 Siying Ren
Siying Ren Yue Chen
Yue Chen Likun Wang1
Likun Wang1 Guofeng Wu
Guofeng Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 05 August 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.966478
This article is part of the Research Topic Ferroptosis in Cancer and Beyond View all 34 articles
Intracerebral hemorrhage (ICH) is a devastating form of stroke with high rates of morbidity, mortality, and disability. It induces cell death that is responsible for the secondary brain injury (SBI). The underlying mechanism of SBI after ICH is still unclear, and whether it is related to iron overload is worthy to be discussed. Ferroptosis is an iron-dependent non-apoptotic modes of cell death and plays a particularly important role in the occurrence and progression of ICH. Many ICH-induced regulators and signalling pathways of ferroptosis have been reported as promising targets for treating ICH. In this article, we review the definition, characteristics, and inhibition methods of neuronal ferroptosis caused by iron deposition after ICH, and review the biomarkers for ferroptosis.
Intracerebral hemorrhage (ICH) is a subtype of stroke with the highest morbidity, mortality, and disability (van Asch et al., 2010). After ICH occurs, in addition to the primary brain injury caused by the hematoma compressing the surrounding brain tissue, blood components, haemoglobin (Hb), iron, and other neurotoxic substances released by the hematoma also contribute to neuroinflammation and oxidative stress, increase the synthesis of reactive oxygen species (ROS), resulting in secondary brain injury (SBI) (Xi et al., 2006; Wang, 2010). Both the primary and SBI trigger significant cell death and loss of neurological functions. In consideration of the physical compression of the hematoma, many studies have explored the effect of surgical removal of hematoma; however, no significant benefits have been found for patients with ICH (Xi et al., 2014). Similarly, there are limited medical therapy available to alleviate SBI effectively (Bai et al., 2020).
The mechanism of SBI after ICH is complex and remains unclear. Modes of cell death which ICH-induced including necroptosis, pyroptosis, ferroptosis, autophagy, and parthanatos (Zhang et al., 2022). These models of cell death may be associated with SBI after ICH simultaneously. Thus, a better understanding of the modes of cell death in ICH should provide new insights to counter the pathology of ICH. It could result in more effective and targeted neuroprotective or neurorestorative therapeutic strategies (Guo et al., 2020; Zhang et al., 2022).
Studies have shown that excess Hb and iron ions released from hematomas accumulate in the brain parenchyma, causing neurotoxicity and accelerating neurodegeneration (Zecca et al., 2004; Wang et al., 2002). Iron-dependent non-apoptotic cell death, namely, ferroptosis, has been identified as a potential therapeutic target for ICH (Dixon et al., 2012). Neuronal ferroptosis plays key roles in SBI caused by ICH (Xu et al., 2018). Limiting ferroptosis caused by ferrotoxicity and excess accumulation of ROS reduces brain damage and improves the clinical outcomes of patients with ICH (Wang, 2010; Wu L et al., 2012). Therefore, we review the research progress of neuronal ferroptosis, and provide new ideas for treating SBI following ICH.
ICH refers to blood entering into the brain parenchyma, ventricle system, or subarachnoid space from a fracturing intracerebral vessel. The mortality rate in ICH patients is 50% approximately, while most of the survivors lose the capability of living independently (Gross et al., 2019). Especially, SBI, which refers to oxidative stress (Yao et al., 2021), inflammation (Xue and Yong, 2020), blood-brain barrier (BBB) hyperpermeability (Keep et al., 2018), and cerebral vasospasm (Athiraman and Zipfel, 2021), further drives brain cell death. Iron is a major product of lysed erythrocytes in hematoma. It can form highly toxic hydroxyl radicals to attack DNA, proteins, and lipid membranes, thereby disrupting cellular functions and causing neuronal death. Iron released from haemoglobin triggers ROS formation, which also induced ferroptosis (Magtanong and Dixon, 2018; Zhang et al., 2022) and is required for the accumulation of lipid peroxides and the execution of ferroptosis (Stockwell et al., 2017). Ferroptosis is iron-dependent non-apoptotic cell death and may play an important role in the development of SBI following ICH.
Ferroptosis, a newly identified regulated cell death (RCD) type. More and more researches in this field have revealed the potential roles of ferroptosis in development, immune system regulation, ischemia-reperfusion injury, and tumor suppression (Stockwell et al., 2017). Ferroptosis was first discovered and reported by Dixon et al. in 2012 (Dixon et al., 2012). Ferroptosis is an iron-dependent non-apoptotic form of cell death, which occurs through excessive accumulation of ROS when glutathione (GSH) peroxidase 4 (GPX4), a lipid peroxide reduction system, is dysregulated or relatively insufficient. The Nomenclature Committee on Cell Death (NCCD) defined ferroptosis as “a form of RCD initiated by oxidative perturbations of the intracellular microenvironment that is under constitutive control by GPX4 and can be inhibited by iron chelators and lipophilic antioxidants” (Galluzzi et al., 2018). Iron, lipid, and ROS are three core components of ferroptosis. Metabolic dysregulation of any one of them may influence ferroptotic cell death. Any molecular change or pharmacological intervention that regulates any of these elements may affect the final consequences of ferroptosis (Liu and Gu, 2022).
Ferroptosis is distinct from apoptosis, necrosis, and autophagy in terms of morphological, biochemical, and genetic properties (Dixon et al., 2012). Electron microscopy has shown that the morphological characteristics of ferroptosis include an obvious reduction in mitochondrial volume, increased density of the mitochondrial double membrane structure, reduced or disappeared cristae formed by the inner membrane, and a near absence of other obvious morphological changes before cell death, particularly the intact nuclei (Yu et al., 2017). The biochemical characteristics of ferroptosis include excessive ROS accumulation, which is dependent on iron ions (Dixon et al., 2012). During the oxidative phosphorylation of mitochondria, cells generate a certain amount of ROS and adenosine triphosphate (ATP). However, a level of ROS that exceeds the antioxidant capacity of cells leads to an enhanced oxidative stress response, which directly or indirectly damages proteins, nucleic acids, lipids, and other macromolecular substances, leading to cell damage or cell death (Abrams et al., 2016). The genetic characteristics of ferroptosis are regulated by a unique set of genes, mainly including ribosomal protein L8 (RPL8), ATP synthase F0 complex subunit C3 (ATP5G3), iron response element binding protein 2 (IREB2), citrate synthase (CS), tetratricopeptide repeat domain 35 (TTC35), and acyl-CoA synthetase family member 2 (ACSF2) genes (Dixon et al., 2012).
Ferroptosis is caused by dysregulated cell metabolism, including iron, lipid, amino acids, and ROS metabolism (Liu and Gu, 2022). Gao et al. showed that autophagy promotes ferroptosis by regulating intracellular iron homeostasis and ROS synthesis (Gao et al., 2016). In vitro experiments showed that the application of Erastin, a synthetic small-molecule compound, which induces ferroptosis and activates autophagy, led to intracellular ferritin degradation to further increase the level of intracellular iron ions through autophagy, resulting in rapid accumulation of intracellular ROS, which promote ferroptosis. Hou et al. also demonstrated that the activation of autophagy further promoted ferroptosis by degrading ferritin in tumor cells (Hou et al., 2016).
Li et al. used a collagenase-induced ICH mouse model to observe morphological changes and showed that the proportion of shrunken mitochondria in the cytoplasm and axon of neurons around the hematoma increased, which strongly confirmed the occurrence of ferroptosis and was the earliest report of neuronal ferroptosis after ICH. They also applied ferrostatin-1 (Fer-1),a specific ferroptosis inhibitor after acute ICH and showed that Fer-1 improved the neurological functions of the mice (Li et al., 2017a). In the same year, Zille et al. showed that the ICH model pretreated with Hb had increased levels of phosphorylated extracellular regulated protein kinases (ERK1/2) (Zille et al., 2017). As ERK1/2 is an important signaling molecule in the RAS-RAF-MEK pathway in the process of ferroptosis induced by the small-molecule Erastin, the finding of Zille et al. further prove from a molecular viewpoint that neurons undergo ferroptosis after ICH. In 2018, Li et al. used transmission electron microscopy (TEM) to monitor the ultra-micromorphological changes of neurons in a collagenase-induced ICH animal model and showed that on the third and sixth day after ICH, both the soma and axon of neurons had an increased proportion of shrunken mitochondria (Li et al., 2018), providing sufficient evidence for the occurrence of neuronal ferroptosis after ICH. Recently, an increasing number of researchers have investigated neuronal ferroptosis after ICH, with the aim to identify new directions and targets for treating SBI after ICH.
Observations of morphological changes by TEM or pharmacological and molecular characteristics are the most common approaches to identify neuronal ferroptosis. Although these are relatively complicated processes, the identification of biomarkers of ferroptosis will bring great convenience to the determination of ferroptosis in the future. Li et al. used Hb to stimulate hippocampal tissue slice culture in vitro and showed high expression of prostaglandin endoperoxide synthase 2 (PTGS2), but no significant changes in the mRNA levels of IREB2, CS, RPL8, and ATP5G3 (Li et al., 2017b). PTGS2 encodes cyclooxygenase-2 (COX-2), which is a key enzyme in prostaglandin biosynthesis. Studies have confirmed that ferroptosis is regulated by PTGS2, a unique and potentially direct downstream gene that induces ferroptosis. COX-2 is highly expressed after ICH and its inhibition alleviates the SBI caused by ICH, suggesting that COX-2 may be a biomarker for neuronal ferroptosis after ICH. However, existing research is limited to the Hb-induced ICH mouse model, and further research is needed to confirm the sensitivity, representativeness, and reproducibility of using COX-2 as a biomarker for neuronal ferroptosis after ICH (Chen et al., 2019).
Iron ions are essential for the occurrence of ferroptosis. However, the specific mechanism by which iron ions promote ferroptosis is not yet fully understood (Cao and Dixon, 2016). After ICH, the excess ferric ions from the red blood cells (RBC) lysis combine with transferrin (TF) in the serum to transport iron ions into cells through receptor-mediated effects (Andrews, 2000). Ferric ions are reduced to ferrous ions by divalent metal transporter 1 (DMT1) and accumulate in neurons, where ferrous ions induce excessive generation of lethal ROS and lipid peroxides (Figure 1). A previous study of the ICH mouse model showed that two iron chelators, deferoxamine (DFX) and VK-28 (5-[4-(2-hydroxyethyl) piperazine- 1-ylmethyl]-quinoline-8-ol), reduced neuronal death, ROS synthesis, accumulation of iron ions around the hematoma, activation of microglia, and improved the neurological function of the mouse model (Li et al., 2017a). Wu et al. demonstrated that fat-soluble iron chelator, 2,2′-dipyridy (DP), reduced iron deposition around the hematoma and ROS synthesis, improving the neuronal function and reducing the neuronal death of the ICH mouse model (Wu L et al., 2012). Pyridoxal isonicotinoyl hydrazine (PIH), a lipophilic iron-chelating agent, has been reported to reduce excess iron-induced cytotoxicity following ICH, which was associated with mitigation of inflammation and ferroptosis (Zhang H et al., 2021). The effect of iron ion chelators on the improvement of neuronal function in mice after ICH indicated that the occurrence of ferroptosis was inseparable from the effect of iron ions (Figure 1). Nevertheless, the specific mechanism still needs further exploration.
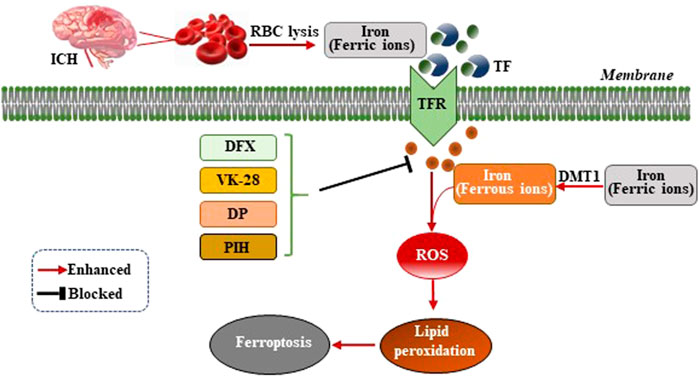
FIGURE 1. Mechanism of ferroptosis following ICH by intracellular iron overload. After ICH occurs, the hematoma releases iron ions from RBC lysis, the concentration of iron ions increase around neurons and combine with transferrin (TF) to transport iron ions into cells through TF receptor (TFR)-mediated effects. Ferric ions are reduced to ferrous ions by divalent metal transporter 1 (DMT1) and accumulate in neurons, where ferrous ions induce excessive generation of lethal ROS and lipid peroxides, therefore, resulting in ferroptosis. Iron chelators (DFX, VK-28, DP, and PIH) form a chelating ferric amine with iron ions to prevent iron ions from donating electrons to oxygen to form ROS.
Studies have confirmed an increased level of glutamate (Glu) in the brain tissue surrounding the hematoma after ICH in mice, rabbits, and patients with ICH (Li et al., 2017b; Wu et al., 2013; Castillo et al., 2002). The addition of Glu to the culture medium of HT22 hippocampal neurons led to a significant increase in cell death, and the application of the 5-lipoxygenase (5-LOX) inhibitor Zileuton suppressed 5-LOX to reduce lipid peroxide production, which inhibits ferroptosis and thereby protects neurons (Liu et al., 2015). Li et al. observed an increase in the level of Glu around the hematoma of the ICH mouse model and used the glutaminase inhibitor 968 to inhibit the decomposition of glutamine into Glu to significantly reduce the number of degenerated neurons around the hematoma, suggesting that the breakdown of glutamine led to neuronal ferroptosis after ICH in vivo (Li et al., 2017a). The reason for this finding may be that excessive Glu around neurons inhibits the activity of cystine/glutamate antiporter (System Xc−) and the transfer of cysteine (Cys). As Cys is the raw material for GSH synthesis, the reduction of Cys leads to a further reduction of GSH synthesis, which reduces the activity of GPX4 (Figure 2). This, results in excessive ROS and lipid peroxide levels which cannot be scavenged, thereby leading to neuronal ferroptosis and aggravating the dysfunction, cerebral edema, oxidative stress, BBB damage, and inflammatory response caused by ICH (Gao et al., 2015). Up-regulating the expression of GPX4 in ICH model can inhibit ferroptosis and treat ICH (Peng et al., 2022).
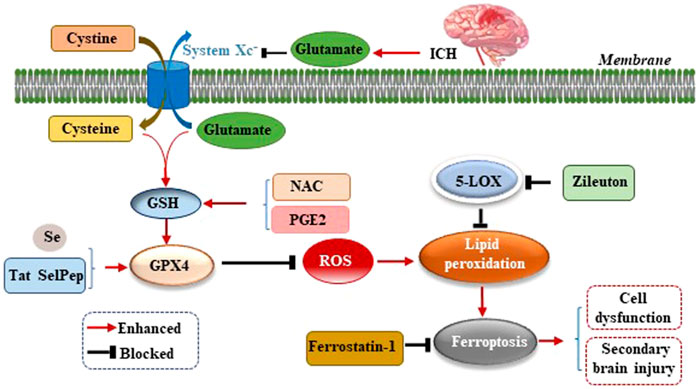
FIGURE 2. Mechanism of ferroptosis following ICH by inhibiting cystine/glutamate antiporter (System Xc−). After ICH occurs, excessive glutamate (Glu) around neurons inhibits the activity of System Xc− and the transfer of cysteine (Cys), leading to reduction of glutathione (GSH) synthesis, which reduces the activity of glutathione peroxidase 4 (GPX4), resulting in excessive reactive oxygen species (ROS) and lipid peroxide levels which cannot be scavenged, thereby leading to neuronal ferroptosis, cell dysfunction, and secondary brain injury (SBI) caused by ICH.
The tumor suppressor p53 as a master regulator of ferroptosis, has been among the most extensively studied genes since its discovery in 1979. A major function of p53 is mediating cellular and systematic metabolism. Interestingly, p53 is tightly associated with all key metabolic pathways involved in ferroptosis. Unlike apoptotic cell death, activation of p53 solely is not sufficient to induce ferroptosis directly; instead, through its metabolic targets, p53 is able to modulate the ferroptosis response in the presence of ferroptosis inducers such as GPX4 inhibitors or high levels of ROS. More and more studies to this day have been revealed that p53 is a key regulator of both canonical and non-canonical ferroptosis pathways via a variety of mechanisms. In most cases, p53 promotes ferroptosis. However, under a certain context, p53 can inhibit ferroptosis (Liu and Gu, 2022).
p53 was shown to promote ferroptosis via its capacity to inhibit the import of cystine into target cells. Mechanistically, p53 was found to suppress the transcription of SLC7A11 solute carrier family 7 member 11 (SLC7A11), which is a core subunit of the System Xc−. The activity of System Xc− is mainly determined by SLC7A11. SLC7A11 was first identified as a direct target gene suppressed by p53 (Jiang et al., 2015). SLC7A11 mediates cellular uptake of extracellular cystine in exchange for intracellular Glu. Interference of cystine absorption reduces downstream GSH bio- synthesis and thus decreases GPX4’s ability to antagonize ferroptosis. p53 also promotes ferroptosis through regulating other metabolic pathways. Spermidine/spermine N1-acetyltrans- ferase 1 (SAT1) is a rate-limiting enzyme in polyamine catabolism. p53 can transactivate SAT1. SAT1 induction leads to lipid peroxidation and ferroptosis. This effect is due to arachidonate-15-lipoxygenase (ALOX15) upregulation after SAT1 induction. Therefore, the p53/SAT1/ALOX15 axis partially contributes to p53-mediated ferroptosis. p53 enhances the activity of SLC25A28 solute carrier family 25 member 28 (SLC25A28), a protein coding gene that causes abnormal accumulation of redox-active iron and promotes ferroptosis (Zhang et al., 2020). p53 can inhibit the serine synthesis pathway and transsulfuration pathway by suppressing phosphoglycerate dehydrogenase (PHGDH) and cystathionine β-synthase (CBS) respectively, limiting GSH production (Ou et al., 2015; Wang et al., 2019). In summary, p53 promotes ferroptosis through its multipotent roles in regulating cellular metabolism, particularly lipid, iron, ROS, and amino acid metabolism (Liu and Gu, 2021). Mechanistically, dipeptidyl peptidase 4 (DPP4) promotes ferroptosis in p53-deficient cells by binding nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (NOX1) and boosting the production of ROS resulting in lipid peroxidation and ferroptosis (Gao et al., 2019). p21 is a target gene of p53 (Hong et al., 2010), and p21 induction redistributes the serine usage from nucleotide biogenesis to GSH synthesis, and GSH is an inhibitor of ROS and ferroptosis (Tarangelo et al., 2018; Mattocks et al., 2013). p53 modulates GPX4-dependent ferroptosis pathways as is shown in the Figure 3.
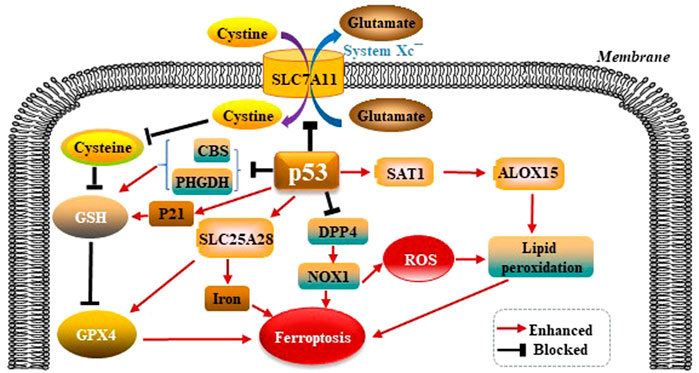
FIGURE 3. p53 modulates GPX4-dependent ferroptosis pathways. p53 suppresses the transcription of SLC7A11, which is a core subunit of the System Xc−. SLC7A11 mediates cellular uptake of extracellular cystine in exchange for intracellular Glu, reduces downstream GSH and GPX4 biosynthesis, thus leading to ferroptosis. p53 also promotes ferroptosis through regulating other metabolic pathways. p53 can upregulate ALOX15 via SAT1-mediated, leads to lipid peroxidation and ferroptosis. p53 enhances the activity of SLC25A28, causes abnormal accumulation of redox-active iron and promotes ferroptosis. p53 also can suppress PHGDH and CBS respectively, limiting GSH production. p53 inhibits DPP4 by binding NOX1, boosting the production of ROS, and resulting in lipid peroxidation and ferroptosis. p21 is a target gene of p53. p21 suppresses GSH synthesis, and promotes ferroptosis.
It is well established that ferroptosis is controlled by GPX4 primarily. Amazingly, Chu et al. observed that p53 modulates ferroptosis without apparent effects on GPX4. By screening the ALOX arachidonate lipoxygenase family to identify potential contributors to p53-mediated ferroptosis, founding that ALOX12 is a critical candidate (Chu et al., 2019). p53 promotes the activity of ALOX12 via inhibiting SLC7A11 which binds and sequesters ALOX12 from its substrate, polyunsaturated fatty acid (PUFA), including those esterified in membranes. When p53 downregulates SLC7A11, ALOX12 is released and subsequently oxidizes membrane PUFAs and initiates ferroptosis. Therefore, the p53/SLC7A11/ALOX12 axis is independent of the reduce in GSH and GPX4 activity, and is a mechanism underlying different than the p53/ SLC7A11/GPX4 pathway. In a recent study, Yang et al. reported that another ALOX family member, ALOXE3, acts in a similar way to ALOX12 in generating ferroptosis (Yang et al., 2021). SLC7A11 also binds and sequesters ALOXE3 from its substrate. p53/SLC7A11/ ALOXE3- mediated ferroptosis is independent of long-chain acyl-CoA synthetases 4 (ACSL4). In consideration of that ALOXE3 is downstream of ALOX12, the p53/ SLC7A11/ALOX12 and p53/SLC7A11 /ALOXE3 axes can both collaboratively and independently in modulating ferroptosis. Phospholipase A2 group VI (iPLA2β) is a calcium-independent phospholipase that gaps acyl tails from the glycerol backbone of lipids and releases oxidized fatty acids, which can be further detoxified by the antioxidants in the cytoplasm (Malley et al., 2018). iPLA2β-mediated detoxification of peroxidated membrane lipids is adequate to inhibit p53/ALOX12-driven ferroptosis in a GPX4-independent pattern (Liu and Gu, 2022). P53 modulates GPX4-independent ferroptosis pathways as is shown in the Figure 4.
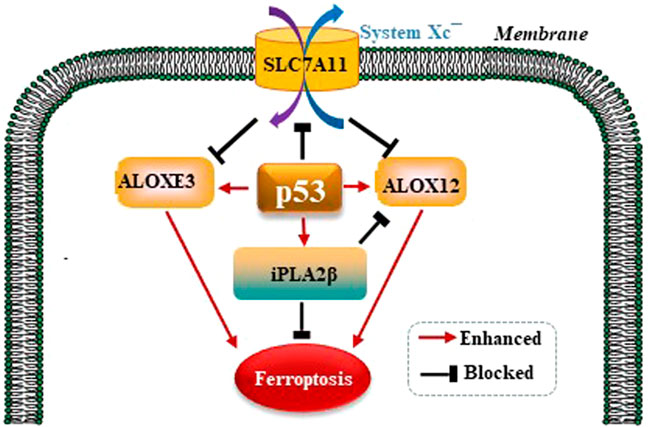
FIGURE 4. p53 modulates GPX4-independent ferroptosis pathways. p53/ SLC7A11/ALOX12 and p53/SLC7A11/ALOXE3 axes can both collaboratively and independently in modulating ferroptosis in a GPX4-independent manner. iPLA2β-mediated detoxification of peroxidated membrane lipids is adequate to inhibit p53/ALOX12-driven ferroptosis also in a GPX4-independent pattern.
Kuang et al.observed ferroptosis characteristics in the cerebral cortex of rats with subarachnoid haemorrhage (SAH) after 24 h, and could be alleviated by Fer-1 treatment. Fer-1 could increase SLC7A11 and GPX4. Similarly, BBB impairment, brain edema, behavioral deficits and neuronal damage were relieved by inhibiting ferroptosis. Moreover, the p53 inhibitor Pifithrin-α could block cortical SAH-induced ferroptosis. These results indicated that ferroptosis aggravated SBI after SAH was partly dependent on p53, and inhibiting ferroptosis might be an effective therapeutic target for SBI (Kuang et al., 2021). Zhang et al. used ICH rats model to explore the mechanism of Ubiquitin-specific protease 11(USP11) regulating neuroinflammation in ICH. It was showed in microglial cells that USP11 stabilized p53 by deubiquitination and p53 targeted Krüppel like factor 2(KLF2) promoter to repress KLF2 transcription, thereby activating the nuclear factor-kappa B (NF-κB) pathway. Further, rescue experiments were conducted in vivo to make sure the function of USP11/p53/KLF2/NF-κB axis in ICH-induced inflammation, which confirmed that USP11 silencing inhibited the release of pro-inflammatory cytokines following ICH by downregulating p53, thus protecting against neurological deficit. (Zhang X et al., 2021). Xu et al. first reported p53 could increase apoptosis-associated protein cysteine aspartate protease-3 (Caspase-3) as well as Bcl-2 associated X protein (Bax) expressions, and decrease B-celllymphoma-2 (Bcl-2) expression conversely. Therefore, p53 plays key roles in SBI caused by ICH (Xu et al., 2018). Isorhynchophylline (IRN), a component of traditional Chinese herb Uncaria rhynchophylla, possesses strong antioxidant activity. A present study showed IRN exhibited neuroprotective effects in vivo and in vitro by inhibiting ferroptosis. In particular, IRN suppressed the p53 expression to promote the transcription of SLC7A11 by upregulating the miR-122–5p expression, thus exerting its anti-ferroptosis activity. The fndings reveal that IRN protects neurocyte from ferric ammonium citrate (FAC) -induced ferroptosis via miR-122–5p/ p53/SLC7A11 pathway, which may provide a potential therapeutic mechanism for ICH (Zhao et al., 2021). p53 modulates ferroptosis in ICH as is shown in the Figure 5.
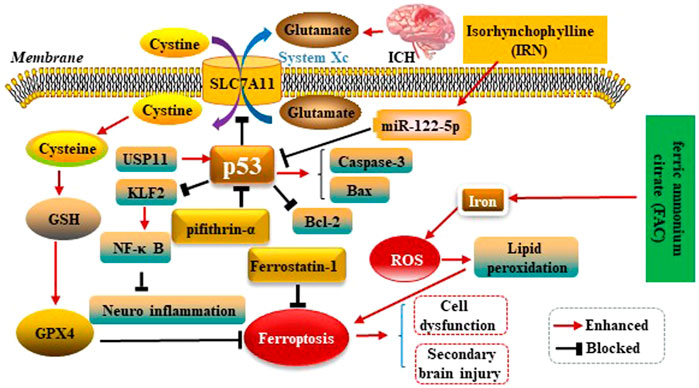
FIGURE 5. p53 modulates ferroptosis in ICH. p53 inhibits SLC7A11 resulting in dysfunction of the cystine/glutamate antiporter (System Xc−) after ICH, leading to decreased synthesis of glutathione (GSH) and activity of glutathione peroxidase 4 (GPX4). p53 could increase Caspase-3 as well as Bax expressions, and decrease Bcl-2 expression conversely. Therefore, P53 plays key roles in SBI caused by ICH. IRN suppressed the p53 expression to anti-ferroptosis via miR-122–5p/p53/SLC7A11 pathway. Ubiquitin-specific protease 11(USP11) stabilized p53 by deubiquitination and p53 targeted Krüppel like factor 2 (KLF2) promoter to repress KLF2 transcription, thereby activating the nuclear factor-kappa B (NF-κB) pathway to anti-ferroptosis. Ferric ammonium citrate (FAC) could be a source of iron to induce iron overload, thus leading to ferroptosis.
In conclusion, how can p53 enhance or block ferroptosis are developing, and many problems remain unknown. Emerging research results have increasingly advanced our knowledge of p53. p53 and ferroptosis fields need much more research to obtain more achievements.
Fer-1 is a specific ferroptosis inhibitor. Dixon et al. showed that Fer-1 inhibited Hb-induced cell death in hippocampal slice culture, confirming that Fer-1 reduced iron ion-induced cell death and the release of lactate dehydrogenase (LDH) induced by Hb and ferrous ions. Additionally, the content of ROS in the cultured hippocampal slices in the experimental group with Fer-1 intervention was significantly lower than that in the control group (Dixon et al., 2012). Zhang et al. (Zhang et al., 2018) also confirmed that Fer-1 significantly reduced the levels of interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) in the serum and cerebrospinal fluid, which in turn alleviated the inflammatory response of rats after ICH, reduced albumin extravasation and BBB damage, and alleviated SBI after ICH. Generally, the in vivo and in vitro experiments have confirmed that the application of Fer-1 inhibits the occurrence of ferroptosis, thereby alleviating the SBI after ICH and providing a theoretical basis for further clinical application of Fer-1. Fer-1 is listed in Supplementary Table S1 which summarize the potential inhibition methods of ferroptosis following ICH.
The iron chelator DFX reduces brain edema, neurological deficit, and brain atrophy in rats after ICH (Okauchi et al., 2010). The sustained release of iron ions from the hematoma after ICH activates local microglia and causes secondary brain damage. DFX forms a chelating ferric amine with iron ions in the brain tissue around the hematoma to prevent iron ions from donating electrons to oxygen to form ROS and inhibits the hyperactivation of microglia (Li et al., 2017b; Dixon and Stockwell, 2014), which is the most important mechanism of neuronal ferroptosis. Another study showed that the application of DFX or VK-28 6 h after ICH in mice effectively reduced ROS synthesis by approximately 50%, thereby reducing neuronal cell death after ICH and improving secondary neuronal damage. Compared to DFX, the iron chelator VK-28 had a greater advantage of penetrating the intact BBB, while DFX failed to do so. Thus, VK-28 may act in the brain at a relatively low concentration, which is more suitable for clinical application (Li et al., 2017a). Generally, iron chelators further inhibit the occurrence of ferroptosis by forming chelating ferric amines with iron ions in the brain tissue around hematoma. VK-28 is more effective and safer than DFX in inhibiting ferroptosis. Additionally, a previous study showed that fat-soluble iron chelator, 2,2′-dipyridy (DP), reduced iron deposition around the hematoma and ROS synthesis, improving the neuronal function and reducing the neuronal death of the ICH mouse model (Wu H et al., 2012).
Pyridoxal isonicotinoyl hydrazine (PIH), a lipophilic iron-chelating agent, has been reported to reduce excess iron-induced cytotoxicity after ICH, which was associated with mitigation of inflammation and ferroptosis (Zhang H et al., 2021). Iron chelators (DFX, VK-28, DP and PIH) are listed in Supplementary Table S1 which summarize the potential inhibition methods of ferroptosis following ICH.
DMT1 is involved in iron ionization in cells and is the only protein that transports iron ions into cells (Gao et al., 2015). After ICH, the expression of DMT1 is significantly increased, and ferric ions are reduced to ferrous irons under the action of DMT1. Ferrous ions induce excessive ROS and lipid peroxide production and are important factors causing neuronal ferroptosis (Xie et al., 2012). Pretreatment with Ebselen, a DMT1 inhibitor, significantly reduces the iron ion transport activity of DMT1 and suppresses ROS synthesis (Wang et al., 2016). A previous study from China showed that Ebselen further reduced neuronal ferroptosis after subarachnoid hemorrhage in rats by inhibiting DMT1 (Zhang et al., 2017). Hence, it is necessary to further strengthen the research on Ebselen in the context of ICH. Ebselen is listed in Supplementary Table S1 which summarize the potential inhibition methods of ferroptosis following ICH.
Epicatechin (EC) is a flavanol compound present in natural plants (e.g., fruits, vegetables, and green tea). EC activates NF-E2-related factor (Nrf2) signaling to scavenge oxidants and free radicals. Existing reports have shown that the application of EC reduced the volume of brain damage and brain edema, improved neurological function and prognosis, and significantly reduced IREB2 mRNA expression in the early stage of ICH in mice. IREB2 is involved in encoding iron-ion regulators and inhibits neuronal ferroptosis by further regulating the intracellular iron-ion metabolism (Chang et al., 2014). After ICH, the neuronal cell viability of IREB2-knockout mice was higher than that of the mice in the IREB2-non-knockout group, showing a relatively strong resistance to Hb toxicity (Regan et al., 2008). Importantly, EC passes through the BBB and is convenient to use (Wu L et al., 2012). Hence, EC has promising clinical research value for inhibiting neuronal ferroptosis and SBI after ICH. EC is listed in Supplementary Table S1 which summarize the potential inhibition methods of ferroptosis following ICH.
NAC is a Cys precursor approved by the Food and Drug Administration of the United States for clinical use for treating paracetamol-induced liver failure (Green et al., 2013). Much preclinical data are available to support the use of NAC in acute and chronic neurological and psychiatric disorders (Ansari et al., 2019; De Rosa et al., 2015; Monti et al., 2016). Cys, as a component of the cellular reducing agent GSH, is necessary to maintain cellular redox homeostasis; hence, Cys deficiency further depletes intracellular GSH, thereby increasing ROS synthesis and inducing ferroptosis. Under these circumstances, supplementation with another biosynthetic precursor of GSH, i.e., NAC, can effectively prevent cells from undergoing ferroptosis (Liu et al., 2015). A study by Zille et al. on ICH in male mice showed that NAC inhibited neuronal ferroptosis in vitro (Zille et al., 2017). Moreover, Karuppagounder et al. confirmed that NAC prevented heme oxygenase-1 induction by neutralizing toxic lipids produced by arachidonic acid-dependent 5- LOX 7 days after ICH, resulting in reduced neuronal cell death after ICH and improved functional recovery. The application of PGE2 combined with NAC may have a synergistic effect and reduce the NAC administration dosage required for the protection and functional recovery of brain tissue. Nevertheless, the application of NAC may cause serious adverse reactions. Thus, further research should focus on controlling NAC dosage and its adverse reactions (Karuppagounder et al., 2018). Combined NAC and PGE2 are listed in Supplementary Table S1 which summarize the potential inhibition methods of ferroptosis following ICH.
Ingold et al. reported that Se effectively inhibited GPX4-dependent ferroptosis and synergistically activated transcription factor activator protein 2C (TFAP2C) and transcription factor Sp1 (TSFP1), which further activated GPX4 transcription (Ingold et al., 2018). Another study confirmed that an intraventricular injection of sodium selenite promoted the expression of TSFP1 and GPX4 in neurons of the ICH mouse model, significantly reducing the neuronal death rate and promoting the recovery of nerve function in the mice (Alim et al., 2019). However, intraventricular injection requires the insertion of a catheter or needle into the brain tissue, which is invasive and carries the risk of developing intracranial infection and exacerbating the disease (Alim et al., 2019; Ding et al., 2015). Additionally, Se has a narrow therapeutic window and causes neurotoxicity if the dose is not titrated within an appropriate range (Fam et al., 2017). To overcome this problem, Alim et al. developed a Se-containing Cys polypeptide, Tat SelPep, to not only penetrate the BBB in a broad therapeutic window, but also to induce high GPX4 expression in various organs including the brain, heart, and liver. Se and its related new drugs provide additional potential treatment options for diseases related to GPX4 deficiency, including ICH. Se or Tat SelPep is listed in Supplementary Table S1 which summarize the potential inhibition methods of ferroptosis following ICH.
ICH is the stroke subtype with the highest mortality rate of acute cerebrovascular disease, accounting for 15–20% of all strokes, with no effective treatments currently available (Feigin et al., 2009; Keep et al., 2012). Neuronal cell death after ICH includes necroptosis, pyroptosis, ferroptosis, autophagy, and parthanatos (Zhang et al., 2022). Ferroptosis may play an important role in the development of SBI after ICH. Recently, research on ferroptosis has become popular, and new mechanisms of ferroptosis and its potential clinical functions are still emerging, which are of great significance for exploring the therapeutic directions and intervention targets after ICH. This review article presents the definition of ferroptosis, the discovery of neuronal ferroptosis after ICH, the mechanism of ferroptosis, and the ways in which ferroptosis can be suppressed. Generally, the occurrence of ferroptosis is closely related to the content of intracellular iron ions, GSH, lipid peroxidases. Fer-1, iron-ion chelators, Ebselen, EC, NAC, PGE2, Se, and others, which may suppress ferroptosis after ICH, providing a new direction for the clinical treatment of ICH. However, more in-depth studies are necessary to determine how to best apply the above substances in clinical practice and reduce their related adverse reactions.
SR and YC drafted the manuscript. LW and GW revised the manuscript for content. All authors approved the final manuscript.
This work was supported by grants from the Science and Technology Foundation of Guizhou Province (No. Qiankehe Foundation (2020) 1Y316), the Cultivation Fund of the Affiliated Hospital of Guizhou Medical University for National Natural Science Foundation of China (No. gyfynsfc (2020)-11), and the Doctoral Research Start-up Fund of the Affiliated Hospital of Guizhou Medical University (No. gyfybsky-2021-30).
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.966478/full#supplementary-material
Abrams, R. P., Carroll, W. L., and Woerpel, K. A. (2016). Five-membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem. Biol. 11, 1305–1312. doi:10.1021/acschembio.5b00900
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279. doi:10.1016/j.cell.2019.03.032
Andrews, N. C. (2000). Iron homeostasis: Insights from genetics and animal models. Nat. Rev. Genet. 1, 208–217. doi:10.1038/35042073
Ansari, S. F., Memon, M., Brohi, N., and Tahir, A. (2019). N-Acetylcysteine in the management of acute exacerbation of chronic obstructive pulmonary disease. Cureus 11, e6073. doi:10.7759/cureus.6073
Athiraman, U., and Zipfel, G. J. (2021). Role of anesthetics and their adjuvants in neurovascular protection in secondary brain injury after aneurysmal subarachnoid hemorrhage. Int. J. Mol. Sci. 22, 6550. doi:10.3390/ijms22126550
Bai, Q., Sheng, Z., Liu, Y., Zhang, R., Yong, V. W., and Xue, M. (2020). Intracerebral haemorrhage:from clinical settings to animal models. Stroke Vasc. Neurol. 5, 388–395. doi:10.1136/svn-2020-000334
Cao, J., and Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209. doi:10.1007/s00018-016-2194-1
Castillo, J., Dávalos, A., Álvarez-Sabín, J., Pumar, J. M., Leira, R., Silva, Y., et al. (2002). Molecular signatures of brain injury after intracerebral hemorrhage. Neurology 58, 624–629. doi:10.1212/WNL.58.4.624
Chang, C., Cho, S., and Wang, J. (2014). (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann. Clin. Transl. Neurol. 1, 258–271. doi:10.1002/acn3.54
Chen, B., Chen, Z., Liu, M., Gao, X., Cheng, Y., Wei, Y., et al. (2019). Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res. Bull. 153, 122–132. doi:10.1016/j.brainresbull.2019.08.013
Chu, B., Kon, N., Chen, D., Li, T., Liu, T., Jiang, L., et al. (2019). ALOX12 is required for p53- mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591. doi:10.1038/s41556-019-0305-6
De Rosa, S. C., Zaretsky, M. D., Dubs, J. G., Roederer, M., Green, A., Mitra, D., et al. (2015). N- acetylcysteine replenishes glutathione in HIV infection. Eur. J. Clin. Invest. 30, 915–929. doi:10.1046/j.1365-2362.2000.00736.x
Ding, D., Przybylowski, C. J., Starke, R. M., Street, R. S., Tyree, A. E., Crowley, R. W., et al. (2015). A minimally invasive anterior skull base approach for evacuation of a basal ganglia hemorrhage. J. Clin. Neurosci. 22, 1816–1819. doi:10.1016/j.jocn.2015.03.052
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17. doi:10.1038/nchembio.1416
Fam, M. D., Hanley, D., Stadnik, A., Zeineddine, H. A., Girard, R., Jesselson, M., et al. (2017). Surgical performance in minimally invasive surgery plus recombinant tissue plasminogen activator for intracerebral hemorrhage evacuation phase III clinical trial. Neurosurgery 81, 860–866. doi:10.1093/neuros/nyx123
Feigin, V. L., Lawes, C. M. M., Bennett, D. A., Barker-Collo, S. L., and Parag, V. (2009). Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet. Neurol. 8, 355–369. doi:10.1016/s1474-4422(09)70025-0
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi:10.1038/s41418-017-0012-4
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., and Jiang, X. (2016). Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032. doi:10.1038/cr.2016.95
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and trans- ferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi:10.1016/j.molcel.2015.06.011
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363. e3. doi:10.1016/j.molcel.2018.10.042
Green, J. L., Heard, K. J., Reynolds, K. M., and Albert, D. (2013). Oral and intravenous acetylcysteine for treatment of acetaminophen toxicity: A systematic review and meta-analysis. West. J. Emerg. Med. 14, 218–226. doi:10.5811/westjem.2012.4.6885
Gross, B. A., Jankowitz, B. T., and Friedlander, R. M. (2019). Cerebral intraparenchymal hemorrhage: A review. JAMA 321, 1295–1303. doi:10.1001/jama.2019.2413
Guo, X., Xue, Q., Zhao, J., Yang, Y., Yu, Y., Liu, D., et al. (2020). Clinical diagnostic and therapeutic guidelines of stroke neurorestoration (2020 China Version). J. Neurorestoratology 8, 241–251. doi:10.26599/JNR.2020.9040026
Hong, L., Zhao, X., and Zhang, H. (2010). p53-mediated neuronal cell death in ischemic brain injury. Neurosci. Bull. 26, 232–240. doi:10.1007/s12264-010-1111-0
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422. e21. doi:10.1016/j.cell.2017.11.048
Jiang, L., Kon, N., Li, T., Wang, S., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53- mediated activity during tumour suppression. Nature 520, 57–62. doi:10.1038/nature14344
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N- acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglan- din E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872. doi:10.1002/ana.25356
Keep, R. F., Andjelkovic, A. V., Xiang, J., Stamatovic, S. M., Antonetti, D. A., Hua, Y., et al. (2018). Brain endothelial cell junctions after cerebral hemorrhage: Changes, mechanisms and therapeutic targets. J. Cereb. Blood Flow. Metab. 38, 1255–1275. doi:10.1177/0271678X18774666
Keep, R. F., Hua, Y., and Xi, G. (2012). Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet. Neurol. 11, 720–731. doi:10.1016/S1474-4422(12)70104-7
Kuang, H., Wang, T., Liu, L., Tang, C., Li, T., Liu, M., et al. (2021). Treatment of early brain injury after subarachnoid hemorrhage in the rat model by inhibiting p53-induced ferroptosis. Neurosci. Lett. 762, 136134. doi:10.1016/j.neulet.2021.136134
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017a). Inhibition of neuronal ferroptosis protects Hemorrhagic brain. JCI Insight 2, e90777. doi:10.1172/jci.insight.90777
Li, Q., Wan, J., Lan, X., Han, X., Wang, Z., and Wang, J. (2017b). Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J. Cereb. Blood Flow. Metab. 37, 3110–3123. doi:10.1177/0271678X17709186
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018). Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage : Coexistence of ferroptosis, autophagy, and necrosis. Front. Neurol. 9, 581. doi:10.3389/fneur.2018.00581
Liu, Y., and Gu, W. (2022). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 29, 895–910. doi:10.1038/s41418-022-00943-y
Liu, Y., and Gu, W. (2021). The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. S1044-579X (21), 00060–00062. doi:10.1016/j.semcancer.2021.03.010
Liu, Y., Wang, W., Li, Y., Xiao, Y., Cheng, J., and Jia, J. (2015). The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239. doi:10.1248/bpb.b15-00048
Magtanong, L., and Dixon, S. J. (2018). Ferroptosis and brain injury. Dev. Neurosci. 40, 382–395. doi:10.1159/000496922
Malley, K. R., Koroleva, O., Miller, I., Sanishvili, R., Jenkins, C. M., Gross, R. W., et al. (2018). The structure of iPLA2β reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 9, 765. doi:10.1038/s41467-018-03193-0
Mattocks, O. D., Berkers, C. R., Mason, S. M., Zheng, L., Blyth, K., Gottlieb, E., et al. (2013). Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546. doi:10.1038/nature11743
Monti, D. A., George, Z., Daniel, K., Liang, T., Wintering, N. A., Cai, J., et al. (2016). N-acetyl cysteine may support dopamine neurons in Parkinson's disease: Preliminary clinical and cell line data. PLoS One 11, e0157602. doi:10.1371/journal.pone.0157602
Okauchi, M., Hua, Y., Keep, R. F., Morgenstern, L. B., Schallert, T., Xi, G., et al. (2010). Deferoxamine treatment for intracerebral hemorrhage in aged rats: Therapeutic time window and optimal duration. Stroke 41, 375–382. doi:10.1161/STRO-KEAHA.109.569830
Ou, Y., Wang, S., Jiang, L., Zheng, B., and Gu, W. (2015). p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J. Biol. Chem. 290, 457–466. doi:10.1074/jbc.M114.616359
Peng, C., Fu, X., Wang, K., Chen, L., Luo, B., Huang, N., et al. (2022). Dauricine alleviated secondary brain injury after intracerebral hemorrhage by upregulating GPX4 expression and inhibiting ferroptosis of nerve cells. Eur. J. Pharmacol. 914, 174461. doi:10.1016/j.ejphar.2021.174461
Regan, R. F., Chen, M., Li, Z., Zhang, X., Benvenisti-Zarom, L., and Chen-Roetling, J. (2008). Neurons lacking iron regulatory protein-2 are highly resistant to the toxicity of hemoglobin. Neurobiol. Dis. 31, 242–249. doi:10.1016/j.nbd.2008.04.008
Stockwell, B. R., Friedmann, A. J., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Tarangelo, A., Magtanong, L., Bieging-Rolett, K. T., Li, Y., Ye, J., Attardi, L. D., et al. (2018). p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22, 569–575. doi:10.1016/j.celrep.2017.12.077
van Asch, C. J., Luitse, M. J., Rinkel, G. J., van der Tweel, I., Algra, A., and Klijn, C. J. (2010). Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: A systematic review and meta-analysis. Lancet. Neurol. 9, 167–176. doi:10.1016/S1474-4422(09)70340-0
Wang, G., Hu, W., Tang, Q., Wang, L., Sun, X., Chen, Y., et al. (2016). Effect comparison of both iron chelators on outcomes, iron deposit, and iron transporters after intracerebral hemorrhage in rats. Mol. Neurobiol. 53, 3576–3585. doi:10.1007/s12035-015-9302-3
Wang, J. (2010). Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 92, 463–477. doi:10.1016/j.pneurobio.2010.08.001
Wang, M., Mao, C., Ouyang, L., Liu, Y., Lai, W., Liu, N., et al. (2019). Long noncoding RNA LINC00 336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 26, 2329–2343. doi:10.1038/s41418-019-0304-y
Wang, X., Mori, T., Sumii, T., and Lo, E. H. (2002). Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: Caspase activation and oxidative stress. Stroke 33, 1882–1888. doi:10.1161/01.str.0000020121.41527.5d
Wu, G., Sun, S., Sheng, F., Wang, L., and Wang, F. (2013). Perihematomal glutamate level is associated with the blood–brain barrier disruption in a rabbit model of intracerebral hemorrhage. Springerplus 2, 358. doi:10.1186/2193-1801-2-358
Wu, H., Wu, T., Li, M., and Wang, J. (2012). Efficacy of the lipid-soluble iron chelator 2, 2'-dipyridyl against hemorrhagic brain injury. Neurobiol. Dis. 45, 388–394. doi:10.1016/j.nbd.2011.08.028
Wu, L., Zhang, Q., Zhang, X., Lv, C., Li, J., Yuan, Y., et al. (2012). Pharmacokinetics and blood-brain barrier penetration of (+)-catechin and (-)-epicatechin in rats by microdialysis sampling coupled to high-performance liquid chromatography with chemiluminescence detection. J. Agric. Food Chem. 60, 9377–9383. doi:10.1021/jf301787f
Xi, G., Keep, R. F., and Hoff, J. T. (2006). Mechanisms of brain injury after intracerebral haemor- rhage. Lancet. Neurol. 5, 53–63. doi:10.1016/S1474-4422(05)70283-0
Xi, G., Strahle, J., Hua, Y., and Keep, R. F. (2014). Progress in translational research on intra- cerebral hemorrhage: Is there an end in sight? Prog. Neurobiol. 115, 45–63. doi:10.1016/j.pneurobio.2013.09.007
Xie, L., Zheng, W., Xin, N., Xie, J., Wang, T., and Wang, Z. (2012). Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem. Int. 61, 334–340. doi:10.1016/j.neuint.2012.05.016
Xu, W., Gao, L., Li, T., Zheng, J., and Shao, A. (2018). Mesencephalic astrocyte-derived neuro- trophic factor (MANF) protects against neuronal apoptosis via activation of akt/ MDM2/p53 signaling pathway in a rat model of intracerebral hemorrhage. Front. Mol. Neurosci. 11, 176. doi:10.3389/fnmol.2018.00176
Xue, M., and Yong, V. W. (2020). Neuroinflammation in intracerebral haemorrhage: Immunotherapies with potential for translation. Lancet. Neurol. 19, 1023–1032. doi:10.1016/S1474-4422(20)30364-1
Yang, X., Liu, J., Wang, C., Cheng, K., Xu, H., Li, Q., et al. (2021). miR-18a promotes glioblastoma development by down-regulating ALOXE3-mediated ferroptotic and anti-migration activities. Oncogenesis 10, 15. doi:10.1038/s41389-021-00304-3
Yao, Z., Bai, Q., and Wang, G. (2021). Mechanisms of oxidative stress and therapeutic targets following intracerebral hemorrhage. Oxid. Med. Cell. Longev. 2021, 8815441. doi:10.1155/2021/8815441
Yu, H., Guo, P., Xie, X., Wang, Y., and Chen, G. (2017). Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 21, 648–657. doi:10.1111/jcmm.13008
Zecca, L., Youdim, M. B. H., Riederer, P., Connor, J. R., and Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873. doi:10.1038/nrn1537
Zhang, H., Jiang, D., Che, X., Zhao, Q., Zhao, J., Xiang, J., et al. (2017). Ebselen relieves ferroptosis induced by divalent mental transporter 1 in rats with subarachnoid hemorrhage. Acta. Acad. med.mil.tert. 39, 1618–1624. (in Chinese). doi:10.16016/j.1000-5404.201701128
Zhang, H., Wen, M., Chen, J., Yao, C., Lin, X., Lin, Z., et al. (2021). Pyridoxal isonicotinoyl hydrazone improves neurological recovery by attenuating ferroptosis and inflammation in cerebral hemorrhagic mice. Biomed. Res. Int. 2021, 9916328. doi:10.1155/2021/9916328
Zhang, X., Liu, T., Xu, S., Gao, P., Dong, W., Liu, W., et al. (2021). A pro-inflammatory mediator USP11 enhances the stability of p53 and inhibits KLF2 in intracerebral hemorrhage. Mol. Ther. Methods Clin. Dev. 21, 681–692. doi:10.1016/j.omtm.2021.01.015
Zhang, Y., Khan, S., Liu, Y., Zhang, R., Li, H., Wu, G., et al. (2022). Modes of brain cell death following intracerebral hemorrhage. Front. Cell. Neurosci. 16, 799753. doi:10.3389/fncel.2022.799753
Zhang, Z., Guo, M., Shen, M., Kong, D., Zhang, F., Shao, J., et al. (2020). The BRD7-P53- SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 36, 101619. doi:10.1016/j.redox.2020.101619
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intra- cerebral hemorrhage. Brain Res. 1701, 112–125. doi:10.1016/j.brainres.2018.09.012
Zhao, H., Li, X., Yang, L., Zhang, L., Jiang, X., Gao, W., et al. (2021). Isorhynchophylline relieves ferroptosis-induced nerve damage after intracerebral hemorrhage via miR-122-5p/ TP53/ SLC7A11 pathway. Neurochem. Res. 46, 1981–1994. doi:10.1007/s11064-021-03320-2
Zille, M., Karuppagounder, S. S., Chen, Y., Gough, P. J., Bertin, J., Finger, J., et al. (2017). Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48, 1033–1043. doi:10.1161/STROKEAHA.116.015609
ATP adenosine triphosphate
ATP5G3 ATP synthase F0 complex subunit C3
ACSF2 acyl-CoA synthetase family member 2
ALOX15 arachidonate-15-lipoxygenase
BBB blood-brain barrier
Bax Bcl-2 associated X protein
Bcl-2 B-celllymphoma-2
Caspase-3 Cysteine aspartate protease-3
CS citrate synthase
COX-2 cyclooxygenase-2
Cys cysteine
CBS cystathionine β-synthase
DMT1 divalent metal transporter 1
DFX deferoxamine
DP 2,2′-dipyridy
DPP4 dipeptidyl peptidase 4
EC Epicatechin
ERK1/2 extracellular regulated protein kinases
Fer-1 ferrostatin-1
FXDR ferredoxin reductase
FAC ferric ammonium citrate
Glu glutamate
GSH glutathione
GPX4 glutathione peroxidase 4
HB hemoglobin
IREB2 Iron response element binding protein 2
ICH intracerebral haemorrhage
IL-1β interleukin-1 beta
iPLA2β Phospholipase A2 group VI
IRN isorhynchophylline
KLF2 Krüppel like factor 2
LDH dehydrogenase
5-LOX 5-lipoxygenase
Nrf2 NF-E2-related factor
NAC N-Acetyl-L-Cys
NOX1 NADPH oxidase 1
NCCD The Nomenclature Committee on Cell Death
NADPH nicotinamide adenine dinucleotide phosphate
NF-κB nuclear factor-kappa B
PTGS2 prostaglandin endoperoxide synthase 2
PGE2 prostaglandin E2
PIH pyridoxal isonicotinoyl hydrazine
PHGDH phosphoglycerate dehydrogenase phosphoglycerate dehydrogenase
PUFA polyunsaturated fatty acid
ROS reactive oxygen species
RPL8 ribosomal protein L8
RCD regulated cell death
SBI secondary brain injury
SLC7A11 solute carrier family 7 member 11
SLC25A28 olute carrier family 25 member 28
System Xc- cystine/glutamate antiporter
SAT1 spermidine/spermine N1- acetyltransferase 1
Se selenium
SAH subarachnoid haemorrhage
TF transferrin
TFR transferrin receptor
TTC35 tetratricopeptide repeat domain 35
TEM transmission electron microscopy
TNF-α tumor necrosis factor-alpha
TSFP1 transcription factor Sp1
TFAP2C transcription factor activator protein 2C
Tat SelPep Se-containing Cys polypeptide
USP11 Ubiquitin-specific protease 11
Keywords: intracerebral hemorrhage, secondary brain injury, cell death, ferroptosis, iron overload, oxidative damage, reactive oxygen species
Citation: Ren S, Chen Y, Wang L and Wu G (2022) Neuronal ferroptosis after intracerebral hemorrhage. Front. Mol. Biosci. 9:966478. doi: 10.3389/fmolb.2022.966478
Received: 11 June 2022; Accepted: 12 July 2022;
Published: 05 August 2022.
Edited by:
Yanqing Liu, Columbia University, United StatesReviewed by:
Qian Wang, College of Staten Island, United StatesCopyright © 2022 Ren, Chen, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siying Ren, NjEwOTA5ODU3QHFxLmNvbQ==; Guofeng Wu, d3VndW9mZW5nMzAxM0BzaW5hLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.