 Sankar Basu
Sankar Basu Devlina Chakravarty
Devlina Chakravarty Qingzhen Hou
Qingzhen Hou Vladimir N. Uversky
Vladimir N. Uversky
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
EDITORIAL article
Front. Mol. Biosci., 06 February 2023
Sec. Structural Biology
Volume 10 - 2023 | https://doi.org/10.3389/fmolb.2023.1151676
This article is part of the Research TopicFrom the Hydrophobic Core to the Globular-Disorder Interface: New Challenges and Insights into Protein DesignView all 7 articles
Editorial on the Research Topic
From the hydrophobic core to the globular-disorder interface: New challenges and insights into protein design
The biophysical foundations of protein folding (Dill and MacCallum, 2012) still remain largely unexplained despite the many years of intensive experimental (Dobson, 2004) and computational research (Callaway, 2020; Jumper et al., 2021). It is rightly believed that the most objective way of probing our understanding of the basic principles of protein folding is reflected in the ability to successfully resolve the ‘inverse protein folding” problem (Yue and Dill, 1992); i.e., the capability to rationally design functional proteins, de novo. In recent years, the field has undoubtedly become diverse and complex, with the design of fold-switch proteins, proteins with targeted functional modulations etc.—one of the unmet goals of the latter of which is to aid ‘protein therapy’ (Dimitrov, 2012; Blazeck et al., 2022; Svilenov et al., 2022).
Furthermore, since the discovery of intrinsically disordered proteins (IDPs), with their characteristic binding promiscuity attributed to their physical flexibility, attempts to understand and design protein disorder transitions (Bandyopadhyay and Basu, 2020; Roy et al., 2022) and to address the “globular/ordered–disordered” evolutionary interface (Nagibina et al., 2020) in proteins are among the most intriguing challenges in this field. It is but natural that designs of ordered proteins and IDPs should use different principles and the field is enriching with ideas and findings on both (Figure 1). For example, in globular proteins, a given protein fold (or a hydrophobic core) can be modulated by the alternative modes of side-chain packing (Basu et al., 2011; Biswas et al., 2022), whereas in IDPs, the disorder-to-order transitions can be triggered by cumulative point mutations (Basu and Biswas, 2018). Although there are not yet general evolutionary rules regarding the origin of disordered and globular proteins, instances of successful design (for example) of folded globular repeats from disordered ancestors (Zhu et al., 2016) have raised much hope. A successful endeavor along these directions is of high biophysical and mechanistic importance (Theillet et al., 2013). Furthermore, findings in this area would also serve great therapeutic benefits in the wide array of IDP-related deadly human diseases (Coskuner-Weber and Uversky, 2018).
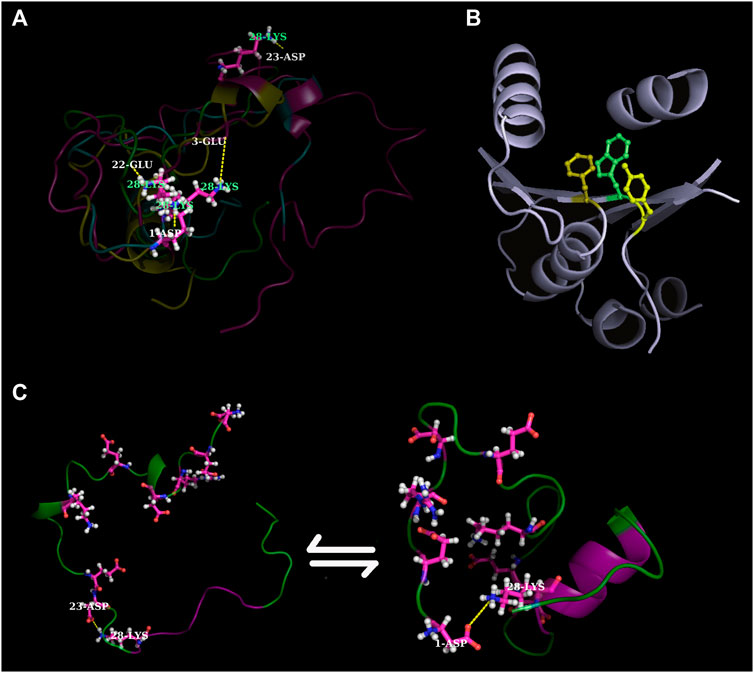
FIGURE 1. Recent findings in contemporary protein design across the globular—disorder evolutionary interface. (A) shows ephemeral salt-bridge dynamics in IDPs (in relation to administering cumulative mutations in probing disorder-to-order transitions in IDPs) (Basu and Biswas, 2018). (B) shows a 3-clique (Basu et al., 2011), a ubiquitous packing motif in densely packed cores that heavily influences the formation and sustenance of alternatively packed hydrophobic cores within the same protein fold (Biswas et al., 2022). (C) shows how Aβ42 temporarily shapes into partial order during its molecular dynamic simulations (Basu and Biswas, 2018). Images in (A,C) have been reconstructed from scratch using suitable MD simulation snapshots. In contrast, the image in (B) have been reproduced with appropriate copyright permissions from BMC Bioinformatics.
Themed on protein designs across globular and disordered proteins as well as their evolutionary interface, this Research Topic (RT) currently comprises of five original research articles and a review on the experimental and computational advances in protein design.
In their study of the concentration- and time-dependent effects of a cholesterol-based detergent Chobimalt on insulin amyloid aggregation, a group of Andrey Musatov utilized a multiparametric experimental approach that included a combination ThT fluorescence assay, CD, AFM, SANS, and SAXS (Siposova et al.). This analysis revealed intriguing dose-dependent effect of this amphiphilic compound. The data indicate that not only kinetics but also the morphology of fibrillar aggregates can be dramatically changed in the presence of specific additives.
A group of Bogdan S. Melnik used their experience in the analysis of protein mutants to understand the effect of various amino acid substitutions on the protein energy landscape (Majorina et al.). This knowledge provides means for the rational design of amino acid substitutions that would have the desired effects on the multistate proteins stability and the rates of their native or intermediate state formation. It also can be used for rational design of protein mutants with the desired properties.
A group of Cheng Wang and Dongdong Qiao provided an up-to-date overview of computational methods for prediction of protein-protein binding sites of intrinsically disordered proteins (IDP-PPIS) (Chen et al.). Analysis of 30 recently published IDP-PPIS predictors indicated that based on their underlying principles, they can be divided into three categories, where the algorithms are grounded on the scoring functions, machine learning-based prediction, or consensus approach. Provided information on the details of algorithms and their performances can be used as a guide for selecting different methods.
A group of Ramanathan Sowdhamini utilized extensive computational docking and multi-ns molecular dynamics (MD) simulations to look at the effects of a plant cyclic disulfide-rich peptide (“cyclotide”) isolated from a medicinal plant Clitoria ternatea on the conformational dynamics and destabilization of the β-amyloid fibrils, which are well-known hallmarks of Alzheimer’s disease (Kalmankar et al.). This study revealed that the anti-aggregation potential of this cyclic disulfide-rich peptide can be attributed to the cyclotide binding-induced unfolding and opening up of the amyloid β-sheet structure of Aβ fibrils and opened a way for the design of novel cyclotide-based drugs against protein aggregation.
Ennist et al. described a designed electron-transport chain with the multi-step electron-transfer activity based on the de novo designed reaction center maquette protein (the RC maquette) that assembles metal ions, tyrosine, a Zn tetrapyrrole, and heme. Although in its apo-form, the RC maquette is characterized by a dynamic and flexible structure, it can predictably fold into a more ordered holo-state upon binding of various cofactors, such as electron donors, pigments, and electron acceptors. The authors believe that the novel and better RC maquettes can be designed with the thermodynamic efficiency that could be substantially higher than that of the natural photosystems.
A group of Raghavan Varadarajan designed a novel high throughput approach for the accurate mapping of interfacial residues based on screening of a query protein against a panel of chemically masked single cysteine mutants of its interacting partner, which are displayed on the surface of yeast cells (Ahmed et al.). In this approach, the binding residues are decrypted by the flow cytometry-based probing of the loss of binding to the labeled cognate partner. This tool was applied for the identification of the interfacial residues for MazEF3, MazEF6 and MazEF9 toxin-antitoxin (TA) systems from Mycobacterium tuberculosis, where the toxin has a conserved fold, while the structurally diverse antitoxins have intrinsically disordered toxin-binding regions.
Based on the response from the scientific community across the globe, the Research Topic has been extended for a second year (Volume II).
SB and VU jointly drafted the editorial. All authors have read and approved the final version.
We thank all the contributors to this Research Topics. We also take the opportunity to acknowledge Abhirup Bandyopadhyay, of Aix-Marseille Université who has acted as coordinator and has contributed to the preparation of the proposal for this Research Topic. We also thank Nalok Dutta, of Washington State University for his participation in author’s searches and key searches.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bandyopadhyay, A., and Basu, S. (2020). Criticality in the conformational phase transition among self-similar groups in intrinsically disordered proteins: Probed by salt-bridge dynamics. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1868, 140474. doi:10.1016/j.bbapap.2020.140474
Basu, S., Bhattacharyya, D., and Banerjee, R. (2011). Mapping the distribution of packing topologies within protein interiors shows predominant preference for specific packing motifs. BMC Bioinforma. 12, 195. doi:10.1186/1471-2105-12-195
Basu, S., and Biswas, P. (2018). Salt-bridge dynamics in intrinsically disordered proteins: A trade-off between electrostatic interactions and structural flexibility. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1866, 624–641. doi:10.1016/j.bbapap.2018.03.002
Biswas, G., Ghosh, S., Basu, S., Bhattacharyya, D., Datta, A. K., and Banerjee, R. (2022). Can the jigsaw puzzle model of protein folding re-assemble a hydrophobic core? Proteins 90, 1390–1412. doi:10.1002/prot.26321
Blazeck, J., Karamitros, C. S., Ford, K., Somody, C., Qerqez, A., Murray, K., et al. (2022). Bypassing evolutionary dead ends and switching the rate-limiting step of a human immunotherapeutic enzyme. Nat. Catal. 5, 952–967. doi:10.1038/s41929-022-00856-6
Callaway, E. (2020). ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 588, 203–204. doi:10.1038/d41586-020-03348-4
Coskuner-Weber, O., and Uversky, V. N. (2018). Insights into the molecular mechanisms of Alzheimer’s and Parkinson’s diseases with molecular simulations: Understanding the roles of artificial and pathological missense mutations in intrinsically disordered proteins related to pathology. Int. J. Mol. Sci. 19, 336. doi:10.3390/ijms19020336
Dill, K. A., and MacCallum, J. L. (2012). The protein-folding problem, 50 Years on. Science 338, 1042–1046. doi:10.1126/science.1219021
Dimitrov, D. S. (2012). Therapeutic proteins. Methods Mol. Biol. 899, 1–26. doi:10.1007/978-1-61779-921-1_1
Dobson, C. M. (2004). Experimental investigation of protein folding and misfolding. Methods 34, 4–14. doi:10.1016/j.ymeth.2004.03.002
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Nagibina, G. S., Glukhova, K. A., Uversky, V. N., Melnik, T. N., and Melnik, B. S. (2020). Intrinsic disorder-based design of stable globular proteins. Biomolecules 10, 64. doi:10.3390/biom10010064
Roy, S., Ghosh, P., Bandyopadhyay, A., and Basu, S. (2022). Capturing a crucial ‘disorder-to-order transition’ at the heart of the coronavirus molecular pathology—triggered by highly persistent, interchangeable salt-bridges. Vaccines 10, 301. doi:10.3390/vaccines10020301
Svilenov, H. L., Bester, R., Sacherl, J., Absmeier, R., Peters, C., Protzer, U., et al. (2022). Multimeric ACE2-IgM fusions as broadly active antivirals that potently neutralize SARS-CoV-2 variants. Commun. Biol. 5, 1237–7. doi:10.1038/s42003-022-04193-z
Theillet, F.-X., Kalmar, L., Tompa, P., Han, K.-H., Selenko, P., Dunker, A. K., et al. (2013). The alphabet of intrinsic disorder: I. Act like a pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins 1, e24360. doi:10.4161/idp.24360
Yue, K., and Dill, K. A. (1992). Inverse protein folding problem: Designing polymer sequences. Proc. Natl. Acad. Sci. U.S.A. 89, 4163–4167. doi:10.1073/pnas.89.9.4163
Keywords: protein design, intrinsically disordered proteins (IDPs), disorder—globular interface, mutational scanning, disorder transitions in proteins, prediction of protein-protein binding sites of IDPs, multi-step electron-transfer activity, high throughput approach
Citation: Basu S, Chakravarty D, Hou Q and Uversky VN (2023) Editorial: From the hydrophobic core to the globular-disorder interface: New challenges and insights into protein design. Front. Mol. Biosci. 10:1151676. doi: 10.3389/fmolb.2023.1151676
Received: 26 January 2023; Accepted: 30 January 2023;
Published: 06 February 2023.
Edited and reviewed by:
Annalisa Pastore, King’s College London, United KingdomCopyright © 2023 Basu, Chakravarty, Hou and Uversky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sankar Basu, c2Fua2FyY2hhbmRyYS5iYXN1QGFzdXRvc2hjb2xsZWdlLmlu; Vladimir N. Uversky, dnV2ZXJza3lAdXNmLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.