 Pamela Stanley*
Pamela Stanley* Ankit Tanwar
Ankit Tanwar- Department of Cell Biology, Albert Einstein College Medicine, New York, NY, United States
Notch signaling via NOTCH1 stimulated by Delta-like ligand 4 (DLL4) is required for the development of T cells in thymus, and NOTCH2 stimulated by Notch ligand DLL1 is required for the development of marginal zone (MZ) B cells in spleen. Notch signaling also regulates myeloid cell production in bone marrow and is an essential contributor to the generation of early hematopoietic stem cells (HSC). The differentiation program in each of these cellular contexts is optimized by the regulation of Notch signaling strength by O-glycans attached to epidermal growth factor-like (EGF) repeats in the extracellular domain of Notch receptors. There are three major types of O-glycan on NOTCH1 and NOTCH2 - O-fucose, O-glucose and O-GlcNAc. The initiating sugar of each O-glycan is added in the endoplasmic reticulum (ER) by glycosyltransferases POFUT1 (fucose), POGLUT1/2/3 (glucose) or EOGT (GlcNAc), respectively. Additional sugars are added in the Golgi compartment during passage through the secretory pathway to the plasma membrane. Of particular significance for Notch signaling is the addition of GlcNAc to O-fucose on an EGF repeat by the Fringe GlcNAc-transferases LFNG, MFNG or RFNG. Canonical Notch ligands (DLL1, DLL4, JAG1, JAG2) expressed in stromal cells bind to the extracellular domain of Notch receptors expressed in hematopoietic stem cells and myeloid and lymphoid progenitors to activate Notch signaling. Ligand-receptor binding is differentially regulated by the O-glycans on Notch. This review will summarize our understanding of the regulation of Notch signaling in myeloid and lymphoid cell development by specific O-glycans in mice with dysregulated expression of a particular glycosyltransferase and discuss how this may impact immune system development and malignancy in general, and in individuals with a congenital defect in the synthesis of the O-glycans attached to EGF repeats.
Introduction
Notch signaling is a highly conserved pathway that plays a pivotal role in the maintenance of tissue development and homeostasis. Defective Notch signaling leads to numerous pathological conditions (Varshney and Stanley, 2018; Zhou et al., 2022). In mammals, there are four Notch receptors (NOTCH1-NOTCH4) and five ligands of the Jagged and Delta-like families (DLL1, DLL3, DLL4 - orthologs to fly Delta; JAG1, JAG2 - orthologs to fly Serrate). Both Notch receptors and the canonical ligands, are transmembrane proteins with an extracellular domain (ECD) that consists primarily of epidermal growth factor-like (EGF) repeats (Figure 1). Many of these EGF repeats harbor consensus sites for the addition of different types of O-glycan to Ser or Thr, which takes place in the endoplasmic reticulum (ER) (Saiki et al., 2021). Classical Notch signaling is induced when Notch receptors interact with Notch ligands in neighboring cells (Kopan, 2012; Hori et al., 2013) (Figure 1). The binding of ligand promotes two proteolytic cleavages of a Notch receptor. The first is catalyzed by an ADAM-family metalloprotease and releases the extracellular domain, which is endocytosed into the ligand-expressing cell (Seib and Klein, 2021). The second cleavage is mediated by γ-secretase, an enzyme complex that contains presenilin, nicastrin, PEN2 and APH1 whose action releases the Notch intracellular domain (ICD), which translocates to the nucleus and cooperates with the DNA-binding protein RBP-Jκ/CSL and a co-activator Mastermind-like 1 (MAML1), to promote transcription and activate Notch target genes (Kovall et al., 2017). There are numerous factors involved in the regulation of Notch signaling, including the expression of Notch ligands in signal-receiving cells that may cause cis-inhibition of Notch receptors, factors affecting secretory pathway trafficking, and the complement of O-glycans attached to the Notch ECD.
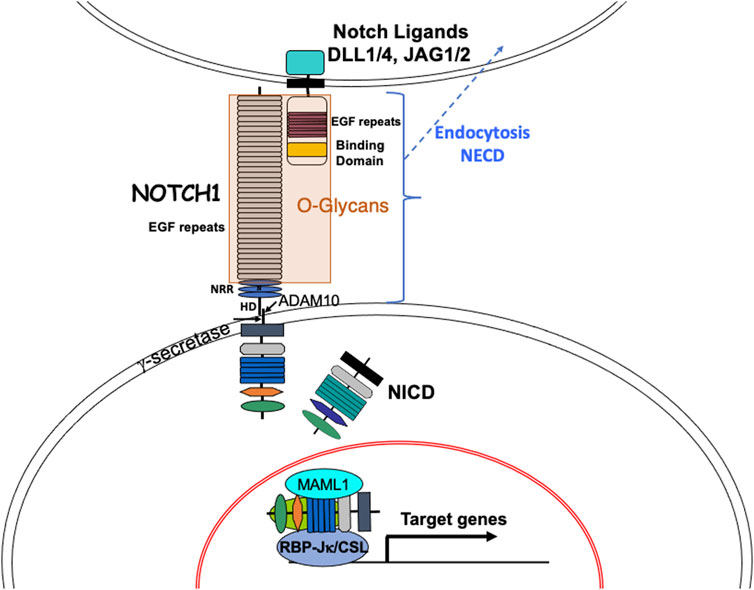
FIGURE 1. The Notch signaling pathway. The basic components of the Notch signaling pathway are shown. Canonical Notch ligands DLL1, DLL4, JAG1 and JAG2 in a signal-sending cell contact heterodimeric Notch receptors (e. g. NOTCH1) in a signal-receiving cell to initiate Notch signaling. Binding of ligand to NOTCH1 is regulated by O-glycans on EGF repeats of the Notch ECD and induces a conformational change that exposes a juxta-membrane cleavage site for the metalloprotease ADAM10. The released Notch ECD is endocytosed into the signal-sending cell. The transmembrane remnant of NOTCH1 is then cleaved within the membrane by gamma-secretase. The released Notch ICD transits to the nucleus where it combines with RBP-Jκ/CSL and transcriptional activators including MAML1 to form a complex that recognizes RBP-Jκ/CSL DNA binding sites in many target genes, thereby activating transcription. Notch signaling target genes include transcriptional suppressors like Hes and Hey family genes and transcriptional activators like c-Myc. NRR, Notch regulatory domain; HD, heterodimerization domain.
The major O-glycans found on EGF repeats of Notch receptors and the canonical Notch ligands are initiated by three different sugars: 1) O-fucose is added to Ser/Thr in the consensus site C2XXXX(S/T)C3 between the second and third Cys residues, where X is any amino acid, and either Ser or Thr accepts a fucose transferred from GDP-fucose by protein O-fucosyltransferase 1 (POFUT1); 2) O-glucose is added to Ser in the consensus site C1XSX(P/A)C2 where Ser accepts glucose from UDP-Glc transferred by protein O-glucosyltransferase 1 (POGLUT1); and 3) O-GlcNAc added to Ser/Thr in the consensus site C5XXGX(S/T)GXXC6 where Ser or Thr accepts GlcNAc transferred from UDP-GlcNAc by EGF domain-specific O-linked N-acetylglucosaminyltransferase (EOGT) (Saiki et al., 2021) (Figures 2A,B). The consensus sequence for POGLUT2 and POGLUT3 is distinct from that of POGLUT1 (Takeuchi et al., 2018a) (Figure 2B). The four mammalian Notch receptors contain large numbers of EGF repeats in their respective ECD (Figure 2C), the Notch ligands contain fewer (Figure 2D). The predicted O-glycan occupation of NOTCH1 based on EGF consensus sequences is shown in Figure 2C. Almost all structural studies to determine O-glycan occupancy have been performed on NOTCH1 constructs expressed in cells overexpressing different O-glycan glycosyltransferases. To date there is no O-glycan analysis of a Notch receptor purified directly from a mammalian physiological source. However, species comparisons of O-glycan consensus sites in EGF repeats reveal that some are highly conserved like the O-fucose site in EGF12, and others may vary between species (Haines and Irvine, 2003; Saiki et al., 2021). Each of the initiating sugars can be extended by the addition of other sugars to form a glycan. O-fucose is extended by the transfer of GlcNAc from UDP-GlcNAc by one of three Fringe GlcNAc-transferases termed LFNG, MFNG and RFNG. The GlcNAc may be extended by Gal, which may in turn be extended by sialic acid (Sia) to generate the O-linked tetra-saccharide Sia-Gal-GlcNAc-Fuc-O-EGF. Structural analyses of NOTCH1 overexpressed in HEK293T cells showed that O-fucose consensus sites almost always have a fucose, but the degree of extension of each fucose is variable (Kakuda and Haltiwanger, 2017). In T cells from mouse spleen activated in culture, Fringe modification of NOTCH1 occurs at few O-fucose residues (Matsumoto et al., 2022). O-glucose may be extended by xylose transferred from UDP-Xyl by xylosyltransferases GXYLT1 or GXYLT2, and a second Xyl may be added by XXYLT1 (Yu and Takeuchi, 2019). O-GlcNAc may be extended by Gal and Sia as well as Fuc (Tsukamoto et al., 2022). Several congenital human diseases arise from mutations in genes encoding the glycosyltransferases that modify EGF repeats in Notch receptors and ligands (Varshney and Stanley, 2018; Matsumoto et al., 2021).
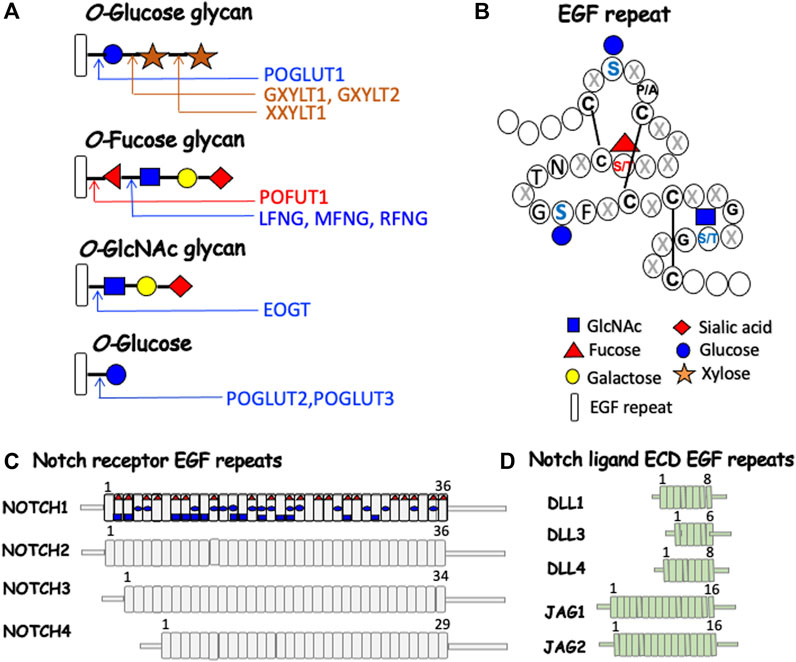
FIGURE 2. Notch receptor and Notch ligand glycosylation in mammals. (A) Each O-glycan predicted to be attached to an EGF repeat with an appropriate consensus sequence is shown. (B) A generic EGF repeat of ∼40 amino acids (Campbell et al., 1990) with the relevant sugar attached at the consensus site (Takeuchi et al., 2018a). Amino acids are shown by single letter code or X for any amino acid. The symbol for each sugar and an EGF repeat is shown in the key. The glycosyltransferases responsible for each sugar transfer are given. (C) NOTCH1-NOTCH4 ECDs are depicted with their respective number of EGF repeats, many of which can be modified by one or more O-glycans. The predicted O-glycan additions to mouse NOTCH1 are indicated by the respective initiating sugar. (D) The ECD of canonical Notch ligands with their respective number of EGF repeats, several of which can be modified by O-glycans.
In this review, we summarize our current understanding of roles for O-glycans in Notch signaling during hematopoiesis, myeloid and lymphoid cell development in the mouse. We also discuss how different O-glycans may impact immune cell development and malignancy, in general, and in individuals with a congenital defect in the synthesis of the O-glycans attached to Notch receptors.
Notch signaling in hematopoiesis
The yolk sac is the first organ with hematopoietic potential in the mouse embryo, producing mainly nucleated red blood cells (RBCs) and a few macrophages and megakaryocytes during primitive hematopoiesis, beginning at embryonic day E7.5 (Pajcini et al., 2011; Bigas et al., 2012). Hematopoiesis continues in the embryo at sites of hemogenic endothelium, initially in the aorta-gonad-mesonephros (AGM), which forms HSCs that migrate to the fetal liver, and from there to the bone marrow (BM), to finally give rise to all erythroid, myeloid and lymphoid cells in the adult. Variations in the strength of Notch signaling are crucial for the switch between the formation of endothelial cells (low Notch signal induced by JAG1), and the formation of HSCs (high Notch signal induced by DLL4) from hemogenic endothelium (Gama-Norton et al., 2015; Porcheri et al., 2020). Abolishing Notch signaling has no effect on hematopoiesis in the yolk sac (Pajcini et al., 2011; Bigas et al., 2012). However, signaling via NOTCH1, but not NOTCH2, is required for the generation of HSCs in the embryo, although not for their proliferation or maintenance in the adult (Kumano et al., 2003; Souilhol et al., 2016). On the other hand, various experimental approaches that increase Notch signaling in HSCs lead to increased proliferation and expansion of the stem cell pool (Lampreia et al., 2017). Most recently, conditional deletion of RBP-Jκ in HSC has shown that Notch signaling, while not required for the generation or maintenance of long- or short-term HSCs, is important for HSC self-renewal after irradiation or chemotherapeutic stress (Lakhan and Rathinam, 2021).
Roles for individual Notch receptors and ligands in hematopoiesis have been identified by conditional knockout strategies in the mouse. Overall requirements of Notch signaling have been investigated by inactivation of downstream transcription factors including RBP-Jκ, that are required for signaling through all canonical Notch receptor/ligand interactions (Kovall et al., 2017). Inactivation of Notch1 in HSCs using Mx1-Cre revealed that NOTCH1 is essential for the development of T cells in the thymus (Radtke et al., 1999; MacDonald et al., 2001). Conditional deletion of Notch1 in thymic T cells using a CD4-Cre revealed requirements for NOTCH1 in the generation of T cells involved in innate immunity in liver, intestine and spleen (Chennupati et al., 2016). A role for NOTCH2 in early T cell development was recently identified in co-culture investigations of Notch1 and Notch2 following inactivation in bone marrow progenitors (Romero-Wolf et al., 2020). In elegant rescue experiments of HSCs conditionally-deleted for RBP-Jκ, the downstream effector of Notch signaling, a block in the development of thymus-seeking T cell progenitors in bone marrow was identified (Chen et al., 2019).
Experiments to determine roles for Notch ligands in thymic stromal cells used Foxn1-Cre to identify DLL4 as essential for stimulating Notch signaling in the thymus (Hozumi et al., 2008; Koch et al., 2008). However, recent rescue experiments have shown that transgenic Dll1 can substitute for Dll4 when Dll4 is deleted, and can stimulate NOTCH1 and NOTCH2 to promote T cell development in thymus (Hirano et al., 2022). Immunochemistry shows that DLL1, DLL4, JAG1 and JAG2 Notch ligands are expressed in thymus in a regio-specific manner which may be important in controlling Notch signaling strength in different regions of the thymus (Felli et al., 1999; Garcia-Leon et al., 2018).
Notch2 is expressed in Pro B and Pre B cells of the bone marrow, and in the different B cell subsets of spleen (Garis and Garrett-Sinha, 2021). However, conditional deletion of Notch2 using Mx1-Cre primarily inhibits development of MZ B cells and their precursors in spleen (Saito et al., 2003). DLL1 in splenic fibroblasts is the Notch ligand responsible for stimulating Notch signaling via NOTCH2 to generate MZ B cells in spleen (Sheng et al., 2008). Plasticity in this differentiation has recently been revealed in the generation of MZ B cells from follicular B (Fo B) cells (Lechner et al., 2021). A diagram of bone marrow HSC differentiation into lymphoid and myeloid cells in thymus and spleen including expression of Notch receptor, Notch ligand and Notch-related glycosyltransferase genes from the ImmGen Project (Immunological Genome Project, 2020) is shown in Figure 3.
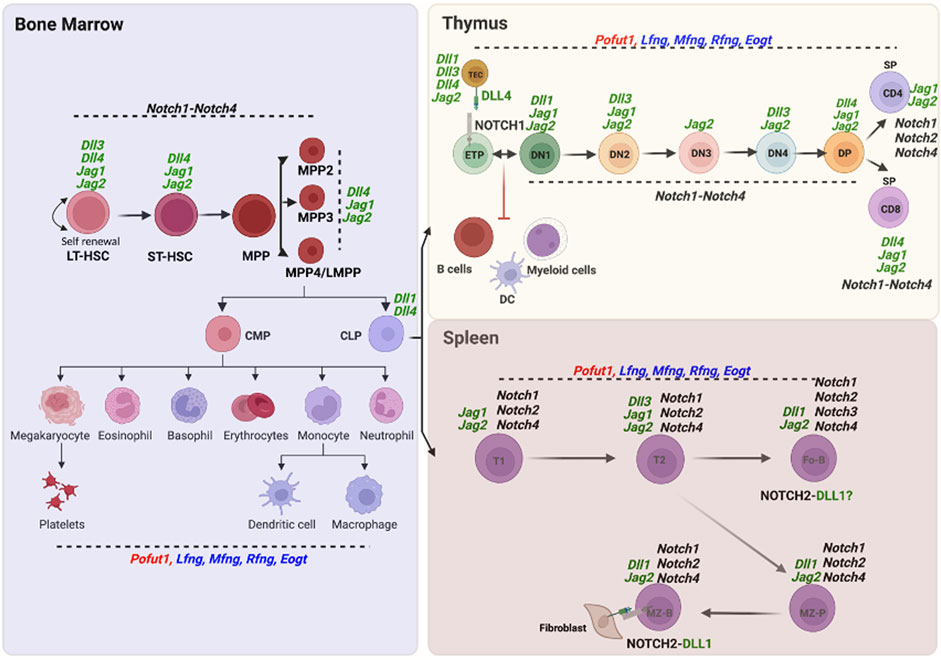
FIGURE 3. Expression of O-glycan glycosyltransferase, Notch receptor and Notch ligand genes in hematopoietic cells. Expression of the O-glycan glycosyltransferase, Notch receptor and canonical Notch ligand genes whose deletion causes developmental changes in lymphopoiesis and myelopoiesis are shown in cells of the three hematopoietic compartments - bone marrow, thymus and spleen - based on data from single cell RNA-seq or microarray experiments reported in the ImmGen Skyline database (http://rstats.immgen.org/Skyline/skyline.html). NOTCH1 in ETP and DLL4 in thymic stroma, as well as NOTCH2 in MZ B and DLL1 in splenic stroma, are also shown as proteins.
Notch target genes in lymphopoiesis and myelopoiesis
Determining genes altered in expression following deletion of Notch pathway members in HSCs or progenitors must be performed in the earliest lineage in which the gene is expressed because an essential requirement for Notch signaling may mean that certain cell populations are not generated or are contaminated by cells that differentiate along aberrant pathways. For example, deletion of Notch1 using Mx1-Cre does not appear to perturb bone marrow cell populations derived from HSCs, but blocks T cell development at an early stage, and causes T cell progenitors to differentiate into B cells and myeloid cells in the thymus (Radtke et al., 1999; Feyerabend et al., 2009). Consequently, gene expression analysis of CD4/CD8 double-negative (DN) thymocytes lacking Notch1 which have become B cells and myeloid cells, would not provide insights into genes related to T cell development. Interestingly, deletion of RBP-Jκ in HSCs using Vav1-Cre or Mx1-Cre does not affect Notch target gene expression in HSCs, and actually increases expression of classic Notch target genes like Hes1 and Hes5 in megakaryocyte, erythroid and myeloid bone marrow progenitors (Duarte et al., 2018). However, investigations of thymus-seeding progenitors in bone marrow by an elegant gene rescue approach revealed requirements for RBP-Jκ and Notch signaling in the generation of the MPP4/LMPP lymphoid progenitor population in bone marrow (Chen et al., 2019). Thus, Notch signaling is required for generation of thymus-seeding progenitors in bone marrow. In the case of Notch target Hes1, conditional knockout with Mx1-Cre does not affect HSC or HSPC numbers but causes a major block in T cell development in thymus, as expected if Notch signaling in inhibited (Wendorff et al., 2010). However, MZ B cell numbers in spleen are not altered following the loss of Hes1 in HSCs (Wendorff et al., 2010). Deletion of Hes1 using Vav1-Cre also showed that hematopoiesis was not obviously altered. However, stress hematopoiesis was ineffective in stem cells lacking Hes1, which exhibit an exhausted phenotype (Ma et al., 2020).
Strategies to identify roles for O-glycans in hematopoiesis
As mentioned above, Notch receptors and ligands are transmembrane glycoproteins with an ECD comprising numerous EGF repeats (Figures 2C,D). Each EGF repeat consists of ∼40 amino acids, including six conserved cysteines which form three disulfide bonds. EGF repeats may contain consensus sequences for the addition of fucose (Fuc), glucose (Glc) or N-acetylglucosamine (GlcNAc) to Ser or Thr to initiate an O-Fuc, O-Glc or O-GlcNAc glycan (Haltiwanger et al., 2022). Glycosyltransferases that transfer an initiating sugar reside in the ER - POFUT1 transfers Fuc from GDP-Fuc, POGLUT1/2/3 transfer Glc from UDP-Glc, and EOGT transfers GlcNAc from UDP-GlcNAc. O-fucose on an EGF repeat may be extended by Fringe GlcNAc-transferases LFNG, MFNG or RFNG, and the GlcNAc may be further extended by Gal and sialic acid (Sia). O-glucose may be extended by xylose (Urata et al., 2020) and O-GlcNAc may be extended by Gal and Sia (Ogawa et al., 2020). To date, experiments aimed at defining roles for O-glycans on EGF repeats in immune cell development have been performed by manipulating the O-fucose glycans by deleting or overexpressing Pofut1, Lfng, Mfng or Rfng (Figure 2A). Only approximately 100 proteins contain EGF repeats that may be modified by these O-glycans (Rampal et al., 2007). The four Notch receptors and five Notch ligands comprise almost 10% of these substrates. However, ascribing phenotypic changes observed after deletion or overexpression of one of these glycosyltransferases to changes in Notch signaling depends on whether the phenotype mimics that of mutants deleted for, or overexpressing, other Notch pathway members; or on the mutation causing defective activation of Notch receptors by ligand-induced cleavage to generate NICD; or on the altered expression of known Notch signaling target genes. To directly determine if loss of one or more O-glycans on a particular Notch receptor or Notch ligand provides the basis of a glycosylation-defective phenotype, it is necessary to mutate individual Ser/Thr residues to preclude glycosylation in a specific EGF repeat. This, however, comes with caveats, since the alternative amino acid chosen can affect the outcome, as described below.
O-Fucose glycans and Notch signaling in lymphopoiesis and myelopoiesis
Deletion of Pofut1 in mice results in embryonic lethality with a phenotype typical of defective Notch signaling (Shi and Stanley, 2003; Okamura and Saga, 2008). Conditional deletion of Pofut1 in bone marrow HSCs using Mx1-Cre or Vav1-Cre causes changes in lymphoid and myeloid cell differentiation (Yao et al., 2011; Yu et al., 2015). HSC progenitors (Lineage minus (Lin-)Sca1+cKit + bone marrow cells termed LSK) lacking Pofut1 exhibit a 3-6-fold reduction in the binding of soluble Notch ligands DLL1-Fc and DLL4-Fc, a major decrease in ligand recognition, and a slight reduction of ∼30% in cell surface expression of NOTCH1 and NOTCH2 (Yao et al., 2011), indicating a minor effect on Notch receptor trafficking. Myeloid cells in bone marrow were increased, whereas T cell production in thymus, and MZ B cells in spleen were decreased, as expected if the loss of O-fucose glycans causes defective Notch signaling via both NOTCH1 and NOTCH2. In addition, expression of Notch target genes Dtx1 and Hes1 was depressed in co-cultures of LSK cells with OP9 stromal cells under conditions that induce differentiation to T cells. Bone marrow transplantation revealed that the reduced lymphopoiesis and altered myelopoiesis following conditional deletion of Pofut1 arose largely from cell-autonomous effects in HSCs and HSC progenitors (HSPC). The overall findings validated conclusions of earlier experiments that characterized defective myelopoiesis in mice with a global knockout of Gmds, an enzyme required for the synthesis of GDP-Fuc, the substrate of POFUT1 (Zhou et al., 2008). Loss of O-fucose on Notch receptors, proposed as the basis of changes in myelopoiesis in the Gmds [−/−] mice, is consistent with the Notch-defective phenotype of mice lacking Pofut1 the enzyme that transfers O-fucose to all Notch receptors and ligands (Yan et al., 2010). Interestingly, conditional deletion of the downstream transcription factor RBP-Jκ that transduces Notch signaling via all combinations of Notch receptors and ligands, gives more severe reductions in lymphoid and myeloid cells than deletion of Pofut1 in HSC (Yu et al., 2015), suggesting that other O-glycans on Notch receptors might support Notch signaling in the absence of O-fucose glycans. We recently addressed this question by generating mice lacking Eogt and O-GlcNAc glycans as well as being conditionally deleted for Pofut1 in HSCs, as described below (Tanwar and Stanley, 2022).
An alternative approach to deleting Pofut1 is to pinpoint functions of O-fucose in NOTCH1 by mutating key O-fucose sites in EGF repeats of the ECD. The first example in mouse was mutation of the O-fucose site in EGF12 of NOTCH1 from Thr to Ala (Ge and Stanley, 2008). EGF12 is within the Notch ligand binding domain, and we now know that the O-fucose in EGF12 of NOTCH1 is recognized directly by Notch ligands (Luca et al., 2015; Luca et al., 2017). Homozygotes with the EGF12 mutation termed Notch1[12f/12f], are viable and fertile in a mixed genetic background. However, they exhibit effects typical of reduced NOTCH1 signaling in T cell development. Thymus size and thymocyte numbers were reduced ∼50%, and T cell progenitors, CD4 or CD8 single-positive (SP) and CD4/CD8 double-positive (DP) T cells were reduced in thymus. In addition, Notch1[12f/12f] thymocytes exhibit reduced expression of activated NOTCH1 (NICD), and reduced levels of Notch targets Hes1 and Dtx1. Binding of Notch ligand DLL1 to thymocytes is also reduced. The defective T cell phenotype is cell autonomous following transplantation of Notch1[12f/12f] bone marrow. Therefore, loss of one critical fucose in EGF12 of NOTCH1 significantly weakens signaling via NOTCH1, even though O-fucose is probably present on 19 other EGF repeats with an O-fucose consensus site (Figure 2C). This hypomorphic phenotype is surprisingly maintained in mice overexpressing Lfng in T cells (Visan et al., 2010).
The question of whether loss of O-Fuc from EGF12 in NOTCH1 due to conversion of Thr to Ala or the amino acid mutation itself causes the T cell phenotype was not addressed in the mouse mutant. However, it was previously investigated in cell-based Notch signaling assays. In these experiments, the Thr in NOTCH1 EGF12 was changed to Ala to abrogate O-Fuc addition or, alternatively, changed to Ser which accepted O-fucose (Shi et al., 2007). The Thr to Ala mutation in EGF12 inactivated NOTCH1 signaling in the cell-based assay, but the Thr to Ser mutation did not, consistent with O-fucose being required in EGF12 for NOTCH1 signaling. The inhibition of NOTCH1 signaling in the Thr to Ala mutant construct was essentially complete in these experiments. Thus, it was surprising that Notch1[12f/12f] homozygous mice were viable (Ge and Stanley, 2008). The explanation turned out to be genetic background. When the Notch1[12f] mutation was backcrossed into the C57BL/6J background for >10 generations, Notch1[12f/12f] homozygotes died at E11.5 (Varshney et al., 2019). It seems that a modifying activity in the initial mixed genetic background rescued Notch1[12f/12f] from embryonic lethality.
These findings highlight the complexities of interpreting the results of point mutations that preclude a glycosylation reaction. In another example, a mutation converting an Asn which receives an N-glycan in the enzyme POFUT1 to Leu was found to reduce POFUT1 activity. However, substitution of the Asn with Gln that also could not receive an N-glycan, restored POFUT1 activity (Takeuchi et al., 2018b). Therefore, certain amino acids at that position are required for POFUT1 activity, but the N-glycan attached to Asn is not required for POFUT1 to be active. In another example, mutation of Thr to Ala in the EGF repeat of Cripto inhibits Cripto signaling. However, mutation to Ser, which could receive an O-fucose, also inhibits Cripto signaling, leading to the conclusion that the Thr, and not the O-fucose, is necessary for Cripto signaling (Shi et al., 2007). The take-home message from these experiments is that all caveats should be explored to enable interpretation of mutant phenotypes in relation to glycosylation status.
Fringe GlcNAc-transferases in lymphopoiesis and myelopoiesis
The first investigation of roles for Fringe GlcNAc-transferases in T cell development showed that overexpression of Lfng under the control of the lck promoter that is initially expressed in T cell progenitors, blocks T cell development and leads to a large population of B cells in the thymus (Koch et al., 2001). This result is consistent with numerous subsequent experiments showing that B cells and myeloid cells are generated from thymic T cell precursors blocked in Notch signaling by a variety of strategies (Brandstadter and Maillard, 2019). Most interesting is the basis of the block caused by overexpressing Lfng in thymocytes, which might be expected to enhance rather than block Notch signaling because LFNG modification of Notch receptors generally enhances signaling induced by DLL ligands (Hicks et al., 2000). Elegant experiments showed that mature thymic T cells overexpressing Lfng compete with early T cell progenitors for binding to DLL4-expressing thymic stroma, and thereby prevent access of T cell progenitors to Notch ligands that stimulate Notch signaling and promote differentiation along the T cell lineage (Visan et al., 2006). Overexpression or loss of Lfng in fetal liver-derived HSC confirmed that Lfng is required for optimal T cell development (Tsukumo et al., 2006). Loss of Lfng from CD4/CD8 double-negative 3 (DN3) T cell progenitors inhibits their proliferation and causes them to differentiate prematurely to CD4/CD8 double-positive (DP) T cells (Yuan et al., 2011). Thus, extension of O-fucose on NOTCH1 in pre-T cells by LFNG enhances Notch ligand binding to Notch receptors and Notch signaling, thereby prolonging proliferation of the pool of DN3 T cell progenitors and promoting the production of mature thymic T cells. The role of LFNG is therefore to enhance the strength of Notch signaling in T cell progenitors, a necessary boost required for optimal production of T cells in thymus (Yuan et al., 2011). However, overexpression of Lfng under the lck promoter could not fully rescue reduced T cell production in the Notch1[12f/12f] thymus (Visan et al., 2010). This result indicates that extension of the O-fucose at EGF12 of NOTCH1, regardless of potentially enhanced extension of O-fucose at other EGF repeats due to prolonged overexpression of Lfng, is required for optimal Notch signaling in T cell progenitors. In these experiments, Lfng expression was manipulated only in Notch receptor-expressing T cells. Requirements of Lfng for Notch ligand functions in thymic stroma were not addressed. However, there are cellular contexts in which effects of Fringe modification of canonical Notch ligands have been identified in other systems (for example (Bochter et al., 2022) and (Kadur Lakshminarasimha Murthy et al., 2018)), and it will be important to examine this issue and the role of each FNG during hematopoiesis in future.
Roles for each of the Fringe GlcNAc-transferases in T and B cell development were revealed in mice inactivated for all three Fng genes compared to mice expressing a single Fng gene from one allele (Song et al., 2016). Loss of all three Fringe GlcNAc-transferases leaves Notch receptors and Notch ligands with EGF repeats carrying O-fucose that cannot be extended (Figure 2A). Triple knockout (Fng tKO) mice have reduced DLL4-Fc binding to DN T cell progenitors, and reduced frequencies of early thymic progenitors (ETP), DN2 T cell progenitors, and double-positive (DP) T cells (Song et al., 2016). Increased frequencies of CD4 and CD8 single-positive (SP) T cells in the thymus were a novel characteristic of Fng tKO thymus that could indicate delayed exit of mature SP T cells from the thymus. The Fng tKO phenotype is transferred cell-autonomously by bone marrow. Substantial rescue of T cell development is achieved in mice expressing only Lfng, only Mfng, or only Rfng from a single allele. Thus, each Fng contributes to Notch signaling in thymic T cells and promotes optimal T cell development.
In spleen, both LFNG and MFNG are required for MZ B cell development (Tan et al., 2009). Deficiency of either one reduces the number of MZ B cells in spleen. In mice expressing no Fng genes, there is a reduced frequency of MZ B cells as expected, and a corresponding increase in Fo B and MZ P precursor B cells. CD4+ and CD8+ splenic T cells are also decreased. By contrast, granulocyte numbers are increased (Song et al., 2016). All changes are consistent with reduced Notch signaling and show that Fringe modification is necessary for optimal T and B cell and granulocyte production. In terms of individual Fng genes, Lfng was able to rescue the frequency of T and MZ B cells in the absence of both Mfng and Rfng. Mfng or Rfng expressed from a single allele accomplished a less complete rescue (Song et al., 2016). There are also functional consequences for T cell responses in mice lacking all Fringe activities. Fng tKO splenocytes stimulated by anti-CD3/CD28 beads or lipopolysaccharide (LPS) or Concanavalin A exhibit reduced proliferation (Song et al., 2016). In addition, naïve T cells and effector memory T cells were reduced in Fng tKO spleen.
O-GlcNAc glycans in hematopoiesis
Mice lacking Eogt and O-GlcNAc are viable and fertile but exhibit mildly defective Notch signaling during retinal angiogenesis (Sawaguchi et al., 2017). This laboratory is investigating effects of deleting Eogt and O-GlcNAc glycans on hematopoiesis, lymphopoiesis and myelopoiesis in Eogt null mice. In Eogt[−/−] thymus, T cell progenitors and DP T cell numbers were reduced, while SP T cells were slightly increased (Tanwar and Stanley, 2022), as observed when all three Fng genes are deleted (Song et al., 2016). Consistent with Notch signaling being inhibited due to the loss of Eogt and O-GlcNAc glycans, B cells and myeloid cells were increased in thymus, and Notch target genes Hes1 and Il2ra were reduced in expression. In spleen, CD4+ and CD8+ T cells were not reduced, but NK T cells were reduced along with myeloid cells. Surprisingly, B cell subsets were slightly increased. The Eogt null hematopoietic phenotype was largely cell-autonomous following bone marrow transfer.
The fact that loss of Eogt caused effects on T and B cell development consistent with reduced Notch signaling suggested that O-GlcNAc glycans might support Notch signaling in the absence of Pofut1 and O-fucose glycans. This hypothesis was tested by deleting Pofut1 via Vav1-Cre in HSCs, in the presence and absence of Eogt. Deletion of Pofut1 in HSCs caused myeloid hyperplasia and a drastic reduction in T cell development and MZ B cell production, as described previously (Yao et al., 2011; Yu et al., 2015). The additional deletion of Eogt enhanced the Notch-defective phenotype in bone marrow, thymus and spleen (Tanwar and Stanley, 2022). Thus, EOGT and O-GlcNAc glycans are required for optimal Notch signaling in the development of myeloid and lymphoid cells. In addition, O-GlcNAc glycans promote Notch signaling in the absence of O-fucose glycans and partially rescue HSCs and descendants that lack Pofut1.
Congenital diseases affected in EGF repeat modification by O-glycans
Congenital disorders of glycosylation (CDG) are rare in human populations, but there are many distinct diseases. At latest count, congenital mutations have been reported in >140 different genes involved in glycosylation (Sosicka et al., 2021; Lefeber et al., 2022). Of the glycosyltransferases that modify Notch EGF repeats and promote hematopoiesis, deleterious mutations compatible with life in humans have been identified in POFUT1, LFNG, and EOGT. Mutations in POFUT1 cause an autosomal dominant disease termed Dowling Degos Disease 2 (DDD2) which causes skin hyperpigmentation and follicular skin lesions (Li et al., 2013). However, genome sequencing and mosaic analysis have identified mutations in POFUT1 that are not classified as a classical DDD2 phenotype. A recessive mutation in a person with microencephaly and developmental delay was found to correlate with weak POFUT1 activity (Takeuchi et al., 2018b). Another recessive POFUT1 mutation expands the DDD2 phenotype with new features such as dermatitis (Atzmony et al., 2019). Given the requirement for POFUT1 for lymphopoiesis and myelopoiesis described above, patients with weak POFUT1 may have deficiencies in innate and adaptive immunity. Skin lesions observed in these patients may reflect reduced activity of the immune system in skin (Nguyen and Soulika, 2019).
Humans with inactivating mutations in LFNG have Spondylocostal Dystosis Type 3 (SCDO3), an autosomal recessive disease that gives rise to malformation of the skeleton during embryogenesis (Sparrow et al., 2006; Dunwoodie, 2009). Deletion of Lfng in the mouse causes severe skeletal damage with loss of the tail (Evrard et al., 1998; Zhang and Gridley, 1998), and may be embryonic lethal, depending on genetic background (unpublished observations). LFNG promotes T and MZ B cell development, but is partially compensated for by MFNG and RFNG (Tan et al., 2009; Song et al., 2016). Consequently, effects of the loss of LFNG activity on immune cell development would be expected to be relatively minor in humans, and thus impacts on the immune response of SCDO3 patients would also presumably be minor.
Autosomal recessive EOGT mutations in humans give rise to a disease termed Adams-Oliver Syndrome type 4 (AOS4), which is characterized by aplasia cutis congenita of the scalp, limb and digit malformations, and in some cases, vascular and cardiac defects (Schröder et al., 2019). The O-GlcNAc-transferase activity of EOGT containing AOS4 mutations is greatly reduced (Ogawa et al., 2015). Considering that AOS4 disease varies in severity and that AOS4 patients may live to adulthood, and given the defective T and altered B cell development observed in Eogt null mice (Tanwar and Stanley, 2022), EOGT mutations would be expected to affect immune cell development in humans with AOS4, and potentially to compromise certain immune responses. Further experiments on innate and adaptive immune responses in Eogt null mice will assist in predicting immune response concerns in AOS4 patients.
Lymphoid and myeloid malignancies with altered Notch signaling
Notch signaling was first discovered in mammals in acute T lymphoblastoid leukemia (T-ALL) which arises in many cases from a chromosomal translocation that causes constitutive activation of NOTCH1 (Ellisen et al., 1991). Subsequently, activating mutations in the NOTCH1 gene were found to cause >50% of T-ALLs (Weng et al., 2004). A proportion of these mutations occur in the NOTCH1 heterodimerization (HD) domain just above the plasma membrane (Figure 1) and render NOTCH1 active independently of Notch ligand stimulation. However, Notch ligands may theoretically stimulate such activated NOTCH1 mutants, since the ECD with its attached O-glycans is present. Another class of NOTCH1 mutations in T-ALL occurs in the PEST domain and functions by prolonging the life of NICD in the nucleus (Weng et al., 2004). In this case, Notch ligand induces Notch signaling which is sustained due to the lack, or inactivation of, the PEST domain on NICD. Several other types of leukemia also arise from PEST domain mutations in NOTCH1, including chronic lymphocytic leukemia (CLL), and Diffuse Large B cell lymphoma (Siebel and Lendahl, 2017). While there have been no reports of an involvement of O-glycan synthesis in leukemogenesis or other blood disorders with disrupted Notch signaling, amplification of the POFUT1 gene has been observed in certain cases of myeloid malignancy (Mackinnon et al., 2010). Thus, loss of the long arm of one copy of chromosome 20, that occurs in cases of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) and is presumed to assist leukemogenesis by removal of a tumor suppressor-like gene, is often accompanied by amplification of the remaining portion of chromosome 20 (20q11.2). The POFUT1 gene resides in the smallest amplified domain at 20q11.21, along with the HCK, TM9SF4 and PLAGL2 genes. Investigation of a functional role for POFUT1 gene amplification in models of AML and MDS might reveal enhanced Notch signaling due to increased O-fucosylation of Notch receptors, or ligands. A similar amplification of chromosome region 20q11.21 is seen in colon cancer (Chabanais et al., 2018). However, the website Tumor Portal sponsored by the BROAD Institute (http://www.tumorportal.org/), which reports expression levels and mutations across many cancer types, notes that neither POFUT1, LFNG nor EOGT are “near significance” for mutation or expression changes in any lymphoid or myeloid tumor type.
Discussion
The development of HSCs, the many HSPCs, myeloid, and lymphoid cells reflects a complicated set of lineage decisions requiring co-ordination at the molecular and cellular levels. Notch signaling is a key regulator of several stages of differentiation through different combinations of Notch receptors, Notch ligands and glycosyltransferases that modify extracellular domain EGF repeats (Figure 1). The focus of this review is on Notch receptors, ligands and glycosyltransferase genes that have been manipulated to uncover specific roles in hematopoiesis, myelopoiesis and lymphopoiesis for regulation by glycosylation. Table 1 summarizes key experimental systems and findings discussed in the text. NOTCH1 is essential for T cell development and DLL4 is required, but Dll4 deletion in thymic stroma can be rescued by Dll1. NOTCH2 is essential for MZ B cell development and DLL1 is also required. It is not known whether DLL4 could substitute for DLL1 in splenic stroma. JAG1 and JAG2 have roles in the generation of HSCs from hemogenic endothelium and the glycosyltransferases have various roles. Deletion of Poglut1 is embryonic lethal (Fernandez-Valdivia et al., 2011), and to date there have been no reports on the consequences of conditionally deleting Poglut1 and O-glucose glycans in HSCs, or other immune cells, or stromal cells. However, the effects of knocking out Pofut1, Lfng, Mfng, Rfng and Eogt on myeloid and lymphoid cell development have been investigated. Deletion of Pofut1 in HSCs causes altered generation of B cells, myeloid cells, and thymus-seeding progenitors in bone marrow. Pofut1 null T cells that enter the thymus are mainly converted to B cells and myeloid cells, and T cell development is blocked. A similarly dramatic phenotype is observed when Lfng is overexpressed in thymic T cells. Loss of Lfng causes a relatively mild reduction in T cell development which is ameliorated by the presence of Mfng and/or Rfng. In spleen, Lfng and Mfng are required together for optimal MZ B-cell development. Mice lacking Eogt exhibit altered cell numbers in bone marrow B cells and myeloid cells, thymic T cells and MZ B cells in spleen. As structural methods develop it should become possible to analyze the actual changes in O-glycans occupancy and extension among Notch receptors in mouse models. For now, the combined data suggest that sugars added to the EGF repeats of Notch receptors and ligands play a part in optimizing the generation of appropriate numbers of cells during hematopoiesis, myelopoiesis and lymphopoiesis. Reduced ligand binding to progenitor cells identifies one mechanism for reduced Notch signaling strength. Consistent with this, reduced ligand binding is observed in DN T cells expressing NOTCH1[12f] in which there is no O-fucose attached to EGF12, or no expression of LFNG, MFNG or RFNG. LSK cells in bone marrow show reduced Notch ligand binding when POFUT1 is absent. However, loss of EOGT does not significantly affect soluble Notch ligand binding to DN T cells, although Notch signaling is reduced, indicating that binding strength may be impaired. Since glycosyltransferases that modify Notch promote immune cell development, people with deleterious mutations in one of these glycosyltransferase genes may have immune response defects. Functional consequences were observed in responses of activated T cells lacking Fringe activity, but many more studies are needed to uncover potential problems with immune responses. In immune cell cancers, inhibition of Notch signaling might be achieved by inhibition of glycosylation. One example of this approach used incorporation of a chemically modified fucose into EGF repeats to block Notch signaling (Schneider et al., 2018). A better understanding of how Notch signaling is regulated and becomes dysregulated will enable promotion or inhibition of signaling in order to optimize immune responses in humans. Other signaling pathways such as those directed by cytokines or NFκB also influence lymphoid and myeloid cell development (Tsaouli et al., 2020). However, these pathways should not be directly affected by inactivation of any of the EGF repeat-modifying glycosyltransferases discussed in this review, unless a critical member of the signaling pathway contains one or more EGF repeats with O-glycans required for the function of the parent molecule.
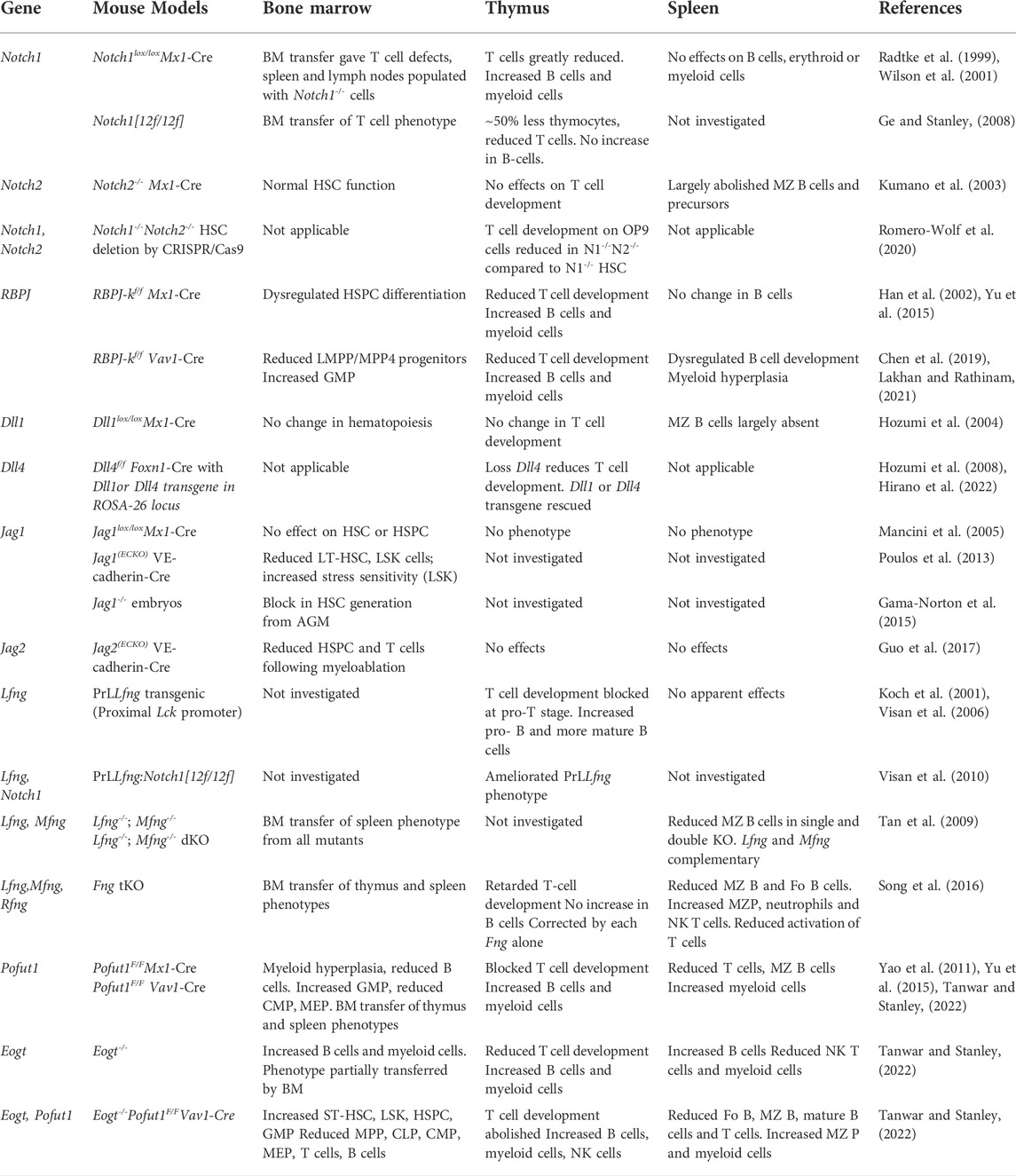
TABLE 1. Notch Signaling-Defective Mutants and Hematopoiesis.
Author contributions
AT wrote a proposed draft and PS wrote the manuscript. AT and PS edited the manuscript.
Funding
This work was supported by grant RO1 GM106417 to PS from the National Institutes of Health.
Acknowledgments
The manuscript by Tanwar and Stanley (2022) is a preprint at the bioRxiv server. https://doi.org/10.1101/2022.03.21.485194.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Atzmony, L., Zaki, T. D., Antaya, R. J., and Choate, K. A. (2019). Phenotypic expansion of POFUT1 loss of function mutations in a disorder featuring segmental dyspigmentation with eczematous and folliculo-centric lesions. Am. J. Med. Genet. A 179, 2469–2473. doi:10.1002/ajmg.a.61362
Bigas, A., D'altri, T., and Espinosa, L. (2012). The Notch pathway in hematopoietic stem cells. Curr. Top. Microbiol. Immunol. 360, 1–18. doi:10.1007/82_2012_229
Bochter, M. S., Servello, D., Kakuda, S., D'amico, R., Ebetino, M. F., Haltiwanger, R. S., et al. (2022). Lfng and Dll3 cooperate to modulate protein interactions in cis and coordinate oscillatory Notch pathway activation in the segmentation clock. Dev. Biol. 487, 42–56. doi:10.1016/j.ydbio.2022.04.004
Brandstadter, J. D., and Maillard, I. (2019). Notch signalling in T cell homeostasis and differentiation. Open Biol. 9, 190187. doi:10.1098/rsob.190187
Campbell, I. D., Baron, M., Cooke, R. M., Dudgeon, T. J., Fallon, A., Harvey, T. S., et al. (1990). Structure-function relationships in epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-alpha). Biochem. Pharmacol. 40, 35–40. doi:10.1016/0006-2952(90)90175-k
Chabanais, J., Labrousse, F., Chaunavel, A., Germot, A., and Maftah, A. (2018). POFUT1 as a promising novel biomarker of colorectal cancer. Cancers (Basel) 10, 411. doi:10.3390/cancers10110411
Chen, E. L. Y., Thompson, P. K., and Zúñiga-Pflücker, J. C. (2019). RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat. Immunol. 20, 1456–1468. doi:10.1038/s41590-019-0518-7
Chennupati, V., Koch, U., Coutaz, M., Scarpellino, L., Tacchini-Cottier, F., Luther, S. A., et al. (2016). Notch signaling regulates the homeostasis of tissue-restricted innate-like T cells. J. Immunol. 197, 771–782. doi:10.4049/jimmunol.1501675
Duarte, S., Woll, P. S., Buza-Vidas, N., Chin, D. W. L., Boukarabila, H., Luís, T. C., et al. (2018). Canonical Notch signaling is dispensable for adult steady-state and stress myelo-erythropoiesis. Blood 131, 1712–1719. doi:10.1182/blood-2017-06-788505
Dunwoodie, S. L. (2009). Mutation of the fucose-specific beta1, 3 N-acetylglucosaminyltransferase LFNG results in abnormal formation of the spine. Biochim. Biophys. Acta 1792, 100–111. doi:10.1016/j.bbadis.2008.11.003
Ellisen, L. W., Bird, J., West, D. C., Soreng, A. L., Reynolds, T. C., Smith, S. D., et al. (1991). TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 66, 649–661. doi:10.1016/0092-8674(91)90111-b
Evrard, Y. A., Lun, Y., Aulehla, A., Gan, L., and Johnson, R. L. (1998). Lunatic fringe is an essential mediator of somite segmentation and patterning. Nature 394, 377–381. doi:10.1038/28632
Felli, M. P., Maroder, M., Mitsiadis, T. A., Campese, A. F., Bellavia, D., Vacca, A., et al. (1999). Expression pattern of notch1, 2 and 3 and Jagged1 and 2 in lymphoid and stromal thymus components: Distinct ligand-receptor interactions in intrathymic T cell development. Int. Immunol. 11, 1017–1025. doi:10.1093/intimm/11.7.1017
Fernandez-Valdivia, R., Takeuchi, H., Samarghandi, A., Lopez, M., Leonardi, J., Haltiwanger, R. S., et al. (2011). Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 138, 1925–1934. doi:10.1242/dev.060020
Feyerabend, T. B., Terszowski, G., Tietz, A., Blum, C., Luche, H., Gossler, A., et al. (2009). Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity 30, 67–79. doi:10.1016/j.immuni.2008.10.016
Gama-Norton, L., Ferrando, E., Ruiz-Herguido, C., Liu, Z., Guiu, J., Islam, A. B., et al. (2015). Notch signal strength controls cell fate in the haemogenic endothelium. Nat. Commun. 6, 8510. doi:10.1038/ncomms9510
Garcia-Leon, M. J., Fuentes, P., De La Pompa, J. L., and Toribio, M. L. (2018). Dynamic regulation of NOTCH1 activation and Notch ligand expression in human thymus development. Development 145, dev165597. doi:10.1242/dev.165597
Garis, M., and Garrett-Sinha, L. A. (2021). Notch signaling in B cell immune responses. Front. Immunol. 11, 609324. doi:10.3389/fimmu.2020.609324
Ge, C., and Stanley, P. (2008). The O-fucose glycan in the ligand-binding domain of Notch1 regulates embryogenesis and T cell development. Proc. Natl. Acad. Sci. U. S. A. 105, 1539–1544. doi:10.1073/pnas.0702846105
Guo, P., Poulos, M. G., Palikuqi, B., Badwe, C. R., Lis, R., Kunar, B., et al. (2017). Endothelial jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J. Clin. Invest. 127, 4242–4256. doi:10.1172/JCI92309
Haines, N., and Irvine, K. D. (2003). Glycosylation regulates Notch signalling. Nat. Rev. Mol. Cell. Biol. 4, 786–797. doi:10.1038/nrm1228
Haltiwanger, R. S., Wells, L., Freeze, H. H., Jafar-Nejad, H., Okajima, T., and Stanley, P. (2022). “Other classes of eukaryotic glycans,” in Essentials of glycobiology. Editors Th, A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebiet al. ((NY): Cold Spring Harbor), 155–164.
Han, H., Tanigaki, K., Yamamoto, N., Kuroda, K., Yoshimoto, M., Nakahata, T., et al. (2002). Inducible gene knockout of transcription factor recombination signal binding protein‐J reveals its essential role in T versus B lineage decision. Int. Immunol. 14, 637–645. doi:10.1093/intimm/dxf030
Hicks, C., Johnston, S. H., Disibio, G., Collazo, A., Vogt, T. F., and Weinmaster, G. (2000). Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell. Biol. 2, 515–520. doi:10.1038/35019553
Hirano, K.-I., Hosokawa, H., Yahata, T., Ando, K., Tanaka, M., Imai, J., et al. (2022). Dll1 can function as a ligand of Notch1 and Notch2 in the thymic epithelium. Front. Immunol. 13, 852427. doi:10.3389/fimmu.2022.852427
Hori, K., Sen, A., and Artavanis-Tsakonas, S. (2013). Notch signaling at a glance. J. Cell. Sci. 126, 2135–2140. doi:10.1242/jcs.127308
Hozumi, K., Mailhos, C., Negishi, N., Hirano, K.-I., Yahata, T., Ando, K., et al. (2008). Delta-like 4 is indispensable in thymic environment specific for T cell development. J. Exp. Med. 205, 2507–2513. doi:10.1084/jem.20080134
Hozumi, K., Negishi, N., Suzuki, D., Abe, N., Sotomaru, Y., Tamaoki, N., et al. (2004). Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat. Immunol. 5, 638–644. doi:10.1038/ni1075
Immunological Genome Project (2020). ImmGen at 15. Nat. Immunol. 21, 700–703. doi:10.1038/s41590-020-0687-4
Kadur Lakshminarasimha Murthy, P., Srinivasan, T., Bochter, M. S., Xi, R., Varanko, A. K., Tung, K. L., et al. (2018). Radical and lunatic fringes modulate notch ligands to support mammalian intestinal homeostasis. Elife 7, e35710. doi:10.7554/eLife.35710
Kakuda, S., and Haltiwanger, R. S. (2017). Deciphering the fringe-mediated notch code: Identification of activating and inhibiting sites allowing discrimination between ligands. Dev. Cell. 40, 193–201. doi:10.1016/j.devcel.2016.12.013
Koch, U., Fiorini, E., Benedito, R., Besseyrias, V., Schuster-Gossler, K., Pierres, M., et al. (2008). Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J. Exp. Med. 205, 2515–2523. doi:10.1084/jem.20080829
Koch, U., Lacombe, T. A., Holland, D., Bowman, J. L., Cohen, B. L., Egan, S. E., et al. (2001). Subversion of the T/B lineage decision in the thymus by lunatic fringe-mediated inhibition of Notch-1. Immunity 15, 225–236. doi:10.1016/s1074-7613(01)00189-3
Kopan, R. (2012). Notch signaling. Cold Spring Harb. Perspect. Biol. 4, a011213. doi:10.1101/cshperspect.a011213
Kovall, R. A., Gebelein, B., Sprinzak, D., and Kopan, R. (2017). The canonical notch signaling pathway: Structural and biochemical insights into shape, sugar, and force. Dev. Cell. 41, 228–241. doi:10.1016/j.devcel.2017.04.001
Kumano, K., Chiba, S., Kunisato, A., Sata, M., Saito, T., Nakagami-Yamaguchi, E., et al. (2003). Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity 18, 699–711. doi:10.1016/s1074-7613(03)00117-1
Lakhan, R., and Rathinam, C. V. (2021). Deficiency of rbpj leads to defective stress-induced hematopoietic stem cell functions and hif mediated activation of non-canonical notch signaling pathways. Front. Cell. Dev. Biol. 8, 622190. doi:10.3389/fcell.2020.622190
Lampreia, F. P., Carmelo, J. G., and Anjos-Afonso, F. (2017). Notch signaling in the regulation of hematopoietic stem cell. Curr. Stem Cell. Rep. 3, 202–209. doi:10.1007/s40778-017-0090-8
Lechner, M., Engleitner, T., Babushku, T., Schmidt-Supprian, M., Rad, R., Strobl, L. J., et al. (2021). Notch2-mediated plasticity between marginal zone and follicular B cells. Nat. Commun. 12, 1111. doi:10.1038/s41467-021-21359-1
Lefeber, D. J., Freeze, H. H., Steet, R., and Kinoshita, T. (2022). “Congenital disorders of glycosylation,” in Essentials of glycobiology. Editors Th, A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebiet al. ((NY): Cold Spring Harbor), 599–614.
Li, M., Cheng, R., Liang, J., Yan, H., Zhang, H., Yang, L., et al. (2013). Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am. J. Hum. Genet. 92, 895–903. doi:10.1016/j.ajhg.2013.04.022
Luca, V. C., Jude, K. M., Pierce, N. W., Nachury, M. V., Fischer, S., and Garcia, K. C. (2015). Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science 347, 847–853. doi:10.1126/science.1261093
Luca, V. C., Kim, B. C., Ge, C., Kakuda, S., Wu, D., Roein-Peikar, M., et al. (2017). Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 355, 1320–1324. doi:10.1126/science.aaf9739
Ma, Z., Xu, J., Wu, L., Wang, J., Lin, Q., Chowdhury, F. A., et al. (2020). Hes1 deficiency causes hematopoietic stem cell exhaustion. Stem Cells 38, 756–768. doi:10.1002/stem.3169
Macdonald, H. R., Wilson, A., and Radtke, F. (2001). Notch1 and T-cell development: Insights from conditional knockout mice. Trends Immunol. 22, 155–160. doi:10.1016/s1471-4906(00)01828-7
Mackinnon, R. N., Selan, C., Wall, M., Baker, E., Nandurkar, H., and Campbell, L. J. (2010). The paradox of 20q11.21 amplification in a subset of cases of myeloid malignancy with chromosome 20 deletion. Genes. Chromosom. Cancer 49, 998–1013. doi:10.1002/gcc.20806
Mancini, S. P. J. C., Mantei, N., Dumortier, A., Suter, U., Macdonald, H. R., and Radtke, F. (2005). Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood 105, 2340–2342. doi:10.1182/blood-2004-08-3207
Matsumoto, K., Kumar, V., Varshney, S., Nairn, A. V., Ito, A., Pennarubia, F., et al. (2022). Fringe GlcNAc-transferases differentially extend O-fucose on endogenous NOTCH1 in mouse activated T cells. J. Biol. Chem. 298, 102064. doi:10.1016/j.jbc.2022.102064
Matsumoto, K., Luther, K. B., and Haltiwanger, R. S. (2021). Diseases related to Notch glycosylation. Mol. Asp. Med. 79, 100938. doi:10.1016/j.mam.2020.100938
Nguyen, A. V., and Soulika, A. M. (2019). The dynamics of the skin's immune system. Int. J. Mol. Sci. 20, 1811. doi:10.3390/ijms20081811
Ogawa, M., Sawaguchi, S., Kawai, T., Nadano, D., Matsuda, T., Yagi, H., et al. (2015). Impaired O-linked N-acetylglucosaminylation in the endoplasmic reticulum by mutated epidermal growth factor (EGF) domain-specific O-linked N-acetylglucosamine transferase found in Adams-Oliver syndrome. J. Biol. Chem. 290, 2137–2149. doi:10.1074/jbc.M114.598821
Ogawa, M., Tashima, Y., Sakaguchi, Y., Takeuchi, H., and Okajima, T. (2020). Contribution of extracellular O-GlcNAc to the stability of folded epidermal growth factor-like domains and Notch1 trafficking. Biochem. Biophys. Res. Commun. 526, 184–190. doi:10.1016/j.bbrc.2020.03.066
Okamura, Y., and Saga, Y. (2008). Pofut1 is required for the proper localization of the Notch receptor during mouse development. Mech. Dev. 125, 663–673. doi:10.1016/j.mod.2008.04.007
Pajcini, K. V., Speck, N. A., and Pear, W. S. (2011). Notch signaling in mammalian hematopoietic stem cells. Leukemia 25, 1525–1532. doi:10.1038/leu.2011.127
Porcheri, C., Golan, O., Calero-Nieto, F. J., Thambyrajah, R., Ruiz-Herguido, C., Wang, X., et al. (2020). Notch ligand Dll4 impairs cell recruitment to aortic clusters and limits blood stem cell generation. EMBO J. 39, e104270. doi:10.15252/embj.2019104270
Poulos, M. G., Guo, P., Kofler, N. M., Pinho, S., Gutkin, M. C., Tikhonova, A., et al. (2013). Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell. Rep. 4, 1022–1034. doi:10.1016/j.celrep.2013.07.048
Radtke, F., Wilson, A., Stark, G., Bauer, M., Van Meerwijk, J., Macdonald, H. R., et al. (1999). Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10, 547–558. doi:10.1016/s1074-7613(00)80054-0
Rampal, R., Luther, K. B., and Haltiwanger, R. S. (2007). Notch signaling in normal and disease States: Possible therapies related to glycosylation. Curr. Mol. Med. 7, 427–445. doi:10.2174/156652407780831593
Romero-Wolf, M., Shin, B., Zhou, W., Koizumi, M., Rothenberg, E. V., and Hosokawa, H. (2020). Notch2 complements Notch1 to mediate inductive signaling that initiates early T cell development. J. Cell. Biol. 219, e202005093. doi:10.1083/jcb.202005093
Saiki, W., Ma, C., Okajima, T., and Takeuchi, H. (2021). Current views on the roles of O-glycosylation in controlling notch-ligand interactions. Biomolecules 11, 309. doi:10.3390/biom11020309
Saito, T., Chiba, S., Ichikawa, M., Kunisato, A., Asai, T., Shimizu, K., et al. (2003). Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 18, 675–685. doi:10.1016/s1074-7613(03)00111-0
Sawaguchi, S., Varshney, S., Ogawa, M., Sakaidani, Y., Yagi, H., Takeshita, K., et al. (2017). O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals. eLife 6, e24419. doi:10.7554/eLife.24419
Schneider, M., Kumar, V., Nordstrom, L. U., Feng, L., Takeuchi, H., Hao, H., et al. (2018). Inhibition of Delta-induced Notch signaling using fucose analogs. Nat. Chem. Biol. 14, 65–71. doi:10.1038/nchembio.2520
Schröder, K. C., Duman, D., Tekin, M., Schanze, D., Sukalo, M., Meester, J., et al. (2019). Adams-Oliver syndrome caused by mutations of the EOGT gene. Am. J. Med. Genet. A 179, 2246–2251. doi:10.1002/ajmg.a.61313
Seib, E., and Klein, T. (2021). The role of ligand endocytosis in notch signalling. Biol. Cell. 113, 401–418. doi:10.1111/boc.202100009
Sheng, Y., Yahata, T., Negishi, N., Nakano, Y., Habu, S., Hozumi, K., et al. (2008). Expression of Delta-like 1 in the splenic non-hematopoietic cells is essential for marginal zone B cell development. Immunol. Lett. 121, 33–37. doi:10.1016/j.imlet.2008.08.001
Shi, S., Ge, C., Luo, Y., Hou, X., Haltiwanger, R. S., and Stanley, P. (2007). The threonine that carries fucose, but not fucose, is required for Cripto to facilitate Nodal signaling. J. Biol. Chem. 282, 20133–20141. doi:10.1074/jbc.M702593200
Shi, S., and Stanley, P. (2003). Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc. Natl. Acad. Sci. U. S. A. 100, 5234–5239. doi:10.1073/pnas.0831126100
Siebel, C., and Lendahl, U. (2017). Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294. doi:10.1152/physrev.00005.2017
Song, Y., Kumar, V., Wei, H.-X., Qiu, J., and Stanley, P. (2016). Lunatic, manic, and radical fringe each promote T and B cell development. J. Immunol. 196, 232–243. doi:10.4049/jimmunol.1402421
Sosicka, P., Ng, B. G., and Freeze, H. H. (2021). Chemical therapies for congenital disorders of glycosylation. ACS Chem. Biol. 10, acschembio.1c00601. doi:10.1021/acschembio.1c00601
Souilhol, C., Lendinez, J. G., Rybtsov, S., Murphy, F., Wilson, H., Hills, D., et al. (2016). Developing HSCs become Notch independent by the end of maturation in the AGM region. Blood 128, 1567–1577. doi:10.1182/blood-2016-03-708164
Sparrow, D. B., Chapman, G., Wouters, M. A., Whittock, N. V., Ellard, S., Fatkin, D., et al. (2006). Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet. 78, 28–37. doi:10.1086/498879
Takeuchi, H., Schneider, M., Williamson, D. B., Ito, A., Takeuchi, M., Handford, P. A., et al. (2018a). Two novel protein O-glucosyltransferases that modify sites distinct from POGLUT1 and affect Notch trafficking and signaling. Proc. Natl. Acad. Sci. U. S. A. 115, E8395–E8402. doi:10.1073/pnas.1804005115
Takeuchi, H., Wong, D., Schneider, M., Freeze, H. H., Takeuchi, M., Berardinelli, S. J., et al. (2018b). Variant in human POFUT1 reduces enzymatic activity and likely causes a recessive microcephaly, global developmental delay with cardiac and vascular features. Glycobiology 28, 276–283. doi:10.1093/glycob/cwy014
Tan, J. B., Xu, K., Cretegny, K., Visan, I., Yuan, J. S., Egan, S. E., et al. (2009). Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity 30, 254–263. doi:10.1016/j.immuni.2008.12.016
Tanwar, A., and Stanley, P. (2022). Synergistic regulation of notch signaling by different O-glycans promotes hematopoiesis. bioRxiv. doi:10.1101/2022.1103.1121.485194
Tsaouli, G., Barbarulo, A., Vacca, A., Screpanti, I., and Felli, M. P. (2020). Molecular mechanisms of notch signaling in lymphoid cell lineages development: NF-κB and beyond. Adv. Exp. Med. Biol. 1227, 145–164. doi:10.1007/978-3-030-36422-9_10
Tsukamoto, Y., Ogawa, M., Yogi, K., Tashima, Y., Takeuchi, H., and Okajima, T. (2022). Glycoproteomics of NOTCH1 EGF repeat fragments overexpressed with different glycosyltransferases in HEK293T cells reveals insights into O-GlcNAcylation of NOTCH1. Glycobiology 32, 616–628. doi:10.1093/glycob/cwac015
Tsukumo, S., Hirose, K., Maekawa, Y., Kishihara, K., and Yasutomo, K. (2006). Lunatic fringe controls T cell differentiation through modulating notch signaling. J. Immunol. 177, 8365–8371. doi:10.4049/jimmunol.177.12.8365
Urata, Y., Saiki, W., Tsukamoto, Y., Sago, H., Hibi, H., Okajima, T., et al. (2020). Xylosyl extension of O-glucose glycans on the extracellular domain of NOTCH1 and NOTCH2 regulates notch cell surface trafficking. Cells 9, 1220. doi:10.3390/cells9051220
Varshney, S., and Stanley, P. (2018). Multiple roles for O‐glycans in Notch signalling. FEBS Lett. 592, 3819–3834. doi:10.1002/1873-3468.13251
Varshney, S., Wei, H.-X., Batista, F., Nauman, M., Sundaram, S., Siminovitch, K., et al. (2019). A modifier in the 129S2/SvPasCrl genome is responsible for the viability of Notch1[12f/12f] mice. BMC Dev. Biol. 19, 19. doi:10.1186/s12861-019-0199-3
Visan, I., Tan, J. B., Yuan, J. S., Harper, J. A., Koch, U., and Guidos, C. J. (2006). Regulation of T lymphopoiesis by Notch1 and Lunatic fringe-mediated competition for intrathymic niches. Nat. Immunol. 7, 634–643. doi:10.1038/ni1345
Visan, I., Yuan, J. S., Liu, Y., Stanley, P., and Guidos, C. J. (2010). Lunatic fringe enhances competition for delta-like Notch ligands but does not overcome defective pre-TCR signaling during thymocyte beta-selection in vivo. J. Immunol. 185, 4609–4617. doi:10.4049/jimmunol.1002008
Wendorff, A. A., Koch, U., Wunderlich, F. T., Wirth, S., Dubey, C., Bruning, J. C., et al. (2010). Hes1 is a critical but context-dependent mediator of canonical Notch signaling in lymphocyte development and transformation. Immunity 33, 671–684. doi:10.1016/j.immuni.2010.11.014
Weng, A. P., Ferrando, A. A., Lee, W., Morris, J. P. T., Silverman, L. B., Sanchez-Irizarry, C., et al. (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271. doi:10.1126/science.1102160
Wilson, A., Macdonald, H. R., and Radtke, F. (2001). Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J. Exp. Med. 194, 1003–1012. doi:10.1084/jem.194.7.1003
Yan, Q., Yao, D., Wei, L. L., Huang, Y., Myers, J., Zhang, L., et al. (2010). O-fucose modulates Notch-controlled blood lineage commitment. Am. J. Pathol. 176, 2921–2934. doi:10.2353/ajpath.2010.090702
Yao, D., Huang, Y., Huang, X., Wang, W., Yan, Q., Wei, L., et al. (2011). Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood 117, 5652–5662. doi:10.1182/blood-2010-12-326074
Yu, H., and Takeuchi, H. (2019). Protein O-glucosylation: Another essential role of glucose in biology. Curr. Opin. Struct. Biol. 56, 64–71. doi:10.1016/j.sbi.2018.12.001
Yu, V. W. C., Saez, B., Cook, C., Lotinun, S., Pardo-Saganta, A., Wang, Y.-H., et al. (2015). Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J. Exp. Med. 212, 759–774. doi:10.1084/jem.20141843
Yuan, J. S., Tan, J. B., Visan, I., Matei, I. R., Urbanellis, P., Xu, K., et al. (2011). Lunatic Fringe prolongs Delta/Notch-induced self-renewal of committed αβ T-cell progenitors. Blood 117, 1184–1195. doi:10.1182/blood-2010-07-296616
Zhang, N., and Gridley, T. (1998). Defects in somite formation in lunatic fringe-deficient mice. Nature 394, 374–377. doi:10.1038/28625
Zhou, B., Lin, W., Long, Y., Yang, Y., Zhang, H., Wu, K., et al. (2022). Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 7, 95. doi:10.1038/s41392-022-00934-y
Keywords: Notch signaling, O-fucose, O-GlcNAc, Fringe, HSCs, lymphoid, myeloid
Citation: Stanley P and Tanwar A (2022) Regulation of myeloid and lymphoid cell development by O-glycans on Notch. Front. Mol. Biosci. 9:979724. doi: 10.3389/fmolb.2022.979724
Received: 27 June 2022; Accepted: 13 October 2022;
Published: 04 November 2022.
Edited by:
Kayvon Pedram, Janelia Research Campus, United StatesReviewed by:
Saran Kumar, Indian Institute of Technology Delhi, IndiaLeonhard Möckl, Max Planck Institute for the Science of Light, Germany
Copyright © 2022 Stanley and Tanwar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela Stanley, cGFtZWxhLnN0YW5sZXlAZWluc3RlaW5tZWQuZWR1