 Yongkai Hong
Yongkai Hong Dantian Chen
Dantian Chen Yaqing Jin
Yaqing Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Mol. Biosci. , 18 October 2022
Sec. Biological Modeling and Simulation
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.971768
This article is part of the Research Topic Revolutionizing Life Sciences: The Nobel Leap in Artificial Intelligence-Driven Biomodeling View all 16 articles
Drug combinations can increase the therapeutic effect by reducing the level of toxicity and the occurrence of drug resistance. Therefore, several drug combinations are often used in the management of complex diseases. However, due to the exponential growth in drug development, it would be impractical to evaluate all combinations through experiments. In view of this, we developed Pathway Interaction Network (PINet) biological model to estimate the optimal drug combinations for various diseases. The random walk with restart (RWR) algorithm was used to capture the “disease state” and “drug state,” while PINet was used to evaluate the optimal drug combinations and the high-order drug combination1. The model achieved a mean area under the curve of a receiver operating characteristic curve of 0.885. In addition, for some diseases, PINet predicted the optimal drug combination. For example, in the case of acute myeloid leukemia, PINet correctly predicted midostaurin and gemtuzumab as effective drug combinations, as demonstrated by the results of a Phase-I clinical trial. Moreover, PINet also correctly predicted the potential drug combinations for diseases that lacked a training dataset that could not be predicted using standard machine learning models.
Compared with the “one disease, one gene” drug paradigm, drug combinations can more effectively cope with multifactorial diseases such as infections, cardiovascular diseases, and tumors (Bayat Mokhtari et al., 2017) (Huffman et al., 2017). Drug combinations can also delay the development of drug resistance and are often used in the treatment of acquired immunodeficiency syndrome (AIDS) and multi-drug resistant bacteria (Liu et al., 2021) (Cihlar and Fordyce, 2016). Network or multi-pharmacology involves the combinations of several drugs used for different targets to create a synergistic effect that can perturb the biological networks and thus increase the clinical benefits (Jia et al., 2009).
The development of optimal drug combinations typically involves three stages: the intuition phase, the clinical trials phase, and the biological data mining phase. However, since the development of the current drug combination is based on the researchers’ intuition and expertise, the process is often inefficient. As a result, it is now gradually being replaced by the high-throughput screening method (Shinn et al., 2019). Nevertheless, as the number of approved drugs increases, the number of drug combinations requiring high-throughput screening verification has increased exponentially, eventually leading to a significant prolongation of the verification process and research costs. Machine learning and deep learning, which can mine the correlation between massive amounts of biological data, are increasingly being used in the discovery of effective drug combinations (Shi et al., 2018) (Li et al., 2020) (Kim et al., 2021) (Zagidullin et al., 2021). Since machine learning depends on training datasets, it is mostly used for tumors. However, for diseases that lack training datasets, the model is difficult to optimize because it is not possible to fit the parameters into the model. In addition, the results provided by the machine learning algorithms are often difficult to explain, and therefore clinicians find it difficult to apply the machine learning solution in clinical practice. An alternative approach to the data-driven machine learning method is to use theory-driven methods based on the knowledge of biological systems and networks (Wang et al., 2021) (Jafari et al., 2022). Compared with data-driven methods, theory-driven methods are more explanatory, and their performance is not affected by the quality of the training dataset. The limitation of theory-driven methods is that they rely on the accurate generation of a theoretical hypothesis.
(Yang et al., 2008) define two network biological states: the disease and normal states. According to (Yang et al., 2008), the transition from the disease state to the normal state is achieved through the perturbation of specific target combinations within the arachidonic acid network (a kind of inflammation-related network). This approach has several limitations. First of all, there is a lack of uniform standards to define the disease and normal states. Therefore, the definition of these states often requires the subjective input of expert professionals. In addition, not all disease targets have corresponding drugs available, and more than one pathway may be involved in the development of a specific disease (Geva-Zatorsky et al., 2010). found that the protein responses to drug combinations can be accurately described by a linear superposition (weighted sum) of each protein’s response to each specific individual drug. Based on this finding (Lee et al., 2012), made use of gene set enrichment analysis to convert the gene expression profile of specific cancers (non-small cell lung cancer and triple-negative breast cancer) into related signaling pathways. The data about the linear drug superposition combinations was combined with the disease pathways data to obtain the optimal drug combination. Through this method (Lee et al., 2012), found two combination drug pairs with a synergistic effect on lung cancer cells. However, this method still has a number of shortcomings since it ignores the relationship between pathways. Moreover, the theory of linear superposition does not fit all kinds of protein. Because drugs acting on the same pathway through different targets or drugs regulating a relatively small number of highly-connected pathways are more likely to produce synergistic effects (Chen et al., 2016), proposed a “pathway to pathway interaction” network model to predict the therapeutic effect of synergistic drug combinations. This model resulted in an area under the curve (AUC) of a receiver operating characteristic curve of 0.75. The method proposed by (Chen et al., 2016) still has some shortcomings. This method ignores the disease condition, and only the pathway associations of gene overlap are retained, while the pathway associations of protein interactions and function associations are discarded. In addition, the drug combinations are evaluated based on the shortest path without considering the global topology features2. Therefore (Cheng et al., 2019) quantifyied the network-based relationship between drug targets and the diseased human protein to protein interaction. Although this method revealed the existence of six distinct potiential drug combinations, only one of these six drug combinations correlated with therapeutic effects. Eventually, a beneficial therapeutic effect was noted when the drug targets hit the same disease module located in separate neighborhoods. Still, the application of this model is limited as it ignores the pathway information and uses the shortest path to evaluate the optimal drug combinations without considering the global topology features.
In view of this, we constructed a Pathway Interaction Network (PINet) model to overcome the limitations of the models described in previous studies (Table 1). This new model abstracts the human body as a two-layer network containing gene and pathway information and describes the influence of a disease or drug on the human as a probability distribution in the network, which is called “disease state” and “drug state.” In addition, it predicts the optimal drug combinations by combining “disease state” and “drug state”.

TABLE 1. Optimization of previous research.
The main advantage of the PINet model over the other models is that it can evaluate 5-drug combinations, while most models can only evaluate 2-drug combinations. In addition, PINet is also sensitive to various diseases.
PINet is composed of four types of entities and eight types of relationships3: The four types of entities include pathways, genes, drugs and diseases, while the eight types of interactions include pathway to pathway, pathway to gene, gene to gene, drug to gene, disease to gene, disease to pathway, drug to disease and drug to pathway. Except for drug to disease, other data come from databases (Table 2). The specific data cleaning and processing methods are described in the Supplementary Material S1.1; Supplementary Material S1.2.

TABLE 2. Data source.
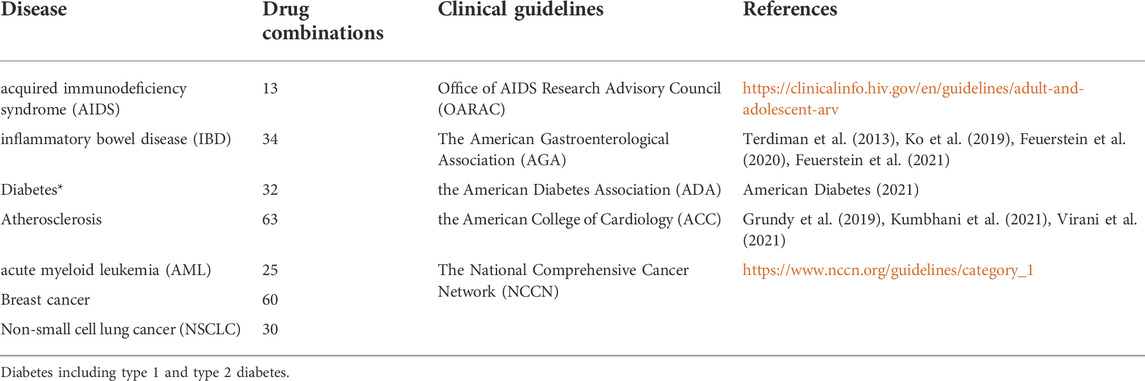
TABLE 3. Disease-specific drug combinations.
Databases include KEGG (Kanehisa et al., 2021), STRING (Szklarczyk et al., 2021), DrugBank (Wishart et al., 2018), BindingDB (Gilson et al., 2016), CTD (Davis et al., 2021) and HVIDB (Yang et al., 2021).
The theoretical basis of the model was built based on the findings of four studies (Yang et al., 2008). Showed that perturbing the targets can shift the disease state to the normal state. Based on this study, we introduce the probability distribution of different drugs or diseases in the network as drug states or disease states and the higher degree of overlap between the drug state and the disease state, the better the efficacy of the drug. Chen et al. (2016). showed that the effect of a disease or drug on the body is achieved through the manipulation of genetic pathways. Therefore, our model included information on the genes and pathways. We also made use of the work of Geva-Zatorsky et al. (2010), which simplified the drug combinations as a linear summation of drug targets. The targets of drug A within our model were denoted as (a1, a2, and a3), and the targets of drug B were denoted as (b1, b2). Based on the study of (Geva-Zatorsky et al., 2010), the drug state of the combination of drugs A and B was deemed to be equivalent to the drug state of the virtual drug V, of which targets are (a1, a2, a3, b1, b2). Finally, to narrow down the scope of potential drug combinations and reduce the computational power costs, we used the research of Cheng et al. (2019), which demonstrated that drug synergy is more likely to occur when the drugs act on different disease targets at the same time.
PINet consists of seven networks4 (pathway to pathway, gene to gene, pathway to gene, drug to gene, drug to pathway, disease to pathway, disease to gene), each stored in an adjacency matrix (Figure 1). The main part of the PINet model was based on the restart random walks (RWR) algorithm built on the pathway to pathway, gene to gene, and pathway to gene networks. Further details about the model constructions are provided in the Supplementary Material S1.3.
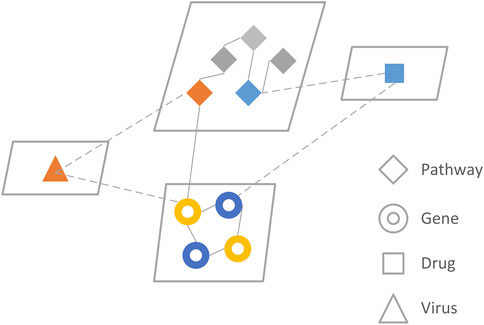
FIGURE 1. The network model of PINet. PINet model consists of four entities and seven relationships. The genes and pathways were directly related to RWR, and the drugs and viruses were integrated with the RWR algorithm through indirect connections.
The effect of a drug or disease on the body can be represented by a vector that contains both pathway and genetic information, which is called a drug state or disease state. These two states were obtained by selecting specific initial nodes on the model to perform the RWR, and the stable probability distribution was defined as the drug or disease state. The specific state capture is described in more detail in the Supplementary Material S2.
Biological systems can be simplified into heterogeneous networks, and the RWR algorithm is widely used in the analysis of heterogeneous networks (Cho et al., 2016) (Luo et al., 2017). The RWR algorithm was developed by determining the initial probability, the transition matrix, and the stable probability distribution threshold as follows. More detail about the RWR algorithm is available in the Supplementary Material S2.
The initial nodes were composed of disease or drug-related genes and pathways. The initial probability in a specific network was composed of the initial gene to gene and pathway to pathway networks and can be calculated according to a specific node. For example, in the case of influenza, the initial gene was associated with influenza, and the initial pathway path: hsa05164 was identified from the KEGG database and was fixed to 1. On the other hand, for a drug, the original gene was considered as the drug target, the initial pathway was identified through pathway enrichment analysis, and the number of potential initial pathways was not fixed.
The initial probability of the pathway to pathway network a0 was formed so that equal probabilities were assigned to the initial nodes in the pathway to pathway network, and the sum of the nodes’ probabilities was equal to 1. Therefore if the probabilities of non-initial nodes are 0, then the initial probability of the gene to gene network b0 is the same. This relationship is summarized by the equation.
Whereby a0 is the pathway initial probability, and b0 is the gene initial probability. Both a0 and b0 are vectors.
The transition matrix describes the transition characteristics of all nodes within the network model. There are four transfer modes in PINet: pathway to pathway, pathway to gene, gene to gene, and gene to the pathway. Each transfer mode requires a transition matrix. The description of the PINet transition node requires a large transition matrix M composed of four small transition matrices Mi.
The (t) th probability distribution was obtained by mapping the (t-1) th probability distribution through the transition matrix as follows:
Whereby M1 is the pathway to pathway, M2 is the gene to pathway, M3 is the pathway to gene, and M4 is the gene to gene. r is the restart probability which is generally equal to 0.5.
The initial node was selected to perform the RWR. As the number of iterations increased, the probability distribution gradually became stable. When the difference in the probability distribution between the (n)th and the (n+1)th was less than the given threshold, the (n)th probability distribution was considered to be a stable probability distribution, and the threshold was generally set to 10–10.
The disease state was then captured through the identification of the initial nodes of the disease in the pathway to pathway network and, subsequently, the gene-gene network. The initial probability p0 of the disease was constructed, and then RWR was performed until the probability distribution became stable. The stable probability of the disease site pn was then captured for the disease state.
The drug state was captured through the identification of the virtual drug corresponding to the drug combination. The initial probability p0 of the drug was determined according to the target and enrichment pathway of the virtual drug. Finally, RWR was performed until the probability distribution became stable, and the stable probability pn was captured for the drug state.
Since the drug combinations have certain indications, we evaluated the drug combinations under specific disease conditions by “drug state” and “disease state.” The same drug combinations have different scores on different disease conditions in PINet. The absolute drug score value was obtained by calculating the difference between the “drug state” and the “disease state”.
Sdi is the disease state, Sdr is the drug state.
A lower score indicates a higher likelihood of a synergistic drug combination. Further details on the calculation of the drug combination score can be found in Supplementary Material S3.
During the development of PINet, it was assumed that the drug combination contained two types of information: the drug composition and the indication. Therefore two tests were performed to evaluate the sensitivity of PINet to detect disease and drug quantity. The disease sensitivity analysis assessed whether PINet can correctly identify the indications for the different drug combinations. For example, whether PINet will wrongly judge a drug designed to treat AIDS as a drug used to treat cancer. The drug quantity sensitivity analysis evaluated the ability of PINet to identify the n-drugs combination (n = 2, 3, 4, and 5).
The drug combination highlighted in the clinical guidelines of each disease was regarded as the positive gold standard treatment. The clinical indications of the drug combinations used to manage a specific disease were then modified to represent a negative example, i.e., another disease. All positive and negative examples were entered into the PINet for scoring, and the AUC under the ROC was calculated for each example. An AUC below 0.5 indicates that the PINet model was not sensitive enough to detect the disease and corresponding drug combinations, and these were therefore excluded from the model. The remaining diseases and drug combinations in the clinical guidelines were evaluated again in the next step.
The drug combinations may include four possible options with 2, 3, 4, or 5 drugs. The sensitivity of PINet to different drug combinations was calculated as follows. First, the drug combination in the clinical guidelines was used as a positive example, and the randomly generated drug combination was used as a negative example. Subsequently, the drug status and disease status were calculated according to the drug composition and indications, respectively, as explained in Section 3.3. Then, the score for each drug combination was calculated, as explained in Section 3.4. Finally, based on the calculated score, the AUC was calculated for each drug combination.
Outliers of disease state are identified by Quartile, and these outliers are key genes and key pathways of the disease. The potential drugs were selected if the target of the drug had an intersection with the key gene of the disease and the enriched pathway of the drug had an intersection with the key pathway of the disease. We assumed that for N potential drugs, there are
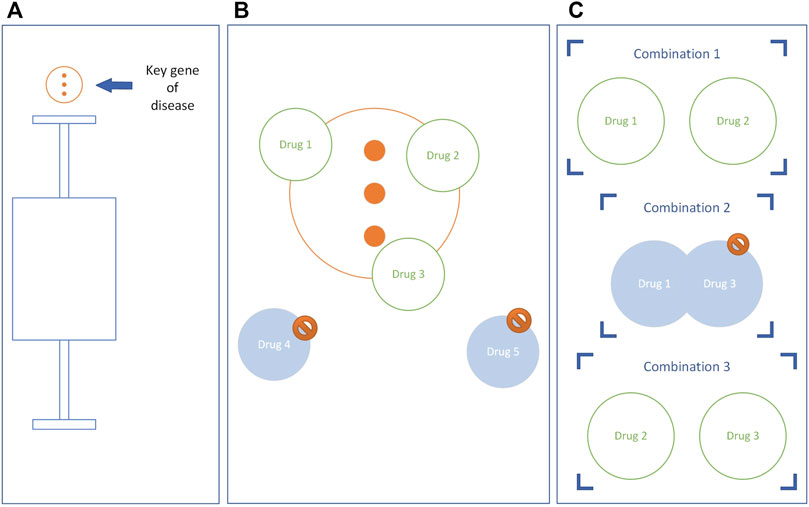
FIGURE 2. The construction of the potential drug combinations. Taking the key genes of diseases as an example, the key pathways are the same. (A) Genes above the upper limit are key genes. (B) Eliminate drugs that do not have intersections with key disease genes. (C) A drug combination is constructed, and if the drugs in the combination have the same target, the combination is eliminated.
The drug combinations with overlapping drug targets were removed from the primary potential drug combination to obtain the secondary potential drug combination (Figure 2C).
To improve the prediction accuracy of the model, we used the score corresponding to the false positive rate of 10% on the ROC of the “Drug quantity sensitivity” as the threshold. The scores of the secondary potential drug combinations were calculated, and those below the threshold were classified as synergistic drug combinations.
The PINet had a high sensitivity for NSCLC, AML, breast cancer, and IBD and low sensitivity for diabetes type 1, diabetes type 2, AIDS, and atherosclerosis (Figure 3).

FIGURE 3. Disease sensitivity of PINet.
Figure 4 illustrates the drug quantity sensitivity after excluding the diseases with a low PINet sensitivity. The sensitivity of PINet increased as the order of drug combinations increased. PINet also achieved good results in the identification of high-order drug combinations. However, since the sample was too small (2 positive cases and 58 negative cases in the fifth-order drug combination), the ROC may not be accurate.
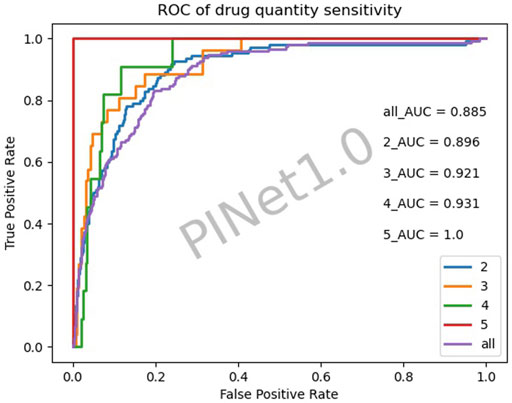
FIGURE 4. Drug quantity sensitivity of PINet.
Since PINet had the highest sensitivity for predicting AML, we decided to use PINet to predict the optimal drug combinations for this disease. PINet was first used to identify the key genes and pathways of AML. Subsequently, the drugs based on these genes and pathways were identified and used to construct the primary drug combinations. This revealed a total of 26,106 possible primary drug combinations. The drug combinations with the same target were eliminated, and the remaining drug combinations (n = 17,713) were scored to identify the optimal drug combinations (n = 2,590). After excluding the unapproved drugs, 1,221 possible drug combinations were identified. The efficacy of two of the drug combinations identified by PINet has been validated in clinical trials or in vivo studies. Röllig et al. (2021) demonstrated the synergy between gemtuzumab ozogamicin and midostaurin in newly diagnosed AML in a phase-I clinical trial. Tian et al. (2018) found that Emricasan and Ponatinib can synergistically reduce ischemia-reperfusion injury in rat brains.
As the development of new drugs continues to increase, there is a need to develop novel methods to identify optimal drug combinations for managing specific diseases. In this study, we proposed a novel model PINet to make it easier for clinicians to identify optimal drug combinations. When compared with other machine learning models, PINet has several advantages and limitations.
PINet is a theory-driven method for evaluating drug combinations based on the assumption that “drugs can correct disease states.” A low PINet score means that the drug combination is more applicable to a specific disease. This simple scoring system used in PINet is easily understood by researchers in the non-data science fields, making PINet easy to generalize.
Unlike machine learning, there is no need to fit all parameters in PINet, and therefore, PINet does not require a training dataset. This is crucial for drug combination prediction for some diseases that lack a training dataset.
Most drug combination prediction models focus on 2-drug combinations since high-order drug combinations are computationally expensive to calculate. PINet takes the same time to evaluate 2-drug combinations as higher-order drug combinations by narrowing the range of candidate drugs based on theory to maintain the computational power consumption within an acceptable range.
A variety of diseases are already included in PINet, and the model’s effectiveness in predicting optimal drug combinations in breast cancer, IBD, AML, and NSCL has already been verified. With the advancement of disease pathway research in KEGG, the applicability of PINet will be extended to more diseases.
The sensitivity of PINet in some diseases, such as AIDS and diabetes, was found to be low in our study. A possible explanation for this could be that the effect of these diseases on genes is expressed as either an up-regulation or down-regulation gene expression. However, PINet simplifies the relationship between diseases and genes to 0 or 1, resulting in the loss of information. Furthermore, most anti-infective drugs target pathogens, and the targets of these drugs do not have corresponding genes in KEGG.
The drug-to-target relationship was simplified to 0 or 1, and the antagonist effects of drug combinations were not considered when assessing the drug sensitivity on PINet. This means that PINet cannot distinguish between synergy and antagonism. Although we avoided competitive antagonism by narrowing down the drug candidates, this does not solve the problem on a theoretical level.
The validation of PINet is not sufficient for the following reasons: Various theoretical models are suitable for different diseases, and there are certain differences in the range of drugs that can be selected, so it is difficult to make an objective comparison (Table 4). In fact, the drug combinations in PINet 1.0 are all derived from clinical guidelines, and many of these drugs lack transcriptome data and cannot be evaluated by the method of (Lee et al., 2012). There are differences between other methods (Cheng et al., 2019) (Chen et al., 2016) (Yang et al., 2008) and PINet1.0 in the indication, which makes it impossible to compare. On the other hand, due to a lack of experimental conditions, it was not possible to validate the accuracy of the PINet predictions.

TABLE 4. Comparison of different models.
Several aspects can be improved on PINet to increase its prediction accuracy and applicability.
In PINet, we evaluate drug combinations by comparing disease states and drug states, considering both synergy and indications of the drug combination together. First, we found that PINet has moderate disease sensitivity but can accurately distinguish synergistic drug combinations from random drug combinations, during the evaluation of the model. In addition, the combination of drugs predicted to treat AML is suitable for ischemia-reperfusion injury, which may be related to the multi-targets phenomenon of drugs and multi-phenotypes phenomenon of diseases (Tian et al., 2018). Furthermore, synergy was identified by relying only on the shortest path in the pathway network without disease information (Chen et al., 2016). Based on the above facts, we suggest that synergy and indication should be two relatively independent attributes of a drug combination and these attributes are relatively independent and may provide a new theoretical basis for the development of a repository for the rapid identification of drug combinations. If the conjecture is correct, PINet could be used in the future to evaluate drug combinations independently of the disease state, eventually increasing the scope of application of the model. As a result, the indications can be isolated and analyzed separately in finer divisions according to the drug function (e.g., anti-inflammatory, or anti-viral) rather than the entire disease.
We plan to elucidate the synergistic effect of drug combinations through information theory. This will enable us to locate key pathways and key genes to define the indications of drug combinations and verify whether the conjecture is correct.
The relationship between diseases and genes can be optimized as −1, 0, and one to achieve differentiation of different diseases, thereby improving the disease sensitivity of PINet.
The drug-to-target relationship can also be optimized to −1, 0, and one to simulate the antagonistic relationship between drugs. In follow-up studies, we will additionally evaluate the ability of PINet to identify antagonistic drug combinations. Chen et al., 2012, Hopkins, 2008, Hsieh et al., 2021, Zhang et al., 2021.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
YH was responsible for the theoretical design and code reproduction. DC took part in data cleaning and model building. MZ was involved in the theoretical design, YJ performed the data collection, and YZ performed the feasibility analysis. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.971768/full#supplementary-material
1Combinations of three or more drugs.
2The regulatory distance of upstream targets to downstream targets may exceed the shortest path (usually 3).
3Relationship between drugs is predicted by the model. So it does not appear in the model. We assume that the patient has only one disease, so the relationship between diseases does not exist in the model.
4The drug-disease relationship is the data used to evaluate the model. So it doesn't appear in the model.
A D A, (2021). 9. Pharmacologic approaches to glycemic treatment: Standards of medical care in diabetes-2021. Diabetes Care 44 (1), S111–S124. doi:10.2337/dc21-S009
Bayat Mokhtari, R., Homayouni, T. S., Baluch, N., Morgatskaya, E., Kumar, S., Das, B., et al. (2017). Combination therapy in combating cancer. Oncotarget 8 (23), 38022–38043. doi:10.18632/oncotarget.16723
Chen, D., Zhang, H., Lu, P., Liu, X., and Cao, H. (2016). Synergy evaluation by a pathway-pathway interaction network: A new way to predict drug combination. Mol. Biosyst. 12 (2), 614–623. doi:10.1039/c5mb00599j
Chen, X., Liu, M.-X., and Yan, G.-Y. (2012). Drug–target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst. 8 (7), 1970–1978. doi:10.1039/c2mb00002d
Cheng, F., Kovacs, I. A., and Barabasi, A. L. (2019). Network-based prediction of drug combinations. Nat. Commun. 10 (1), 1197. doi:10.1038/s41467-019-09186-x
Cho, H., Berger, B., and Peng, J. (2016). Compact integration of multi-network topology for functional analysis of genes. Cell Syst. 3 (6), 540–548. e545. doi:10.1016/j.cels.2016.10.017
Cihlar, T., and Fordyce, M. (2016). Current status and prospects of HIV treatment. Curr. Opin. Virol. 18, 50–56. doi:10.1016/j.coviro.2016.03.004
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D., Wiegers, J., Wiegers, T. C., et al. (2021). Comparative toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 49 (D1), D1138–D1143. doi:10.1093/nar/gkaa891
Feuerstein, J. D., Ho, E. Y., Shmidt, E., Singh, H., Falck-Ytter, Y., Sultan, S., et al. (2021). AGA clinical practice guidelines on the medical management of moderate to severe luminal and perianal fistulizing crohn’s disease. Gastroenterology 160 (7), 2496–2508. doi:10.1053/j.gastro.2021.04.022
Feuerstein, J. D., Isaacs, K. L., Schneider, Y., Siddique, S. M., Falck-Ytter, Y., Singh, S., et al. (2020). AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology 158 (5), 1450–1461. doi:10.1053/j.gastro.2020.01.006
Geva-Zatorsky, N., Dekel, E., Cohen, A. A., Danon, T., Cohen, L., and Alon, U. (2010). Protein dynamics in drug combinations: A linear superposition of individual-drug responses. Cell 140 (5), 643–651. doi:10.1016/j.cell.2010.02.011
Gilson, M. K., Liu, T., Baitaluk, M., Nicola, G., Hwang, L., and Chong, J. (2016). BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44 (D1), D1045–D1053. doi:10.1093/nar/gkv1072
Grundy, S. M., Stone, N. J., Bailey, A. L., Beam, C., Birtcher, K. K., Blumenthal, R. S., et al. (2019). 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American college of cardiology/American heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 73 (24), e285–e350. doi:10.1016/j.jacc.2018.11.003
Hopkins, A. L. (2008). Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 4 (11), 682–690. doi:10.1038/nchembio.118
Hsieh, K., Wang, Y., Chen, L., Zhao, Z., Savitz, S., Jiang, X., et al. (2021). Drug repurposing for COVID-19 using graph neural network and harmonizing multiple evidence. Sci. Rep. 11 (1). doi:10.1038/s41598-021-02353-5
Huffman, M. D., Xavier, D., and Perel, P. (2017). Uses of polypills for cardiovascular disease and evidence to date. Lancet 389 (10073), 1055–1065. doi:10.1016/s0140-6736(17)30553-6
Jafari, M., Mirzaie, M., Bao, J., Barneh, F., Zheng, S., Eriksson, J., et al. (2022). Bipartite network models to design combination therapies in acute myeloid leukaemia. Nat. Commun. 13 (1), 2128. doi:10.1038/s41467-022-29793-5
Jia, J., Zhu, F., Ma, X., Cao, Z., Cao, Z. W., Li, Y., et al. (2009). Mechanisms of drug combinations: Interaction and network perspectives. Nat. Rev. Drug Discov. 8 (2), 111–128. doi:10.1038/nrd2683
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., and Tanabe, M. (2021). Kegg: Integrating viruses and cellular organisms. Nucleic Acids Res. 49 (D1), D545–D551. doi:10.1093/nar/gkaa970
Kim, Y., Zheng, S., Tang, J., Jim Zheng, W., Li, Z., and Jiang, X. (2021). Anticancer drug synergy prediction in understudied tissues using transfer learning. J. Am. Med. Inf. Assoc. 28 (1), 42–51. doi:10.1093/jamia/ocaa212
Ko, C. W., Singh, S., Feuerstein, J. D., Falck-Ytter, C., Falck-Ytter, Y., Cross, R. K., et al. (2019). AGA clinical practice guidelines on the management of mild-to-moderate ulcerative colitis. Gastroenterology 156 (3), 748–764. doi:10.1053/j.gastro.2018.12.009
Kumbhani, D. J., Cannon, C. P., Beavers, C. J., Bhatt, D. L., Cuker, A., Gluckman, T. J., et al. (2021). 2020 acc expert consensus decision pathway for anticoagulant and antiplatelet therapy in patients with atrial fibrillation or venous thromboembolism undergoing percutaneous coronary intervention or with atherosclerotic cardiovascular disease: A report of the American college of cardiology solution set oversight committee. J. Am. Coll. Cardiol. 77 (5), 629–658. doi:10.1016/j.jacc.2020.09.011
Lee, J. H., Kim, D. G., Bae, T. J., Rho, K., Kim, J. T., Lee, J. J., et al. (2012). Cda: Combinatorial drug discovery using transcriptional response modules. PLoS One 7 (8). doi:10.1371/journal.pone.0042573
Li, J., Tong, X. Y., Zhu, L. D., and Zhang, H. Y. (2020). A machine learning method for drug combination prediction. Front. Genet. 11, 1000. doi:10.3389/fgene.2020.01000
Liu, Y., Tong, Z., Shi, J., Li, R., Upton, M., and Wang, Z. (2021). Drug repurposing for next-generation combination therapies against multidrug-resistant bacteria. Theranostics 11 (10), 4910–4928. doi:10.7150/thno.56205
Luo, Y., Zhao, X., Zhou, J., Yang, J., Zhang, Y., Kuang, W., et al. (2017). A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8 (1), 573. doi:10.1038/s41467-017-00680-8
Röllig, C., Schliemann, C., Mikesch, J.-H., Fransecky, L., Baldus, C. D., Heydrich, B.-N., et al. (2021). Gemtuzumab ozogamicin plus midostaurin in combination with standard intensive induction therapy in newly diagnosed AML: Results from a phase-I study. Blood 138 (1), 2324. doi:10.1182/blood-2021-150069
Shi, J. Y., Huang, H., Li, J. X., Lei, P., Zhang, Y. N., Dong, K., et al. (2018). Tmfuf: A triple matrix factorization-based unified framework for predicting comprehensive drug-drug interactions of new drugs. BMC Bioinforma. 19 (14), 411. doi:10.1186/s12859-018-2379-8
Shinn, P., Chen, L., Ferrer, M., Itkin, Z., Klumpp-Thomas, C., McKnight, C., et al. (2019). High-throughput screening for drug combinations. Methods Mol. Biol. 1939, 11–35. doi:10.1007/978-1-4939-9089-4_2
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49 (D1), D605–D612. doi:10.1093/nar/gkaa1074
Terdiman, J. P., Gruss, C. B., Heidelbaugh, J. J., Sultan, S., Falck-Ytter, Y. T., Practice, A. G. A. I. C., et al. (2013). American Gastroenterological Association Institute guideline on the use of thiopurines, methotrexate, and anti-TNF-alpha biologic drugs for the induction and maintenance of remission in inflammatory Crohn's disease. Gastroenterology 145 (6), 1459–1463. doi:10.1053/j.gastro.2013.10.047
Tian, J., Guo, S., Chen, H., Peng, J. J., Jia, M. M., Li, N. S., et al. (2018). Combination of emricasan with Ponatinib synergistically reduces ischemia/reperfusion injury in rat brain through simultaneous prevention of apoptosis and necroptosis. Transl. Stroke Res. 9 (4), 382–392. doi:10.1007/s12975-017-0581-z
Virani, S. S., Morris, P. B., Agarwala, A., Ballantyne, C. M., Birtcher, K. K., Kris-Etherton, P. M., et al. (2021). 2021 acc expert consensus decision pathway on the management of ascvd risk reduction in patients with persistent hypertriglyceridemia: A report of the American college of cardiology solution set oversight committee. J. Am. Coll. Cardiol. 78 (9), 960–993. doi:10.1016/j.jacc.2021.06.011
Wang, Y., Yang, H., Chen, L., Jafari, M., and Tang, J. (2021). Network-based modeling of herb combinations in traditional Chinese medicine. Brief. Bioinform. 22 (5), bbab106. doi:10.1093/bib/bbab106
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46 (D1), D1074–D1082. doi:10.1093/nar/gkx1037
Yang, K., Bai, H., Ouyang, Q., Lai, L., and Tang, C. (2008). Finding multiple target optimal intervention in disease-related molecular network. Mol. Syst. Biol. 4, 228. doi:10.1038/msb.2008.60
Yang, X., Lian, X., Fu, C., Wuchty, S., Yang, S., and Zhang, Z. (2021). Hvidb: A comprehensive database for human-virus protein-protein interactions. Brief. Bioinform. 22 (2), 832–844. doi:10.1093/bib/bbaa425
Zagidullin, B., Wang, Z., Guan, Y., Pitkanen, E., and Tang, J. (2021). Comparative analysis of molecular fingerprints in prediction of drug combination effects. Brief. Bioinform. 22 (6), bbab291. doi:10.1093/bib/bbab291
Keywords: pathway, gene, drug combination, network pharmacology, random walk with restart
Citation: Hong Y, Chen D, Jin Y, Zu M and Zhang Y (2022) PINet 1.0: A pathway network-based evaluation of drug combinations for the management of specific diseases. Front. Mol. Biosci. 9:971768. doi: 10.3389/fmolb.2022.971768
Received: 17 June 2022; Accepted: 03 October 2022;
Published: 18 October 2022.
Edited by:
Fuhai Li, Washington University in St. Louis, United StatesReviewed by:
Nenad Filipovic, University of Kragujevac, SerbiaCopyright © 2022 Hong, Chen, Jin, Zu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mian Zu, cmFiYml0enVtaWFuQG91dGxvb2suY29t; Yin Zhang, YXJpbmcyMDEwQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.