 Jaeyoun Lee
Jaeyoun Lee Soohyun Um
Soohyun Um Seung Hyun Kim
Seung Hyun Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 02 September 2022
Sec. Metabolomics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.967945
This article is part of the Research TopicApplications of Metabolomics to the Discovery of Biomolecules from Natural ProductsView all 9 articles
In this study, the Salinivibrio costicola strain was isolated from Suaeda maritima (L.) Dumort. collected in Sinan, Republic of Korea. The endophytic characteristics of the Gram-negative bacterium S. costicola were verified with metagenomics sequencing of S. maritima. S. costicola was cultivated for 3 days in a liquid medium with 3.3% sea salt and analyzed the metabolites produced by the strain cultured in five different bacterial cultivation media. From the bacterial cultures, polyhydroxybutyrate derivatives were detected using high-resolution mass spectrometry, and three major compounds were isolated by high-performance liquid chromatography. The chemical structures of the compounds were elucidated using nuclear magnetic resonance and MS analyses. The relationship between the compounds was confirmed with Global Natural Product Social Molecular Networking, which showed clustering of the compounds. From the S. maritima extract, polyhydroxybutyrate derivatives produced by S. costicola were detected as being accumulated in the host plant.
The close relationship between plants and their endophytes indicates that individual host plants and microbial communities are essential for thriving and survival (Kratz, 1991; Compant et al., 2010; White et al., 2019). Despite the pervasiveness of these mutualistic interactions, the molecular underpinnings of most of them either remain unexplored or are only being understood. In recent years, several studies have emphasized the importance of plant-microbe relationships as particularly attractive sources for new bioactive small molecules, many of which may have potential applications in the pharmaceutical industry (Helfrich et al., 2018; Newman, 2018). The discovery of natural products from holobiont phenotype analysis through metagenomics, the study of the collective genome of a microbial community in environmental samples, has been established over the past few years (Fuhrman, 2012; Iquebal et al., 2022; Liu et al., 2022). Metagenomic research on plant microbiomes has yielded further insights into functional information beyond the genome information of particular bacterial strains (Lucaciu et al., 2019; Liu et al., 2022). In addition, it is possible to consider both the computational and experimental aspects of molecular plant-microbiome interactions (Lucaciu et al., 2019).
This study hypothesized that the native coastal annual halophyte, S. maritima (Amaranthaceae), has habitat-specific microbial communities to overcome osmotic pressure stress, which may have evolved adaptive traits in response to high-salt environments (Clipson, 1987; Cherian and Reddy, 2000). Known to be a salt-marsh herb, it has leaves that are pale green, which changes to blackish red in autumn, and are fleshy and 1–5 cm long. The whole plant is 30–60 cm long, with branches spreading from near the ground. S. maritima is most common in lower marshes, where it grows in single colonies with scattered individuals (Chapman, 1947). We assumed that the microorganisms associated with S. maritima endosphere might be beneficial to the host. The purpose of this study was to investigate the assembly and structure of in situ bacterial microbiomes associated with the endosphere using metagenomic analysis. In addition, we isolated endophytes of S. maritima exhibiting microbial features associated with salt adaptation and estimated functional extensions of plant phenotypes within a phylogenetic framework (Yeo and Flowers, 1980; Lee et al., 2003).
The plant material, Suaeda maritima (L.) Dumort. which is a halophyte, was collected from the Taepyeong saltern (34° 59′ 53.0″ N 126° 10′ 19.4″ E) in Sinan, South Korea in October 2021.
DNA preparation was performed using the FastDNA® Spin Kit for soil (MP Biomedicals), and the quantity of DNA was measured with an Epoch™ Spectrometer (BioTek). Hypervariable regions V3-V4 of the bacterial 16S rRNA gene were amplified using the primer pair 341F [5′-TCGTCGGCAGCGTC-AGATGTGTATAAGAGACAG-CCTACGGGNGGCWGCAG-3' (nextera consensus - sequencing adaptor - target sequence), 50mer] and 805R [5′- GTCTCGTGGGCTCGG-AGATGTGTATAAGAGACAG-GACTACHVGGGTATCTAATCC-3' (nextera consensus - sequencing adaptor - target sequence), 55mer] containing the appropriate Illumina adapters following Illumina’s 16S Metagenomics Protocol for the first amplicon PCR. The second PCR (index PCR) was performed with forward index i5 [AATGATACGGCGACCACCGAGATCTACAC-XXXXXXXX-TCGTCGGCAGCGTC, composition: left–i5 (XXXXXXXX)–right, 51mer] and reverse index i7 [CAAGCAGAAGACGGCATACGAGAT-GTCTCGTGGGCTCGG, composition: left–i5 (XXXXXXXX)–right, 47mer]. PCR reagents (25 μL in total) were 10X buffer (2.5 μL), dNTP (2.5 μL), forward primer (10 pmole/μL, 1 μL), reverse primer (10 pmole/μL, 1 μL), Taq polymerase (TaKaRa Ex Taq DNA polymerase, 1000 U), DNA (2 μL), and D.W. (15.75 μL). The quantity and quality of a single library were determined using the Quanti-iT PicoGreen dsDNA Assay Kit (Invitrogen) and Agilent 2,100 Bioanalyzer System (Agilent Technologies, US.). The samples were sequenced paired-end using a MiSeq® Reagent Kit v2 (500 cycles; Illumina, Inc.).
To obtain Q-values by sequence, sequences that could be misidentified as ‘new species’ by Q-value were removed. Illumina next-generation sequencing (NGS) data were analyzed using Trimmomatic software, and other NGS data were analyzed using self-developed code. The Pandaseq software was used for bioinformatics, but this was not performed for single-end reads or PacBio’s CCS data (Bartram et al., 2011). CJ Bioscience pipeline adopting in-house EzBioCloud 16S rRNA database was used with in-house algorithms to remove 16S rRNA PCR primer sequences, as these primer regions originated from the annealing process. To reduce computational cost and increase accuracy, different versions of the reference database were used to match various 16S rRNA regions. A combination of in-house algorithms and HMMER software was used for further analyses. Raw NGS data contained approximately 0.5% sequencing errors, which may occur randomly. However, as the same gene was sequenced several times during NGS experiments, it was possible to correct these errors through adequate modeling. Quality-controlled sequences were used for taxonomic assignment. The pipeline uses proven identification algorithms implemented in EzBioCloud to identify all the sequences. This two-step algorithm consists of (i) searching reference databases using the USEARCH program, and (ii) calculating similarity with robust pairwise alignment. Query sequences that matched the reference sequences in EzBioCloud by ≥ 97% similarity were considered to be at the species level. Lower sequence similarity cutoffs were used for genus or higher taxonomic levels. *x = sequence similarity to reference sequences; species (x ≥ 97%), genus (97 > x ≥ 94.5%), family (94.5 > x ≥ 86.5%), order (86.5 > x ≥ 82%), class (82 > x ≥ 78.5%), phylum (78.5 > x ≥ 75%). Cutoff values from Yarza et al. were considered for this study (Yarza et al., 2014). However, during the detection of chimeric sequences, a sequence with a similarity of ≥97% in the identification step that was previously been identified at the species level was found; therefore, it was removed from the detection target. Operational taxonomic units (OTUs) are arbitrary classification units that are often referred to as a ‘species’in microbial community analysis.
All the sequences in the samples were grouped into multiple OTUs using various algorithms and programs. 1) Except for chimeras, sequences with ≥97% similarity were classified as species and each was assigned an OTU. 2) For sequences with <97% similarity, CD-hit and UCLUST programs were used to perform cluster analysis, and each cluster was assigned an OTU. 3) The OTUs from steps 1 and 2 were combined to obtain the final OTU information. 4) The OTU formed in step 2 as a single sequence was removed from the diversity analysis because it caused the diversity index to increase more than the actual value. It calculates various alpha diversity indices within a single sample using OTU information (the number of OTUs and the number of sequences in an OTU). Shannon, Simpson, and other species abundance and evenness statistics were included in alpha diversity.
To remove exterior contaminants, whole parts of S. maritima were disinfected by soaking in 5 percent sodium hypochlorite for 5 minutes and wiping with 80 percent aqueous ethanol. The sterile leaves, stems, and roots of S. maritima were sliced into flakes and added to a chitin medium with sea salt (6 g of chitin, 0.75 g of K2HPO4, 0.5 g of MgSO4.7H2O, and 3.5 g of K2HPO4, 10 mg of FeSO4.7H2O, 10 mg of MnCl2.4H2O, 10 mg of ZnSO4.7H2O, 100 mg of cycloheximide, 33 g of sea salt, and 36 g of agar per 1 L of sterilized water), Czapek-Dox medium with sea salt (30 g of sucrose, 2 g of NaNO3, 1 g of K2HPO4, 0.5 g of MgCl2, 0.5 g of KCl, 0.01 g of FeCl2, 100 mg of cycloheximide, 33 g of sea salt and 18 g of agar per 1 L of sterilized water), and A1 medium with sea salt (10 g of starch, 4 g of yeast extract, 2 g of peptone, 100 mg of cycloheximide, 33 g of sea salt, and 18 g of agar per 1 L of sterilized water) for 14 days to isolate endophytes from S. maritima. Each isolated endophyte was cultured in a modified K medium with sea salt [12 g of Luria-Bertani (LB), 12 g of potato dextrose broth (PDB), 1 g of tryptic soy broth (TSB), 33 g of sea salt, and 18 g of agar per 1 L of sterilized water at 28 °C for 6 days].
For identification of the isolated bacteria, DNA was extracted using the MagAttract HMW DNA Kit (Qiagen) according to the manufacturer’s instructions. The DNA extract was amplified with the following conditions: initial denaturation at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C, and extension at 68°C for 1.5 min, and a final extension at 68°C for 10 min. The purified PCR products were sequenced in the forward and reverse directions in separate reactions and duplicates. The PCR amplification primer sequences were as follows: 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 1492R (5′-GGT TAC CTT GTT ACG ACT T-3′) for the 16S rRNA. The purified PCR products were sequenced using two primer pairs: 785F (5′-GGA TTA GAT ACC CTG GTA-3′) and 907R (5′-CCG TCA ATT CMT TTR AGT TT-3′) for 16S rRNA and NS1 (5′-GTA GTC ATA TGC TTG TCT C-3′). Sequencing was performed using a Big Dye Terminator 3.1 cycle Sequencing Kit (Applied BioSystems, United States ).
After isolating the single bacterial strains from a chitin medium with sea salt, the isolated Gram-negative bacteria (see Supplementary Figure S1), S. costicola YSL5, were cultivated in a modified K liquid medium with sea salt for 3 days (28°C, 180 rpm). Then, 10 ml of the bacterial culture was inoculated into diverse bacterial culture media, including K medium with salt (12 g of LB medium, 12 g of PDB, 1 g of TSB, 33 g of sea salt per 1 L of sterilized water), potato dextrose broth (PDB) medium with salt (27 g of potato extract and 33 g of sea salt per 1 L of sterilized water), potato dextrose yeast (PDY) media with salt (26.5 g of potato extract, 5 g of yeast extract, and 33 g of sea salt per 1 L of sterilized water), yeast extract-malt extract (YEME) medium with salt (5 g of malt extract, 2 g of yeast extract, and 33 g of sea salt per 1 L of sterilized water), and yeast peptone mannitol (YPM) medium with salt (25 g of mannitol, 5 g of yeast extract, 3 g of peptone, and 33 g of sea salt per 1 L of sterilized water) in 250 ml Erlenmeyer flask (28°C, 3 days). Each culture was extracted with ethyl acetate/water layer separation using a separatory funnel, and the ethyl acetate layer was concentrated in vacuo. The dried crude extract was dissolved in methanol at a concentration of 250 μg/ml and analyzed using liquid chromatography-mass spectrometry (LC-MS). High-performance liquid chromatography (HPLC) measurements were performed with an Agilent 1,290 ultra-high-performance liquid-chromatography (UHPLC) system (Agilent Technologies, Santa Clara, United States ) equipped with a 1,290 Infinity binary pump and a YMC-triart C18 column (150 × 2.0 mm, 1.9 μm; YMC KOREA Co., Seongnam, Korea). The mobile phases were 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) with the following gradients: 10–100% B (0–20 min), 100% B (20.1–25 min), 100–10% B (25.1–30 min), and 10% B (30.1–35 min) with an injection in six replicates at a volume of 10 μL. Mass spectrometry was performed using an Agilent 6,530 quadrupole time-of-flight mass spectrometer (Q-TOF-MS, Agilent Technologies, Santa Clara, United States ) equipped with an electrospray ionization (ESI) source. The MS experiment was performed under the following conditions: source temperature, 100 °C; desolvation temperature, 300°C; desolvation gas flow rate, 500 L/h; cone gas flow rate, 30 L/h; capillary voltage +3.0 kV; positive and negative modes; cone voltage, 60 V; extractor voltage, 3 V.
The modified K medium was used for high-resolution tandem mass spectrometry (HRMS/MS)-based Global Natural Product Social Molecular Networking (GNPS) analysis of the crude ethyl acetate extracts obtained from 3 days cultures of S. costicola YSL5 and the methanol extract of S. maritima. Based on the tandem mass spectrometry (MS/MS) data from an Agilent 6530 Q-TOF-MS, molecular networks of the secondary metabolites were created using the GNPS platform (http://gnps.ucsd.edu) (Wang et al., 2016; Nothias et al., 2020). The data were converted to mzML format using MS-Convert. Using the GNPS Web server, the converted files were used to generate an MS/MS molecular network. Both the precursor ion mass tolerance and the fragment ion mass tolerance were adjusted to 2.0 Da. The default value for both was 0.5 Da. Molecular networks were generated using a cosine score of 0.7, a minimum of six matched peaks, and a minimum cluster size of 2. After analysis, data were visualized using Cytoscape 3.9.0 software (Shannon et al., 2003).
With mzML files of six replicates of each medium, mass feature detection was performed using MZmine 2.53 (Pluskal et al., 2010). In the first step of the feature detection process using MZmine, each mass spectrum was processed separately to detect individual ion peaks. The mzML file was imported to a retention time range of 0.03–30.32 min and the mass detection noise level was 1.0E1 for MS2 with Centroid mass detector. Chromatograms were then constructed for each m/z value found in the mass list across the entire retention period. Chromatograms were built with a minimum time span of 0.01 min, a minimum height of 5.0E3, and an m/z tolerance of 0.001 m/z or 5.0 ppm. Finally, each chromatogram was deconvoluted into its individual features. Chromatogram deconvolution, which separates each detected chromatogram into individual peaks, was achieved using a baseline cut-off algorithm with a minimum peak height of 2.0E3, a peak duration range of 0.00–0.4 min, and a baseline level of 5.0E2. Deconvoluted peaks were de-isotoped using the isotopic peaks grouper algorithm with an m/z tolerance of 0.006 or 10.0 ppm and a retention time tolerance of 0.15 min. MZmine selects different algorithms for each of these steps depending on the nature of the MS data (Weng, 2020). The peaks were aligned in a peak table by joint alignment with an m/z tolerance of 0.001 or 5.0 ppm. The aligned data were statistically analyzed using MetaboAnalyst 5.0 (http://www.metaboanalyst.ca) (Pang et al., 2021).
After the matrix data were normalized, the multivariate analysis comprised principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), and the building of a heatmap showing the correlation between variables and samples. Finally, a hierarchical clustering dendrogram was obtained using a Euclidian distance measure and Ward clustering algorithm.
A 10 ml liquid culture of S. costicola YSL5 preculture was transferred to 100 ml of modified K medium in a 250 ml Erlenmeyer flask. With continuous shaking at 28°C and 180 rpm for 3 days, 10 ml of the liquid culture was inoculated in a 2 L Erlenmeyer flask containing 1.2 L of the medium (28°C, 180 rpm, 3 days, 12 L in total). The culture was extracted using ethyl acetate/water layer separation, and the ethyl acetate layer was subsequently concentrated in vacuo. The dried plant extracts were purified using a semi-preparative HPLC equipped with a 1,260 Infinity binary pump and YMC-triart C18 column (150 × 10.0 mm, 5 μm; YMC KOREA Co., Seongnam, Korea) with 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The conditions were 10% B (0–10 min), 10–50% B (10–30 min), 50–100% B (30–35 min), and UV detection at 210 nm. Nuclear magnetic resonance (NMR) data of the separated single compounds were recorded on a JEOL 600 MHz NMR spectrometer equipped with a 600 MHz ECZR narrow bore spectrometer. 1H NMR chemical shifts at δH 2.50 (DMSO-d6) and 13C NMR chemical shifts at δC 39.51 (DMSO-d6) were set as reference peaks, and the chemical structures were assigned by interpreting combinational data from two-dimensional (2D) NMR such as correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond correlation (HMBC) experiments.
Whole parts of S. maritima were dried at 45°C for 1 day with a dehydrator (SHINIL, Cheonan, Korea). Ground dried samples were extracted with methanol at 35°C for 120 min using an ultrasound-assisted extraction method. Methanol was used as the extraction solvent to obtain a diverse variety of compounds from plants. The extract was concentrated in vacuo and dissolved in methanol to a final concentration of 250 μg/ml. Metabolite analysis was conducted with MS/Q-TOF-MS with 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The conditions were 10–100% B (0–20 min), 100% B (20.1–25 min), 100–10% B (25.1–30 min), and 10% B (30.1–35 min), with an injection volume of 10 μL and a flow rate of 0.3 ml/min. Mass spectrometry was performed using electrospray ionization (ESI) source in the positive mode. Extracted-ion chromatogram (m/z [M + H]+ and m/z [M + Na]+) and fragmentation patterns were analyzed to compare and detect compounds in both the methanol extract of S. maritima and liquid culture of S. costicola YSL5. The ethyl acetate extract was dissolved in methanol at a concentration of 1,000 μg/ml and the compound was isolated using a semi-preparative LC-MS system. HPLC measurements were performed using an Agilent 1,100 Series (Agilent Technologies, Santa Clara, CA, United States) equipped with a YMC-Triart C18 column (150 × 10.0 mm, 5 μm; YMC KOREA Co., Seongnam, Korea). The mobile phases were 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B), with the following gradients: 50–100% B (0–10 min), 100% B (10.1–15 min), 100–10% B (15.1–20 min), and 10% B (20.1–25 min). Mass spectrometry was performed using a single quadrupole mass spectrometer (Waters Micromass ZQ, Waters, Massachusetts, United States ) equipped with an ESCi multimode ionization source. The MS experiment was performed under the conditions same as the analyses of S. costicola YSL5.
About 411 bp read lengths (min = 111, max = 446) of V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified using the genomic DNA extracted from the plant material. A total of 64772 amplicons were sequenced using the Illumina MiSeq platform, and 24004 quality-filtered reads were obtained (low-quality amplicons = 1,664, non-target amplicons = 39688, chimeric amplicons = 416). The number of reads that were identified at the species level based on 97% similarity was 59942 (92.5%), and 364 bacterial species were identified. Good’s coverage, calculated as the ratio of OTUs excluding singleton in the entire read, reflects the database coverage rate of the sample, and its value for S. maritima was 99.6%, indicating that the sample reached sufficient sampling depth (cutoff = 97%, number of OTUs found in the sample = 454). The values of alpha diversity, the species diversity existing in a particular location and a particular sample, for S. maritima were 522.6 ± 29.9 for ACE, 49.0 ± 27.0 for Chao1, and 552.0 ± 0 for Jackknife. The Shannon value, an index that reflects the number of species and the effect of evenness, for S. maritima was 2.544 ± 0.028. The phylogenetic diversity of S. maritima was calculated to be 879.0 by summing the shortest distance between the system diagram’s nodes. The detected OTUs were assigned to 15 phyla (24004 OTUs in total), 38 classes, 67 orders, 127 families, 248 genera, and 383 species. Of the identified 15 phyla, the most prominent phyla were Proteobacteria (79.4%), followed by Actinobacteria (15.6%), Bacteroidetes (2.0%), and Firmicutes (1.7%). All ten major phyla have been described as the dominant taxa in soil studies. In the Proteobacteria phylum, the Gammaproteobacteria group (68.2%) was the dominant bacterial group, followed by Alphaproteobacteria and Betaproteobacteria with a total percentage of reads of 6.4 and 4.6% at the class level (Figure 1; Supplementary Tables S1–4).
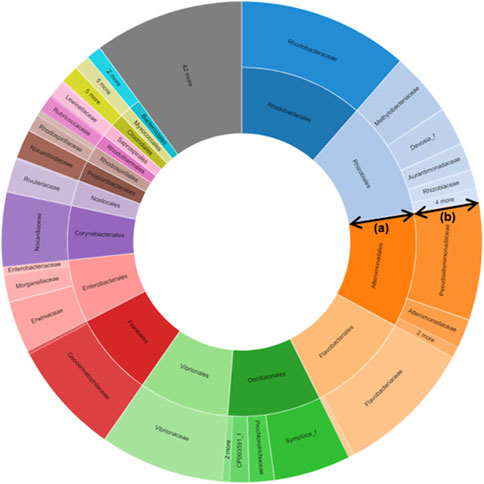
FIGURE 1. Metagenomics-targeted bacteria taxon of Suaeda maritima (L.) Dumort. The data shows that Vibrionaceae, the family of Salinivibrio costicola YSL5, was detected from S. maritima. (A) Order; (B) Family.
From the fleshy leaves and roots of S. maritima on the Chitin and Czapek-Dox media, four endophytes were isolated including Streptomyces xinghaiensis (YSL1, GenBank accession No. OM992324), Nocardiopsis aegyptia (YSL2; GenBank accession No. OM992320), and two Gram-negative bacteria, Salinivibrio costicola (YSL5, GenBank accession No. OM992322), and Alcaligenes aquatilis (YSL9; GenBank accession No. ON013930).
S. costicola YSL5, an endophyte of S. maritima, was cultivated for 3 days in modified K, PDB, PDY, YEME, and YPM liquid media with sea salt (Supplementary Figure S1), and the extract was analyzed using LC-MS/MS. The diversity and quantity of secondary metabolites produced in each medium were quantified using GNPS pie charts (Figure 2). The bacterial strain S. costicola YSL5 cultured in K medium produced twelve related metabolites between m/z 543.2–1,059.4 with an increase of 86 (Supplementary Figure S3), displaying identical UV absorption (λmax at 210). HRMS2-based analysis of the YSL5 extract clustered structurally related molecules with GNPS, and the results were visualized using Cytoscape 3.9.0. (Figure 3). The clustered nodes showed eleven analogs and other metabolites (Figure 3A). Eleven analogs: m/z 543.2 [M + Na]+ (1), m/z 629.2 [M + Na]+ (2), m/z 715.3 [M + Na]+ (3), m/z 801.3 [M + Na]+ (4), m/z 887.3 [M + Na]+ (5), m/z 973.4 [M + Na]+ (6), m/z 1,059.4 [M + Na]+ (7), m/z 1,145.5 [M + Na]+ (8), m/z 1,231.5 [M + Na]+ (10), m/z 1,317.5 [M + Na]+ (11), m/z 1,403.6 [M + Na]+ (12) made a large cluster.
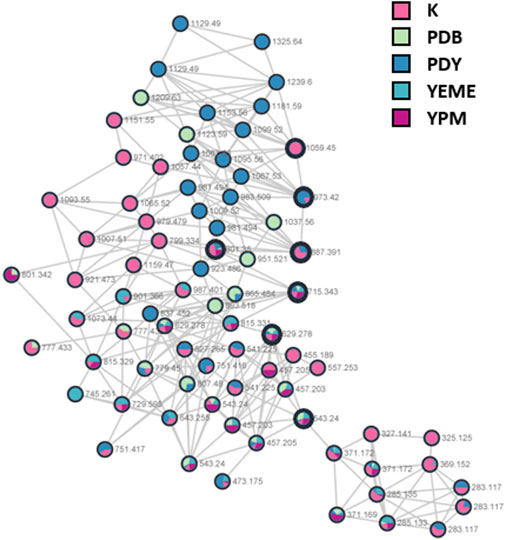
FIGURE 2. A hydroxybutyrate derivatives (bold black circles) cluster based on LC-MS/MS data of the ethyl acetate extract of S. costicola YSL5 in five broth media for 3 days. Red nodes, the metabolites produced by YSL5 in K medium; green nodes, PDB medium; blue nodes, PDY medium; light blue nodes, YEME medium; purple nodes, YPM medium.
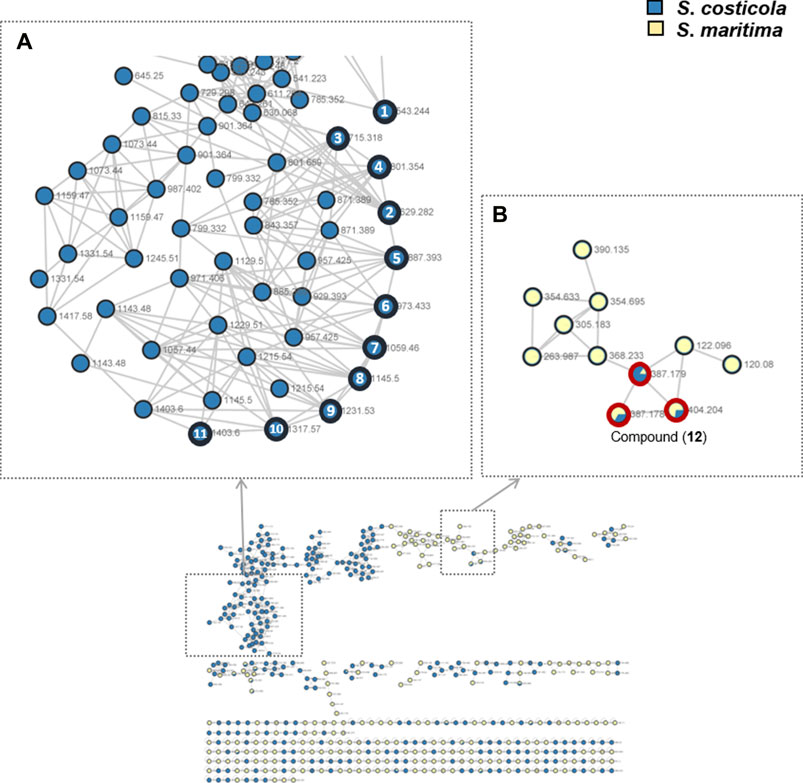
FIGURE 3. GNPS-clusters based with LC-MS/MS data of ethyl acetate extract of S. costicola YSL5 (blue nodes) and methanol extract of S. maritima (yellow nodes). (A) Eleven metabolites annotated as hydroxybutyrate analogs (marked with bold black circles); m/z 543.24 [M + Na]+ (1), m/z 629.28 [M + Na]+ (2), m/z 715.32 [M + Na]+ (3), m/z 801.35 [M + Na]+ (4), m/z 887.39 [M + Na]+(5), m/z 973.41 [M + Na]+ (6), m/z 1,059.46 [M + Na]+ (7), m/z 1,145.50 [M + Na]+ (8), m/z 1,231.53 [M + Na]+ (9), m/z 1,317.57 [M + Na]+ (10), and m/z 1,403.60 [M + Na]+ (11) are clustered. (B) One metabolite m/z 387.18 [M + H]+, m/z 404.20 [M + Na]+ (12) was detected both of S. costicola YSL5 extract and S. maritima extract (bold red circles).
Preprocessing was performed for statistical processing with data from six replicates. After necessary data preprocessing, a total of 971 metabolites in positive ion mode were identified from the raw data using the LC-MS/MS system. The covariance for the first five components was evaluated using unsupervised PCA. The explained variance of compounds produced by YSL5 in each of the five different media (K, PDB, PDY, YEME, and YPM) was 23.7% (PC1) and 18.9% (PC2), with a total variance of 42.6% (Figure 4A). The discrimination among the five groups was better represented by PLS-DA (covariance of 29.7%; Figure 4B), and the PLS-DA model classified K medium and others. Box and whisker plots (Figure 5A) represent differences in the relative abundance of metabolites, with m/z increasing by 86 among the media. A heatmap was generated based on the top 25 differentially expressed metabolites with the highest statistical difference and also exhibited distinct patterns of metabolites produced by S. costicola YSL5 in the five bacterial culture media (Figure 4C) (Pryke et al., 2007). Box and whisker plots of the normalized values of seven metabolites produced by S. costicola YSL5 in K broth with sea salt (Figure 5A) (Hssaini et al., 2020). The hierarchical clustering dendrogram shows similarities between the secondary metabolites produced in each medium (Figure 5B). With the PCA, PLS-DA, heatmap, and hierarchical clustering dendrogram, it can be observed that there are differences between the secondary metabolites produced in each medium. The K medium had particularly different metabolites, and the other four media showed more similarities among them than the K medium.
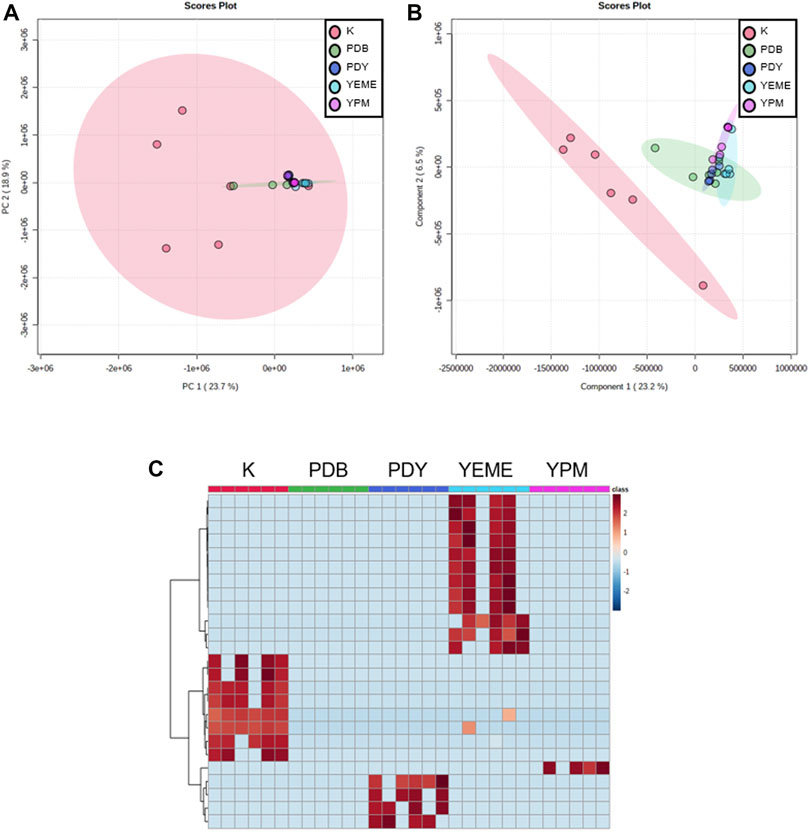
FIGURE 4. Metabolic profiling obtained using LC-MS of secondary metabolites produced by S. costicola YSL5 in five broth media with sea salt. (A) Partial least square discriminant analysis (PCA) plot based on the metabolic statistics analysis with the data obtained using LC-MS. (B) Partial least square discriminant analysis (PLS-DA) score plot based on the metabolic statistics analysis. PLS-DA model shows metabolic profile differences among metabolites produced by S. costicola YSL5 in K broth medium and metabolites produced in other four broth media (C) Heat map of the top 25 differential metabolites selected based on analysis of variance. Red colors indicate upregulation; blue colors indicate downregulation.
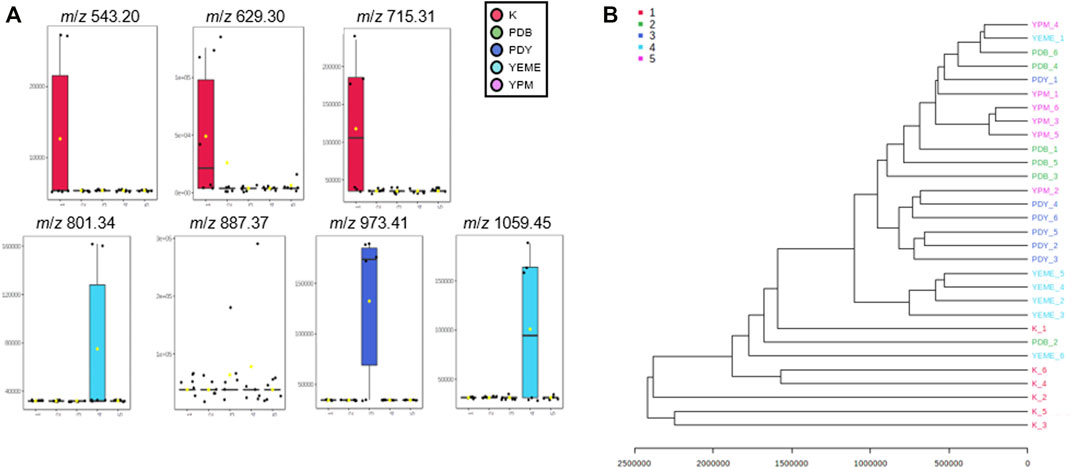
FIGURE 5. (A) Box and whisker plots of the normalized values of seven metabolites produced by S. costicola YSL5 in K broth medium with sea salt. Seven metabolites were m/z 543.20 [M + Na]+, m/z 629.30 [M + Na]+, m/z 715.31 [M + Na]+, m/z 801.34 [M + Na]+, m/z 887.37 [M + Na]+, m/z 973.41 [M + Na]+, and m/z 1,059.45 [M + Na]+. The metabolites increase m/z by 86 (B) Hierarchical clustering dendrogram based on the metabolic statistics analysis. The distance measure was Euclidean and the dendrogram was clustered with the Ward algorithm. The dendrogram shows distances among metabolites produced by S. costicola in five broth media.
2-hydroxy-3.7,11,15-tetramethyl-5.9,13,17-tetraoxo-4.8,12,16-tetraoxaicosan-19-yl 3-hydroxybutanoate (1) was obtained at tR 33 min using semi-preparative HPLC. Compound 1 was isolated as a yellow oil; UV (MeOH) λmax (log ε) 218 and 290 nm. The molecular formula of compound 1 was determined to have a molecular formula of C24H40O12Na, based on high-resolution electrospray ionization (HR-ESI) mass spectrometry, which indicated five degrees of unsaturation with a molecular ion at m/z 543.2481 [M + Na]+. The structure was confirmed by 1H, 13C NMR, and 2D data (Supplementary Figure S4–8).
2-hydroxy-3.7,11,15-tetramethyl-5.9,13,17-tetraoxo-4.8,12,16-tetraoxaicosan-19-yl 3-hydroxybutanoate (1): 1H NMR (600 MHz, DMSO-d6) δH 5.17-5.05 (5H, m), 4.75 (1H, br s), 4.67 (2H, qt, J = 6.5), 3.95 (1H, q, J = 6.5), 3.60 (1H, m), 2.64-2.51 (9H, m), 2.32 (1H, dd, J = 14.5), 2.26 (1H, dd, J = 14.5), 1.20 (2H, s), 1.19 (3H, d, J = 1.4), 1.19-1.18 (9H, m), 1.17 (3H, d, J = 1.2)
13C NMR (150 MHz, DMSO-d6) five carbonyl carbons [δC 170.25, 169.36, 169.05, 168.99, 168.98], four methoxy and two alcohol carbons [δC 74.82, 73.77, 67.40, 67.23, 67.18, 66.69, 63.28], five methylene carbons [δC 44.06, 40.21, 40.10, 40.01, 39.99], seven methyl carbons [δC 23.21, 19.39, 19.37, 19.31 (2C), 18.34, 15.05]; HR-ESI-MS m/z 543.2481 [M + Na]+ (calcd. for C24H40O12Na, 543.2423).
PHB, a similar structure to 2-hydroxy-3.7,11,15-tetramethyl-5.9,13,17-tetraoxo-4.8,12,16-tetraoxaicosan-19-yl 3-hydroxybutanoate (1), is known to be an energy source for bacteria and plays important role in plant growth (Okon and Itzigsohn, 1992; Reddy et al., 2019). However, the mechanism of PHB as a plant growth-promoting factor remains unclear (Trainer and Charles, 2006). The endophytic bacterium Herbaspirillum seropedicae produces PHB, which is stored in insoluble granules. PHB production showed that H. seropedicae significantly increased the growth of a variety of plants, including rice, corn, and sugarcane. In that study, Setaria viridis and five mutants in the PHB metabolism of H. seropedicae revealed several results regarding the importance of PHB for bacterial colonization in S. viridis roots (Alves et al., 2019). In another study, PHB-producing Pseudomonas fluorescens was isolated from the leaves of abiotic stress-tolerant Pangola grass. P. fluorescens is known as a plant growth-promoting bacterium, and a study revealed that a novel extracellular enzyme related to PHB production could have positive effects on plant growth (Stritzler et al., 2018).
Based on the metagenomic analysis, S. costicola YSL5 is thought to be an endophyte of S. maritima. To provide additional evidence, we investigated whether the metabolites produced by S. costicola YSL5 accumulated in S. maritima. S. maritima methanol extract was analyzed by LC-MS/MS under the same conditions as S. costicola YSL5 extract. The cluster showed that PHB derivatives (12) (m/z 387.1 [M + H]+, m/z 404.2 [M + Na]+) were detected in both S. costicola YSL5 culture broth extract and S. maritima methanol extract using GNPS analysis (Figure 3B). In the LC-MS chromatogram, EIC (m/z 387.1 [M + H]+) demonstrated that a compound (12) was detected at the same retention time and mass pattern as YSL5 (Figures 6C, E). Compound 12 was isolated as an oil with a molecular ion at m/z 387.1787 [M + H]+ at tR = 6 min by semi-preparative LC-MS. Based on 1H, COSY, HSQC, HMBC, and NOESY data, the molecular formula of compound 12 was determined to be a PHB derivative (Supplementary Figure S9–11).
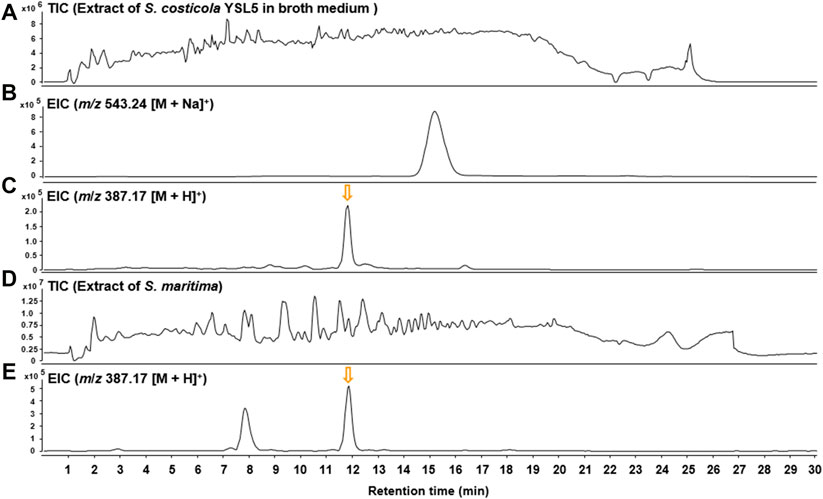
FIGURE 6. LC-MS chromatograms of m/z 387.17 [M + H]+ were obtained from the extract of S. costicola YSL5 in broth medium and S. maritima in positive ion mode. (A) Total ion current (TIC) chromatogram of the ethyl acetate extract of the S. costicola YSL5 liquid culture broth. (B) Extracted-ion chromatogram (EIC) of the S. costicola YSL5 liquid broth (2-hydroxy-3.7,11,15-tetramethyl-5.9,13,17-tetraoxo-4.8,12,16-tetraoxaicosan-19-yl 3-hydroxybutanoate, m/z 387.17 [M + H]+). (C) Extracted-ion chromatogram (EIC) of the S. costicola YSL5 liquid broth (polyhydroxybutyrate derivatives, m/z 387.17 [M + H]+). (D) Total ion current (TIC) chromatogram of the methanol extract of S. maritima. (E) Extracted-ion chromatogram (EIC) of S. maritima (polyhydroxybutyrate derivatives, m/z 387.17 [M + H]+). The arrows indicate the exact elution times of the extracted ions.
Plant endophytes utilize the endosphere of plants to stimulate plant development, elicit a defense response against pathogen attack, and mitigate abiotic stresses induced by drastic changes in extreme climates under harsh conditions. Although salinity is one of the most influential environmental factors on microbial evolution and community composition, little is known about the ecological functioning of microbial communities associated with halophytes growing in extremely saline environments such as seawater, salterns, salt mines, and mud flats. They have developed distinct morphological, anatomical, and physiological mechanisms to thrive in high-salt conditions during the course of evolution (Grigore et al., 2014). Microorganisms living in a high-salinity environment must consider the high energy loss in metabolic processes due to insufficient energy sources (Oren, 2008). The metabolic mechanisms that reduce the energy required to sustain life in microorganisms are encouraged. These mechanisms are characterized by modest growth rates, and the majority of the organism’s energy is directed towards maintenance-related activities (Shoemaker et al., 2021). These activities include osmotic equilibration, oxygen stress defense, motility, and more sustainable metabolic pathways. For instance, certain types of bacteria can store lipid droplets within their cells in the form of polyhydroxyalkanoates, triglycerides, or wax esters when subjected to stress (Alvarez et al., 1997; Wältermann and Steinbüchel, 2005; Thomas et al., 2019).
In this study, we used metagenomics analysis to track the bacterial endophytes of S. maritima and their secondary metabolites to advance plant microbiome research. Also, we used it to identify specific bacterial strains that generate biologically active or functional secondary metabolites that accumulate in host plants. When constructing agricultural systems for progressive and sustainable agricultural production, information regarding the association between plants and the microbial population in the soil would be useful, and this knowledge may be gained through the application of metagenomics. The utilization of metagenomic techniques to investigate soils that have been infected with organic manure would be beneficial when designing strategies for fertilization and would lessen the reliance of farmers on chemical fertilizers (Manjula and Narsimha, 2015). Goel et al. reported that metagenomics has the potential to be utilized for the development of sustainable agriculture. These programs can constantly evaluate microorganisms that are beneficial for plant growth (Goel et al., 2017). Similarly, functional metagenomics can be used to reshape and redirect rhizosphere microbial activity (Louca et al., 2018). Investigations into metagenomics, when combined with assessments of efficiency, may thus assist in improving bioponic systems and urban agriculture, such as smart farms, in a way that is more environmentally friendly (Bao et al., 2021). To achieve sustainable agriculture on a global scale and to reclaim soil through phytoremediation and bioremediation, a growing number of countries are now utilizing rhizospheric bacteria that are capable of producing plant growth promoters in the presence of salt stress (Tank and Saraf, 2010). Our investigation of the bacteria isolated from S. maritima resulted in the identification of four bacterial strains (YSL1, 2, 5, and 9), with YSL5 being the only strain matching the metagenomic results of S. maritima at the family level. While moderate salinity of 15% is generally regarded to be a transitional salinity for marine microbes and moderate halophiles, the bacterial strain YSL5 grew in a non-saline bacterial culture medium. We cultivated YSL5 in diverse bacterial cultivation media to investigate secondary metabolites by upregulating the growth of the bacterium, and identified major secondary metabolites, PHB, using spectroscopic analysis. The crude extracts of YSL5 were compared with methanol extracts of S. maritima through GNPS analysis to detect accumulated secondary metabolites in the host produced by the endophytic bacterium YSL5. Using LC-MS and GNPS chemical profiling, two secondary metabolites were detected at identical retention times, showing the identical MS/MS patterns (m/z 387.1787 [M + H]+, m/z 404.2029 [M + Na]+). After cultivating YSL5 on a large scale, the metabolite was identified as PHB with molecular weight (m/z 387.1787 [M + H]+). PHB, a high-molecular-weight polyester, is found in plants as a plant growth-promoting agent and accumulates as storage carbon in many bacterial species. In a hyperosmotic environment, PHB in the cytoplasm promotes the survival of bacterial cells exposed to osmotic stress. Although the mechanism of its protective action is unknown, the primary strategy of most bacteria under hyperosmotic stress is the accumulation of a diverse range of compatible solutes. The ability to adapt to changes in external osmolarity is critical for survival. Endophytic bacteria have developed various strategies to deal with osmotic pressure and changes in extracellular osmolarity in halophytes. Owing to the high osmotic potential of the soil solution and nutrient absorption limitations resulting from an oversupply of sodium and chloride ions, the high salinity of the water directly and effectively inhibits plant development (Sairam and Tyagi, 2004). Nutrient uptake inadequacies are induced by an excess of sodium and chloride in the soil solution (Nabti, 2015). Halophytes and halotolerant rhizospheric and endophytic microorganisms are uniquely suited to combat saline stress conditions. Functional interactions between halotolerant hosts and endophytic microorganisms can reduce the effects of salt pressure in the rhizosphere (Biró et al., 2002). In addition, the halophilic or halotolerant plant growth-promoting bacteria are characterized by their capacity to stimulate anti-oxidant enzymes that are involved in the tolerance of plants against salt stress (Kohler et al., 2009). According to Kadouri et al., compounds including PHB may also play a role in the resistance of Azospirillum brasilense to osmotic stress and root colonization (Kadouri et al., 2003). Arora et al. reported that the maximum PHB content was detected in salt-tolerant strains at higher salt concentrations, which defines the critical function of PHB in cell protection in saline environments, while the halotolerant strains accumulated the least amount of PHB when the salinity was either low or nil. However, the role of the accumulated PHB remains unclear (Arora et al., 2006). Based on the hypothesis regarding PHB accumulation in the host plant, we observed that S. costicola YSL5 produces PHB derivatives in the host halophyte S. maritima. These PHB derivatives potentially interact with one another to counter the osmotic stress, suggesting that PHB plays a role in cell protection under saline conditions.
In conclusion, we explored the bacterial communities associated with the halophyte plant S. maritima using metagenomics and functionally associated traits of the endophytic bacterium YSL5 and its secondary metabolites. The results from this study may form the basis for the future development of new natural product discovery and their function using metagenomic techniques. Furthermore, this study demonstrates that halophytes in high-salinity environments, such as salterns, are an enormous and untapped resource of new microorganisms that could have practical benefits in environments with a high level of salinity.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, OM992322; https://www.ncbi.nlm.nih.gov/, OM992320; https://www.ncbi.nlm.nih.gov/, OM992324; https://www.ncbi.nlm.nih.gov/, ON013930.
SU and SK conceptualized this study. SU collected the plant material and performed metagenomic analyses. JL performed bacterial isolation and fermentation. SU and JL purified the compounds and identified the hydroxybutyrate derivatives using NMR spectroscopic data. SU, JL, and SK wrote and edited the manuscript. SU and JL contributed equally to the study.
We acknowledge the financial support from the National Research Foundation of Korea (NRF-2018R1A6A1A03023718 and NRF-2020R1A2C1012632), funded by the Ministry of Education, Science and Technology, Republic of Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed, or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.967945/full#supplementary-material
Alvarez, H. M., Pucci, O. H., and Steinbüchel, A. (1997). Lipid storage compounds in marine bacteria. Appl. Microbiol. Biotechnol. 47 (2), 132–139. doi:10.1007/s002530050901
Alves, L. P. S., do Amaral, F. P., Kim, D., Bom, M. T., Gavídia, M. P., Teixeira, C. S., et al. (2019). Importance of poly-3-hydroxybutyrate metabolism to the ability of Herbaspirillum seropedicae to promote plant growth. Appl. Environ. Microbiol. 85 (6). doi:10.1128/aem.02586-18
Arora, N. K., Singhal, V., and Maheshwari, D. K. (2006). Salinity-induced accumulation of poly-β-hydroxybutyrate in rhizobia indicating its role in cell protection. World J. Microbiol. Biotechnol. 22 (6), 603–606. doi:10.1007/s11274-005-9077-1
Bao, Y., Feng, Y., Qiu, C., Zhang, J., Wang, Y., and Lin, X. (2021). Organic matter- and temperature-driven deterministic assembly processes govern bacterial community composition and functionality during manure composting. Waste Manag. 131, 31–40. doi:10.1016/j.wasman.2021.05.033
Bartram, A. K., Lynch, M. D., Stearns, J. C., Moreno-Hagelsieb, G., and Neufeld, J. D. (2011). Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77 (11), 3846–3852. doi:10.1128/AEM.02772-10
Biró, B., Villányi, P., and Köves-Péchy, K. (2002). Abundance and adaptation level of some soil microbes in salt-affected soils. Agrokémia és Talajt. 51, 99–106. doi:10.1556/Agrokem.51.2002.1-2.12
Cherian, S., and Reddy, M. (2000). Salt tolerance in the halophyte Suaeda nudiflora moq: effect of NaCl on growth, ion accumulation and oxidative enzymes. Indian J. Plant Physiology 5 (1), 32–37.
Clipson, N. (1987). Salt tolerance in the halophyte Suaeda maritima L. Dum. growth, ion and water relations and gas exchange in response to altered salinity. J. Exp. Bot. 38 (12), 1996–2004. doi:10.1093/jxb/38.12.1996
Compant, S., Clément, C., and Sessitsch, A. (2010). Plant growth-promoting bacteria in the rhizo-and endosphere of plants: their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 42 (5), 669–678. doi:10.1016/j.soilbio.2009.11.024
Fuhrman, J. A. (2012). Metagenomics and its connection to microbial community organization. F1000 Biol. Rep. 4, 15. doi:10.3410/B4-15
Goel, R., Suyal, D. C., NarayanDash, B. K., and Soni, R. (2017). Soil metagenomics: A tool for sustainable agriculture. Singapore: Springer.
Grigore, M., Ivanescu, L., and Toma, C. (2014). Halophytes: An integrative anatomical study. Cham: Springer, 33–37.
Helfrich, E. J., Vogel, C. M., Ueoka, R., Schäfer, M., Ryffel, F., Müller, D. B., et al. (2018). Bipartite interactions, antibiotic production and biosynthetic potential of the Arabidopsis leaf microbiome. Nat. Microbiol. 3 (8), 909–919. doi:10.1038/s41564-018-0200-0
Hssaini, L., Charafi, J., Razouk, R., Hernández, F., Fauconnier, M. L., Ennahli, S., et al. (2020). Assessment of morphological traits and fruit metabolites in eleven fig varieties (Ficus carica L.). Int. J. Fruit Sci. 20, 8–28. doi:10.1080/15538362.2019.1701615
Iquebal, M. A., Jagannadham, J., Jaiswal, S., Prabha, R., Rai, A., and Kumar, D. (2022). Potential use of microbial community genomes in various dimensions of agriculture productivity and its management: a review. Front. Microbiol. 13 708335. doi:10.3389/fmicb.2022.708335
Kadouri, D., Jurkevitch, E., and Okon, Y. (2003). Involvement of the reserve material poly-beta-hydroxybutyrate in Azospirillum brasilense stress endurance and root colonization. Appl. Environ. Microbiol. 69 (6), 3244–3250. doi:10.1128/aem.69.6.3244-3250.2003
Kohler, J., Caravaca, F., del Mar Alguacil, M., and Roldán, A. (2009). Elevated CO2 increases the effect of an arbuscular mycorrhizal fungus and a plant-growth-promoting rhizobacterium on structural stability of a semiarid agricultural soil under drought conditions. Soil Biol. Biochem. 41 (8), 1710–1716. doi:10.1016/j.soilbio.2009.05.014
Kratz, R. F. (1991). Symbiosis as a source of evolutionary innovation: speciation and morphogenesis. Cambridge: MIT press.
Lee, S.-H., Ji, K.-J., An, Y., and Ro, H.-M. (2003). Soil salinity and vegetation distribution at four tidal reclamation project areas. Korean J. Environ. Agric. 22 (2), 79–86.
Liu, S., Moon, C. D., Zheng, N., Huws, S., Zhao, S., and Wang, J. (2022). Opportunities and challenges of using metagenomic data to bring uncultured microbes into cultivation. Microbiome 10 (1), 76–14. doi:10.1186/s40168-022-01272-5
Louca, S., Doebeli, M., and Parfrey, L. W. (2018). Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6, 1–12. doi:10.1186/s40168-018-0420-9
Lucaciu, R., Pelikan, C., Gerner, S. M., Zioutis, C., Köstlbacher, S., Marx, H., et al. (2019). A bioinformatics guide to plant microbiome analysis. Front. Plant Sci. 10, 1313. doi:10.3389/fpls.2019.01313
Manjula, A., and Narsimha, G. (2015). “"XCYPF: A flexible and extensible framework for agricultural crop yield prediction,” in 2015 IEEE 9th International Conference on Intelligent Systems and Control (ISCO), Taiwan, 9-10 Jan. 2015, 1–5.
Nabti, E. (2015). Application of halotolerant bacteria to restore plant growth under salt stress. Cham: Springer, 235–259.
Newman, D. J. (2018). Are microbial endophytes the ‘actual’producers of bioactive antitumor agents? Trends Cancer 4 (10), 662–670. doi:10.1016/j.trecan.2018.08.002
Nothias, L.-F., Petras, D., Schmid, R., Dührkop, K., Rainer, J., Sarvepalli, A., et al. (2020). Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 17 (9), 905–908. doi:10.1038/s41592-020-0933-6
Okon, Y., and Itzigsohn, R. (1992). Poly-β-hydroxybutyrate metabolism in Azospirillum brasilense and the ecological role of PHB in the rhizosphere. FEMS Microbiol. Lett. 9 (2-4), 131–139. doi:10.1016/0378-1097(92)90302-5
Oren, A. (2008). Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst. 4, 2. doi:10.1186/1746-1448-4-2
Pang, Z., Chong, J., Zhou, G., de Lima Morais, D. A., Chang, L., Barrette, M., et al. (2021). MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49 (W1), W388–W396. doi:10.1093/nar/gkab382
Pluskal, T., Castillo, S., Villar-Briones, A., and Orešič, M. (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinforma. 11 (1), 395–411. doi:10.1186/1471-2105-11-395
Pryke, A., Mostaghim, S., and Nazemi, A. (2007). “Heatmap visualization of population based multi objective algorithms,” in International conference on evolutionary multi-criterion optimization (Berlin: Springer), 361–375.
Reddy, M. V., Mawatari, Y., Onodera, R., Nakamura, Y., Yajima, Y., and Chang, Y.-C. (2019). Bacterial conversion of waste into polyhydroxybutyrate (PHB): a new approach of bio-circular economy for treating waste and energy generation. Bioresour. Technol. Rep. 7, 100246. doi:10.1016/j.biteb.2019.100246
Sairam, R. K., and Tyagi, A. (2004). Physiology and molecular biology of salinity stress tolerance in plants. Curr. Sci. 86, 407–421.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504. doi:10.1101/gr.1239303
Shoemaker, W. R., Jones, S. E., Muscarella, M. E., Behringer, M. G., Lehmkuhl, B. K., and Lennon, J. T. (2021). Microbial population dynamics and evolutionary outcomes under extreme energy limitation. Proc. Natl. Acad. Sci. U. S. A. 118(33), e2101691118. doi: doi:doi:10.1073/pnas.2101691118
Stritzler, M., Diez Tissera, A., Soto, G., and Ayub, N. (2018). Plant growth-promoting bacterium Pseudomonas fluorescens FR1 secrets a novel type of extracellular polyhydroxybutyrate polymerase involved in abiotic stress response in plants. Biotechnol. Lett. 40 (9), 1419–1423. doi:10.1007/s10529-018-2576-6
Tank, N., and Saraf, M. (2010). Salinity-resistant plant growth promoting rhizobacteria ameliorates sodium chloride stress on tomato plants. J. Plant Interact. 5 (1), 51–58. doi:10.1080/17429140903125848
Thomas, F., Morris, J. T., Wigand, C., and Sievert, S. M. (2019). Short-term effect of simulated salt marsh restoration by sand-amendment on sediment bacterial communities. PLOS ONE 14 (4), e0215767. doi:10.1371/journal.pone.0215767
Trainer, M. A., and Charles, T. C. (2006). The role of PHB metabolism in the symbiosis of rhizobia with legumes. Appl. Microbiol. Biotechnol. 71 (4), 377–386. doi:10.1007/s00253-006-0354-1
Wältermann, M., and Steinbüchel, A. (2005). Neutral lipid bodies in prokaryotes: recent insights into structure, formation, and relationship to eukaryotic lipid depots. J. Bacteriol. 187 (11), 3607–3619. doi:10.1128/jb.187.11.3607-3619.2005
Wang, M., Carver, J. J., Phelan, V. V., Sanchez, L. M., Garg, N., Peng, Y., et al. (2016). Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 34 (8), 828–837. doi:10.1038/nbt.3597
White, J. F., Kingsley, K. L., Zhang, Q., Verma, R., Obi, N., Dvinskikh, S., et al. (2019). Review: Endophytic microbes and their potential applications in crop management. Pest Manag. Sci. 75 (10), 2558–2565. doi:10.1002/ps.5527
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K.-H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12 (9), 635–645. doi:10.1038/nrmicro3330
Keywords: secondary metabolites, halophytic endophyte, Salinivibrio costicola, Suaeda maritima (L.) dumort., polyhydroxybuthyrate (PHB)
Citation: Lee J, Um S and Kim SH (2022) Metabolomic analysis of halotolerant endophytic bacterium Salinivibrio costicola isolated from Suaeda maritima (L.) dumort. Front. Mol. Biosci. 9:967945. doi: 10.3389/fmolb.2022.967945
Received: 13 June 2022; Accepted: 09 August 2022;
Published: 02 September 2022.
Edited by:
Michel Frederich, University of Liège, BelgiumReviewed by:
R. Nischitha, Kuvempu University, IndiaCopyright © 2022 Lee, Um and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seung Hyun Kim, a2ltc2gxMUB5b25zZWkuYWMua3I=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.