 Heng-Chang Chen
Heng-Chang Chen- Epigenetics of Infectious Diseases Research Group, Center for Population Diagnostics, Łukasiewicz–PORT Polish Center for Technology Development, Wrocław, Poland
The outbreak of SARS-CoV-2 has made us more alert to the importance of viral diagnostics at a population level to rapidly control the spread of the disease. The critical question would be how to scale up testing capacity and perform a diagnostic test in a high-throughput manner with robust results and affordable costs. Here, the latest 26 articles using barcoding technology for COVID-19 diagnostics and biologically-relevant studies are reviewed. Barcodes are molecular tags, that allow proceeding an array of samples at once. To date, barcoding technology followed by high-throughput sequencing has been made for molecular diagnostics for SARS-CoV-2 infections because it can synchronously analyze up to tens of thousands of clinical samples within a short diagnostic time. Essentially, this technology can also be used together with different biotechnologies, allowing for investigation with resolution of single molecules. In this Mini-Review, I first explain the general principle of the barcoding strategy and then put forward recent studies using this technology to accomplish COVID-19 diagnostics and basic research. In the meantime, I provide the viewpoint to improve the current COVID-19 diagnostic strategy with potential solutions. Finally, and importantly, two practical ideas about how barcodes can be further applied in studying SARS-CoV-2 to accelerate our understanding of this virus are proposed.
1 Introduction: the general principle of the barcoding strategy
Barcoding strategy has first proposed to solve the problems of PCR duplications and to improve the accuracy of next-generation sequencing quantification (Casbon et al., 2011; Kinde et al., 2011). In the past, barcodes have been given various names, such as unique molecular identifier (UMI) (Kivioja et al., 2012), primer ID (Jabara et al., 2011), and duplex barcodes. Barcodes are usually in the string form of random nucleotides, partially degenerate nucleotides, or defined nucleotides. The concept of the barcoding strategy is that individual original DNA or RNA fragments within the same pool of samples are tagged with a unique sequence of a molecular tag (Peng et al., 2015). Sequence reads that contain different barcodes illustrate different origins of molecules, whereas sequence reads with the same barcodes are the result of PCR duplication from the same original molecule (Peng et al., 2015). In general, the workflow of studies using barcoding technology consists of several main steps including 1) tag samples of interest with unique barcodes, 2) multiplex samples, 3) proceed barcoded samples by sequencers or other high-throughput techniques, and 4) demultiplex readouts and assign each sample to the corresponding barcode. Barcodes can be introduced in at least three ways. In the first approach, barcodes are embedded into molecular adaptors while constructing sequencing libraries. A classic example was given by (Schmitt et al., 2012). They first generated a pair of double-stranded and Y-shaped adaptors embedded with unique barcodes and ligated them to both ends of amplicons. This sequencing library is made to correct sequencing errors shown in sequencing reads (Figure 1A). Several commercial kits already provide the option of a PCR-free barcoding procedure with the same logistic strategy (so-called direct ligation approach shown in Table 1). In the second approach (so-called primer-associated approach in the following context and Table 1), barcodes are embedded in target-specific primers and introduced on a template by reverse transcription (RT) or PCR amplification (Figure 1B). The third approach is to use molecular inversion probes (MIP) carrying barcodes. A classic example was shown in the study from Hiatt et al. (2013) (Hiatt et al., 2013), where molecular tags (the same as barcodes discussed here) were introduced to the reverse complement strand of the gene of interest using polymerase and ligase, allowing distinguishing reads derived from different genomic equivalents within individual DNA samples (Figure 1C). Other methods that are not frequently used for barcoding will also be briefly discussed in the latter section (summarized in Table 1). The following contents will focus on barcoding applied to diagnostics of SARS-CoV-2 infections and biologically-relevant studies.
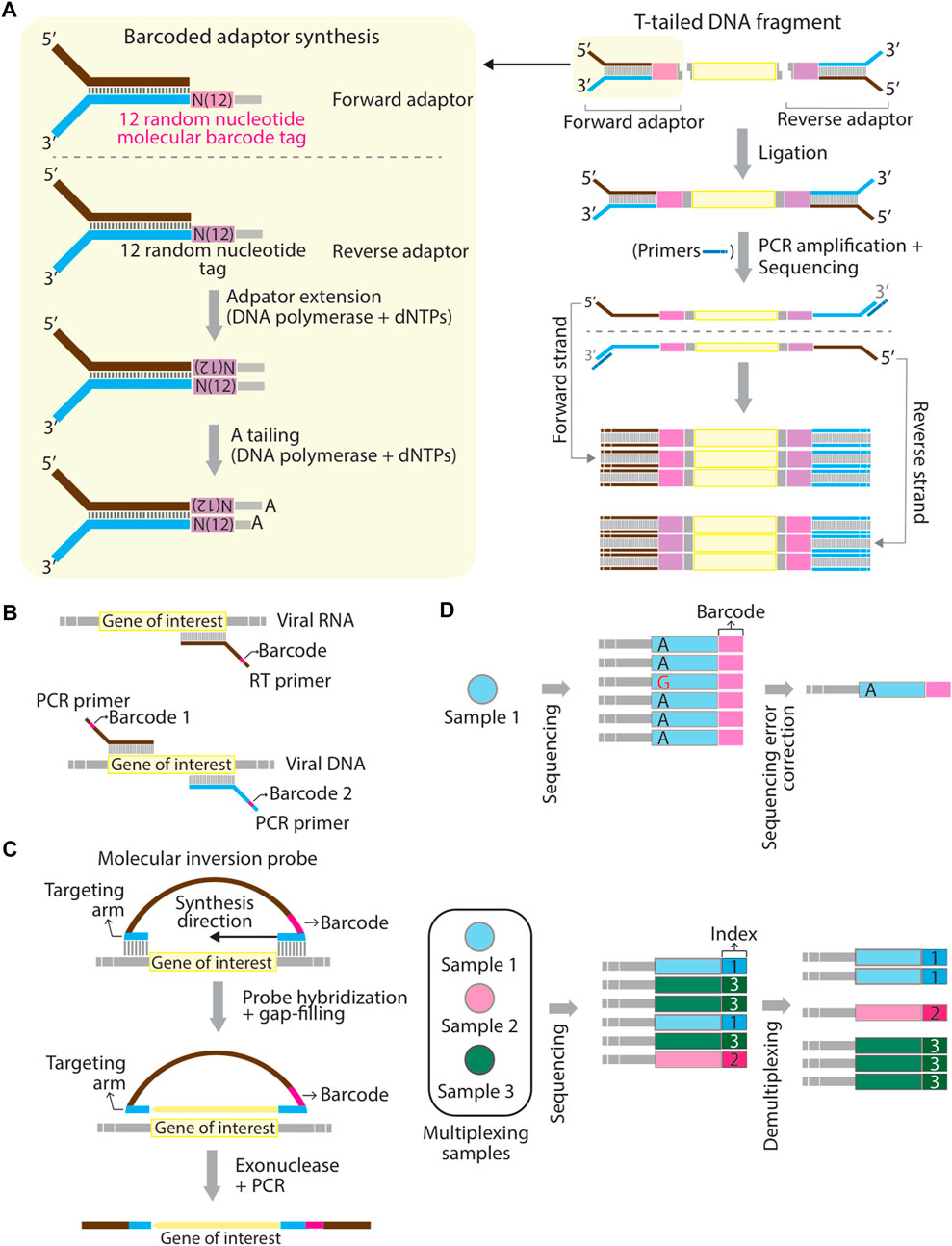
FIGURE 1. Schematic representation of mechanistic strategies of barcoding. (A–C) Barcodes can be introduced to a template using adaptors through direct ligation (A), using RT- or PCR primers at the reverse transcription or PCR amplification step (B), and using hybridizing molecular inversion probes (C). (D) Schematic representation of the difference between “barcodes” and “sample indexes”. Barcodes aim to correct sequencing errors. For example, a misreading nucleotide, guanosine (G) can be corrected in final consensus sequences for a pool of Sample 1 (top panel). Sample indexes are used to multiplex different sequencing amplicons generated from different pools of samples (Sample 1, 2, and 3) (bottom panel). Panel (A) is modified based on Figure 1 in (Schmitt et al., 2012) and panel (C) is modified based on Figure 1 in (Hiatt et al., 2013).
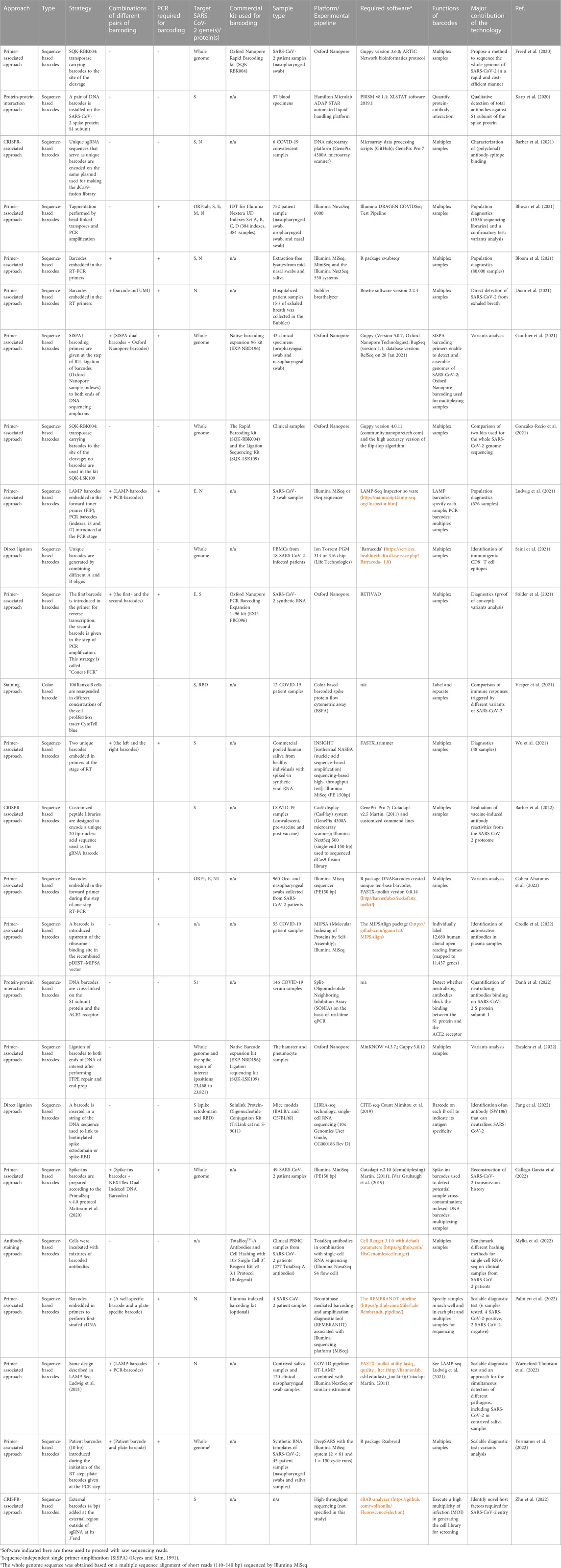
TABLE 1. Systematic comparison of barcoding strategies used in the category of molecular barcodes.
2 Subsections
2.1 Background information about SARS-CoV-2 and COVID-19
The outbreak of novel coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) occurred in early December 2019 and has quickly spread worldwide and turned into a global pandemic. Although the origin of SARS-CoV-2 has been the topic of substantial debate (a natural origin through zoonosis or the introduction from a laboratory source), molecular evidence indicates that coronaviruses originated in bats (Drexler et al., 2014) and then transmitted to civets and several wildlife species as potential intermediate hosts, and then to humans. Coronaviruses, like other RNA viruses that can frequently undergo host switching under different selection pressures, are genetically heterogeneous, in part due to the highly error-prone and low-fidelity RNA-dependent RNA polymerases that replicate their genomes (Vignuzzi et al., 2006; Peck and Lauring, 2018; Jones et al., 2021), resulting in this virus possibly infecting a broad spectrum of hosts.
The genome of SARS-CoV-2 is composed of 29,881 nucleotides (Lu et al., 2020), making this virus one of the largest known single-stranded RNA-enveloped viruses. Its genome encodes four structural proteins, including spike (S), small protein (E), matrix (M), nucleocapsid (N) (Chan et al., 2020), and other accessory or non-structural proteins. In SARS-CoV-2, the S protein is the main structural protein to ensure the attachment of the virion to the target cell and mediate membrane fusion, thereby achieving successful viral entry (Ou et al., 2020) and being a key protein in determining the infectivity of this virus and the transmissibility in the host (Hulswit et al., 2016). Additionally, this protein is also the major antigen inducing protective immune responses (He et al., 2004; Du et al., 2009; Li, 2016; Walls et al., 2020). Pathologically speaking, it is suggested that severe COVID-19 results from virus-driven perturbations in the immune system and tissue injury, including neutrophil extracellular traps, and thrombosis even though the mechanisms that lead to manifestations of viral infection are not fully understood.
2.2 Literature search strategy
A rigorous literature search was done using PubMed with the keywords ((SARS-CoV-2) OR (COVID-19)) AND (barcode). Research articles were searched from 2019 till the time of writing (end of November 2022), with the limitation of solely selecting the research articles published in the English language and the exclusion of the review articles and preprints, and news features. In the first place, 345 articles were released from PubMed searching with the keywords. A careful examination was then performed throughout all articles and removed the ones that do not match the scope of this Mini-Review. Eventually, 45 articles fit the criteria. Based on the function and the type of barcodes described in these 45 articles, the barcodes were classified into three categories: molecular barcodes (26 articles), genetic barcodes (10 articles), and digital barcodes (9 articles). Molecular barcodes refer to sequence-based barcodes, which are often implemented with different biotechnologies, such as PCR, RT-PCR, flow cytometry, CRISPR/Cas9 and so on. In contrast, in this Mini-Review genetic barcodes refer to either the unique viral genomic regions, enabling to classify SARS-CoV-2 variants or host cellular genetic signatures (Fischer et al., 2021). It is worth noting that although the concept of viral genetic barcodes is indeed fascinating for tracking and discriminating variants of SARS-CoV-2 and perhaps can also be beneficial for COVID-19 diagnostics, computational methods/algorithms used to retrieve genetic barcodes are presently not optimized and how frequently that currently known genetic barcodes still remain in the latest variant of SARS-CoV-2 is required to be evaluated. Here I summarize sequences of known genetic barcodes present in major clades, and their corresponding variants, and SARS-CoV-2 genes in Table 2. Genetic barcodes were collected from (Guan et al., 2020) and Zhao et al. (2020) (Zhao et al., 2020). Digital barcodes refer to 2D QR barcodes used to store information. In this Mini-Review, the focus will be placed on molecular barcodes. A comparison of the articles using molecular barcodes is summarized in Table 1.
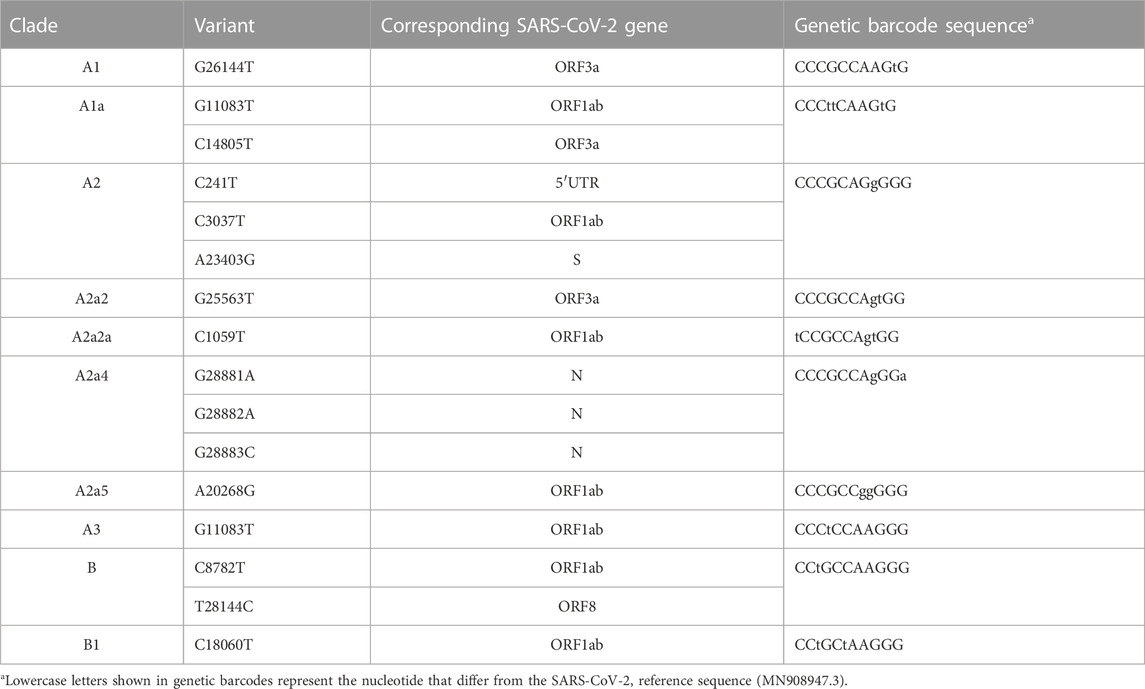
TABLE 2. Known genetic barcodes present in main clades of SARS-CoV-2.
2.3 The barcoding strategy for studying SARS-CoV-2
Barcodes used in these 26 articles are sequence-based, except the study from (Vesper et al., 2021), in which the authors used different concentrations of the cell proliferation tracer, CytoTell blue, as color-based barcodes read by the flow cytometry. The barcoding step can be achieved either using commercial kits, like the kits from Illumina and Oxford Nanopore Technologies, or a customized design (Table 1). In the latter case, a sequence of a barcode is often embedded in primers as an overhang at the step of reverse transcription of viral RNA or PCR amplification of the RT product (Figure 1B) (Bhoyar et al., 2021; Bloom et al., 2021; Duan et al., 2021; Gauthier et al., 2021; Ludwig et al., 2021; Stüder et al., 2021; Wu et al., 2021; Cohen-Aharonov et al., 2022; Credle et al., 2022; Gallego-García et al., 2022; Palmieri et al., 2022; Warneford-Thomson et al., 2022; Yermanos et al., 2022). Barcoded primers used in SwabSeq (Bloom et al., 2021) and by Cohen-Aharonoc et al. (2022) (Cohen-Aharonov et al., 2022) are compatible with one-step RT-PCR. The workflow of barcoding here resembles the primer-associated approach described in the first section that yields a final product of the amplicons carrying barcodes when the procedure of RT-PCR is complete. The length of barcodes can vary (generally between 4–20 base pairs): the longer length of a barcode is, the lower probability that multiple reads contain the same barcode. Of note, “barcodes” and “sample indexes” are conceptually two different molecular tags even though they both consist of a string of a DNA sequence. There are indeed some functional overlaps. However, precisely speaking, “barcodes” resolve to correct sequencing errors, thereby increasing sequencing accuracy, whereas “sample indexes” are used to multiplex sequencing libraries into the same lane of flow cells (Figure 1D). It is noteworthy that while reviewing these articles, I notice that it presently appears to be ambiguous between the usage of the term “barcodes” and “sample indexes”.
Using a combination of multiple barcodes or different layers of barcoding appears to be popular to increase the sequencing capacity and make the readouts more informative. For example, amplicons from SwabSeq (Bloom et al., 2021) are subjected to barcodes (i5 and i7) used to maximize the specificity and avoid false-positive results. Amplicons from LAMP-Seq (Ludwig et al., 2021) and COV-ID (Warneford-Thomson et al., 2022) contain one LAMP barcode (10 bp used in LAMP-Seq and 5 bp used in COV-ID) and two standard PCR barcodes (Illumina i5 and i7) to scale up the deep sequencing capacity. Gauthier et al. (2021) (Gauthier et al., 2021) employed SISPA barcoded primers (Reyes and Kim, 1991) to detect and assemble genomes of SARS-CoV-2 and Oxford Nanopore barcodes to multiplex samples. Stüder et al. (2021) used two sets of barcoded primers to track variants of viruses and multiplex samples for sequencing. Wu et al. (2021) embedded a left and a right barcode (5 bp of each) in the forward- and the reserve primer, respectively, to specify patient samples. Gallego-García et al. (2022) spiked in a string of 20 random nucleotide barcode sequences inserted in the forward- and reverse primer to minimize cross-sample contamination. Palmieri et al. (2022) and Yermanos et al. (2022) applied two-dimensional barcoding primers to specify samples pooled in wells and plates. Similarly, Duan et al. (2021) directly included a known sequence of a barcode (8 bp) and a UMI with three random nucleotides in RT primers at the same time to multiplex samples and correct sequencing reads. In addition to the PCR- or RT-PCR-based method, barcodes can also be introduced using different ways. For example, Danh et al. (2022) used chemical cross-linkers to install DNA barcodes. Studies from Fang et al. (2022), Saini et al. (2021), and Karp et al. (2020)) directly ligated a DNA sequence of a barcode to the protein of interest (peptide-MHC complex multimers or the spike protein), which is a PCR-free approach. Mylka et al. (2022) used barcoded-labeled antibodies or lipid anchors to stain a pool of cells individually. More importantly, the spectrum of its application can be broadened when barcoding is adapted to other biotechnologies. For example, designed unique sgRNA to serve as identifiers (unique barcodes), which are co-expressed with the Cas9 protein Barber et al. (2021); Barber et al., 2022). Zhu et al. (2022) included an additional sequence-based barcode adjacent to the 3’ end of sgRNA in addition to unique guide sequences. As mentioned previously, Vesper et al. (2021) applied the cell proliferation tracer dye with different dilutions to label samples, allowing samples to be separated using flow cytometry.
2.4 The barcoding strategy for current COVID-19 diagnostics, fundamental research, and future perspectives
One of the main contributions of barcoding is to scale up testing capacity for population diagnostics. Diagnostics at a population level has become one of the essential strategies to control the outbreak of COVID-19 because it allows the detection of people with SARS-CoV-2 infections in the first place and immediately places them in quarantine. Available and mature methods, which have been benchmarked for COVID-19 diagnostics at a population level include DRAGEN COVIDSeq (Bhoyar et al., 2021), SwabSeq (Bloom et al., 2021), and LAMP-Seq (Ludwig et al., 2021) (Table 1). Amplicons prepared based on these methods are sequenced using Illumina sequencing platforms (iSeq, MiniSeq, MiSeq, NextSeq, NovaSeq). One strong advantage of Illumina sequencing is that Illumina adapter sequences are made public, benefiting researchers to implement barcodes adapted to their experimental designs subtly. These methods are made to diagnose a small region of a gene, thereby shortening the duration of diagnostic time. Most importantly, these methods appear to be less labor-intensive and cost-effective. Other potential methods for COVID-19 population diagnostics are listed in Table 1. Although nowadays public health policy in many countries tends to coexist with viruses, COVID-19 diagnostics is still crucial to control the spread of the disease in countries where medical resources are insufficient. Since around 33% of people with SARS-CoV-2 infection are estimated to be asymptomatic Oran and Topol (2021) the accurate assessment of COVID-19 diagnostic capacity remains important in first place for strategic planning, public health control measures, and patient management.
In addition to multiplexing samples, several groups apply barcoding to identify new variants of concern (Bhoyar et al., 2021; Gauthier et al., 2021; Stüder et al., 2021; Cohen-Aharonov et al., 2022; Escalera et al., 2022; Gallego-García et al., 2022; Yermanos et al., 2022) (Table 1). Indeed, SARS-CoV-2 is a typical zoonotic RNA virus that enables itself to complete infection across different species. The appearance of viruses that evolve to adapt to a new living niche often reflects on viral sequence changes. Fixation of these changes may require a long time through repeated transmission, eventually resulting in a reduced size of an effective population harboring dominant alterations in their sequence spaces. Investigation of how the virus genetically evolves to achieve host jumps could therefore be essential to understand the molecular basis of this process, benefitting developing better antiviral strategies. One of the methodologies to study virus cross-species transmission is to use the reverse genetics approach, allowing elucidation of the consequence of genetic mutations by examining changes to phenotypes. Here, I propose that barcodes could be implemented in the in vitro or in vivo system and used as tracers for reconstructing individual evolutionary transmission routes over a large experimental timescale. Practically, unique barcodes could be used to tag the genome of SARS-CoV-2 or embedded in SARS-CoV-2 pseudotyped virus. Barcoded viruses then infect an appropriate model system with multiple rounds of infection. Since barcodes distinguish individual viral infections, it becomes feasible to monitor the genetic alteration of individual viruses from different lineages of evolutionary paths.
Barcoding has also been applied to characterize specific antibody-epitope binding (Karp et al., 2020; Barber et al., 2021; Saini et al., 2021; Vesper et al., 2021; Barber et al., 2022; Credle et al., 2022; Danh et al., 2022; Fang et al., 2022), and identify novel host factors required for viral entry (Zhu et al., 2022) (Table 1). A typical feature of an RNA virus is high rates of mutations due to the high error-prone and low fidelity of the RNA-dependent RNA polymerase, thereby exhausting our immune system and weakening the efficacy of antiviral drugs. For this reason, an effective strategy to develop a broad spectrum of SARS-CoV-2 neutralizing antibodies and antiviral drugs that cover variants of SARS-CoV-2 is a requisite shortly. Another idea proposed here is to high-throughput select drug-resistant variants of SARS-CoV-2 in vitro. Barcoded SARS-CoV-2 will be used for in vitro infections in the presence of different antiviral drugs. After multiple rounds of infections, barcoded viruses that remain vivid are collected and sequenced. Based on unique barcodes, it thus becomes possible to unveil mutations, which are essential to resist the killing of corresponding antiviral drugs with resolution of individual viruses at a single-nucleotide level.
3 Discussion
In the past 10 years, technological progress in barcoding has been made to reach the resolution at a single-molecule level and detect low-frequency and subclonal variations. Such advantages are now applied to elevate diagnostic capacity and study or track variations of individual viruses in a pool of samples. Collectively, the advantages of barcoding strategy toward COVID-19 diagnosis include 1) increasing the throughput of diagnostic samples, 2) shortening the processing time, 3) diminishing the risk of technical batch effects, 4) lowing library preparation costs and per-sample cost, and 5) increasing accuracy of diagnostic results. Furthermore, the potential application of the barcoding strategy in SARS-CoV-2 research can be extended to track variants over a large timescale and perform SARS-CoV-2 progression surveillance beyond the usage in COVID-19 diagnosis. Nevertheless, critical issues (shortcomings of barcoding strategy), such as barcode collisions and barcode hopping are still required to pay attention. These problems could be solved at the experimental- and analytical level. The potential solution worked out at the bench could be by increasing the complexity of unique barcodes in a pooled library, thereby minimizing the probability that multiple molecules initially receive the same barcode (barcode collisions) or barcodes are incorrectly assigned to other molecules (barcode hopping) at the amplification step. The complexity of barcodes can be lifted either by increasing the abundance of a pool of barcodes (quantity) or adjusting a minimum Levenshtein distance (Yujian and Bo, 2007) among barcodes (quality). Alternatively, errors could also be corrected using better error-correcting algorithms and quantification methods.
In this Mini-Review, recent 26 research articles using the barcoding strategy, which mainly contributes to COVID-19 diagnostics and biological research of SARS-CoV-2 were systematically reviewed (Table 1). In addition to increased diagnostic capacity, rapid duration of diagnostic time, and low costs, the accuracy of diagnostic results is another factor that should be well considered. Several of the studies (Bloom et al., 2021; Gauthier et al., 2021; Ludwig et al., 2021) reviewed here already discussed and proposed possible solutions to correct false-positive results caused by barcode swapping. Importantly, it has been documented that up to 58% of COVID-19 patients may face initial false-negative diagnostics results (Pecoraro et al., 2022). One of the risks is due to frequent mutations in the genome of SARS-CoV-2, rendering primers used for detecting viruses ineffective. A potential solution could be to perform population-scale long-read sequencing. Although this idea has been put forward (Freed et al., 2020; Gauthier et al., 2021; González-Recio et al., 2021; Stüder et al., 2021; Escalera et al., 2022), the current methods are required to be further optimized. Essentially, two practical ideas to expand the power of barcoding are proposed in this Mini-Review. In the first idea, barcodes can be used as a tracer to depict the history of genome alterations in every lineage of variants of SARS-CoV-2 over time. It is beneficial to screen potential mutations that are required for cross-species transmission. The second idea would then benefit medical doctors to adjust antiviral regimens for treatments to satisfy the need of individual patients. Collectively, barcoding is one of the molecular tools that assist to read a massive array of samples in parallel and the onset of investigating variations at a population level.
Author contributions
Conceptualization, H-CC; literature search, H-CC; writing—original draft preparation, H-CC; writing—review and editing, H-CC; funding acquisition, H-CC. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by institutional funding; no extramural funding was received.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Barber, K. W., Shrock, E., and Elledge, S. J. (2022). CasPlay provides a gRNA-barcoded CRISPR-based display platform for antibody repertoire profiling. Cell. Rep. Methods 2, 100318. doi:10.1016/j.crmeth.2022.100318
Barber, K. W., Shrock, E., and Elledge, S. J. (2021). CRISPR-based peptide library display and programmable microarray self-assembly for rapid quantitative protein binding assays. Mol. Cell. 81, 3650–3658. doi:10.1016/j.molcel.2021.07.027
Bhoyar, R. C., Jain, A., Sehgal, P., Divakar, M. K., and Sharma, D. (2021). High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS One 16, 0247115. doi:10.1371/journal.pone.0247115
Bloom, J. S., Sathe, L., Munugala, C., Jones, E. M., Gasperini, M., Lubock, N. B., et al. (2021). Massively scaled-up testing for SARS-CoV-2 RNA via next-generation sequencing of pooled and barcoded nasal and saliva samples. Nat. Biomed. Eng. 5, 657–665. doi:10.1038/s41551-021-00754-5
Casbon, J. A., Osborne, R. J., Brenner, S., and Lichtenstein, C. P. (2011). A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Res. 39, 81. doi:10.1093/nar/gkr217
Chan, J. F., Kok, K. H., Zhu, Z., Chu, H., To, K. K., Yuan, S., et al. (2020). Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 9, 221–236. doi:10.1080/22221751.2020.1719902
Cohen-Aharonov, L. A., Rebibo-Sabbah, A., Yaacov, A., Granit, R. Z., Strauss, M., Colodner, R., et al. (2022). High throughput SARS-CoV-2 variant analysis using molecular barcodes coupled with next generation sequencing. PLoS One 17, 0253404. doi:10.1371/journal.pone.0253404
Credle, J. J., Gunn, J., Sangkhapreecha, P., Monaco, D. R., Zheng, X. A., Tsai, H. J., et al. (2022). Unbiased discovery of autoantibodies associated with severe COVID-19 via genome-scale self-assembled DNA-barcoded protein libraries. Nat. Biomed. Eng. 6, 992–1003. doi:10.1038/s41551-022-00925-y
Danh, K., Karp, D. G., Singhal, M., Tankasala, A., Gebhart, D., de Jesus Cortez, F., et al. (2022). Detection of neutralizing antibodies against multiple SARS-CoV-2 strains in dried blood spots using cell-free PCR. Nat. Commun. 13, 4212. doi:10.1038/s41467-022-31796-1
Drexler, J. F., Corman, V. M., and Drosten, C. (2014). Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 101, 45–56. doi:10.1016/j.antiviral.2013.10.013
Du, L., He, Y., Zhou, Y., Liu, S., Zheng, B. J., and Jiang, S. (2009). The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7, 226–236. doi:10.1038/nrmicro2090
Duan, C., Buerer, L., Wang, J., Kaplan, S., Sabalewski, G., Jay, G. D., et al. (2021). Efficient detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from exhaled breath. J. Mol. Diagn 23, 1661–1670. doi:10.1016/j.jmoldx.2021.09.005
Escalera, A., Gonzalez-Reiche, A. S., Aslam, S., Mena, I., Laporte, M., Pearl, R. L., et al. (2022). Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell. Host Microbe 30, 373–387.e7. doi:10.1016/j.chom.2022.01.006
Fang, Y., Sun, P., Xie, X., Du, M., Du, F., Ye, J., et al. (2022). An antibody that neutralizes SARS-CoV-1 and SARS-CoV-2 by binding to a conserved spike epitope outside the receptor binding motif. Sci. Immunol. 7, 9962. doi:10.1126/sciimmunol.abp9962
Fischer, D. S., Ansari, M., Wagner, K. I., Jarosch, S., Huang, Y., Mayr, C. H., et al. (2021). Single-cell RNA sequencing reveals ex vivo signatures of SARS-CoV-2-reactive T cells through ‘reverse phenotyping. Nat. Commun. 12, 4515. doi:10.1038/s41467-021-24730-4
Freed, N. E., Vlková, M., Faisal, M. B., and Silander, O. K. (2020). Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 5, 014. doi:10.1093/biomethods/bpaa014
Gallego-García, P., Varela, N., Estévez-Gómez, N., De Chiara, L., Fernández-Silva, I., Valverde, D., et al. (2022). Limited genomic reconstruction of SARS-CoV-2 transmission history within local epidemiological clusters. Virus Evol. 8, 008. doi:10.1093/ve/veac008
Gauthier, N. P. G., Nelson, C., Bonsall, M. B., Locher, K., Charles, M., MacDonald, C., et al. (2021). Nanopore metagenomic sequencing for detection and characterization of SARS-CoV-2 in clinical samples. PLoS One 16, 0259712. doi:10.1371/journal.pone.0259712
González-Recio, O., Gutiérrez-Rivas, M., Peiró-Pastor, R., Aguilera-Sepúlveda, P., Cano-Gómez, C., Jiménez-Clavero, M. Á., et al. (2021). Sequencing of SARS-CoV-2 genome using different nanopore chemistries. Appl. Microbiol. Biotechnol. 105, 3225–3234. doi:10.1007/s00253-021-11250-w
Grubaugh, N. D., Gangavarapu, K., Quick, J., Matteson, N. L., De Jesus, J. G., Main, B. J., et al. (2019). An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8. doi:10.1186/s13059-018-1618-7
Guan, Q., Sadykov, M., Mfarrej, S., Hala, S., Naeem, R., Nugmanova, R., et al. (2020). A genetic barcode of SARS-CoV-2 for monitoring global distribution of different clades during the COVID-19 pandemic. Int. J. Infect. Dis. 100, 216–223. doi:10.1016/j.ijid.2020.08.052
He, Y., Zhou, Y., Wu, H., Luo, B., Chen, J., Li, W., et al. (2004). Identification of immunodominant sites on the spike protein of severe acute respiratory syndrome (SARS) coronavirus: Implication for developing SARS diagnostics and vaccines. J. Immunol. 173, 4050–4057. doi:10.4049/jimmunol.173.6.4050
Hiatt, J. B., Pritchard, C. C., Salipante, S. J., O’Roak, B. J., and Shendure, J. (2013). Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 23, 843–854. doi:10.1101/gr.147686.112
Hulswit, R. J. G., de Haan, C. A. M., and Bosch, B. J. (2016). Coronavirus spike protein and tropism changes. Adv. Virus Res. 96, 29–57. doi:10.1016/bs.aivir.2016.08.004
Jabara, C. B., Jones, C. D., Roach, J., Anderson, J. A., and Swanstrom, R. (2011). Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. U. S. A. 108, 20166–20171. doi:10.1073/pnas.1110064108
Jones, J. E., Le Sage, V., and Lakdawala, S. S. (2021). Viral and host heterogeneity and their effects on the viral life cycle. Nat. Rev. Microbiol. 19, 272–282. doi:10.1038/s41579-020-00449-9
Karp, D. G., Cuda, D., Tandel, D., Danh, K., Robinson, P. V., Seftel, D., et al. (2020). Sensitive and specific detection of SARS-CoV-2 antibodies using a high-throughput, fully automated liquid-handling robotic system. SLAS Technol. 25, 545–552. doi:10.1177/2472630320950663
Kinde, I., Wu, J., Papadopoulos, N., Kinzler, K. W., and Vogelstein, B. (2011). Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. U. S. A. 108, 9530–9535. doi:10.1073/pnas.1105422108
Kivioja, T., Vähärautio, A., Karlsson, K., Bonke, M., Enge, M., Linnarsson, S., et al. (2012). Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 9, 72–74. doi:10.1038/nmeth.1778
Li, F. (2016). Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261. doi:10.1146/annurev-virology-110615-042301
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395, 565–574. doi:10.1016/S0140-6736(20)30251-8
Ludwig, K. U., Schmithausen, R. M., Li, D., Jacobs, M. L., Hollstein, R., Blumenstock, K., et al. (2021). LAMP-Seq enables sensitive, multiplexed COVID-19 diagnostics using molecular barcoding. Nat. Biotechnol. 39, 1556–1562. doi:10.1038/s41587-021-00966-9
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17, 10–12. doi:10.14806/ej.17.1.200
Matteson, N., Grubaugh, N., Gangavarapu, K., Quick, J., Loman, N., and Andersen, K. (2020). PrimalSeq: Generation of tiled virus amplicons for MiSeq sequencing. Protocols. doi:10.17504/protocols.io.bez7jf9n
Mimitou, E. P., Cheng, A., Montalbano, A., Hao, S., Stoeckius, M., Legut, M., et al. (2019). Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412. doi:10.1038/s41592-019-0392-0
Mylka, V., Matetovici, I., Poovathingal, S., Aerts, J., Vandamme, N., Seurinck, R., et al. (2022). Comparative analysis of antibody- and lipid-based multiplexing methods for single-cell RNA-seq. Genome Biol. 23, 55. doi:10.1186/s13059-022-02628-8
Oran, D. P., and Topol, E. J. (2021). The proportion of SARS-CoV-2 infections that are asymptomatic: A systematic review. Ann. Intern Med. 174, 655–662. doi:10.7326/M20-6976
Ou, X., Liu, Y., Lei, X., Li, P., Mi, D., Ren, L., et al. (2020). Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 11, 1620. doi:10.1038/s41467-020-15562-9
Palmieri, D., Javorina, A., Siddiqui, J., Gardner, A., Fries, A., Chapleau, R. R., et al. (2022). Mass COVID-19 patient screening using UvsX and UvsY mediated DNA recombination and high throughput parallel sequencing. Sci. Rep. 12, 4082. doi:10.1038/s41598-022-08034-1
Peck, K. M., and Lauring, A. S. (2018). Complexities of viral mutation rates. J. Virol. 92, 010311–e1117. doi:10.1128/JVI.01031-17
Pecoraro, V., Negro, A., Pirotti, T., and Trenti, T. (2022). Estimate false-negative RT-PCR rates for SARS-CoV-2. A systematic review and meta-analysis. Eur. J. Clin. Invest. 52, 13706. doi:10.1111/eci.13706
Peng, Q., Satya, R. V., Lewis, M., Randad, P., and Wang, Y. (2015). Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genomics 16, 589. doi:10.1186/s12864-015-1806-8
Reyes, G. R., and Kim, J. P. (1991). Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol. Cell. Probes 5, 473–481. doi:10.1016/s0890-8508(05)80020-9
Saini, S. K., Hersby, D. S., Tamhane, T., Povlsen, H. R., Amaya Hernandez, S. P., Nielsen, M., et al. (2021). SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8+ T cell activation in COVID-19 patients. Sci. Immunol. 6, 7550. doi:10.1126/sciimmunol.abf7550
Schmitt, M. W., Kennedy, S. R., Salk, J. J., Fox, E. J., Hiatt, J. B., and Loeb, L. A. (2012). Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 109, 14508–14513. doi:10.1073/pnas.1208715109
Stüder, F., Petit, J. L., Engelen, S., and Mendoza-Parra, M. A. (2021). Real-time SARS-CoV-2 diagnostic and variants tracking over multiple candidates using nanopore DNA sequencing. Sci. Rep. 11, 15869. doi:10.1038/s41598-021-95563-w
Vesper, N., Ortiz, Y., Bartels-Burgahn, F., Yang, J., de la Rosa, K., Tenbusch, M., et al. (2021). A barcoded flow cytometric assay to explore the antibody responses against SARS-CoV-2 spike and its variants. Front. Immunol. 12, 730766. doi:10.3389/fimmu.2021.730766
Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E., and Andino, R. (2006). Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348. doi:10.1038/nature04388
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., and Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 181, 281–292. doi:10.1016/j.cell.2020.02.058
Warneford-Thomson, R., Shah, P. P., Lundgren, P., Lerner, J., Morgan, J., Davila, A., et al. (2022). A LAMP sequencing approach for high-throughput co-detection of SARS-CoV-2 and influenza virus in human saliva. Elife 11, 69949. doi:10.7554/eLife.69949
Wu, Q., Suo, C., Brown, T., Wang, T., Teichmann, S. A., and Bassett, A. R. (2021). Insight: A population-scale COVID-19 testing strategy combining point-of-care diagnosis with centralized high-throughput sequencing. Sci. Adv. 7, 5054. doi:10.1126/sciadv.abe5054
Yermanos, A., Hong, K. L., Agrafiotis, A., Han, J., Nadeau, S., Valenzuela, C., et al. (2022). DeepSARS: Simultaneous diagnostic detection and genomic surveillance of SARS-CoV-2. BMC Genomics 23, 289. doi:10.1186/s12864-022-08403-0
Yujian, L., and Bo, L. (2007). A normalized Levenshtein distance metric. IEEE Trans. Pattern Anal. Mach. Intell. 29, 1091–1095. doi:10.1109/tpami.2007.1078
Zhao, Z., Sokhansanj, B. A., Malhotra, C., Zheng, K., and Rosen, G. L. (2020). Genetic grouping of SARS-CoV-2 coronavirus sequences using informative subtype markers for pandemic spread visualization. PLoS Comput. Biol. 16, 1008269. doi:10.1371/journal.pcbi.1008269
Keywords: barcoding technology, SARS-CoV-2, COVID-19, COVID-19 diagnostics, population diagnostics
Citation: Chen H-C (2023) A systematic review of the barcoding strategy that contributes to COVID-19 diagnostics at a population level. Front. Mol. Biosci. 10:1141534. doi: 10.3389/fmolb.2023.1141534
Received: 10 January 2023; Accepted: 30 June 2023;
Published: 11 July 2023.
Edited by:
Hem Chandra Jha, Indian Institute of Technology Indore, IndiaCopyright © 2023 Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heng-Chang Chen, aGVuZy1jaGFuZy5jaGVuQHBvcnQubHVrYXNpZXdpY3ouZ292LnBs