 Vivian Robin
Vivian Robin Antoine Bodein
Antoine Bodein Marie-Pier Scott-Boyer
Marie-Pier Scott-Boyer Mickaël Leclercq
Mickaël Leclercq Olivier Périn2
Olivier Périn2 Arnaud Droit
Arnaud Droit- 1Molecular Medicine Department, CHU de Québec Research Center, Université Laval, Québec, QC, Canada
- 2Digital Sciences Department, L'Oréal Advanced Research, Aulnay-sous-bois, France
At the heart of the cellular machinery through the regulation of cellular functions, protein–protein interactions (PPIs) have a significant role. PPIs can be analyzed with network approaches. Construction of a PPI network requires prediction of the interactions. All PPIs form a network. Different biases such as lack of data, recurrence of information, and false interactions make the network unstable. Integrated strategies allow solving these different challenges. These approaches have shown encouraging results for the understanding of molecular mechanisms, drug action mechanisms, and identification of target genes. In order to give more importance to an interaction, it is evaluated by different confidence scores. These scores allow the filtration of the network and thus facilitate the representation of the network, essential steps to the identification and understanding of molecular mechanisms. In this review, we will discuss the main computational methods for predicting PPI, including ones confirming an interaction as well as the integration of PPIs into a network, and we will discuss visualization of these complex data.
Introduction
Proteins are essential to life, controlling molecular and cellular mechanisms. Their main role is to carry out cellular biological functions through interactions with molecules or macromolecules (Pellegrini et al., 1999; Vinayagam et al., 2014; Fionda, 2019). These interactions are organized in networks (Bersanelli et al., 2016) of various molecular elements (e.g., protein–DNA and protein–drug) involved in physical and biochemical processes in structured environments. Biological networks have been highlighted by the work of Barabási and Oltvai (2004), who showed that cellular networks are governed by universal laws. This new concept revolutionized the vision of system biology, initiating creation and analysis of the first protein–protein interaction (PPI) network of yeast Saccharomyces cerevisiae (Dezso, Oltvai and Barabási, 2003).
In the PPI network, proteins are represented by nodes, and interactions between proteins by edges (Gursoy, Keskin and Nussinov, 2008; Zou et al., 2018). The size of the network and the amount of information (e.g., discovered node) varies between species (Kotlyar, Rossos and Jurisica, 2017; Wang and Jin, 2017). The number of PPIs is constantly changing due to complexity of the genome and many interactions remain undiscovered (Safari-Alighiarloo et al., 2014; Thanasomboon et al., 2020). PPIs can be determined by high-throughput experiments such as co-immunoprecipitation, two-hybrid screening, pull-down assays (MacDonald, 1998; Lin and Lai, 2017; Louche, Salcedo and Bigot, 2017), or by computational methods. Experimental methods are time-consuming, relatively expensive, and difficult to reproduce (von Mering et al., 2002; Piehler, 2005; Browne et al., 2010; Ngounou Wetie et al., 2013). In response to these challenges, computational methods have emerged, showing promising results in terms of performance to integrate functional (i.e., same biochemical reaction) and physical interactions. A physical interaction describes a physical contact between proteins, as a result of biochemical events steered by interactions including electrostatic forces, hydrogen bonding, and the hydrophobic effect (Berne, Weeks and Zhou, 2009; Nitzan, Casadiego and Timme, 2017). These computational methods allow a more specific identification of interactions than experimental prediction methods (Droit, Poirier and Hunter, 2005; Shoemaker and Panchenko, 2007; Zhou, Li and Wang, 2016).
Although PPIs from computational methods provide a better prediction of physical interactions, PPI databases contain a few false positive interactions (Peng et al., 2017; Luck et al., 2020). One way to remove these false interactions is through integration methods (as can be seen in session integration of a PPI network). Following the integration of the data, it becomes possible to filter PPI. To observe the resulting network and the proteins having a role in mechanisms, visualization is a key step.
Visual representation allows to understanding PPIs and to analyze networks (Iranzo, Krupovic and Koonin, 2016; Armanious et al., 2020; Schneider et al., 2021; Sejdiu and Tieleman, 2021). However, due to complexity of proteomes of different organisms, visualization is a challenge (Crowther, Wipat and Goñi-Moreno, 2021). Moreover, the density of the graph representing the proportion of interactions in the network compared to the total number of possible interactions makes representation more difficult (Ren et al., 2013; Franzese et al., 2019; Wu et al., 2019). To facilitate representation, the network is divided into sub-networks (He and Chan, 2018; Farahani, Karwowski and Lighthall, 2019). These sub-networks are obtained by filtration or by decomposing the network according to proteins of interest, with the concept of ego network (Liu et al., 2019; Tian, Ju and Yang, 2019). Ego networks are subgraphs centered on a seed node and comprise all nodes connected at a defined distance from the ego (seed node) (Zhou, Miao and Yuan, 2018; Malek, Zorzan and Ghoniem, 2020). Sub-networks facilitate representation and allow identification and understanding of cellular mechanisms, core proteins, or biomarkers (Gehlenborg et al., 2010; Laniau, 2017; Hao et al., 2019).
In this review, we will discuss computational methodologies for construction of PPI networks as well as integration and validation of these networks. Next, we will discuss the visualization aspect of a network by discussing its roles and advantages and disadvantages of different visualization tools.
Computational methods for PPI construction
Computational methods for predicting PPIs can be classified into three prediction methods: based on the genomic context, machine learning algorithm, and text mining (Table 1).
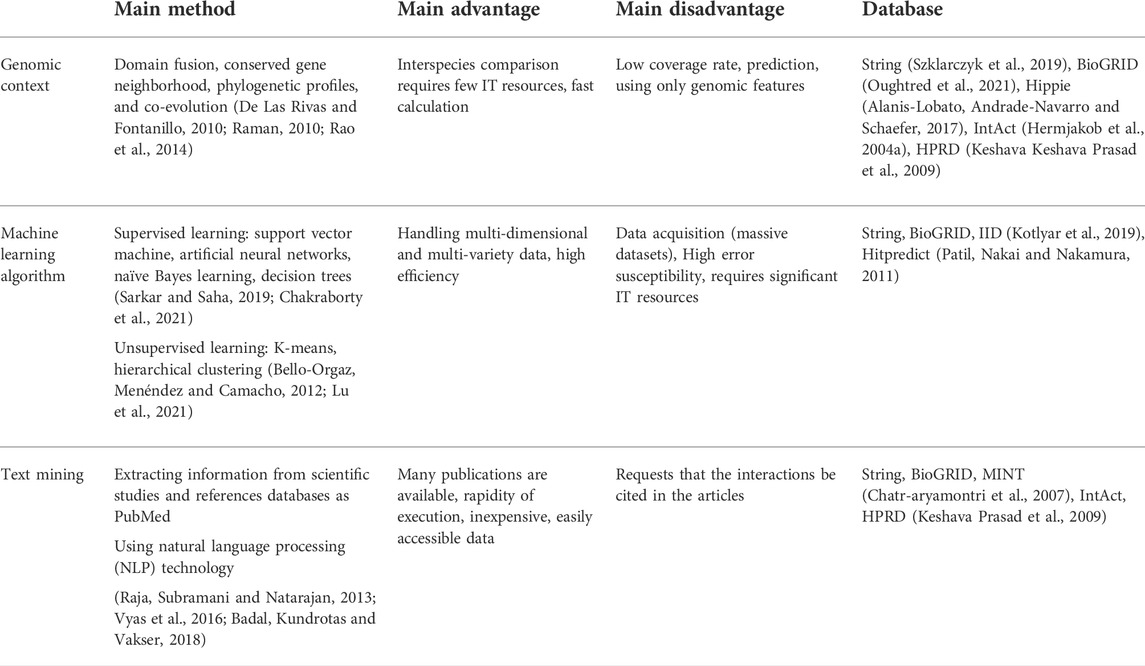
TABLE 1. Summary table of computational methods for the prediction of a protein–protein interaction. Computational methods for predicting PPIs are grouped into three distinct categories: genomic context–based methods, machine learning, and text mining. Within each of these approaches, several sub-methods exist. A database can be composed of interactions obtained by several prediction methods.
The methods can be combined to refine the prediction of PPIs. Alachram et al. (2021) exploited text mining algorithms mixed with machine learning algorithms to capture biologically significant relationships between entities, including PPIs.
Methods based on genomic context
The genomic context refers to the structure of genomic data (e.g., genes), as well as the statistical or mathematical methods to test for gene, protein set association (Dimitrieva and Bucher, 2012; Mooney et al., 2014). Genomic context methods are usually based on gene sequences, structure, and organization of genes on the chromosome (Skrabanek et al., 2008; De Las Rivas and Fontanillo, 2010; Reimand et al., 2012; Rao et al., 2014).
Domain fusion interaction prediction method
Gene fusion leads to fusion proteins, which are an assembly of several proteins encoded by different genes created by joining (fusion) of one or more genes (Morilla et al., 2010; Latysheva et al., 2016). This fusion results in a single or multiple polypeptides that takes on the functional properties of each in original proteins. The existence of a functional interaction between protein A and protein B is based on the hypothesis that if protein domains A and B of one species have fused homologs in a single AB polypeptide in another species, then domains A and B are functionally linked (Truong and Ikura, 2003; Chia and Kolatkar, 2004). The gene fusion method marked a major turning point in methods for predicting PPIs. This computational method, developed by Eisenberg et al. (2000), was the first computational method to find PPIs from the genome of distinct species based on polypeptides (Marcotte et al., 1999).
The comparison of inter-species sequences can show AB sequences, which are also called Rosetta stones because they allow the interaction between A and B to be deciphered (Date, 2007). This method assumes that if the affinity of A and B increases as B increases when A is fused to B, then pairs of proteins may have evolved from proteins with A and B interaction domains on the same polypeptide (Chia and Kolatkar, 2004; Kamisetty et al., 2011). To improve this method, Veitia, (2002) integrated eukaryotic gene sequences. This incorporation increases robustness of AB polypeptide prediction due to the larger volume of sequences in eukaryotes. A question of equilibrium explains this increase in robustness: the required concentrations of proteins A and B cannot be higher than the equilibrium concentration of AB polypeptides, proteins A and B cannot be separated. Despite the addition of these sequences, few PPIs are found explaining a limited interactome or many PPIs are missing (Latysheva et al., 2016). This method is usually combined with other methods such as machine learning methods (De Braekeleer, Douet-Guilbert and De Braekeleer, 2014; Birtles and Lee, 2021). The accuracy values, therefore, take several methods and are not specific to the domain fusion method. Tagore et al. (2019) have developed the ProtFus tool which combines machine learning, protein fusion, and text mining methods to obtain accuracy values between 75% and 83% to predict PPIs.
Conserved gene neighborhood
This method relies on neighbor gene conservation at the genomic scale. This method compares the position of genes from different genomes to predict potential interactions (Dandekar et al., 1998). For example, a gene is always next to the B gene. Two direct neighboring genes in different genomes suggest interactions. This method is widely used in the prediction of PPIs in eukaryotes (Rogozin et al., 2002). Nomenclature discrepancies in ortholog genes, as well as the search of orthologs that are adjacent on chromosome, explain the low predictive coverage of PPIs (Raman, 2010; Lv et al., 2021). Recently, this method in multi-omics integration has confirmed that bacterial genomes are not randomly organized and can form clusters depending on the local genomic context (Esch and Merkl, 2020). They obtained an accuracy value of 55%. As they mention, this type of method is not intended for the discovery of direct interactions. Recently, a new tool: GENPPI (Anjos et al., 2021), allowing the generation of PPI networks by taking into account evolutionary relationships that can only be annotated from genomes, namely, conserved gene neighborhoods (CN), phylogenetic profiles (PPs), and gene fusions, has been introduced, showing that these three methods mainly allow the annotation of missing data and thus the understanding of a limited number of interactions. At present, the tool is being tested in their laboratory.
Phylogenetic profiles
This method is based on the comparison of phylogenetic data between gene families of different organisms (Pellegrini et al., 1999; Škunca and Dessimoz, 2015). The phenotypic profile is represented by a binary vector composed of values 0 and 1, corresponding to the absence and presence of proteins in an organism, respectively. Proteins with close or similar phylogenetic profiles tend to be strongly functionally related (Pellegrini, 2019). Ding and Kihara (2018) recently implemented this approach to predict new interactions from known Arabidopsis thaliana interactions. The phylogenetic profile approach is combined with machine learning approaches. This method allowed the detection of PPIs with high precision and accuracy. In their work, the performance values range from 75% to 93.2% accuracy.
Coevolution
Coevolution is a fundamental principle of evolutionary theory. Coevolution is defined as the chain of transformation events during the evolution of two species in a mutually dependent manner (de Juan, Pazos and Valencia, 2013). Coevolution results from selective pressure between two or more species (Anderson and de Jager, 2020; Takagi et al., 2020). The interactions of coevolved proteins can be kept either by direct binding or by functional associations (Tillier and Charlebois, 2009). If there is an interaction between two proteins, when one protein mutates, the other protein might have a compensatory mutation, otherwise; two proteins cannot support stability or functions of the interaction during evolution. The evolutionary pressure resulted in the elaboration of co-evolutionary protein pairs in cells that keep the interaction and therefore the function of the protein (Pazos et al., 1997; Goh and Cohen, 2002; Xia et al., 2008).
The global advantage of methods based on the genomic context is the interspecies comparison that requires high computing resources (Sun et al., 2008; Pattin and Moore, 2009). The limitations of these methods are a limited number of predicted PPIs, using only genomic features (Chiang et al., 2007; Raman, 2010; Rao et al., 2014). Recent work by Green et al. (2021) using coevolution had accuracy values of the order of 80% showing promising results for the prediction of protein interaction structures and interfaces. The work of Croce et al. (2019) offered similar results in terms of accuracy for the prediction of protein domain interactions.
The methods based on the genomic context are relevant for evolutionary history analysis, small proteome size, or for experimental verification, agronomic analysis on mutations, or other variants (Koh et al., 2012; Zahiri, Bozorgmehr and Masoudi-Nejad, 2013; Malik, Sharma and Khatri, 2017). On the other hand, these prediction methods are less appropriate for medical data analysis, especially for the search of driving proteins in mechanisms due to the high complexity of the human proteome (Kuzmanov and Emili, 2013; Zhong et al., 2019; Swamy, Schuyler and Leu, 2021).
Methods based on the machine learning algorithm
Machine learning (ML) belongs to the field of artificial intelligence (AI) and computer science. ML algorithms learn from already obtained data to predict outcomes in a specific context (El Naqa and Murphy, 2015; Murdoch et al., 2019). This field has undergone a considerable revolution in the last 10 years with the emergence of promising new methods for PPI prediction (Ding and Kihara, 2018; Kotlyar et al., 2019; Das et al., 2020). ML can be classified into two subclasses: supervised and unsupervised learning. Supervised learning can be defined as a machine learning task that learns to predict from labeled data, conversely; unsupervised learning will learn to predict an outcome on unlabeled data (Zhao, Wang and Wu, 2017; Sarkar and Saha, 2019; Razaghi-Moghadam and Nikoloski, 2020).
Supervised learning method for PPI prediction
Support vector machines
Support vector machines, developed by Vapnik, (1963); (Cortes and Vapnik, 1995), build the best hyperplane to separate training sample classes by a maximal margin, with all positive samples lying on one side and all negative samples lying on the other side. Hyperplane, in the framework of a PPI network, will classify the protein pairs as a binary problem. Protein pairs serve as input, and it classifies if an interaction is possible or not. Protein pairs that are close to the hyperplane are called support vectors and predict an interaction between that pair of proteins (Sarkar and Saha, 2019; Chakraborty et al., 2021).
Ma et al. (2020) developed a method called ACT-SVM for predicting PPIs. This model maps protein sequences to numerical features. Extraction of numerical features is performed twice on the protein sequence to obtain two vectors: a vector and descriptor CT (composition and transformation) are combined to form a single vector. Feature vectors of a protein pair will be the input of the SVM. The closer these feature vectors of a pair of proteins are to to hyperplane, the higher the probability of an interaction between these proteins.
Dunham and Ganapathiraju, (2021) benchmarked different PPI prediction algorithms, and show how well they perform on realistically proportioned datasets. Based on verified interactions and a known false interaction rate, 16 datasets using the SVM method are generated. Accuracy values ranged from 51 to 96%, which highlights false interactions predicted or not predicted by the SVM methods.
Artificial neural networks
Artificial neural networks (ANNs) are inspired by neural networks in the brain (Wang, 2003; Zhang, 2018). An artificial neural network is composed of different layers with a variable number of neurons, and each layer is connected between them (Yann Lecun, 1986). To simplify, an ANN network works like an artificial neuron that can receive and send information as a signal to the neurons connected to it. This signal is represented by a real number calculated by a non-linear function of the sum of the inputs to a neuron. Neurons and edges can be weighted, and the weighting is adjusted during the learning process. Weight varies according to the intensity of the signal. Signals travel from the first to the last layer, and this results in the output of active neurons (those with a high intensity) (Baxt, 1995; Krogh, 2008; Dongare, Kharde, and Kachare, 2012).
In the context of PPI prediction, artificial neurons represent pairs of proteins. The signal propagates between different artificial neurons. Neurons and edges with high intensity suggest a connection between proteins. A suggested input for these algorithms is the protein sequences of two proteins, other inputs can be put such as 3D structures of proteins (Xie, Deng and Shu, 2020; Pan et al., 2021). The prediction of PPIs based on their amino acid sequences as well as their physiochemical properties is of great interest to understand the probabilistic constraints of the prediction (Ahmed, Witbooi and Christoffels, 2018; Tang et al., 2021). Sharma and Shrivastava (2015) applied an ANN approach that takes the animated acidic sequences of protein pairs as inputs and returns as output whether the pair interacts or not.
The ANN method had quite similar results to the SVM methods. The accuracy values are variable, Hu et al. (2021) showed an accuracy of 71.5% for the prediction of hot spots in a PPI while Pan et al. (2022) observed an accuracy of about 90% in predicting protein interactions in Arabidopsis thaliana as a result of this work.
ANNs are exploited as a reference method in several classification tasks (Rohani and Eslahchi, 2019; Baek et al., 2021), but they suffer from some limitations. Artificial neurons that are interaction pairs are checked to limit the introduction of bias during the prediction step (H. Li et al., 2018a; Wu et al., 2021).
Naïve Bayes classifier
A naïve Bayes classifier (NBC) relies on the simple probability of the Bayes’ theorem (Bayes et al., 1763). NBC classifies an item by taking each feature of the item independently (e.g., color and shape). To predict a PPI interaction, protein sequences are split into several sub-sequences of n residues. Bayes classifier establishes a probability matrix allowing to classify the different residues; residues that will interact with each other and the non-interface residues. This method is based on conditional probabilities, the probability that is an interaction knowing that an interaction has already occurred. This method will predict interaction sites from protein sequence information alone (Murakami and Mizuguchi, 2010; Geng, Chen and Wang, 2021). Accuracy values are generally lower than those of the SVM and ANN methods, due to the difference in the amount of information available on the proteins.
In PPI prediction, each observation is represented by a vector Z (X1; X2;X3;….; Xm,Y), where X{X1,X2,X3,….,Xm} is the m-dimensional input variable and Y is the output variable taking {0,1}. As input, this method can take either protein interaction datasets or genomic interaction datasets (Jansen et al., 2003; Alashwal, Deris and Othman, 2009; Lin et al., 2021). In the end, the classifier gives a binary response, a zero indicating the interaction is not verified, and a one when there is a potential interaction. Geng et al. (2015) adopted naive Bayes classification to predict site interactions between two proteins. Each pair of proteins is split into several residues, with two residues of two proteins in the same cluster interacting. In terms of performance, they achieved an accuracy value of 60%, which is generally lower than those of the SVM and ANN methods, due to the difference in the amount of information available on the proteins (Ahmed, 2020; Jonathan et al., 2021; Lin et al., 2021).
Identification of interface residues by this method is less expensive and gives results comparable to experimental methods for the prediction of interactions (Murakami and Mizuguchi, 2010; Amirkhah et al., 2015).
Decision trees
A decision tree is a statistical tool that will represent a set of choices as a hierarchical tree. According to different choices made, the algorithm ranks the input elements according to distinctive features: domain presence, spatial folding, site fixation, etc. The decision tree will classify the pair of proteins either as interacting (the proteins in the pair interact with each other) or as non-interacting. Each pair of proteins is characterized by several information and subdomains forming a vector. An interaction is predicted as true if the probability of interactions between two different protein domains is high (Chen and Liu, 2005).
Lee and Oh, (2014) exploited the decision tree method to find discriminating biological features that allow the identification and identify true positive interaction. They have acquired accuracy averages of 97%. This classification helps to understand the biological context of an interaction. The performance of these methods is dependent on the amount of information available for a biological entity and the projection of low-dimensional features (Xuan et al., 2019; Blassel et al., 2021; Zhou et al., 2021). Li et al. (2021) presented challenges of these methods in terms of performance.
Within supervised methods, a sub-class of methods has emerged in recent years: self-supervised learning methods (Chen et al., 2022; Murphy, Jegelka and Fraenkel, 2022), able to train themselves to learn and predict the output of one part of the input data from another part of the data (Wang et al., 2021; Guo et al., 2022). A graph neural network is a self-supervised method for predicting interactions and in particular PPIs (Mahdipour and Ghasemzadeh, 2021; Jha, Saha and Singh, 2022; Y. Wu et al., 2022b). They are based on machine learning algorithms that extract important information from graphs and use this information to make predictions (Li et al., 2020b; Shen et al., 2021). Jha, Saha, and Singh (2022) developed a method for predicting PPI interactions based on structural information contained in the PDB (Burley et al., 2021) and the sequence characteristics of proteins. The molecular graph of a protein has nodes representing the amino acids (also called residues) of which proteins are made up of. A PPI is formed when pairs of atoms contained in two different residues, have a Euclidean distance less than the threshold distance set, here 6 angstroms. They obtained accuracy values after training of 99.5%. The results of this work show better prediction effectiveness than traditional machine learning methods such as SVM and ANN. Although this method is recent, the resulting accuracy values for interaction prediction are promising such as the prediction of drug–target interactions with an average accuracy value of 89.76% (Zhao et al., 2021), and the prediction of ncRNA–protein interactions with an accuracy value of 93.3% (Shen et al., 2021).
Unsupervised learning method for PPI prediction
The unsupervised analysis includes several methods. The most widely used method is clustering, which aimed to group data into clusters. We will focus on two main clustering methods in the context of creating PPI networks (Malouche, 2013; Creusier and Biétry, 2014).
Clustering methods
K-means clustering and hierarchical clustering methods are unsupervised learning techniques, the most used in the prediction of PPIs (Johansson-Åkhe, Mirabello and Wallner, 2019; Nath and Leier, 2020; Wang et al., 2020; Shirmohammady, Izadkhah and Isazadeh, 2021). Proteins will be clustered according to common characteristics (Ou-Yang, Yan and Zhang, 2017). Clustering steps are repeated to refine the clusters and improve prediction of PPIs (Bello-Orgaz, Menéndez and Camacho, 2012; Lu et al., 2021). Proteins in the same cluster have a high probability of interaction (Geng, Chen and Wang, 2021).
The input data can be of various nature for the prediction of PPIs (Krause, Stoye and Vingron, 2005; Zhao, Wang and Wu, 2017; Wang et al., 2020). Sun et al. (2008) relied on the phylogenetic profile of a protein as input. The phylogenetic profile is a comparative genomic method that predicts the large-scale biological molecule function through evolution information (Mikkelsen, Galagan and Mesirov, 2005). Liu et al. (2018) resorted to hot spot residues databases and in particular the Alanine Thermodynamic Scanning Database. Hot spot residues are functional sites in protein interaction interfaces, and these sites allow the understanding of the type of interactions and are highly conserved in proteins to ensure the functions. Itraq (K. Wang et al., 2018a) used protein sequences as input and hierarchical clustering to identify age-related biomarkers of dental caries. Protein interactions were then successfully validated by multiple reaction control mass spectrometry.
Each of these two clustering methods has sub-methods. For example, hierarchical clustering methods can be divided into two sub-families: “bottom-up” and “top-down” methods (Maimon and Rokach, 2006; Wang et al., 2010; S Bhowmick and Seah, 2015).
Clustering methods are known to be sensitive to noisy data due to experimental bias during acquisition of protein sequences (Arnau, Mars and Marín, 2005; Brohée and van Helden, 2006; Wang et al., 2008). As a result, false-positive interactions appear in the clusters (Sloutsky et al., 2013; Pizzuti and Rombo, 2014; Aghakhani, Qabaja and Alhajj, 2018; Stacey, Skinnider and Foster, 2021).
The global advantage of methods based on machine learning is the processing of multidimensional and multivariate data from several omics or horizontal omics (Das et al., 2020; Jamasb et al., 2021). Prediction of interactions is highly efficient (Terayama et al., 2019; Balogh et al., 2022), but machine learning requires large computational resources and large datasets of good quality (Hashemifar et al., 2018; Y. Wang et al., 2018b).
Machine learning–based approaches are approaches that will be scalable in different domains, these approaches offer very promising results (Casadio, Martelli and Savojardo, 2022; Huang et al., 2022; Pan et al., 2022). However, as we have seen in the articles, many sequences or interactions are necessary to train the model (Li M. et al, 2022; Hu et al., 2022; Jha, Saha and Singh, 2022). So, these approaches will be preferred for large-scale omics approaches, prediction of new interactions, or identification of clusters or hubs (protein with many interactions) (Pei et al., 2021; Song et al., 2022; Stringer et al., 2022). Different studies on PPI by You et al. (2013), Shirmohammady, Izadkhah, and Isazadeh (2021), and Kusuma et al. (2019), respectively, showed an accuracy of 88%, 63.8%, and 84.6% for clustering methods. This difference in accuracy is explained by the fact that clustering methods depend on the annotations and missing data contained in them (Wang et al., 2010; Zhou et al., 2022).
Methods based on text mining
Text mining is a technique for exploring and transforming unstructured text into structured data (e.g., tables). In PPI prediction, text mining allowed to extracting information about proteins and their interactions from scientific studies and reference databases. Text mining techniques try to automate the extraction of sentence-related proteins from abstracts or paragraphs of text corpora (Papanikolaou et al., 2015). Several text mining methods exist, some are based on statistical matches between gene names, protein names in public repositories, and online resources. Links and types of interactions between proteins are defined by action verbs, for example, interact, interfering, and reacting. He, Wang and Li (2009) benefited from this technique through the PPI finder tool that was developed to extract human PPIs from PubMed abstracts based on their co-occurrences and interaction words, the retrieved interactions are then validated by the occurrence of Gene Ontology (GO) terms. More complex text mining methodologies use advanced dictionaries and generate natural language processes (NLPs) to build networks. The networks generated by these methods have as nodes the names of the genes or proteins, and as edges the verbs found. By these methods, a semantic notion is added (Raja, Subramani and Natarajan, 2013; Badal, Kundrotas and Vakser, 2018; Roth, Subramanian and Ganapathiraju, 2018). Newer methods utilized kernel methods, a class of algorithms for pattern analysis, to predict PPIs from the text. Vyas et al. (2016) applied this method and data mining for disease-related protein identification, functional annotation, and other proteomic studies. The overall advantage of text mining–based methods is the amount of information available and the extremely low cost to acquire PPIs (Alanis-Lobato, 2015; Zhu and Schmotzer, 2017). The main limitation is that the interactors must be close together or in the same sentence (Badal, Kundrotas and Vakser, 2015; Bajpai et al., 2020). Text mining methods have generally high accuracies because PPIs come from the text published as a result of experiments, thus reducing false interactions. For example, the InfersentPPI (Li X. et al, 2022) tool gave an accuracy value of 0.89 for humans, and the ModEx (Farahmand, Riley and Zarringhalam, 2020) tool gave an assurance value of 0.88.
Interaction prediction methods based on text mining are highlighted in the literature because of the large amount of data available in all domains (Jia et al., 2018; Khashan, Tropsha and Zheng, 2022). These methods are recommended for the study of molecular mechanisms and for a large and fast statistical analysis. But in the context of new experiments where little information is available, these methods do not seem to be very suitable (Elangovan, Davis and Verspoor, 2020; Piereck et al., 2020; Shi et al., 2021).
Integration of a PPI network
A set of interactions between different biological entities that allows the study of biological systems is called an interactome (Cusick et al., 2005; Tieri et al., 2014; Guney et al., 2016; Pinu et al., 2019; Halder et al., 2020; Castillo-Arnemann et al., 2021; Wörheide et al., 2021). Understanding molecular interactions and how they give rise to higher-level functions or diseases is important, especially for repositioning drugs, finding new biomarkers, and potentially developing new therapies or elucidating biological and functional processes (Tieri et al., 2014; Guney et al., 2016; Zhou, Miao and Yuan, 2018; Halder et al., 2020; Castillo-Arnemann et al., 2021; Dimitrakopoulos et al., 2021; L. Wu et al., 2022a). These PPI networks can be integrated horizontally and/or vertically (Lercher and Pál, 2008; Ma and Zhang, 2019). Horizontal integration aimed to create a PPI network from different PPI databases for many interactions (Hibbs et al., 2007; Subramanian et al., 2020), whereas vertical integration will assemble information from different omics (genomics, proteomics, metabolomics, etc.) databases for a given interaction (Wang and Jin, 2017; Ulfenborg, 2019; Das et al., 2020; Welch et al., 2021). All interactions can be modeled into a multi-layered graph structure (Kinsley et al., 2020) where each layer represents a network associated with omic-specific information (Hammoud and Kramer, 2020). PPI networks are a central layer in the multi-omics integration process (Mosca and Milanesi, 2013; Hammoud and Kramer, 2020; Dugourd, Christoph Kuppe and Marco Sciacovelli, 2021) (Figure 1).
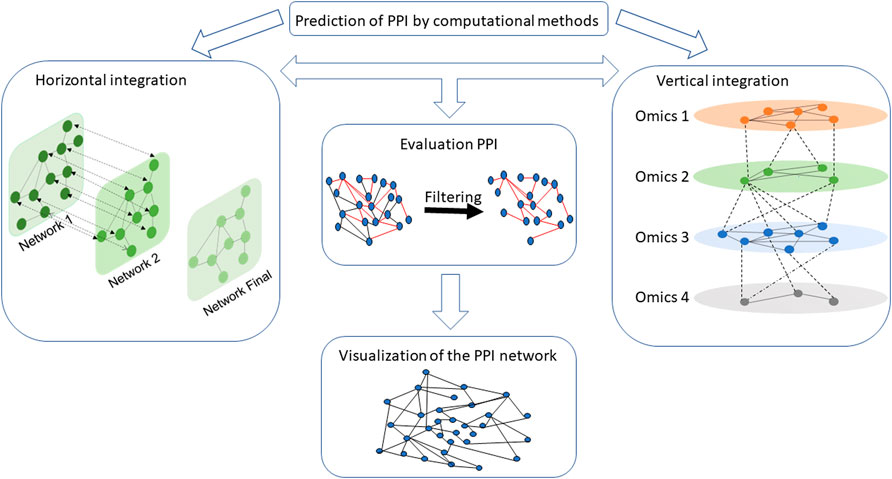
FIGURE 1. Workflow of key steps to design a PPI network assembly. PPI networks can be integrated horizontally and/or vertically. Horizontal integration creates a PPI network by concatenating interaction information from different PPI databases (here networks 1 and 2 represent two PPI networks from two different databases), while vertical integration gathered information from different omics databases for a given interaction. In the vertical integration box, each omics network represents different interactomes such as protein–protein, drug–protein, and RNA–protein. Once the networks are generated, it is necessary to evaluate its interactions confidence to filter the network. Interactions in red are interactions with a high confidence score. After narrowing the network, specialized tools can be used to visualize the network and information about the connected entities (e.g., identify proteins with a central role in the mechanisms).
Horizontal and vertical integration took advantage of topological properties of the network to facilitate construction of different interactomes, to improve classification and evaluation of a PPI (Peng et al., 2017; Kim, Jeong and Sohn, 2019; Halder et al., 2020; Novkovic et al., 2020). Network topology helps in understanding inter/intracellular interactions and functionality, identifying sub-networks (Banerjee et al., 2020; Pournoor et al., 2020; Mishra, Kumar and Mukhtar, 2021). Thus, the topological properties of a PPI network give insight into dynamics of the network and sub-networks and allow the detection of proteins whose roles can be key in complex central biological mechanisms (Yu et al., 2004; Chen et al., 2019; Wahab Khattak et al., 2021). Filtering the network on topological properties allows the acquisition of highly connected nodes and thus facilitates analysis against the topological data. For example, it is possible to filter network by keeping only proteins of a certain degree (Wu et al., 2009; Navlakha et al., 2014; Azevedo and Moreira-Filho, 2015), or by other topological properties from the graph theory such as, degree distribution (Han et al., 2004; Pablo Porras et al., 2020), shortest path (Du et al., 2014), and transitivity (Hakes et al., 2008; Lynn and Bassett, 2021).
Integration of a PPI network in a multi-omics context is nowadays an essential issue in the understanding of biological mechanisms (Hawe, Theis and Heinig, 2019; Bodein et al., 2021; Dimitrakopoulos et al., 2021). To integrate an interaction into a network, it must first be estimated by a so-called confidence score (Stelzl and Wanker, 2006; Li et al., 2016; Xu et al., 2021), representing probability that the interaction is accurately identified by algorithms and is expressed as a percentage (Kamburov et al., 2012; Peng et al., 2017). This score is usually a ratio of the measured value to the total number of the measured value for each interaction. For example, the Mi-score measures the number of publications observed for an interaction out of the overall number of publications available to the network (Villaveces et al., 2015a). Sub-networks represent a part of the network retaining only interactions with a high confidence score (Flórez et al., 2010; Pietrosemoli and Dobay, 2018; Hao et al., 2019), which can also be extracted to facilitate visualizations. Proteins forming groups called clusters in the sub-networks are recovered. By modifying the threshold of the confidence score, we can better define new clusters and the impact size of the sub-network.
Horizontal integration of a PPI network
Horizontal integration is a solution to eliminate these false interactions and allows to find missing data, thus adjusting the resulting confidence score (Everson et al., 2019; Gebreyesus et al., 2022). Horizontal integration methods have contributed to development of various types of databases based on organism-specific diseases, biological processes, and detection methods, such as the Integrated Interactions Database (IID) (Kotlyar et al., 2019), IntAct (Hermjakob et al., 2004a), and StringDB (Szklarczyk et al., 2019). PPI is usually redundant in different databases. A PPI found in one database may also be found in others such as BioGRID (Oughtred et al., 2021) or Reactome (Gillespie et al., 2022). This communication between the different databases corresponds to horizontal data integration (Zitnik and Leskovec, 2017; Cowman et al., 2020).
Assembly and merging are the main algorithms for horizontal integrations (De Las Rivas, Alonso-López and Arroyo, 2018; Amanatidou and Dedoussis, 2021). Two PPI networks are assembled by alignment algorithms. Alignment of PPI networks aimed at finding topological and functional similarities between different PPI networks (Kazemi et al., 2016; Ma and Liao, 2020). In a first step, the alignment algorithm looks for overlapping regions in two networks. These regions form clusters that will be assembled to make a local alignment. Then, using local interactions between clusters, a second alignment is performed: global alignment (Malod-Dognin, Ban and Pržulj, 2017; Alcalá et al., 2020; Chow et al., 2021). Other horizontal integration algorithms applied propagation algorithms as the random walk with restart (RWR) process (detailed in vertical integration of a PPI network). Xu et al. (2018) drawled on these propagation methods to reconstruct a multi-level PPI network and identify protein complexes.
Through these different network alignment algorithms, many PPI databases have been updated or created. The most exploited are BioGRID (Oughtred et al., 2021), IntAct (Hermjakob et al., 2004b), String (Szklarczyk et al., 2019), and UniprotKB (The UniProt Consortium, 2019). A large set of databases is referenced in startbioinfo.org (Kshitish et al., 2013) and pathguide.org (Bader, Cary and Sander, 2006). Following the revolution in NGS technology and the increase in PPI datasets, the integration of a single cell with PPI networks is showing promising results. Indeed, the single-cell method coupled PPI network will allow the understanding of gene regulation, cellular heterogeneity (Cha and Lee, 2020), tissue-specific networks, identification of ligand–receptor interactions, functional interactions, and cell–cell communication (Armingol et al., 2021; Johnson et al., 2021; F. Ma et al., 2021a). Cell–cell interactions mediated by ligand–receptor complexes are essential for the coordination of various biological processes, such as development, differentiation, and inflammation. These interactions subsequently ensure that physiological processes are carried out (Vento-Tormo et al., 2018; Efremova et al., 2020). Using single-cell data and PPI networks, it will be possible to understand this crucial interaction and thus to create new therapies targeting these ligand–receptor interactions in future (Ji et al., 2020; Lee et al., 2021). The applications of single cell PPI are numerous and in many fields such as health (Qi et al., 2022) and agronomy (Zhang et al., 2019). These methods will help in the understanding of cellular mechanisms, regulation according to the environment, and in the development of new therapy (Ryu et al., 2019; Mahdessian et al., 2021). Single-cell data can also be used to filter and weight the PPI network following a differential analysis or by filtering according to fluorescence (Dünkler et al., 2015; Wu et al., 2017). Recently, Klimm et al. (2020) have developed SCPPIN, a method of integrating single-cell RNA-seq data with protein–protein interaction networks. By filtering the network by differentially expressed genes and maximum subgraph weight, they detected active modules in cells of different transcriptional states.
However, horizontal integration faces problems such as uniformity of protein interaction identifiers and redundancy of information, data structure, and organization (Dohrmann, Puchin and Singh, 2015; L. Liu et al., 2020a).
Vertical integration of a PPI network
Vertical integration of networks is generally represented by multi-layer networks (Lv et al., 2021; Watson, Schwartz and Francavilla, 2021). Each layer represents an interactome (protein, gene, and drug). Biological relationships between biological entities and types of interactions form the relationships between different omics layers (Lee and Nam, 2018). Network propagation (or diffusion) algorithms are commonly promoted in omics vertical integration (Di Nanni et al., 2020; Pak et al., 2021). By integrating the information from the different omics and by diffusion algorithms, it is possible to understand the most probable interactions where the diffusion signal has strongly transited (Zhao et al., 2018). Propagation algorithms are a class of algorithms that integrate input data information across connected nodes of a given network. Propagation is usually performed by random walk with restart (RWR) algorithms, inspired by the work of Page et al. (1999) to classify web pages in an objective and mechanical way. RWR is the state-of-the-art approach to infer the relationship: as the name suggests, a random walker, starting from a set of nodes of interest (starting nodes), jumps to neighboring nodes, or nodes in another layer according to a certain probability assigned to the edges of the nodes (Lee and Yoon, 2018). In addition, the walker has a certain probability, known as the damping factor, such that for each step taken in any direction, there is a probability associated with returning to one of the original sets of nodes (Valdeolivas et al., 2019; Nguyen et al., 2021; Qu et al., 2021; Wen et al., 2021). The probability is calculated from a transition matrix from one node to the other, allowing to obtain a weight for each interaction. This node-dependent weight will reflect an interaction between two omics layers (Bhatia, 2019; Dupré, 2022). Lei et al. (2019a) adjusted this method to detect essential proteins. In this method, PPIs are weighted according to network topology, gene expression, and GO annotation data. Then, an initial score is assigned to each protein in a PPI network by exploiting information on subcellular localization and protein complexes. Then the RWR algorithm is applied to the weighted PPI networks to iteratively score the proteins, allowing the filtration of interactions with high weight.
The main other algorithms based on topological properties use integration strategies from two classes: empirical methods and machine learning method (Jin et al., 2014; Haas et al., 2017; Eicher et al., 2020). Empirical methods simply assembled different layers of the network, whereas machine learning methods tried to find missing information about how information flows between the omics layers (Picard et al., 2021; Santiago-Rodriguez and Hollister, 2021). MoGCN (Li X. et al., 2022) is a tool for multi-omics integration based on a convolutional graph network. This tool allows the classification and analysis of cancer subtypes. MoGCN can extract the most significant topological features and properties of each omic layer for downstream biological knowledge discovery.
Integration of PPI networks into multi-layer networks has a central role (Liang et al., 2019; Huang and Zitnik, 2021). Indeed, projection of PPI and layer connectivity allows improvement of the mechanistic and functional knowledge of a cell, identifying key proteins and repositioning drugs (F. Li et al., 2020c). Silverbush and Sharan (2019) created an approach to direct the human PPI network using the drug response and cancer genomic data. A directed graph is a graph in which the edges have a direction. The direction of the relationships or edges is found by diffusion methods. The oriented network allows the detection of key genes in cancers.
In vertical or horizontal integration, the PPI layer must be reliable. The topological properties of the network can allow the establishment of a confidence score for a given interaction. It is essential to understand these properties to build the most robust network possible (Zhang, Xu and Xiao, 2013; Sardiu et al., 2019).
Validation of PPI
An important question persists in network analysis: can we trust on the network of interactions to be a true biological interaction? PPIs from these methods have supplied insights into functions of individual proteins, regulatory pathways, molecular mechanisms, and entire biological systems. Noise inherent in the interactome information hinders evaluation of PPI data (Correia et al., 2019). Several PPIs are, in fact, false positives in these methods and even in methods using strict criteria to define a positive (Yu et al., 2004; Scott and Barton, 2007). It should be noted that the coverage of the interactome is also incomplete and uneven, so we cannot always filter out the less reliable evidence (Han et al., 2005; Stelzl and Wanker, 2006). Many different methods exist for finding reliability and giving a measure of confidence. These techniques can be classified into three main categories.
Contextual biological information
This strategy for assessing the veracity of an interaction looked for different information, for example, overlapping patterns of co-expression, conservation of structure, and sequences (Aytuna, Gursoy and Keskin, 2005; Tirosh and Barkai, 2005). As an example, Schaefer et al. (2013) seek biological information based on influenza virus knowledge to validate PPIs.
Scores based on the literature
Acts as an orthogonal validation and analyzed how often a PPI is cited in publications. The main problem with implementing this method is the application of thresholds, so that only interactions with a sufficiently high score are retained (Bozhilova et al., 2019). Well-studied proteins will have a greater number of interactions and associated publications than proteins that are new or have little information. Hence, thresholds need to be standardized. In order to normalize thresholds among different databases, the MI-score method was created (Villaveces et al., 2015a). This method allows to merge data from different databases that are in the PSI–MI(Proteomics Standards Initiative–Molecular Interaction) format (Hermjakob et al., 2004a; Bader, et al., 2006; Kerrien et al., 2007), and link an interaction to a notation system. This method generates three different scores: publication score (number of different publications on an interaction), method score (considers the different methods of detecting an interaction), and the type of score which refers to the type of interaction. The type of interaction follows the nomenclature of the PSI-MI controlled vocabulary, for example, genetic interaction, physical association, and co-location.
Aggregated methods
Use different score calculation strategies and combine these strategies into a single score. Several scoring methods exist, including the toolkit developed by Braun et al. (2009) that includes four statistical tests to verify a PPI from a high-throughput experiment. The results of the four tests are then combined to calculate the probability that a new pair of interactions is a true biophysical interaction. Intscore is a reference aggregation tool, which calculates confidence scores for user-specified sets of interactions. Its scoring system is based on network topology and annotations. The aggregated score can be computed by machine learning approaches (Kamburov et al., 2012). Recently, Paul and Anand (2022) developed several similarity measures using GO to create a confidence score for PPIs.
Apart from these three distinct categories, to measure the confidence of PPIs, robust measures resulting from data provenance and network topology are needed, such as the average redundancy difference between various sources, natural connectivity of the PPI network as well as the number of edges in a protein-centered sub-arrays (ego networks) (Bozhilova et al., 2019; Wang et al., 2019). The main problem with all these methods is that a score is mainly specific to one database, so threshold values are highly database dependent (Kamburov et al., 2012; Dahiya et al., 2019; Xu et al., 2019). To address this issue, consensus networks appeared such as HugGan (Huang et al., 2022) which is a tool that gathers 31 data sources using deep learning approaches to keep only interactions with a high confidence score resulting in a network with high coverage and quality.
Visualization of protein–protein networks
Networks are a powerful way to visualize complex systems (Charitou, Bryan and Lynn, 2016; Mlecnik, Galon and Bindea, 2018). Visualization of PPI networks is crucial for the understanding of pathways, sub-graphs, sub-network, and central proteins (Sharan and Ideker, 2006; Fionda et al., 2009; Snider et al., 2015; De Las Rivas, Alonso-López and Arroyo, 2018; Vella et al., 2018; Marai et al., 2019). The simplistic and rapid visualization of networks makes it a tool of choice (Gillis, Ballouz and Pavlidis, 2014; Chung et al., 2015; Xu et al., 2021). This has led to the development of methods and tools that allow visualization. The integration of PPI networks and their visualizations in a multi-omics context has helped in the modeling of complex systems such as Parkinson’s disease (Tomkins and Manzoni, 2021), identifying central proteins in diseases (Narayanan et al., 2011; Deng, Xu and Wang, 2019), understanding protein clusters linked to cellular function (Zhao, Wang and Wu, 2017; Amanatidou and Dedoussis, 2021), understanding mechanisms of action (Jia et al., 2021; Yuan et al., 2021), and drug repositioning (Lee and Yoon, 2018; Soleimani Zakeri, Pashazadeh and MotieGhader, 2021).
Larger and complex networks are more difficult to visualize. This is the case of the most popular source offering a representation of PPI networks such as StringDB (Szklarczyk et al., 2019). This online database is intended for the inspection of small networks or sub-networks (less than 500 interactions). Therefore, because of their size and topology, the PPI network requires specialized tools (Bosque et al., 2014; Freilich et al., 2018; Aihaiti et al., 2021).
The methods for visualizing a network can be divided into three categories (Table 2).

TABLE 2. Summary table of tool for visualizing of protein–protein interaction network. Visualization methods to analyze network are grouped into three distinct categories: visualization through downloadable tools, visualization by libraries integrated with languages, and visualization through graph-oriented databases. The user has to choose his tools according to his study context. For analysis of high dimensional data containing a large amount of information, it is advisable to manipulate tools based on graph databases. Conversely, if the user wants to have a quick representation, we recommend the user to turn more to visualization libraries or downloadable software.
The methods can be combined to take advantage of each of the benefits of these categories. This is the case with cyNeo4j (Summer et al., 2015) which combines Cytoscape (Otasek et al., 2019) and Neo4j (Gong et al., 2018) for fast visualization of large networks based on a graph-oriented database. Cytoscape is the most widely used tool for the visualization of large networks (Shannon et al., 2003). Other visualization systems do not fit into these categories and are based on web-based visualization interfaces and on a relational database (Salazar-Ciudad and Jernvall, 2013; Salazar et al., 2014; Hayashi et al., 2018). This is the case of the PINA 3.0 (Du et al., 2021) tool, which is a consensus database containing five interactomes and offering a web visualization service allowing the identification of interacting protein pairs in different cancer types. The weaknesses of these methods are the size of the networks, the execution time of a query, and their limited applicability (Jeanquartier, Jean-Quartier and Holzinger, 2015; Zhou and Xia, 2018; Perlasca et al., 2020).
Visualization tools are evaluated by four criteria: compatibility (available on which OS (operating systems): Windows, Mac Os, and Linux, analytic functions (presence of functions measuring the topological properties of the network, weak interactions of external data, etc.), visualizations (graph layout, dynamics, and parallel implementation), and the extensibility of the tool (addition of plugins, type of input, and output file) forming distinct classes (Sanz-Pamplona et al., 2012; Agapito, Guzzi and Cannataro, 2013; Dallago et al., 2020). In the context of biological network analysis and in particular protein networks, one of the essential criteria is dynamic visualization tools (Xia, Benner and Hancock, 2014; Zhou and Xia, 2018). PPI networks have a dynamic organization of biological sub-networks (Yang, Wagner and Beli, 2015). In other words, the molecular interactions in a cell vary in time, as do the signals from the environment surrounding an interaction (Przytycka, Singh and Slonim, 2010; M. Li et al., 2018b).
In order to overcome the limitation of network size and consider the dynamics of the networks, several tools have been developed over the last decades (Sanz-Pamplona et al., 2012; Winkler et al., 2021). The success of Cytoscape is due to the large number of plugins/features that can be added directly from the tool (Saito et al., 2012; Lotia et al., 2013). The calculation of overrepresented GO terms in a network can be performed by Bingo (Maere, Heymans and Kuiper, 2005), a widely downloaded Cystoscape plugin. Through Cytoscape, we also find plugins allowing the understanding of the dynamic organization of biological networks such as TVNViewer (Curtis et al., 2011), KDDN (Tian et al., 2015), and Dynetviewer. Another downloadable software offering a visual representation of PPI networks is the Gephi (Bastian, Heymann and Jacomy, 2009). Downloadable network visualization tools have difficulties with the implementation of data (Villaveces et al., 2015b; Li et al., 2016). Visualization libraries such as igraph (Csárdi and Nepusz, 2006) and NetworkX (Hagberg et al., 2008) will make it easier to import and export networks but are limited in terms of adding new functionality and graphic possibilities (Pandey, 2018; L. Wu et al., 2022a).
Network visualization tools are specific to the detection method (Ashtiani et al., 2018). HPIminer (Subramani et al., 2015) extracts information from human PPIs and PPI pairs in biomedical literature and provides a visualization of interactions, networks, and associated pathways using two databases, namely, HPRD (Goel et al., 2012) and KEGG (Kanehisa et al., 2016). Another area of improvement for online or general-purpose visualization tools and libraries is the addition of a visualization engine or search engine (Chisanga et al., 2017). Tools integrating visualization engines such as NAViGaTOR (Brown et al., 2009) and MIST (Hu et al., 2018) have been developed. These tools allow the acceleration of the visualization of large PPI networks (Yu and Zhang, 2008; Gerasch et al., 2014; Zaki and Tennakoon, 2017). It is also possible to improve the speed of visualizations by connecting directly to graph databases such as Neo4j (Gong et al., 2018, p. 4) and ArangoDB (Touré et al., 2016; Timón-Reina, Rincón and Martínez-Tomás, 2021; ArangoDB NoSQL Multi-Model Database: Graph, Document, Key/Value, 2022). Since graph databases store data directly in a graph form, they are becoming a preferred resource for storing complex relationships of heterogeneous biological data (Yoon, Kim and Kim, 2017; Jupe et al., 2018; Castillo-Arnemann et al., 2021). Flexibility of multi-omics integration offered by graph databases facilitates data mining to support different hypotheses (Lysenko et al., 2016; Brandizi et al., 2018; Wandy and Daly, 2021).
All these tools for the visualization of PPI networks are based on different visualization algorithms (Koutrouli et al., 2020; Sandoval and Orlando, 2021). Visualization algorithms can be based on simplistic approaches such as adjacent matrices (Fekete, 2009), circular layouts (Suderman and Hallett, 2007), or complex approaches such as force-directed algorithms (Liu et al., 2021). The main differences between simple and complex algorithms for visualization depend on the size of the network, the topology of the network, and the dimensionality of the information (Heberle et al., 2017; Becker et al., 2020; Raja et al., 2020). The selection of the appropriate visualization algorithm will depend on the nature of the network. In the context of single networks, in particular PPI networks, visualization algorithms focus on the identification of protein sub-clusters or hub proteins (Li et al., 2020b; H. Ma et al., 2021b). Cytoscape’s Cytohubba (Chin et al., 2014) plugin is commonly dedicated for sub-network identification and central protein identification. The most powerful method of Cytohubba for better sub-network visualization is the maximum clique centrality (MCC) method. This algorithm allows the visualization of groups of proteins called clusters, based on the assumption that essential proteins tend to be grouped together (Lu et al., 2010; Lei et al., 2019b; Kim, Jeong and Sohn, 2019). Recently, Zu et al. (2017) used this plugin’s method to visualize six target genes for quercetin (an organic compound of the flavonoid family), suggesting a therapeutic potential in type 2 diabetes mellitus (T2DM) and Alzheimer’s disease.
However, in a multi-omics integrations context one seeks above all to connect information from different omics fields (transcriptomics, proteomics, metabolomics, lipidomics, and metabolomics (Haas et al., 2017; Fan, Zhou and Ressom, 2020; Cansu Demirel, Kaan Arici and Tuncbag, 2022). In this context, multi-layer algorithms for visualization are preferable to force-directed algorithms (Bodein et al., 2021; Dursun, Kwitek and Bozdag, 2021; Marín-Llaó et al., 2021). There are several algorithms for implementing multi-layer networks, in the context of multi-omics integration, the most highlighted implementation is the one named by Hammoud and Kramer, (2020): “Interactive/Interconnected/Interdependent Networks and Networks of Networks Implementation.” This implementation has as input a set of monoplex networks (single layer networks, e.g., PPI network). Each network interacts with the other networks. The different monoplex networks will form distinct layers which will be connected by the inter-side nodes (Rappoport and Shamir, 2018; Yan et al., 2018; Zoppi et al., 2021; Cuenca et al., 2022). Recently Arena3dweb (Karatzas et al., 2021), a web application incorporating these algorithms and offering a visualization of multi-layer graphs in a 3D space, has enabled GPCR signaling pathways implicated in melanoma.
Summary and outlook
In this review, different computational strategies for predicting PPI, from integration to visualization to methods for validating interactions have been studied. Many computational prediction approaches rely on experimental methods to predict a PPI interaction (Rao et al., 2014; Peng et al., 2017; Ding and Kihara, 2018; Tanwar and George Priya Doss, 2018). Although this increases the coverage of the network, it can disrupt the horizontal integration process (Browne et al., 2010; Ngounou Wetie et al., 2013). Sets of PPI interactions from different datasets are constructed and transformed independently, which can lead to information gaps, redundant information, and poor identifier compatibility when aligning two PPI networks. Ideally, at any point in the overall integration process (including vertical and horizontal), each omics data set should be evaluated in the context of the other datasets, so that complementary information can be fully exploited, and added information can be identified (Bozhilova et al., 2019; Bajpai et al., 2020). Implementation of validation scores based on topological properties allows to limit the redundancy of edges and will allow to filter the PPI network (Pietrosemoli and Dobay, 2018; Sardiu et al., 2019).
Information redundancy is the repetition of information without adding additional information in different databases. The increase in omics data and PPI integration methods has contributed to the growth of many PPI databases. However, this increase in the number of databases increases the redundancy of information, making it difficult for the user to choose a PPI database (Rabbani et al., 2018; Hawe, Theis and Heinig, 2019; Zahiri et al., 2020). In addition, information redundancy slows down the calculation time for the construction and visualization of networks (Chen et al., 2019, 2019). To limit and remove redundancy, different information scores have been set up (Silverbush and Sharan, 2019; Mahdipour and Ghasemzadeh, 2021). The Mi-score (Villaveces et al., 2015b) consisting of three scores, is increasingly used to validate a PPI.
The study of PPI networks is a growing field of systems biology. Due to their significant role, PPI networks are used to understand cellular functions or biological mechanisms (Stelzl and Wanker, 2006; Jordán, Nguyen and Liu, 2012; Safari-Alighiarloo et al., 2014). The integration of these networks, both vertically and horizontally, can highlight clusters of proteins with central roles, aiding the understanding of drug action mechanisms (Martin, Roe and Faulon, 2005; Dimitrakopoulos et al., 2021; Marín-Llaó et al., 2021; Tomkins and Manzoni, 2021). PPI networks offer prospects in many fields, such as medicine, health and also in agri-food (Hao et al., 2019; Hasan et al., 2020; Thanasomboon et al., 2020; Charmpi et al., 2021). Vertical and horizontal integration algorithms are mainly based on propagation and alignment algorithms but are often combined with machine learning methods to predict the probability of reliability of an interaction (Li and Ilie, 2017; Lee and Nam, 2018; Zhang et al., 2018; Das and Chakrabarti, 2021). These propagation algorithms will allow to focus on sub-networks, keeping only the interactions where the propagation signal is high (Gehlenborg et al., 2010; Laniau, 2017).
By focusing on sub-networks as opposed to complete networks, visualization is facilitated allowing the identification of sub-groups of interactions (Tian, Ju and Yang, 2019; T.-H. Liu et al., 2020b). The visualization of networks is a problematic issue for networks and especially for PPI networks (Du et al., 2021). Visualization tools depend mainly on the size of our networks (Summer et al., 2015; Zou et al., 2017). Currently, multilayer network visualization is limited to small networks and requires a consequent pre-formatting of the data (Smith-Aguilar et al., 2019; Hammoud and Kramer, 2020; Sebestyén, Domokos and Abonyi, 2020).The study of multilayer networks based on the PPI network is constantly evolving and will become more powerful with advancement of more powerful mathematical models offering better predictions (Kapadia et al., 2019; Karatzas et al., 2021; Cuenca et al., 2022). Different perspectives on the integration of PPI networks can be imagined. The visualization of multilayer multi-omics networks and creation of consensus networks for each omics dimension to understanding new mechanisms of multi-omics integration. A consensus network is the result of the horizontal integration of different databases (Berto et al., 2016; Mosca et al., 2021). Through this network, it will be possible to homogenize the different thresholds of the different databases and to eliminate the recurrence of information (Leblanc et al., 2013; Affeldt et al., 2016; Zohra Smaili et al., 2021). Recently, Woo and Yoon (2021) created a Monaco aligner that can find multiple alignments with high accuracy to identify functional modules. In the era of big data and NGS (next generating sequencing) technologies, it is difficult to know which information is needed to build a PPI network. Machine learning and deep learning methods offer novel perspectives in the prediction and standardization of information in PPI networks (Gligorijević and Pržulj, 2015; Borhani et al., 2022; Cervantes-Gracia, Chahwan and Husi, 2022). Standardizing and evaluating the relevance of interactions will facilitate integration of PPI networks (Fiorentino et al., 2021; Nadeau, Byvsheva and Lavallée-Adam, 2021).
On the visualization side, several perspectives can be imagined, a tool to visualize each layer independently and globally in a multilayer network (Kanai, Maeda and Okada, 2018; McGee et al., 2019). As the size and complexity of PPI networks increases, more efficient visualization algorithms are needed (Chong, Wishart and Xia, 2019; Koutrouli et al., 2020). Augmented reality technologies and virtual reality (VR) remove the constraints of 2D/3D space constraints (Pirch et al., 2021; Hütter et al., 2022). Moreover, the notable advances in the prediction of the structure of proteins from their sequence in amino acids with alphafold (Jumper et al., 2021), which could lead to a revolution in the PPI prediction algorithm. In view of the generous size of PPI networks, visualization tools focus on specific networks, including Mechnetor (González-Sánchez et al., 2021), a tool for visualization of biological mechanisms. At the moment, there are no tools available to visualize the interactome protein specific to a tissue, but there are different databases on this subject (Islam et al., 2013; Basha et al., 2018).
Author contributions
VR wrote the manuscript, VR designed the figures and tables, and VR, AB, MPSB, ML, OP and AD revised the manuscript. AD supervised the research.
Funding
This work was supported by Research and Innovation chair L'Oréal in Digital Biology.
Conflict of interest
Author OP is employed by company L'Oréal.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Affeldt, S., Sokolovska, N., Prifti, E., and Zucker, J. D. (2016). Spectral consensus strategy for accurate reconstruction of large biological networks. BMC Bioinforma. 17 (16), 493. doi:10.1186/s12859-016-1308-y
Agapito, G., Guzzi, P. H., and Cannataro, M. (2013). Visualization of protein interaction networks: Problems and solutions. BMC Bioinforma. 14 (1), S1. doi:10.1186/1471-2105-14-S1-S1
Aghakhani, S., Qabaja, A., and Alhajj, R. (2018). Integration of k-means clustering algorithm with network analysis for drug-target interactions network prediction. Int. J. Data Min. Bioinform. 20, 185. doi:10.1504/IJDMB.2018.10016075
Ahmed, I., Witbooi, P., and Christoffels, A. (2018). Prediction of human-Bacillus anthracis protein-protein interactions using multi-layer neural network. Bioinforma. Oxf. Engl. 34 (24), 4159–4164. doi:10.1093/bioinformatics/bty504
Ahmed, M. (2020). Modified naive Bayes classifier for classification of protein- protein interaction sites. J. Biosci. Agric. Res. 26, 2177–2184. doi:10.18801/jbar.260220.266
Aihaiti, Y., Song Cai, Y., Tuerhong, X., Ni Yang, Y., Ma, Y., Shi Zheng, H., et al. (2021). Therapeutic effects of naringin in rheumatoid arthritis: Network pharmacology and experimental validation. Front. Pharmacol. 12, 672054. doi:10.3389/fphar.2021.672054
Alachram, H., Chereda, H., BeiBbarth, T., Wingender, E., and Stegmaier, P. (2021). Text mining-based word representations for biomedical data analysis and protein-protein interaction networks in machine learning tasks. PloS One 16 (10), e0258623. doi:10.1371/journal.pone.0258623
Alanis-Lobato, G., Andrade-Navarro, M. A., and Schaefer, M. H. (2017). HIPPIE v2.0: Enhancing meaningfulness and reliability of protein–protein interaction networks. Nucleic Acids Res. 45, D408–D414. doi:10.1093/nar/gkw985
Alanis-Lobato, G. (2015). Mining protein interactomes to improve their reliability and support the advancement of network medicine. Front. Genet. 6. Availableat: https://www.frontiersin.org/article/10.3389/fgene.2015.00296 (Accessed: February 25, 2022).
Alashwal, H., Deris, S., and Othman, R. M. (2009). A bayesian kernel for the prediction of protein- protein interactions, 6.
Alcalá, A., Alberich, R., Llabres, M., Rossello, F., and Valiente, G. (2020). AligNet: Alignment of protein-protein interaction networks. BMC Bioinforma. 21 (6), 265. doi:10.1186/s12859-020-3502-1
Amanatidou, A. I., and Dedoussis, G. V. (2021). Construction and analysis of protein-protein interaction network of non-alcoholic fatty liver disease. Comput. Biol. Med. 131, 104243. doi:10.1016/j.compbiomed.2021.104243
Amirkhah, R., Farazmand, A., Gupta, S. K., Ahmadi, H., Wolkenhauer, O., and Schmitz, U. (2015). Naïve Bayes classifier predicts functional microRNA target interactions in colorectal cancer. Mol. Biosyst. 11 (8), 2126–2134. doi:10.1039/c5mb00245a
Anjos, W. F., Lanes, G. C., Azevedo, V. A., and Santos, A. R. (2021). Genppi: Standalone software for creating protein interaction networks from genomes. BMC Bioinforma. 22 (1), 596. doi:10.1186/s12859-021-04501-0
ArangoDB NoSQL Multi-Model Database: Graph, Document, Key/Value (2022). ArangoDB. Availableat: https://www.arangodb.com/(Accessed March 2, 2022).
Armanious, D., Schuster, J., Tollefson, G. A., Agudelo, A., DeWan, A. T., Istrail, S., et al. (2020). Proteinarium: Multi-sample protein-protein interaction analysis and visualization tool. Genomics 112 (6), 4288–4296. doi:10.1016/j.ygeno.2020.07.028
Armingol, E., Officer, A., Harismendy, O., and Lewis, N. E. (2021). Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22 (2), 71–88. doi:10.1038/s41576-020-00292-x
Arnau, V., Li, S., and Marín, I. (2005). MarsIterative cluster Analysis of protein interaction data. Bioinformatics 21 (3), 364–378. doi:10.1093/bioinformatics/bti021
Ashtiani, M., Salehzadeh-Yazdi, A., Razaghi-Moghadam, Z., Hennig, H., Wolkenhauer, O., Mirzaie, M., et al. (2018). A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst. Biol. 12, 80. doi:10.1186/s12918-018-0598-2
Auber, D., Archambault, D., and Bourqui, R. (2017). “Tulip 5,” in Encyclopedia of social network analysis and mining. Editors R. Alhajj, and J. Rokne (Springer), 1–28. doi:10.1007/978-1-4614-7163-9_315-1
Aytuna, A., Gursoy, A., and Keskin, O. (2005). Prediction of protein-protein interactions by combining structure and sequence conservation in protein interfaces. Bioinformatics, 21. Oxford, England, 2850–2855. doi:10.1093/bioinformatics/bti443
Azevedo, H., and Moreira-Filho, C. A. (2015). Topological robustness analysis of protein interaction networks reveals key targets for overcoming chemotherapy resistance in glioma. Sci. Rep. 5 (1), 16830. doi:10.1038/srep16830
Badal, V. D., Kundrotas, P. J., and Vakser, I. A. (2018). Natural language processing in text mining for structural modeling of protein complexes. BMC Bioinforma. 19 (1), 84. doi:10.1186/s12859-018-2079-4
Badal, V. D., Kundrotas, P. J., and Vakser, I. A. (2015). Text mining for protein docking. PLoS Comput. Biol. 11 (12), e1004630. doi:10.1371/journal.pcbi.1004630
Bader, G. D., Cary, M. P., and Sander, C. (2006). Pathguide: A pathway resource list. Nucleic Acids Res. 34, D504–D506. doi:10.1093/nar/gkj126
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Sci. (New York, N.Y.) 373 (6557), 871–876. doi:10.1126/science.abj8754
Bajpai, A. K., Davuluri, S., Tiwary, K., Narayanan, S., Oguru, S., Basavaraju, K., et al. (2020). Systematic comparison of the protein-protein interaction databases from a user’s perspective. J. Biomed. Inf. 103, 103380. doi:10.1016/j.jbi.2020.103380
Balogh, O. M., Benczik, B., Horvath, A., Petervari, M., Csermely, P., Ferdinandy, P., et al. (2022). Efficient link prediction in the protein–protein interaction network using topological information in a generative adversarial network machine learning model. BMC Bioinforma. 23 (1), 78. doi:10.1186/s12859-022-04598-x
Banerjee, K., Jana, T., Ghosh, Z., and Saha, S. (2020). PSCRIdb: A database of regulatory interactions and networks of pluripotent stem cell lines. J. Biosci. 45, 53. doi:10.1007/s12038-020-00027-4
Barabási, A.-L., and Oltvai, Z. N. (2004). Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 5 (2), 101–113. doi:10.1038/nrg1272
Basha, O., Shpringer, R., Argov, C. M., and Yeger-Lotem, E. (2018). The DifferentialNet database of differential protein–protein interactions in human tissues. Nucleic Acids Res. 46 (1), D522–D526. doi:10.1093/nar/gkx981
Bastian, M., Heymann, S., and Jacomy, M. (2009). Gephi : An open source software for exploring and manipulating networks, 2.
Baxt, W. G. (1995). Application of artificial neural networks to clinical medicine. Lancet 346 (8983), 1135–1138. doi:10.1016/S0140-6736(95)91804-3
Bayes, M., Moivre, A., and Price, M. (1763). An essay towards solving a problem in the doctrine of chances. By the late rev. Mr. Bayes, F. R. S. Communicated by mr. Price, in a letter to john canton, A. M. F. R. S.’. Philos. Trans. 53, 370–418.
Becker, M., Lippel, J., Stuhlsatz, A., and Zielke, T. (2020). Robust dimensionality reduction for data visualization with deep neural networks. Graph. Models 108, 101060. doi:10.1016/j.gmod.2020
Bello-Orgaz, G., Menéndez, H. D., and Camacho, D. (2012). Adaptive k-means algorithm for overlapped graph clustering. Int. J. Neural Syst. 22 (5), 1250018. doi:10.1142/S0129065712500189
Berne, B. J., Weeks, J. D., and Zhou, R. (2009). Dewetting and hydrophobic interaction in physical and biological systems. Annu. Rev. Phys. Chem. 60, 85–103. doi:10.1146/annurev.physchem.58.032806.104445
Bersanelli, M., Mosca, E., Remondini, D., Giampieri, E., Sala, C., Castellani, G., et al. (2016). Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinforma. 17 (2), S15. doi:10.1186/s12859-015-0857-9
Berto, S., Perdomo-Sabogal, A., Gerighausen, D., Qin, J., and Nowick, K. (2016). A consensus network of gene regulatory factors in the human frontal lobe. Front. Genet. 7, 31. doi:10.3389/fgene.2016.00031
Bhatia, C. (2019). ‘Random walk with restart and its applications’. Medium 8. Availableat: https://medium.com/@chaitanya_bhatia/random-walk-with-restart-and-its-applications-f53d7c98cb9 (Accessed: April 4, 2022).
Bhowmick, S., and Seah, B.-S. (2015). Clustering and summarizing protein-protein interaction networks: A survey. IEEE Trans. Knowl. Data Eng. 28, 638–658. doi:10.1109/TKDE.2015.2492559
Birtles, D., and Lee, J. (2021). Identifying distinct structural features of the SARS-CoV-2 spike protein fusion domain essential for membrane interaction. Biochemistry 60 (40), 2978–2986. doi:10.1021/acs.biochem.1c00543
Blassel, L., Tostevin, A., Villabona-Arenas, C. J., Peeters, M., Hue, S., Gascuel, O., et al. (2021). Using machine learning and big data to explore the drug resistance landscape in HIV. PLoS Comput. Biol. 17 (8), e1008873. doi:10.1371/journal.pcbi.1008873
Bodein, A., Scott-Boyer, M. P., Perin, O., Le Cao, K. A., and Droit, A. (2021). Interpretation of network-based integration from multi-omics longitudinal data. Nucleic Acids Res. 50, e27. doi:10.1093/nar/gkab1200
Borhani, N., Ghaisari, J., Abedi, M., Kamali, M., and Gheisari, Y. (2022). A deep learning approach to predict inter-omics interactions in multi-layer networks. BMC Bioinforma. 23 (1), 53. doi:10.1186/s12859-022-04569-2
Bosque, G., Folch-Fortuny, A., Pico, J., Ferrer, A., and Elena, S. F. (2014). Topology analysis and visualization of Potyvirus protein-protein interaction network. BMC Syst. Biol. 8, 129. doi:10.1186/s12918-014-0129-8
Bozhilova, L. V., Whitmore, A. V., Wray, J., Reinert, G., and Deane, C. M. (2019). Measuring rank robustness in scored protein interaction networks. BMC Bioinforma. 20 (1), 446. doi:10.1186/s12859-019-3036-6
Brandizi, M., Singh, A., Rawlings, C., and Hassani-Pak, K. (2018). Towards FAIRer biological knowledge networks using a hybrid linked data and graph database approach. J. Integr. Bioinform. 15 (3). doi:10.1515/jib-2018-0023
Braun, P., Tasan, M., Dreze, M., Barrios-Rodiles, M., Lemmens, I., Yu, H., et al. (2009). An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 6 (1), 91–97. doi:10.1038/nmeth.1281
Brohée, S., and van Helden, J. (2006). Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinforma. 7 (1), 488. doi:10.1186/1471-2105-7-488
Brown, K. R., Otasek, D., Ali, M., McGuffin, M. J., Xie, W., Devani, B., et al. (2009). NAViGaTOR: Network analysis, visualization and graphing toronto. Bioinformatics 25 (24), 3327–3329. doi:10.1093/bioinformatics/btp595
Browne, F., Wang, H., and Zheng, H. (2010). From experimental approaches to computational techniques: A review on the prediction of protein-protein interactions. Adv. Artif. Intell. 2010, e924529. doi:10.1155/2010/924529
Burley, S. K., Bhikadiya, C., Bi, C., Bittrich, S., Chen, L., Crichlow, G. V., et al. (2021). RCSB protein data bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49 (1), D437–D451. doi:10.1093/nar/gkaa1038
Cansu Demirel, H., Kaan Arici, M., and Tuncbag, N. (2022). Computational approaches leveraging integrated connections of multi-omic data toward clinical applications. Mol. Omics 18 (1), 7–18. doi:10.1039/D1MO00158B
Casadio, R., Martelli, P. L., and Savojardo, C. (2022). Machine learning solutions for predicting protein–protein interactions. WIREs Comput. Mol. Sci., e1618. doi:10.1002/wcms.1618
Castillo-Arnemann, J. J., Solodova, O., Dhillon, B. K., and Hancock, R. E. W. (2021). PaIntDB: Network-based omics integration and visualization using protein–protein interactions in Pseudomonas aeruginosa. Bioinformatics 37 (22), btab363–4281. doi:10.1093/bioinformatics/btab363
Cervantes-Gracia, K., Chahwan, R., and Husi, H. (2022). Integrative OMICS data-driven procedure using a derivatized meta-analysis approach. Front. Genet. 13, 828786. doi:10.3389/fgene.2022.828786
Cha, J., and Lee, I. (2020). Single-cell network biology for resolving cellular heterogeneity in human diseases. Exp. Mol. Med. 52 (11), 1798–1808. doi:10.1038/s12276-020-00528-0
Chakraborty, A., Mitra, S., De, D., Pal, A. J., Ghaemi, F., Ahmadian, A., et al. (2021). Determining protein–protein interaction using support vector machine: A review. IEEE Access 9, 12473–12490. doi:10.1109/ACCESS.2021.3051006
Charitou, T., Bryan, K., and Lynn, D. J. (2016). Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 48 (1), 27. doi:10.1186/s12711-016-0205-1
Charmpi, K., Chokkalingam, M., Johnen, R., and Beyer, A. (2021). Optimizing network propagation for multi-omics data integration. PLoS Comput. Biol. 17 (11), e1009161. doi:10.1371/journal.pcbi.1009161
Chatr-aryamontri, A., Ceol, A., Palazzi, L. M., Nardelli, G., Schneider, M. V., Castagnoli, L., et al. (2007). Mint: The molecular INTeraction database. Nucleic Acids Res. 35, D572–D574. doi:10.1093/nar/gkl950
Chen, J., Zhang, L., Cheng, K., Jin, B., Lu, X., and Che, C. (2022). Predicting drug-target interaction via self-supervised learning. IEEE/ACM Trans. Comput. Biol. Bioinform., 1. doi:10.1109/TCBB.2022.3153963
Chen, S.-J., Liao, D. L., Chen, C. H., Wang, T. Y., and Chen, K. C. (2019). Construction and analysis of protein-protein interaction network of heroin use disorder. Sci. Rep. 9 (1), 4980. doi:10.1038/s41598-019-41552-z
Chen, X.-W., and Liu, M. (2005). Prediction of protein–protein interactions using random decision forest framework. Bioinformatics 21 (24), 4394–4400. doi:10.1093/bioinformatics/bti721
Chia, J.-M., and Kolatkar, P. R. (2004). Implications for domain fusion protein-protein interactions based on structural information. BMC Bioinforma. 5, 161. doi:10.1186/1471-2105-5-161
Chiang, T., Scholtens, D., Sarkar, D., Gentleman, R., and Huber, W. (2007). Coverage and error models of protein-protein interaction data by directed graph analysis. Genome Biol. 8 (9), R186. doi:10.1186/gb-2007-8-9-r186
Chin, C.-H., Chen, S. H., Wu, H. H., Ho, C. W., Ko, M. T., and Lin, C. Y. (2014). cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8 (4), S11. doi:10.1186/1752-0509-8-S4-S11
Chisanga, D., Keerthikumar, S., and Mathivanan, S. (2017). Network tools for the analysis of proteomic data. Methods Mol. Biol. 1549, 177–197. doi:10.1007/978-1-4939-6740-7_14
Chong, J., Wishart, D. S., and Xia, J. (2019). Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinforma. 68 (1), e86. doi:10.1002/cpbi.86
Chow, K., Sarkar, A., Elhesha, R., Cinaglia, P., Ay, A., and Kahveci, T. (2021). Anca: Alignment-based network construction algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 18 (2), 512–524. doi:10.1109/TCBB.2019.2923620
Chung, S. S., Pandini, A., Annibale, A., Coolen, A. C. C., Thomas, N. S. B., and Fraternali, F. (2015). Bridging topological and functional information in protein interaction networks by short loops profiling. Sci. Rep. 5 (1), 8540. doi:10.1038/srep08540
Correia, F. B., Coelho, E. D., and Oliveira, J. L. (2019). Handling noise in protein interaction networks. BioMed Res. Int. 2019, 8984248. doi:10.1155/2019/8984248
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20 (3), 273–297. doi:10.1007/BF00994018
Cowman, T., Coskun, M., Grama, A., and Koyuturk, M. (2020). Integrated querying and version control of context-specific biological networks. Database., 2020, baaa018. doi:10.1093/database/baaa018
Creusier, J., and Biétry, F. (2014). Analyse comparative des méthodes de classifications. RIMHE Revue Interdiscip. Manag. Homme & Entreprise 103 (1), 105–123. doi:10.3917/rimhe.010.0105
Croce, G., Gueudre, T., Ruiz Cuevas, M. V., Keidel, V., Figliuzzi, M., Szurmant, H., et al. (2019). A multi-scale coevolutionary approach to predict interactions between protein domains. PLoS Comput. Biol. 15 (10), e1006891. doi:10.1371/journal.pcbi.1006891
Crowther, M., Wipat, A., and Goñi-Moreno, Á. (2021). Network visualisation of synthetic biology designs. bioRxiv, 2021. doi:10.1101/2021.09.14.460206
Csárdi, G., and Nepusz, T. (2006). The igraph software package for complex network research. Availableat: https://www.semanticscholar.org/paper/The-igraph-software-package-for-complex-network-Cs%C3%A1rdi-Nepusz/1d2744b83519657f5f2610698a8ddd177ced4f5c (Accessed January 29, 2022).
Cuenca, E., Sallaberry, A., Ienco, D., and Poncelet, P. (2022). VERTIGo: A visual platform for querying and exploring large multilayer networks. IEEE Trans. Vis. Comput. Graph. 28 (3), 1634–1647. doi:10.1109/TVCG.2021.3067820
Curtis, R. E., Yuen, A., Song, L., Goyal, A., and Xing, E. P. (2011). TVNViewer: An interactive visualization tool for exploring networks that change over time or space. Bioinforma. Oxf. Engl. 27 (13), 1880–1881. doi:10.1093/bioinformatics/btr273
Cusick, M. E., Klitgord, N., Vidal, M., and Hill, D. E. (2005). Interactome: Gateway into systems biology. Hum. Mol. Genet. 14, R171–R181. doi:10.1093/hmg/ddi335
Dahiya, S., Saini, V., Kumar, P., and Kumar, A. (2019). Protein-Protein interaction network analyses of human WNT proteins involved in neural development. Bioinformation 15 (5), 307–314. doi:10.6026/97320630015307
Dallago, C., Goldberg, T., Andrade-Navarro, M. A., Alanis-Lobato, G., and Rost, B. (2020). Visualizing human protein-protein interactions and subcellular localizations on cell images through CellMap. Curr. Protoc. Bioinforma. 69 (1), e97. doi:10.1002/cpbi.97
Dandekar, T., Snel, B., HuynenM., , and Bork, P. (1998). Conservation of gene order: A fingerprint of proteins that physically interact. Trends biochem. Sci. 23 (9), 324–328. doi:10.1016/s0968-0004(98)01274-2
Das, S., and Chakrabarti, S. (2021). Classification and prediction of protein–protein interaction interface using machine learning algorithm. Sci. Rep. 11 (1), 1761. doi:10.1038/s41598-020-80900-2
Das, T., Andrieux, G., and Ahmed, M. (2020). Integration of online omics-data resources for cancer research. Front. Genet. 11, 578345. doi:10.3389/fgene.2020.578345
Date, S. V. (2007). “Estimating protein function using protein-protein relationships,” in Gene function analysis. Editor M. F. Ochs (Totowa, NJ: Humana Press), 109–127. doi:10.1007/978-1-59745-547-3_7
De Braekeleer, E., Douet-Guilbert, N., and De Braekeleer, M. (2014). RARA fusion genes in acute promyelocytic leukemia: A review. Expert Rev. Hematol. 7 (3), 347–357. doi:10.1586/17474086.2014.903794
de Juan, D., Pazos, F., and Valencia, A. (2013). Emerging methods in protein co-evolution. Nat. Rev. Genet. 14 (4), 249–261. doi:10.1038/nrg3414
De Las Rivas, J., Alonso-López, D., and Arroyo, M. M. (2018). “Chapter nine - human interactomics: Comparative analysis of different protein interaction resources and construction of a cancer protein–drug bipartite network,” in Advances in protein chemistry and structural biology. Editor R. Donev (Academic Press (Protein-Protein Interactions in Human Disease, Part B), 263–282. doi:10.1016/bs.apcsb.2017.09.002
De Las Rivas, J., and Fontanillo, C. (2010). Protein–protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 6 (6), e1000807. doi:10.1371/journal.pcbi.1000807
Deng, J.-L., Xu, Y.-H., and Wang, G. (2019). Identification of potential crucial genes and key pathways in breast cancer using bioinformatic analysis. Front. Genet. 10, 695. doi:10.3389/fgene.2019.00695
Dezso, Z., Oltvai, Z. N., and Barabási, A.-L. (2003). Bioinformatics analysis of experimentally determined protein complexes in the yeast Saccharomyces cerevisiae. Genome Res. 13 (11), 2450–2454. doi:10.1101/gr.1073603
Di Nanni, N., Bersanelli, M., and Milanesi, L. (2020). Network diffusion promotes the integrative analysis of multiple omics. Front. Genet. 11, 106. doi:10.3389/fgene.2020.00106
Dimitrakopoulos, C., Hindupur, S. K., Colombi, M., Liko, D., Ng, C. K. Y., Piscuoglio, S., et al. (2021). Multi-omics data integration reveals novel drug targets in hepatocellular carcinoma. BMC genomics 22 (1), 592. doi:10.1186/s12864-021-07876-9
Dimitrieva, S., and Bucher, P. (2012). Genomic context analysis reveals dense interaction network between vertebrate ultraconserved non-coding elements. Bioinformatics 28 (18), i395–i401. doi:10.1093/bioinformatics/bts400
Ding, Z., and Kihara, D. (2018). Computational methods for predicting protein-protein interactions using various protein features. Curr. Protoc. Protein Sci. 93 (1), e62. doi:10.1002/cpps.62
Dohrmann, J., Puchin, J., and Singh, R. (2015). Global multiple protein-protein interaction network alignment by combining pairwise network alignments. BMC Bioinforma. 16 (13), S11. doi:10.1186/1471-2105-16-S13-S11
Droit, A., Poirier, G. G., and Hunter, J. M. (2005). Experimental and bioinformatic approaches for interrogating protein-protein interactions to determine protein function. J. Mol. Endocrinol. 34 (2), 263–280. doi:10.1677/jme.1.01693
Du, Y., Cai, M., Xing, X., Ji, J., Yang, E., and Wu, J. (2021). Pina 3.0: Mining cancer interactome. Nucleic Acids Res. 49 (D1), D1351–D1357. doi:10.1093/nar/gkaa1075
Du, Z.-P., Wu, B. L., Wang, S. H., Shen, J. H., Lin, X. H., Zheng, C. P., et al. (2014). Shortest path analyses in the protein-protein interaction network of NGAL (neutrophil gelatinase-associated lipocalin) overexpression in esophageal squamous cell carcinoma. Asian pac. J. Cancer Prev. 15 (16), 6899–6904. doi:10.7314/apjcp.2014.15.16.6899
Dugourd, C., Sciacovelli, M., Gjerga, E., Gabor, A., and Emdal, K. B. (2021). Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. Mol. Syst. Biol. 17 (1), e9730. doi:10.15252/msb.20209730
Dunham, B., and Ganapathiraju, M. K. (2021). Benchmark evaluation of protein–protein interaction prediction algorithms. Molecules 27 (1), 41. doi:10.3390/molecules27010041
Dünkler, A., Rösler, R., and Kestler, H. A. (2015). “Spliff: A single-cell method to map protein-protein interactions in time and space,” in Single cell protein analysis: Methods and protocols. Editors A. K. Singh, and A. Chandrasekaran (New York, NY: Springer), 151–168. doi:10.1007/978-1-4939-2987-0_11
Dupré, X. (2022). Random walk with restart (système de recommandations) — Papierstat. Availableat: http://www.xavierdupre.fr/app/papierstat/helpsphinx/notebooks/tinygraph_rwr.html (Accessed: April 4, 2022).
Dursun, C., Kwitek, A., and Bozdag, S. (2021). PhenoGeneRanker: Gene and phenotype prioritization using multiplex heterogeneous networks. IEEE/ACM Trans. Comput. Biol. Bioinform., 1. doi:10.1109/TCBB.2021.3098278
Efremova, M., Vento-Tormo, M., Teichmann, S. A., and Vento-Tormo, R. (2020). CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15 (4), 1484–1506. doi:10.1038/s41596-020-0292-x
Eicher, T., Kinnebrew, G., Patt, A., Spencer, K., Ying, K., Ma, Q., et al. (2020). Metabolomics and multi-omics integration: A survey of computational methods and resources. Metabolites 10 (5), E202. doi:10.3390/metabo10050202
Eisenberg, D., Marcotte, E. M., XenarIos, I., and Yeates, T. O. (2000). Protein function in the post-genomic era. Nature 405 (6788), 823–826. doi:10.1038/35015694
El Naqa, I., and Murphy, M. J. (2015). “What is machine learning?,” in Machine learning in radiation oncology: Theory and applications. Editors I. El Naqa, R. Li, and M. J. Murphy (Cham: Springer International Publishing), 3–11. doi:10.1007/978-3-319-18305-3_1
Elangovan, A., Davis, M., and Verspoor, K. (2020). Assigning function to protein-protein interactions: A weakly supervised BioBERT based approach using PubMed abstracts, 6.
Ellson, J., Gansner, E., and Koutsofios, L. (2001). “Graphviz — Open source graph drawing tools,” in Lecture notes in computer science (Springer-Verlag), 483–484. doi:10.1007/3-540-45848-4_57
Esch, R., and Merkl, R. (2020). Conserved genomic neighborhood is a strong but no perfect indicator for a direct interaction of microbial gene products. BMC Bioinforma. 21, 5. doi:10.1186/s12859-019-3200-z
Everson, J., Richards, M. R., and Buntin, M. B. (2019). Horizontal and vertical integration’s role in meaningful use attestation over time. Health Serv. Res. 54 (5), 1075–1083. doi:10.1111/1475-6773.13193
Fan, Z., Zhou, Y., and Ressom, H. W. (2020). Mota: Network-based multi-omic data integration for biomarker discovery. Metabolites 10 (4), 144. doi:10.3390/metabo10040144
Farahani, F. V., Karwowski, W., and Lighthall, N. R. (2019). Application of graph theory for identifying connectivity patterns in human brain networks: A systematic review. Front. Neurosci. 13, 585. doi:10.3389/fnins.2019.00585
Farahmand, S., Riley, T., and Zarringhalam, K. (2020). ModEx: A text mining system for extracting mode of regulation of transcription factor-gene regulatory interaction. J. Biomed. Inf. 102, 103353. doi:10.1016/j.jbi.2019.103353
Fekete, J.-D. (2009). “Visualizing networks using adjacency matrices: Progresses and challenges,” in 2009 11th IEEE International Conference on Computer-Aided Design and Computer Graphics, 32. doi:10.1109/CADCG.2009.5246808
Fionda, V. (2019). “Networks in biology,” in Encyclopedia of bioinformatics and computational biology. Editor S. Ranganathan (Oxford: Academic Press), 915–921. doi:10.1016/B978-0-12-809633-8.20420-2
Fionda, V., Palopoli, L., and Panni, S. (2009). “Extracting similar sub-graphs across PPI networks,” in 2009 24th International Symposium on Computer and Information Sciences, 183–188. doi:10.1109/ISCIS.2009.5291845
Fiorentino, G., Visintainer, R., Domenici, E., Lauria, M., and Marchetti, L. (2021). Mousse: Multi-omics using subject-specific SignaturEs. Cancers 13 (14), 3423. doi:10.3390/cancers13143423
Flórez, A. F., Park, D., Bhak, J., Kim, B. C., Kuchinsky, A., Morris, J. H., et al. (2010). Protein network prediction and topological analysis in Leishmania major as a tool for drug target selection. BMC Bioinforma. 11, 484. doi:10.1186/1471-2105-11-484
Franzese, N., Groce, A., Murali, T. M., and Ritz, A. (2019). Hypergraph-based connectivity measures for signaling pathway topologies. PLoS Comput. Biol. 15 (10), e1007384. doi:10.1371/journal.pcbi.1007384
Freilich, R., Arhar, T., Abrams, J. L., and Gestwicki, J. E. (2018). Protein-protein interactions in the molecular chaperone network. Acc. Chem. Res. 51 (4), 940–949. doi:10.1021/acs.accounts.8b00036
Gebreyesus, S. T., Siyal, A. A., Kitata, R. B., Chen, E. S. W., Enkhbayar, B., Angata, T., et al. (2022). Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat. Commun. 13 (1), 37. doi:10.1038/s41467-021-27778-4
Gehlenborg, N., O'Donoghue, S. I., Baliga, N. S., Goesmann, A., Hibbs, M. A., Kitano, H., et al. (2010). Visualization of omics data for systems biology. Nat. Methods 7, S56–S68. doi:10.1038/nmeth.1436
Geng, H., Chen, X., and Wang, C. (2021). Systematic elucidation of the pharmacological mechanisms of Rhynchophylline for treating epilepsy via network pharmacology. BMC Complement. Med. Ther. 21, 9. doi:10.1186/s12906-020-03178-x
Geng, H., Lu, T., and Lin, X. (2015). Prediction of protein-protein interaction sites based on naive bayes classifier. Biochem. Res. Int. 2015, 978193. doi:10.1155/2015/978193
Gerasch, A., Faber, D., Kuntzer, J., Niermann, P., Kohlbacher, O., Lenhof, H. P., et al. (2014). BiNA: A visual analytics tool for biological network data. PLOS ONE 9 (2), e87397. doi:10.1371/journal.pone.0087397
Gillespie, M., Jassal, B., Stephan, R., Milacic, M., Rothfels, K., Senff-Ribeiro, A., et al. (2022). The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50 (1), D687–D692. doi:10.1093/nar/gkab1028
Gillis, J., Ballouz, S., and Pavlidis, P. (2014). Bias tradeoffs in the creation and analysis of protein-protein interaction networks. J. Proteomics 100, 44–54. doi:10.1016/j.jprot.2014.01.020
Giovanni, T., and Renaud, D. (2015). Siren investigate. Availableat: https://siren.io/.
Gligorijević, V., and Pržulj, N. (2015). Methods for biological data integration: Perspectives and challenges. J. R. Soc. Interface 12 (112), 20150571. doi:10.1098/rsif.2015.0571
Goel, R., Harsha, H. C., Pandey, A., and Prasad, T. S. K. (2012). Human protein reference database and human proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 8 (2), 453–463. doi:10.1039/c1mb05340j
Goh, C.-S., and Cohen, F. E. (2002). Co-evolutionary analysis reveals insights into protein-protein interactions. J. Mol. Biol. 324 (1), 177–192. doi:10.1016/s0022-2836(02)01038-0
Gong, F., Ma, Y., Gong, W., Li, X., Li, C., and Yuan, X. (2018). Neo4j graph database realizes efficient storage performance of oilfield ontology. PloS One 13 (11), e0207595. doi:10.1371/journal.pone.0207595
González-Sánchez, J. C., Ibrahim, M. F. R., Leist, I. C., Weise, K. R., and Russell, R. B. (2021). Mechnetor: A web server for exploring protein mechanism and the functional context of genetic variants. Nucleic Acids Res. 49 (W1), W366–W374. doi:10.1093/nar/gkab399
Green, A. G., Elhabashy, H., Brock, K. P., Maddamsetti, R., Kohlbacher, O., and Marks, D. S. (2021). Large-scale discovery of protein interactions at residue resolution using co-evolution calculated from genomic sequences. Nat. Commun. 12 (1), 1396. doi:10.1038/s41467-021-21636-z
Guney, E., Menche, J., Vidal, M., and Barabasi, A. L. (2016). Network-based in silico drug efficacy screening. Nat. Commun. 7 (1), 10331. doi:10.1038/ncomms10331
Guo, Y., Wu, J., and Ma, H. (2022). “Self-supervised pre-training for protein embeddings using tertiary structures,”, Proceedings of the AAAI Conference on Artificial Intelligence, 9. doi:10.1609/aaai.v36i6.20636
Gursoy, A., Keskin, O., and Nussinov, R. (2008). Topological properties of protein interaction networks from a structural perspective. Biochem. Soc. Trans. 36 (6), 1398–1403. doi:10.1042/BST0361398
Haas, R., Zelezniak, A., Iacovacci, J., Kamrad, S., Townsend, S., and Ralser, M. (2017). Designing and interpreting “multi-omic” experiments that may change our understanding of biology. Curr. Opin. Syst. Biol. 6, 37–45. doi:10.1016/j.coisb.2017.08.009
Hagberg, A., Swart, P., and S Chult, D. (2008). Exploring network structure, dynamics, and function using networkx. LA-UR-08-05495; LA-UR-08-5495. Los Alamos National Lab. LANL, Los Alamos, NM United States. Availableat: https://www.osti.gov/biblio/960616-exploring-network-structure-dynamics-function-using-networkx (Accessed March 14, 2022).
Hakes, L., Pinney, J. W., Robertson, D. L., and Lovell, S. C. (2008). Protein-protein interaction networks and biology—what’s the connection? Nat. Biotechnol. 26 (1), 69–72. doi:10.1038/nbt0108-69
Halder, A. K., Denkiewicz, M., Sengupta, K., Basu, S., and Plewczynski, D. (2020). Aggregated network centrality shows non-random structure of genomic and proteomic networks. Methods (San Diego, Calif.) 181–182, 5–14. doi:10.1016/j.ymeth.2019.11.006
Hammoud, Z., and Kramer, F. (2020). Multilayer networks: Aspects, implementations, and application in biomedicine. Big Data Anal. 5 (1), 2. doi:10.1186/s41044-020-00046-0
Han, J.-D., Bertin, N., Hao, T., Goldberg, D. S., Berriz, G. F., Zhang, L. V., et al. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93. doi:10.1038/nature02555
Han, J.-D. J., Dupuy, D., Bertin, N., Cusick, M. E., and Vidal, M. (2005). Effect of sampling on topology predictions of protein-protein interaction networks. Nat. Biotechnol. 23 (7), 839–844. doi:10.1038/nbt1116
Hao, T., Zhao, L., Wu, D., Wang, B., Feng, X., Wang, E., et al. (2019). The protein-protein interaction network of Litopenaeus vannamei haemocytes. Front. Physiol. 10, 156. doi:10.3389/fphys.2019.00156
Hasan, Md.R., Paul, B. K., Ahmed, K., and Bhuyian, T. (2020). Design protein-protein interaction network and protein-drug interaction network for common cancer diseases: A bioinformatics approach. Inf. Med. Unlocked 18, 100311. doi:10.1016/j.imu.2020.100311
Hashemifar, S., Neyshabur, B., Khan, A. A., and Xu, J. (2018). Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics 34 (17), i802i802–i810. doi:10.1093/bioinformatics/bty573
Hawe, J. S., Theis, F. J., and Heinig, M. (2019). Inferring interaction networks from multi-omics data. Front. Genet. 10, 535. doi:10.3389/fgene.2019.00535
Hayashi, T., Matsuzaki, Y., Yanagisawa, K., Ohue, M., and Akiyama, Y. (2018). MEGADOCK-web: An integrated database of high-throughput structure-based protein-protein interaction predictions. BMC Bioinforma. 19 (4), 62. doi:10.1186/s12859-018-2073-x
He, M., Wang, Y., and Li, W. (2009). PPI finder: A mining tool for human protein-protein interactions. PLOS ONE 4 (2), e4554. doi:10.1371/journal.pone.0004554
He, T., and Chan, K. C. C. (2018). Evolutionary graph clustering for protein complex identification. IEEE/ACM Trans. Comput. Biol. Bioinform. 15 (3), 892–904. doi:10.1109/TCBB.2016.2642107
Heberle, H., Carazzolle, M. F., Telles, G. P., Meirelles, G. V., and Minghim, R. (2017). CellNetVis: A web tool for visualization of biological networks using force-directed layout constrained by cellular components. BMC Bioinforma. 18 (10), 395. doi:10.1186/s12859-017-1787-5
Hermjakob, H., Montecchi-Palazzi, L., Bader, G., Wojcik, J., Salwinski, L., Ceol, A., et al. (2004b). The HUPO PSI’s molecular interaction format—A community standard for the representation of protein interaction data. Nat. Biotechnol. 22 (2), 177–183. doi:10.1038/nbt926
Hermjakob, H., Montecchi-Palazzi, L., Lewington, C., Mudali, S., Kerrien, S., Orchard, S., et al. (2004a) ‘IntAct: An open source molecular interaction database’, Nucleic Acids Res., 32pp. D452, D455. doi:10.1093/nar/gkh052
Hibbs, M., Wallace, G., and Li, K., (2007) ‘Viewing the larger context of genomic data through horizontal integration’, in 2007 11th International Conference Information Visualization (IV ’07), 04-06 July 2007, Zurich, Switzerland, IEEEpp. 326–334. doi:10.1109/IV.2007.120
Hu, J., Zhou, L., Li, B., Zhang, X., and Chen, N. (2021). Improve hot region prediction by analyzing different machine learning algorithms. BMC Bioinforma. 22 (3), 522. doi:10.1186/s12859-021-04420-0
Hu, X., Feng, C., Zhou, Y., Harrison, A., and Chen, M. (2022). DeepTrio: A ternary prediction system for protein–protein interaction using mask multiple parallel convolutional neural networks. Bioinformatics 38 (3), 694–702. doi:10.1093/bioinformatics/btab737
Hu, Y., Vinayagam, A., Nand, A., Comjean, A., Chung, V., Hao, T., et al. (2018). Molecular interaction search tool (MIST): An integrated resource for mining gene and protein interaction data. Nucleic Acids Res. 46 (1), D567–D574. doi:10.1093/nar/gkx1116
Huang, K., and Zitnik, M. (2021). Graph meta learning via local subgraphs. arXiv:2006.07889. Availableat: http://arxiv.org/abs/2006.07889 (Accessed March 29, 2021).
Huang, X.-T., Jia, S., Gao, L., and Wu, J. (2022). Reconstruction of human protein-coding gene functional association network based on machine learning. Brief. Bioinform. 23, bbab552. doi:10.1093/bib/bbab552
Hütter, C. V. R., Sin, C., Muller, F., and Menche, J. (2022). Network cartographs for interpretable visualizations. Nat. Comput. Sci. 2 (2), 84–89. doi:10.1038/s43588-022-00199-z
Iranzo, J., Krupovic, M., and Koonin, E. V. (2016). The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. Mbio 7 (4), e0097816. doi:10.1128/mBio.00978-16
Islam, M. F., Hoque, M. M., Banik, R. S., Roy, S., Sumi, S. S., Hassan, F. M. N., et al. (2013). Comparative analysis of differential network modularity in tissue specific normal and cancer protein interaction networks. J. Clin. Bioinforma. 3 (1), 19. doi:10.1186/2043-9113-3-19
Jamasb, A. R., Day, B., Cangea, C., Lio, P., and Blundell, T. L. (2021). Deep learning for protein-protein interaction site prediction. Methods Mol. Biol. 2361, 263–288. doi:10.1007/978-1-0716-1641-3_16
Jansen, R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N. J., Chung, S., et al. (2003). A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302 (5644), 449–453. doi:10.1126/science.1087361
Jeanquartier, F., Jean-Quartier, C., and Holzinger, A. (2015). Integrated web visualizations for protein-protein interaction databases. BMC Bioinforma. 16 (1), 195. doi:10.1186/s12859-015-0615-z
Jha, K., Saha, S., and Singh, H. (2022). Prediction of protein–protein interaction using graph neural networks. Sci. Rep. 12 (1), 8360. doi:10.1038/s41598-022-12201-9
Ji, A. L., Rubin, A. J., Thrane, K., Jiang, S., Reynolds, D. L., Meyers, R. M., et al. (2020). Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182 (2), 497–514. doi:10.1016/j.cell.2020.05.039
Jia, D., Li, S., Li, D., Xue, H., Yang, D., and Liu, Y. (2018). Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging 10 (4), 592–605. doi:10.18632/aging.101415
Jia, Y., Zou, J., Wang, Y., Zhang, X., Shi, Y., Liang, Y., et al. (2021). Action mechanism of Roman chamomile in the treatment of anxiety disorder based on network pharmacology. J. Food Biochem. 45 (1), e13547. doi:10.1111/jfbc.13547
Jin, N., Wu, D., and Gong, Y. (2014). Integration strategy is a key step in network-based analysis and dramatically affects network topological properties and inferring outcomes. BioMed Res. Int. 2014, e296349. doi:10.1155/2014/296349
Johansson-Åkhe, I., Mirabello, C., and Wallner, B. (2019). Predicting protein-peptide interaction sites using distant protein complexes as structural templates. Sci. Rep. 9 (1), 4267. doi:10.1038/s41598-019-38498-7
Johnson, K. L., Qi, Z., Yan, Z., Wen, X., Nguyen, T. C., Zaleta-Rivera, K., et al. (2021). Revealing protein-protein interactions at the transcriptome scale by sequencing. Mol. Cell 81 (19), 4091–4103. doi:10.1016/j.molcel.2021.07.006
Jonathan, J., Sanga, C., Mwita, M., and Mgode, G. (2021). Visual analytics of tuberculosis detection rat performance. Online J. Public Health Inf. 13 (2), e12. doi:10.5210/ojphi.v13i2.11465
Jordán, F., Nguyen, T.-P., and Liu, W. (2012). Studying protein–protein interaction networks: A systems view on diseases. Brief. Funct. Genomics 11 (6), 497–504. doi:10.1093/bfgp/els035
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Jupe, S., Ray, K., Roca, C. D., Varusai, T., Shamovsky, V., Stein, L., et al. (2018). Interleukins and their signaling pathways in the Reactome biological pathway database. J. Allergy Clin. Immunol. 141 (4), 1411–1416. doi:10.1016/j.jaci.2017.12.992
Kamburov, A., Grossmann, A., Herwig, R., and Stelzl, U. (2012). Cluster-based assessment of protein-protein interaction confidence. BMC Bioinforma. 13 (1), 262. doi:10.1186/1471-2105-13-262
Kamisetty, H., Ramanathan, A., Bailey-Kellogg, C., and Langmead, C. J. (2011). Accounting for conformational entropy in predicting binding free energies of protein-protein interactions: Entropy and Protein-Protein Interactions. Proteins 79 (2), 444–462. doi:10.1002/prot.22894
Kanai, M., Maeda, Y., and Okada, Y. (2018). Grimon: Graphical interface to visualize multi-omics networks. Bioinformatics 34 (22), 3934–3936. doi:10.1093/bioinformatics/bty488
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (1), D457–D462. doi:10.1093/nar/gkv1070
Kapadia, P., Khare, S., and Priyadarshini, P. (2019). “Predicting protein-protein interaction in multi-layer blood cell PPI networks,” in Advanced informatics for computing research. Editor A. K. Luhach (Singapore: Springer), 240–251. doi:10.1007/978-981-15-0111-1_22
Karatzas, E., Baltoumas, F. A., Panayiotou, N. A., Schneider, R., and Pavlopoulos, G. A. (2021). Arena3Dweb: Interactive 3D visualization of multilayered networks. Nucleic Acids Res. 49 (W1), W36–W45. doi:10.1093/nar/gkab278
Kazemi, E., Hassani, H., Grossglauser, M., and Pezeshgi Modarres, H. (2016). Proper: Global protein interaction network alignment through percolation matching. BMC Bioinforma. 17 (1), 527. doi:10.1186/s12859-016-1395-9
Kerrien, S., Orchard, S., Montecchi-Palazzi, L., Aranda, B., Quinn, A. F., Vinod, N., et al. (2007). Broadening the horizon--level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol. 5, 44. doi:10.1186/1741-7007-5-44
Keshava Prasad, T. S., Goel, R., Kandasamy, K., Keerthikumar, S., Kumar, S., Mathivanan, S., et al. (2009). Human protein reference database--2009 update. Nucleic Acids Res. 37, D767–D772. doi:10.1093/nar/gkn892
Khashan, R., Tropsha, A., and Zheng, W. (2022). Data mining meets machine learning: A novel ANN-based multi-body interaction docking scoring function (MBI-score) based on utilizing frequent geometric and chemical patterns of interfacial atoms in native protein-ligand complexes. Mol. Inf., e2100248. doi:10.1002/minf.202100248
Kim, T. R., Jeong, H.-H., and Sohn, K.-A. (2019). Topological integration of RPPA proteomic data with multi-omics data for survival prediction in breast cancer via pathway activity inference. BMC Med. Genomics 12 (5), 94. doi:10.1186/s12920-019-0511-x
Klimm, F., Toledo, E. M., Monfeuga, T., Zhang, F., Deane, C. M., and Reinert, G. (2020). Functional module detection through integration of single-cell RNA sequencing data with protein–protein interaction networks. BMC Genomics 21 (1), 756. doi:10.1186/s12864-020-07144-2
Koh, G. C. K. W., Porras, P., Aranda, B., Hermjakob, H., and Orchard, S. E. (2012). Analyzing protein–protein interaction networks. J. Proteome Res. 11 (4), 2014–2031. doi:10.1021/pr201211w
Kotlyar, M., Pastrello, C., Malik, Z., and Jurisica, I. (2019). IID 2018 update: Context-specific physical protein-protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 47 (1), D581–D589. doi:10.1093/nar/gky1037
Kotlyar, M., Rossos, A. E. M., and Jurisica, I. (2017). Prediction of protein-protein interactions. Curr. Protoc. Bioinforma. 60, 821–8. doi:10.1002/cpbi.38
Koutrouli, M., Karatzas, E., and Espino, D. (2020). A guide to conquer the biological network era using graph theory. Front. Bioeng. Biotechnol. 8, 31. doi:10.3389/fbioe.2020.0003
Krause, A., Stoye, J., and Vingron, M. (2005). Large scale hierarchical clustering of protein sequences. BMC Bioinforma. 6 (1), 15. doi:10.1186/1471-2105-6-15
Krogh, A. (2008). What are artificial neural networks? Nat. Biotechnol. 26 (2), 195–197. doi:10.1038/nbt1386
Kshitish, A., Salaingambi, S., Raksha, H. N., Deepika, T. S., and Preeti, G. (2013). Startbioinfo contributors. Availableat: https://startbioinfo.org/contributors.html (Accessed February 23, 2022).
Kusuma, W., F Ahmad, H., and Suryono, M. (2019). Clustering of protein-protein interactions (PPI) and gene ontology molecular function using Markov clustering and fuzzy K partite algorithm. IOP Conf. Ser. Earth Environ. Sci. 299 (1), 012034. doi:10.1088/1755-1315/299/1/012034
Kuzmanov, U., and Emili, A. (2013). Protein-protein interaction networks: Probing disease mechanisms using model systems. Genome Med. 5 (4), 37. doi:10.1186/gm441
Laniau, J. (2017). Structure de réseaux biologiques : Rôle des nøeuds internes vis-à-vis de la production de composés. Theses. Inria Rennes - Bretagne Atlantique. Availableat: https://hal.archives-ouvertes.fr/tel-01656474 (Accessed: January 26, 2022).
Latysheva, N. S., Oates, M. E., Maddox, L., Flock, T., Gough, J., Buljan, M., et al. (2016). Molecular principles of gene fusion mediated rewiring of protein interaction networks in cancer. Mol. Cell 63 (4), 579–592. doi:10.1016/j.molcel.2016.07.008
Leblanc, H. J., Zhang, H., Koutsoukos, X., and Sundaram, S. (2013). Resilient asymptotic consensus in robust networks. IEEE J. Sel. Areas Commun. 31 (4), 766–781. doi:10.1109/jsac.2013.130413
Lee, I., and Nam, H. (2018). Identification of drug-target interaction by a random walk with restart method on an interactome network. BMC Bioinforma. 19 (8), 208. doi:10.1186/s12859-018-2199-x
Lee, J. J., Bernard, V., Semaan, A., Monberg, M. E., Huang, J., Stephens, B. M., et al. (2021). Elucidation of tumor-stromal heterogeneity and the ligand-receptor interactome by single-cell transcriptomics in real-world pancreatic cancer biopsies. Clin. Cancer Res. 27 (21), 5912–5921. doi:10.1158/1078-0432.CCR-20-3925
Lee, M. S., and Oh, S. (2014). Alternating decision tree algorithm for assessing protein interaction reliability. Vietnam J. Comput. Sci. 1 (3), 169–178. doi:10.1007/s40595-014-0018-5
Lee, T., and Yoon, Y. (2018). Drug repositioning using drug-disease vectors based on an integrated network. BMC Bioinforma. 19 (1), 446. doi:10.1186/s12859-018-2490-x
Lei, X., Wang, S., and Wu, F. (2019a). Identification of essential proteins based on improved HITS algorithm. Genes 10 (2), 177. doi:10.3390/genes10020177
Lei, X., Yang, X., and Fujita, H. (2019b). Random walk based method to identify essential proteins by integrating network topology and biological characteristics. Knowledge-Based Syst. 167, 53–67. doi:10.1016/j.knosys.2019.01.012
Lercher, M. J., and Pál, C. (2008). Integration of horizontally transferred genes into regulatory interaction networks takes many million years. Mol. Biol. Evol. 25 (3), 559–567. doi:10.1093/molbev/msm283
Li, F., Zhu, F., Ling, X., and Liu, Q. (2020c). Protein interaction network reconstruction through ensemble deep learning with attention mechanism. Front. Bioeng. Biotechnol. 8, 390. doi:10.3389/fbioe.2020.00390
Li, H., Gong, X. J., Yu, H., and Zhou, C. (2018a). Deep neural network based predictions of protein interactions using primary sequences. Molecules 23 (8), 1923. doi:10.3390/molecules23081923
Li, M., Jiang, Y., and Ryu, K. H. (2022b). InfersentPPI: Prediction of protein-protein interaction using protein sentence embedding with gene ontology information. Front. Genet. 13. doi:10.3389/fgene.2022.82754
Li, M., Yang, J., Wu, F. X., Pan, Y., and Wang, J. (2018b). DyNetViewer: A cytoscape app for dynamic network construction, analysis and visualization. Bioinformatics 34 (9), 1597–1599. doi:10.1093/bioinformatics/btx821
Li, X., Ma, J., Han, M., and He, F. (2022a). MoGCN: A multi-omics integration method based on graph convolutional network for cancer subtype Analysis. Front. Genet. 13. doi:10.3389/fgene.2022.806842
Li, X., Tran, K. M., Aziz, K. E., Sorokin, A. V., Chen, J., and Wang, W. (2016). Defining the protein-protein interaction network of the human protein tyrosine phosphatase family. Mol. Cell. Proteomics 15 (9), 3030–3044. doi:10.1074/mcp.M116.060277
Li, Y., Liang, Y., Ma, T., and Yang, Q. (2020b). Identification of DGUOK-AS1 as a prognostic factor in breast cancer by bioinformatics analysis. Front. Oncol. 10, 1092. doi:10.3389/fonc.2020.01092
Li, Y., Qian, B., Zhang, X., and Liu, H. (2020a). Graph neural network-based diagnosis prediction. Big Data 8 (5), 379–390. doi:10.1089/big.2020.0070
Li, Y., and Ilie, L. (2017). Sprint: Ultrafast protein-protein interaction prediction of the entire human interactome. BMC Bioinforma. 18 (1), 485. doi:10.1186/s12859-017-1871-x
Li, Y., Wang, Z., Li, L. P., You, Z. H., Huang, W. Z., Zhan, X. K., et al. (2021). Robust and accurate prediction of protein-protein interactions by exploiting evolutionary information. Sci. Rep. 11 (1), 16910. doi:10.1038/s41598-021-96265-z
Liang, L., Chen, V., Zhu, K., Fan, X., Lu, X., and Lu, S. (2019). Integrating data and knowledge to identify functional modules of genes: A multilayer approach. BMC Bioinforma. 20, 225. doi:10.1186/s12859-019-2800-y
Lin, H.-H., Zhang, Q. R., Kong, X., Zhang, L., Zhang, Y., Tang, Y., et al. (2021). Machine learning prediction of antiviral-HPV protein interactions for anti-HPV pharmacotherapy. Sci. Rep. 11 (1), 24367. doi:10.1038/s41598-021-03000-9
Lin, J.-S., and Lai, E.-M. (2017). Protein-protein interactions: Co-immunoprecipitation. Methods Mol. Biol. 1615, 211–219. doi:10.1007/978-1-4939-7033-9_17
Liu, L., Zhu, X., Ma, Y., Piao, H., Yang, Y., Hao, X., et al. (2020a). Combining sequence and network information to enhance protein–protein interaction prediction. BMC Bioinforma. 21 (16), 537. doi:10.1186/s12859-020-03896-6
Liu, Q., Chen, P., Wang, B., Zhang, J., and Li, J. (2018). Hot spot prediction in protein-protein interactions by an ensemble system. BMC Syst. Biol. 12 (9), 132. doi:10.1186/s12918-018-0665-8
Liu, T.-H., Chen, W. H., Chen, X. D., Liang, Q. E., Tao, W. C., Jin, Z., et al. (2020b). Network pharmacology identifies the mechanisms of action of TaohongSiwu decoction against essential hypertension. Med. Sci. Monit. 26, e920682. doi:10.12659/MSM.920682
Liu, X., Chang, C., Han, M., Yin, R., Zhan, Y., Li, C., et al. (2019). PPIExp: A web-based platform for integration and visualization of protein-protein interaction data and spatiotemporal proteomics data. J. Proteome Res. 18 (2), 633–641. doi:10.1021/acs.jproteome.8b00713
Liu, Y., Zhu, Y., and He, C. (2021). BENviewer: A gene interaction network visualization server based on graph embedding model, Database. 2021, baab033. doi:10.1093/database/baab033
Lotia, S., Montojo, J., Dong, Y., Bader, G. D., and Pico, A. R. (2013). Cytoscape app store. Bioinforma. Oxf. Engl. 29 (10), 1350–1351. doi:10.1093/bioinformatics/btt138
Louche, A., Salcedo, S. P., and Bigot, S. (2017). Protein-protein interactions: Pull-down assays. Methods Mol. Biol. 1615, 247–255. doi:10.1007/978-1-4939-7033-9_20
Lu, C., Hu, X., Wang, G., Leach, L. J., Yang, S., Kearsey, M. J., et al. (2010). Why do essential proteins tend to be clustered in the yeast interactome network? Mol. Biosyst. 6 (5), 871–877. doi:10.1039/b921069e
Lu, X., Liu, F., Miao, Q., Liu, P., Gao, Y., and He, K. (2021). A novel method to identify gene interaction patterns. BMC Genomics 22 (1), 436. doi:10.1186/s12864-021-07628-9
Luck, K., Kim, D. K., Lambourne, L., Spirohn, K., Begg, B. E., Bian, W., et al. (2020). A reference map of the human binary protein interactome. Nature 580 (7803), 402–408. doi:10.1038/s41586-020-2188-x
Lv, Y., Huang, S., and Zhang, T. (2021). Application of multilayer network models in bioinformatics. Front. Genet. 12. doi:10.3389/fgene.2021.664860
Lynn, C. W., and Bassett, D. S. (2021). Quantifying the compressibility of complex networks. Proc. Natl. Acad. Sci. U. S. A. 118 (32), e2023473118. doi:10.1073/pnas.2023473118
Lysenko, A., Roznovat, I. A., Saqi, M., Mazein, A., Rawlings, C. J., and Auffray, C. (2016). Representing and querying disease networks using graph databases. BioData Min. 9 (1), 23. doi:10.1186/s13040-016-0102-8
Ma, C.-Y., and Liao, C.-S. (2020). A review of protein–protein interaction network alignment: From pathway comparison to global alignment. Comput. Struct. Biotechnol. J. 18, 2647–2656. doi:10.1016/j.csbj.2020.09.011
Ma, F., Zhang, S., Song, L., Wang, B., Wei, L., and Zhang, F. (2021a). Applications and analytical tools of cell communication based on ligand-receptor interactions at single cell level. Cell Biosci. 11 (1), 121. doi:10.1186/s13578-021-00635-z
Ma, H., He, Z., Chen, J., Zhang, X., and Song, P. (2021b). Identifying of biomarkers associated with gastric cancer based on 11 topological analysis methods of CytoHubba. Sci. Rep. 11 (1), 1331. doi:10.1038/s41598-020-79235-9
Ma, T., and Zhang, A. (2019). Integrate multi-omics data with biological interaction networks using Multi-view Factorization AutoEncoder (MAE). BMC Genomics 20 (11), 944. doi:10.1186/s12864-019-6285-x
Ma, W., Cao, Y., and Bao, W. (2020). ACT-SVM: Prediction of protein-protein interactions based on support vector basis model. Sci. Program. 2020, e8866557. doi:10.1155/2020/8866557
MacDonald, P. N. (1998). “A two-hybrid protein interaction system to identify factors that interact with retinoid and vitamin D receptors,” in Retinoid protocols. Editor C. P. F. Redfern (Totowa, NJ: Humana Press), 359–375. doi:10.1385/0-89603-438-0:359
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: A cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21 (16), 3448–3449. doi:10.1093/bioinformatics/bti551
Mahdessian, D., Cesnik, A. J., Gnann, C., Danielsson, F., Stenstrom, L., Arif, M., et al. (2021). Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590 (7847), 649–654. doi:10.1038/s41586-021-03232-9
Mahdipour, E., and Ghasemzadeh, M. (2021). The protein-protein interaction network alignment using recurrent neural network. Med. Biol. Eng. Comput. 59 (11), 2263–2286. doi:10.1007/s11517-021-02428-5
Maimon, O., and Rokach, L. (2006). Data mining and knowledge discovery handbook. Springer Science & Business Media.
Malek, M., Zorzan, S., and Ghoniem, M. (2020). A methodology for multilayer networks analysis in the context of open and private data: Biological application. Appl. Netw. Sci. 5 (1), 41–28. doi:10.1007/s41109-020-00277-z
Malik, S., Sharma, D., and Khatri, S. K. (2017). Reconstructing phylogenetic tree using a protein-protein interaction technique. IET Nanobiotechnol. 11 (8), 1005–1016. doi:10.1049/iet-nbt.2016.0177
Malod-Dognin, N., Ban, K., and Pržulj, N. (2017). Unified alignment of protein-protein interaction networks. Sci. Rep. 7 (1), 953. doi:10.1038/s41598-017-01085-9
Marai, G. E., Pinaud, B., Buhler, K., Lex, A., and Morris, J. H. (2019). Ten simple rules to create biological network figures for communication. PLoS Comput. Biol. 15 (9), e1007244. doi:10.1371/journal.pcbi.1007244
Marcotte, E. M., Pellegrini, M., and Ng, H. L. (1999). ‘Detecting protein function and protein-protein interactions from genome sequences’. Science 285. doi:10.1126/science.285.5428.751
Marín-Llaó, J., Mubeen, S., Perera-Lluna, A., Hofmann-Apitius, M., Picart-Armada, S., and Domingo-Fernandez, D. (2021). MultiPaths: A Python framework for analyzing multi-layer biological networks using diffusion algorithms. Bioinformatics 37 (1), 137–139. doi:10.1093/bioinformatics/btaa1069
Martin, S., Roe, D., and Faulon, J.-L. (2005). Predicting protein–protein interactions using signature products. Bioinformatics 21 (2), 218–226. doi:10.1093/bioinformatics/bth483
McGee, F., Ghoniem, M., Melancon, G., Otjacques, B., and Pinaud, B. (2019). The state of the art in multilayer network visualization. Comput. Graph. Forum 38 (6), 125–149. doi:10.1111/cgf.13610
Mikkelsen, T. S., Galagan, J. E., and Mesirov, J. P. (2005). Improving genome annotations using phylogenetic profile anomaly detection. Bioinformatics 21 (4), 464–470. doi:10.1093/bioinformatics/bti027
Mishra, B., Kumar, N., and Mukhtar, M. S. (2021). Network biology to uncover functional and structural properties of the plant immune system. Curr. Opin. Plant Biol. 62, 102057. doi:10.1016/j.pbi.2021.102057
Mlecnik, B., Galon, J., and Bindea, G. (2018). Comprehensive functional analysis of large lists of genes and proteins. J. Proteomics 171, 2–10. doi:10.1016/j.jprot.2017.03.016
Mooney, M. A., Nigg, J. T., McWeeney, S. K., and Wilmot, B. (2014). Functional and genomic context in pathway analysis of GWAS data. Trends Genet. 30 (9), 390–400. doi:10.1016/j.tig.2014.07.004
Morilla, I., Lees, J. G., Reid, A. J., Orengo, C., and Ranea, J. A. G. (2010). Assessment of protein domain fusions in human protein interaction networks prediction: Application to the human kinetochore model. N. Biotechnol. 27 (6), 755–765. doi:10.1016/j.nbt.2010.09.005
Mosca, E., Bersanelli, M., Matteuzzi, T., Di Nanni, N., Castellani, G., Milanesi, L., et al. (2021). Characterization and comparison of gene-centered human interactomes. Brief. Bioinform. 22 (6), bbab153. doi:10.1093/bib/bbab153
Mosca, E., and Milanesi, L. (2013). Network-based analysis of omics with multi-objective optimization. Mol. Biosyst. 9 (12), 2971–2980. doi:10.1039/c3mb70327d
Mrvar, A., and Batagelj, V. (2016). Analysis and visualization of large networks with program package Pajek. Complex adapt. Syst. Model. 4 (1), 6. doi:10.1186/s40294-016-0017-8
Murakami, Y., and Mizuguchi, K. (2010). Applying the Naïve Bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites. Bioinformatics 26 (15), 1841–1848. doi:10.1093/bioinformatics/btq302
Murdoch, W. J., Singh, C., Kumbier, K., Abbasi-Asl, R., and Yu, B. (2019). Definitions, methods, and applications in interpretable machine learning. Proc. Natl. Acad. Sci. U. S. A. 116 (44), 22071–22080. doi:10.1073/pnas.1900654116
Murphy, M., Jegelka, S., and Fraenkel, E. (2022). Self-supervised learning of cell type specificity from immunohistochemical images. Bioinformatics 38 (1), i395–i403. doi:10.1093/bioinformatics/btac263
Nadeau, R., Byvsheva, A., and Lavallée-Adam, M. (2021). Pignon: A protein-protein interaction-guided functional enrichment analysis for quantitative proteomics. BMC Bioinforma. 22 (1), 302. doi:10.1186/s12859-021-04042-6
Narayanan, T., Gersten, M., Subramaniam, S., and Grama, A. (2011). Modularity detection in protein-protein interaction networks. BMC Res. Notes 4 (1), 569. doi:10.1186/1756-0500-4-569
Nath, A., and Leier, A. (2020). Improved cytokine-receptor interaction prediction by exploiting the negative sample space. BMC Bioinforma. 21 (1), 493. doi:10.1186/s12859-020-03835-5
Navlakha, S., He, X., Faloutsos, C., and Bar-Joseph, Z. (2014). Topological properties of robust biological and computational networks. J. R. Soc. Interface 11 (96), 20140283. doi:10.1098/rsif.2014.0283
Neuditschko, M., Khatkar, M. S., and Raadsma, H. W. (2012). NetView: A high-definition network-visualization approach to detect fine-scale population structures from genome-wide patterns of variation. PLOS ONE 7 (10), e48375. doi:10.1371/journal.pone.0048375
Ngounou Wetie, A. G., Sokolowska, I., Woods, A. G., Roy, U., Loo, J. A., and Darie, C. C. (2013). Investigation of stable and transient protein–protein interactions: Past, present, and future. PROTEOMICS 13 (3–4), 538–557. doi:10.1002/pmic.201200328
Nguyen, V. T., Le, T. T. K., Than, K., and Tran, D. H. (2021). Predicting miRNA-disease associations using improved random walk with restart and integrating multiple similarities. Sci. Rep. 11 (1), 21071. doi:10.1038/s41598-021-00677-w
Nitzan, M., Casadiego, J., and Timme, M. (2017). Revealing physical interaction networks from statistics of collective dynamics. Sci. Adv. 3 (2), e1600396. doi:10.1126/sciadv.1600396
Novkovic, M., Onder, L., Bocharov, G., and Ludewig, B. (2020). Topological structure and robustness of the lymph node conduit system. Cell Rep. 30 (3), 893–904. doi:10.1016/j.celrep.2019.12.070
Otasek, D., Morris, J. H., Boucas, J., Pico, A. R., and Demchak, B. (2019). Cytoscape automation: Empowering workflow-based network analysis. Genome Biol. 20 (1), 185. doi:10.1186/s13059-019-1758-4
Ou-Yang, L., Yan, H., and Zhang, X.-F. (2017). A multi-network clustering method for detecting protein complexes from multiple heterogeneous networks. BMC Bioinforma. 18 (13), 463. doi:10.1186/s12859-017-1877-4
Oughtred, R., Rust, J., Chang, C., Breitkreutz, B. J., Stark, C., Willems, A., et al. (2021). The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 30 (1), 187–200. doi:10.1002/pro.3978
Pablo Porras, M., Ochoa, D., and Rogon, M. (2020). Network analysis of protein interaction data: An introduction. Hinxton, Cambridgeshire, UK: EBI : european Bioinformatic Institute. doi:10.6019/TOL.Networks_t.2016.00001.1
Page, L., Brin, S., and Motwani, R. (1999). The PageRank citation ranking: Bringing order to the web. Stanf. InfoLab. Availableat: http://ilpubs.stanford.edu:8090/422/(Accessed: March 21, 2022).
Pak, M., Jeong, D., and Moon, J. (2021). Network propagation for the analysis of multi-omics data. Recent Adv. Biol. Netw. Analysis, 185–217. doi:10.1007/978-3-030-57173-3_9
Pan, J., Li, P., and Hong, Z. (2021). Prediction of protein–protein interactions in Arabidopsis, maize, and rice by combining deep neural network with discrete hilbert transform. Front. Genet. 12. doi:10.3389/fgene.2021.745228
Pan, J., You, Z. H., and Li, L. P. (2022). Dwppi: A deep learning approach for predicting protein–protein interactions in plants based on multi-source information with a large-scale biological network. Front. Bioeng. Biotechnol. 10. doi:10.3389/fbioe.2022.807522
Pandey, B. (2018). Analysis of protein-protein interaction networks using high performance scalable tools, 33.
Papanikolaou, N., Pavlopoulos, G. A., Theodosiou, T., and Iliopoulos, I. (2015). Protein–protein interaction predictions using text mining methods. Methods 74, 47–53. doi:10.1016/j.ymeth.2014.10.026
Patil, A., Nakai, K., and Nakamura, H. (2011). HitPredict: A database of quality assessed protein-protein interactions in nine species. Nucleic Acids Res. 39, D744–D749. doi:10.1093/nar/gkq897
Pattin, K. A., and Moore, J. H. (2009). Role for protein–protein interaction databases in human genetics. Expert Rev. Proteomics 6 (6), 647–659. doi:10.1586/epr.09.86
Paul, M., and Anand, A. (2022). A new family of similarity measures for scoring confidence of protein interactions using gene ontology. IEEE/ACM Trans. Comput. Biol. Bioinform. 19 (1), 19–30. doi:10.1109/TCBB.2021.3083150
Pazos, F., HelMer-Citterich, M., Ausiello, G., and VAlenciA, A. (1997). Correlated mutations contain information about protein-protein interaction. J. Mol. Biol. 271 (4), 511–523. doi:10.1006/jmbi.1997.1198
Pei, F., Shi, Q., Zhang, H., and Bahar, I. (2021). Predicting protein–protein interactions using symmetric logistic matrix factorization. J. Chem. Inf. Model. 61 (4), 1670–1682. doi:10.1021/acs.jcim.1c00173
PeixotoTiago, P. (2014). The graph-tool python library. Availableat: http://figshare.com/articles/graph_tool/1164194.
Pellegrini, M. (2019). “Community detection in biological networks,” in Encyclopedia of bioinformatics and computational biology. Editor S. Ranganathan (Oxford: Academic Press), 978–987. doi:10.1016/B978-0-12-809633-8.20428-7
Pellegrini, M., Marcotte, E. M., Thompson, M. J., Eisenberg, D., and Yeates, T. O. (1999). Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. U. S. A. 96 (8), 4285–4288. doi:10.1073/pnas.96.8.4285
Peng, X., Wang, J., Peng, W., Wu, F. X., and Pan, Y. (2017). Protein–protein interactions: Detection, reliability assessment and applications. Brief. Bioinform. 18 (5), 798–819. doi:10.1093/bib/bbw066
Perlasca, P., Frasca, M., Ba, C. T., Gliozzo, J., Notaro, M., Pennacchioni, M., et al. (2020). Multi-resolution visualization and analysis of biomolecular networks through hierarchical community detection and web-based graphical tools. PloS One 15 (12), e0244241. doi:10.1371/journal.pone.0244241
Picard, M., Scott-Boyer, M. P., Bodein, A., Perin, O., and Droit, A. (2021). Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 19, 3735–3746. doi:10.1016/j.csbj.2021.06.030
Piehler, J. (2005). New methodologies for measuring protein interactions in vivo and in vitro. Curr. Opin. Struct. Biol. 15 (1), 4–14. doi:10.1016/j.sbi.2005.01.008
Piereck, B., Oliveira-Lima, M., Benko-Iseppon, A. M., Diehl, S., Schneider, R., Brasileiro-Vidal, A. C., et al. (2020). LAITOR4HPC: A text mining pipeline based on HPC for building interaction networks. BMC Bioinforma. 21 (1), 365. doi:10.1186/s12859-020-03620-4
Pietrosemoli, N., and Dobay, M. P. (2018). “Optimized protein–protein interaction network usage with context filtering,” in Computational cell biology: Methods and protocols. Editors L. von Stechow, and A. Santos Delgado (New York, NY: Springer), 33–50. doi:10.1007/978-1-4939-8618-7_2
Pinu, F. R., Beale, D. J., Paten, A. M., Kouremenos, K., Swarup, S., Schirra, H. J., et al. (2019). Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 9 (4), E76. doi:10.3390/metabo9040076
Pirch, S., Muller, F., Iofinova, E., Pazmandi, J., Hutter, C. V. R., Chiettini, M., et al. (2021). The VRNetzer platform enables interactive network analysis in Virtual Reality. Nat. Commun. 12 (1), 2432. doi:10.1038/s41467-021-22570-w
Pizzuti, C., and Rombo, S. E. (2014). Algorithms and tools for protein–protein interaction networks clustering, with a special focus on population-based stochastic methods. Bioinformatics 30 (10), 1343–1352. doi:10.1093/bioinformatics/btu034
Pournoor, E., Mousavian, Z., Dalini, A. N., and Masoudi-Nejad, A. (2020). Identification of key components in colon adenocarcinoma using transcriptome to interactome multilayer framework. Sci. Rep. 10 (1), 4991. doi:10.1038/s41598-020-59605-z
Przytycka, T. M., Singh, M., and Slonim, D. K. (2010). Toward the dynamic interactome: it’s about time. Brief. Bioinform. 11 (1), 15–29. doi:10.1093/bib/bbp057
Qi, J., Sun, H., Zhang, Y., Wang, Z., Xun, Z., Li, Z., et al. (2022). Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 13, 1742. doi:10.1038/s41467-022-29366-6
Qu, J., Wang, C. C., Cai, S. B., Zhao, W. D., Cheng, X. L., and Ming, Z. (2021). Biased random walk with restart on multilayer heterogeneous networks for MiRNA-disease association prediction. Front. Genet. 12, 720327. doi:10.3389/fgene.2021.720327
Rabbani, G., Baig, M. H., Ahmad, K., and Choi, I. (2018). Protein-protein interactions and their role in various diseases and their prediction techniques. Curr. Protein Pept. Sci. 19 (10), 948–957. doi:10.2174/1389203718666170828122927
Raja, K., Natarajan, J., and Kuusisto, F. (2020). Automated extraction and visualization of protein-protein interaction networks and beyond: A text-mining protocol. Methods Mol. Biol. 2074, 13–34. doi:10.1007/978-1-4939-9873-9_2
Raja, K., Subramani, S., and Natarajan, J. (2013). PPInterFinder—A mining tool for extracting causal relations on human proteins from literature. Database, 2013, bas052. doi:10.1093/database/bas052
Raman, K. (2010). Construction and analysis of protein–protein interaction networks. Autom. Exp. 2, 2. doi:10.1186/1759-4499-2-2
Rao, V. S., Srinivas, K., Sujini, G. N., and Kumar, G. N. S. (2014). Protein-protein interaction detection: Methods and analysis. Int. J. Proteomics, 2014, 147648. doi:10.1155/2014/147648
Rappoport, N., and Shamir, R. (2018). Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 46 (20), 10546–10562. doi:10.1093/nar/gky889
Razaghi-Moghadam, Z., and Nikoloski, Z. (2020). Supervised learning of gene regulatory networks. Curr. Protoc. Plant Biol. 5 (2), e20106. doi:10.1002/cppb.20106
Reimand, J., Hui, S., Jain, S., Law, B., and Bader, G. D. (2012). Domain-mediated protein interaction prediction: From genome to network. FEBS Lett. 586 (17), 2751–2763. doi:10.1016/j.febslet.2012.04.027
Ren, J., Wang, J., Li, M., and Wang, L. (2013). Identifying protein complexes based on density and modularity in protein-protein interaction network. BMC Syst. Biol. 7 (4), S12. doi:10.1186/1752-0509-7-S4-S12
Rogozin, I. B., Makarova, K. S., Murvai, J., Czabarka, E., Wolf, Y. I., Tatusov, R. L., et al. (2002). Connected gene neighborhoods in prokaryotic genomes. Nucleic Acids Res. 30 (10), 2212–2223. doi:10.1093/nar/30.10.2212
Rohani, N., and Eslahchi, C. (2019). Drug-drug interaction predicting by neural network using integrated similarity. Sci. Rep. 9 (1), 13645. doi:10.1038/s41598-019-50121-3
Roth, A., Subramanian, S., and Ganapathiraju, M. K. (2018). Towards extracting supporting information about predicted protein-protein interactions. IEEE/ACM Trans. Comput. Biol. Bioinform. 15 (4), 1239–1246. doi:10.1109/TCBB.2015.2505278
Ryu, J. Y., Kim, J., Shon, M. J., Sun, J., Jiang, X., Lee, W., et al. (2019). Profiling protein–protein interactions of single cancer cells with in situ lysis and co-immunoprecipitation. Lab. Chip 19 (11), 1922. doi:10.1039/C9LC00139E
Safari-Alighiarloo, N., Taghizadeh, M., Rezaei-Tavirani, M., Goliaei, B., and Peyvandi, A. A. (2014). Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol. Hepatol. Bed Bench 7 (1), 17–31.
Saito, R., Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., Lotia, S., et al. (2012). A travel guide to Cytoscape plugins. Nat. Methods 9 (11), 1069–1076. doi:10.1038/nmeth.2212
Salazar, G. A., Meintjes, A., Mazandu, G. K., Rapanoel, H. A., Akinola, R. O., and Mulder, N. J. (2014). A web-based protein interaction network visualizer. BMC Bioinforma. 15 (1), 129. doi:10.1186/1471-2105-15-129
Salazar-Ciudad, I., and Jernvall, J. (2013). The causality horizon and the developmental bases of morphological evolution. Biol. Theory 8 (3), 286–292. doi:10.1007/s13752-013-0121-3
Sandoval, O., and Orlando, O. (2021). Analysis and visualization of signal execution in network-driven biological processes. Thesis. Availableat: https://ir.vanderbilt.edu/handle/1803/16761 (Accessed: March 7, 2022).
Santiago-Rodriguez, T. M., and Hollister, E. B. (2021). Multi ‘omic data integration: A review of concepts, considerations, and approaches. Semin. Perinatol. 45 (6), 151456. doi:10.1016/j.semperi.2021.151456
Sanz-Pamplona, R., Berenguer, A., Sole, X., Cordero, D., Crous-Bou, M., Serra-Musach, J., et al. (2012). Tools for protein-protein interaction network analysis in cancer research. Clin. Transl. Oncol. 14 (1), 3–14. doi:10.1007/s12094-012-0755-9
Sardiu, M. E., Gilmore, J. M., Groppe, B. D., Dutta, A., Florens, L., and Washburn, M. P. (2019). Topological scoring of protein interaction networks. Nat. Commun. 10, 1118. doi:10.1038/s41467-019-09123-y
Sarkar, D., and Saha, S. (2019). Machine-learning techniques for the prediction of protein–protein interactions. J. Biosci. 44 (4), 104. doi:10.1007/s12038-019-9909-z
Schaefer, M. H., Lopes, T. J. S., Mah, N., Shoemaker, J. E., Matsuoka, Y., Fontaine, J. F., et al. (2013). Adding protein context to the human protein-protein interaction network to reveal meaningful interactions. PLoS Comput. Biol. 9 (1), e1002860. doi:10.1371/journal.pcbi.1002860
Schneider, L., Guo, Y. K., Birch, D., and Sarkies, P. (2021). Network-based visualisation reveals new insights into transposable element diversity. Mol. Syst. Biol. 17 (6), e9600. doi:10.15252/msb.20209600
Scott, M. S., and Barton, G. J. (2007). Probabilistic prediction and ranking of human protein-protein interactions. BMC Bioinforma. 8, 239. doi:10.1186/1471-2105-8-239
Sebestyén, V., Domokos, E., and Abonyi, J. (2020). Multilayer network based comparative document analysis (MUNCoDA). MethodsX 7, 100902. doi:10.1016/j.mex.2020
Sejdiu, B. I., and Tieleman, D. P. (2021). ProLint: A web-based framework for the automated data analysis and visualization of lipid-protein interactions. Nucleic Acids Res. 49 (W1), W544–W550. doi:10.1093/nar/gkab409
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504. doi:10.1101/gr.1239303
Sharan, R., and Ideker, T. (2006). Modeling cellular machinery through biological network comparison. Nat. Biotechnol. 24 (4), 427–433. doi:10.1038/nbt1196
Sharma, P., and Shrivastava, N. (2015). “Artificial neural network to prediction of protein-protein interactions in yeast,” in 2015 International Conference on Computer, Communication and Control, 1–5. doi:10.1109/IC4.2015.73756384
Sharp, A. (2017). Janusgraph. Availableat: https://janusgraph.org/.
Shay Banon, B. (2014). Elastic. Availableat: https://www.elastic.co/.
Shen, Z.-A., Luo, T., Zhou, Y. K., Yu, H., and Du, P. F. (2021). NPI-GNN: Predicting ncRNA–protein interactions with deep graph neural networks. Brief. Bioinform. 22 (5), bbab051. doi:10.1093/bib/bbab051
Shi, Q., Chen, W., Huang, S., Wang, Y., and Xue, Z. (2021). Deep learning for mining protein data. Brief. Bioinform. 22 (1), 194–218. doi:10.1093/bib/bbz156
Shirmohammady, N., Izadkhah, H., and Isazadeh, A. (2021). PPI-GA: A novel clustering algorithm to identify protein complexes within protein-protein interaction networks using genetic algorithm. Complexity 2021, e2132516. doi:10.1155/2021/2132516
Shoemaker, B. A., and Panchenko, A. R. (2007). Deciphering protein–protein interactions. Part I. Experimental techniques and databases. PLoS Comput. Biol. 3 (3), e42. doi:10.1371/journal.pcbi.0030042
Silverbush, D., and Sharan, R. (2019). A systematic approach to orient the human protein–protein interaction network. Nat. Commun. 10 (1), 3015. doi:10.1038/s41467-019-10887-6
Skrabanek, L., Saini, H. K., Bader, G. D., and Enright, A. J. (2008). Computational prediction of protein-protein interactions. Mol. Biotechnol. 38 (1), 1–17. doi:10.1007/s12033-007-0069-2
Škunca, N., and Dessimoz, C. (2015). Phylogenetic profiling: How much input data is enough? PLOS ONE 10 (2), e0114701. doi:10.1371/journal.pone.0114701
Sloutsky, R., Jimenez, N., Swamidass, S. J., and Naegle, K. M. (2013). Accounting for noise when clustering biological data. Brief. Bioinform. 14 (4), 423–436. doi:10.1093/bib/bbs057
Smith-Aguilar, S. E., Aureli, F., Busia, L., Schaffner, C., and Ramos-Fernandez, G. (2019). Using multiplex networks to capture the multidimensional nature of social structure. Primates. 60 (3), 277–295. doi:10.1007/s10329-018-0686-3
Snider, J., Kotlyar, M., Saraon, P., Yao, Z., Jurisica, I., and Stagljar, I. (2015). Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 11 (12), 848. doi:10.15252/msb.20156351
Soleimani Zakeri, N. S., Pashazadeh, S., and MotieGhader, H. (2021). ‘Drug repurposing for alzheimer’s disease based on protein-protein interaction network’. BioMed Res. Int. 2021, 1280237. doi:10.1155/2021/1280237
Song, B., Luo, X., Luo, X., Liu, Y., Niu, Z., and Zeng, X. (2022). Learning spatial structures of proteins improves protein-protein interaction prediction. Brief. Bioinform. 23 (2), bbab558. doi:10.1093/bib/bbab558
Stacey, R. G., Skinnider, M. A., and Foster, L. J. (2021). On the robustness of graph-based clustering to random network alterations. Mol. Cell. Proteomics. 20, 100002. doi:10.1074/mcp.RA120.002275
Stelzl, U., and Wanker, E. E. (2006). The value of high quality protein–protein interaction networks for systems biology. Curr. Opin. Chem. Biol. 10 (6), 551–558. doi:10.1016/j.cbpa.2006.10.005
Stringer, B., de Ferrante, H., Abeln, S., Heringa, J., Feenstra, K. A., and Haydarlou, R. (2022). Pipenn: Protein interface prediction from sequence with an ensemble of neural nets. Bioinformatics 38 (8), 2111–2118. doi:10.1093/bioinformatics/btac071
Subramani, S., Kalpana, R., Monickaraj, P. M., and Natarajan, J. (2015). HPIminer: A text mining system for building and visualizing human protein interaction networks and pathways. J. Biomed. Inf. 54, 121–131. doi:10.1016/j.jbi.2015.01.006
Subramanian, I., Verma, S., Kumar, S., Jere, A., and Anamika, K. (2020). Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 14, 1177932219899051. doi:10.1177/1177932219899051
Suderman, M., and Hallett, M. (2007). Tools for visually exploring biological networks. Bioinformatics 23 (20), 2651–2659. doi:10.1093/bioinformatics/btm401
Summer, G., Kelder, T., Ono, K., Radonjic, M., Heymans, S., and Demchak, B. (2015). cyNeo4j: connecting Neo4j and Cytoscape. Bioinformatics 31 (23), 3868–3869. doi:10.1093/bioinformatics/btv460
Sun, P., Ma, Y., Lu, L., and Ma, Z. (2008). “Application of improved K-mean clustering in predicting protein-protein interactions,” in 2008 International Conference on BioMedical Engineering and Informatics, Sanya, China, 83–86. doi:10.1109/BMEI.2008.82
Swamy, K. B. S., Schuyler, S. C., and Leu, J.-Y. (2021). Protein complexes form a basis for complex hybrid incompatibility. Front. Genet. 12. doi:10.3389/fgene.2021.609766
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi:10.1093/nar/gky1131
Tagore, S., Gorohovski, A., Jensen, L. J., and Frenkel-Morgenstern, M. (2019). ProtFus: A comprehensive method characterizing protein-protein interactions of fusion proteins. PLoS Comput. Biol. 15 (8), e1007239. doi:10.1371/journal.pcbi.1007239
Tang, M., Wu, L., and Yu, X. (2021). Prediction of protein–protein interaction sites based on stratified attentional mechanisms. Front. Genet. 12. doi:10.3389/fgene.2021.784863
Tanwar, H., and George Priya Doss, C. (2018). Computational resources for predicting protein-protein interactions. Adv. Protein Chem. Struct. Biol. 110, 251–275. doi:10.1016/bs.apcsb.2017.07.006
Terayama, K., Shinobu, A., Tsuda, K., Takemura, K., and Kitao, A. (2019). evERdock Bai: Machine-learning-guided selection of protein-protein complex structure. J. Chem. Phys. 151 (21), 215104. doi:10.1063/1.5129551
Tesoriero, C. (2013). Getting started with OrientDB. Availableat: http://orientdb.com/docs/last/index.html.
Thanasomboon, R., Kalapanulak, S., Netrphan, S., and Saithong, T. (2020). Exploring dynamic protein-protein interactions in cassava through the integrative interactome network. Sci. Rep. 10 (1), 6510. doi:10.1038/s41598-020-63536-0
The UniProt Consortium (2019). UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47 (1), D506–D515. doi:10.1093/nar/gky1049
Tian, X., Ju, H., and Yang, W. (2019). An ego network analysis approach identified important biomarkers with an association to progression and metastasis of gastric cancer. J. Cell. Biochem. 120 (9), 15963–15970. doi:10.1002/jcb.28873
Tian, Y., Zhang, B., Hoffman, E. P., Clarke, R., Zhang, Z., Shih, I. M., et al. (2015). Kddn: An open-source cytoscape app for constructing differential dependency networks with significant rewiring. Bioinforma. Oxf. Engl. 31 (2), 287–289. doi:10.1093/bioinformatics/btu632
Tieri, P., Zhou, X., Zhu, L., and Nardini, C. (2014). Multi-omic landscape of rheumatoid arthritis: Re-evaluation of drug adverse effects. Front. Cell Dev. Biol. 2, 59. doi:10.3389/fcell.2014.00059
Tillier, E. R. M., and Charlebois, R. L. (2009). The human protein coevolution network. Genome Res. 19 (10), 1861–1871. doi:10.1101/gr.092452.109
Timón-Reina, S., Rincón, M., and Martínez-Tomás, R. (2021). An overview of graph databases and their applications in the biomedical domain. Database. 2021, baab026. doi:10.1093/database/baab026
Tirosh, I., and Barkai, N. (2005). Computational verification of protein-protein interactions by orthologous co-expression. BMC Bioinforma. 6 (1), 40. doi:10.1186/1471-2105-6-40
Tomkins, J. E., and Manzoni, C. (2021). Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiol. Dis. 155, 105395. doi:10.1016/j.nbd.2021.105395
Touré, V., Mazein, A., Waltemath, D., Balaur, I., Saqi, M., Henkel, R., et al. (2016). Ston: Exploring biological pathways using the SBGN standard and graph databases. BMC Bioinforma. 17 (1), 494. doi:10.1186/s12859-016-1394-x
Truong, K., and Ikura, M. (2003). Domain fusion analysis by applying relational algebra to protein sequence and domain databases. BMC Bioinforma. 4 (1), 16. doi:10.1186/1471-2105-4-16
Ulfenborg, B. (2019). Vertical and horizontal integration of multi-omics data with miodin. BMC Bioinforma. 20 (1), 649. doi:10.1186/s12859-019-3224-4
Valdeolivas, A., Tichit, L., Navarro, C., Perrin, S., Odelin, G., Levy, N., et al. (2019). Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics 35 (3), 497–505. doi:10.1093/bioinformatics/bty637
Vapnik, V. (1963). Pattern recognition using generalized portrait method. Availableat: https://www.semanticscholar.org/paper/Pattern-recognition-using-generalized-portrait-Vapnik/7cabbdf6a7288d15e26fa6ea504009bab3d1edf4 (Accessed January 16, 2022).
Veitia, R. A. (2002). Rosetta stone proteins: “chance and necessity”. Genome Biol. 3 (2). doi:10.1186/gb-2002-3-2-interactions1001
Vella, D., Marini, S., Vitali, F., Di Silvestre, D., Mauri, G., and Bellazzi, R. (2018). Mtgo: PPI network analysis via topological and functional module identification. Sci. Rep. 8 (1), 5499. doi:10.1038/s41598-018-23672-0
Vento-Tormo, R., Efremova, M., Botting, R. A., Turco, M. Y., Vento-Tormo, M., Meyer, K. B., et al. (2018). Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563 (7731), 347–353. doi:10.1038/s41586-018-0698-6
Villaveces, J. M., Jimenez, R. C., Porras, P., Del-ToroN., , DuesburyM., , DuMousseauM., , et al. (2015a). Merging and scoring molecular interactions utilising existing community standards: Tools, use-cases and a case study. Database. 2015, bau131. doi:10.1093/database/bau131
Villaveces, J. M., Koti, P., and Habermann, B. H. (2015b). Tools for visualization and analysis of molecular networks, pathways, and -omics data. Adv. Appl. Bioinform. Chem. 8, 11–22. doi:10.2147/AABC.S63534
Vinayagam, A., Zirin, J., Roesel, C., Hu, Y., Yilmazel, B., Samsonova, A. A., et al. (2014). Integrating protein-protein interaction networks with phenotypes reveals signs of interactions. Nat. Methods 11 (1), 94–99. doi:10.1038/nmeth.2733
von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S. G., Fields, S., et al. (2002). Comparative assessment of large-scale data sets of protein–protein interactions. Nature 417 (6887), 399–403. doi:10.1038/nature750
Vyas, R., Bapat, S., Jain, E., Karthikeyan, M., Tambe, S., and Kulkarni, B. D. (2016). Building and analysis of protein-protein interactions related to diabetes mellitus using support vector machine, biomedical text mining and network analysis. Comput. Biol. Chem. 65, 37–44. doi:10.1016/j.compbiolchem.2016.09.011
Wahab Khattak, F., Salamah Alhwaiti, Y., Ali, A., Faisal, M., and Siddiqi, M. H. (2021). Protein-protein interaction analysis through network topology (oral cancer). J. Healthc. Eng. 2021, 6623904. doi:10.1155/2021/6623904
Wandy, J., and Daly, R. (2021). GraphOmics: An interactive platform to explore and integrate multi-omics data. BMC Bioinforma. 22 (1), 603. doi:10.1186/s12859-021-04500-1
Wang, J., Li, M., Deng, Y., and Pan, Y. (2010). Recent advances in clustering methods for protein interaction networks. BMC Genomics 11 (3), S10. doi:10.1186/1471-2164-11-S3-S10
Wang, K., Wang, X., Zheng, S., Niu, Y., Zheng, W., Qin, X., et al. (2018a). iTRAQ-based quantitative analysis of age-specific variations in salivary proteome of caries-susceptible individuals. J. Transl. Med. 16 (1), 293. doi:10.1186/s12967-018-1669-2
Wang, R.-S., Zhang, S., Wang, Y., Zhang, X. S., and Chen, L. (2008). Clustering complex networks and biological networks by nonnegative matrix factorization with various similarity measures. Neurocomputing 72 (1), 134–141. doi:10.1016/j.neucom.2007.12.043
Wang, S.-C. (2003). “Artificial neural network,” in Interdisciplinary computing in java programming. Editor S.-C. Wang (Boston, MA: Springer US (The Springer International Series in Engineering and Computer Science), 81–100. doi:10.1007/978-1-4615-0377-4_5
Wang, X., and Jin, Y. (2017). Predicted networks of protein-protein interactions in Stegodyphus mimosarum by cross-species comparisons. BMC Genomics 18, 716. doi:10.1186/s12864-017-4085-8
Wang, X., Yang, Y., Li, K., Li, W., Li, F., and Peng, S. (2021). BioERP: Biomedical heterogeneous network-based self-supervised representation learning approach for entity relationship predictions. Bioinforma. Oxf. Engl. 37, 4793–4800. doi:10.1093/bioinformatics/btab565
Wang, X., Yu, B., Ma, A., Chen, C., Liu, B., and Ma, Q. (2019). Protein–protein interaction sites prediction by ensemble random forests with synthetic minority oversampling technique. Bioinformatics 35 (14), 2395–2402. doi:10.1093/bioinformatics/bty995
Wang, Y., Li, L., and Li, C. (2018b). Predicting protein interactions using a deep learning method-stacked sparse autoencoder combined with a probabilistic classification vector machine. Complexity 2018, e4216813. doi:10.1155/2018/4216813
Wang, Z., Li, Y., and Pan, J. (2020). Prediction of protein-protein interactions from protein sequences by combining MatPCA feature extraction algorithms and weighted sparse representation models. Math. Problems Eng. 2020, e5764060. doi:10.1155/2020/5764060
Watson, J., Schwartz, J.-M., and Francavilla, C. (2021). Using multilayer heterogeneous networks to infer functions of phosphorylated sites. J. Proteome Res. 20 (7), 3532–3548. doi:10.1021/acs.jproteome.1c00150
Welch, N., Singh, S. S., Kumar, A., Dhruba, S. R., Mishra, S., Sekar, J., et al. (2021). Integrated multiomics analysis identifies molecular landscape perturbations during hyperammonemia in skeletal muscle and myotubes. J. Biol. Chem. 297 (3), 101023. doi:10.1016/j.jbc.2021.101023
Wen, Y., Song, X., Yan, B., Yang, X., Wu, L., Leng, D., et al. (2021). Multi-dimensional data integration algorithm based on random walk with restart. BMC Bioinforma. 22 (1), 97. doi:10.1186/s12859-021-04029-3
Winkler, J., Mylle, E., De Meyer, A., Pavie, B., Merchie, J., Grones, P., et al. (2021). Visualizing protein–protein interactions in plants by rapamycin-dependent delocalization. Plant Cell 33 (4), 1101–1117. doi:10.1093/plcell/koab004
Woo, H.-M., and Yoon, B.-J. (2021). Monaco: Accurate biological network alignment through optimal neighborhood matching between focal nodes. Bioinforma. Oxf. Engl. 37 (10), 1401–1410. doi:10.1093/bioinformatics/btaa962
Wörheide, M. A., Krumsiek, J., Kastenmuller, G., and Arnold, M. (2021). Multi-omics integration in biomedical research – a metabolomics-centric review. Anal. Chim. Acta 1141, 144–162. doi:10.1016/j.aca.2020.10.038
Wu, J., Khodaverdian, A., Weitz, B., and Yosef, N. (2019). Connectivity problems on heterogeneous graphs. Algorithms Mol. Biol. 14, 5. doi:10.1186/s13015-019-0141-z
Wu, L., Wang, X., Zhang, J., Luan, T., Bouveret, E., and Yan, X. (2017). Flow cytometric single-cell analysis for quantitative in vivo detection of protein–protein interactions via relative reporter protein expression measurement. Anal. Chem. 89 (5), 2782–2789. doi:10.1021/acs.analchem.6b03603
Wu, L., Xie, X., Liang, T., Ma, J., Yang, L., Yang, J., et al. (2022a). Integrated multi-omics for novel aging biomarkers and antiaging targets. Biomolecules 12 (1), 39. doi:10.3390/biom12010039
Wu, X., Zeng, W., Lin, F., and Zhou, X. (2021). NeuRank: Learning to rank with neural networks for drug–target interaction prediction. BMC Bioinforma. 22 (1), 567. doi:10.1186/s12859-021-04476-y
Wu, Y., Gao, M., Zeng, M., Zhang, J., and Li, M. (2022b). BridgeDPI: A novel graph neural network for predicting drug-protein interactions. Bioinforma. Oxf. Engl. 38, 2571–2578. doi:10.1093/bioinformatics/btac155
Wu, Y., Zhang, X., Yu, J., and Ouyang, Q. (2009). Identification of a topological characteristic responsible for the biological robustness of regulatory networks. PLoS Comput. Biol. 5 (7), e1000442. doi:10.1371/journal.pcbi.1000442
Xia, J., Benner, M. J., and Hancock, R. E. W. (2014). NetworkAnalyst - integrative approaches for protein–protein interaction network analysis and visual exploration. Nucleic Acids Res. 42 (W1), W167–W174. doi:10.1093/nar/gku443
Xia, K., Fu, Z., Hou, L., and Han, J. D. J. (2008). Impacts of protein–protein interaction domains on organism and network complexity. Genome Res. 18 (9), 1500–1508. doi:10.1101/gr.068130.107
Xie, Z., Deng, X., and Shu, K. (2020). Prediction of protein–protein interaction sites using convolutional neural network and improved data sets. Int. J. Mol. Sci. 21 (2), 467. doi:10.3390/ijms21020467
Xu, B., Guan, J., Wang, Y., and Wang, Z. (2019). Essential protein detection by random walk on weighted protein-protein interaction networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 16 (2), 377–387. doi:10.1109/TCBB.2017.2701824
Xu, B., Liu, Y., and Dong, J. (2018). Reconstruction of the protein-protein interaction network for protein complexes identification by walking on the protein pair fingerprints similarity network. Front. Genet. 9. doi:10.3389/fgene.2018.00272
Xu, W., Gao, Y., Wang, Y., and Guan, J. (2021). Protein-protein interaction prediction based on ordinal regression and recurrent convolutional neural networks. BMC Bioinforma. 22 (6), 485. doi:10.1186/s12859-021-04369-0
Xuan, P., Sun, C., Zhang, T., Ye, Y., Shen, T., and Dong, Y. (2019). Gradient boosting decision tree-based method for predicting interactions between target genes and drugs. Front. Genet. 10, 459. doi:10.3389/fgene.2019.00459
Yan, J., Risacher, S. L., Shen, L., and Saykin, A. J. (2018). Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data. Brief. Bioinform. 19 (6), 1370–1381. doi:10.1093/bib/bbx066
Yang, J., Wagner, S. A., and Beli, P. (2015). Illuminating spatial and temporal organization of protein interaction networks by mass spectrometry-based proteomics. Front. Genet. 6. Availableat: https://www.frontiersin.org/article/10.3389/fgene.2015.00344 (Accessed: January 28, 2022).
Yann Lecun, L. (1986). A Learning Scheme for assymetric threshold network. Availableat: http://yann.lecun.com/exdb/publis/pdf/lecun-85.pdf (Accessed: February 18, 2022).
Yoon, B.-H., Kim, S.-K., and Kim, S.-Y. (2017). Use of graph database for the integration of heterogeneous biological data. Genomics Inf. 15 (1), 19–27. doi:10.5808/GI.2017.15.1.19
You, Z.-H., Lei, Y. K., Zhu, L., Xia, J., and Wang, B. (2013). Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinforma. 14 (8), S10. doi:10.1186/1471-2105-14-S8-S10
Yu, H., Zhu, X., Greenbaum, D., Karro, J., and Gerstein, M. (2004). TopNet: A tool for comparing biological sub-networks, correlating protein properties with topological statistics. Nucleic Acids Res. 32 (1), 328–337. doi:10.1093/nar/gkh164
Yuan, C., Wang, M. H., and Wang, F. (2021). Network pharmacology and molecular docking reveal the mechanism of Scopoletin against non-small cell lung cancer. Life Sci. 270, 119105. doi:10.1016/j.lfs.2021.119105
Zahiri, J., Bozorgmehr, J. H., and Masoudi-Nejad, A. (2013). Computational prediction of protein–protein interaction networks: Algo-rithms and resources. Curr. Genomics 14 (6), 397–414. doi:10.2174/1389202911314060004
Zahiri, J., Emamjomeh, A., Bagheri, S., Ivazeh, A., Mahdevar, G., Sepasi Tehrani, H., et al. (2020). Protein complex prediction: A survey. Genomics 112 (1), 174–183. doi:10.1016/j.ygeno.2019.01.011
Zaki, N., and Tennakoon, C. (2017). BioCarian: Search engine for exploratory searches in heterogeneous biological databases. BMC Bioinforma. 18 (1), 435. doi:10.1186/s12859-017-1840-4
Zhang, J., Suo, Y., Liu, M., and Xu, X. (2018). Identification of genes related to proliferative diabetic retinopathy through RWR algorithm based on protein-protein interaction network. Biochim. Biophys. Acta. Mol. Basis Dis. 1864 (6), 2369–2375. doi:10.1016/j.bbadis.2017.11.017
Zhang, X., Xu, J., and Xiao, W. (2013). A new method for the discovery of essential proteins. PloS One 8 (3), e58763. doi:10.1371/journal.pone.0058763
Zhang, Y., Natale, R., Domingues, A. P., Toleco, M. R., Siemiatkowska, B., Fabregas, N., et al. (2019). Rapid identification of protein-protein interactions in plants. Curr. Protoc. Plant Biol. 4 (4), e20099. doi:10.1002/cppb.20099
Zhang, Z. (2018). “Artificial neural network,” in Multivariate time series analysis in climate and environmental research. Editor Z. Zhang (Cham: Springer International Publishing), 1–35. doi:10.1007/978-3-319-67340-0_1
Zhao, B., Wang, J., and Wu, F.-X. (2017). Computational methods to predict protein functions from protein-protein interaction networks. Curr. Protein Pept. Sci. 18 (11), 1120–1131. doi:10.2174/1389203718666170505121219
Zhao, Q., Zhang, Y., and Hu, H. (2018). Irwnrlpi: Integrating random walk and neighborhood regularized logistic matrix factorization for lncRNA-protein interaction prediction. Front. Genet. 9. doi:10.3389/fgene.2018.00239
Zhao, T., Hu, Y., Valsdottir, L. R., Zang, T., and Peng, J. (2021). Identifying drug-target interactions based on graph convolutional network and deep neural network. Brief. Bioinform. 22 (2), 2141–2150. doi:10.1093/bib/bbaa044
Zhong, M., Lee, G. M., Sijbesma, E., Ottmann, C., and Arkin, M. R. (2019). Modulating protein-protein interaction networks in protein homeostasis. Curr. Opin. Chem. Biol. 50, 55–65. doi:10.1016/j.cbpa.2019.02.012
Zhou, G., and Xia, J. (2018). OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 46 (1), W514–W522. doi:10.1093/nar/gky510
Zhou, J., Guo, Y., Fu, J., and Chen, Q. (2022). Construction and validation of a glioma prognostic model based on immune microenvironment, Neuroimmunomodulation, 30. 1–12. doi:10.1159/000522529
Zhou, L., Wang, Z., Tian, X., and Peng, L. (2021). LPI-deepGBDT: A multiple-layer deep framework based on gradient boosting decision trees for lncRNA–protein interaction identification. BMC Bioinforma. 22, 479. doi:10.1186/s12859-021-04399-8
Zhou, M., Li, Q., and Wang, R. (2016). Current experimental methods for characterizing protein–protein interactions. Chemmedchem 11 (8), 738–756. doi:10.1002/cmdc.201500495
Zhou, W.-Z., Miao, L.-G., and Yuan, H. (2018). Identification of significant ego networks and pathways in rheumatoid arthritis. J. Cancer Res. Ther. 14 (1), S1024–S1028. doi:10.4103/0973-1482.189250
Zhu, M.-L., and Schmotzer, C. (2017). Writing the genome: Are we ready? Clin. Chem. 63 (4), 929–930. doi:10.1373/clinchem.2016.270066
Keywords: interactome, biological network, computational prediction, integrated strategies, graphic view, protein-protein interaction
Citation: Robin V, Bodein A, Scott-Boyer M-P, Leclercq M, Périn O and Droit A (2022) Overview of methods for characterization and visualization of a protein–protein interaction network in a multi-omics integration context. Front. Mol. Biosci. 9:962799. doi: 10.3389/fmolb.2022.962799
Received: 06 June 2022; Accepted: 16 August 2022;
Published: 08 September 2022.
Edited by:
Ornella Cominetti, Nestlé Research Center, SwitzerlandReviewed by:
Bharat Mishra, University of Alabama at Birmingham, United StatesHan Wang, Northeast Normal University, China
Copyright © 2022 Robin, Bodein, Scott-Boyer, Leclercq, Périn and Droit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arnaud Droit, YXJuYXVkLmRyb2l0QGNyY2h1cS51bGF2YWwuY2E=