 Yumeng Zhang
Yumeng Zhang Xiaorong Liu
Xiaorong Liu Jianhan Chen
Jianhan Chen- Department of Chemistry, University of Massachusetts, Amherst, MA, United States
Gram-positive pathogenic bacteria Staphylococcus express and secret staphylococcal peroxidase inhibitor (SPIN) proteins to help evade neutrophil-mediated immunity by inhibiting the activity of the main oxidative-defense player myeloperoxidase (MPO) enzyme. SPIN contains a structured 3-helix bundle C-terminal domain, which can specifically bind to MPO with high affinity, and an intrinsically disordered N-terminal domain (NTD), which folds into a structured β-hairpin and inserts itself into the active site of MPO for inhibition. Mechanistic insights of the coupled folding and binding process are needed in order to better understand how residual structures and/or conformational flexibility of NTD contribute to the different strengths of inhibition of SPIN homologs. In this work, we applied atomistic molecular dynamics simulations on two SPIN homologs, from S. aureus and S. delphini, respectively, which share high sequence identity and similarity, to explore the possible mechanistic basis for their different inhibition efficacies on human MPO. Direct simulations of the unfolding and unbinding processes at 450 K reveal that these two SPIN/MPO complexes systems follow surprisingly different mechanisms of coupled binding and folding. While coupled binding and folding of SPIN-aureus NTD is highly cooperative, SPIN-delphini NTD appears to mainly utilize a conformational selection-like mechanism. These observations are in contrast to an overwhelming prevalence of induced folding-like mechanisms for intrinsically disordered proteins that fold into helical structures upon binding. Further simulations of unbound SPIN NTDs at room temperature reveal that SPIN-delphini NTD has a much stronger propensity of forming β-hairpin like structures, consistent with its preference to fold and then bind. These may help explain why the inhibition strength is not well correlated with binding affinity for different SPIN homologs. Altogether, our work establishes the relationship between the residual conformational stability of SPIN-NTD and their inhibitory function, which can help us develop new strategies towards treating Staphylococcal infections.
Introduction
Staphylococcus is a group of gram-positive pathogenic bacteria that can lead to a broad range of infections including pneumonia and toxic shock syndrome (Lowy, 1998; Tong et al., 2015). Staphylococcal infections are becoming an increasingly severe threat to public health, with an estimate of ∼3 million cases in the United States every year and expanding incidence of antibiotic resistance (Tong et al., 2015). To defend against the invasions of staphylococcus, neutrophils are critical innate immune response components in hosts and serve as the first defensive line by releasing the anti-bacterium hypochlorous acid (Ploscariu et al., 2018) and other reactive oxidant species (ROS) (Rigby and DeLeo, 2012; de Jong et al., 2017). Particularly, myeloperoxidase (MPO) is one of the most abundant granule enzymes in neutrophils that can catalyze the production of ROSs from hydrogen peroxide (H2O2) to help kill the bacterium. However, Staphylococcus has been found to be able to evade the neutrophil-mediated innate immune defense and sometimes turn host cells into “Trojan Horses” for bacterial dissemination in vivo (Lambris et al., 2008; Thwaites and Gant, 2011; Kim et al., 2012; Spaan et al., 2013; Garcia et al., 2016). In particular, the bacterium can secret Staphylococcal Peroxidase INhibitor (SPIN) proteins, which bind MPO with nanomolar affinity and inhibit its enzymatic activity (de Jong et al., 2017; Ploscariu et al., 2018). SPIN consists of an intrinsically disordered N-terminal domain (NTD) and a structured 3-helix bundle C-terminal domain (CTD) (de Jong et al., 2017; de Jong et al., 2018; Ploscariu et al., 2018). The inhibitory activity requires the disordered SPIN NTD and can be largely abolished with deletion or certain mutations of the NTD region (de Jong et al., 2018). Structural studies have revealed that SPIN NTD folds into a β-hairpin and inserts itself into MPO’s active site in the complex (de Jong et al., 2018), which prevents the substrate H2O2 from accessing the catalytic heme in MPO’s active pocket. As a result, the enzyme becomes incapable of producing ROSs, thus protecting Staphylococcus from killing by neutrophils (de Jong et al., 2017).
Recently, multiple SPIN homologs that share high sequence identity and conformational similarity have been identified with various inhibitory capacities towards human MPO (Ploscariu et al., 2018). Interestingly, their inhibitory capacities show little correlation with their binding affinities to MPO (Ploscariu et al., 2018). For example, while SPIN-agnetis binds human MPO with a KD of ∼42 nM, it has little measurable inhibitory effect on MPO activity. The implication is that, the folded SPIN CTD largely determines the binding affinity to MPO, while the disordered NTD dictates the inhibitory efficacy. Furthermore, structural studies suggest that all SPN NTD homologs likely fold into essentially the same β-hairpin conformation in the bound state (Ploscariu et al., 2018). Therefore, functional differences between SPIN homologs may be directly related to the disordered unbound state and/or the coupled binding and folding processes themselves. Specifically, two key questions are: 1) how residual structures or conformational plasticity contribute to the facile folding and binding of SPIN NTD, thus potentially impacting the inhibition strength, and 2) whether SPIN homologs show different mechanisms of coupled binding and folding.
Intrinsically disordered proteins/regions (IDPs/IDRs) like SPIN NTD are prevalent in biology and frequently play key roles in cellular regulation and signal transduction (Wright and Dyson, 1999; Dunker et al., 2001; Tsai et al., 2001; Dyson and Wright, 2005; Uversky et al., 2005; Click et al., 2010). IDPs also frequently undergo coupled binding and folding for function (Koshland, 1960; Agarwal et al., 2002; Antikainen et al., 2005; Hammes et al., 2009; Wright and Dyson, 2009; Zhou, 2010). Two classes of mechanisms have been generally invoked in studies of IDP coupled binding and folding. In so-called conformational selection-like mechanisms (Fuxreiter et al., 2004; Shammas et al., 2016; Crabtree et al., 2017; Bonetti et al., 2018; Troilo et al., 2019), residual structures in unbound state of an IDP may resemble the folded complex and serve as initial binding sites to facilitate efficient molecular recognition (that is, fold and then bind). On the other hand, an IDP could undergo rapid folding upon non-specific encountering with its target, following the so-called induced folding-like mechanism (Levy et al., 2007; Huang and Liu, 2009; Liu and Huang, 2014). Here, structural plasticity plays a more important role, such as to enable facile IDP folding on the target surface (Pontius, 1993; Oldfield et al., 2005; Turjanski et al., 2008; Gsponer and Babu, 2009; Trizac et al., 2010; Ganguly et al., 2012a; Ganguly et al., 2012b; Chu et al., 2012; Ganguly et al., 2013; Rogers et al., 2014; Liu et al., 2019). For the cases where the binding pocket is deep and rugged, induced fitting can direct the peptide to reach the spot and then fold to the energetically favored states (Sugase et al., 2007; Dosnon et al., 2015; Gianni et al., 2016; Sen and Udgaonkar, 2019; Yang et al., 2019; Toto et al., 2020). It should be noted that existing mechanistic studies have mainly involved IDPs that fold into α-helices, ordered loops or a single β-strand upon binding and that induced folding has been found to be prevalent in these IDPs (Wright and Dyson, 2009; Chen, 2012; Chen et al., 2020). SPIN NTD is notably different from these existing studies; it represents the first case study of coupled binding and folding of an IDP into a β-hairpin. Folding of β-hairpin structures involves cooperative formation of long-range contacts and has been shown to be much slower than helix-coil transitions with substantial entropy-dominant free energy barriers (Munoz et al., 1997; Klimov and Thirumalai, 2000; Chen et al., 2018). It remains unclear if SPIN-NTD will display similar mechanistic features to IDPs with simple folded structures.
In this work, we focus on two SPIN homologs, SPIN-aureus and SPIN-delphini. They share 53% sequence identity and 80% sequence similarity, and both bind to human MPO with nanomolar affinities and fold into essentially identical β-hairpin structures (Lambris et al., 2008; Thwaites and Gant, 2011; Kim et al., 2012; Spaan et al., 2013; Garcia et al., 2016). Interestingly, although SPIN-delphini binds to MPO ∼19 times weaker than SPIN-aureus, its half maximal inhibitory concentration (IC50) is only ∼6 times higher. We will mainly utilize atomistic simulations in explicit solvent to probe the conformational properties of unbound NTDs from SPIN-aureus and SPIN-delphini and to investigate their coupled binding and folding processes. We note that several coarse-grained protein models have also been recently proposed for IDPs, such as AWSEM-IDP (Wu et al., 2018), SOP-IDP (Baul et al., 2019), a modifield MARTINI model (Benayad et al., 2021) and HyRes II (Zhang et al., 2022). However, none of these models is capable of describing both folded and disordered protein states required for studies of coupled binding and folding. Instead, atomistic simulations have significantly benefited from recent advances in both GPU-enabled MD algorithms (Phillips et al., 2005; Brooks et al., 2009; Eastman et al., 2012; Gotz et al., 2012; Abraham et al., 2015; Case et al., 2017), which can provide over 100-fold acceleration compared to traditional CPU-based approaches, and accurate general-purposed protein force fields (Nerenberg et al., 2012; Piana et al., 2015; Robertson et al., 2015; Huang et al., 2017; Robustelli et al., 2018; Liu and Chen, 2019), which have been extensively rebalanced for describing both folded and disordered proteins. Simulations of temperature-driven dissociation process of two SPIN/MPO complexes at 450 K recapitulate that SPIN CTD dominates specific binding to MPO and further reveal surprising differences in coupled binding and folding of NTD of these two SPIN homologs. The binding and folding are highly cooperative for SPIN-aureus NTD, while SPIN-delphini NTD prefers to be partially folded before binding to the MPO active site. Further simulations at the room temperature show that unbound SPIN-delphini NTD is much more structured. These results suggest an important role of residual structures of SPIN NTD in its facile recognition and inhibition of MPO, which may help us better understand the sequence-structure-function relationship of SPIN.
Methods
High temperature simulations of SPIN/MPO complexes
All simulations were performed with the GPU accelerated CHARMM/OpenMM interface (Brooks et al., 2009; Lee et al., 2016; Eastman et al., 2017) in CHARMM36m force field (Huang et al., 2017), which is one of the latest general-purpose protein force field specifically optimized for both IDPs and structured proteins. The initial structures of SPIN-aureus and SPIN-delphini in complex with human MPO were taken from the crystal structures [PDB 5UZU SPIN-aureus (de Jong et al., 2018) and 6BMT for SPIN-delphini (Ploscariu et al., 2018)] (see Figure 1A). To reduce the computational cost, only segments of MPO that are within 12 Å of SPIN are included in the current simulations, which consist of residues 167–200, 255–444, 490–506, 526–540, and 566–596 for MPO (Supplemetary Figure S1). To prevent the unfolding of MPO, all backbone heavy atoms of structured MPO segments (excluding loop residues 268–288, 380–395, and 317–328) and the bound heme group were restrained by harmonic potentials with a force constant of 1.0 kcal/(mol Å2) in all simulations. Proper amount of Na+ and Cl− ions were added to neutralize the systems and to reach a NaCl concentration of 50 mM in accord with the experimental conditions (Ploscariu et al., 2018). The final solvated box contains about ∼30,000 TIP3P water molecules and has a dimension of ∼9.2nm3 × 9.4nm3 × 11.3 nm3.
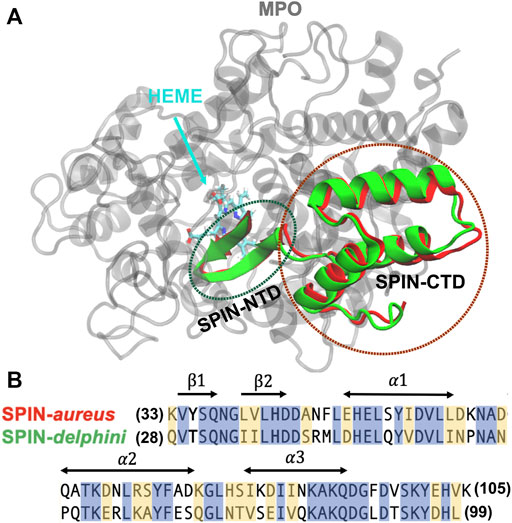
FIGURE 1. (A) Overlay of structures SPIN-aureus and SPIN-delphini in complex with human MPO. The structures were taken from PDB 5UZU and 6BMT for SPIN-aureus and SPIN-delphini, respectively. MPO is colored grey and SPIN-aureus and SPIN-delphini are colored red and green, respectively. Heme was shown in sticks. (B) NTD sequences of two SPIN homologs with conserved residues are highlighted in blue and similar residues in yellow. The sequence alignment is calculated using BLAST (Park et al., 2012) that shows 53% identity and 80% similarity. The secondary structures in the bound state are marked with arrows.
Each solvated system was first energetically minimized for 500 steps using steepest decent and another 500 steps using the adopted basis Newton-Raphson algorithm. The system was then slowly heated up from 100 K to 300 K in 10 ps under the constant volume condition. Equilibration simulations were then performed at 300 K and 1 atm for a total of 1 ns, during which all protein heavy atoms were additionally restrained using harmonic potentials with force constants slowly decreasing from 5.0 kcal/(mol Å2) to 0.0 kcal/(mol Å2). Langevin thermostat was used to control the temperature and Monte Carlo barostat with volume move attempt every 25 steps was used to control the pressure. Lengths of all bonds involving hydrogen atoms were constrained using the SHAKE algorithm (Ryckaert et al., 1977) to allow for an integration time step of 2 fs. Long-range electrostatic interactions were treated using the particle mesh Ewald method (Darden et al., 1993), and the short-range van der Waals (vdW) interactions were treated with the twin-range cutoff at 12 and 14 Å.
To identify the optimal temperatures for unbinding/unfolding simulations, a series of pilot simulations were performed at temperatures ranging from 400 K to 500 K at 1 atm. Note that the complex was found to be highly stable below 400 K. Once an optimal temperature was identified (450 K), two sets of simulations were performed for each complex to probe temperature-induced SPIN unfolding and unbinding process. In one set, three additional simulations were performed at 450 K with different initial velocities to better characterize the dissociation of SPIN from MPO. These simulations were run until the NTD dissociated from the active pocket (i.e., with the fraction of native contacts between two molecules Qinter < 0.3), which all occurred within 400 ns. In the second set, 40 independent replicas were performed for each complex at 450 K for 250 ns each, with the helical region of SPIN CTD (Figure 1B) harmonically restrained with a force constant of 1.0 kcal/(mol Å2). The purpose of the second set is to directly examine the unfolding and unbinding of the NTDs.
Room temperature simulations of free NTDs
The initial folded hairpin structures of SPIN-NTDs were taken from the same complex structures (Figure 1A). Both systems contain a 13-residue fragment (SPIN-aureus residues 33–45 and SPIN-delphini residues 28–40; see Figure 1B for sequences). 20 replicas were used to simulate the unfolding events for two SPIN-NTDs at 300 K. The solvated systems contain ∼3,500 TIP3P waters and have dimensions of ∼4.2nm3 × 4.3nm3 × 5.4 nm3. Similar protocols as described above were applied to minimize and equilibrate the system. For each system, 20 independent production simulations were performed for 50 ns each at 300 K, which was sufficient to observe spontaneous unfolding of the β-hairpin structure.
Analysis
All the analyses were carried out using CHARMM and additional in-house scripts. All molecular visualizations were prepared using VMD (Humphrey et al., 1996). The fractions of intermolecular and intramolecular native contacts, Qinter and Qintra, are calculated to monitor the unfolding and unbinding process. The native contacts are first identified from the crystal structure of two complexes if the minimum heavy atom distance between two residues is no greater than 4.2 Å (Supplemetary Tables S1, S2). Note that for intramolecular native contacts, we exclude residue pairs that are close in sequence space and only consider those whose residue IDs are different by at least three. The contacts in simulation trajectories were then calculated using the same criterion. Based on protein folding funnel theory, native interactions dominate the overall pathway (Onuchic et al., 1996; Tsai et al., 1999; Shoemaker et al., 2000; Best et al., 2013). Therefore, only native contacts were considered here. The unbinding and unfolding kinetics were analyzed using a double exponential approximation of the decay of Qinter and Qintra averaged over all replica runs (40 for the complexes and 20 for free NTDs). The first 50 ns trajectories were considered in unfolding and unbinding kinetic analysis, which were sufficient to capture the dissociation events. Pseudo free energy surfaces were also calculated to better characterize the baseline mechanisms of coupled binding and folding, derived directly from two-dimensional (2D) probability distributions along Qinter and Qintra. For the data used to construct contact probabilities, we specifically focused on short segments of the trajectories where actual dissociation transitions occurred. For example, only the first 15 ns trajectory in replica one of SPIN-aureus/MPO simulation was considered, which included the entire unbinding and unfolding transition (see Supplemetary Figure S2. By doing this, the results will not be interfered by the transient refolding events observed after complete dissociation (see Supplemetary Figure S2 replica 40 at 200 ns for example). The segments for each trajectory that were selected to calculate the contact maps can be found in Supplemetary Table S3. Note that for replicas where NTD remains bound and folded at the end of the 250 ns-simulation, we only selected the first 50 ns of trajectories to compute contact maps. In this way, we could avoid masking important details about the transition pathways by over-representing data of the bound and folded state.
Results and discussion
High temperature simulations reveal step-wise binding of SPIN NTD and CTD
High temperature simulations have been shown to be capable of providing reliable mechanistic insights in to folding of structured proteins as well as coupled binding and folding of IDPs (Verkhivker et al., 2003; Daggett, 2006; Chen and Luo, 2007; Schaeffer et al., 2008; Zhang et al., 2012). The assumption here is that unfolding and unbinding is largely a reverse of coupled binding and folding. While many mechanistic details derived from high-temperature simulations have compared well to experiments (Verkhivker et al., 2003; Daggett, 2006; Chen and Luo, 2007; Schaeffer et al., 2008; Zhang et al., 2012), it is also known that the most probable transition pathways may depend on the temperature (Dinner and Karplus, 1999). Therefore, it is important to find the lowest temperature to drive the unfolding and unbinding process within a given simulation timeframe. The pilot simulations suggest that the NTD of SPIN-aureus only starts to dissociate from the active pocket of MPO at 450 K within ∼100 ns timescale, which becomes much faster at higher temperatures (Supplemetary Figure S1B). Note that rapid dissociation (e.g., at 475 K) is not always preferred due to the risks of missing important details under non-physiological conditions and activating pathways not generally accessible under the physiological conditions. For example, the three-helix bundle of SPIN CTD would melt rapidly at 475 K and above, leading to premature disassociation from MPO within 10 s of ns. This is consistent with the experimental observation that SPIN CTD largely dictates MPO binding (de Jong et al., 2018). Instead, simulations at 450 K seem to depict a more realistic dissociation process, where NTD unbinds first while the CTD remains largely fold and bound (Figure 2). The apparent decoupling and step-wise nature of the binding of SPIN CTD and NTD could explain why there is little correlation between the inhibition strength and binding affinity for different SPIN homologs. It’s likely that two domains of SPIN bind and function almost independently when interacting with MPO. As such, some SPIN homologs, e.g., SPIN-agnetis, show comparable nanomolar binding affinity as SPIN-aureus, but have no detectable inhibitory ability to human MPO (Ploscariu et al., 2018). Based on these observations, we will focus on the coupled binding and folding of SPIN NTD while the CTD is harmonically restrained to the bound state in subsequent simulation and analysis.
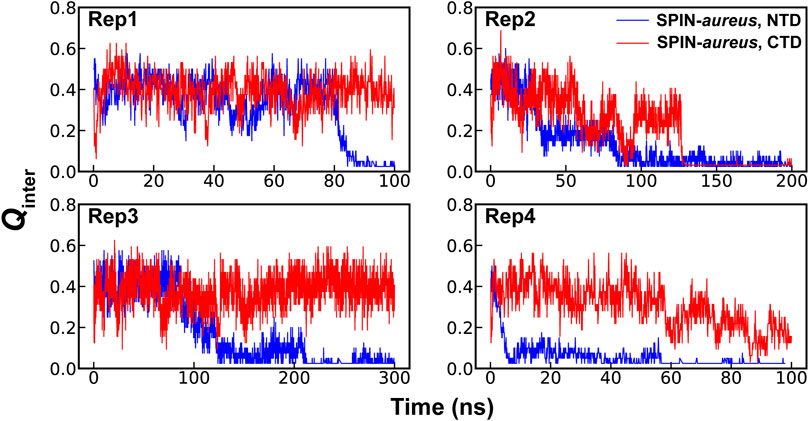
FIGURE 2. Fractions of native intermolecular contacts between the NTD (blue) and CTD (red) of SPIN during four independent simulations of the SPIN-aureus/MPO complex at 450 K.
Cooperative binding and folding of SPIN-aureus NTD
A total of 40 independent 250-ns simulations were performed at 450 K to explore the conformational fluctuations, dynamic interactions and dissociation pathways of SPIN-aureus NTD with human MPO. As summarized in Supplemetary Figure S2, SPIN-aureus NTD tends to dissociate rapidly and its unfolding and unbinding often happen simultaneously. For example, in 36 out of 40 replicas (except for replicas 6, 8, 16 and 25), NTD fully dissociated (with Qinter < 0.2) within 200 ns. Particularly, among 30 out of the 36 runs (except for replicas 10, 11, 17, 23, 29 and 35) unbind/unfold occurred within the first 50 ns, or sometimes even more rapidly within 15 ns. To quantitively describe the dissociation process and probe the mechanisms of coupled binding and folding, we calculated the average fractions of intermolecular and intramolecular native contacts formed by NTD, denoted Qinter and Qintra, respectively, from all replicas. The results were then fitted with a double exponential function (Figure 3A). Not surprisingly, the unbinding and unfolding kinetics of SPIN-aureus NTD are similar, consistent with the observation that they appear highly correlated. As shown in Figure 3A, the initial fast phase
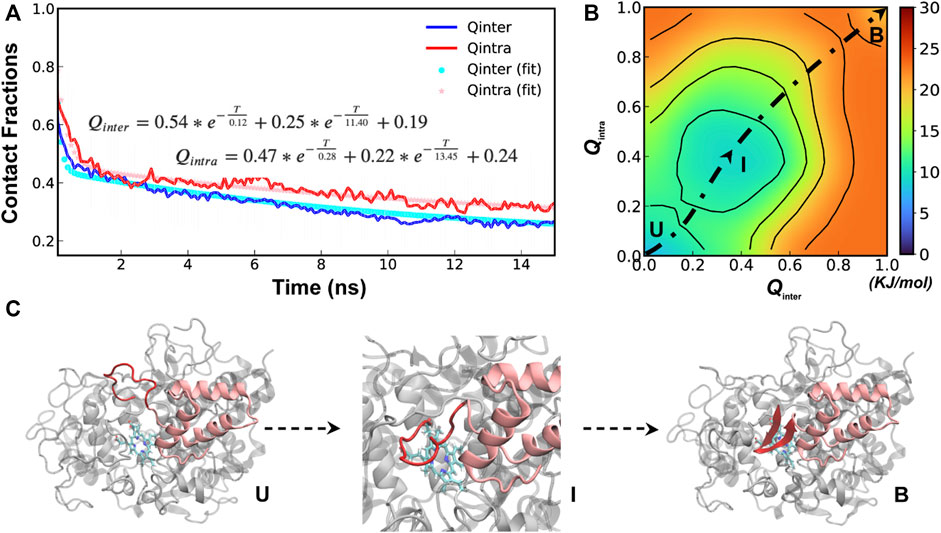
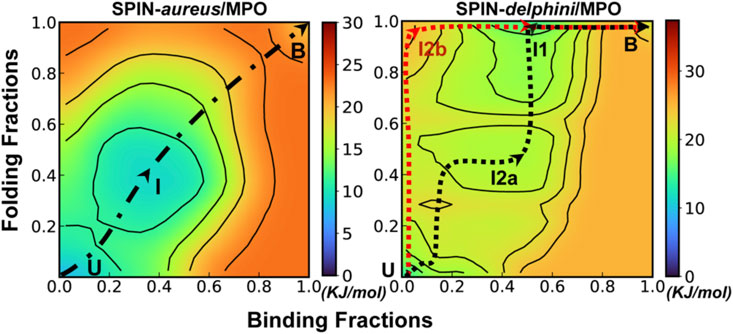
FIGURE 3. Cooperative binding and folding of SPIN-aureus NTD. (A) Average intramolecular and intermolecular native contact fractions (Qinter and Qintra) as a function of simulation time at 450 K. The double exponential fits are plot using dotted lines, with the actual parameters also shown. (B) Pseudo free energy surface as a function of Qinter and Qintra derived from the transition paths (see Methods). The dashed line indicates the minimum free energy pathway. Key states (U, I and B) are also labeled. (C) Representative conformations for key states along the minimum free energy path, with SPIN-aureus and MPO shown in red and light grey, respectively.
The free energy surface also reveals three major conformational states of NTD folding and binding to MPO. State B (bound), with both Qinter and Qintra above 0.8, is the fully folded and bound state, and State U (unbound), with both Qinter and Qintra below 0.2, is the fully unfolded and unbound state. In addition, there is a partially bound and folded substate, I (intermediate state), where the values of Qinter and Qintra are around 0.4. Representative conformations for the three states of the complex (Figure 3C) illustrate that SPIN-aureus NTD does not tend to pre-fold into some “native-like” β-hairpins conformations prior to binding to the active site of MPO, and vice versa. The cooperative nature of SPIN-aureus NTD is in contrast to previous experimental and computational studies of coupled binding and folding of IDPs into non-β-hairpin structures (Chen, 2012), where induced folding-like mechanisms are prevalent. However, this may not be surprising given the cooperative nature of folding of isolated β-hairpins (Munoz et al., 1997; Klimov and Thirumalai, 2000; Chen et al., 2018). In particular, the “speed-limit” of β-hairpin folding usually is ∼μs−1, much slower compared to helix-coil transitions (∼100 ns), due to the requirement of forming long-range interactions and the presence of entropy-dominant barriers. Therefore, once SPIN-aureus CTD is tightly bound, native-like interactions with the MPO surface play a direct role to facilitate the rapid folding of NTD and achieve a facile blockage of the MPO active site for inhibition.
Conformational selection-like mechanism for SPIN-delphini NTD
Compared to SPIN-aureus, which is secreted by S. aureus that appears to be particularly adapted to survive the neutrophil-mediated immunity with the highest binding affinity (KD = 15.9 nM) and inhibition strength (IC50 = 4.6 nM) to human MPO, SPIN-delphini has a moderate binding affinity (KD = 310 nM) but the second strong inhibitory ability (IC50 = 29.7 nM) among nine SPIN homologs previously analyzed (Ploscariu et al., 2018). A possible explanation is that SPIN-delphini NTD may have evolved to be less dependent on the tight binding of CTD. Interestingly, high-temperature simulations indeed reveal significant differences between coupled binding and folding of NTDs from SPIN-aureus and SPIN-delphini. As summarized in Supplemetary Figure S3, ∼40% of the 40 (17/40) replicas failed to observe full dissociation of SPIN-delphini NTD’s during the 250 ns simulations, which is about 3-fold of ∼10% for SPIN-aureus. The implication is that SPIN-delphini NTD fits the active site of MPO tighter than SPIN-aureus NTD, which would be consistent with disproportionally strong inhibitory function of SPIN-delphini despite weakened overall binding affinity.
Further analysis of unfolding and unbinding kinetics and free energy surface reveal that SPIN-delphini NTD mainly follow a distinct conformational selection-like mechanism (Figure 4), where the NTD tends to gain substantial native β-hairpin structures prior to forming intermolecular interactions with MPO. This is well reflected in unbinding and unfolding kinetics. On average, the unbinding rates of SPIN-delphini NTD ((
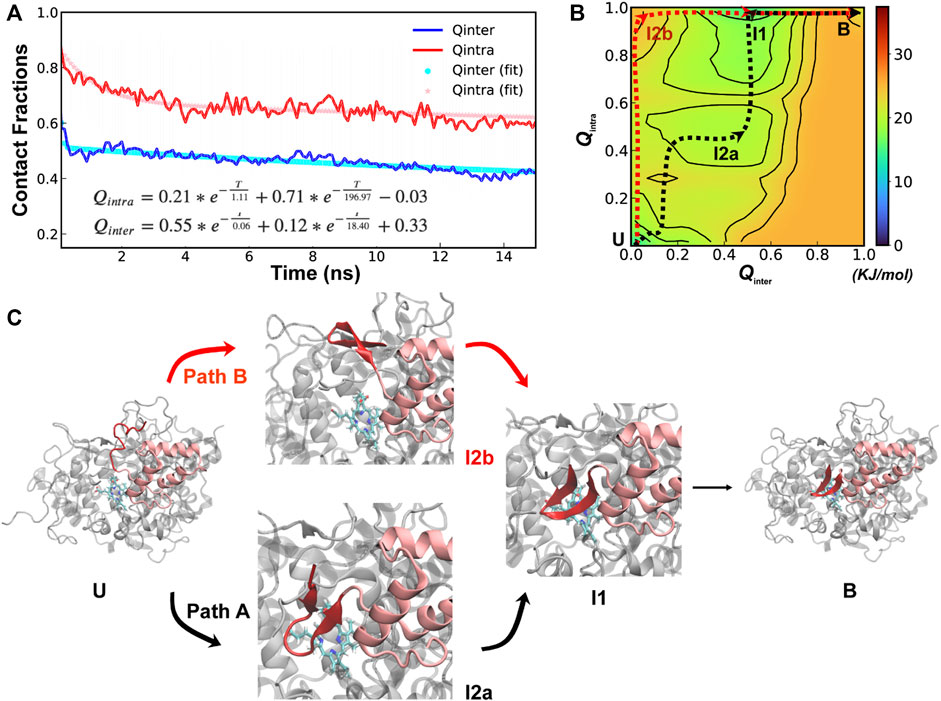
FIGURE 4. Cooperative binding and folding of SPIN-delphini. (A) Average intramolecular and intermolecular native contact fractions (Qinter and Qintra) as a function of simulation time at 450 K. The double exponential fits are plot using dotted lines, with the actual parameters also shown. (B) Pseudo free energy surface as a function of Qinter and Qintra derived from the transition paths (see Methods). The dashed lines indicate the minimum free energy pathways, with key states labeled. (C) Two major parallel dissociation pathways and key intermediate states for coupled binding and folding of SPIN-delphini NTD to human MPO, with SPIN and MPO shown in red and light grey, respectively.
The minimal free energy paths, indicated by the dash lines in Figure 4B, demonstrate that SPIN-delphini NTD coupled binding and folding is not cooperative and follows two major routes with multiple intermediate states. Both routes go through an intermediate state I1, where there Qinter drops below 0.7 while the β-hairpin structure is essentially intact with Qintra ∼ 1.0. Overlay of representative structures from B (fully bound) and I1, shown in Supplemetary Figure S4, illustrates that how SPIN-delphini NTD becomes mobile within the active site pocket of MPO without unfolding. From state I1, the major pathway (path A) goes through another intermediate state I2a, which mainly has similar level of residual intermolecular native contacts (Qinter ∼ 0.5) but the hairpin conformation becomes partially unfolded (Qintra ∼ 0.5). From state I2a, SPIN-delphini NTD would further unbind and then unfold to reach the fully disassociated state (U). In the parallel pathway B, SPIN-delphini NTD would continue to become fully unbound from MPO without significant unfolding (I2b, Qintra > 0.8, Qinter < 0.2), before unfold outside of the MPO active site. The observed conformational selection-like mechanism of SPIN-delphini NTD interaction with MPO is summarized in Figure 4C. It shows that the disordered segment could become fully folded before inserting into the MPO active site (Path B), which is an ideally conformational selection mechanism. Such a process is best represented by high-temperature simulation run 24 (Supplemetary Figure S3, Rep24). Path A, which is more prevalent, involves multi-step conformational selections. In each step (U to I2a to B), the NTD first fold and then bind to MPO. The later pathway is best illustrated in Supplemetary Figure S3 Re33. The distinct mechanisms of SPIN-aureus and SPIN-delphini NTD coupled binding and folding may help explain why the inhibition strength doesn’t fully correlate with binding affinity among different SPIN homologs.
Elevated pre-folding in unbound SPIN-delphini NTD
For conformational selection to be an efficient mechanism for coupled binding and folding, there should be high levels residual structures in unbound IDPs (Liu et al., 2019). Since SPIN-aureus NTD follows cooperative binding and folding while SPIN-delphini NTD prefers a conformational selection-like mechanism, we further characterized the stability of hairpin-like structures in their unbound states under the physiological conditions. As shown in Figure 5, although SPIN-delphini NTD showed slightly faster unfolding rates, it remained more structured than SPIN-aureus NTD. The limiting NTD Qintra decayed to 0.57 and 0.40 for SPIN-delphini and SPIN-aureus, respectively (see Figure 5A). Importantly, the probability distributions of Qintra show that there is a very high probability for SPIN-delphini NTD to remain partially folded (Qintra > 0.5). Such an elevated residual β-hairpin structures in unbound SPIN-delphini NTD is consistent with the observation of conformational selection-like mechanism of its coupled binding and folding (see above). The more dynamic nature of SPIN-aureus NTD suggests that it depends on specific MPO interactions to facilitate its folding into the β-hairpin structure, thus following a cooperative binding and folding mechanism (Figure 3B).
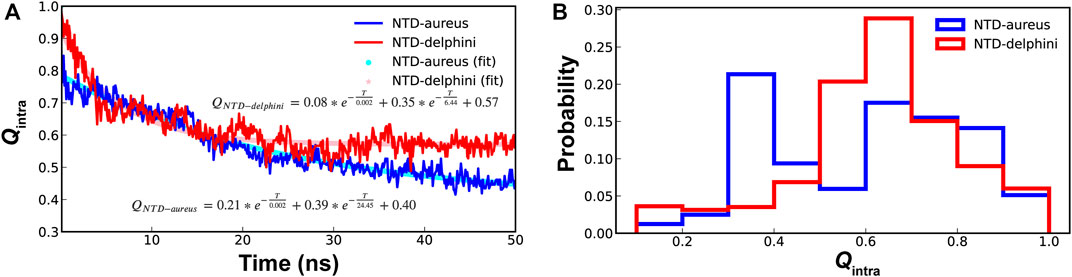
FIGURE 5. (A) Decay of NTD Qintra for SPIN-aureus (blue) and SPIN-delphini (red) at 300 K starting with the fully folded conformation, averaged over 20 replicas of 50-ns simulations. (B) Distributions of NTD Qintra for two SPIN homologs.
Conclusion
Extensive atomistic simulations have been performed in explicit solvent to gain a deeper understanding of the structural basis of how SPIN, a protein secreted by Staphylococcus, inhibits the activity of human MPO to help evade the neutrophil-mediated host innate immunity. It has been shown that the folded SPIN CTD can bind to MPO even in the absence of the disordered NTD, but the latter is required for the MPO inhibition function. Structural studies further revealed that SPIN NTDs folded into similar β-hairpins upon binding and inserted into the MPO active site for inhibition. Curiously, there is a poor correlation between the MPO binding affinity and inhibition efficacy among different SPIN analogs. The implication is that the conformational properties of unbound SPIN NTDs and their coupled binding and folding likely play central roles in their MPO inhibitory activity.
To further address these questions, we carried out extensive atomistic simulations in explicit solvent using the CHARMM36m force field and studied the structures and interactions of two SPIN homologs, namely, SPIN-aureus and SPIN-delphini. At an optimal temperature of 450 K, high-temperature simulations reveal that SPIN CTD and NTD binding to MPO follows a decoupled step-wise mechanism, consistent with the experimental observation that CTD is mainly responsible for specific MPO binding (de Jong et al., 2018). Further 450 K simulations of the unbinding and unfolding of SPIN NTD with CTD restrained in the bound state revealed striking difference in SPIN-aureus and SPIN-delphini. While coupled binding and folding SPIN-aureus NTD during interaction with MPO is highly cooperative, that of SPIN-delphini mainly follows a conformational selection-like mechanism. Both are in contrast to a prevalence of induced folding-like mechanism previously observed in experimental and computational studies of IDPs that fold into relatively simple structures such as helices and ordered loops (Chen, 2012). This is an important new insight on coupled binding and folding of IDPs that is likely applicable to other IDPs that require the formation of long-range interactions for specific binding. Non-etheless, we caution that not all mechanistic details generated at high temperature will be true at room or physiological temperature. Additional experimental and/or computational studies will be required to further validate the above predictions. For example, steered MD approaches (Lu and Schulten, 1999) could provide a viable alternative that may introduce less perturbation to the unfolding/unbinding pathway. A caveat is to find an appropriate pulling speed that is gentle enough but computationally feasible.
Atomistic simulations at room temperature further reveal that the mechanistic difference between SPIN-aureus and SPIN-delphini may be related to the intrinsic conformational properties of their NTDs in the unbound state. Specifically, SPIN-aureus NTD is more dynamic and less structured, requiring MPO binding to facilitate its folding and thus a cooperative binding and folding mechanism. On the other hand, SPIN-delphini NTD has a much higher propensity to adopt pre-folded hairpin-like conformations, allowing it to follow a conformational selection-like mechanism. As such, SPIN-delphini NTD is less dependent on CTD binding to MPO for specific interaction and MPO inhibition. These structural and mechanistic differences could explain why SPIN-delphini binds to MPO ∼19 times weaker than SPIN-aureus, but its IC50 is only ∼6 times higher. Taken together, the current atomistic simulations do not only provide new mechanistic principles on coupled binding and folding of IDPs into non-trivial β-hairpins, but also help to establish the structure-dynamics-function relationship of SPIN homologs. Moreover, it may suggest a new strategy to combating Staphylococcus infection, such as by designing drug molecules that could destabilize residual structures in SPIN NTD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
YZ, XL, and JC conception and design of the study; YZ and XL performing the simulation and analysis; YZ, XL, and JC analysis and interpretation of data, drafting and revising the manuscript.
Funding
This work was supported by National Institutes of Health Grant R35 GM144045 (JC).
Acknowledgments
We thank Brian Geisbrecht for insightful early discussions that led to this work. All simulations were performed on the Pikes GPU cluster housed in the Massachusetts Green High-Performance Computing Cluster (MGHPCC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1130189/full#supplementary-material
References
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. doi:10.1016/j.softx.2015.06.001
Agarwal, P. K., Billeter, S. R., Rajagopalan, P. T., Benkovic, S. J., and Hammes-Schiffer, S. (2002). Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. U. S. A. 99 (5), 2794–2799. doi:10.1073/pnas.052005999
Antikainen, N. M., Smiley, R. D., Benkovic, S. J., and Hammes, G. G. (2005). Conformation coupled enzyme catalysis: Single-molecule and transient kinetics investigation of dihydrofolate reductase. Biochemistry 44 (51), 16835–16843. doi:10.1021/bi051378i
Baul, U., Chakraborty, D., Mugnai, M. L., Straub, J. E., and Thirumalai, D. (2019). Sequence effects on size, shape, and structural heterogeneity in intrinsically disordered proteins. J. Phys. Chem. B 123 (16), 3462–3474. doi:10.1021/acs.jpcb.9b02575
Benayad, Z., von Bulow, S., Stelzl, L. S., and Hummer, G. (2021). Simulation of FUS protein condensates with an adapted coarse-grained model. J. Chem. Theory Comput. 17 (1), 525–537. doi:10.1021/acs.jctc.0c01064
Best, R. B., Hummer, G., and Eaton, W. A. (2013). Native contacts determine protein folding mechanisms in atomistic simulations. Proc. Natl. Acad. Sci. U. S. A. 110, 17874–17879. doi:10.1073/pnas.1311599110
Bonetti, D., Troilo, F., Brunori, M., Longhi, S., and Gianni, S. (2018). How robust is the mechanism of folding-upon-binding for an intrinsically disordered protein? Biophys. J. 114 (8), 1889–1894. doi:10.1016/j.bpj.2018.03.017
Brooks, B. R., Brooks, C. L., Mackerell, A. D., Nilsson, L., Petrella, R. J., Roux, B., et al. (2009). Charmm: The biomolecular simulation program. J. Comput. Chem. 30 (10), 1545–1614. doi:10.1002/jcc.21287
Case, D. A. D. S. C., Cheatham, T. E., Darden, T. A., Duke, R. E., Giese, T. J., Gohlke, H., et al. (2017). Amber 2017. San Francisco: University of California.
Chen, H. F., and Luo, R. (2007). Binding induced folding in p53-MDM2 complex. J. Am. Chem. Soc. 129 (10), 2930–2937. doi:10.1021/ja0678774
Chen, J., Liu, X., and Chen, J. (2018). Atomistic peptide folding simulations reveal interplay of entropy and long-range interactions in folding cooperativity. Sci. Rep. 8 (1), 13668. doi:10.1038/s41598-018-32028-7
Chen, J. L., Liu, X. R., and Chen, J. H. (2020). Targeting intrinsically disordered proteins through dynamic interactions. Biomolecules 10 (5), 743. ARTN 743. doi:10.3390/biom10050743
Chen, J. (2012). Towards the physical basis of how intrinsic disorder mediates protein function. Arch. Biochem. Biophys. 524 (2), 123–131. doi:10.1016/j.abb.2012.04.024
Chu, X., Wang, Y., Gan, L., Bai, Y., Han, W., Wang, E., et al. (2012). Importance of electrostatic interactions in the association of intrinsically disordered histone chaperone Chz1 and histone H2A.Z-H2B. PLoS Comput. Biol. 8 (7), e1002608. doi:10.1371/journal.pcbi.1002608
Click, T. H., Ganguly, D., and Chen, J. (2010). Intrinsically disordered proteins in a physics-based world. Int. J. Mol. Sci. 11 (12), 5292–5309. doi:10.3390/ijms11125292
Crabtree, M. D., Borcherds, W., Poosapati, A., Shammas, S. L., Daughdrill, G. W., and Clarke, J. (2017). Conserved helix-flanking prolines modulate intrinsically disordered protein:target affinity by altering the lifetime of the bound complex. Biochemistry 56 (18), 2379–2384. doi:10.1021/acs.biochem.7b00179
Daggett, V. (2006). Protein folding-simulation. Chem. Rev. 106 (5), 1898–1916. doi:10.1021/cr0404242
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: An N-log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
de Jong, N. W. M., Ploscariu, N. T., Ramyar, K. X., Garcia, B. L., Herrera, A. I., Prakash, O., et al. (2018). A structurally dynamic N-terminal region drives function of the staphylococcal peroxidase inhibitor (SPIN). J. Biol. Chem. 293 (7), 2260–2271. doi:10.1074/jbc.RA117.000134
de Jong, N. W. M., Ramyar, K. X., Guerra, F. E., Nijland, R., Fevre, C., Voyich, J. M., et al. (2017). Immune evasion by a staphylococcal inhibitor of myeloperoxidase. Proc. Natl. Acad. Sci. U. S. A. 114 (35), 9439–9444. doi:10.1073/pnas.1707032114
Dinner, A. R., and Karplus, M. (1999). Is protein unfolding the reverse of protein folding? A lattice simulation analysis. J. Mol. Biol. 292 (2), 403–419. doi:10.1006/jmbi.1999.3051
Dosnon, M., Bonetti, D., Morrone, A., Erales, J., di Silvio, E., Longhi, S., et al. (2015). Demonstration of a folding after binding mechanism in the recognition between the measles virus NTAIL and X domains. ACS Chem. Biol. 10 (3), 795–802. doi:10.1021/cb5008579
Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., et al. (2001). Intrinsically disordered protein. J. Mol. Graph. Modell. 19 (1), 26–59. doi:10.1016/s1093-3263(00)00138-8
Dyson, H. J., and Wright, P. E. (2005). Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6 (3), 197–208. doi:10.1038/nrm1589
Eastman, P., Friedrichs, M. S., Chodera, J. D., Radmer, R. J., Bruns, C. M., Ku, J. P., et al. (2012). OpenMM 4: A reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 9 (1), 461–469. doi:10.1021/ct300857j
Eastman, P., Swails, J., Chodera, J. D., McGibbon, R. T., Zhao, Y., Beauchamp, K. A., et al. (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 13 (7), e1005659. doi:10.1371/journal.pcbi.1005659
Fuxreiter, M., Simon, I., Friedrich, P., and Tompa, P. (2004). Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 338 (5), 1015–1026. doi:10.1016/j.jmb.2004.03.017
Ganguly, D., Otieno, S., Waddell, B., Iconaru, L., Kriwacki, R. W., and Chen, J. (2012). Electrostatically accelerated coupled binding and folding of intrinsically disordered proteins. J. Mol. Biol. 422, 674–684. doi:10.1016/j.jmb.2012.06.019
Ganguly, D., Zhang, W., and Chen, J. (2013). Electrostatically accelerated encounter and folding for facile recognition of intrinsically disordered proteins. PLoS Comput. Biol. 9 (11), e1003363. doi:10.1371/journal.pcbi.1003363
Ganguly, D., Zhang, W., and Chen, J. (2012). Synergistic folding of two intrinsically disordered proteins: Searching for conformational selection. Mol. Biosyst. 8 (1), 198–209. Research Support, U.S. Gov't, Non-P.H.S. doi:10.1039/c1mb05156c
Garcia, B. L., Zwarthoff, S. A., Rooijakkers, S. H., and Geisbrecht, B. V. (2016). Novel evasion mechanisms of the classical complement pathway. J. Immunol. 197 (6), 2051–2060. doi:10.4049/jimmunol.1600863
Gianni, S., Dogan, J., and Jemth, P. (2016). Coupled binding and folding of intrinsically disordered proteins: What can we learn from kinetics? Curr. Opin. Struct. Biol. 36, 18–24. doi:10.1016/j.sbi.2015.11.012
Gotz, A. W., Williamson, M. J., Xu, D., Poole, D., Le Grand, S., and Walker, R. C. (2012). Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 8 (5), 1542–1555. doi:10.1021/ct200909j
Gsponer, J., and Babu, M. M. (2009). The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 99 (2-3), 94–103. doi:10.1016/j.pbiomolbio.2009.03.001
Hammes, G. G., Chang, Y. C., and Oas, T. G. (2009). Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. U. S. A. 106 (33), 13737–13741. doi:10.1073/pnas.0907195106
Huang, J., Rauscher, S., Nawrocki, G., Ran, T., Feig, M., de Groot, B. L., et al. (2017). CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14 (1), 71–73. doi:10.1038/nmeth.4067
Huang, Y., and Liu, Z. (2009). Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: A critical assessment of the "fly-casting" mechanism. J. Mol. Biol. 393 (5), 1143–1159. S0022-2836(09)01117-6 [pii]. doi:10.1016/j.jmb.2009.09.010
Humphrey, W., Dalke, A., and Schulten, K. (1996). Vmd: Visual molecular dynamics. J. Mol. Graph. 14 (1), 33–38. Article. doi:10.1016/0263-7855(96)00018-5
Kim, H. K., Thammavongsa, V., Schneewind, O., and Missiakas, D. (2012). Recurrent infections and immune evasion strategies of Staphylococcus aureus. Curr. Opin. Microbiol. 15 (1), 92–99. doi:10.1016/j.mib.2011.10.012
Klimov, D. K., and Thirumalai, D. (2000). Mechanisms and kinetics of beta-hairpin formation. Proc. Natl. Acad. Sci. U. S. A. 97 (6), 2544–2549. doi:10.1073/pnas.97.6.2544
Koshland, D. E. (1960). The active site and enzyme action. Adv. Enzymol. Relat. Subj. Biochem. 22, 45–97. doi:10.1002/9780470122679.ch2
Lambris, J. D., Ricklin, D., and Geisbrecht, B. V. (2008). Complement evasion by human pathogens. Nat. Rev. Microbiol. 6 (2), 132–142. doi:10.1038/nrmicro1824
Lee, J., Cheng, X., Swails, J. M., Yeom, M. S., Eastman, P. K., Lemkul, J. A., et al. (2016). CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12 (1), 405–413. doi:10.1021/acs.jctc.5b00935
Levy, Y., Onuchic, J. N., and Wolynes, P. G. (2007). Fly-casting in protein-DNA binding: Frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 129 (4), 738–739. doi:10.1021/ja065531n
Liu, X., Chen, J., and Chen, J. (2019). Residual structure accelerates binding of intrinsically disordered ACTR by promoting efficient folding upon encounter. J. Mol. Biol. 431 (2), 422–432. doi:10.1016/j.jmb.2018.12.001
Liu, X., and Chen, J. (2019). Residual structures and transient long-range interactions of p53 transactivation domain: Assessment of explicit solvent protein force fields. J. Chem. Theory Comput. 15 (8), 4708–4720. doi:10.1021/acs.jctc.9b00397
Liu, Z. R., and Huang, Y. Q. (2014). Advantages of proteins being disordered. Protein Sci. 23 (5), 539–550. doi:10.1002/Pro.2443
Lowy, F. D. (1998). Staphylococcus aureus infections. N. Engl. J. Med. 339 (8), 520–532. doi:10.1056/NEJM199808203390806
Lu, H., and Schulten, K. (1999). Steered molecular dynamics simulations of force-induced protein domain unfolding. Proteins-Structure Funct. Genet. 35 (4), 453–463. doi:10.1002/(sici)1097-0134(19990601)35:4<453::aid-prot9>3.0.co;2-m
Munoz, V., Thompson, P. A., Hofrichter, J., and Eaton, W. A. (1997). Folding dynamics and mechanism of beta-hairpin formation. Nature 390 (6656), 196–199. doi:10.1038/36626
Nerenberg, P. S., Jo, B., So, C., Tripathy, A., and Head-Gordon, T. (2012). Optimizing solute-water van der Waals interactions to reproduce solvation free energies. J. Phys. Chem. B 116 (15), 4524–4534. doi:10.1021/jp2118373
Oldfield, C. J., Cheng, Y. G., Cortese, M. S., Romero, P., Uversky, V. N., and Dunker, A. K. (2005). Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 44 (37), 12454–12470. doi:10.1021/bi050736e
Onuchic, J. N., Socci, N. D., Luthey-Schulten, Z., and Wolynes, P. G. (1996). Protein folding funnels: The nature of the transition state ensemble. Fold. Des. 1 (6), 441–450. doi:10.1016/S1359-0278(96)00060-0
Park, Y., Sheetlin, S., Ma, N., Madden, T. L., and Spouge, J. L. (2012). New finite-size correction for local alignment score distributions. BMC Res. Notes 5, 286. doi:10.1186/1756-0500-5-286
Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., et al. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26 (16), 1781–1802. doi:10.1002/jcc.20289
Piana, S., Donchev, A. G., Robustelli, P., and Shaw, D. E. (2015). Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119 (16), 5113–5123. doi:10.1021/jp508971m
Ploscariu, N. T., de Jong, N. W. M., van Kessel, K. P. M., van Strijp, J. A. G., and Geisbrecht, B. V. (2018). Identification and structural characterization of a novel myeloperoxidase inhibitor from Staphylococcus delphini. Arch. Biochem. Biophys. 645, 1–11. doi:10.1016/j.abb.2018.03.007
Pontius, B. W. (1993). Close encounters: Why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem. Sci. 18 (5), 181–186. doi:10.1016/0968-0004(93)90111-y
Rigby, K. M., and DeLeo, F. R. (2012). Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 34 (2), 237–259. doi:10.1007/s00281-011-0295-3
Robertson, M. J., Tirado-Rives, J., and Jorgensen, W. L. (2015). Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 11 (7), 3499–3509. doi:10.1021/acs.jctc.5b00356
Robustelli, P., Piana, S., and Shaw, D. E. (2018). Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A. 115 (21), E4758–E4766. doi:10.1073/pnas.1800690115
Rogers, J. M., Oleinikovas, V., Shammas, S. L., Wong, C. T., De Sancho, D., Baker, C. M., et al. (2014). Interplay between partner and ligand facilitates the folding and binding of an intrinsically disordered protein. Proc. Natl. Acad. Sci. U. S. A. 111 (43), 15420–15425. Research Support, Non-U.S. Gov't. doi:10.1073/pnas.1409122111
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. C. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23 (3), 327–341. doi:10.1016/0021-9991(77)90098-5
Schaeffer, R. D., Fersht, A., and Daggett, V. (2008). Combining experiment and simulation in protein folding: Closing the gap for small model systems. Curr. Opin. Struct. Biol. 18 (1), 4–9. doi:10.1016/j.sbi.2007.11.007
Sen, S., and Udgaonkar, J. B. (2019). Binding-induced folding under unfolding conditions: Switching between induced fit and conformational selection mechanisms. J. Biol. Chem. 294 (45), 16942–16952. doi:10.1074/jbc.RA119.009742
Shammas, S. L., Crabtree, M. D., Dahal, L., Wicky, B. I., and Clarke, J. (2016). Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem. 291 (13), 6689–6695. doi:10.1074/jbc.R115.692715
Shoemaker, B. A., Portman, J. J., and Wolynes, P. G. (2000). Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. U. S. A. 97 (16), 8868–8873. doi:10.1073/pnas.160259697
Spaan, A. N., Surewaard, B. G., Nijland, R., and van Strijp, J. A. (2013). Neutrophils versus Staphylococcus aureus: A biological tug of war. Annu. Rev. Microbiol. 67, 629–650. doi:10.1146/annurev-micro-092412-155746
Sugase, K., Dyson, H. J., and Wright, P. E. (2007). Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447 (7147), 1021–1025. doi:10.1038/nature05858
Thwaites, G. E., and Gant, V. (2011). Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9 (3), 215–222. doi:10.1038/nrmicro2508
Tong, S. Y., Davis, J. S., Eichenberger, E., Holland, T. L., and Fowler, V. G. (2015). Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28 (3), 603–661. doi:10.1128/CMR.00134-14
Toto, A., Malagrino, F., Visconti, L., Troilo, F., Pagano, L., Brunori, M., et al. (2020). Templated folding of intrinsically disordered proteins. J. Biol. Chem. 295 (19), 6586–6593. doi:10.1074/jbc.REV120.012413
Trizac, E., Levy, Y., and Wolynes, P. G. (2010). Capillarity theory for the fly-casting mechanism. Proc. Natl. Acad. Sci. U. S. A. 107 (7), 2746–2750. doi:10.1073/pnas.0914727107
Troilo, F., Bonetti, D., Bignon, C., Longhi, S., and Gianni, S. (2019). Understanding intramolecular crosstalk in an intrinsically disordered protein. ACS Chem. Biol. 14 (3), 337–341. doi:10.1021/acschembio.8b01055
Tsai, C. J., Kumar, S., Ma, B. Y., and Nussinov, R. (1999). Folding funnels, binding funnels, and protein function. Protein Sci. 8 (6), 1181–1190. doi:10.1110/ps.8.6.1181
Tsai, C. J., Ma, B., Sham, Y. Y., Kumar, S., and Nussinov, R. (2001). Structured disorder and conformational selection. Proteins 44 (4), 418–427. [pii]. doi:10.1002/prot.1107
Turjanski, A. G., Gutkind, J. S., Best, R. B., and Hummer, G. (2008). Binding-induced folding of a natively unstructured transcription factor. PLoS Comput. Biol. 4 (4), e1000060. doi:10.1371/journal.pcbi.1000060
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2005). Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18 (5), 343–384. doi:10.1002/jmr.747
Verkhivker, G. M., Bouzida, D., Gehlhaar, D. K., Rejto, P. A., Freer, S. T., and Rose, P. W. (2003). Simulating disorder-order transitions in molecular recognition of unstructured proteins: Where folding meets binding. Proc. Natl. Acad. Sci. U. S. A. 100 (9), 5148–5153. doi:10.1073/pnas.0531373100
Wright, P. E., and Dyson, H. J. (1999). Intrinsically unstructured proteins: Re-Assessing the protein structure-function paradigm. J. Mol. Biol. 293 (2), 321–331. doi:10.1006/jmbi.1999.3110
Wright, P. E., and Dyson, H. J. (2009). Linking folding and binding. Curr. Opin. Struct. Biol. 19 (1), 31–38. doi:10.1016/j.sbi.2008.12.003
Wu, H., Wolynes, P. G., and Papoian, G. A. (2018). AWSEM-IDP: A coarse-grained force field for intrinsically disordered proteins. J. Phys. Chem. B 122 (49), 11115–11125. doi:10.1021/acs.jpcb.8b05791
Yang, J., Gao, M., Xiong, J., Su, Z., and Huang, Y. (2019). Features of molecular recognition of intrinsically disordered proteins via coupled folding and binding. Protein Sci. 28 (11), 1952–1965. doi:10.1002/pro.3718
Zhang, W., Ganguly, D., and Chen, J. (2012). Residual structures, conformational fluctuations, and electrostatic interactions in the synergistic folding of two intrinsically disordered proteins. PLoS Comput. Biol. 8 (1), e1002353. doi:10.1371/journal.pcbi.1002353
Zhang, Y., Liu, X., and Chen, J. (2022). Toward accurate coarse-grained simulations of disordered proteins and their dynamic interactions. J. Chem. Inf. Model. 62 (18), 4523–4536. From NLM Medline. doi:10.1021/acs.jcim.2c00974
Keywords: instrinsically disordered region, innate immunity, atomistic simulaitons, conformational selection and induced fit, residual structures, transition path
Citation: Zhang Y, Liu X and Chen J (2023) Coupled binding and folding of disordered SPIN N-terminal region in myeloperoxidase inhibition. Front. Mol. Biosci. 10:1130189. doi: 10.3389/fmolb.2023.1130189
Received: 22 December 2022; Accepted: 27 January 2023;
Published: 09 February 2023.
Edited by:
Arvind Ramanathan, Argonne National Laboratory (DOE), United StatesReviewed by:
Debabani Ganguly, JIS Institute of Advanced Studies and Research, IndiaAdam Liwo, University of Gdansk, Poland
Copyright © 2023 Zhang, Liu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhan Chen, amlhbmhhbmNAdW1hc3MuZWR1
‡Present address: Xiaorong Liu, Department of Chemistry, University of Michigan, Ann Arbor, MI, United States
†These authors have contributed equally to this work