
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 07 October 2022
Sec. Molecular Diagnostics and Therapeutics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.956095
This article is part of the Research Topic Molecular Chaperones and Human Disease View all 11 articles
 Alexander Mrozek1
Alexander Mrozek1 Tetyana Antoshchenko1Yun Chen1Carlos Zepeda-Velázquez2
Tetyana Antoshchenko1Yun Chen1Carlos Zepeda-Velázquez2 David Smil2Nirbhay Kumar3
David Smil2Nirbhay Kumar3 Hua Lu1
Hua Lu1 Hee-Won Park1*
Hee-Won Park1*Drug resistance to front-line malarial treatments represents an ongoing threat to control malaria, a vector borne infectious disease. The malarial parasite, Plasmodium falciparum has developed genetic variants, conferring resistance to the current standard therapeutic artemisinin and its derivatives commonly referred to as artemisinin-combination therapies (ACTs). Emergence of multi-drug resistance parasite genotypes is a warning of potential treatment failure, reaffirming the urgent and critical need to find and validate alternate drug targets to prevent the spread of disease. An attractive and novel drug target includes glucose-regulated protein 78 kDa (GRP78, or BiP), an essential molecular chaperone protein involved in the unfolded protein response that is upregulated in ACT treated P. falciparum parasites. We have shown that both sequence and structure are closely related to human GRP78 (hGRP78), a chaperone belonging to the HSP70 class of ATPase proteins, which is often upregulated in cellular stress responses and cancer. By screening a library of nucleoside analogues, we identified eight ‘hit’ compounds binding at the active site of the ATP binding domain of P. falciparum GRP78 using a high-throughput ligand soaking screen using x-ray crystallography. These compounds were further evaluated using protein thermal shift assays to assess target binding activity. The nucleoside analogues identified from our screen provide a starting point for the development of more potent and selective antimalarial inhibitors. In addition, we have established a well-defined, high-throughput crystal-based screening approach that can be applied to many crystallizable P. falciparum proteins for generating anti-Plasmodium specific compounds.
Malaria is a vector borne infectious disease caused by the protozoan parasite of the Plasmodium genus, infecting both the host liver and blood cells causing an estimated 627,000 deaths annually (World Health Organization, 2021). There are five species known to infect humans: P. falciparum, P. vivax, P. ovalae, P. malariae and P. knowelsi, however P. falciparum is by far the most dangerous resulting in the highest associated mortality and morbidity (World Health Organization, 2021). Despite numerous therapeutic and preventative efforts leading to an overall reduction in death and disease over the past decade, the spread of partial resistance to front-line malarial therapeutics such as artemisinin and related artemisinin combination therapies (ACTs) has emerged and continues to spread (Visser et al., 2014; Suresh and Haldar, 2018). Malaria multidrug resistance highlights the critical and urgent need to discover new targets and chemical scaffolds.
One class of druggable targets is the endoplasmic reticulum (ER) resident molecular heat shock chaperone GRP78, which is also referred to as Immunoglobulin Binding Protein (BiP), an HSP70-like stress response protein implicated in the cellular unfolded protein response (UPR). GRP78 is essential in the maintenance of ER homeostasis, where it facilitates proper protein folding, mitigating physiological and pathological damages that may cause an accumulation of misfolded proteins and ER stress (Ibrahim et al., 2019). Moreover, it has been shown that GRP78 from P. falciparum (pfGRP78) is associated with numerous protein families such as: transcriptional and translational machinery, the proteosome and proteolytic enzymes, as well as proteins involved in physiological and metabolic pathways (Gardner et al., 2013). There is growing evidence pfGRP78 is cytoprotective, aiding in adaptation between physiological and environmental changes occurring during transmission from the host vector to the infected cell (Kumar et al., 1988; Przyborski et al., 2015; Day et al., 2019). We hypothesize that up-regulated expression of pfGRP78 allows P. falciparum infected cells to withstand and adapt to host conditions and have improved rates of survival.
The presence of the ER stress-response chaperone pfGRP78 in the malaria parasite suggest that this protein could be used as a rational therapeutic target, and various studies including the previous work from our group have explored small molecule inhibitors against P. falciparum and human HSP70-like chaperones (Frame et al., 2015; Jones et al., 2016; Chen et al., 2018). pfGRP78 shares an 84.4% sequence identity to the human orthologue of GRP78 and this value increased to 87.8% when evaluating the nucleotide-binding domain (NBD) only (Chen et al., 2018). GRP78 is structurally divided into two distinct domains: the nucleotide-binding domain at the amino terminus followed by a flexible linker to the substrate-binding domain at the carboxyl-terminus (Chakafana et al., 2019). ATP hydrolysis and ADP/ATP exchange within the NBD of GRP78 play an essential role in the chaperone activity (Pobre et al., 2019). Therefore, the disruption of the interaction of pfGRP78-NBD with ATP using nucleoside analogues with varying structural modifications could inhibit pfGRP78 functions requiring ATP hydrolysis.
In this present study, we expressed and purified recombinant pfGRP78-NBD and the human orthologue hGRP78-NBD from bacteria. We then grew crystals of pfGRP78-NBD to perform a novel compound screening assay using adenine-based nucleoside analogues with distinct modifications to interrogate the active site of pfGRP78-NBD in effort to identify compounds that bind the Plasmodium protein with enhanced selectivity when compared to the human ortholog hGRP78-NBD. Our crystal-based assay identified eight distinct nucleoside analogues present at the active site of pfGRP78-NBD in the complex crystal structures. We then used a protein Thermal Shift Assay (TSA) to further characterize and identify potential differences between the malaria and human chaperone ATP binding sites.
All reagents, materials and nucleoside analogues were purchased from commercial suppliers and were used as received unless otherwise noted. HP1, HP2, HP3, HP4, HP5, HP13, HP14 were purchased from Carbosynth. HP18 was purchased from Acros Organics. HP19, HP20, HP21, HP22, HP23, HP24, HP25, HP26, HP27 were purchased from TargetMol. HP28, HP31, HP32 and HP33 were purchased from Selleck Chemicals. HP9, HP10, HP11 and HP12 were synthesized as detailed below.
The plasmids used in this study have been described previously (Chen et al., 2018). Briefly, the gene encoding for pfGRP78-NBD (residues 26-404) and hGRP78-NBD (residues 26-407) were amplified from full-length pfGRP78 (residues 26-652) and hGRP78 (residues 1-654) by PCR and ligated into a pET28-MHL vector containing an N-terminal His6-tag (GenBank accession EF456735) and expressed in E. coli BL21 (DE3) competent cells (Thermo Scientific). Bacterial starter cultures were grown in LB medium containing 50 μg/ml kanamycin for 16 h at 37°C and then transferred to Terrific Broth containing 50 μg/ml kanamycin. The cells were further grown until the OD600nm reached ∼2.0 and expression was induced with 0.2 mM isopropyl β-D-1-thiogalactopyranoside at 17°C. Eighteen hours post-induction, the cells were harvested by centrifugation at 10,000 rpm for 20 min at 4°C, resuspended in a binding buffer (20 mM TRIS-HCl pH 7.5, 200 mM NaCl, 1 mM β-mercaptoethanol) supplemented with (1 mM PMSF, 1 mM Benzamidine, 0.1% IGEPAL CA-630) and lysed by sonification. Lysates were clarified by centrifugation at 10,000 rpm for 20 min at 4°C and the supernatant was loaded on to 5 ml column of Ni-NTA agarose beads (Thermo Scientific) pre-equilibrated with the binding buffer. The matrix was washed in a stepwise manner using the binding buffer containing increasing concentrations of imidazole (5 mM, 20 mM, 50 mM) and the protein was eluted with the same buffer containing 250 mM imidazole. The eluate was loaded on to a HiLoad 26/600 Superdex 200 pg column (Cytiva) pre-equilibrated with the binding buffer. Target protein-containing fractions were pooled and concentrated using 10 kDa MWCO centrifugal filters (Amicon), flash frozen and stored at −80°C. Expression and purification of pfGRP78 was identical to hGRP78. The N-terminal His6-tag was cleaved using a 1:50 ratio of TEV protease at 4°C overnight. TEV-cut pfGRP78-NBD was mixed with a 10-fold molar excess of 8-bromoadenosine and was concentrated to ∼30 mg/ml for crystallization.
Initial crystallization trials were conducted using the sitting-drop, vapor diffusion method using in-house crystallization screening kits, with each drop containing a solution of the complex of pfGRP78-NBD with 8-bromoadenosine and a reservoir solution by high throughput crystallization robot Mosquito Xtal3 (SPT Labtech). Drops were set in 96-well Intelliplates (Art Robbins Instruments). Crystals appeared within a week in the reservoir solution containing 0.7 M (NH4)2SO4, 1.2 M Li2SO4, 0.1 M NaCitrate, pH 5.6 at a room temperature. These crystals were then used for crystal-ligand replacement screening using DMSO. Crystals were first subjected to a DMSO soaking test by titrating them with various concentrations of DMSO to determine a critical threshold between surviving and dissolving (5% addition of DMSO to the drop volume dissolved crystals completely, but 4% DMSO kept crystals intact without change in morphology). Subsequent experiments added 5% of DMSO and 5 mM of each nucleoside analogue into the crystal-containing drop for DMSO-driven crystal-ligand replacement screening. Survived crystals for eight different nucleoside analogues were cryoprotected using paratone-N and flash-frozen in liquid nitrogen until data collection. These survived crystals come from bound 8-bromoadenosine being replaced by the added nucleoside analog, resulting in stabilization of proteins in the crystal lattice. The diffraction data were collected at the Canadian Light Source 08B1-1 beamline in Saskatoon, Canada, as well as the Advanced Photon Source 19-ID-D and 23-ID-D beamlines at the Argonne National Laboratory in Chicago, Illinois. Data were processed using XDS (Kabsch, 2010) and all structures were solved by molecular replacement using MOLREP (Vagin and Teplyakov, 2010) in the CCP4 crystallographic suite (Winn et al., 2011). Structures were solved using the ADP bound pfGRP78-NBD structure excluding ADP (PDB ID: 5UMB) as a search model. After molecular replacement, each model was subjected to multiple rounds of model building and refinement using COOT (Emsley and Cowtan, 2004) and REFMAC (Murshudov et al., 2011), respectively. Coordinate files for all eight pfGRP78-NBD structures have been deposited in the Protein Data Bank with accession codes: 8DIQ (HP2), 8DIP (HP3), 8DIS (HP5), 8D1W (HP10), 8DIY (HP19), 8D20 (HP20), 8D22 (HP22) AND 8D24 (HP23). Structural figures were generated using PyMOL Molecular Graphics System (Version 2.5.0 Schrödinger, LLC) as well as LIGPLOT (Laskowski and Swindells, 2011).
Thermal shift assay measurements were performed on a QuantStudio 6 Flex Real-Time PCR instrument (Applied Biosystems) in 96-well plates (Axygen) sealed with transparent adhesive film (BioRad). pfGRP78-NBD and hGRP78-NBD were diluted to a final concentration of 5 µM in DSF assay buffer (25 mM HEPES pH 7.5, 250 mM NaCl, 1 mM β-mercaptoethanol) and nucleoside analogues were added at a final concentration of 0.5 mM (5% DMSO final concentration). Protein-compound samples were aliquoted in four replicates followed by the addition of the fluorescent probe bis-ANS (Invitrogen) diluted to 50 µM in a 20 µL total reaction volume. Temperature was continuously increased from 10°C to 95°C at an increment of 1°C/min. Melting curves were analyzed using the software Thermal Shift Assay—Curve Rapid and Automatic Fitting Tool (Lee et al., 2019). Tm was defined as the temperature corresponding to the maximum value of the first derivative of fluorescence. Error bars shown in Figure 4 represent standard error of the mean and were calculated using GraphPad Prism (version 9.3.1) for macOS (GraphPad Software, San Diego, California, United States, www.graphpad.com/).
A series of nucleoside analogues (HP9, HP10, HP11, HP12) with modifications at the C8 position of the adenine moiety were synthesized from a previously described method (Tatani et al., 2015) and all characterizations by LRMS and 1H NMR were consistent with reported values.
HP9 was synthesized according to the synthesis procedure above from a from a mixture of 8-bromoadenosine (100 mg, 0.289 mmol, 1 eq.), N-methyl-1-phenylmethanamine (0.867 mmol, 3 eq.), and N,N-diisopropylethylamine (1.73 mmol, 0.302 ml, 6 eq.) was heated to 140°C under microwave irradiation for 10 h. The reaction mixture was then concentrated under reduced pressure, and the residue purified by column chromatography on silica gel (Biotage SNAP 10 g column, 0-40% MeOH/EtOAc as the eluent, 28 CV) to afford the desired product in 56% yield (64 mg). LRMS (M + H)+: 387.5 Purity: 98% (UV/254 nm).
HP10 was synthesized according to the synthesis procedure above from a mixture of 8-bromoadenosine (100 mg, 0.289 mmol, 1 eq.), (2-chlorophenyl)methanamine (0.867 mmol, 3 eq.), and N,N-diisopropylethylamine (1.73 mmol, 0.302 ml, 6 eq.) was heated to 140°C under microwave irradiation for 10 h. The reaction mixture was then concentrated under reduced pressure, and the residue purified by column chromatography on silica gel (Biotage SNAP 10 g column, 0-40% MeOH/EtOAc as the eluent, 28 CV) to afford the desired product in 59% yield (71 mg). LRMS (M + H)+: 407.4. Purity: 98% (UV/254 nm).
HP11 was synthesized according to the synthesis procedure above from a mixture of 8-bromoadenosine (100 mg, 0.289 mmol, 1 eq.), (4-methoxyphenyl)methanamine (0.867 mmol, 3 eq.), and N,N-diisopropylethylamine (1.73 mmol, 0.302 ml, 6 eq.) was heated to 140°C under microwave irradiation for 10 h. The reaction mixture was then concentrated under reduced pressure, and the residue purified by column chromatography on silica gel (Biotage SNAP 10 g column, 0-40% MeOH/EtOAc as the eluent, 28 CV) to afford the desired product in 67% yield (78 mg). LRMS (M + H)+: 403.5. Purity: 100% (UV/254 nm).
HP12 was synthesized according to the synthesis procedure above from a mixture of 8-bromoadenosine (100 mg, 0.289 mmol, 1 eq.), naphthalen-1-ylmethanamine (0.867 mmol, 3 eq.), and N,N-diisopropylethylamine (1.73 mmol, 0.302 ml, 6 eq.) was heated to 140°C under microwave irradiation for 10 h. The reaction mixture was then concentrated under reduced pressure, and the residue purified by column chromatography on silica gel (Biotage SNAP 10 g column, 0-40% MeOH/EtOAc as the eluent, 28 CV) to afford the desired product in 61% yield (74 mg). LRMS (M + H)+: 423.4. Purity: 96% (UV/254 nm).
In order to structurally characterize nucleoside analogue as potential anti-plasmodium chemical scaffolds in complexes with pfGRP78-NBD, we crystallized pfGRP78-NBD in complex with HP1 (8-bromoadenosine). We then soaked crystals with nucleoside analogues (Figure 1) using our novel DMSO-driven crystal-ligand replacement screening method. Certain compounds stabilized proteins in the crystal lattice, leading to the survival of the soaked crystals, resulting in the identification of protein-binding compounds later confirmed by x-ray crystallography. On the other hand, crystals soaked with non-binding compounds were dissolved within 24 h (Figure 2A). These results demonstrate our novel crystal-ligand DMSO soaking assay reduces the total number of crystal samples sent out for data collection and can be used for potential drug discovery efforts at large. Synchrotron beam time is a rate-limiting step in compound screening by conventional soaking approaches, thus the reduced number of samples sent out for data collection leads to a reduction in false-positives and expedited data analysis, representing a significant improvement in throughput. Using this approach, we successfully identified eight pfGRP78-NBD inhibitor complex structures to high resolutions. PDB entries for these pfGRP78-NBD nucleoside analogue complex structures are summarized in Table 1. These hit nucleoside analogs are HP2: 8-Aminoadenosine, HP3: 7-Deaza-2′-C-methyladenosine, HP5: Toyocamycin, HP10: (2R,3R,4S,5R)-2-(6-amino-8-((2-chlorobenzyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol), HP19: Trans-Zeatin Riboside, HP20: 5′-Methylthioadenosine, HP22: Piclidenoson and HP23: VER155008. In the active sites of all pfGRP78-NBD and nucleoside analogue complexes, electron density is well shaped for unambiguous modeling of eight hit compounds that replaced the 8-bromoadenosine compound (Figure 2B).
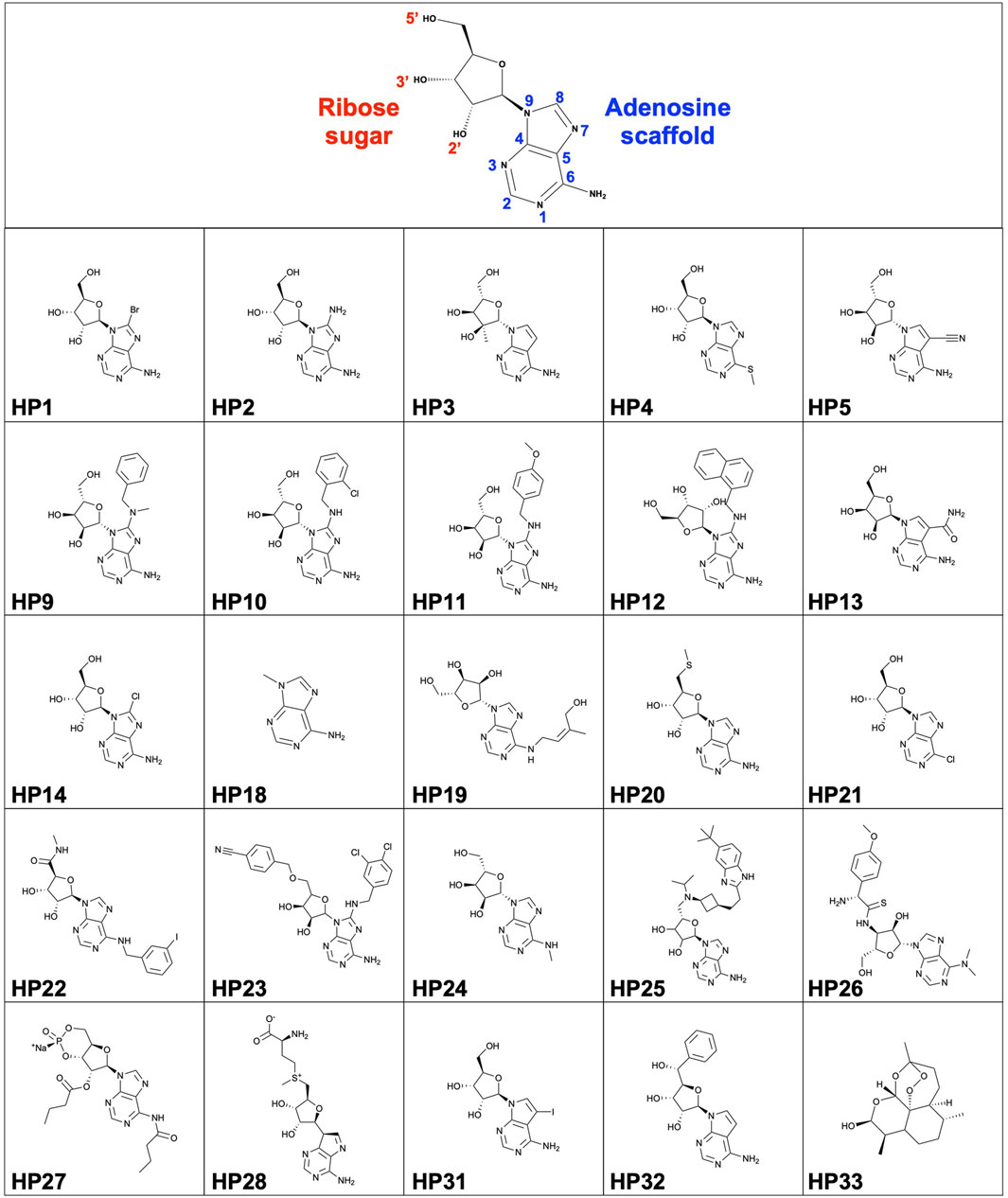
FIGURE 1. Structure of nucleoside scaffold and HP compound library (HP1-HP33).
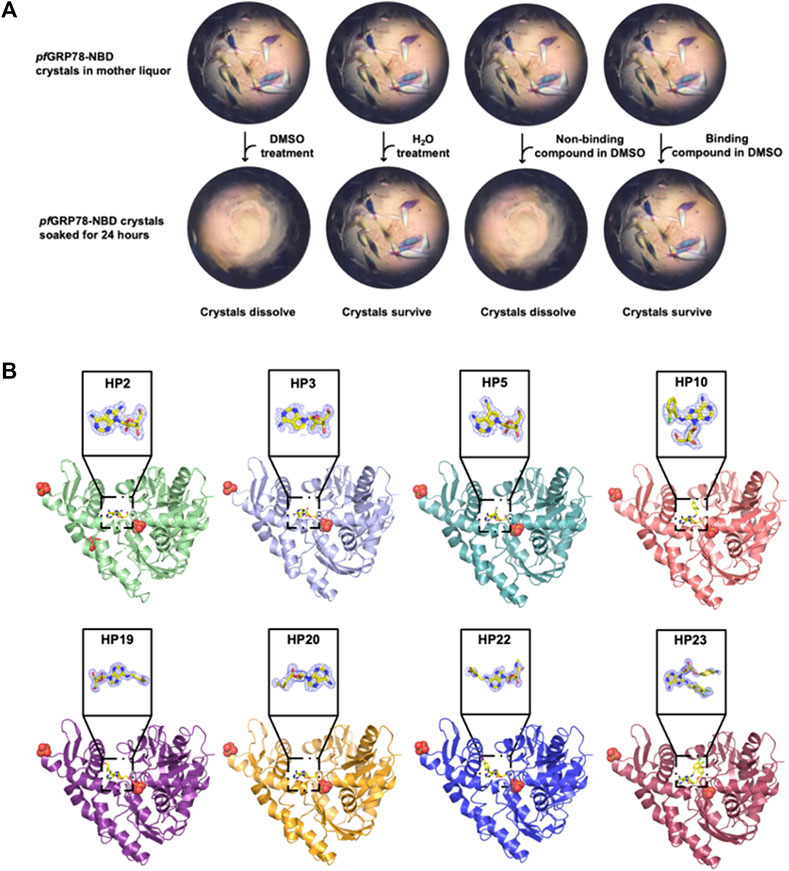
FIGURE 2. Crystal structures of pfGRP78-NBD bound with eight nucleoside analogues. (A) DMSO-driven Crystal-ligand replacement assay schematic. (B) Protein-ligand complex structures of pfGRP78-NBD with nucleoside analogues (HP2, HP3, HP5, HP10, HP19, HP20, HP22 and HP23) and corresponding electron density maps contoured to 2Fo-Fc at the 1 σ level showing molecular replacement of 8-bromoadenosine at the active site of pfGRP78-NBD.
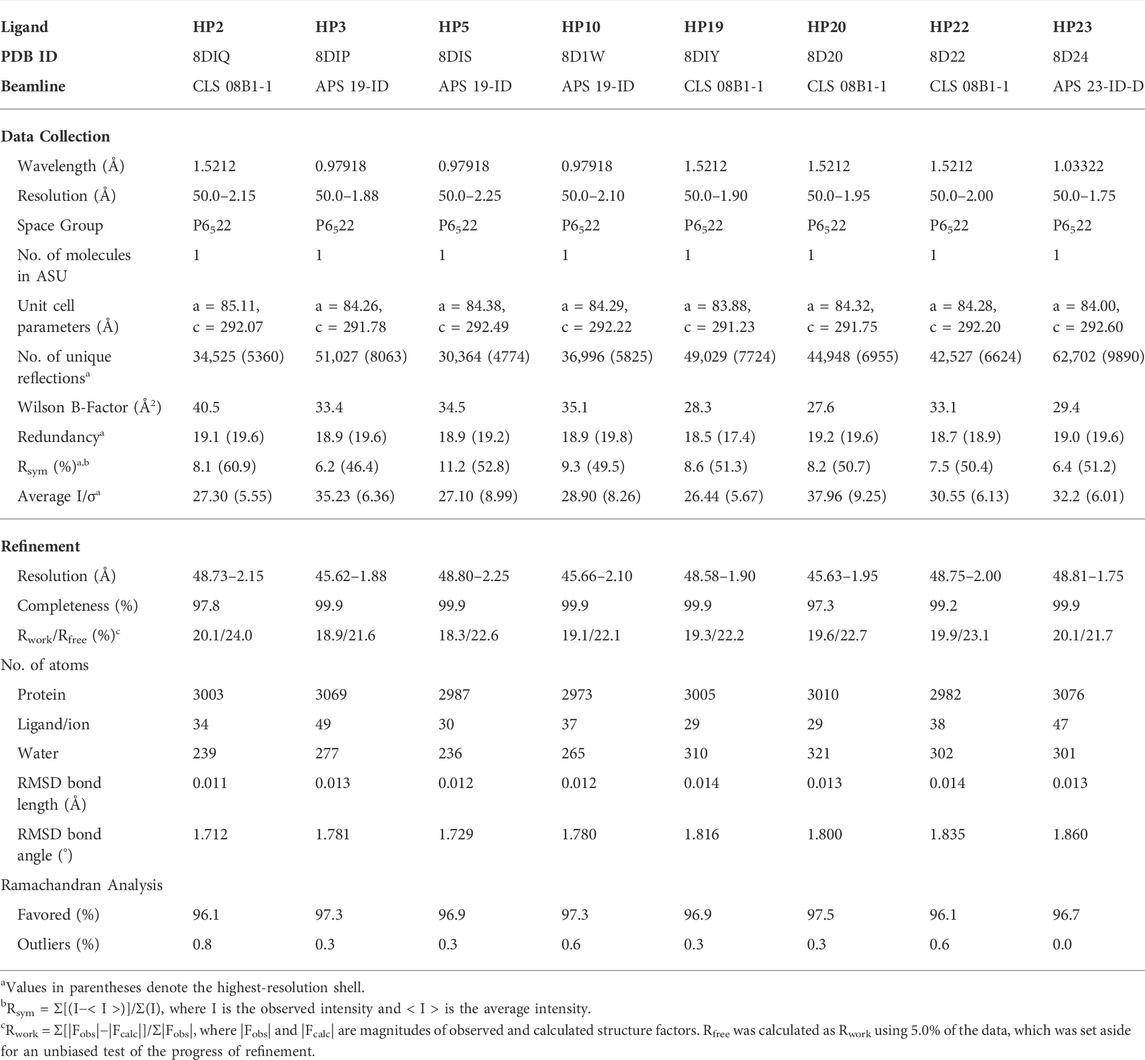
TABLE 1. X-ray data collection, refinement and validation statistics.
To identify specific modifications of adenosine advantageous for designing plasmodium specific compounds, we analyzed the complex structures of pfGRP78-NBD with the eight hit compounds identified through the crystal-ligand replacement screening method and compared the interactions to those present in hGRP78-NBD. Here we discuss the relevance of chemical modifications of the eight hit nucleoside analogues at the C8, C6, N7, 2′ and 5’ positions.
In Figure 3A, HP2 (8-Aminoadenosine) forms several hydrogen bonds with pfGRP78-NBD active site residues. HP2 forms direct hydrogen bonds with Lys293, Ser297 and Arg364. Four water molecules provide water-mediated hydrogen bonding with Asp256, Glu290, Arg294 and Ser362, further stabilizing HP2 to pfGRP78-NBD. Hydrophobic contacts are present with Gly224, Gly252, Gly361 and Ile365. The amino substituent at the C8 position directly interacts with Asp388, forming a hydrogen bond and has minimal hydrophobic interactions with Arg294. In comparison, the HP2-bound structure of hGRP78-NBD shows the conservation of most residues involved in HP2 binding (e.g., pfLys293 and hLys296, pfGly252 and hGly255). Specifically, a hydrogen bond and hydrophobic interactions with equivalent residues of hGRP78-NBD (hAsp391 and hArg297, respectively) are conserved, although the hydrogen bond is water-mediated in hGRP78-NBD. This suggests that the C8 amino substituent may not enhance specificity for pfGRP78-NBD, despite the improved stabilization this modification provides.
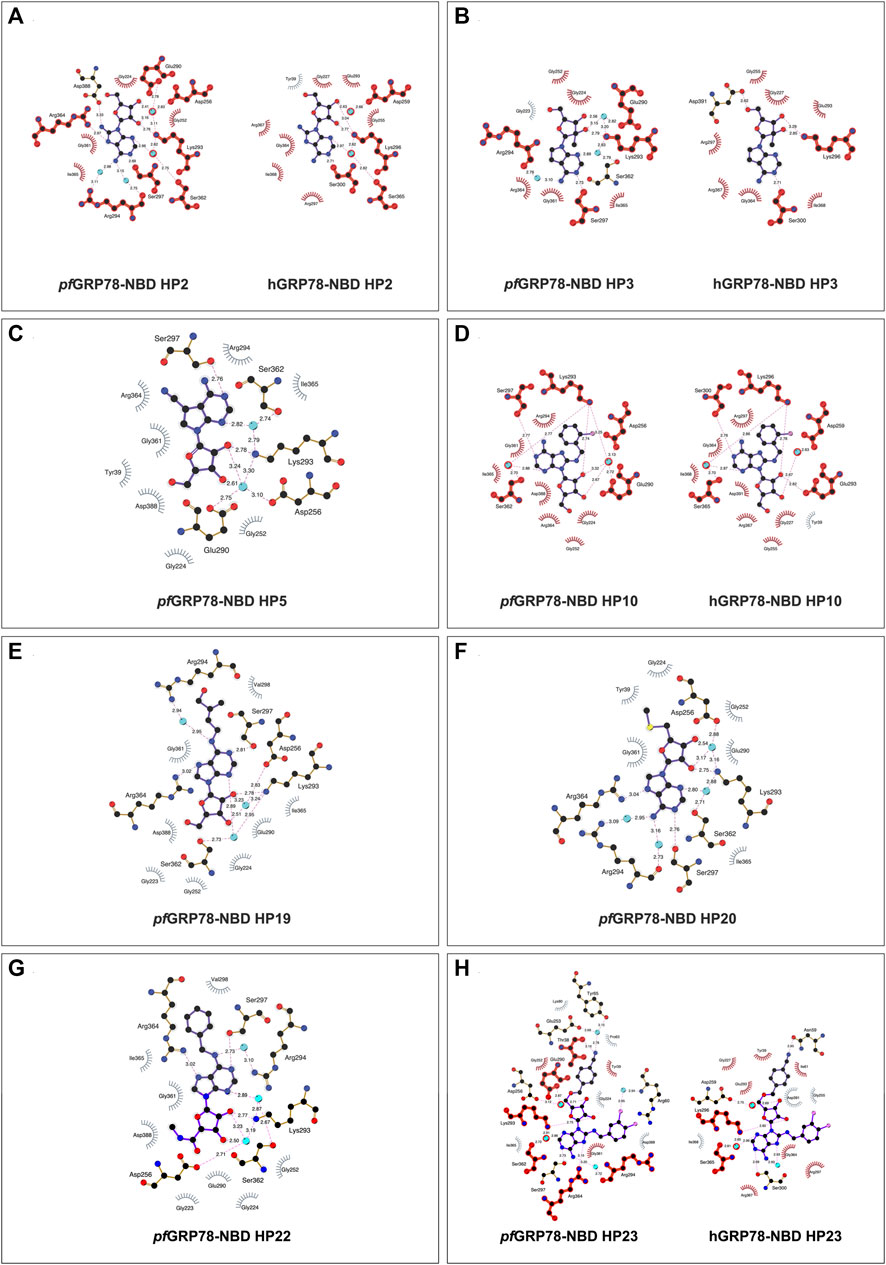
FIGURE 3. 2D representation of protein-ligand interactions of hit nucleoside analogues identified from crystal-ligand soaking. (A) HP2, (B) HP3, (C) HP5, (D) HP10, (E) HP19, (F) HP20, (G) HP22 and (H) HP23. Ligand bonds are shown in purple, non-ligand bonds are shown in brown. C, N, O, S and Cl atoms are represented in black, blue, red, yellow and pink, respectively. Water atoms are represented in turquoise. Hydrophobic interactions are represented by grey spokes radiating towards the contacting ligand atoms and hydrogen bonds are represented by pink dashed lines with bond-lengths in Å. Red highlight represents amino acid equivalence between pfGRP78-NBD and hGRP78-NBD structure.
As depicted in Figure 3B, HP3 (7-Deaza-2′-C-methyladenosine) was used to assess how chemical modification at the 2′ hydroxyl group of the ribose sugar and the deaza-modified adenine moiety (i.e., N7 to C5) affect protein-ligand interactions at the ATP binding site of pfGRP78-NBD. It is evident that HP3 interacts with pfGRP78-NBD, but lacks hydrogen bonding with Arg364 due to the deaza substitution. Similarly, the HP3-bound structure of hGRP78-NBD shows no hydrogen bonding between hArg367 and HP3 either. In other nucleoside analogue-bound pfGRP78-NBD structures, a hydrogen bond is clear present between N7 and Arg364 (Table 2). The 2′ methyl substituent directs the 2′ hydroxyl group to form a hydrogen bond with Lys293, but it itself is involved in hydrophobic interactions with Glu290. In the HP3-bound structure of hGRP78-NBD, the N7 deaza substituent of HP3 is not involved in hydrogen binding, similar to that of pfGRP78-NBD. In addition, the 2′ methyl substituent of HP3 helps orient the 2′ hydroxyl group to form a hydrogen bond with hLys296 and has hydrophobic interactions with hGlu293 conserved in pfGRP78-NBD. The finding that the hydrogen bonding and hydrophobic contacts are conserved and indistinguishable between the P. falciparum and the human orthologue suggest that neither the deaza modification at the N7 position nor the 2′ methylation enhance selectivity for pfGRP78-NBD.
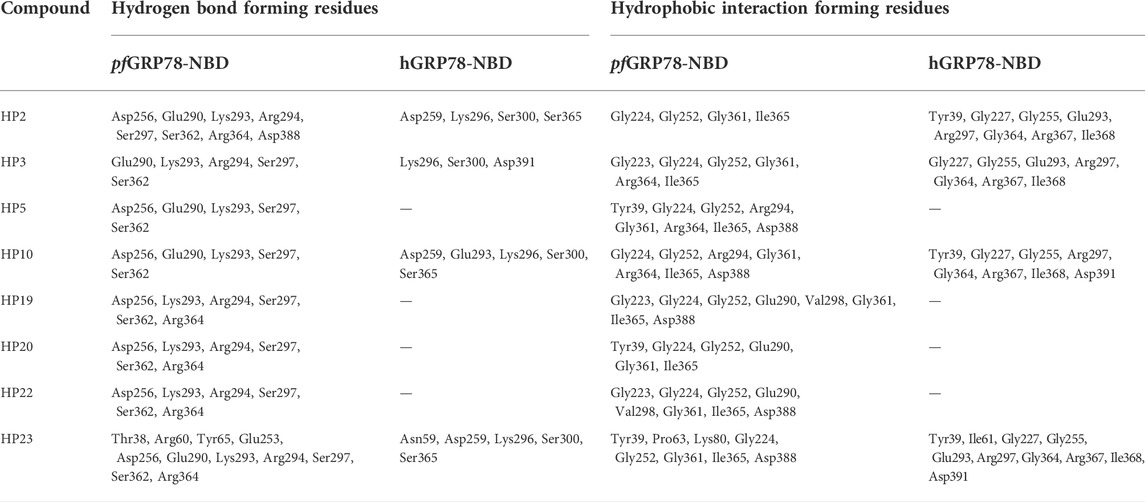
TABLE 2. pfGRP78-NBD & hGRP78-NBD Ligand Interaction Profile with Hit Nucleoside Analogues.
The active site architecture of pfGRP78-NBD with HP5 (Toyocamycin) is shown in Figure 3C. The HP5 compound also has deaza modification at the N7 position, which becomes C5. In addition, a cyano substituent is attached at the C5 position. Direct hydrogen bonds are made between Lys293 and the 2’ hydroxyl on the ribose moiety as well as Ser297 and the N1 position on the adenine moiety. Two water molecules provide water-mediated hydrogen bonds with Asp256, Glu290 and Ser362. Eight hydrophobic interactions are observed with Tyr39, Gly224, Gly252, Arg294, Gly361, Arg364, Ile365 and Asp388. The cyano substituent at the C5 position does not form direct hydrogen bonding with pfGRP78-NBD, but forms hydrophobic interactions with the aliphatic side chain of Arg294. Direct structural comparison with hGRP78-NBD cannot be made at this time due to the lack of structural data for the human orthologue in complex with HP5. However, most of the residues of pfGRP78-NBD interacting with HP5 are conserved in hGRP78-NBD, (e.g., hArg297, hSer300, hLys296) suggesting a similar binding mode for HP5, which involves no direct hydrogen bonding of the cyano substituent of HP5 with hGRP78-NBD.
The structure of HP10 ((2R,3R,4S,5R)-2-(6-amino-8-((2-chlorobenzyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol) in complex with pfGRP78-NBD is shown in Figure 3D. This compound has a modification at the C8 position, by adding an amino group followed by a chlorobenzyl moiety. Direct hydrogen bonding is observed with Lys293 and Ser297. Water-mediated hydrogen bonding is shown with Asp256, Glu290 and Ser362. Hydrophobic interactions are found with Gly224, Gly252, Arg294, Gly361, Arg364, Ile365 and Asp388. The C8 substituent shows minimal hydrophobic interactions with the hydrocarbon side chain of Arg294, although multiple hydrophobic interactions at the C8 substituent are observed in hGRP78-NBD involving Glu293, Lys296 and Ser300 (Figure 3D). This loose hydrophobic interactions at the active site of pfGRP78-NBD can be tightened by additional chemical modifications at this position to further occupy the open active site pocket, allowing for the development of more potent inhibitors specific to the P. falciparum protein.
Interactions of HP19 (Trans-Zeatin Riboside) with pfGRP78-NBD is shown in Figure 3E. This compound has a methylbutenol substituent at the C6 position. Direct hydrogen bonding of HP19 occurs with Lys293, Ser297 and Arg364. Three water molecules provide indirect hydrogen bonding with Asp256, Ser362 and Arg294. Hydrophobic interactions are also shown with Gly223, Gly224, Gly252, Glu290, Val298, Gly361, Ile365 and Asp388. The C6 substituent does not have any direct hydrogen bonding interactions with pfGRP78-NBD, although there is a water-mediated hydrogen bond that may provide additional stability. The C6 substituent forms hydrophobic interactions with Arg294 and Val298. Although the HP19-bound structure of hGRP78-NBD is not available, many active site residues of pfGRP78-NBD involved in HP19 interactions are conserved in hGRP78-NBD (e.g., hArg297, hArg367, hSer365). Interestingly, the C6 substituent forms hydrophobic interactions with pfVal298, which may not occur in hGRP78-NBD since pfVal298 is not conserved in hGRP78-NBD (hGln301). This difference can therefore be further probed for the development of compounds specific to pfGRP78-NBD.
Figure 3F shows the interactions between HP20 (5′-Methylthioadenosine) and pfGRP78-NBD. This compound has a modification at the 5′ position of the ribose sugar, with the deletion of the 5′ hydroxyl group and the addition of a methylated sulfur moiety. It is clear that no direct hydrogen bonding is observed at this modified 5′ position. Direct hydrogen bonding is however shown at other sites on HP20, which interact with Lys293, Ser297 and Arg364. Four water molecules provide water-mediated hydrogen bonding with Asp256, Arg294 and Ser362. There are six hydrophobic interactions shown with Try39, Gly224, Gly252, Glu290, Gly361 and Ile365. In pfGRP78-NBD, there are hydrophobic interactions of the 5′ substituent with Tyr39. Without the HP20-bound structure of hGRP78-NBD, we predict a similar binding mode of P. falciparum and human GRP78 for HP20 because of the conservation of HP20 binding residues between the two species including pfTyr39 and hTyr39.
HP22 (Piclidenoson) interacts with the active site residues of pfGRP78-NBD in Figure 3G. This compound has modifications at both the 5′ position and the C6 position. Direct hydrogen bonds of HP22 are formed with Ser297, Lys293 and Arg364. Three water molecules provide water-mediated hydrogen bonds with Asp256, Arg294 and Ser362. Neither the 5′ modified group nor C6 modified group form direct hydrogen bonds with pfGRP78-NBD active site residues. However, there is a water-mediated hydrogen bond between the nitrogen atom of the C6 substituent and Arg294. There are eight hydrophobic interactions which include Gly223, Gly224, Gly252, Glu290, Val298, Gly361, Ile365 and Asp388. The 5′ modified substituent has hydrophobic interactions with Asp388 and Gly223 and the ring structure of the C6 substituent forms hydrophobic interactions with Val298. In the absence of the HP22-bound structure of hGRP78-NBD, we still can predict the binding mode of hGRP78-NBD for HP22. The 5’ substituent of HP22 can form hydrophobic interactions with hAsp391, equivalent to pfAsp388. However, the C6 substituent may not form hydrophobic interactions with the pfVal298-equivalent residue of hGRP78-NBD (hGln301). This suggests that further modification at the C6 position could be used in the development of the next generation of C6-modified nucleoside analogues that are more selective to pfGRP78-NBD.
The structure of pfGRP78-NBD with HP23 (VER155008) is shown in Figure 3H. This compound has modifications at the C8 and 5′ positions. Direct hydrogen bonding of HP23 is shown with Thr38, Lys293, Ser297 and Arg364. Five water molecules provide water-mediated hydrogen bonds with Tyr65, Arg60, Glu253, Asp256, Arg294, Ser362. Hydrophobic interactions are shown with Tyr39, Pro63, Lys80, Gly224, Gly252, Gly361, Asp388 and Ile365. Especially, the 5’ substituent makes direct interactions with Thr38 and water-mediated contacts with Glu253 and Tyr65. Moreover, a chlorine atom of the C8 substituent makes water-mediated hydrogen bonding with Arg60. In hGRP78-NBD, HP23 has direct hydrogen bonding with Asn59 and Lys296. Three water molecules mediate hydrogen bonds are present between N7 with Asp259, Lys296 and Ser365. When comparing pfGRP78-NBD and hGRP78-NBD, many interactions at the active site are conserved (e.g., pfLys293 and hLys296, pfSer362 to hSer365). These findings suggest HP23 binds to both pfGRP78 and hGRP78 indistinguishably. Different modification will be required to generate nucleoside analogues specific for pfGRP78-NBD.
To further evaluate the interaction of pfGRP78-NBD with the hit nucleoside analogues, we performed thermal shift assays to measure the stability of protein-ligand complexes. Recombinant pfGRP78-NBD and hGRP78-NBD were used to evaluate potential preferential binding differences between the parasite versus host chaperone. We speculated that the limited structural variation between the highly conserved P. falciparum and human NBD of GRP78 may be an obstacle to developing selective antimalarial drugs, however discernable differences were noted between the thermal melting profiles. In this assay, the melting temperature (Tm; temperature at which protein denaturation = 50%) is monitored by a fluorescent molecular probe binding to exposed hydrophobic regions on the protein and protein-ligand interactions are characterized by an observed shift in the melting temperature (ΔTm; difference in Tm between a sample treated with ligand minus the Tm of a sample in the absence of ligand). Thermal shifts values above 2°C were considered significant and are indicative of protein-compound interaction.
The extent of thermal stabilization of pfGRP78-NBD and hGRP78-NBD in the presence of various nucleoside analogues to determine their binding to GRP78-NBDs (Figure 4). As a positive control, we used ATP as a model binding substrate. Both pfGRP78-NBD and hGRP78-NBD showed a positive thermal shift in the presence of ATP, compared to protein only. Although, the increase in melting temperature was more noticeable for hGRP78-NBD with a thermal shift of 5.6°C compared to only 0.5°C for pfGRP78-NBD. It is evident that numerous compounds interact with pfGRP78-NBD with different ΔTm values, generating a unique chemical fingerprint. Eight out of the twenty-five screened compounds showed a significant effect on the pfGRP78-NBD thermal stability, these include: HP2, HP10, HP11, HP12, HP13, HP23, HP31 and HP32. Three of the eight compounds that showed a notable difference in the thermal stability were also identified using our DMSO crystal-ligand replacement screening, those being HP2, HP10 and HP23. Relative to the ATP control, the TSA data demonstrated stabilizing effects for many of the nucleoside analogues, with changes in melting temperature higher than ATP. These results are consistent with our hypothesis that the adenosine structural moiety common among compounds in this library would elevate the overall melting temperature and provides justification for further investigation.
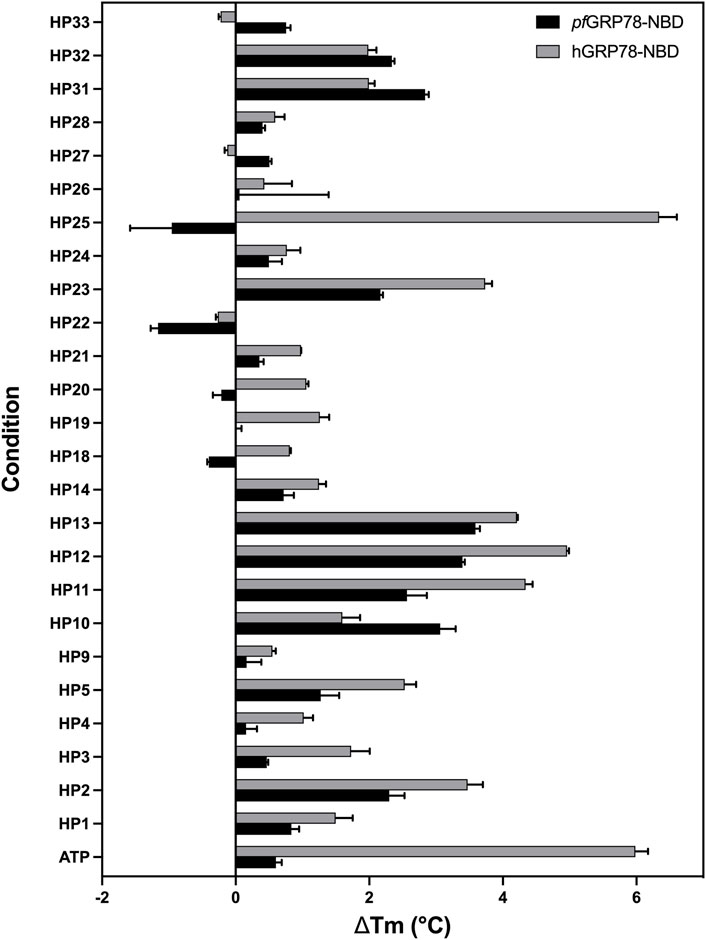
FIGURE 4. Interaction of nucleoside analogues on thermal stability of pfGRP78-NBD and hGRP78-NBD. ΔTm is defined as the difference in Tm between protein with ligand minus the Tm of protein without ligand. Error bars represent standard error of the mean of four repeats.
In addition, screening the human homologue of GRP78-NBD generated different fingerprints indicative of differences in chemical sensitivity between the P. falciparum and human GRP78 chaperones that might be exploited in the design of selective inhibitors. Most notably, this can be seen in the case of HP25, which had a more noticeable (ΔTm > 2 °C) shift for hGRP78, whereas the plasmodium chaperone was destabilized by HP25. These finding suggest the potential for designing inhibitors specific for pfGRP78. Interestingly, HP22 destabilized both pfGRP78-NBD and hGRP78-NBD in TSA, however our DMSO crystal-ligand replacement assay results clearly provide the crystal structure of pfGRP78-NBD with this nucleoside analogue bound at the active site. This result stresses the importance of performing orthogonal approaches to validate putative hit compounds.
In summary, we purified recombinantly expressed pfGRP78-NBD and hGRP78-NBD and performed biophysical assays such as TSA to identify chemical moieties that could be used to generate more specific therapeutics to P. falciparum. We further characterized the active site of the NBD by providing new crystal structures of the P. falciparum GRP78 in complex with eight different nucleoside analogues using our innovative, high-throughput DMSO crystal-ligand replacement screening method. The results from the TSA and DMSO crystal-ligand replacement screening are in agreement and provide further validation of the hit compounds we identified in this study. Moreover, we provided proof of concept that protein crystals can tolerate DMSO soaking and stabilizing compounds remain intact, whereas non-binding compounds dissolve. Taken together, this study established an excellent platform for a rapid, stream-lined approach to compound screening, data collection and identification of protein-ligand complexes using x-ray crystallography. It is our belief that the development of our DMSO crystal-ligand screening approach can be applied in the larger context of drug discovery by minimizing time, cost and hours spent working to obtain protein-ligand co-crystal structures and will complement existing drug discovery approaches to identify and validate candidate drug targets. The research outcomes presented here will play a fundamental role in future studies to generate a refined adenosine chemotype containing structure-driven modifications on the adenosine scaffold specific and selective to pfGRP78.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Conceived and designed the experiments: AM and H-WP. Performed the experiments: AM, TA, YC, CZ-V and DS. Analyzed the data: AM, H-WP and HL. Contributed reagents and analysis tools: H-WP, HL, DS, NK. Wrote the paper: AM and H-WP.
Research described in this paper used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 (APS Beamlines 19-ID-D & 23-ID-D). Part of the research described in this paper was performed using beamline CMCF-ID at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan (CLS Beamlines 08B1-1). HL was supported by NIH-NCI grant R01 CA234605.
We kindly thank the following current and previous lab members for their work and support on this project including: Claire Pierce, Yuhua Huang and Matthew Moreida for help with diffraction data collection. We also thank Jia Fan for mass spectrometry validation of synthesized HP compounds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Chakafana, G., Zininga, T., and Shonhai, A. (2019). Comparative structure-function features of Hsp70s of Plasmodium falciparum and human origins. Biophys. Rev. 11, 591–602. doi:10.1007/s12551-019-00563-w
Chen, Y., Murillo-Solano, C., Kirkpatrick, M. G., Antoshchenko, T., Park, H.-W., and Pizarro, J. C. (2018). Repurposing drugs to target the malaria parasite unfolding protein response. Sci. Rep. 8 (1), 10333. doi:10.1038/s41598-018-28608-2
Day, J., Passecker, A., Beck, H. P., and Vakonakis, I. (2019). The Plasmodium falciparum Hsp70-x chaperone assists the heat stress response of the malaria parasite. FASEB J. 33 (12), 14611–14624. doi:10.1096/fj.201901741R
Emsley, P., and Cowtan, K. (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60 (12), 2126–2132. doi:10.1107/S0907444904019158
Frame, I. J., Deniskin, R., Arora, A., and Akabas, M. H. (2015). Purine import into malaria parasites as a target for antimalarial drug development. Ann. N. Y. Acad. Sci. 1342, 19–28. doi:10.1111/nyas.12568
Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M., and Walter, P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5 (3), a013169. doi:10.1101/cshperspect.a013169
Ibrahim, I. M., Abdelmalek, D. H., and Elfiky, A. A. (2019). GRP78: A cell's response to stress. Life Sci. 226, 156–163. doi:10.1016/j.lfs.2019.04.022
Jones, A. M., Westwood, I. M., Osborne, J. D., Matthews, T. P., Cheeseman, M. D., Rowlands, M. G., et al. (2016). A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Sci. Rep. 6, 34701. doi:10.1038/srep34701
Kabsch, W. (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D. Biol. Crystallogr. 66 (2), 133–144. doi:10.1107/S0907444909047374
Kumar, N., Syin, C., Carter, R., Quakyi, I., and Miller, L. H. (1988). Plasmodium falciparum gene encoding a protein similar to the 78-kDa rat glucose-regulated stress protein. Proc. Natl. Acad. Sci. U. S. A. 85, 6277–6281. doi:10.1073/pnas.85.17.6277
Laskowski, R. A., and Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51 (10), 2778–2786. doi:10.1021/ci200227u
Lee, P. H., Huang, X. X., Teh, B. T., and Ng, L. M. (2019). TSA-CRAFT: A free software for automatic and robust thermal shift assay data analysis. SLAS Discov. 24 (5), 606–612. doi:10.1177/2472555218823547
Murshudov, G. N., Skubak, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67 (4), 355–367. doi:10.1107/S0907444911001314
Pobre, K. F. R., Poet, G. J., and Hendershot, L. M. (2019). The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 294 (6), 2098–2108. doi:10.1074/jbc.REV118.002804
Przyborski, J. M., Diehl, M., and Blatch, G. L. (2015). Plasmodial HSP70s are functionally adapted to the malaria parasite life cycle. Front. Mol. Biosci. 2, 34. doi:10.3389/fmolb.2015.00034
Suresh, N., and Haldar, K. (2018). Mechanisms of artemisinin resistance in Plasmodium falciparum malaria. Curr. Opin. Pharmacol. 42, 46–54. doi:10.1016/j.coph.2018.06.003
Tatani, K., Hiratochi, M., Nonaka, Y., Isaji, M., and Shuto, S. (2015). Identification of 8-aminoadenosine derivatives as a new class of human concentrative nucleoside transporter 2 inhibitors. ACS Med. Chem. Lett. 6 (3), 244–248. doi:10.1021/ml500343r
Vagin, A., and Teplyakov, A. (2010). Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66 (1), 22–25. doi:10.1107/S0907444909042589
Visser, B. J., van Vugt, M., and Grobusch, M. P. (2014). Malaria: An update on current chemotherapy. Expert Opin. Pharmacother. 15 (15), 2219–2254. doi:10.1517/14656566.2014.944499
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67 (4), 235–242. doi:10.1107/S0907444910045749
Keywords: Plasmodium falciparum, malaria, GRP78, chaperone, unfolded protein response, nucleoside analogues, drug discovery & development
Citation: Mrozek A, Antoshchenko T, Chen Y, Zepeda-Velázquez C, Smil D, Kumar N, Lu H and Park H-W (2022) A non-traditional crystal-based compound screening method targeting the ATP binding site of Plasmodium falciparum GRP78 for identification of novel nucleoside analogues. Front. Mol. Biosci. 9:956095. doi: 10.3389/fmolb.2022.956095
Received: 29 May 2022; Accepted: 03 August 2022;
Published: 07 October 2022.
Edited by:
Xolani Henry Makhoba, University of Fort Hare, South AfricaReviewed by:
Tawanda Zininga, Stellenbosch University, South AfricaCopyright © 2022 Mrozek, Antoshchenko, Chen, Zepeda-Velázquez, Smil, Kumar, Lu and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hee-Won Park, aHBhcmsxQHR1bGFuZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.