1 cause multiple morphological abnormalities of the sperm flagella resulting in male infertility. Transl. Androl. Urol. 10, 1656–1664. doi:10.21037/tau-20-1434
Kon, T., Nishiura, M., Ohkura, R., Toyoshima, Y. Y., and Sutoh, K. (2004). Distinct functions of nucleotide-binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry 43, 11266–11274. doi:10.1021/bi048985a
Kosaki, K., and Casey, B. (1998). Genetics of human left-right axis malformations. Semin. Cell Dev. Biol. 9, 89–99. doi:10.1006/scdb.1997.0187
Levin, M. (2004). The embryonic origins of left-right asymmetry. Crit. Rev. Oral Biol. Med. 15, 197–206. doi:10.1177/154411130401500403
Li, Y., Yagi, H., Onuoha, E. O., Damerla, R. R., Francis, R., Furutani, Y., et al. (2016). DNAH6 and its interactions with PCD genes in heterotaxy and primary ciliary dyskinesia. PLoS Genet. 12, e1005821. doi:10.1371/journal.pgen.1005821
Little, R. B., and Norris, D. P. (2021). Right, left and cilia: How asymmetry is established. Semin. Cell Dev. Biol. 110, 11–18. doi:10.1016/j.semcdb.2020.06.003
Liu, M., Huang, S., Zhao, X., Wu, F., Zhu, D., Zhai, X., et al. (2021). Successful live birth following natural cycle oocyte retrieval in a woman with primary infertility and atypical primary ovarian insufficiency with a DNAH1 gene deletion mutation. Genet. Test. Mol. Biomarkers 25, 668–673. doi:10.1089/gtmb.2020.0303
Lobo, J., Zariwala, M. A., and Noone, P. G. (2015). Primary ciliary dyskinesia. Semin. Respir. Crit. Care Med. 36, 169–179. doi:10.1055/s-0035-1546748
Marion, V., Stutzmann, F., Gérard, M., De Melo, C., Schaefer, E., Claussmann, A., et al. (2012). Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet--Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317–321. doi:10.1136/jmedgenet-2012-100737
McGrath, J., and Brueckner, M. (2003). Cilia are at the heart of vertebrate left-right asymmetry. Curr. Opin. Genet. Dev. 13, 385–392. doi:10.1016/s0959-437x(03)00091-1
McGrath, J., Somlo, S., Makova, S., Tian, X., and Brueckner, M. (2003). Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 114, 61–73. doi:10.1016/s0092-8674(03)00511-7
Mocz, G., and Gibbons, I. R. (2001). Model for the motor component of dynein heavy chain based on homology to the AAA family of oligomeric ATPases. Structure 9, 93–103. doi:10.1016/s0969-2126(00)00557-8
Neesen, J., Kirschner, R., Ochs, M., Schmiedl, A., Habermann, B., Mueller, C., et al. (2001). Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum. Mol. Genet. 10, 1117–1128. doi:10.1093/hmg/10.11.1117
Offen, S., Jackson, D., Canniffe, C., Choudhary, P., and Celermajer, D. S. (2016). Dextrocardia in adults with congenital heart disease. Heart Lung Circ. 25, 352–357. doi:10.1016/j.hlc.2015.09.003
Okada, Y., Takeda, S., Tanaka, Y., Belmonte, J. I., and Hirokawa, N. (2005). Mechanism of nodal flow: A conserved symmetry breaking event in left-right axis determination. Cell 121, 633–644. doi:10.1016/j.cell.2005.04.008
Olbrich, H., Häffner, K., Kispert, A., Völkel, A., Volz, A., Sasmaz, G., et al. (2002). Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 30, 143–144. doi:10.1038/ng817
Oud, M. S., Houston, B. J., Volozonoka, L., Mastrorosa, F. K., Holt, G. S., Alobaidi, B. K. S., et al. (2021). Exome sequencing reveals variants in known and novel candidate genes for severe sperm motility disorders. Hum. Reprod. 36, 2597–2611. doi:10.1093/humrep/deab099
Peeters, H., and Devriendt, K. (2006). Human laterality disorders. Eur. J. Med. Genet. 49, 349–362. doi:10.1016/j.ejmg.2005.12.003
Perles, Z., Moon, S., Ta-Shma, A., Yaacov, B., Francescatto, L., Edvardson, S., et al. (2015). A human laterality disorder caused by a homozygous deleterious mutation in MMP21. J. Med. Genet. 52, 840–847. doi:10.1136/jmedgenet-2015-103336
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Schmidt, H., and Carter, A. P. (2016). Review: Structure and mechanism of the dynein motor ATPase. Biopolymers 105, 557–567. doi:10.1002/bip.22856
Schmidt, H., Zalyte, R., Urnavicius, L., and Carter, A. P. (2015). Structure of human cytoplasmic dynein-2 primed for its power stroke. Nature 518, 435–438. doi:10.1038/nature14023
Sha, Y., Yang, X., Mei, L., Ji, Z., Wang, X., Ding, L., et al. (2017). DNAH1 gene mutations and their potential association with dysplasia of the sperm fibrous sheath and infertility in the Han Chinese population. Fertil. Steril. 107, 1312–1318.e2. doi:10.1016/j.fertnstert.2017.04.007
Shingyoji, C., Higuchi, H., Yoshimura, M., Katayama, E., and Yanagida, T. (1998). Dynein arms are oscillating force generators. Nature 393, 711–714. doi:10.1038/31520
Shiraishi, I., and Ichikawa, H. (2012). Human heterotaxy syndrome-from molecular genetics to clinical features, management, and prognosis. Circ. J. 76, 2066–2075. doi:10.1253/circj.cj-12-0957
Supp, D. M., Brueckner, M., Kuehn, M. R., Witte, D. P., Lowe, L. A., McGrath, J., et al. (1999). Targeted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left-right asymmetries. Development 126, 5495–5504. doi:10.1242/dev.126.23.5495
Supp, D. M., Witte, D. P., Potter, S. S., and Brueckner, M. (1997). Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature 389, 963–966. doi:10.1038/40140
Sutherland, M. J., and Ware, S. M. (2009). Disorders of left-right asymmetry: Heterotaxy and situs inversus. Am. J. Med. Genet. C Semin. Med. Genet. 151C, 307–317. doi:10.1002/ajmg.c.30228
Wambergue, C., Zouari, R., Fourati Ben Mustapha, S., Martinez, G., Devillard, F., Hennebicq, S., et al. (2016). Patients with multiple morphological abnormalities of the sperm flagella due to DNAH1 mutations have a good prognosis following intracytoplasmic sperm injection. Hum. Reprod. 31, 1164–1172. doi:10.1093/humrep/dew083
Wang, C. C., Wu, M. H., Wu, E. T., Lu, F., and Chen, S. J. (2022). Clinical implications of airway anomalies and stenosis in patients with heterotaxy syndrome. Pediatr. Pulmonol. 57, 2074–2081. doi:10.1002/ppul.25981
Wang, X., Jin, H., Han, F., Cui, Y., Chen, J., Yang, C., et al. (2017). Homozygous DNAH1 frameshift mutation causes multiple morphological anomalies of the sperm flagella in Chinese. Clin. Genet. 91, 313–321. doi:10.1111/cge.12857
Wells, J. R., Padua, M. B., and Ware, S. M. (2022). The genetic landscape of cardiovascular left-right patterning defects. Curr. Opin. Genet. Dev. 75, 101937. doi:10.1016/j.gde.2022.101937
Wendler, P., Ciniawsky, S., Kock, M., and Kube, S. (2012). Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14. doi:10.1016/j.bbamcr.2011.06.014
Winer-Muram, H. T. (1995). Adult presentation of heterotaxic syndromes and related complexes. J. Thorac. Imaging 10, 43–57. doi:10.1097/00005382-199501010-00004
Xia, H., Huang, X., Xu, H., Guo, Y., Hu, P., Deng, X., et al. (2018). An OTOF frameshift variant associated with auditory neuropathy spectrum disorder. Curr. Genomics 19, 370–374. doi:10.2174/1389202919666171113152951
Xia, H., Huang, X., Xu, H., Zhou, Y. A., Gong, L., Yang, Z., et al. (2019). GJB2 c.235delC variant associated with autosomal recessive nonsyndromic hearing loss and auditory neuropathy spectrum disorder. Genet. Mol. Biol. 42, 48–51. doi:10.1590/1678-4685-gmb-2017-0318
Xiang, Q., Cao, Y., Xu, H., Guo, Y., Yang, Z., Xu, L., et al. (2019). Identification of novel pathogenic ABCA4 variants in a Han Chinese family with Stargardt disease. Biosci. Rep. 39, BSR20180872. doi:10.1042/BSR20180872
Xiang, Q., Guo, Y., Cao, Y., Xiong, W., Deng, X., Xu, H., et al. (2018). Identification of a CNGB1 frameshift mutation in a Han Chinese family with retinitis pigmentosa. Optom. Vis. Sci. 95, 1155–1161. doi:10.1097/OPX.0000000000001305
Xiao, H., Huang, X., Xu, H., Chen, X., Xiong, W., Yang, Z., et al. (2019). A novel splice-site mutation in the ATP2C1 gene of a Chinese family with Hailey-Hailey disease. J. Cell. Biochem. 120, 3630–3636. doi:10.1002/jcb.27640
Xiao, H., Yuan, L., Xu, H., Yang, Z., Huang, F., Song, Z., et al. (2018). Novel and recurring disease-causing NF1 variants in two Chinese families with neurofibromatosis type 1. J. Mol. Neurosci. 65, 557–563. doi:10.1007/s12031-018-1128-9
Xiong, Y., Xia, H., Yuan, L., Deng, S., Ding, Z., and Deng, H. (2021). Identification of compound heterozygous DNAH11 variants in a Han-Chinese family with primary ciliary dyskinesia. J. Cell. Mol. 25, 9028–9037. doi:10.1111/jcmm.16866
Yang, X., Zhu, D., Zhang, H., Jiang, Y., Hu, X., Geng, D., et al. (2018). Associations between DNAH1 gene polymorphisms and male infertility: A retrospective study. Medicine 97, e13493. doi:10.1097/MD.0000000000013493
Yu, W., An, M., Xu, Y., Gao, Q., Lu, M., Li, Y., et al. (2021). Mutational landscape of DNAH1 in Chinese patients with multiple morphological abnormalities of the sperm flagella: Cohort study and literature review. J. Assist. Reprod. Genet. 38, 2031–2038. doi:10.1007/s10815-021-02201-5
Yu, X., Yuan, L., Deng, S., Xia, H., Tu, X., Deng, X., et al. (2022). Identification of DNAH17 variants in Han-Chinese patients with left-right asymmetry disorders. Front. Genet. 13, 862292. doi:10.3389/fgene.2022.862292
Zhu, X., Shi, C., Zhong, Y., Liu, X., Yan, Q., Wu, X., et al. (2020). Cilia-driven asymmetric Hedgehog signalling determines the amphioxus left-right axis by controlling Dand5 expression. Development 147, dev182469. doi:10.1242/dev.182469
Keywords: DNAH1, left-right asymmetry disorder, whole exome sequencing, novel variant, genetic analysis
Citation: Yuan L, Yu X, Xiao H, Deng S, Xia H, Xu H, Yang Y and Deng H (2023) Identification of novel compound heterozygous variants in the DNAH1 gene of a Chinese family with left-right asymmetry disorder. Front. Mol. Biosci. 10:1190162. doi: 10.3389/fmolb.2023.1190162
Received: 20 March 2023; Accepted: 19 June 2023;
Published: 29 June 2023.
Edited by:
Bruno Carmona, Escola Superior de Tecnologia da Saúde de Lisboa (ESTeSL), PortugalReviewed by:
Matthias Rath, Medical School Hamburg, GermanyHemakumar M. Reddy, Brown University, United States
Copyright © 2023 Yuan, Yu, Xiao, Deng, Xia, Xu, Yang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Deng, aGRlbmcwMDhAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship