
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci., 14 November 2022
Sec. Metabolomics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.967205
This article is part of the Research TopicAdvances in Methods and Tools for Multi-Omics Data AnalysisView all 11 articles
 Francis E. Agamah1,2
Francis E. Agamah1,2 Jumamurat R. Bayjanov3
Jumamurat R. Bayjanov3 Anna Niehues3
Anna Niehues3 Kelechi F. Njoku1Michelle Skelton2
Kelechi F. Njoku1Michelle Skelton2 Gaston K. Mazandu1,2,4
Gaston K. Mazandu1,2,4 Thomas H. A. Ederveen3*
Thomas H. A. Ederveen3* Nicola Mulder2Emile R. Chimusa5*
Nicola Mulder2Emile R. Chimusa5* Peter A. C. 't Hoen3*
Peter A. C. 't Hoen3*Advances in omics technologies allow for holistic studies into biological systems. These studies rely on integrative data analysis techniques to obtain a comprehensive view of the dynamics of cellular processes, and molecular mechanisms. Network-based integrative approaches have revolutionized multi-omics analysis by providing the framework to represent interactions between multiple different omics-layers in a graph, which may faithfully reflect the molecular wiring in a cell. Here we review network-based multi-omics/multi-modal integrative analytical approaches. We classify these approaches according to the type of omics data supported, the methods and/or algorithms implemented, their node and/or edge weighting components, and their ability to identify key nodes and subnetworks. We show how these approaches can be used to identify biomarkers, disease subtypes, crosstalk, causality, and molecular drivers of physiological and pathological mechanisms. We provide insight into the most appropriate methods and tools for research questions as showcased around the aetiology and treatment of COVID-19 that can be informed by multi-omics data integration. We conclude with an overview of challenges associated with multi-omics network-based analysis, such as reproducibility, heterogeneity, (biological) interpretability of the results, and we highlight some future directions for network-based integration.
Studies that implement large-scale molecular profiling techniques (-omics technologies) have increased our understanding of disease mechanisms and led to the discovery of new biological pathways, genetic loci underpinning disease progression, biomarkers, and targets for therapeutic development (Horgan and Kenny, 2011; Sun and Hu, 2016; Karczewski and Snyder, 2018). Until recently, these studies have mostly relied on single omics investigations. Dependencies between biological features and the relationships between different molecular layers (for example transcriptome, proteome, metabolome, microbiome, and lipidome) remain mostly elusive. The holistic understanding of the molecular and cellular bases of disease phenotypes and normal physiological processes requires integrated investigations of the contributions and associations between multiple (different but parallel) molecular layers driving the observed outcome. Most importantly, genetic information flows from the genome to traits and involves several molecular layers (Sun and Hu, 2016; Hasin et al., 2017). Thus, understanding the genetic architecture of complex phenotypes would involve integrating and investigating the interactions between different molecular layers (Buescher and Driggers, 2016; Hasin et al., 2017; Chakravorty et al., 2018; Zapalska-Sozoniuk et al., 2019).
Multi-omics datasets require appropriate computational methods for data integration and analysis. These methods/models implement statistical, network-based, and/or machine learning (ML) techniques on different omics layers to elucidate key omics features associated with diseases at various molecular levels and predict phenotypic traits and outcomes with increased accuracy (Ritchie et al., 2015; Bersanelli et al., 2016; Zeng and Lumley, 2018).
Based on the hypothesis that molecular features within a system establish functional connections or are part of modules to carry out processes, network-based methods offer a framework to conceptualize the complex interactions in a system as a collection of connected nodes (molecular features). They further suggest possible connections (e.g., genotype to phenotype relationships) and/or subnetworks (e.g., biological pathways) that are informative of an observed phenotype (Chakravorty et al., 2018). Therefore, network-based methods are particularly useful to assess complex interactions within multi-omics datasets and illustrate dependencies among multiple features. In addition, some network-based methods can incorporate prior information to guide the integrative analysis. For this reason, network-based methods have attracted considerable attention in multi-omics data integration around understanding disease mechanisms and drug discovery (Wu et al., 2018; Agamah et al., 2021). Previous reviews have mostly focused on the network-based analysis of single-omics data (Camacho et al., 2018; Yan et al., 2018; Zitnik et al., 2019) or different approaches toward multi-omics data integration (Cavill et al., 2016; Duruflé et al., 2021). Here, we review different integrative network-based approaches and some tools for multi-omics data analysis.
The outline of the review is as follows; we begin with a discussion on integrative multi-omics approaches, where we highlight the approaches for network-based analyses. We then discuss the different classes of methods for multi-modal network analysis. Next, we describe several network-based integrative multi-omics tools. This is followed by a discussion on the application of network-based tools to pertinent biological questions. This section provides guidance on the choice of the most appropriate network-based tools to answer a given biological question. As further examples, we show how some tools have been applied to COVID-19 research, which is currently one of the research areas benefiting from multi-omics integration approaches. Finally, we conclude with a discussion on some challenges associated with multi-omics analysis and the possible directions to mitigate such challenges.
After initial data selection, processing, and quality assurance, an appropriate data analysis approach needs to be selected. We categorize integrative multi-omics analysis approaches into two main categories, multi-stage and multi-dimensional (multi-modal) analytical approaches (Figure 1) (Holzinger and Ritchie, 2012; Wen et al., 2021). The multi-stage integration involves integrating data from different technologies using a stepwise approach. In this approach, omics layers are analysed separately before investigating statistical correlations between different biological features from the datasets under consideration. This analytical approach puts an initial emphasis on the relationships of features within an omics layer and how they relate to the phenotype of interest (Ritchie et al., 2015). The multi-modal analytical approach involves integrating multiple omics profiles in a simultaneous analysis (Holzinger and Ritchie, 2012; Ritchie et al., 2015; Karczewski and Snyder, 2018; Ulfenborg, 2019).
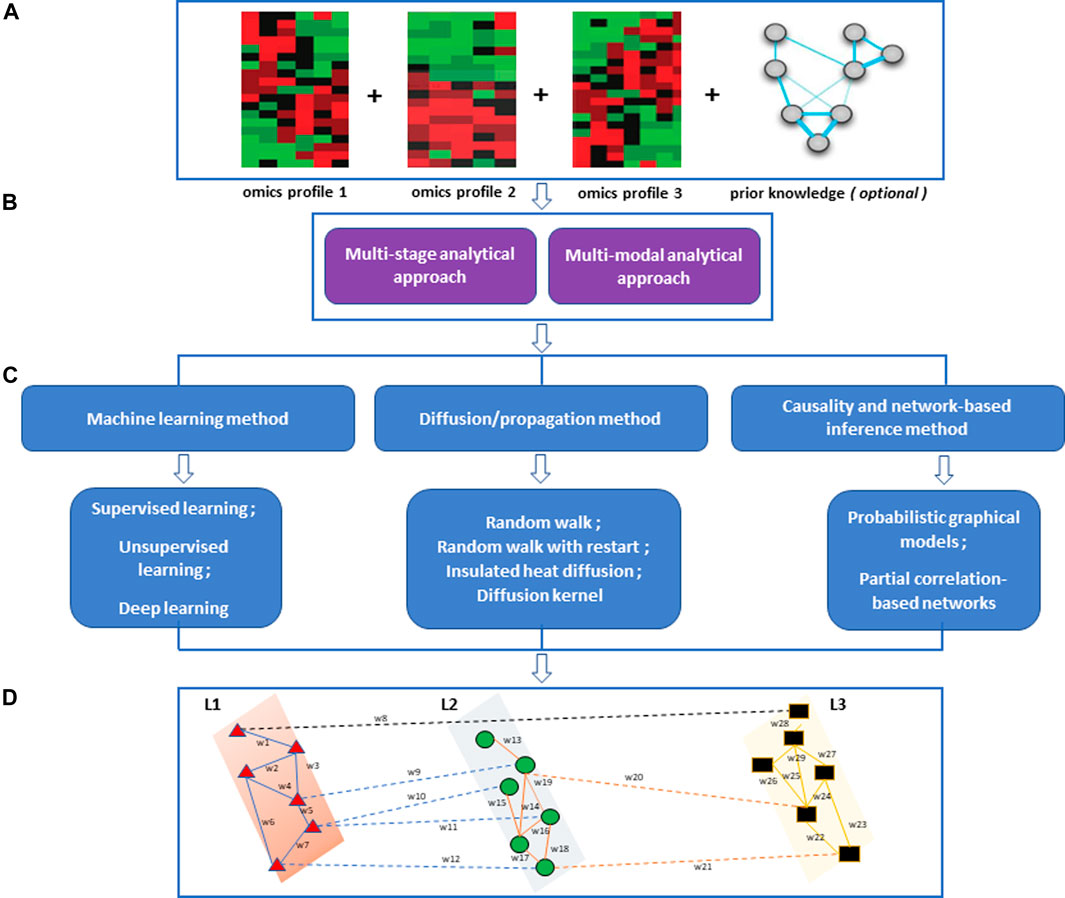
FIGURE 1. An overview of the multi-omics integration approach and the methods for network-based integration. (A) Processed omics data and prior knowledge for integrative analysis. (B) An integrative multi-omics approach that could be implemented. (C) Integrative network-based methods (D) Multi-layered network showing intra-layer interaction (solid lines) and crosstalk (dashed lines) across different layers (L1, L2, L3). The nodes are shaped and coloured to represent different omics features within the omics layers they are involved in. The edges are coloured to show different interactions within and between omics layers.
In this review, we focus on (i) machine learning-driven network-based methods, (ii) network-based diffusion/propagation methods, and (iii) causality- and network-based inference methods. The selection criteria were based on the fact that these multi-omics/multi-modal network-based methods implement network architectures together with statistical and mathematical models for integrative multi-omics data analysis. Most of these methods can be implemented in both multi-stage and multi-dimensional multi-omics analysis (Figure 1).
ML is a collection of data-driven techniques for fitting an analytical model to a given dataset. ML methods do not only provide the framework to automatically learn models from large multi-omics data and make accurate predictions but also implement network architectures to exploit interaction across the different omics layers e.g., for exploring omics-phenotype associations (Reel et al., 2021). ML comprises mainly supervised and unsupervised learning methods. Supervised learning uses labelled datasets to train models to yield the desired output and emphasizes predictions by inferring discriminating rules from the data. Supervised learning model training requires comprehensive data and can be time-consuming, while unsupervised learning uses unlabelled data, to find latent structures or patterns in the data.
Classical graph-based ML methods (e.g., label propagation, a method for assigning labels to unlabelled points) can be used for a variety of tasks including generating graph edges, estimating node weights (quantitative measure of node importance) as well as estimating and optimizing edge weights (quantitative measure of the importance of the pairwise interaction between nodes) in a network to exploit the structure of graphs and learn models from the data (Karasuyama and Mamitsuka, 2017). Subsequent network optimization techniques introduce perturbations into the network and identify highly perturbed subnetworks to prioritize the most relevant features that correlate with the biological processes under study.
Multiview/multi-modal ML is an emerging method for multi-omics data integration used to exploit information captured in each omics dataset and infer from the associations between the different data types (Nguyen and Wang, 2020). Multi-view learning implements the alignment-based framework and the factorization-based framework (Nguyen and Wang, 2020). The alignment-based framework is a method based on the supervised setting for seeking pairwise alignment among different omics data whereas the factorization-based framework is based on an unsupervised setting for seeking a common representation of features across different omics layers. Deep learning methods, an example of multiview/multi-modal learning, have become one of the more promising integration methods not only because of their ability to exploit the structure of graph neural networks/graph or convolutional networks in both supervised and unsupervised settings with high sensitivity, specificity, and efficiency compared to classical ML methods but also, the predictive performance and capability to capture nonlinear and hierarchical representative features (Martorell-Marugán et al., 2019; Kang et al., 2022). The hierarchical feature processing can capture complex nonlinear associations in a multi-layered manner. The architecture of deep learning models consists of the input layer, hidden layer(s), and output layer. From the perspective of multiomics data integration, most deep learning methods follow the steps of (i) feature selection, (ii) transforming high dimensional multiomics data into low-ranked latent variables, (iii) concatenating multi-omics features into a larger dataset and (iv) analysing the data for the desired task such as node ranking, link prediction, node classification and clustering (Figure 2) (Kang et al., 2022). It is worth noting that the deeper the hidden layer, the more it can learn complex patterns in the data. A major challenge for deep learning methods is the problem of overfitting due to large features and the small sample size of multi-omics data. In addition, a large amount of cleaned data is required to train and validate the model, thus influencing how the model is interpreted (Kang et al., 2022). We refer the reader to a current review on deep learning in multi-omics data integration by Kang et al. (Kang et al., 2022).
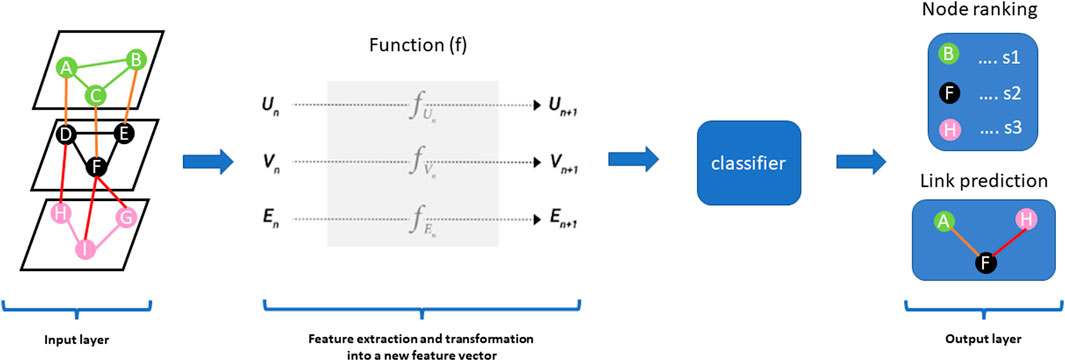
FIGURE 2. Graph Neural Networks (GNNs) are a class of deep learning methods designed to perform inference and predictions on graph data by learning embeddings for graph attributes (nodes, edges, global-context). The concept behind the architecture of these methods is such that it accepts graph data as input and produces the same input graph with updated embeddings before making predictions. GNN uses a function (f) on each graph component vector [nodes vector (Vn), edge vector (En), global-context vector (Un)] in the input graph to learn abstract feature representations of the graph to compute a new feature vector for nodes (Vn+1), edges (En+1) and global-context (Un+1)). The output layer could predict nodes ranked according to a particular score (s1, s2, s3) and also predict edges (links) in the input network.
Network-based diffusion/propagation is a technique for detecting the spread of biological information throughout the network along network edges, thanks to its ability to amplify feature associations based on the hypothesis that node proximity within a network is a measure of their relatedness and contribution to biological processes (Cowen et al., 2017; Di Nanni et al., 2020). The method has been exploited in many network-based analysis pipelines and is suitable for analysing patient-level molecular profiles with different aims including disease subtyping because of its label propagation (Di Nanni et al., 2020). Propagation methods, including random walk, random walk with restart, insulated heat diffusion, and diffusion kernel networks, provide a quantitative estimation of proximity between features associated with different data types by considering all possible paths beyond the shortest paths (Figure 3) (Cowen et al., 2017; Di Nanni et al., 2020).
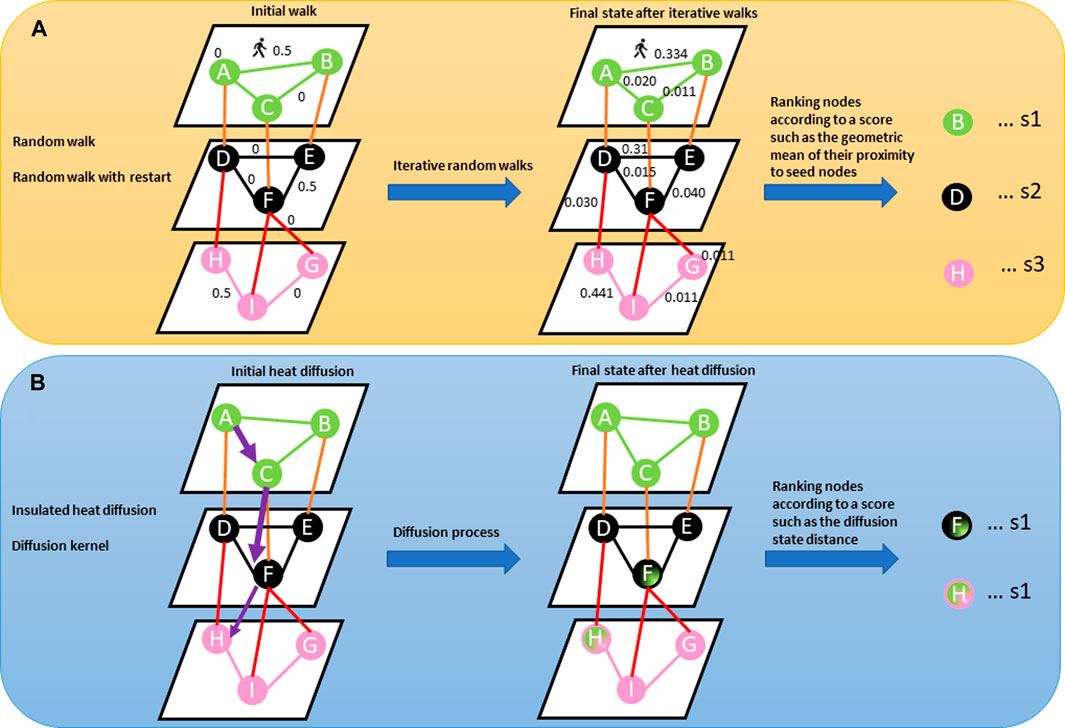
FIGURE 3. (A) Describes a random walk from the seed node (e.g., node A). The concept behind random walk is a guilt-by-association approach where an imaginary particle explores the network structure from seed nodes. The direction of movement of the particle is completely independent of the previous directions moved. At each step, the particle transition from any node in the graph with a certain probability (shown on the edges). The probability flow of random walks on a network is used as a proxy for information flows in the network to study the function of features, subnetworks, and prioritize features in the network. After several iterations, we are interested in the distribution of our position (Stationary distribution) in the graph (final state after iterative walks). The stationary probability distribution can be seen as a measure of the proximity between the seed(s) and all the other nodes in the graph. Nodes within the network can be prioritized using a specific metric (s1, s2, s3) such as the geometric mean of their proximity to seed nodes. (B) Describes heat diffusion from a reference query (e.g., node A). The concept behind heat diffusion in biological networks is perturbing nodes and simulating how the disturbance flows across edges within the network. Node disturbance means adding a scalar value (e.g., log fold changes from gene expression experiment, copy number variations) to node(s). Within a biological network, heat diffusion allows for the assessment of connectivity and topology of features which can allow the identification of relevant/dysregulated pathways and/or mutational effects across edges to neighbouring nodes. The purple arrow means diffusion jumps across different layers. The thickness of the purple arrow signifies the effect of query node (A) on nodes (F) and (H) as shown in nodes (F) and (H) in the final state graph after diffusion. Nodes within the network can be prioritized using a specific metric such as diffusion state distance.
From a data analysis perspective, the network diffusion (ND) methods require omics data and network data. The network data could be obtained from a priori knowledge, inferred from omics data, or generated using a mixed approach of a priori and novel knowledge (Di Nanni et al., 2020). Omics data information, e.g., genetic aberration events underlying differential expression and/or a biological phenotype, are superimposed on the nodes (source nodes) within the network before the information is propagated via the edges until convergence and consensus features are found (Cowen et al., 2017; Di Nanni et al., 2020).
ND methods transform input vectors of scores obtained from the omics data into dense vectors to eliminate missing values and ties. This transformation process can be applied before, after, or during the integration step to refine the results based on molecular network data (Di Nanni et al., 2020). In the ND-before integration approach, the diffusion method is applied to a collection of scores (scores obtained from the omics data) that represent the multi-omics data. The ND-after integration approach is implemented when the various multi-omics data have been initially integrated into a unique structure. The ND-during integration approach is implemented in an instance where each layer exchanges information during the diffusion process. Box 1 provides a summary of the equations related to the diffusion methods.
BOX 1 Summary equations of the network propagation/diffusion methods
The mechanism of action within a biological system is fundamental to understanding such a system. For this reason, biological network inference and causal learning can be used to investigate the direct and indirect multi-layer associations and possible causal relations between omics data features in the system (Griffin et al., 2018).
Causal networks are generally graphical representations that demonstrate likely causal relations between nodes by capturing directional interactions and modelling dependencies between biological variables. The method enables researchers to put directionality between features in a network as well as decipher modules (subnetworks) and/or features associated with patient survival, disease processes, or pinpoint sources of perturbations within multi-omics biological network data (Hawe et al., 2019).
Partial correlation-based networks enable the inference of features regulating co-expression or the activities of other features within the network by estimating conditional dependencies (partial correlations) (Hawe et al., 2019). Partial correlation corrects for spurious associations among features that are mediated by other variables measured in the dataset, thereby reducing the density of the network and enhancing its interpretability (Hawe et al., 2019). These methods have been implemented to infer mechanistic regulatory interactions or predict markers in biological networks (Hawe et al., 2019).
Alternatively, network-based computational frameworks that implement probabilistic graphical models offer attractive solutions for causal reasoning and inference over multi-omics data (Friedman, 2004; Koller and Friedman, 2009; Griffin et al., 2018). A probabilistic graphical model (PGM) is a graph technique for modelling joint probability distributions and (in)dependencies over a set of random variables (Koller and Friedman, 2009). From a data analysis perspective, PGM uses graph-based representation (nodes as features and edges as direct probabilistic interactions between node pairs) as the basis to encode the complex distribution of the data for probabilistic reasoning and inference (Koller and Friedman, 2009). The framework of probabilistic graphical models includes a variety of directed and undirected models (Koller and Friedman, 2009). Directed models (e.g., Bayesian networks) require pre-defined directionality or capture conditional (in)dependencies to assert an influence on features. Undirected models (e.g., Markov networks) are undirected graphical models that offer a simpler perspective on directed models, especially in instances where the directionality of the interactions between features cannot be determined. Compared to directed models which can be used for causal reasoning and inference, undirected models are limited to inference tasks because they fail to capture the influence of nodes on neighbouring nodes.
In addition to partial correlation and probabilistic graphical models, advanced ML models and frameworks that are more computationally efficient have been explored for inferring causal relationships between multi-modal data (Peters et al., 2017; Badsha and Fu, 2019; Luo et al., 2020; Wein et al., 2021). Also, new methods that extend Bayesian networks have been developed for causal inference. For instance, Zheng et al. (Zheng et al., 2018) developed a new method to estimate the structure and inference from a Bayesian network by transforming the structure learning problem into a continuous optimization formulation that does not impose any structural assumptions on the graph. In another instance, Lachapelle et al. (Lachapelle et al., 2019) proposed a novel score-based approach to learning from Bayesian networks via the edge weights of neural networks. The approach developed by the authors adapts the optimization method presented by Zheng et al. (Zheng et al., 2018) to allow for non-linear relationships between variables using neural networks. Box 2 provides a summary of the equations related to the Bayesian and Markov methods. Given that the underlying principles behind network-based approaches for analysis vary, combining such approaches is feasible and may increase prediction accuracy as shown by Zheng et al. (Zheng et al., 2018) and Lacapelle et al. (Lachapelle et al., 2019).
BOX 2 Summary equations of the Bayesian and Markov network.
We systematically reviewed literature primarily published between 2010 and 2022 that report on ML-driven network-based tools, network-based diffusion/propagation tools, and causality- and network-based inference tools. We further highlight the tool’s uniqueness in terms of (i) input data types, (ii) method/algorithm implemented, (iii) most important analytical steps, (iv) potential node and/or edge weighting, and (v) predicted outcome (crosstalk, disease subtypes, biomarkers, subnetworks, and patient survival). The tools presented in this review (Table 1) (i) have broad biomedical data applications and are not restricted to specific (disease) research topics only, (ii) are implemented as standalone software like R, MATLAB, Python libraries, or as part of a pipeline and, (iii) account for the weight of nodes and/or edges within the network.
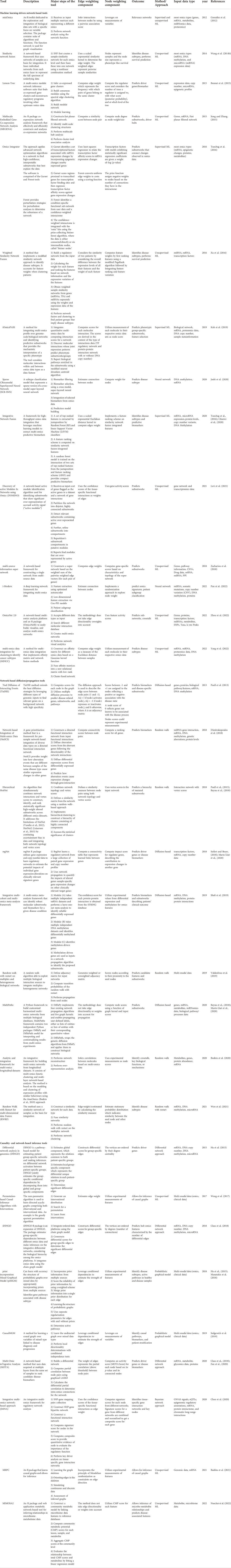
TABLE 1. Network-based multi-omics integrative tools for predicting biomarkers, crosstalk, disease subtypes, and subnetworks/enriched modules.
A perturbed biological system is characterized by deviations in the behaviour of the molecules (omics data features) causing changes in crosstalk (Figure 1). These changes could become apparent in multiple (connected and dependent) omics levels and may represent a wide range of molecular events responsible for disease phenotype or impaired biological processes.
Network-based diffusion/propagation tools (described in Table 1) offer a framework to identify aberrant omics features (e.g., gene expression, somatic mutations, copy number variations, molecular subnetworks informative of disease subtype) and how their presence and activities within the network induce possible (downstream) changes that might underpin disease phenotype.
In a study to understand the molecular function of SARS-CoV-2 and SARS-CoV proteins and their interaction with the human host, Stukalov et al. (Stukalov et al., 2021) profiled the interactomes of both virus groups and investigated the effect of viral infection on the transcriptome, proteome, ubiquitinome, and phosphoproteome of a lung-derived human cell line. Functional analysis of the various biomolecules within a molecular network revealed crosstalk between the cellular processes during perturbations taking place upon infection at different omics layers and pathway levels. The authors (Stukalov et al., 2021) implemented the Hierarchical HotNet ND method to explore host-SARS-CoV-2 protein interactions during viral infection and its impact on omics levels and cell lines to understand how that could influence molecular pathways. Importantly, the group observed that the transforming growth factor beta (TGF-β) signalling pathway, known for its involvement in tissue fibrosis as one of the hallmarks of COVID-19 (Mo et al., 2020), was specifically dysregulated by SARS-CoV-2 ORF8. Further results revealed that autophagy, one of the mechanisms for controlling SARS-CoV-2 replication and monitoring the progression of viral infection (Sargazi et al., 2021), was specifically dysregulated by SARS-CoV-2 ORF3. These findings highlight the biological relevance of crosstalk and the insights it provides to understanding disease mechanisms.
Modular organizations within a network, characterized by clusters of neighbouring nodes highlight features that are functionally related or involved in similar activities within the system. In contrast to identifying (crosstalk of) features informative of disease mechanism, the focus here is on identifying different omics data features that cluster together to inform molecular transitions that describe disease severity level and/or disease subtypes.
Network-based tools that predict disease subtypes or subnetworks informative of a phenotype or a phenotypic group (described in Table 1) are useful for answering such questions and can help in e.g., estimating survival rates across different patient groups. Tools that implement ML and ND-based methods are useful to identify clusters in a network (see Table 1). It is noteworthy that the approach or steps, algorithms, and input data types implemented by such tools to predict subnetworks vary (as described in Table 1). In a recent application of a network-based method to COVID-19 research, Sun et al. (Sun et al., 2021a), employed MEGENA (Song and Zhang, 2015), an unsupervised ML method, to perform protein-metabolite-lipid multi-omics network analysis based on the differential co-expression (correlation between pair of omics features) of these omics data features. The network analysis indicated that tryptophan metabolism and melatonin, a metabolite related to tryptophan metabolism may contribute to molecular transitions in critical COVID-19 patients. Studies have shown that tryptophan and melatonin can improve the immune system and reduce inflammation in COVID-19, suggesting that function disorder may cause impairment to tryptophan metabolism and immune response (Essa et al., 2020; Shneider et al., 2020). Interestingly, activation of tryptophan metabolism has been clinically shown to be selectively enhanced in severe patients (Takeshita and Yamamoto, 2022). The authors further identified pathologically-relevant lipid modules which are being altered among mild COVID-19 patients.
Interestingly, connections between clusters/modules in the omics data may explain the crosstalk of biological features which are specific to the disease state and may serve as biomarkers for monitoring disease progression, treatment, and management (Yan et al., 2016; Overmyer et al., 2020; Su et al., 2020).
The contributory effect of features (nodes) within a system varies and depends on factors including but not limited to the level of feature expression or abundance, the level of interaction with other features, and the (background) state of the system. While some of these omics data features are passive (i.e., have little or no effect on system stability), others may have a significant effect on the observed phenotype.
In many biological disease-related problems, exploring relationships between multi-omics data extends beyond measuring marginal associations between features. Thus, identifying biologically relevant nodes that influence changes within the system could serve as candidate disease-related nodes responsible for an underlying phenotype (Dimitrakopoulos et al., 2018). Causal and network inference methods described in Table 1 can be implemented to explore likely causal features, potential causal relationships, and infer networks that differentiate severe disease from mild in a multi-modal network. Although causal methods provide insights into likely causal agents, investigating and confirming true causality extends beyond computational analysis to experimental validation in relevant models. Also, ML and diffusion-based methods can be used to explore candidate drivers. We describe in Table 1 some network-based tools that predict candidate disease-related nodes. In a recent COVID-19-related study, Tomazou et al. (Tomazou et al., 2021) implemented a network-based multi-omics data integration approach based on a multi-source information super-network scheme (described in Table 1) to prioritize COVID-19-related genes that could be useful as drug targets. The super network was constructed based on the weighted sum of the pairwise weighted edge vectors (for each pair of features) obtained from different sources. The method then prioritizes genes in the network by calculating a characteristic score known as the Multi-source Information Gain (MIG). Some of the genes identified by the authors include Serum Amyloid A (SAA1, SAA2, SAA3) which has been clinically verified as a sensitive biomarker in evaluating the severity and prognosis of COVID-19 (Li et al., 2020), C-reactive protein (CRP) clinically shown to be a marker of systemic inflammation associated with adverse outcomes in COVID-19 patients (Smilowitz et al., 2021), Serine proteinase inhibitor A3 (SERPINA3) shown to be a biomarker for COVID-19-related organ damage (coronary artery disease) and erythropoiesis impairment (Demichev et al., 2021), and vascular cell adhesion molecule (VCAM1) shown to be a vascular and inflammatory implicated in the inflammatory response to sever COVID-19 (Birnhuber et al., 2021).
Network-based methods that employ systematic integration of disease-specific omics profiles coupled with drug-related data (e.g., FDA-approved, experimental drugs, drug-target interactions) into a heterogeneous network have been shown to provide answers to biological questions related to drug development (Wang et al., 2014a; Vitali et al., 2016; Luo et al., 2017). In this type of network analysis, nodes could represent both omics data features and non-omics data features such as drugs, diseases, and drug targets. The edges represent the functional association between the data types such as pharmacological or phenotypic information.
The network-based view of drug discovery and development may involve multiple methods or tools at different steps. ND and ML methods have been widely implemented in this research area to make predictions (Luo et al., 2017; Tomazou et al., 2021). Predictions from such methods present an effective way to complement experimental methods with the aim of, (i) identifying drug targets, (ii) understanding the disease-drug relationship, (iii) investigating drug-target interactions, (iv) identifying potential drug candidates, (v) drug response prediction, (vi) drug-drug relations, and (vii) predict effective drug combinations. Of note, driver nodes or subnetworks as predicted by tools described in Table 1 might also inform on drug targets. An interesting application of network-based methods for drug discovery is the COVID-19 study by Tomazou et al. (Tomazou et al., 2021), whereby some of the predicted candidate compounds including dexamethasone, atorvastatin, beta-estradiol, cyclosporin-A, imatinib, and remdesivir have been found to generate promising results in clinical trials (https://clinicaltrials.gov/). We describe in Table 2, some useful integrative multi-modal network-based tools that are specifically for drug discovery.

TABLE 2. Useful network-based integrative multi-omics tools for drug discovery.
The choice of a network-based integration method does not only depend on the biological question but also the experimental design. Certain network-based methods can only deal with paired data, whereas others can also deal with sparse datasets where there is no or only partial overlap between the samples profiled with the different omics layers. Importantly, the scope of the research will inform the type of data that should be generated. For instance, the paired data, herein referring to different omics data measurements from the same biological sample, is preferred when establishing a holistic picture of systems biology underpinning molecular mechanisms linked to disorders, whereas non-paired data (data generated from different biological samples) is more appropriate for comparative (meta)analysis of samples or omics data measurements. It is therefore recommended to consider the scope of research and the network-based methods that fit.
Researchers routinely expect that results generated by applying network models are reproducible. For network-based methods, the key issues related to reproducibility are non-harmonized data, biased model evaluation, and lack of transferable code or software. First, multi-omics network-based integration involves the use of heterogeneous data, and some sort of data harmonization is required. A promising approach to harmonize multi-omics research is to ensure that the data comply with FAIR data principles (findability, accessibility, interoperability, and reusability). The data FAIRification process ensures that a (meta)data schema/method which captures relations between (omics) measurements, data structure, and concepts are clearly defined and easily interpretable by both humans and computers. The metadata schema provides information about the omics data structure and facilitates easy mapping of measured features onto persistent identifiers and established biological networks to investigate the connection between network elements (Krassowski et al., 2020). Second, confidence in multi-omics network-based methods requires systematic evaluation and validation of both datasets and models as a prerequisite for benchmarking toward reproducibility (Krassowski et al., 2020). This approach requires harmonized datasets of quality and quantity that provide unbiased ground truth to ensure that the model at least predicts biologically verified features or edges. Given that there is no gold standard metric for validation, it is critical to validate on a variety of data sources and use metrics that are robust to the level of missing data. Third, to replicate results from previous studies, a detailed report of the analysis together with executable analysis code is important to achieve this purpose. The report and code could be hosted in repositories (e.g., GitHub, Bitbucket, GitLab), reproducible scientific workflow management systems (e.g., Nextflow, Galaxy), environment sharing avenues (e.g., Conda, Docker), or packaged as libraries for programming languages (Canzler et al., 2020). In addition to the key issues, adapting general best practices in the computational analysis will aid reproducibility.
Heterogeneity (a measure of variation) of multi-omics datasets, characterized by diverse data sources, data types, and data structure results in computational complexity, analysis bias, and hampers a robust and reproducible integrative network analysis (Lee et al., 2021). There is an increasing awareness of controlling heterogeneity across multi-omics integrative analysis, but most of them are focused on paired data rather than non-paired data.
In the context of network-based integrative analysis developing models and algorithms that could account for non-uniformity by identifying the most robust signals encompassing data, heterogeneity is important. This could be in the form of variable selection models to identify important covariates with the strength of multiple datasets, and yet maintain the flexibility of variable selection between the datasets to account for the data heterogeneity (Lima et al., 2020).
Interpreting results from an integrative multi-omics analysis is a process of disentangling multiple functional relationships. Primarily, the systematic interpretation of results depends on the kind of biological question and the type of omics measurements used for the analysis. Different omics technologies may have different levels of completeness and sensitivity in terms of detecting biological features. This might result in some omics data types containing more information than others as well as impact the results significantly (Jung et al., 2020). It is important to consider the inherent relationship between the omics profiles used during the interpretation of the results. More often functional annotation of features is based on generalized information which allows a less comprehensive understanding of the molecular mechanisms underlying a phenotype. For this reason, incorporating relevant contextualized pathway information (e.g., tissue-specific or cell-specific) in the analysis has been useful to assess the functional relevance of nodes and subnetworks on the disease/phenotypic landscape, thereby facilitating interpretation.
The capacity to interpret predicted features and interactions of known biological relevance may take the form of deductive reasoning or semantic similarities to support a hypothesis (Guo et al., 2022). In the context of algorithms, robust node weighting and edge weighting metrics measured based on known evidence (e.g., text mining, contextualized pathway information) is important to make an inference that is potentially biologically grounded and experimentally confirmable, knowing that the association between omics layers extends from one-to-one and one-to-many to many-to-many.
There is sparsity at the sample level (not all samples have been profiled with the same assays) and at the feature level. The latter is far more prominent in metabolomics and proteomics than in DNA and RNA sequencing. This is mainly due to the selection of peaks (intensities observed in MS1 survey scans) for fragmentation by data-dependent acquisition (DDA) or data-independent acquisition (DIA) tandem mass spectrometry (LC-MS/MS) approaches (Guo and Huan, 2020; Davies et al., 2021). Typically, an ideal acquisition mode ought to produce spectra of high quality for as many of the ions present in the sample as possible, however, that is not the case, resulting in sparsity at the feature level. This issue is partly but not completely resolved in the newer DIA and integrated DDA-DIA modes which operate in a less-selective manner and have higher coverage as compared to the older DDA mode (Sun et al., 2021b; Davies et al., 2021).
Another contributing factor to sparsity in omics data in tandem with omics technologies is the absence of accumulation of a molecule to a detectable level by omics platforms (evidenced even across platforms of the same omics technology (e.g., next-generation RNA and DNA sequencing). This is partly associated with experimental design, poor biological sample quality, and sample processing.
For computational analysis purposes, mputation can be used to solve missing value problems; however, imputation does not apply to all omics data types (Folch-Fortuny et al., 2015). In addition to imputation, sample similarity measurement methods such as matrix calibration (Li, 2015) and the Mahalanobis distance approach (Sitaram et al., 2015) could be useful to extrapolate for missing values, however, these methods are also limited to specific omics data types. Thus, a feature may have values only in a small percentage of samples leading to sparse matrices, where features may have a wide variety of distributions. Some multi-omics data integration methods can handle sparse data and also feature reduction methods; however, skewed estimates might result in a biased interpretation of results (Greenland et al., 2016). To address the issue of sparsity in the context of networks, network integration aggregates independent data sources to form a more comprehensive attributed interactome, where the edges are qualified by specific semantic relations or similarity correlation, and the level of confidence in the node pair relationship based on evidence from similarity scores, literature and graph databases (Guo et al., 2022). Also, incorporating autoencoders, a deep learning approach, and its denoising and variational variants autoencoders (e.g., sparse autoencoders) have been used to address this issue in graph neural networks (Ng, 2011). Autoencoders learn a representation of the data from the input layer, enforce sparsity constraints and try to reproduce it at the output layer. During this process, the model can learn from incomplete data and generate new plausible values for imputation (Pereira et al., 2020).
An area of prospect for integrative multi-omics network-based research, which remains an important opportunity, is making efforts to limit the challenges linked with network-based multi-omics integration in the context of heterogeneity, reproducibility, sparsity, and interpretation of results as discussed above. Another area of importance is building hybrid integrative models which are capable of handling paired and non-paired omics data, as well as other biomedical data. Furthermore, efforts to develop a framework tool or metadata schema that standardizes or harmonizes various multi-omics approaches for data integration could be useful. For example, such a framework may leverage an optimized approach to weigh and prioritize genes, pathways, biological processes, drug targets, and relationships between various other biological features from the multi-omics datasets. However, such framework tools will also require the expertise of domain experts, as well as the detailed and uniform characterization of statistical and technical attributes of the data (Krassowski et al., 2020).
Network-based integrative multi-omics analysis offers the opportunity to elucidate interactions that can occur among all classes of molecules in a biological system as well as information flow between and within multiple omics levels. In addition, it potentially provides substantial improvement of biological understanding by helping in the interpretation of results, as compared to single omics analysis, although collecting multi-omics data from different sources does not guarantee that it will be possible to learn about (all of) the relationships present.
Various graph-based multi-omics methods have been developed for network analysis; however, their application is dependent on the scope of the research question of interest and the (omics) data types available. Consequently, this will inform the choice of an integrative analytical approach and tools. The network-based methods discussed use different scoring metrics, algorithms, and data types which together translate into a comprehensive data source/graph to be employed for interpretation into biological knowledge. The overviewand description of the tools for network-based integrative analysis (Table 1) show that different approaches can be implemented in different ways to achieve similar results. Additionally, the classification of tools (Figure 4) highlights that some tools can be applied to more than one research question. However, due to the difference in approaches of these methods, we recommend the use of multiple analytical and methodological approaches during integrative data analysis, to compare and validate the study results in different ways before interpretation for further downstream tests or follow-up studies.
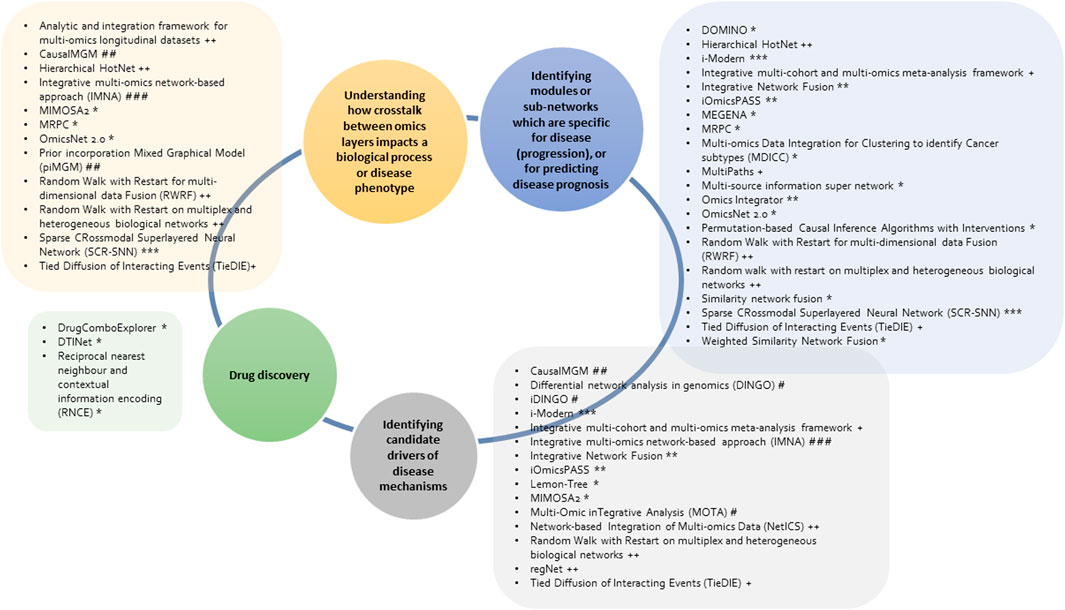
FIGURE 4. Overview of the discussed network-based multi-omics integrative tools and research questions (in the circle) that they can be applied to. The tools implement different methods including unsupervised machine learning (*), supervised machine learning (**), neural networks (***), diffusion-based (+), random walk (++), differential network (#), probabilistic graphical model (##) and Bayesian methods (###).
FA conceived the study and prepared the first draft. JB, AN, KN, MS, GM, NM, EC, TE, and PH contributed to the revision of the article. GM, EC, TE, and PH supervised the work. All authors contributed to the article and approved the submitted version.
This work was partially funded by an LSH HealthHolland grant to the TWOC consortium, a large-scale infrastructure grant from the Dutch Organization of Scientific Research (NWO) to the Netherlands X-omics initiative (184.034.019), and a Horizon2020 research grant from the European Union to the EATRIS-Plus infrastructure project (grant agreement: No 871096).
We acknowledge members of the Trusted World of Corona (TWOC) Consortium. We also acknowledge the staff and colleagues from the Division of Human Genetics and Division of Computational Biology, University of Cape Town, and colleagues from the Center for Molecular and Biomolecular Informatics (CMBI), Radboud University Medical Center, Nijmegen. In memorial of GM who passed away due to COVID-19 complications before the submission of this manuscript. His contribution as supervisor of FA, supervising this work, writing, and revising multi sections of this manuscript will never be forgotten.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agamah, F. E., Damena, D., Skelton, M., Ghansah, A., Mazandu, G. K., and Chimusa, E. R. (2021). Network-driven analysis of human–plasmodium falciparum interactome: Processes for malaria drug discovery and extracting in silico targets. Malar. J. 20 (1), 421. doi:10.1186/s12936-021-03955-0
Badsha, M., and Fu, A. Q. (2019). Learning causal biological networks with the principle of Mendelian randomization. Front. Genet. 10, 460. doi:10.3389/fgene.2019.00460
Badsha, M. B., Martin, E. A., and Fu, A. Q. (2021). Mrpc: An R package for inference of causal graphs. Front. Genet. 12, 460. doi:10.3389/fgene.2019.00460
Bersanelli, M., Mosca, E., Remondini, D., Giampieri, E., Sala, C., Castellani, G., et al. (2016). Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinforma. 17 (2), 15–77. doi:10.1186/s12859-015-0857-9
Birnhuber, A., Fliesser, E., Gorkiewicz, G., Zacharias, M., Seeliger, B., David, S., et al. (2021). Between inflammation and thrombosis: Endothelial cells in COVID-19. Eur. Respir. J. 58 (3), 2100377. doi:10.1183/13993003.00377-2021
Bodein, A., Chapleur, O., Droit, A., and Lê Cao, K-A. (2019). A generic multivariate framework for the integration of microbiome longitudinal studies with other data types. Front. Genet. 10, 963. doi:10.3389/fgene.2019.00963
Bodein, A., Scott-Boyer, M. P., Perin, O., Le Cao, K-A., and Droit, A. (2020). Interpretation of network-based integration from multi-omics longitudinal data. Nucleic acids Res. 50, e27. doi:10.1093/nar/gkab1200
Bonnet, E., Calzone, L., and Michoel, T. (2015). Integrative multi-omics module network inference with Lemon-Tree. PLoS Comput. Biol. 11 (2), e1003983. doi:10.1371/journal.pcbi.1003983
Buescher, J. M., and Driggers, E. M. (2016). Integration of omics: More than the sum of its parts. Cancer Metab. 4 (1), 4–8. doi:10.1186/s40170-016-0143-y
Camacho, D. M., Collins, K. M., Powers, R. K., Costello, J. C., and Collins, J. J. (2018). Next-generation machine learning for biological networks. Cell. 173 (7), 1581–1592. doi:10.1016/j.cell.2018.05.015
Canzler, S., Schor, J., Busch, W., Schubert, K., Rolle-Kampczyk, U. E., Seitz, H., et al. (2020). Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 94 (2), 371–388. doi:10.1007/s00204-020-02656-y
Cavill, R., Jennen, D., Kleinjans, J., and Briedé, J. J. (2016). Transcriptomic and metabolomic data integration. Brief. Bioinform. 17 (5), 891–901. doi:10.1093/bib/bbv090
Chakravorty, D., Banerjee, K., and Saha, S. (2018). Integrative omics for interactomes. Synthetic Biology. Berlin, Germany: Springer, 39–49.
Chen, J., and Wong, K-C. R. N. C. E. (2021). RNCE: Network integration with reciprocal neighbors contextual encoding for multi-modal drug community study on cancer targets. Brief. Bioinform. 22 (3), bbaa118. doi:10.1093/bib/bbaa118
Chen, Y. X., Chen, H., Rong, Y., Jiang, F., Chen, J. B., Duan, Y. Y., et al. (2020). An integrative multi-omics network-based approach identifies key regulators for breast cancer. Comput. Struct. Biotechnol. J. 18, 2826–2835. doi:10.1016/j.csbj.2020.10.001
Chierici, M., Bussola, N., Marcolini, A., Francescatto, M., Zandonà, A., Trastulla, L., et al. (2020). Integrative network fusion: A multi-omics approach in molecular profiling. Front. Oncol. 10, 1065. doi:10.3389/fonc.2020.01065
Class, C. A., Ha, M. J., Baladandayuthapani, V., and Do, K-A. (2018). iDINGO—integrative differential network analysis in genomics with Shiny application. Bioinformatics 34 (7), 1243–1245. doi:10.1093/bioinformatics/btx750
Cowen, L., Ideker, T., Raphael, B. J., and Sharan, R. (2017). Network propagation: A universal amplifier of genetic associations. Nat. Rev. Genet. 18 (9), 551–562. doi:10.1038/nrg.2017.38
Davies, V., Wandy, J., Weidt, S., Van Der Hooft, J. J., Miller, A., Daly, R., et al. (2021). Rapid development of improved data-dependent acquisition strategies. Anal. Chem. 93 (14), 5676–5683. doi:10.1021/acs.analchem.0c03895
Demichev, V., Tober-Lau, P., Lemke, O., Nazarenko, T., Thibeault, C., Whitwell, H., et al. (2021). A time-resolved proteomic and prognostic map of COVID-19. Cell. Syst. 12 (8), 780–794.e7. doi:10.1016/j.cels.2021.05.005
Di Nanni, N., Bersanelli, M., Milanesi, L., and Mosca, E. (2020). Network diffusion promotes the integrative analysis of multiple omics. Front. Genet. 11, 106. doi:10.3389/fgene.2020.00106
Dimitrakopoulos, C., Hindupur, S. K., Häfliger, L., Behr, J., Montazeri, H., Hall, M. N., et al. (2018). Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics 34 (14), 2441–2448. doi:10.1093/bioinformatics/bty148
Duruflé, H., Selmani, M., Ranocha, P., Jamet, E., Dunand, C., and Déjean, S. (2021). A powerful framework for an integrative study with heterogeneous omics data: From univariate statistics to multi-block analysis. Brief. Bioinform. 22 (3), bbaa166. doi:10.1093/bib/bbaa166
Essa, M. M., Hamdan, H., Chidambaram, S. B., Al-Balushi, B., Guillemin, G. J., Ojcius, D. M., et al. (2020). Possible role of tryptophan and melatonin in COVID-19. London, England: SAGE Publications Sage UK, 1178646920951832.
Fan, Z., Zhou, Y., and Ressom, H. W. (2020). MOTA: Network-based multi-omic data integration for biomarker discovery. Metabolites 10 (4), 144. doi:10.3390/metabo10040144
Folch-Fortuny, A., Villaverde, A. F., Ferrer, A., and Banga, J. R. (2015). Enabling network inference methods to handle missing data and outliers. BMC Bioinforma. 16 (1), 283. doi:10.1186/s12859-015-0717-7
Friedman, N. (2004). Inferring cellular networks using probabilistic graphical models. Science 303 (5659), 799–805. doi:10.1126/science.1094068
González, I., Lê Cao, K-A., Davis, M. J., and Déjean, S. (2012). Visualising associations between paired ‘omics’ data sets. BioData Min. 5 (1), 19–23. doi:10.1186/1756-0381-5-19
Greenland, S., Mansournia, M. A., and Altman, D. G. (2016). Sparse data bias: A problem hiding in plain sight. BMJ 352, i1981. doi:10.1136/bmj.i1981
Griffin, P. J., Zhang, Y., Johnson, W. E., and Kolaczyk, E. D. (2018). Detection of multiple perturbations in multi-omics biological networks. Biometrics 74 (4), 1351–1361. doi:10.1111/biom.12893
Guo, J., and Huan, T. (2020). Comparison of full-scan, data-dependent, and data-independent acquisition modes in liquid chromatography–mass spectrometry based untargeted metabolomics. Anal. Chem. 92 (12), 8072–8080. doi:10.1021/acs.analchem.9b05135
Guo, M. G., Sosa, D. N., and Altman, R. B. (2022). Challenges and opportunities in network-based solutions for biological questions. Brief. Bioinform. 23 (1), bbab437. doi:10.1093/bib/bbab437
Ha, M. J., Baladandayuthapani, V., and Do, K. A. (2015). DINGO: Differential network analysis in genomics. Bioinformatics 31 (21), 3413–3420. doi:10.1093/bioinformatics/btv406
Hasin, Y., Seldin, M., and Lusis, A. (2017). Multi-omics approaches to disease. Genome Biol. 18 (1), 83–15. doi:10.1186/s13059-017-1215-1
Hawe, J. S., Theis, F. J., and Heinig, M. (2019). Inferring interaction networks from multi-omics data. Front. Genet. 10, 535. doi:10.3389/fgene.2019.00535
Holzinger, E. R., and Ritchie, M. D. (2012). Integrating heterogeneous high-throughput data for meta-dimensional pharmacogenomics and disease-related studies. Pharmacogenomics 13 (2), 213–222. doi:10.2217/pgs.11.145
Horgan, R. P., and Kenny, L. C. (2011). ‘Omic’ technologies: Genomics, transcriptomics, proteomics and metabolomics. Obstetrician Gynaecol. 13 (3), 189–195. doi:10.1576/toag.13.3.189.27672
Huang, L., Brunell, D., Stephan, C., Mancuso, J., Yu, X., He, B., et al. (2019). Driver network as a biomarker: Systematic integration and network modeling of multi-omics data to derive driver signaling pathways for drug combination prediction. Bioinformatics 35 (19), 3709–3717. doi:10.1093/bioinformatics/btz109
Joshi, P., Jeong, S., and Park, T. (2020). Sparse superlayered neural network-based multi-omics cancer subtype classification. Int. J. Data Min. Bioinform. 24 (1), 58–73. doi:10.1504/ijdmb.2020.109500
Jung, G. T., Kim, K-P., and Kim, K. (2020). How to interpret and integrate multi-omics data at systems level. Anim. Cells Syst. 24 (1), 1–7. doi:10.1080/19768354.2020.1721321
Kang, M., Ko, E., and Mersha, T. B. (2022). A roadmap for multi-omics data integration using deep learning. Brief. Bioinform. 23 (1), bbab454. doi:10.1093/bib/bbab454
Karasuyama, M., and Mamitsuka, H. (2017). Adaptive edge weighting for graph-based learning algorithms. Mach. Learn. 106 (2), 307–335. doi:10.1007/s10994-016-5607-3
Karczewski, K. J., and Snyder, M. P. (2018). Integrative omics for health and disease. Nat. Rev. Genet. 19 (5), 299–310. doi:10.1038/nrg.2018.4
Koh, H. W., Fermin, D., Vogel, C., Choi, K. P., Ewing, R. M., and Choi, H. (2019). iOmicsPASS: network-based integration of multiomics data for predictive subnetwork discovery. NPJ Syst. Biol. Appl. 5 (1), 22–10. doi:10.1038/s41540-019-0099-y
Koller, D., and Friedman, N. (2009). Probabilistic graphical models: Principles and techniques. Cambridge, MA, USA: MIT press.
Kotiang, S., and Eslami, A. (2020). A probabilistic graphical model for system-wide analysis of gene regulatory networks. Bioinformatics 36 (10), 3192–3199. doi:10.1093/bioinformatics/btaa122
Krassowski, M., Das, V., Sahu, S. K., and Misra, B. B. (2020). State of the field in multi-omics research: From computational needs to data mining and sharing. Front. Genet. 11, 610798. doi:10.3389/fgene.2020.610798
Lachapelle, S., Brouillard, P., Deleu, T., and Lacoste-Julien, S. (2019). Gradient-based neural dag learning. arXiv preprint arXiv:02226.
Lee, D., Park, Y., and Kim, S. (2021). Towards multi-omics characterization of tumor heterogeneity: A comprehensive review of statistical and machine learning approaches. Brief. Bioinform. 22 (3), bbaa188. doi:10.1093/bib/bbaa188
Leiserson, M. D., Vandin, F., Wu, H-T., Dobson, J. R., Eldridge, J. V., Thomas, J. L., et al. (2015). Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 47 (2), 106–114. doi:10.1038/ng.3168
Levi, H., Elkon, R., and Shamir, R. (2021). DOMINO: A network-based active module identification algorithm with reduced rate of false calls. Mol. Syst. Biol. 17 (1), e9593. doi:10.15252/msb.20209593
Li, H., Xiang, X., Ren, H., Xu, L., Zhao, L., Chen, X., et al. (2020). Serum Amyloid A is a biomarker of severe Coronavirus Disease and poor prognosis. J. Infect. 80 (6), 646–655. doi:10.1016/j.jinf.2020.03.035
Li, W., (2015). “Estimating jaccard index with missing observations: A matrix calibration approach,” in Proceedings of the 28th International Conference on Neural Information Processing Systems-Volume 2.
Lima, E., Davies, P., Kaler, J., Lovatt, F., and Green, M. (2020). Variable selection for inferential models with relatively high-dimensional data: Between method heterogeneity and covariate stability as adjuncts to robust selection. Sci. Rep. 10 (1), 8002–8011. doi:10.1038/s41598-020-64829-0
Luo, Y., Peng, J., and Ma, J. (2020). When causal inference meets deep learning. Nat. Mach. Intell. 2 (8), 426–427. doi:10.1038/s42256-020-0218-x
Luo, Y., Zhao, X., Zhou, J., Yang, J., Zhang, Y., Kuang, W., et al. (2017). A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8 (1), 573. doi:10.1038/s41467-017-00680-8
Manatakis, D. V., Raghu, V. K., and Benos, P. V. (2018). piMGM: incorporating multi-source priors in mixed graphical models for learning disease networks. Bioinformatics 34 (17), i848–i856. doi:10.1093/bioinformatics/bty591
Marín-Llaó, J., Mubeen, S., Perera-Lluna, A., Hofmann-Apitius, M., Picart-Armada, S., and Domingo-Fernández, D. (2020). MultiPaths: A Python framework for analyzing multi-layer biological networks using diffusion algorithms. Bioinformatics 37, 137. doi:10.1093/bioinformatics/btaa1069
Martorell-Marugán, J., Tabik, S., Benhammou, Y., del Val, C., Zwir, I., Herrera, F., et al. (2019). Deep learning in omics data analysis and precision medicine. Brisbane City, Australia: Exon Publications, 37–53.
Mo, X., Jian, W., Su, Z., Chen, M., Peng, H., Peng, P., et al. (2020). Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur. Respir. J. 55 (6), 2001217. doi:10.1183/13993003.01217-2020
Nguyen, N. D., and Wang, D. (2020). Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 16 (4), e1007677. doi:10.1371/journal.pcbi.1007677
Noecker, C., Eng, A., Muller, E., and Borenstein, E. (2022). MIMOSA2: A metabolic network-based tool for inferring mechanism-supported relationships in microbiome-metabolome data. Bioinformatics 38 (6), 1615–1623. doi:10.1093/bioinformatics/btac003
Overmyer, K. A., Shishkova, E., Miller, I. J., Balnis, J., Bernstein, M. N., Peters-Clarke, T. M., et al. (2020). Large-scale multi-omic analysis of COVID-19 severity. Cell. Syst. 12, 23–40.e7. doi:10.1016/j.cels.2020.10.003
Pan, X., Burgman, B., Wu, E., Huang, J. H., Sahni, N., and Yi, S. S. (2022). i-Modern: Integrated multi-omics network model identifies potential therapeutic targets in glioma by deep learning with interpretability. Comput. Struct. Biotechnol. J. 20, 3511–3521. doi:10.1016/j.csbj.2022.06.058
Paull, E. O., Carlin, D. E., Niepel, M., Sorger, P. K., Haussler, D., and Stuart, J. M. (2013). Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion through Interacting Events (TieDIE). Bioinformatics 29 (21), 2757–2764. doi:10.1093/bioinformatics/btt471
Pereira, R. C., Santos, M. S., Rodrigues, P. P., and Abreu, P. H. (2020). Reviewing autoencoders for missing data imputation: Technical trends, applications and outcomes. J. Artif. Intell. Res. 69, 1255–1285. doi:10.1613/jair.1.12312
Peters, J., Janzing, D., and Schölkopf, B. (2017). Elements of causal inference: Foundations and learning algorithms. Cambridge, MA, USA: The MIT Press.
Reel, P. S., Reel, S., Pearson, E., Trucco, E., and Jefferson, E. (2021). Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 49, 107739. doi:10.1016/j.biotechadv.2021.107739
Reyna, M. A., Leiserson, M. D., and Raphael, B. J. (2018). Hierarchical HotNet: Identifying hierarchies of altered subnetworks. Bioinformatics 34 (17), i972–i980. doi:10.1093/bioinformatics/bty613
Ritchie, M. D., Holzinger, E. R., Li, R., Pendergrass, S. A., and Kim, D. (2015). Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 16 (2), 85–97. doi:10.1038/nrg3868
Sargazi, S., Sheervalilou, R., Rokni, M., Shirvaliloo, M., Shahraki, O., and Rezaei, N. (2021). The role of autophagy in controlling SARS-CoV-2 infection: An overview on virophagy-mediated molecular drug targets. Cell. Biol. Int. 45 (8), 1599–1612. doi:10.1002/cbin.11609
Sedgewick, A. J., Buschur, K., Shi, I., Ramsey, J. D., Raghu, V. K., Manatakis, D. V., et al. (2019). Mixed graphical models for integrative causal analysis with application to chronic lung disease diagnosis and prognosis. Bioinformatics 35 (7), 1204–1212. doi:10.1093/bioinformatics/bty769
Seifert, M., and Beyer, A. (2018). regNet: An R package for network-based propagation of gene expression alterations. Bioinformatics 34 (2), 308–311. doi:10.1093/bioinformatics/btx544
Shafi, A., Nguyen, T., Peyvandipour, A., Nguyen, H., and Draghici, S. (2019). A multi-cohort and multi-omics meta-analysis framework to identify network-based gene signatures. Front. Genet. 10, 159. doi:10.3389/fgene.2019.00159
Shneider, A., Kudriavtsev, A., and Vakhrusheva, A. (2020). Can melatonin reduce the severity of COVID-19 pandemic? Int. Rev. Immunol. 39 (4), 153–162. doi:10.1080/08830185.2020.1756284
Sitaram, D., Dalwani, A., Narang, A., Das, M., and Auradkar, P. (2015). “A measure of similarity of time series containing missing data using the mahalanobis distance,” in 2015 second international conference on advances in computing and communication engineering, 01-02 May 2015 (Dehradun, India: IEEE). doi:10.1109/ICACCE.2015.14
Smilowitz, N. R., Kunichoff, D., Garshick, M., Shah, B., Pillinger, M., Hochman, J. S., et al. (2021). C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Heart J. 42 (23), 2270–2279. doi:10.1093/eurheartj/ehaa1103
Song, W. M., and Zhang, B. (2015). Multiscale embedded gene co-expression network analysis. PLoS Comput. Biol. 11 (11), e1004574. doi:10.1371/journal.pcbi.1004574
Stukalov, A., Girault, V., Grass, V., Karayel, O., Bergant, V., Urban, C., et al. (2021). Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 594 (7862), 246–252. doi:10.1038/s41586-021-03493-4
Su, Y., Chen, D., Yuan, D., Lausted, C., Choi, J., Dai, C. L., et al. (2020). Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell. 183, 1479–1495. doi:10.1016/j.cell.2020.10.037
Sun, C., Sun, Y., Wu, P., Ding, W., Wang, S., Li, J., et al. (2021). Longitudinal multi-omics transition associated with fatality in critically ill COVID-19 patients. Intensive Care Med. Exp. 9 (1), 13–14. doi:10.1186/s40635-021-00373-z
Sun, F., Tan, H., Li, Y., De Boevre, M., Zhang, H., Zhou, J., et al. (2021). An integrated data-dependent and data-independent acquisition method for hazardous compounds screening in foods using a single UHPLC-Q-Orbitrap run. J. Hazard. Mat. 401, 123266. doi:10.1016/j.jhazmat.2020.123266
Sun, Y. V., and Hu, Y-J. (2016). Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases. Adv. Genet. 93, 147–190. doi:10.1016/bs.adgen.2015.11.004
Takeshita, H., and Yamamoto, K. (2022). Tryptophan metabolism and COVID-19-induced skeletal muscle damage: Is ACE2 a key regulator? Front. Nutr. 9, 868845. doi:10.3389/fnut.2022.868845
Tomazou, M., Bourdakou, M. M., Minadakis, G., Zachariou, M., Oulas, A., Karatzas, E., et al. (2021). Multi-omics data integration and network-based analysis drives a multiplex drug repurposing approach to a shortlist of candidate drugs against COVID-19. Briefings Bioinforma. 22, bbab114. doi:10.1093/bib/bbab114
Tuncbag, N., Gosline, S. J., Kedaigle, A., Soltis, A. R., Gitter, A., and Fraenkel, E. (2016). Network-based interpretation of diverse high-throughput datasets through the omics integrator software package. PLoS Comput. Biol. 12 (4), e1004879. doi:10.1371/journal.pcbi.1004879
Ulfenborg, B. (2019). Vertical and horizontal integration of multi-omics data with miodin. BMC Bioinforma. 20 (1), 649. doi:10.1186/s12859-019-3224-4
Valdeolivas, A., Tichit, L., Navarro, C., Perrin, S., Odelin, G., Levy, N., et al. (2019). Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics 35 (3), 497–505. doi:10.1093/bioinformatics/bty637
Vandin, F., Clay, P., Upfal, E., and Raphael, B. J. (2012). Discovery of mutated subnetworks associated with clinical data in cancer. Pac Symp. Biocomput 2012, 55–66.
Vitali, F., Cohen, L. D., Demartini, A., Amato, A., Eterno, V., Zambelli, A., et al. (2016). A network-based data integration approach to support drug repurposing and multi-target therapies in triple negative breast cancer. PloS one 11 (9), e0162407. doi:10.1371/journal.pone.0162407
Wang, B., Mezlini, A. M., Demir, F., Fiume, M., Tu, Z., Brudno, M., et al. (2014). Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 11 (3), 333–337. doi:10.1038/nmeth.2810
Wang, W., Yang, S., Zhang, X., and Li, J. (2014). Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 30 (20), 2923–2930. doi:10.1093/bioinformatics/btu403
Wang, Y., Solus, L., Yang, K. D., and Uhler, C. (2017). Permutation-based causal inference algorithms with interventions. Adv. Neural Inf. Process. Syst.
Wein, S., Malloni, W. M., Tomé, A. M., Frank, S. M., Henze, G-I., Wüst, S., et al. (2021). A graph neural network framework for causal inference in brain networks. Sci. Rep. 11 (1), 8061. doi:10.1038/s41598-021-87411-8
Wen, Y., Song, X., Yan, B., Yang, X., Wu, L., Leng, D., et al. (2021). Multi-dimensional data integration algorithm based on random walk with restart. BMC Bioinforma. 22 (1), 97–22. doi:10.1186/s12859-021-04029-3
Wu, Z., Li, W., Liu, G., and Tang, Y. (2018). Network-based methods for prediction of drug-target interactions. Front. Pharmacol. 9, 1134. doi:10.3389/fphar.2018.01134
Xu, T., Le, T. D., Liu, L., Wang, R., Sun, B., and Li, J. (2016). Identifying cancer subtypes from mirna-tf-mrna regulatory networks and expression data. PloS one 11 (4), e0152792. doi:10.1371/journal.pone.0152792
Yan, J., Risacher, S. L., Shen, L., and Saykin, A. J. (2018). Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data. Brief. Bioinform. 19 (6), 1370–1381. doi:10.1093/bib/bbx066
Yan, W., Xue, W., Chen, J., and Hu, G. (2016). Biological networks for cancer candidate biomarkers discovery. Cancer Inf. 15, S39458. doi:10.4137/CIN.S39458
Yang, Y., Tian, S., Qiu, Y., Zhao, P., and Zou, Q. (2022). MDICC: Novel method for multi-omics data integration and cancer subtype identification. Brief. Bioinform. 23 (3), bbac132. doi:10.1093/bib/bbac132
Zachariou, M., Minadakis, G., Oulas, A., Afxenti, S., and Spyrou, G. M. (2018). Integrating multi-source information on a single network to detect disease-related clusters of molecular mechanisms. J. Proteomics 188, 15–29. doi:10.1016/j.jprot.2018.03.009
Zapalska-Sozoniuk, M., Chrobak, L., Kowalczyk, K., and Kankofer, M. (2019). Is it useful to use several “omics” for obtaining valuable results? Mol. Biol. Rep. 46 (3), 3597–3606. doi:10.1007/s11033-019-04793-9
Zeng, I. S. L., and Lumley, T. (2018). Review of statistical learning methods in integrated omics studies (an integrated information science). Bioinform. Biol. Insights 12, 1177932218759292. doi:10.1177/1177932218759292
Zheng, X., Aragam, B., Ravikumar, P., and Xing, E. P. (2018). Dags with no tears: Continuous optimization for structure learning. Adv. Neural Inf. Process. Syst.
Zhou, G., Pang, Z., Lu, Y., Ewald, J., and Xia, J. (2022). OmicsNet 2.0: A web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 50, W527–W533. doi:10.1093/nar/gkac376
Keywords: multi-omics, data integration, multi-modal network, machine learning, network diffusion/propagation, network causal inference
Citation: Agamah FE, Bayjanov JR, Niehues A, Njoku KF, Skelton M, Mazandu GK, Ederveen THA, Mulder N, Chimusa ER and 't Hoen PAC (2022) Computational approaches for network-based integrative multi-omics analysis. Front. Mol. Biosci. 9:967205. doi: 10.3389/fmolb.2022.967205
Received: 12 June 2022; Accepted: 20 October 2022;
Published: 14 November 2022.
Edited by:
Sumeet Agarwal, Indian Institute of Technology Delhi, IndiaReviewed by:
Eve Syrkin Wurtele, Iowa State University, United StatesCopyright © 2022 Agamah, Bayjanov, Niehues, Njoku, Skelton, Mazandu, Ederveen, Mulder, Chimusa and 't Hoen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas H. A. Ederveen, VG9tLkVkZXJ2ZWVuQHJhZGJvdWR1bWMubmw=; Emile R. Chimusa, ZW1pbGUuY2hpbXVzYUBub3J0aHVtYnJpYS5hYy51aw==; Peter A. C. 't Hoen, UGV0ZXItQnJhbS50SG9lbkByYWRib3VkdW1jLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.